 Bogdan Iorga1,2*
Bogdan Iorga1,2* Kristin Schwanke3
Kristin Schwanke3 Natalie Weber1Meike Wendland1Stephan Greten1
Natalie Weber1Meike Wendland1Stephan Greten1 Birgit Piep1Cristobal G. dos Remedios4
Birgit Piep1Cristobal G. dos Remedios4 Ulrich Martin3
Ulrich Martin3 Robert Zweigerdt3
Robert Zweigerdt3 Theresia Kraft1
Theresia Kraft1 Bernhard Brenner1
Bernhard Brenner1- 1Department of Molecular and Cell Physiology, Hannover Medical School, Hannover, Germany
- 2Department of Physical Chemistry, Faculty of Chemistry, University of Bucharest, Bucharest, Romania
- 3Department of Cardiac, Thoracic, Transplantation and Vascular Surgery, Leibniz Research Laboratories for Biotechnology and Artificial Organs, REBIRTH-Center for Regenerative Medicine, Hannover Medical School, Hannover, Germany
- 4Department of Anatomy, Bosch Institute, University of Sydney, Sydney, NSW, Australia
Characterizing the contractile function of human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) is key for advancing their utility for cellular disease models, promoting cell based heart repair, or developing novel pharmacological interventions targeting cardiac diseases. The aim of the present study was to understand whether steady-state and kinetic force parameters of β-myosin heavy chain (βMyHC) isoform-expressing myofibrils within human embryonic stem cell-derived cardiomyocytes (hESC-CMs) differentiated in vitro resemble those of human ventricular myofibrils (hvMFs) isolated from adult donor hearts. Contractile parameters were determined using the same micromechanical method and experimental conditions for both types of myofibrils. We identified isoforms and phosphorylation of main sarcomeric proteins involved in the modulation of force generation of both, chemically demembranated hESC-CMs (d-hESC-CMs) and hvMFs. Our results indicate that at saturating Ca2+ concentration, both human-derived contractile systems developed forces with similar rate constants (0.66 and 0.68 s−1), reaching maximum isometric force that was significantly smaller for d-hESC-CMs (42 kPa) than for hvMFs (94 kPa). At submaximal Ca2+-activation, where intact cardiomyocytes normally operate, contractile parameters of d-hESC-CMs and hvMFs exhibited differences. Ca2+ sensitivity of force was higher for d-hESC-CMs (pCa50 = 6.04) than for hvMFs (pCa50 = 5.80). At half-maximum activation, the rate constant for force redevelopment was significantly faster for d-hESC-CMs (0.51 s−1) than for hvMFs (0.28 s−1). During myofibril relaxation, kinetics of the slow force decay phase were significantly faster for d-hESC-CMs (0.26 s−1) than for hvMFs (0.21 s−1), while kinetics of the fast force decay were similar and ~20x faster. Protein analysis revealed that hESC-CMs had essentially no cardiac troponin-I, and partially non-ventricular isoforms of some other sarcomeric proteins, explaining the functional discrepancies. The sarcomeric protein isoform pattern of hESC-CMs had features of human cardiomyocytes at an early developmental stage. The study indicates that morphological and ultrastructural maturation of βMyHC isoform-expressing hESC-CMs is not necessarily accompanied by ventricular-like expression of all sarcomeric proteins. Our data suggest that hPSC-CMs could provide useful tools for investigating inherited cardiac diseases affecting contractile function during early developmental stages.
Introduction
In vitro differentiation of human pluripotent stem cells (hPSCs) toward cardiomyocytes provides a model system of human cardiac myogenesis (Murry and Keller, 2008; Birket and Mummery, 2015; Kempf et al., 2016b). Given the in principal unlimited availability of human embryonic (hESC) and induced pluripotent stem cells (hiPSC) (Kempf et al., 2016a), hPSC-derived cardiomyocytes also hold great promise for the treatment of cardiovascular diseases by cell transplantation or engineered cardiac tissue (Kensah et al., 2013; Zimmermann, 2017), for assessing efficiency and toxicity of pharmacological compounds (Burridge et al., 2016), or to be used as cellular disease models in vitro (Moretti et al., 2013). Particularly, cardiomyocytes (CMs) derived from patient-specific hPSCs have facilitated studies of the consequences of hereditary diseases in vitro (Jung and Bernstein, 2014; Kamdar et al., 2015; Pioner et al., 2016). For instance, in hypertrophic cardiomyopathy (HCM), mutations of specific sarcomeric proteins can impair the contractile function of CMs. The related abnormal changes in force generation of CMs may subsequently result in disorder of other cellular functions and further pathological alterations (Kraft et al., 2013; Brenner et al., 2014; Fatkin et al., 2014). Detailed analysis of the contractile function of hPSC-CMs is therefore crucial to further characterize hPSC-CMs as model systems for investigation of disease mechanisms and future therapies.
Whether the contractile function of myofibrils within hPSC-CMs matches that of ventricular myofibrils determining the pump function in the adult human heart is an important question. Previous studies have shown that hPSC-CMs contract periodically, thus revealing the presence of functional myofibrils, which are typically less well-aligned compared to adult human CMs (Bedada et al., 2014; Jung and Bernstein, 2014; Yang et al., 2014a; Pioner et al., 2016; Weber et al., 2016). Achieving more matured hPSC-CMs seems favorable for the in vitro modeling of pathological conditions of the adult heart. On the other hand, an ongoing maturation of early hPSC-CMs in vitro may support molecular and cellular investigations on the disease onset at developmental stages. In this view, very little is known about the in utero onset of cardiomyopathies (Fatkin et al., 2014) and congenital heart diseases (Elhamine et al., 2016), as well as the demise of fetuses and neonates survival in such conditions (Mongiovì et al., 2010; MacColl et al., 2014).
The contractile function in muscle cells is driven by the acto-myosin interaction, where cross-bridges cycle between strongly bound, force-generating and weakly bound, non-force generating conformations (Huxley, 1957; Brenner, 1991a). Kinetic parameters of the force generated by sarcomeres depend mainly on the myosin isoform. Usually, after typical differentiation protocols, sarcomeres of hPSC-CMs are composed of a mixture of α and β isoforms of the myosin heavy chain (MyHC) (Weber et al., 2016), while in the adult, neonate and developing human ventricle the βMyHC isoform predominates (Bouvagnet et al., 1987; Miyata et al., 2000; Reiser et al., 2001; Elhamine et al., 2016; Racca et al., 2016). In addition, most known HCM-related myosin-mutations are expressed in the βMyHC rather than in the αMyHC isoform (Fatkin et al., 2014; Burke et al., 2016). We recently showed that long-term cultivation of human embryonic stem cell-derived cardiomyocytes (hESC-CMs) on stiff isotropic substrates (e.g., laminin-coated glass cover-slips) shifts myosin expression exclusively toward the βMyHC isoform (Weber et al., 2016).
In the present study, we addressed the question whether βMyHC-expressing myofibrils within hESC-derived CMs have the same contractile properties as adult human ventricular myofibrils (hvMFs). Therefore, we aimed to identify which steady-state and kinetic force parameters of myofibrils within hESC-CMs differentiated in vitro resemble the corresponding force parameters generated by adult hvMFs. To directly assess the contractile function of myofibrillar bundles of single cells, chemically demembranated hESC-CMs (d-hESC-CMs) were used. For comparison with healthy human heart, small hvMFs-bundles were isolated from chemically demembranated tissues of adult donor hearts. Biomechanical assessment of such myofibrils at defined Ca2+ concentrations ([Ca2+]) allows direct examination of the cycling cross-bridge-driven contractile performance of myofibrils, including relaxation kinetics, in the absence of Ca2+ handling systems and of upstream signaling modulated by hormonal activities (Poggesi et al., 2005; Stehle et al., 2009; Walker et al., 2011). We measured steady-state and kinetic parameters of isometric forces generated by myofibrils of d-hESC-CMs and by hvMFs using the same micromechanical method and experimental conditions (Stehle et al., 2002b; Weber et al., 2016), and characterized the isoforms and phosphorylation status of main sarcomeric proteins involved in modulation of force generation. Observed similarities and subtle differences in force kinetics are discussed using modeling based on the cross-bridge theory (Huxley, 1957; Brenner, 1988, 1990, 1991a; Brenner and Chalovich, 1999).
Materials and Methods
Solutions Composition
Composition of solutions used to prepare d-hESC-CMs and hvMFs (Na-solution, K-solution, pCa > 9, pH 7.0), protease inhibitor cocktail (PIC) and solutions used to activate (Ca2+-activating-solution, pCa 4.18, pH 7.1) and relax (Relaxing-solution, pCa > 8, pH 7.1) myofibrils for the assessment of their contractile function was described in Weber et al. (2016). Ca2+-activating solutions at intermediate [Ca2+] were obtained by mixing the relaxing and activating solutions in the appropriate ratio as in Kraft et al. (2013).
Preparation of d-hESC-CMs and hvMFs
Demembranated Human Embryonic Stem Cell-Derived Cardiomyocytes (d-hESC-CMs)
Experimental details regarding differentiation and enrichment of hESC-CMs in 12–20 days of suspension culture using defined differentiation media supplemented with Wnt-pathway modulators were previously published (Kempf et al., 2014) (see Supplementary Material). Such suspension culture-derived “cardiac bodies” consisting of essentially pure hESC-CMs were enzymatically dissociated and ~118 cells/mm2 were plated for long-term cultivation on laminin-coated glass cover-slips. This resulted in a majority of hESC-CMs expressing essentially only the βMyHC isoform at the protein level (Weber et al., 2016). We used hESC-CMs from three different cell batches. In this study, hESC-CM (cf. online Supplementary Material, Video 1) were used after 35–56 days of cultivation on glass cover-slips. hESC-CMs were chemically demembranated using 0.5% Triton-X-100 in the presence of 20 mM BDM as previously described (Weber et al., 2016), and then equilibrated with relaxing-solution containing PIC and 5 mM DTT (without BDM) before the experiment.
Human Ventricular Myofibrils (hvMFs)
hvMFs were isolated from two different non-transplanted donor hearts for which no suitable recipient was found. Long-term cryopreservation of muscular tissues was previously described (Kraft et al., 2013). After quick thawing in Na-solution including 20 mM BDM (~20°C), ventricular fragments were cut in small pieces removing connective tissues and fat traces in cold Na-solution (+BDM), and then demembranated in cold K-solution containing 0.5% Triton-X-100 and 20 mM BDM (~50 min, 5°C). Detergent was rinsed twice using cold K-solution without Triton (~50 min). hvMFs-bundles were prepared freshly before the micromechanical experiments by homogenizing the demembranated ventricular pieces equilibrated with relaxing-solution (+PIC, +DTT 5 mM, on ice) for 5–10 s with a blender (Ultra-Turrax T8, IKA Labortechnik, Germany) at 25,000 rpm. The resulting myofibrillar suspension was filtered through a mesh (pore size 21 μm) to remove the thick myofibrillar aggregates. The homogenate was further centrifuged (400 × g, 10 min, 5°C; Biofuge Primo-R, Thermo-Fisher, Massachusetts, USA) and the pellet re-suspended in fresh relaxing-solution (+PIC, +DTT) or subjected to further treatments.
Approval of Hannover Medical School Ethics Committee was obtained for anonymized use of the different muscle biopsies and the hESC-CMs in our study (approval numbers 2729–2001, 507–2009, 1751–2013). All subjects gave written informed consent at the different institutions in accordance with the Declaration of Helsinki.
PKA Treatment, Immunostaining, and Analysis of Sarcomeric Proteins
Details regarding PKA-treatment, double and single immunostaining, and analysis of sarcomeric proteins of d-hESC-CMs and hvMFs are described in Supplementary Material.
Micromechanical Investigations
Basic features and technical details of the custom-built micromechanical setup used to assess the contractile function of either single d-hESC-CMs or small hvMFs-bundles were previously described (Colomo et al., 1998; Stehle et al., 2002a,b; Weber et al., 2016).
hvMFs
Three to five hundred microliters of myofibrillar homogenate in relaxing-solution was pipetted in the pre-cooled chamber of the micromechanical setup and myofibrils allowed to sediment for ~1 h. Then the chamber was filled with relaxing-solution (15°C).
d-hESC-CMs
A laminin-coated glass cover-slip containing plated, chemically demembranated d-hESC-CMs was transferred into the chamber of the micromechanical setup, previously filled with relaxing-solution (15°C).
Contractile function of a single d-hESC-CM containing mainly few bundled myofibrils or a small hvMFs-bundle was investigated (Videos 2–4) with a custom-built micromechanical setup mounted on an inverted microscope (Olympus IX-71). Experiments with d-hESC-CMs and hvMFs were performed with solutions from the same batch at 15°C. Working at this temperature allows comparison of the functional results to several biomechanical studies with hvMFs or skinned cardiomyocytes which were previously performed at 15°C (Stehle et al., 2002b; van der Velden et al., 2003b; Piroddi et al., 2007; Walker et al., 2011; Kraft et al., 2013; van Dijk et al., 2014; Pioner et al., 2016; Racca et al., 2016). Some details about the micromechanical setup are described in Supplementary Material.
Sarcomere length (SL) and cross-sectional area (CSA) of myofibrils within d-hESC-CMs and of hvMF-bundles were determined in bright field (BF) or phase contrast (PhC) at 96-fold magnification (Figures 1A,B), using a CCD camera (Hamamatsu Photonics, Herrsching am Ammersee, Germany) attached to microscope. To calculate specific forces (force/CSA, kPa = nN/μm2) the mean diameter of the myofibrils or myofibrillar bundles within d-hESC-CMs and of entire hvMF-bundles was assessed prior to force measurements as previously described (Stehle et al., 2002b; Weber et al., 2016). In particular, for d-hESC-CMs total CSA of all myofibrils observed within the demembranated cells as darker traces on brighter background (Figure 1A1) was calculated assuming a cylindrical shape of the myofibrils. As a control for myofibrillar content and diameter in d-hESC-CMs, some cells were immunostained against α-actinin prior to Ca2+-activation (Figures 1B1−3), as previously described for isolated myofibrils (Telley et al., 2006). This allowed visualizing myofibrillar thickness and distribution within the cardiomyocytes and to further compare these features with the corresponding ones assigned to the darker traces observed either in PhC (e.g., Figures 1B1−3) within the same cardiomyocyte or in BF (e.g., Figure 1A1). Such analysis suggested essentially the same thickness for the fluorescently labeled myofibrils at their Z-disks and for the dark fascicles within d-hESC-CMs (in BF or PhC). This is also indicated by Video 5 where visualization of sarcomeres by PhC, fluorescence (FL), or overlapped (PhC and FL) is shown. Based on this we measured myofibrillar CSA directly from the dark fascicles in BF or PhC, and thus, reduced the overestimation of CSA which occurs if we would take the total width of the mounted d-hESC-CMs. An overestimated myofibrillar CSA would yield smaller specific force values.
