 Haibo Ni
Haibo Ni- 1Biological Physics Group, University of Manchester, Manchester, United Kingdom
- 2Space Institute of Southern China, Shenzhen, China
- 3Key Laboratory of Medical Electrophysiology, Ministry of Education, Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease/Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
- 4Faculties of Kinesiology and Medicine, University of Calgary, Calgary, AB, Canada
- 5Department of Medicine, Stanford University School of Medicine, Stanford, CA, United States
- 6School of Computer Science and Technology, Harbin Institute of Technology, Harbin, China
Atrial fibrillation (AF) is the most common cardiac arrhythmia. Developing effective and safe anti-AF drugs remains an unmet challenge. Simultaneous block of both atrial-specific ultra-rapid delayed rectifier potassium (K+) current (IKur) and the Na+ current (INa) has been hypothesized to be anti-AF, without inducing significant QT prolongation and ventricular side effects. However, the antiarrhythmic advantage of simultaneously blocking these two channels vs. individual block in the setting of AF-induced electrical remodeling remains to be documented. Furthermore, many IKur blockers such as acacetin and AVE0118, partially inhibit other K+ currents in the atria. Whether this multi-K+-block produces greater anti-AF effects compared with selective IKur-block has not been fully understood. The aim of this study was to use computer models to (i) assess the impact of multi-K+-block as exhibited by many IKur blokers, and (ii) evaluate the antiarrhythmic effect of blocking IKur and INa, either alone or in combination, on atrial and ventricular electrical excitation and recovery in the setting of AF-induced electrical-remodeling. Contemporary mathematical models of human atrial and ventricular cells were modified to incorporate dose-dependent actions of acacetin (a multichannel blocker primarily inhibiting IKur while less potently blocking Ito, IKr, and IKs). Rate- and atrial-selective inhibition of INa was also incorporated into the models. These single myocyte models were then incorporated into multicellular two-dimensional (2D) and three-dimensional (3D) anatomical models of the human atria. As expected, application of IKur blocker produced pronounced action potential duration (APD) prolongation in atrial myocytes. Furthermore, combined multiple K+-channel block that mimicked the effects of acacetin exhibited synergistic APD prolongations. Synergistically anti-AF effects following inhibition of INa and combined IKur/K+-channels were also observed. The attainable maximal AF-selectivity of INa inhibition was greatly augmented by blocking IKur or multiple K+-currents in the atrial myocytes. This enhanced anti-arrhythmic effects of combined block of Na+- and K+-channels were also seen in 2D and 3D simulations; specially, there was an enhanced efficacy in terminating re-entrant excitation waves, exerting improved antiarrhythmic effects in the human atria as compared to a single-channel block. However, in the human ventricular myocytes and tissue, cellular repolarization and computed QT intervals were modestly affected in the presence of actions of acacetin and INa blockers (either alone or in combination). In conclusion, this study demonstrates synergistic antiarrhythmic benefits of combined block of IKur and INa, as well as those of INa and combined multi K+-current block of acacetin, without significant alterations of ventricular repolarization and QT intervals. This approach may be a valuable strategy for the treatment of AF.
Introduction
Despite recent advances in the management of Atrial fibrillation (AF), the world's most common cardiac arrhythmia (Dobrev et al., 2012; Nattel and Dobrev, 2017), developing effective and safe antiarrhythmic drugs for treatment of AF remains challenging (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015). Frequently these antiarrhythmic agents promote ventricular arrhythmias (Dobrev et al., 2012; Woods and Olgin, 2014; Voigt and Dobrev, 2016) by prolonging cellular action potential durations (APDs). The associated QT-interval prolongation can lead to life-threatening consequences. Developing atrial-selective drugs is acknowledged to be a current strategy for the treatment of AF (Burashnikov et al., 2007).
Atrial and ventricular tissues show intrinsic regional differences in their cellular ion channel properties, thus suggesting a basis for developing atrial-selective drugs. For example, the atrial and ventricular fast sodium (Na+) channel currents (INa) exhibit different voltage-dependent inactivation properties, opening the opportunity for atrial-selective Na+ channel blockade (Burashnikov et al., 2007; Antzelevitch and Burashnikov, 2009; Zygmunt et al., 2011). Previous simulation studies have demonstrated that by optimizing state-dependent Na+-channel blocking dynamics (i.e., drug-channel interaction parameters), atrial-selective block of INa could be achieved and that could maximize pharmaceutical effects on the atria while minimizing their proarrhythmic actions in the ventricles (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015).