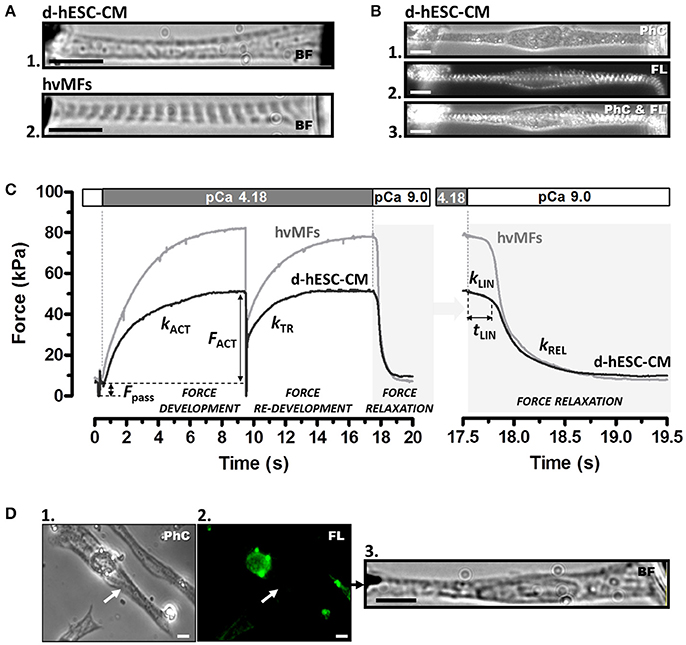
Figure 1. (A) Contractile function of myofibrils of a single demembranated human stem cell-derived cardiomyocyte (d-hESC-CM) (A1) and contractile function of a single bundle of adult human ventricular myofibrils (hvMFs) (A2) were investigated using the same micromechanical setup and experimental conditions. Microphotographs (A1,A2) were taken in bright field (BF). (B) A single elongated d-hESC-CM immunostained against α-actinin was observed in phase contrast (PhC; B1), fluorescence (FL; B2), or simultaneously in PhC and FL (B3). Note the predominant axial alignment of myofibrils within d-hESC-CMs (A1,B1−3). (C) Representative force transients of a single d-hESC-CM (black trace; d-hESC-CMs shown in A1 and Video 3) and of a hvMFs-bundle (gray trace; hvMFs-bundle shown in A2 and Video 4) mounted and held isometrically between the tips of a stiff needle (left) and a nN-sensitive force probe (right). Isometric active (FACT) at saturating [Ca2+] (pCa 4.18) and passive (Fpass) forces, rate constant kACT of Ca2+-induced force development, rate constant kTR of force re-development and the three kinetic parameters (kLIN, tLIN, kREL) characterizing force relaxation upon rapid Ca2+-removal (detailed in the right panel) are indicated for this d-hESC-CM (T = 15°C, pCa 4.18): FACT = 43 kPa, Fpass = 7 kPa, kACT = 0.60 s−1, kTR = 0.65 s−1, kLIN = 0.30 s−1, tLIN = 243 ms, kREL = 4.2 s−1; and for the single hvMF-bundle: FACT = 72 kPa, Fpass = 9 kPa, kACT = 0.50 s−1, kTR = 0.56 s−1, kLIN = 0.23 s−1, tLIN = 216 ms, kREL = 5.3 s−1. (D) d-hESC-CM (indicated by arrow in D1) immunostained solely against αMyHC isoform and exhibiting no sarcomeric fluorescence (i.e., βMyHC-positive d-hESC-CM; D2) was micromechanically investigated (D3). Force parameters from several immunostained d-hESC-CMs are given in Table 1. Images were taken in PhC (D1), FL (D2), or BF (D3). All scale bars = 10 μm.
Considerations Based on the Two-State Cross-Bridge Model
For the interpretation of steady-state and kinetic force parameters emerging from micromechanical measurements with d-hESC-CMs and hvMFs (see “Discussion”), we used a Huxley-Brenner two-state model (Huxley, 1957; Brenner, 1988, 1990, 1991a,b; Brenner and Chalovich, 1999). During muscle activation, one state represents cross-bridges in strongly-binding configurations, while the other state corresponds to cross-bridges in weakly-binding configurations. Each of the two states accommodates many intermediate cross-bridge sub-states of the myosin ATPase reaction pathway. The apparent rate constant fapp describes the probability of cross-bridge transition from the non-force generating (weakly-binding) states to the force-generating (strongly-binding) states upon phosphate release. The apparent rate constant gapp describes the probability of the opposite transition of cross-bridges from the force-generating to the non-force generating states via ADP-release and ATP-binding steps. Both transitions occur through dynamic equilibria of cross-bridges attachment/detachment (Brenner, 1991b) (e.g., cross-bridge detachment does not rate-limit gapp). Probability fapp is dependent on the Ca2+-regulated thin filament status (i.e., modulated by the variation of [Ca2+]), while the probability gapp is independent of [Ca2+] (a first-order rate constant). Both parameters may influence isometric force response, i.e., regulation of the steady-state force response through turnover kinetics (Brenner, 1988). The measured rate constant of force re-development kTR (see below) is given by fapp+gapp (Brenner, 1988, 1990, 1991a). The measured rate constant kLIN of the slow force decay during the first phase of the myofibrillar relaxation (see below) allows estimation of gapp (Stehle et al., 2002a,b; Poggesi et al., 2005).
Force Measurements and Cross-Bridge Kinetics
d-hESC-CMs and hvMFs in relaxing solution were pre-stretched to SL = 2.3 μm from their different individual slack SL (SL0). Then, a slack-restretch maneuver (amplitude 30% of the length of pre-stretched myofibrils) was applied to cancel transiently the passive force (Fpass) stored in the elastic elements of myofibrils. Therefore, this maneuver allows determination of Fpass. The passive stretch further increased the parallel arrangement of myofibrils within the elongated d-hESC-CMs.
A sudden change to Ca2+-activating-solution with defined [Ca2+] determined the force to rise mono-exponentially in both d-hESC-CMs and hvMFs (Figure 1C) with the rate constant kACT (rate constant of Ca2+-induced force development), which depends on Ca2+-induced thin filament activation, cross-bridge formation and force generating conformational change, and cross-bridge turnover. When the developing force reached the maximum steady-state level, a quick (50 ms) release-restretch maneuver (amplitude 30% of the length of pre-stretched myofibrils) was applied which mechanically unloads the contracted myofibrils. The quick release allows a redistribution of the cross-bridges in the non-force-generating states before restretch and force re-development. This maneuver enables, at a given [Ca2+], the measurement of the total generated isometric force and of the rate constant of force re-development kTR (rate constant of mechanically-induced force re-development) re-approaching steady-state force level, while the thin filament is already Ca2+-activated (Figure 1C) (Brenner, 1988). kTR reflects the kinetics of the cross-bridges re-distribution from the non-force- to the force-generating states. Passive force (Fpass) was subtracted from the recorded total force values (Figure 1C), thus yielding the isometric force (FACT) generated by cycling cross-bridges.
Upon rapidly switching back to relaxing-solution, d-hESC-CMs and hvMFs undergo a biphasic force relaxation behavior (Figure 1C-right panel): first, force decays slowly and almost linearly during the period tLIN with the rate constant kLIN (early relaxation phase) (Stehle et al., 2002b; Stehle and Iorga, 2010). In the second, mono-exponential relaxation phase, force drops faster with the rate constant kREL (late relaxation phase; Videos 2–4). Observations of the video streams immediately after rapid Ca2+ removal revealed that SL of d-hESC-CMs and hvMFs remained quasi-constant during the first (slow) relaxation phase, whereas the second (fast) relaxation phase was dominated by the dynamics of the sarcomeres, as previously described (Stehle et al., 2002b; Stehle and Iorga, 2010). Therefore, kLIN is the rate constant determined by strained cross-bridges leaving the force-generating states during the isometric condition at sarcomeric level, while kREL is the rate constant of less strained remaining cross-bridges leaving the force-generating states during lengthening of the sarcomeres along the myofibrils (Stehle et al., 2002a,b; Poggesi et al., 2005).
Data Analysis and Statistics
The trace of force rise after activation and after quick release-restretch of the myofibrils was fitted by a mono-exponential function yielding kACT and kTR, respectively. For the relaxation, force decay was fitted by a function consisting of a linear and mono-exponential term yielding the parameters kLIN, tLIN, and kREL of the force relaxation kinetics (Stehle et al., 2002b). Ca2+ concentration was given as pCa = −log10[Ca2+]. Data of normalized force-pCa relation, resulting from each measured sample, was fitted by the Hill-type equation, where Fn is the fractional force at a given pCa. Fn = FACT/FACT,max where FACT,max is the maximum force recorded at pCa 4.18 (Fn = 1), pCa50 (indicating Ca2+-sensitivity of force) is the pCa at half-maximum force (Fn = 0.5), and nH is the steepness of the force-pCa relation (Hill-coefficient):
If not stated otherwise, steady-state and kinetic parameters were indicated as mean ± SD (Standard Deviation). Mean values were compared using unpaired Student t-test with significant differences when p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***).
From each donor ventricle 29 hvMFs-bundles were investigated for their contractile function (–PKA: 14 and 11 bundles, +PKA: 15 and 18 bundles, respectively), and data of hvMFs were pooled together.
Results
Micromechanical Investigations of d-hESC-CMs vs. hvMFs Reveal Differences of Their Sarcomeric Contractile Function at Submaximal Ca2+-Activation Levels
The main function of cardiomyocytes is to generate force and shorten their length, thus determining the pump function of the heart. Therefore, we focused on the function of the contractile elements of the cardiomyocytes, the myofibrils. Our main aim was to identify whether steady-state and kinetic force parameters of myofibrils within d-hESC-CMs resemble the corresponding force parameters of hvMFs.
We used elongated demembranated hESC-CMs (e.g., Figure 1A1) enabling measurements of the force generated predominantly axially. Single d-hESC-CMs had as mean diameter of their myofibrillar-bundles d = 2.0-5.0 μm, length was L = 30-100 μm, slack sarcomere length was SL0 = 1.75–2.10 μm, and hvMFs-bundles (e.g., Figure 1A2) had d = 1.9–5.8 μm, L = 20–50 μm, and SL0 = 1.8–2.0 μm. Both d-hESC-CMs and hvMFs were initially exposed to relaxing-solution at 15°C and pre-stretched to SL = 2.3 μm. The comparison of the resulting passive forces (Fpass) stored in the elastic elements of myofibrils in relaxing solution suggested that d-hESC-CMs (Fpass = 9 ± 3 kPa, n = 12) were, in average, more compliant (p < 0.0001) than hvMFs (Fpass = 18 ± 7 kPa, n = 25) (Figure S1). Fpass was not responsive (p = 0.712) to PKA-treatment for d-hESC-CMs (Fpass = 10 ± 4 kPa, n = 14; + PKA), but it was slightly reduced (p = 0.029) for hvMFs from 18 ± 7 kPa (n = 25; −PKA) to 15 ± 4 kPa (n = 33; + PKA) (Figure S1).
Steady-State and Kinetic Force Parameters at Saturating [Ca2+]
When activated at saturating [Ca2+], kinetics of force development (kACT) and re-development (kTR) of d-hESC-CMs (kACT = 0.66 ± 0.14 s−1, n = 11; kTR = 0.74 ± 0.10 s−1, n = 11) and hvMFs (kACT = 0.68 ± 0.14 s−1, n = 37; kTR = 0.68 ± 0.10 s−1, n = 37) were similar, respectively, with values as previously reported for hvMFs (Stehle et al., 2002b; Piroddi et al., 2007; Walker et al., 2011; Racca et al., 2016). Maximum isometric force (FACT,max) for d-hESC-CMs (FACT,max = 42 ± 10 kPa, n = 12) was significantly (p < 0.001) smaller than the FACT,max generated by hvMFs (FACT,max = 94 ± 25 kPa, n = 39) (Figure 1C).
However, we cannot exclude the presence of a very low amount of residual αMyHC in d-hESC-CMs [however, under detection limit in gel analysis; see section Overview of Sarcomeric Proteins by Gel Electrophoresis]. To exclude such CMs from the functional analysis, some d-hESC-CMs were immunostained against αMyHC prior micromechanical measurements. Then, FACT,max, kTR, and kLIN were determined at pCa 4.18 for d-hESC-CMs exhibiting null fluorescence, i.e., βMyHC positive cells which had no αMyHC-fluorescence (Figure 1D). These values were taken as reference values for all d-hESC-CMs that were characterized micromechanically without preceding immunostaining (Table 1). It has been shown previously that kTR of d-hESC-CMs which also have αMyHC-positive sarcomeres is significantly faster than for purely βMyHC-positive CMs (Weber et al., 2016). Here we included all d-hESC-CMs measured without preceding immunostaining which had individual kTR and kLIN values that were smaller than the upper limit set for kTR to 0.87 s−1 (= mean + 2 × SD = 0.70 + 0.17) and for kLIN to 0.47 s−1 (= mean + 2 × SD = 0.29 + 0.18), respectively (Table 1). Therefore, there is a 95% probability that d-hESC-CMs included in the analysis without preceding immunostaining are also negative for αMyHC.
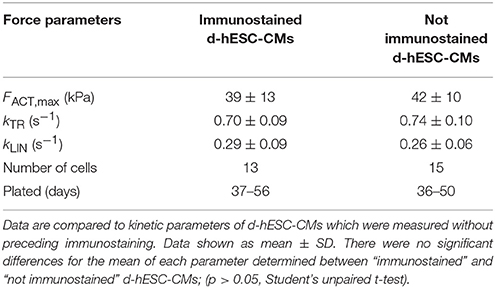
Table 1. Force kinetic parameters (kTR, kLIN) and maximum isometric force (FACT,max) generated at saturating Ca2+ concentration (pCa 4.18, 15°C) by d-hESC-CMs which were identified as purely βMyHC positive CMs by immunostaining against αMyHC.
PKA-treatment did not significantly affect FACT,max, kACT, and kTR force parameters at pCa 4.18 of either myofibrils within d-hESC-CMs (FACT,max = 37 ± 11 kPa, n = 15; kACT = 0.62 ± 0.13 s−1, n = 15; kTR = 0.69 ± 0.11 s−1, n = 16) or hvMFs (FACT,max = 95 ± 27 kPa, n = 33; kACT = 0.64 ± 0.11 s−1, n = 33; kTR = 0.67 ± 0.09 s−1, n = 33). This is consistent with previous observations using human ventricular myofibrils and skinned cardiomyocytes (Walker et al., 2011; Kraft et al., 2013).
The results show that at saturating [Ca2+], cycling βMyHC cross-bridges are able to develop force with similar rate constants, independent of PKA-treatment, for both contractile systems (d-hESC-CMs and hvMFs). Yet, considering the lower maximum force level reached by d-hESC-CMs, the rates by which both contractile systems reach maximum force were significantly different (p < 0.001), being slower for d-hESC-CMs (28.2 ± 8.8 kPa/s, n = 11) than for hvMFs (64.8 ± 26.9 kPa/s, n = 37).
Steady-State Force Response at Intermediate [Ca2+]
Cardiomyocytes in vivo operate at intermediate [Ca2+]. Therefore, it is important also to evaluate and compare myofibrillar contractile function of d-hESC-CMs and hvMFs at submaximal [Ca2+]. d-hESC-CMs responded to intermediate [Ca2+] with significantly (p < 0.001) higher isometric force when compared to hvMFs (ΔpCa50 = +0.24; Figure 2A). pCa determined at half of the maximum generated force (pCa50) was 6.04 ± 0.08 for d-hESC-CMs and 5.80 ± 0.05 for hvMFs (Figure 2C). The force-pCa curve determined for d-hESC-CMs had similar steepness (nH) as the curve determined for hvMFs (Figures 2A–C).
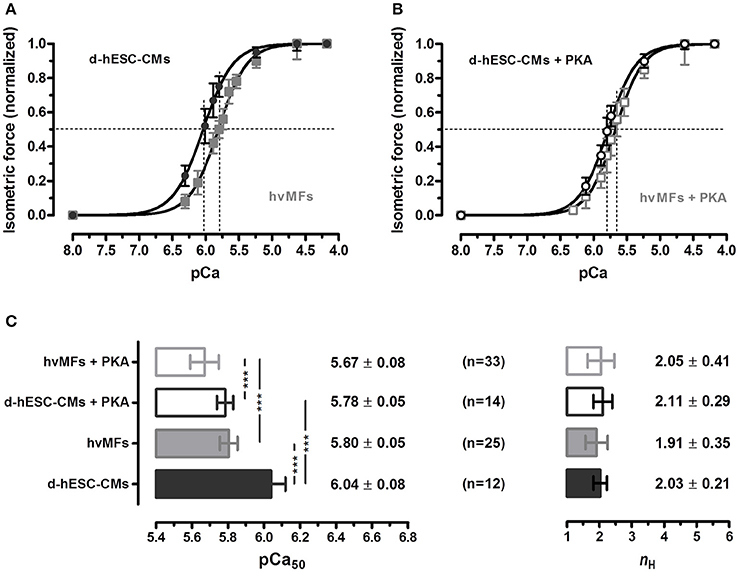
Figure 2. (A,B) Normalized isometric force response at different Ca2+ concentrations (given as pCa) of d-hESC-CMs (black circles and bars) and hvMFs (gray squares and bars), with (open symbols and bars) and without (filled symbols and bars) PKA-treatment prior to force measurements. Measured force at different pCa were normalized to the maximum force generated at saturating [Ca2+] (pCa 4.18; 15°C) and fitted by a dose-response Hill-type equation given in “Material and Methods.” (C) pCa at half maximum force generation (pCa50; indicating Ca2+-sensitivity of force), and the steepness of the force-pCa relationship (nH or Hill coefficient). Data are given as mean ± SD; ***p < 0.001. n, number of d-hESC-CMs and hvMFs, respectively. Force parameters of hvMFs isolated from two different adult donor hearts were pooled together.
Phosphorylation levels of some sarcomeric proteins can persist even after chemical skinning of myocardial samples. In studies with human cardiomyocytes isolated from failing and non-failing hearts (van der Velden et al., 2003b), PKA-treatment shifted the force-pCa curves to the right-side. Here, we found that PKA-treatment also shifted the isometric force-pCa relationships of both d-hESC-CMs (ΔpCa50 = −0.26) and hvMFs (ΔpCa50 = −0.13) distinctly to higher [Ca2+], while the steepness (nH) of the curves was not significantly affected (Figure 2C).
Kinetics of the Force Rise to Different Ca2+-Dependent Force Levels
The force response at a given [Ca2+] (considering both absolute and relative values) was different in d-hESC-CMs than in hvMFs. We compared at different intermediate [Ca2+] the rate constants of force re-development (kTR) of d-hESC-CMs toward the same final fractional force level Fn as in hvMFs (Fn = FACT/FACT,max), and plotted kTR as function of Fn (Figures 3A–D). Relationships between kTR and Fn obtained for d-hESC-CMs and hvMFs before and after PKA were fitted using the equation kTR = gapp/(1−Fn·fapp,max/(fapp,max+gapp)), where fapp,max (fapp at Fn = 1) and gapp are the probabilities (rate constants) of cross-bridges to enter and leave the force-generating states, respectively (Brenner, 1991a; Poggesi et al., 2005). Plots of Fn vs. kTR curves are shown with 95% confidence intervals (95%-CI) to compare separately d-hESC-CMs ± PKA (Figure 3A), hvMFs ± PKA (Figure 3B), d-hESC-CMs with hvMFs (Figure 3C), and d-hESC-CMs + PKA with hvMFs + PKA (Figure 3D).
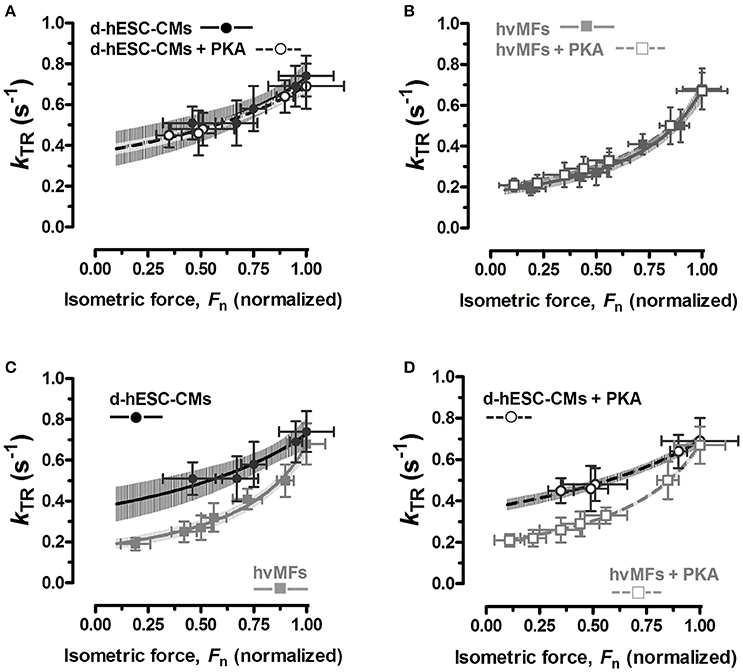
Figure 3. (A–D) Relationships between measured rate constants kTR of force re-development at different [Ca2+] and the fractional isometric force (Fn = FACT/FACT,max) for d-hESC-CMs (black symbols) and hvMFs (gray symbols), with (open symbols), and without (closed symbols) PKA-treatment prior to measurements. The equation kTR = gapp/(1−Fn·fapp,max/(fapp,max+gapp)), which was used to fit relations of kTR vs. Fn (Poggesi et al., 2005), is based on the two-state cross-bridge model (Huxley, 1957; Brenner, 1990). 95%-confidence intervals of the relations are shown, in distinct gray levels, for d-hESC-CMs ± PKA (A), hvMFs ± PKA (B), and for both d-hESC-CMs and hvMFs either not treated (C), or treated (D) with PKA, respectively. n, number of d-hESC-CMs and hvMFs as in Figure 2C.
For d-hESC-CMs at force levels below ~33% of FACT,max (and below ~15–20% of FACT,max for hvMFs), it was not possible to reliably measure kTR, because the laminar solutions flow exerted a hydrodynamic pressure on d-hESC-CMs (and on hvMFs) which perturbed the slow exponential rise of the recorded force signal.
We found that for both types of myofibrils receiving or not the PKA-treatment, the Fn vs. kTR curves (Figures 3A,B) were similar and the 95%-CIs were almost completely overlapped along the entire range of recorded fractional force levels (0 < Fn≤1). At pCa50 (Fn = 0.5), PKA had no effect on kACT and kTR (Figures 4B,D), as at saturating [Ca2+] (Fn = 1; Figures 4A,C).
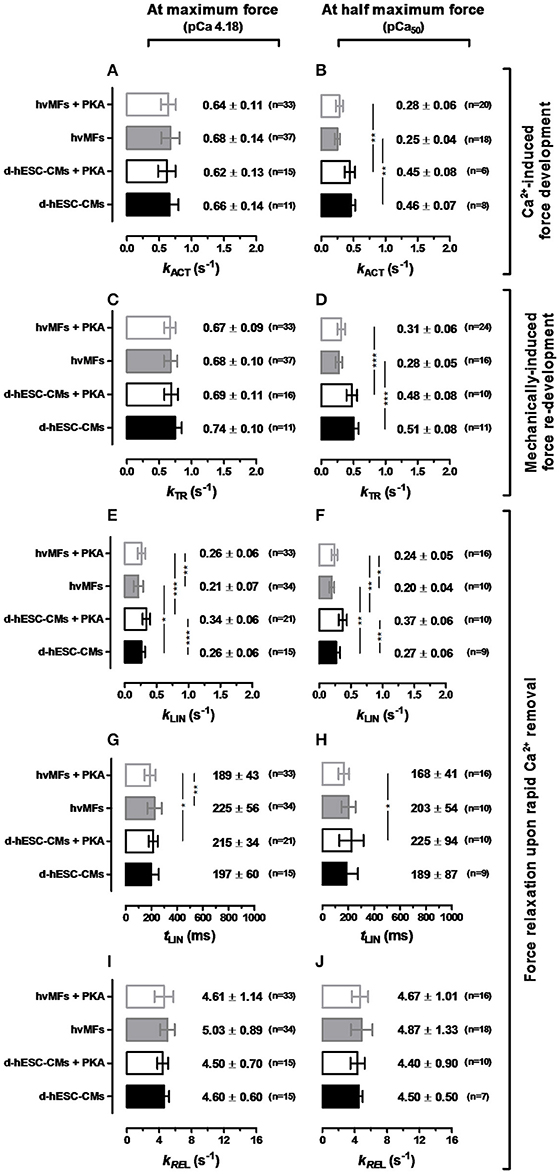
Figure 4. Contractile force kinetic parameters (kACT in A,B, kTR in C,D, kLIN in E,F, tLIN in G,H, kREL in I,J) of d-hESC-CMs (black bars) and hvMFs (gray bars) in the absence (filled bars) or presence (open bars) of PKA-treatment applied prior to measurements. Left panel, at saturating [Ca2+] (pCa 4.18). Right panel, at [Ca2+] where FACT was half of the maximum force (pCa50). Data shown as mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001. n = 7–21 for d-hESC-CMs; n = 10–37 for hvMFs.
We also compared d-hESC-CMs and hvMFs before and after PKA-treatment (Figures 3C,D), respectively. Despite the fact that at saturating [Ca2+] kACT and kTR values were similar when d-hESC-CMs were compared to hvMFs (Figures 4A,C), at submaximal force levels they were different (Figures 3C,D, 4B,D). 95%-CIs of the kTR vs. Fn curves for d-hESC-CMs and hvMFs did not overlap at intermediate fractional force levels (0 < Fn < 1) before and after PKA-treatment, respectively (Figures 3C,D).
These results indicate that cross-bridge kinetics, determining the force rise toward submaximal force levels, were different for d-hESC-CMs and hvMFs, while PKA-mediated phosphorylation had no significant effect.
Kinetics of Force Decay during Relaxation
Here we determined kLIN (the rate constant of the first relaxation phase) which is related to tension cost (= ATPase/Force ~ gapp) (Brenner, 1988; Weber et al., 2016), while the second relaxation phase (described by the rate constant kREL) is related to the rapid drop of force and cardiomyocyte re-lengthening that contributes to the rapidity of the ventricular pressure decay (i.e., during diastole) (Stehle and Iorga, 2010).
kLIN values corresponding to the relaxation from full Ca2+-activation and from pCa50 to pCa > 8 were compared (Figures 4E,F), revealing that kLIN was the same for both activation levels either in d-hESC-CMs or in hvMFs. This suggests that kinetics of cross-bridges leaving the force-generating states are not linked to the Ca2+-dependent occupancy of the previous force levels (Stehle et al., 2003). PKA-treatment accelerated the first relaxation phase of both d-hESC-CMs and hvMFs. kLIN of d-hESC-CMs treated with PKA had the highest value (Figures 4E,F). Independent of PKA-treatment (± PKA) and of the previous force level (at pCa 4.18 or pCa50), kLIN for d-hESC-CMs was significantly faster than for hvMFs (Figures 4E,F).