Another tissue-specific difference between the atria and ventricles is that the ultra-rapid delayed rectifier potassium current (IKur, carried by the KV1.5 channel) contributes to repolarization in the atria but plays little role in the ventricles (Tamargo et al., 2009; Ravens and Wettwer, 2011). Recent studies suggest that atrial-selective blockade of IKur may be an effective pharmacological treatment of AF (Li et al., 2008; Pavri et al., 2012; Loose et al., 2014; Ford et al., 2016). Although the efficacy of IKur block in the treatment of AF remains controversial (Burashnikov and Antzelevitch, 2008), multiple IKur blockers have been developed (Tamargo et al., 2009; Loose et al., 2014; Wettwer and Terlau, 2014; Ford et al., 2016). Interestingly, these IKur blockers actually target multiple channels, and are known to inhibit other K+ currents including Ito and IK, ACh in the atria (Burashnikov and Antzelevitch, 2008). Examples of such blockers include AVE0118 (Gögelein et al., 2004), AVE1231 (Wirth et al., 2007), AZD7009 (Persson et al., 2005), and acacetin (Li et al., 2008). Among these channel blockers, acacetin, a natural flavone initially isolated from a traditional Chinese medicine Xuelianhua, potently blocks IKur, Ito, and IK,ACh, and has a smaller potency in inhibiting IKr and IKs (Li et al., 2008), similar to AVE0118 (Gögelein et al., 2004; Haan et al., 2006). Acacetin is regarded as a promising atrial-selective agent for the treatment of AF (Li et al., 2008). However, the actions of acacetin on atrial electrophysiology, especially its effects following AF-induced electrical remodeling of atrial electrophysiological properties (Dobrev et al., 2012), remain to be elucidated. Furthermore, since most IKur blockers inhibit other K+ channels, the question whether the “additional” inhibitive actions produce favorable antiarrhythmic effects has not been addressed thoroughly. A better understanding of these effects of modulating multiple ion channels on atrial excitation and recovery/repolarization may provide insights into evaluating and developing antiarrhythmic drugs.
Interestingly, simultaneous multiple-channel blocking of both depolarization and repolarization currents is attracting more attention since empirical observations suggest that such multi-channel blockers generally mediate more effective antiarrhythmic effects (Kirchhoff et al., 2015; Reiffel et al., 2015; Hartmann et al., 2016). A recent numerical and experimental study on the canine heart (Aguilar et al., 2015) suggested that blocking K+ currents enhanced the anti-arrhythmic effects and AF-selectivity of INa blockade. In their study, IKur block was modeled using a simple pore block scheme by reducing the conductance of the channel. As the kinetics of drug action plays an important role in the effects of IKur blockers (Scholz et al., 2013; Ellinwood et al., 2017), in simulating IKur block a state-dependent block model reproducing a realistic blocker is more favorable. Once again, the effects of combined INa and IKur block on the human atria, especially in the setting of AF-induced electrical remodeling which reduced IKur, remain to be elucidated. It is also unclear how multiple-channel blockade may affect QT interval.
In the present study, it was hypothesized that combined block of INa and K+-currents (predominantly IKur) could produce antiarrhythmic benefits compared with the application of either blocker alone in the setting of AF-induced electrical remodeling. We have tested the hypothesis with the following three aims: (i) to identify and illustrate the effects of the realistic IKur blocker, acacetin, on atrial electrophysiology following AF-related remodeling; (ii) to assess whether combined INa and IKur block produce synergistic antiarrhythmic effects; and (iii) to investigate the action of such drug combinations on ventricular electrophysiology.
Methods
Modeling Electrophysiology of the Human Heart
To simulate human atrial electrophysiology, an updated Colman et al. model for atrial electrophysiology (Colman et al., 2013, 2017) was used. For in silico study of effects of chronic AF- (cAF) induced electrophysiological remodeling on the atria, we incorporated the cAF model parameters from our previous study (Colman et al., 2013) into the updated atrial single cell model (for details please see Online Supplement Material 1.1).
To assess the effects of the anti-AF drugs on the human ventricles, simulations were performed to investigate the actions of the anti-AF drugs on the ventricular AP, INa and QT intervals in the electrograms. In these simulations, the mathematical model developed by O'Hara et al. (2011) was used to represent the ventricular electrophysiology. Additionally, the INa formulation in the model was replaced by the one in the Luo–Rudy model (Luo and Rudy, 1994), which enabled electrical excitation to propagate in the tissue model.
More detailed descriptions of the electrophysiological models of human atrial and ventricular cells are given in Online Supplementary Material 1.1.
Modeling State-Dependent INa Block
As in previous studies (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015), INa block was simulated using a guarded receptor model with dynamical drug-channel interactions. This approach allows for investigations of the role of the specified parameters for selected INa blockers, and effects of combined IKur block on the atrial selectivity of Na+-channel block. The guarded receptor model considers the binding and unbinding kinetics of the drug to INa channels in a drug concentration-dependent manner. They can be described by first-order transition equations (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015). It was also assumed that the drug predominantly binds to the activated and/or inactivated states of INa. The blockade of INa is given by Aguilar-Shardonofsky et al. (2012) and Aguilar et al. (2015):
where gNa is the maximum conductance of INa; BA and BI are the fractional blockade of activation and inactivation channels; m is the activation gate state variable, h and j are the inactivation gate state variables; Vm the transmembrane potential; ENa the reversal potential of Na+; KA, KI the binding constants and LA, LI the unbinding constants; [DNa+] is the concentration of a Na+-blocker. As in previous studies (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015), a concentration of 60 μM was utilized unless otherwise stated; this concentration was chosen based on previous experimental and modeling studies (Zhu et al., 2006; Moreno et al., 2011; Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015); a parameter set (KA = 100 ms−1· M−1, KI = 100 ms−1· M−1, LA = 1 ms−1, LI = 0.01 ms−1) was first used to represent the kinetics of an INa-selective blocker.