Duration of the first relaxation phase (tLIN) after rapid Ca2+ removal, i.e., the time until the sequential sarcomere lengthening (Videos 2–4) inducing the fast relaxation phase started (Stehle et al., 2002a,b; Poggesi et al., 2005), was similar in d-hESC-CMs and in hvMFs (Figures 4G,H). With PKA-treatment, it became significantly shorter only for hvMFs compared to d-hESC-CMs.
Due to the reduction in strain on the cross-bridges during the second relaxation phase (Figure 1C-right panel, Videos 2–4), the redistribution of cross-bridges toward the non-force-generating states was accelerated by ~10–20 times from the kLIN values (<0.4 s−1) to the values of kREL of ~4–5 s−1 for both d-hESC-CMs and hvMFs (Figures 4I,J). Whereas, strained cross-bridges during the first relaxation phase left the force-generating states faster (p < 0.05) after PKA-treatment (higher kLIN), kinetics of the second relaxation phase (kREL) were not significantly affected by PKA-treatment for both d-hESC-CMs and hvMFs (Figures 4I,J), as it was previously reported for hvMFs (Walker et al., 2011).
Protein Analysis of d-hESC-CMs Reveals a Sarcomeric Protein Isoform Pattern Which Is Different from hvMFs
The observed differences in myofibrillar force kinetics between d-hESC-CMs and hvMFs suggest a distinct isoform pattern of some sarcomeric proteins between these two contractile systems. Therefore, we have next focused on the identification of the isoforms of the myosin essential light chain (MLC-1), myosin regulatory light chain (MLC-2), myosin binding protein C (MyBP-C), troponin I (TnI), troponin T (TnT) and tropomyosin (Tm) of d-hESC-CMs compared to hvMFs. Since different phosphorylation of some sarcomeric proteins could also contribute to the observed differences in force kinetics, we also investigated sarcomeric protein phosphorylation.
Immunofluorescence
Myosin heavy chain
Double immunostaining against βMyHC and αMyHC cardiac isoforms of thin slices of human adult ventricular tissue from which hvMFs were isolated revealed only small myofibrillar regions with a mixture of both isoforms, while the rest of sarcomeres were only βMyHC positive. One example with αMyHC-positive CMs is shown in Figure 5A. With long-term (>35 days) cultivation of hESC-CMs on laminin-coated stiff isotropic glass surfaces, MyHC switches to essentially only the βMyHC isoform, as previously described (Weber et al., 2016) and shown here by double immunostaining against both cardiac MyHC isoforms (Figure 5B, Figure S2).
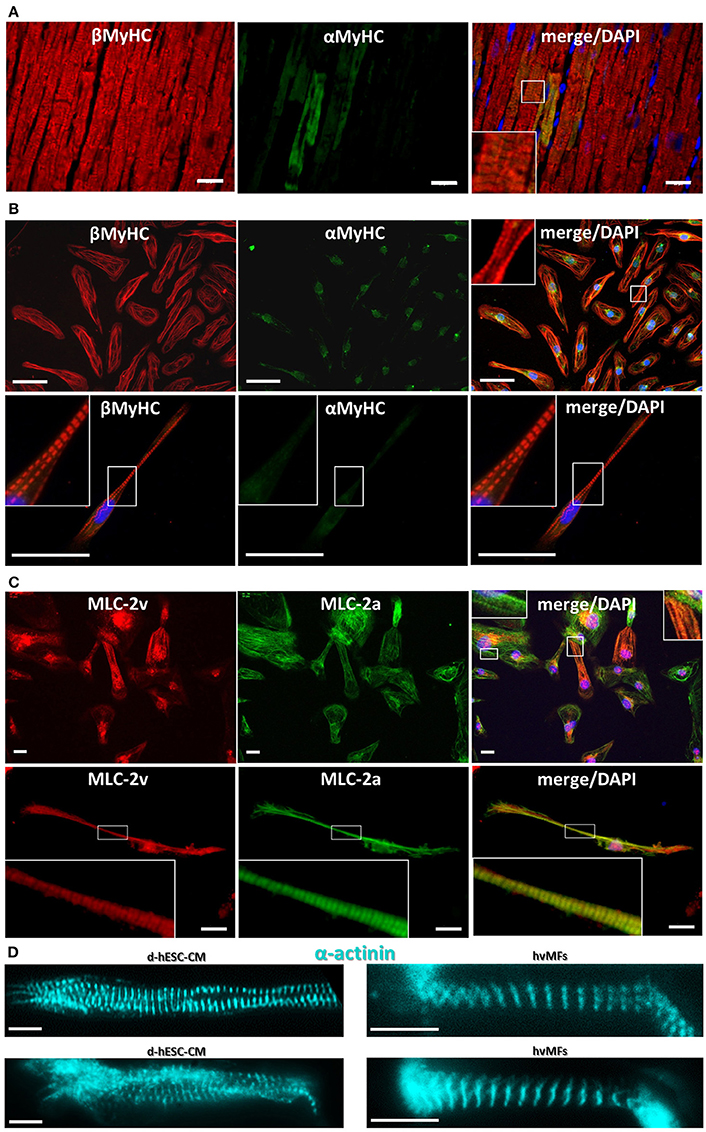
Figure 5. (A) Cryosection from human adult ventricular donor tissue immunostained against αMyHC (green fluorescence) and βMyHC (red fluorescence). A few cardiomyocytes show sarcomeres with some αMyHC expressed among the predominant βMyHC isoform. (B) Double stained (as in A) d-hESC-CMs expressing essentially only βMyHC isoform in their sarcomeres. Some non-sarcomere specific green fluorescence is seen in the αMyHC staining. Images in first row show multiple cells; second row, single cell. (C) d-hESC-CMs immunostained against MLC-2v (red) and MLC-2a (green) show a heterogeneous expression of both MLC-2 isoforms. Images in first row show multiple cells; second row, single cell. (D) d-hESC-CMs (left) and hvMFs (right) immunostained against α-actinin prior to Ca2+-activation in the micromechanical setup. Blue, DAPI for staining of nuclei. Scale bars: 20 μm (A,C), 50 μm (B), 10 μm (D). Insets represent the digital zoom of the selected regions.
Myosin light chain-2
Some d-hESC-CMs plated up to 60 days were immunostained against ventricular (MLC-2v) and atrial (MLC-2a) isoforms of MLC-2 protein (Figure 5C, Figure S3). From 47 analyzed double immunostained d-hESC-CMs (considering only MLC-2a/v incorporated into sarcomeres), ~43% were positive only for MLC-2a, while ~57% of them were positive for both MLC-2a/v isoforms, showing heterogeneous expression of both MLC-2 isoforms in the cells, with a possible predominance of MLC-2a.
Fluorescent immunostaining against MyHC (Figures 5B, Figure S2), MLC-2 (Figures 5C and Figure S3), or α-actinin (Figures 1B, 5D, Video 2) provides information regarding the distribution of myofibrils and sarcomeres within single hESC-CMs compared to adult ventricular tissue (Figure 5A) and single hvMFs-bundles (Figure 5D). Despite morphological (Figures 5B,C, Figures S2, S3) and ultrastructural (Weber et al., 2016) maturation of long-term (>35 days) plated hESC-CMs, myofibrillar density, overall alignment (in particular, the transversal alignment of Z-disks along the myofibrillar bundle) and cardiomyocytes elongation were apparently reduced in single hESC-CMs (Figures 1A1,D3, 5B–D) than in ventricular CMs (Weber et al., 2016) or within single hvMFs-bundles (Figures 1A2, 5A,D).
Overview of Sarcomeric Proteins by Gel Electrophoresis
In d-hESC-CMs, we could clearly identify the bands of MyHC, α-actinin, desmin, actin, TnT, α and β isoforms of Tm, TnI, atrial (MLC-1a, MLC-2a) and ventricular (MLC-1v, MLC-2v) isoforms of the MLC-1 and MLC-2 proteins (Figure 6A, lane 1), and compared them to those in atrial (lane 2), ventricular (lane 3), and skeletal (lane 4, M. gastrocnemius) adult human muscles.
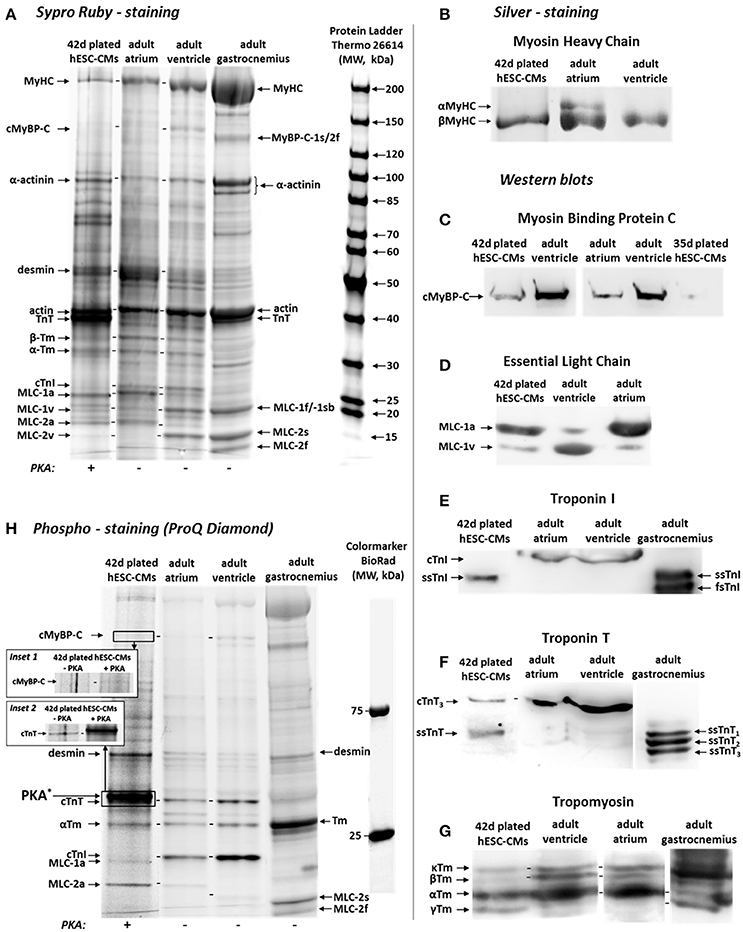
Figure 6. (A) Sypro Ruby-stained SDS-gel showing the bands corresponding to different sarcomeric protein isoforms of 42 days plated d-hESC-CMs (lane 1, +PKA), human adult atrial (lane 2, −PKA), ventricular (lane 3, −PKA), and skeletal (M. gastrocnemius) muscle (lane 4, −PKA) samples. Donor atrial, ventricular, and gastrocnemius adult muscle samples and the 5th lane loaded with protein ladder were used to identify the sarcomeric protein isoforms observed in d-hESC-CMs. (B) Analysis of silver-stained gels revealed an essentially exclusive expression of the βMyHC isoform in both d-hESC-CMs and hvMFs showing not detectable αMyHC isoform. For comparison, demembranated atrial tissue (with αMyHC isoform detected) is shown. (C–G) Western Blot analysis showing the presence of the following sarcomeric protein isoforms in d-hESC-CMs: (C) cMyBP-C (as detected in adult human ventricular and atrial samples); (D) MLC-1a and MLC-1v essential light chains; (E) Slow skeletal TnI (ssTnI); note, cardiac TnI (cTnI) was detectable only in adult ventricular and atrial samples; (F) Adult cardiac TnT (cTnT3) and slow skeletal TnT (ssTnT); (G) Tropomyosin isoforms: αTm-, βTm-, κTm-, and γTm-isoforms. (H) Phospho-stain (ProQ Diamond) of the gels in (A), showing phosphorylation of some sarcomeric proteins. Insets 1 (cMyBP-C) and 2 (cTnT): d-hESC-CMs treated (+PKA) or not treated (−PKA) with protein kinase A. Lane 1 (+PKA) and inset 2, right lane (+PKA): intense band corresponds to the PKA enzyme itself. Lanes 2, 3, 4: demembranated tissues of adult muscles shown here were not treated with PKA.
In d-hESC-CMs and in the homogenized ventricular tissue from which hvMFs were prepared for functional investigations, αMyHC isoform was below the detection limit (Figure 6B). MLC-1a (~84%; Figure 6A) and MLC-2a (81.5 ± 2.1%, n = 3; Figure 6A, Figure S4D) were the predominant protein isoforms expressed in d-hESC-CMs, similar to the atrial sample (~89% for MLC-1a, ~95% for MLC-2a; Figure 6A), while MLC-2v was 18.2 ± 2.5% (n = 3) (Figure 6A, Figure S4D).
Western Blot Analysis
Myosin binding protein C
The cardiac isoform of this protein (cMyBP-C) was clearly identified in d-hESC-CMs with its corresponding band at the same position as in ventricular and in atrial samples (Figure 6C, Figure S4A), being slightly above to the position of skeletal MyBP-C-1s/2f isoforms observed in the sample of M. gastrocnemius (Figure 6A).
Myosin light chain-1
Western blot analysis confirmed the presence of both MLC-1a (86.3 ± 3.3%, n = 3) and MLC-1v (13.7 ± 3.3%, n = 3) isoforms in d-hESC-CMs (Figure 6D, Figure S4B), but the MLC-1a isoform predominated.
Troponin I
The cardiac isoform of TnI (cTnI) was below the detection limit in d-hESC-CMs, while it was the only TnI-isoform in both adult ventricular and atrial samples. Instead, the slow skeletal (ssTnI) isoform was present in d-hESC-CMs, corresponding to the position of the ssTnI isoform identified in the M. gastrocnemius (Figure 6E, Figure S4C).
Troponin T
In d-hESC-CMs, both cardiac cTnT (~33%) and slow skeletal ssTnT (~67%) isoforms were detected, while in the adult ventricular and atrial samples, only the cTnT isoform was present (Figure 6F).
Tropomyosin
For d-hESC-CMs, we observed four bands, as the antibody was specific for all tropomyosin isoforms (Figure 6G). αTm, βTm, and κTm isoforms provided distinct bands, while αTm predominated in all cardiac samples. For d-hESC-CMs, additionally a farther migrating band appeared. This band was less visible in adult cardiac samples and seems to correspond to the last band observed in M. gastrocnemius sample, which could be the γTm isoform (Figure 6G; Jagatheesan et al., 2010; Marston et al., 2013).
Phosphorylated Sarcomeric Proteins
Overall, phospho-staining (ProQ Diamond; Figure 6H) revealed that the following sarcomeric proteins were phosphorylated in d-hESC-CMs (Figure 6H, lane 1; + PKA): cMyBP-C (inset 1; ± PKA), desmin, cTnT (inset 2; ± PKA), αTm, MLC-1a, and MLC-2a. For desmin, contributions of cytoskeletal desmin in d-hESC-CMs to the detected band should be considered, because the cytoskeletal network most likely was not completely removed from detergent-treated d-hESC-CMs. Phospho-stained bands corresponding to the phosphorylated cTnT and αTm isoforms were present in all samples [Figure 6H, lane 1 (+PKA), lanes 2,3 (−PKA)]. In adult ventricular and atrial samples, the phosphorylated form of cTnI was clearly visible. Yet, in d-hESC-CMs (±PKA) this band was missing, likely because the cTnI isoform was below detection limit. In d-hESC-CMs, we observed a weak band corresponding to the phosphorylated MLC-1a, which was absent or under the detection limit in adult atrial and ventricular samples (Figure 6H). MLC-2a isoform was also phosphorylated in d-hESC-CMs and more intense than the MLC-1a isoform.
In conclusion, d-hESC-CMs contain sarcomeric protein isoforms found not only in human adult ventricular samples, but also in the atrial and skeletal muscles of the adult human.
Discussion
In the present study, contractile function of βMyHC isoform-expressing myofibrils within d-hESC-CMs was characterized in comparison to hvMFs. We found that maximum generated force was reduced and Ca2+-sensitivity of force was increased, and that kinetics of force re-development at submaximal activation levels and kinetics of the slow relaxation phase were faster in d-hESC-CMs compared to hvMFs. Analysis of sarcomeric protein isoform patterns in both types of myofibrils revealed that d-hESC-CMs express slow skeletal TnI instead of the cTnI isoform normally found in adult ventricles. For other sarcomeric proteins involved in force regulation, non-ventricular isoforms were detected in d-hESC-CMs. The sarcomeric protein isoform pattern of hESC-CMs rather corresponds to cardiomyocytes in the developing human ventricle. The different protein isoforms can explain the differences in functional parameters.
Developmental Aspects Related to Sarcomeric Protein Isoforms that Modulate Contractile Function
The βMyHC isoform is predominantly expressed in adult, neonates, and fetal ventricles (Bouvagnet et al., 1987; Miyata et al., 2000; Reiser et al., 2001; Elhamine et al., 2016; Racca et al., 2016). Therefore, it is an indicator for ventricular-like cardiomyocytes in humans during developmental and adult stages. βMyHC-expression can be important for disease modeling in hESC-CMs, e.g., when questions regarding HCM or DCM-related βMyHC-mutations and their functional effects are addressed (Jung and Bernstein, 2014; Pioner et al., 2016).
Both, MLC-1a and MLC-1v isoforms are present during fetal development of the human ventricle (at mid-gestation, ~50% MLC-1a). During the months after birth, MLC-1a is being downregulated in the ventricle to low or even undetectable amounts (Elhamine et al., 2016). In adulthood, MLC-1a remains present only in the atria (Cummins et al., 1980; Cummins and Lambert, 1986). Under chronic hemodynamic overload, MLC-1a becomes re-expressed in the adult ventricular myocardium, affecting contractile properties by forming βMyHC·MLC-1a/v heterodimers (Schaub et al., 1998). In the present study, hESC-CMs cultured on a stiff surface expressed both MLC-1a/v isoforms, but the atrial (fetal) isoform predominated (Figures 6A,D, Figure S4B).
The MLC-2v isoform is predominantly expressed in the human developing fetal (e.g., 18–21 weeks gestational age) and in the adult (young or aged) ventricles, and it remains predominantly expressed even in some diseased ventricles (Cummins et al., 1980; Cummins and Lambert, 1986). Therefore, the MLC-2v isoform was previously used as chamber-specific marker to sort hPSC-CMs (Bizy et al., 2013). Nevertheless, during early embryonic stages of cardiogenesis, both atrial and ventricular isoforms of MLC-2 coexist in sarcomeres, while their genes are differentially regulated (Chien et al., 1993; Vestergaard et al., 2017). For instance, in the embryonic heart tube, MLC-2v isoform predominates only in the part of the cTnI isoform-expressing area (Fijnvandraat et al., 2003). In the hESC-CMs differentiated by Wnt-pathway modulators (Kempf et al., 2014), both MLC-2a/v isoforms were expressed (Figure 6A), as also previously reported for some hPSC-CMs (Bedada et al., 2014; Vestergaard et al., 2017). Yet, in the present study, the atrial isoform predominated in hESC-CMs (Figures 5C, 6A, Figure S4D). Notably, in our previous study, hESC-CMs cultured on a stiff surface (>35 days) that were differentiated using a p38-MAPK inhibitor (Xu et al., 2009; Kempf et al., 2011), expressed predominantly the MLC-2v isoform (Weber et al., 2016). This suggests that specific differentiation and cultivation conditions of hPSC-CMs could influence the transcriptional activity of MLC-1/-2 genes, and that the ventricular isoform expression of MLC-1/-2 is uncoupled from βMyHC-expression. This view is supported by previous findings showing that during the development of human ventricles and atria, mixed isotype assemblies occur in vivo (Cummins and Lambert, 1986; Vestergaard et al., 2017). It was suggested that from the pool of various genes related to these proteins, those are preferably expressed during development which build heteromeric myosin assemblies with functional advantages optimally adapted to a specific contractile task (Schaub et al., 1998).
For cMyBP-C no change in isoform expression has been observed during development of cardiomyocytes (i.e., lack of transcomplementation) (Gautel et al., 1998). In our work, western blot analysis of d-hESC-CMs confirmed the presence of cMyBP-C (Figure 6C, Figure S4A).
Regarding cTnT, four cardiac isoforms (cTnT1−4) can be expressed in human cardiomyocytes through combinatorial alternative mRNA splicing in a developmentally regulated manner (Gomes et al., 2002). cTnT1,2,4 and ssTnT isoforms are usually expressed in early fetal heart (Barton et al., 2004). In the adult heart, only cTnT3 is present and ssTnT might be expressed at low levels, as response to myocardial stress. In this study, hESC-CMs express both adult cTnT3 and ssTnT isoforms (Figure 6F), while the other fetal cTnT1 and cTnT2 isoforms, previously reported in early stages of fetal ventricles (Racca et al., 2016), were not detected. In another study with hPSC-CMs, only one cTnT isoform corresponding to the 130 days gestational stage of the human fetal ventricle was reported (Pioner et al., 2016).
TnT has several isoforms and different genes control its expression in cardiac, slow- and fast-twitch skeletal muscles. TnT isoform genes are connected in chromosomal DNA to TnI isoform genes, e.g., the cTnT-gene is paired with the ssTnI-gene and the ssTnT-gene is paired with cTnI-gene, respectively (Wei and Jin, 2011). Such pairing reflects original functional linkages, because in embryonic cardiac muscles the cTnT-gene is expressed together with the ssTnI-gene (Gomes et al., 2002; Wei and Jin, 2011). Further developmental transition of cTnT and cTnI isoforms seems to be regulated by different mechanisms in mammals (Gao et al., 1995). Interestingly, the ssTnT-to-cTnT isoform shift occurs earlier than the ssTnI-to-cTnI shift (Siedner et al., 2003; Elhamine et al., 2016), which may explain the TnI- and TnT-isoform patterns of the d-hESC-CMs observed here (Figures 6E,F, Figure S4C).
In the adult healthy human ventricle, essentially only cTnI is found. Fetal ventricles express both cTnI and ssTnI, which are regulated at the level of gene transcription. ssTnI is progressively downregulated within the first year after birth, similar to the fetal MLC-1a isoform (Elhamine et al., 2016; Racca et al., 2016). In the d-hESC-CMs analyzed here only ssTnI was detected (Figure 6E, Figure S4C), while in other studies with hPSC-CMs, both ssTnI and cTnI isoforms were reported (Bedada et al., 2014; Pioner et al., 2016).
Previous studies indicated that the thyroid growth hormone (T3) increased the cTnI/ssTnI ratio in developing cultured cardiomyocytes (Riedel et al., 2005), promoting also different other maturation aspects of hPSC-CMs (Yang et al., 2014b). However, T3 also causes a shift from the slow βMyHC to the fast αMyHC isoform in hESC-CMs, enhancing their sarcomeric force kinetics (Weber et al., 2016). Therefore, further studies are needed to clarify how cTnT/ssTnT and cTnI/ssTnI ratios can be increased in hESC-CMs while maintaining the predominant expression of βMyHC. Raising cTnT/ssTnT ratio in hPSC-CMs might be particularly important, because the cTnT isoform is commonly used as a cardiac lineage differentiation marker during cells selection procedure. However, to our knowledge, this criterion does not exclude selection of cardiomyocytes expressing both cTnT and ssTnT isoforms and it does not distinguish between the adult cTnT3 and the other fetal cardiac isoforms of TnT.
The αTm and βTm isoforms are both expressed in developing and adult heart. Although the expression of the βTm isoform increases somewhat in transition from fetus to adult, αTm remains the predominant isoform (Muthuchamy et al., 1993; Rajan et al., 2010; Marston et al., 2013). The κTm isoform results from alternative splicing of the TPM1-gene encoding for αTm and it is expressed only at low protein levels (Rajan et al., 2010). The γTm isoform seems to be less expressed or even absent in adult human ventricles, while it is usually found in slow-twitch skeletal muscles (Jagatheesan et al., 2010; Rajan et al., 2010; Marston et al., 2013). In this study, all four α, β, κ, and γ isoforms of Tm were detected in d-hESC-CMs and the band of γTm was even stronger than that of either βTm or κTm isoform (Figure 6G). Most importantly, in d-hESC-CMs the αTm isoform was predominant, as in the adult ventricular sample (Figure 6G).
In the present study, hESC-CMs cultivated on coverslips exhibited a sarcomeric protein isoform pattern similar to cardiomyocytes of the early fetal stage or even embryonic stage of the developing human ventricle. The deviation from the adult ventricular sarcomeric protein isoform pattern of d-hESC-CMs likely affected contractile function of their myofibrils, as discussed in the following section.
Differences in Contractile Function between d-hESC-CMs and hvMFs Are Related to Protein Isoform Expression
Passive Force
The significantly lower passive force (Fpass) for d-hESC-CMs vs. hvMFs (Figure S1), could be due to the presence of the more compliant and longer fetal-N2BA titin isoform (Kruger and Linke, 2009). This is supported by other studies which showed that the fetal-N2BA isoform predominated in hiPSC-CMs (Hinson et al., 2015). Fpass for hvMFs was slightly, but significantly (p < 0.05) diminished following PKA-treatment, while for d-hESC-CMs, PKA had no effect (Figure S1). It was previously shown that adult donor ventricular tissue contains the shorter compliant N2BA and the stiffer N2B titin isoforms in the ratio N2BA:N2B~0.5 and only N2B is target for PKA-mediated phosphorylation (Kruger and Linke, 2009). To determine FACT we subtracted Fpass from the recorded active force.
Contractile Function at Saturating [Ca2+]
We found that maximum isometric force was significantly smaller for d-hESC-CMs (~42 kPa) than for hvMFs (~94 kPa), presumably due to less well-aligned myofibrils within d-hESC-CMs compared to more compact hvMFs-bundles (e.g., Figures 1A, 5D), but differences in sarcomeric protein isoform composition (Figure 6, Figure S4) could also contribute to the lower isometric force.
Upon sudden increase of [Ca2+] from relaxation (>pCa 8) to pCa 4.18, force raised mono-exponentially without lag (Figure 1C) and with very similar kACT in both d-hESC-CMs and hvMFs. This suggests that myofibrils within d-hESC-CMs, as previously shown for hvMFs (Stehle et al., 2002b; Poggesi et al., 2005), rapidly equilibrate with the surrounding Ca2+-defined solution and that the measured kinetics are not rate-limited by diffusional events. Because kACT in both types of myofibrils is similar to kTR, kinetics of the force rise upon Ca2+-activation of d-hESC-CMs, as shown before for hvMFs (Stehle et al., 2002b), seem to be primarily governed by cycling cross-bridges rather than by the much faster Ca2+-controlled switch on-and-off of the regulatory proteins of the thin filaments. kTR for d-hESC-CMs and hvMFs was very similar to kTR previously determined for hvMFs (Stehle et al., 2002b; Piroddi et al., 2007). This suggests that cross-bridge cycling kinetics at full calcium-activation seems not to be affected by the observed differences in thin filaments regulatory protein- and myosin light chain-isoforms.
Contractile Function at Intermediate [Ca2+]
Cardiomyocytes in the living heart and in cell culture are operating at submaximal intracellular [Ca2+]. At intermediate [Ca2+], development-dependent isoform differences of some sarcomeric proteins may influence the steady-state force and cross-bridge cycling kinetics (Metzger et al., 2003; Siedner et al., 2003; Racca et al., 2016).
Ventricular myofibrils and cardiomyocytes expressing the ssTnI isoform exhibit an increased Ca2+-sensitivity of force compared to those containing cTnI, and even a partial expression of ssTnI, co-expressed with cTnI, seemed to have a dominant Ca2+-sensitizing effect (Metzger et al., 1994, 2003; Siedner et al., 2003; Elhamine et al., 2016; Racca et al., 2016). Therefore, it is very likely that the ssTnI, solely detected in the d-hESC-CMs, contributes to the observed higher Ca2+-sensitivity of force when compared to adult hvMFs (Figures 2A,C) containing only cTnI (Figure 6E, Figure S4C). In addition, cTnI was phosphorylated in hvMFs (Figure 6H), which reduces Ca2+-sensitivity and thus, this may add to the large difference in Ca2+-sensitivity of force between d-hESC-CMs and hvMFs (ΔpCa50 = +0.24) (Figures 2A,B). ssTnI does not have the PKA-dependent phosphorylatable N-terminal extension of cTnI. Therefore, effects of PKA-treatment of d-hESC-CMs on pCa50 cannot be mediated through TnI phosphorylation.
In contrast to the Ca2+-sensitizing effect of ssTnI, the ssTnT isoform, detected together with cTnT in d-hESC-CMs (Figure 6F), was shown to reduce the Ca2+-sensitivity of force in cardiac fibers (Pinto et al., 2012). However, the effect of ssTnT apparently does not dominate the Ca2+-sensitivity of force for d-hESC-CMs compared to hvMFs.
αTm is the predominant tropomyosin isoform expressed in both d-hESC-CMs and hvMFs. βTm and κTm isoforms were shown to exert increasing and decreasing effects on Ca2+-sensitivity of force, respectively (Jagatheesan et al., 2010; Rajan et al., 2010). Therefore, the effects may be compensatory in both, d-hESC-CMs and hvMFs. d-hESC-CMs seem to express additionally the γTm isoform (Figure 6G). Co-expression of γTm in cardiac muscle decreases Ca2+-sensitivity of force, and importantly, γTm apparently has a functional dominance over the other Tm isoforms in the regulation of striated muscle performance (Pieples et al., 2002; Jagatheesan et al., 2010). Therefore, the presence of γTm in d-hESC-CMs would argue for a decrease of Ca2+-sensitivity of force compared to hvMFs.
Isoform differences of TnI, TnT and Tm observed in d-hESC-CMs vs. hvMFs may affect the fast switch-on/-off equilibrium of the thin filament, determining in d-hESC-CMs a particular thin filament status which might be different from that in hvMFs. γTm is more negatively charged and less flexible than the αTm (Jagatheesan et al., 2010) and, together with the ssTnT/cTnT and ssTnI isoforms in d-hESC-CMs, may alter dynamics of the thin filaments to expose their strong binding sites to myosin heads and to the other actin-interacting partners, e.g., N-terminus of MLC-1 (Rarick et al., 1996) and of cMyBP-C (Craig et al., 2014). All these may provide a feedback to the dynamical status of thin filaments, especially at lower [Ca2+], as previously proposed considering the cross-bridge-mediated cooperative activation of thin filaments to further recruit new cross-bridges (Campbell, 1997).
MLC-1 interacts in an isoform dependent manner with the lever arm of MyHC (via non-covalent interactions) and with actin (via electrostatic interactions), providing a possible tether for the myosin head between thick and thin filaments. MLC-1a has a higher affinity for the MyHC lever arm and a lower affinity for actin than the MLC-1v isoform (Morano and Haase, 1997; Hernandez et al., 2007). Therefore, the MyHC·MLC-1a may leave the force-generating states faster than MyHC·MLC-1v. It was shown that a higher MLC-1a content by ~20% in human ventricular fibers increases Ca2+-sensitivity of force (pCa50) by +0.36 pCa-units (Morano and Haase, 1997; Schaub et al., 1998; Hernandez et al., 2007). MLC-1 can be phosphorylated (Arrell et al., 2001), and this is believed to increase cross-bridge detachment rates, thus shifting the equilibrium of cross-bridges more toward the non-force-generating states, i.e., increasing gapp. Altogether, a predominant expression of MLC-1a in d-hESC-CMs compared to hvMFs (Figures 6A,D, Figure S4B), which seems to be partially phosphorylated (Figure 6H), may contribute to the higher Ca2+-sensitivity of force (Figure 2A) and to a distinct modulation of cross-bridge cycling kinetics at submaximal force levels. This is supported here by significantly faster kLIN (Figures 4E,F) and kTR (Table 2, Figures 3C, 4D) at pCa50 for d-hESC-CMs compared to hvMFs.
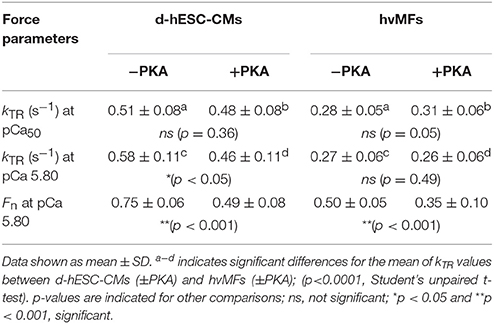
Table 2. Measured force kinetic parameter kTR determined at pCa50 (Fn = 0.5) and at a given [Ca2+] of pCa 5.80 and the fractional force Fn (Fn = FACT/FACT,max) at pCa 5.80 for d-hESC-CMs and hvMFs, which were either treated (+PKA) or not (−PKA) with protein kinase A.
Phosphorylation of MLC-2 was shown to increase kTR and Ca2+-sensitivity of force (van der Velden et al., 2003a,b). In d-hESC-CMs, both MLC-2a/v isoforms co-exist, with MLC-2a being predominant (Figure 6A, Figure S4D) and phosphorylated (Figure 6H). This may contribute to the higher pCa50 (Figure 2) and faster kTR at pCa50 (Table 2, Figures 3C, 4D) or at a given [Ca2+] (e.g., at pCa 5.80 shown in Table 2) for d-hESC-CMs compared to hvMFs.
Altogether, in d-hESC-CMs, functional effects of MLC-1a and ssTnI (both increasing pCa50) seem to dominate the effects of ssTnT and γTm (both decreasing pCa50) at submaximal forces (Table 2), because d-hESC-CMs had a higher Ca2+-sensitivity of force compared to hvMFs, when both were either treated (ΔpCa50 = +0.11) or not treated (ΔpCa50 = +0.24) with PKA, respectively (Figures 2A,B). However, possible differences between the phosphorylation status of other sarcomeric proteins than cTnI of d-hESC-CMs compared to hvMFs may also affect Ca2+-sensitivity.
cMyBP-C is a target for PKA and it is expressed in both types of myofibrils studied here. Even though phospho-staining analysis showed a weak signal for phosphorylation of cMyBP-C (Figure 6H, inset 1), the force-pCa relation of d-hESC-CMs was clearly shifted to higher [Ca2+] upon PKA-treatment (ΔpCa50 = −0.26; Figure 2C). It would be possible that a change in cMyBP-C phosphorylation contributes significantly to this right-shift of the force-pCa relation, because cTnI was undetected in d-hESC-CMs. It was previously shown that in cardiomyocytes lacking cTnI, the phosphorylation of cMyBP-C is able to decrease Ca2+-sensitivity of force (Chen et al., 2010). PKA-treatment reduced less, but significantly Ca2+-sensitivity of force in hvMFs vs. d-hESC-CMs (Figure 2C), suggesting that hvMFs were already more phosphorylated than d-hESC-CMs.
For both d-hESC-CMs and hvMFs at any given submaximal fractional force level (0 < Fn < 1, Fn = FACT/FACT,max), PKA-treatment had no significant effect on measured kTR (Figures 3A,B, 4D) and thus, not on fapp+gapp (= kTR). fapp (which depends on [Ca2+] modulating the thin filament status) and gapp (which is independent of [Ca2+]) are the rate constants (probabilities) of cross-bridges entering and leaving the force-generating states, respectively (Brenner, 1988). At the same given [Ca2+], small differences could eventually exist between kTR values determined before and after PKA-treatment, because fapp is Ca2+-sensitive and PKA-treatment decreased Ca2+-sensitivity of force. For example, at pCa50 (Fn = 0.5) or at a given [Ca2+] (pCa 5.80), kTR values are shown in Table 2. It was previously reported with adult hvMFs, involving the same investigation method to determine kTR as in the present study, that a shift in Ca2+-sensitivity of force following the PKA-treatment did not significantly affect kTR at different fractional force levels (Walker et al., 2011). Using the same methodological approach with human donor cardiomyocytes, kTR at different [Ca2+] was not significantly affected by PKA-treatment (van Dijk et al., 2014). In studies with murine ventricular skinned myocardium, which may have lower endogenous phosphorylation levels than human donor samples, PKA-treatment accelerated kTR and this was attributed mainly to cMyBP-C phosphorylation (Stelzer et al., 2006). It was suggested that the acceleration of kTR occurred by increasing the rate constants of both cross-bridges detachment and recruitment (Stelzer et al., 2006). In d-hESC-CMs expressing cMyBP-C and lacking cTnI, PKA-treatment enhanced the probability of strained cross-bridges to leave the force-generating states, as the differences (p < 0.01) in kLIN for d-hESC-CMs ± PKA suggest (Figures 4E,F), but it had no significant impact on less strained cross-bridges, as kREL values were similar (Figures 4I,J). kLIN was accelerated by PKA-treatment also in hvMFs, but apparently less than in d-hESC-CMs (Figures 4E,F). Therefore, during mechanically loaded cross-bridge turnover at submaximal Fn, the kTR, which was essentially unaffected by PKA-treatment in d-hESC-CMs (Figures 3A, 4D), would likely result from a relatively small increase of gapp and decrease of fapp (to keep fapp+gapp = kTR relatively constant). This could be due to a subtle alteration of thin filaments turning-on/off dynamics in the context of different protein isoform pattern (e.g., lack of cTnI) in d-hESC-CMs compared to hvMFs. However, the potential effects caused by PKA-treatment on gapp (increasing) and fapp (decreasing) could not be accurately predicted (large 95%-CI) from kTR vs. Fn dependencies determined at submaximal force levels for d-hESC-CMs (Figure 3A), but such predictions can be used to compare d-hESC-CMs with hvMFs (see below Figures 7A,B).
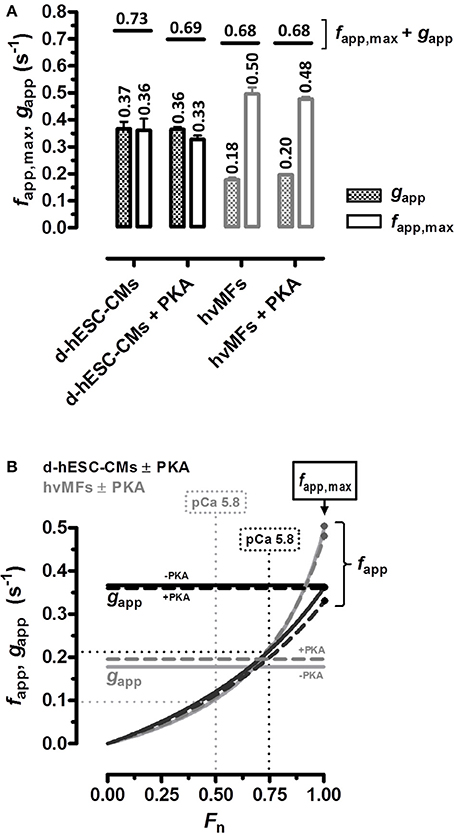
Figure 7. (A) kTR vs. Fn data (plotted in Figure 3) was fitted to the equation given in the legend of Figure 3 and yielded the cross-bridge kinetics-related predicted parameters gapp (patterned bars) and fapp,max at Fn = 1 (open bars) for d-hESC-CMs (black bars) and for hvMFs (gray bars) which were either treated or not with PKA. Horizontal short lines correspond to the sum fapp,max + gapp predicting kTR at saturating [Ca2+] (pCa 4.18). Numbers, mean values. Error bars, standard errors (SE). (B) From all four curves (black, d-hESC-CMs; gray, hvMFs; solid, −PKA; dashed, +PKA) fitting kTR vs. Fn data shown in Figure 3, fapp was calculated by subtracting the predicted gapp values given in A and then plotted against Fn (0 < Fn ≤ 1). gapp is Ca2+-independent (horizontal lines); fapp was positive, monotonically increasing from zero (Fn = 0) to fapp,max (Fn = 1) (Brenner, 1988).
Why Maximum Isometric Force and the Rate Constant of Force Re-development at Submaximal [Ca2+] Might Be Different for d-hESC-CMs Compared to hvMFs?
From the curves (Figure 3) fitting the kTR vs. Fn data, the apparent rate constants gapp at Fn = 0 and fapp,max at Fn = 1 can be predicted (Figure 7A) (Brenner, 1988). In d-hESC-CMs, fapp,max and gapp are essentially equal, different from the hvMFs where we calculated 2 to 3-fold larger values for fapp,max compared to gapp (Figure 7A). Such a difference between fapp,max and gapp, and even larger ones, were also previously shown for hvMFs (Stehle et al., 2002b; Poggesi et al., 2005; Piroddi et al., 2007). Based on these calculations, the decrease in FACT,max generated by d-hESC-CMs (~42 kPa) vs. hvMFs (~94 kPa), partly results from a decreased occupancy of force-generating cross-bridge states determined by a reduced duty-ratio fapp,max/(fapp,max+gapp) (Brenner, 1988), because fapp,max is lower and gapp is higher in d-hESC-CMs compared to corresponding parameters in hvMFs. Yet, if FACT,max is estimated considering only the duty-ratio from the predicted values of fapp,max and gapp (Figure 7A), FACT,max for d-hESC-CMs would be ~67% of FACT,max for hvMFs, while the measured force of d-hESC-CMs (42 kPa) was ~45% of the force generated by hvMFs (94 kPa). Therefore, other factors may contribute to the estimated differences, for instance, a less ordered and less compact sarcomere alignment within d-hESC-CMs than that observed in hvMFs-bundles (Figures 1A, 5D) and in healthy cardiomyocytes (Kraft et al., 2013; van Dijk et al., 2014). In developing hESC-CMs, structural maturation of sarcomeres could play an important role for force generation, as previously described in human fetal cardiac muscles (Racca et al., 2016).
While at full Ca2+-activation kTR is similar for both contractile systems, at submaximal force levels (0 < Fn < 1), kTR was significantly faster for d-hESC-CMs than for hvMFs (Figures 3C, 4D). This is mostly due to a higher gapp in d-hESC-CMs than in hvMFs (Figure 7A). Higher gapp of d-hESC-CMs vs. hvMFs may determine an increase of the tension cost (= ATPase/force) that is proportional to gapp (Brenner, 1988). At lower [Ca2+], fapp/gapp may modulate contractile function in addition to modulation through changes within the regulatory proteins (Brenner, 1988). Predicted curves fitting kTR vs. Fn data (Figure 3C) and gapp parameters (Figure 7A) were further used to calculate fapp and then fapp was plotted against Fn (Figure 7B). Independent on gapp, fapp decreases monotonically with decreasing Fn (Figure 7B). Even if predicted fapp vs. Fn curves seem to be similar below Fn~0.75 for both contractile systems (Figure 7B), each fractional force level Fn is reached at lower [Ca2+] in d-hESC-CMs than in hvMFs, because d-hESC-CMs are more Ca2+-sensitive than hvMFs (Figure 2). For example, at a given [Ca2+] of pCa 5.8, d-hESC-CMs generated 75% of maximum force FACT,max (Table 2) and predicted fapp = 0.22 s−1 (Figure 7B), while hvMFs generated 50% of their maximum force FACT,max (Table 2) and predicted fapp = 0.10 s−1 (Figure 7B). Independent on [Ca2+], predicted gapp was 0.37 s−1 for d-hESC-CMs and 0.18 s−1 for hvMFs (Figures 7A,B). Therefore, kTR (= fapp+gapp) is larger for d-hESC-CMs (0.59 s−1) than for hvMFs (0.28 s−1) at pCa 5.8.
The aspects discussed here resulted solely from the comparison of hESC-CMs [differentiated and cultivated as previously described (Kempf et al., 2014; Weber et al., 2016)] with hvMFs (isolated from adult human ventricles). Variability in the outcomes cannot be excluded if d-hPSC-CMs would be used which were differentiated in another way and subjected to different maturation conditions.
Conclusions
In the present study, myofibrillar contractile function was linked to sarcomeric protein isoform pattern of adult hvMFs and of hESC-CMs after several weeks of cultivation on a stiff surface (Weber et al., 2016). Our present results suggest that more mature morphological and ultrastructural aspects (Weber et al., 2016) of hESC-CMs may not necessarily correspond to an overall adult ventricular-like sarcomeric protein isoform pattern and contractile function at the myofibrillar level (Pioner et al., 2016; Racca et al., 2016). Therefore, one might consider not only cTnI (Bedada et al., 2014; Pioner et al., 2016) or MLC-2v (Bizy et al., 2013; Vestergaard et al., 2017) as sarcomeric protein markers for hPSC-CMs differentiated in vitro, but also the adult ventricle-specific protein isoforms βMyHC, MLC-1v and cTnT3. It would be of great interest to identify the appropriate chemo-mechanical factors to be applied to in vitro differentiated hPSC-CMs to speed-up shifting the expression of MLC-1, MLC-2, TnT, and TnI proteins toward the full spectrum of adult ventricular-like sarcomeric protein isoforms in βMyHC expressing hPSC-CMs in a cost-effective manner. On the other hand, in vitro differentiated cardiomyocytes with sarcomeric protein isoform composition closer to early stages of the developing human heart, may well provide a model to study e.g., basic mechanistic aspects of contraction in developing human cardiomyocytes, mechanisms of heart failure onset during in utero heart development, or inherited cardiac diseases affecting contractile function.
Author Contributions
BI, project design, establishing the micromechanical setup, micromechanical experiments, and functional data analysis and modeling with hESC-CMs and hvMFs, manuscript concept design and manuscript writing; KS, generation, differentiation, and purification of hESC-CMs; NW, MW, and SG, cultivation of hESC-CMs, immunostaining, data analysis, and interpretation; BP, gel-electrophoretic analysis of sarcomeric proteins; CdR, substantial contributions to conception of the project, acquisition of data, and human samples preparation; UM, RZ, TK, and BB, supervising and feedback on the project, intellectual improvement of the manuscript content and structure, manuscript writing. All authors approved the final version of the manuscript.
Funding
This work was supported by grants of Deutsche-Forschungsgemeinschaft BR849/31-1, KR1187/21-1, MA2331/16-1, ZW64/4-1, and Cluster of Excellence REBIRTH DFG EXC62/3; BMBF-grant 13N12606 and StemBANCC (Innovative Medicines Initiative joint undertaking, grant agreement n°115439-2, resources of which are composed of financial contribution from the European Union (FP7/2007-2013) and EFPIA companies' in kind contribution).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RM declared a past co-authorship with one of the authors CdR to the handling Editor.
Acknowledgments
The authors thank Tim Holler, Alexander Lingk, Torsten Beier, and Uwe Krumm (Molecular and Cell Physiology, Hannover Medical School) for their excellent technical assistance and Anja Wenzl for reading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2017.01111/full#supplementary-material
Abbreviations
95%-CI(s), 95% Confidence interval(s); BDM, 2, 3-Butanedione monoxime; BF, Bright field; [Ca2+], Calcium concentration(s); CM(s), Cardiomyocyte(s); CSA, Cross section area; DCM, Dilated cardiomyopathy; DTT, Dithiothreitol; HCM, Hypertrophic cardiomyopathy; FL, Fluorescence; d-hESC-CM(s), demembranated human embryonic stem cell-derived cardiomyocyte(s); hESC-CM(s), human embryonic stem cell-derived cardiomyocyte(s); hPSC-CM(s), human pluripotent stem cell-derived cardiomyocyte(s); hvMFs, human ventricular myofibrils; PhC, Phase contrast; PIC, Protease inhibitor cocktail; PKA, Protein kinase A; SL, Sarcomere length (SL0: SL of slack myofibrils).
References
Arrell, D. K., Neverova, I., Fraser, H., Marbán, E., and Van Eyk, J. E. (2001). Proteomic analysis of pharmacologically preconditioned cardiomyocytes reveals novel phosphorylation of myosin light chain 1. Circ. Res. 89, 480–487. doi: 10.1161/hh1801.097240
Barton, P. J., Felkin, L. E., Koban, M. U., Cullen, M. E., Brand, N. J., and Dhoot, G. K. (2004). The slow skeletal muscle troponin T gene is expressed in developing and diseased human heart. Mol. Cell. Biochem. 263, 91–97. doi: 10.1023/B:MCBI.0000041851.53074.72
Bedada, F. B., Chan, S. S., Metzger, S. K., Zhang, L., Zhang, J., Garry, D. J., et al. (2014). Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Rep. 3, 594–605. doi: 10.1016/j.stemcr.2014.07.012
Birket, M. J., and Mummery, C. L. (2015). Pluripotent stem cell derived cardiovascular progenitors–a developmental perspective. Dev. Biol. 400, 169–179. doi: 10.1016/j.ydbio.2015.01.012
Bizy, A., Guerrero-Serna, G., Hu, B., Ponce-Balbuena, D., Willis, B. C., Zarzoso, M., et al. (2013). Myosin light chain 2-based selection of human iPSC-derived early ventricular cardiac myocytes. Stem Cell Res. 11, 1335–1347. doi: 10.1016/j.scr.2013.09.003
Bouvagnet, P., Neveu, S., Montoya, M., and Leger, J. J. (1987). Development changes in the human cardiac isomyosin distribution: an immunohistochemical study using monoclonal antibodies. Circ. Res. 61, 329–336. doi: 10.1161/01.RES.61.3.329
Brenner, B. (1988). Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269. doi: 10.1073/pnas.85.9.3265
Brenner, B. (1990). “Muscle mechanics and Biochemical Kinetics,” in Molecular Mechanics in Muscular Contraction, ed J. M. Squire (London: Macmillan Press Ltd.), 13, 77–149.
Brenner, B. (1991a). A new concept for the mechanism of Ca+(+)-regulation of muscle contraction. Implications for physiological and pharmacological approaches to modulate contractile function of myocardium. Basic Res. Cardiol. 86(Suppl. 3), 83–92.
Brenner, B. (1991b). Rapid dissociation and reassociation of actomyosin cross-bridges during force generation: a newly observed facet of cross-bridge action in muscle. Proc. Natl. Acad. Sci. U.S.A. 88, 10490–10494.
Brenner, B., and Chalovich, J. M. (1999). Kinetics of thin filament activation probed by fluorescence of N-((2-(iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole-labeled troponin I incorporated into skinned fibers of rabbit psoas muscle: implications for regulation of muscle contraction. Biophys. J. 77, 2692–2708. doi: 10.1016/S0006-3495(99)77103-1
Brenner, B., Seebohm, B., Tripathi, S., Montag, J., and Kraft, T. (2014). Familial hypertrophic cardiomyopathy: functional variance among individual cardiomyocytes as a trigger of FHC-phenotype development. Front. Physiol. 5:392. doi: 10.3389/fphys.2014.00392
Burke, M. A., Cook, S. A., Seidman, J. G., and Seidman, C. E. (2016). Clinical and mechanistic insights into the genetics of cardiomyopathy. J. Am. Coll. Cardiol. 68, 2871–2886. doi: 10.1016/j.jacc.2016.08.079
Burridge, P. W., Li, Y. F., Matsa, E., Wu, H., Ong, S. G., Sharma, A., et al. (2016). Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556. doi: 10.1038/nm.4087
Campbell, K. (1997). Rate constant of muscle force redevelopment reflects cooperative activation as well as cross-bridge kinetics. Biophys. J. 72, 254–262. doi: 10.1016/S0006-3495(97)78664-8
Chen, P. P., Patel, J. R., Rybakova, I. N., Walker, J. W., and Moss, R. L. (2010). Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. J. Gen. Physiol. 136, 615–627. doi: 10.1085/jgp.201010448
Chien, K. R., Zhu, H., Knowlton, K. U., Miller-Hance, W., van-Bilsen, M., O'Brien, T. X., et al. (1993). Transcriptional regulation during cardiac growth and development. Annu. Rev. Physiol. 55, 77–95. doi: 10.1146/annurev.ph.55.030193.000453
Colomo, F., Nencini, S., Piroddi, N., Poggesi, C., and Tesi, C. (1998). Calcium dependence of the apparent rate of force generation in single striated muscle myofibrils activated by rapid solution changes. Adv. Exp. Med. Biol. 453, 373–381. discussion: 381–372. doi: 10.1007/978-1-4684-6039-1_42
Craig, R., Lee, K. H., Mun, J. Y., Torre, I., and Luther, P. K. (2014). Structure, sarcomeric organization, and thin filament binding of cardiac myosin-binding protein-C. Pflugers Arch. 466, 425–431. doi: 10.1007/s00424-013-1426-6
Cummins, P., and Lambert, S. J. (1986). Myosin transitions in the bovine and human heart. A developmental and anatomical study of heavy and light chain subunits in the atrium and ventricle. Circ. Res. 58, 846–858.
Cummins, P., Price, K. M., and Littler, W. A. (1980). Foetal myosin light chain in human ventricle. J. Muscle Res. Cell Motil. 1, 357–366. doi: 10.1007/BF00711936
Elhamine, F., Iorga, B., Krüger, M., Hunger, M., Eckhardt, J., Sreeram, N., et al. (2016). Postnatal development of right ventricular myofibrillar biomechanics in relation to the sarcomeric protein phenotype in pediatric patients with conotruncal heart defects. J. Am. Heart Assoc. 5:e003699. doi: 10.1161/JAHA.116.003699
Fatkin, D., Seidman, C. E., and Seidman, J. G. (2014). Genetics and disease of ventricular muscle. Cold Spring Harb. Perspect. Med. 4:a021063. doi: 10.1101/cshperspect.a021063
Fijnvandraat, A. C., van Ginneken, A. C., Schumacher, C. A., Boheler, K. R., Lekanne Deprez, R. H., Christoffels, V. M., et al. (2003). Cardiomyocytes purified from differentiated embryonic stem cells exhibit characteristics of early chamber myocardium. J. Mol. Cell. Cardiol. 35, 1461–1472. doi: 10.1016/j.yjmcc.2003.09.011
Gao, L., Kennedy, J. M., and Solaro, R. J. (1995). Differential expression of TnI and TnT isoforms in rabbit heart during the perinatal period and during cardiovascular stress. J. Mol. Cell. Cardiol. 27, 541–550. doi: 10.1016/S0022-2828(08)80049-1
Gautel, M., Fürst, D. O., Cocco, A., and Schiaffino, S. (1998). Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: lack of isoform transcomplementation in cardiac muscle. Circ. Res. 82, 124–129. doi: 10.1161/01.RES.82.1.124
Gomes, A. V., Guzman, G., Zhao, J., and Potter, J. D. (2002). Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J. Biol. Chem. 277, 35341–35349. doi: 10.1074/jbc.M204118200
Hernandez, O. M., Jones, M., Guzman, G., and Szczesna-Cordary, D. (2007). Myosin essential light chain in health and disease. Am. J. Physiol. Heart Circ. Physiol. 292, H1643–H1654. doi: 10.1152/ajpheart.00931.2006
Hinson, J. T., Chopra, A., Nafissi, N., Polacheck, W. J., Benson, C. C., Swist, S., et al. (2015). HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. doi: 10.1126/science.aaa5458
Huxley, A. F. (1957). Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7, 255–318.
Jagatheesan, G., Rajan, S., Ahmed, R. P., Petrashevskaya, N., Boivin, G., Arteaga, G. M., et al. (2010). Striated muscle tropomyosin isoforms differentially regulate cardiac performance and myofilament calcium sensitivity. J. Muscle Res. Cell Motil. 31, 227–239. doi: 10.1007/s10974-010-9228-3
Jung, G., and Bernstein, D. (2014). hiPSC Modeling of Inherited Cardiomyopathies. Curr. Treat. Options Cardiovasc. Med. 16:320. doi: 10.1007/s11936-014-0320-7
Kamdar, F., Klaassen Kamdar, A., Koyano-Nakagawa, N., Garry, M. G., and Garry, D. J. (2015). Cardiomyopathy in a dish: using human inducible pluripotent stem cells to model inherited cardiomyopathies. J. Card. Fail. 21, 761–770. doi: 10.1016/j.cardfail.2015.04.010
Kempf, H., Andree, B., and Zweigerdt, R. (2016a). Large-scale production of human pluripotent stem cell derived cardiomyocytes. Adv. Drug Deliv. Rev. 96, 18–30. doi: 10.1016/j.addr.2015.11.016
Kempf, H., Lecina, M., Ting, S., Zweigerdt, R., and Oh, S. (2011). Distinct regulation of mitogen-activated protein kinase activities is coupled with enhanced cardiac differentiation of human embryonic stem cells. Stem Cell Res. 7, 198–209. doi: 10.1016/j.scr.2011.06.001
Kempf, H., Olmer, R., Haase, A., Franke, A., Bolesani, E., Schwanke, K., et al. (2016b). Bulk cell density and Wnt/TGFbeta signalling regulate mesendodermal patterning of human pluripotent stem cells. Nat. Commun. 7:13602. doi: 10.1038/ncomms13602
Kempf, H., Olmer, R., Kropp, C., Rückert, M., Jara-Avaca, M., Robles-Diaz, D., et al. (2014). Controlling expansion and cardiomyogenic differentiation of human pluripotent stem cells in scalable suspension culture. Stem Cell Rep. 3, 1132–1146. doi: 10.1016/j.stemcr.2014.09.017
Kensah, G., Roa Lara, A., Dahlmann, J., Zweigerdt, R., Schwanke, K., Hegermann, J., et al. (2013). Murine and human pluripotent stem cell-derived cardiac bodies form contractile myocardial tissue in vitro. Eur. Heart J. 34, 1134–1146. doi: 10.1093/eurheartj/ehs349
Kraft, T., Witjas-Paalberends, E. R., Boontje, N. M., Tripathi, S., Brandis, A., Montag, J., et al. (2013). Familial hypertrophic cardiomyopathy: functional effects of myosin mutation R723G in cardiomyocytes. J. Mol. Cell. Cardiol. 57, 13–22. doi: 10.1016/j.yjmcc.2013.01.001
Kruger, M., and Linke, W. A. (2009). Titin-based mechanical signalling in normal and failing myocardium. J. Mol. Cell. Cardiol. 46, 490–498. doi: 10.1016/j.yjmcc.2009.01.004
MacColl, C. E., Manlhiot, C., Page, C., McCrindle, B. W., Miner, S. E., Jaeggi, E. T., et al. (2014). Factors associated with in utero demise of fetuses that have underlying cardiac pathologies. Pediatr. Cardiol. 35, 1403–1414. doi: 10.1007/s00246-014-0943-1
Marston, S. B., Copeland, O., Messer, A. E., MacNamara, E., Nowak, K., Zampronio, C. G., et al. (2013). Tropomyosin isoform expression and phosphorylation in the human heart in health and disease. J. Muscle Res. Cell Motil. 34, 189–197. doi: 10.1007/s10974-013-9347-8
Metzger, J. M., Lin, W. I., and Samuelson, L. C. (1994). Transition in cardiac contractile sensitivity to calcium during the in vitro differentiation of mouse embryonic stem cells. J. Cell Biol. 126, 701–711. doi: 10.1083/jcb.126.3.701
Metzger, J. M., Michele, D. E., Rust, E. M., Borton, A. R., and Westfall, M. V. (2003). Sarcomere thin filament regulatory isoforms. Evidence of a dominant effect of slow skeletal troponin I on cardiac contraction. J. Biol. Chem. 278, 13118–13123. doi: 10.1074/jbc.M212601200
Miyata, S., Minobe, W., Bristow, M. R., and Leinwand, L. A. (2000). Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 86, 386–390. doi: 10.1161/01.RES.86.4.386
Mongiovì, M., Fesslova, V., Fazio, G., Barbaro, G., and Pipitone, S. (2010). Diagnosis and prognosis of fetal cardiomyopathies: a review. Curr. Pharm. Des. 16, 2929–2934. doi: 10.2174/138161210793176428
Morano, I., and Haase, H. (1997). Different actin affinities of human cardiac essential myosin light chain isoforms. FEBS Lett. 408, 71–74. doi: 10.1016/S0014-5793(97)00390-6
Moretti, A., Laugwitz, K. L., Dorn, T., Sinnecker, D., and Mummery, C. (2013). Pluripotent stem cell models of human heart disease. Cold Spring Harb. Perspect. Med. 3:a014027. doi: 10.1101/cshperspect.a014027
Murry, C. E., and Keller, G. (2008). Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132, 661–680. doi: 10.1016/j.cell.2008.02.008
Muthuchamy, M., Pajak, L., Howles, P., Doetschman, T., and Wieczorek, D. F. (1993). Developmental analysis of tropomyosin gene expression in embryonic stem cells and mouse embryos. Mol. Cell. Biol. 13, 3311–3323. doi: 10.1128/MCB.13.6.3311
Pieples, K., Arteaga, G., Solaro, R. J., Grupp, I., Lorenz, J. N., Boivin, G. P., et al. (2002). Tropomyosin 3 expression leads to hypercontractility and attenuates myofilament length-dependent Ca2+ activation. Am. J. Physiol. Heart Circ. Physiol. 283, H1344–H1353. doi: 10.1152/ajpheart.00351.2002
Pinto, J. R., Gomes, A. V., Jones, M. A., Liang, J., Nguyen, S., Miller, T., et al. (2012). The functional properties of human slow skeletal troponin T isoforms in cardiac muscle regulation. J. Biol. Chem. 287, 37362–37370. doi: 10.1074/jbc.M112.364927
Pioner, J. M., Racca, A. W., Klaiman, J. M., Yang, K. C., Guan, X., Pabon, L., et al. (2016). Isolation and mechanical measurements of myofibrils from human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rep. 6, 885–896. doi: 10.1016/j.stemcr.2016.04.006
Piroddi, N., Belus, A., Scellini, B., Tesi, C., Giunti, G., Cerbai, E., et al. (2007). Tension generation and relaxation in single myofibrils from human atrial and ventricular myocardium. Pflugers Arch. 454, 63–73. doi: 10.1007/s00424-006-0181-3
Poggesi, C., Tesi, C., and Stehle, R. (2005). Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch. 449, 505–517. doi: 10.1007/s00424-004-1363-5
Racca, A. W., Klaiman, J. M., Pioner, J. M., Cheng, Y., Beck, A. E., Moussavi-Harami, F., et al. (2016). Contractile properties of developing human fetal cardiac muscle. J. Physiol. 594, 437–452. doi: 10.1113/JP271290
Rajan, S., Jagatheesan, G., Karam, C. N., Alves, M. L., Bodi, I., Schwartz, A., et al. (2010). Molecular and functional characterization of a novel cardiac-specific human tropomyosin isoform. Circulation 121, 410–418. doi: 10.1161/CIRCULATIONAHA.109.889725
Rarick, H. M., Opgenorth, T. J., von Geldern, T. W., Wu-Wong, J. R., and Solaro, R. J. (1996). An essential myosin light chain peptide induces supramaximal stimulation of cardiac myofibrillar ATPase activity. J. Biol. Chem. 271, 27039–27043. doi: 10.1074/jbc.271.43.27039
Reiser, P. J., Portman, M. A., Ning, X. H., and Schomisch Moravec, C. (2001). Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 280, H1814–H1820. doi: 10.1152/ajpheart.2001.280.4.H1814
Riedel, B., Jia, Y., Du, J., Akerman, S., and Huang, X. (2005). Thyroid hormone inhibits slow skeletal TnI expression in cardiac TnI-null myocardial cells. Tissue Cell 37, 47–51. doi: 10.1016/j.tice.2004.10.002
Schaub, M. C., Hefti, M. A., Zuellig, R. A., and Morano, I. (1998). Modulation of contractility in human cardiac hypertrophy by myosin essential light chain isoforms. Cardiovasc. Res. 37, 381–404. doi: 10.1016/S0008-6363(97)00258-7
Siedner, S., Krüger, M., Schroeter, M., Metzler, D., Roell, W., Fleischmann, B. K., et al. (2003). Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J. Physiol. 548(Pt 2), 493–505. doi: 10.1113/jphysiol.2002.036509
Stehle, R., and Iorga, B. (2010). Kinetics of cardiac sarcomeric processes and rate-limiting steps in contraction and relaxation. J. Mol. Cell. Cardiol. 48, 843–850. doi: 10.1016/j.yjmcc.2009.12.020
Stehle, R., Krüger, M., and Pfitzer, G. (2002a). Force kinetics and individual sarcomere dynamics in cardiac myofibrils after rapid Ca2+ changes. Biophys. J. 83, 2152–2161. doi: 10.1016/S0006-3495(02)73975-1
Stehle, R., Krüger, M., and Pfitzer, G. (2003). “Does cross-bridge activation determine the time course of myofibrillar relaxation?,” in Molecular and Cellular Aspects of Muscle Contraction. Advances in Experimental Medicine and Biology, Vol. 538, ed H. Sugi (Boston, MA: Springer), 469–479. doi: 10.1007/978-1-4419-9029-7_43
Stehle, R., Krüger, M., Scherer, P., Brixius, K., Schwinger, R. H. G., and Pfitzer, G. (2002b). Isometric force kinetics upon rapid activation and relaxation of mouse, guinea pig and human heart muscle studied on the subcellular myofibrillar level. Basic Res. Cardiol. 97(Suppl. 1), I127–I135. doi: 10.1007/s003950200041
Stehle, R., Solzin, J., Iorga, B., and Poggesi, C. (2009). Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch. 458, 337–357. doi: 10.1007/s00424-008-0630-2
Stelzer, J. E., Patel, J. R., and Moss, R. L. (2006). Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ. Res. 99, 884–890. doi: 10.1161/01.RES.0000245191.34690.66
Telley, I. A., Denoth, J., Stüssi, E., Pfitzer, G., and Stehle, R. (2006). Half-sarcomere dynamics in myofibrils during activation and relaxation studied by tracking fluorescent markers. Biophys. J. 90, 514–530. doi: 10.1529/biophysj.105.070334
van der Velden, J., Papp, Z., Boontje, N. M., Zaremba, R., de Jong, J. W., Janssen, P. M. L., et al. (2003a). The effect of myosin light chain 2 dephosphorylation on Ca2+ -sensitivity of force is enhanced in failing human hearts. Cardiovasc. Res. 57, 505–514. doi: 10.1016/S0008-6363(02)00662-4
van der Velden, J., Papp, Z., Zaremba, R., Boontje, N. M., de Jong, J. W., Owen, V. J., et al. (2003b). Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 57, 37–47. doi: 10.1016/S0008-6363(02)00606-5
van Dijk, S. J., Boontje, N. M., Heymans, M. W., Ten Cate, F. J., Michels, M., Dos Remedios, C., et al. (2014). Preserved cross-bridge kinetics in human hypertrophic cardiomyopathy patients with MYBPC3 mutations. Pflugers Arch. 466, 1619–1633. doi: 10.1007/s00424-013-1391-0
Vestergaard, M. L., Grubb, S. J., Rasmussen, K. K., Anderson-Jenkins, Z., Grunnet-Lauridsen, K., Calloe, K., et al. (2017). Human embryonic stem cell-derived Cardiomyocytes self-arrange with areas of different subtypes during differentiation. Stem Cells Dev. 26, 1566–1577. doi: 10.1089/scd.2017.0054
Walker, J. S., Walker, L. A., Margulies, K., Buttrick, P., and de Tombe, P. (2011). Protein kinase A changes calcium sensitivity but not crossbridge kinetics in human cardiac myofibrils. Am. J. Physiol. Heart Circ. Physiol. 301, H138–H146. doi: 10.1152/ajpheart.00838.2010
Weber, N., Schwanke, K., Greten, S., Wendland, M., Iorga, B., Fischer, M., et al. (2016). Stiff matrix induces switch to pure beta-cardiac myosin heavy chain expression in human ESC-derived cardiomyocytes. Basic Res. Cardiol. 111:68. doi: 10.1007/s00395-016-0587-9
Wei, B., and Jin, J. P. (2011). Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch. Biochem. Biophys. 505, 144–154. doi: 10.1016/j.abb.2010.10.013
Xu, X. Q., Soo, S. Y., Sun, W., and Zweigerdt, R. (2009). Global expression profile of highly enriched cardiomyocytes derived from human embryonic stem cells. Stem Cells 27, 2163–2174. doi: 10.1002/stem.166
Yang, X., Pabon, L., and Murry, C. E. (2014a). Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 114, 511–523. doi: 10.1161/CIRCRESAHA.114.300558
Yang, X., Rodriguez, M., Pabon, L., Fischer, K. A., Reinecke, H., Regnier, M., et al. (2014b). Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 72, 296–304. doi: 10.1016/j.yjmcc.2014.04.005
Keywords: human pluripotent stem cell-derived cardiomyocytes, human embryonic stem cell-derived cardiomyocytes, hiPSC-CMs maturation, human cardiomyocytes development, adult human ventricular myofibrils, cardiac contractile function, β-myosin isoform cross-bridge, cross-bridge kinetics
Citation: Iorga B, Schwanke K, Weber N, Wendland M, Greten S, Piep B, dos Remedios CG, Martin U, Zweigerdt R, Kraft T and Brenner B (2018) Differences in Contractile Function of Myofibrils within Human Embryonic Stem Cell-Derived Cardiomyocytes vs. Adult Ventricular Myofibrils Are Related to Distinct Sarcomeric Protein Isoforms. Front. Physiol. 8:1111. doi: 10.3389/fphys.2017.01111
Received: 14 October 2017; Accepted: 15 December 2017;
Published: 19 January 2018.
Edited by:
Kenneth S. Campbell, University of Kentucky, United StatesReviewed by:
Laurin Michelle Hanft, University of Missouri, United StatesCorrado Poggesi, University of Florence, Italy
Ranganath Mamidi, Case Western Reserve University, United States
Copyright © 2018 Iorga, Schwanke, Weber, Wendland, Greten, Piep, dos Remedios, Martin, Zweigerdt, Kraft and Brenner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bogdan Iorga, aW9yZ2EuYm9nZGFuQG1oLWhhbm5vdmVyLmRl