In our investigations of the AF-selectivity of INa block following AF-remodeling, the binding and unbinding constants of the INa blockers were varied to evaluate the dependence of INa block on these parameters, and whether an atrial-selective anti-AF action could be achieved in cAF-remodeled myoctes. The AF-selectivity of Na+-channel blockade was defined as the product of atrial-selectivity, rate-selectivity and block efficacy. With fractional block (Bf) by Na+-channel blockers being measured as the relative reduction in the peak of INa, the rate-selectivity was defined as the ratio of Bf measured in an atrial myocyte paced at 6 Hz to that paced at 1 Hz (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015). Atrial-selectivity was used to determine the extent of atrial-ventricular difference in response to each drug. This was represented by the ratio of Bf observed from an atrial myocyte to that of a ventricular cell both paced at 1 Hz. In this study, we defined block efficacy (E) as:
where Bf,6Hz is the fractional block of INa measured in an atrial cell paced at 6 Hz. Different from Aguilar et al. (2015), we introduced block efficacy to constrain the measure of AF-selectivity when the fractional block observed in a ventricular cell paced was minimal (and could result in a great atrial-selectivity), which otherwise could give a great value in AF-selectivity regardless of a small Bf,6Hz.
To assess the dependence of the AF-selectivity of INa block on the drug action kinetics, the unbinding constants LA and LI were first varied over a parameter space from 10−5 to 100 ms−1, while KA and KI were fixed (see Figure S6 of online Supplementary Materials for more details). The resultant unbinding constants were used in subsequent optimizations varying KA and KI. The parameter space was {1, 10, 100, 500, 2,500, 10,000} for KA and {1, 10, 100, 200, 500, 2,500} for KI. The parameter space fell into a likely range of INa blockers as summarized in Aguilar-Shardonofsky et al. (2012).
Modeling Effects of Acacetin on Atrial and Ventricular Electrophysiology
Acacetin was the chosen IKur blocker in the present study. To reveal the functional effects of (i) pure IKur block vs. (ii) the effects of combined K+ currents block by acacetin on human atrial electrophysiology, the actions of acacetin were modeled by considering its effects on (a) IKur only, and (b) all the respective K+ currents as detailed in Table 1. This approach allows for modeling the effects of the selective IKur block as well as uncovering the role of “additional” inhibitory effects of acacetin on other K+ currents.
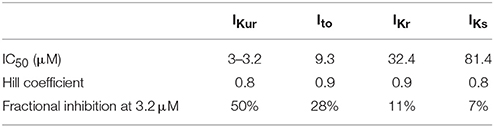
Table 1. Concentration-dependent block of K+-currents by acacetin (Li et al., 2008; Wu et al., 2011).
Modeling Effect of Acacetin on IKur
Previous modeling studies have demonstrated the important role of the kinetic properties of drug actions in IKur block (Tsujimae et al., 2008; Almquist et al., 2010; Scholz et al., 2013). In addition, the pharmaceutical effects of acacetin on IKur are characterized by use- and rate-dependencies (Wu et al., 2011), which have also been observed in other IKur blockers (Pavri et al., 2012; Ford et al., 2016). Therefore, it was necessary to adopt a state-dependent block model (Brennan et al., 2009) for simulating the blockade of IKur by acacetin. Similar to our approach for modeling INa block, the binding and unbinding kinetics of a drug was described by a first-order transition equation, in contrast to simulating IKur block by reducing its conductance in Aguilar et al. (2015). Experimental studies revealed that acacetin binds to both the open and closed gates of KV1.5 (Wu et al., 2011). Therefore, following the guarded receptor formulas given in Equations (1–3), the formulation of inactivation-state binding and unbinding kinetics in INa block was modified to simulate the closed-state block of IKur by acacetin. The guarded receptor model of IKur block by acacetin is given by:
where gKur is the conductance of IKur; Bo and BC are the fractional block on open and closed state variables, respectively; a and i are the activation and inactivation gate variables; EK is the reversal potential of potassium; F, R, and T are the Faraday's constant, universal gas constant and temperature respectively. KO and Kc are the binding constants; LO and Lc are the unbinding constants; ZKO, ZLO, ZKc and ZLc are the drug charge parameters for the corresponding binding or unbinding processes; [DK+] is the concentration of acacetin applied. The binding and unbinding parameters were obtained by fitting the model to the experimental data on the rate-dependent blockade of IKur by acacetin (Wu et al., 2011), as detailed in Online Supplementary Material 1.1.
Figure 1 shows a simulated frequency-dependent block of IKur by acacetin, and this is compared to the experimental data (Figures 1A,B). As shown, repeating the voltage command (Figure 1B, insert) at 0.5 Hz resulted in an approximately 50% blockade in this current after application of 3 μM acacetin. Increasing the voltage command rate to 4 Hz significantly increased the relative fractional block to approximately 63% (Figure 1C).
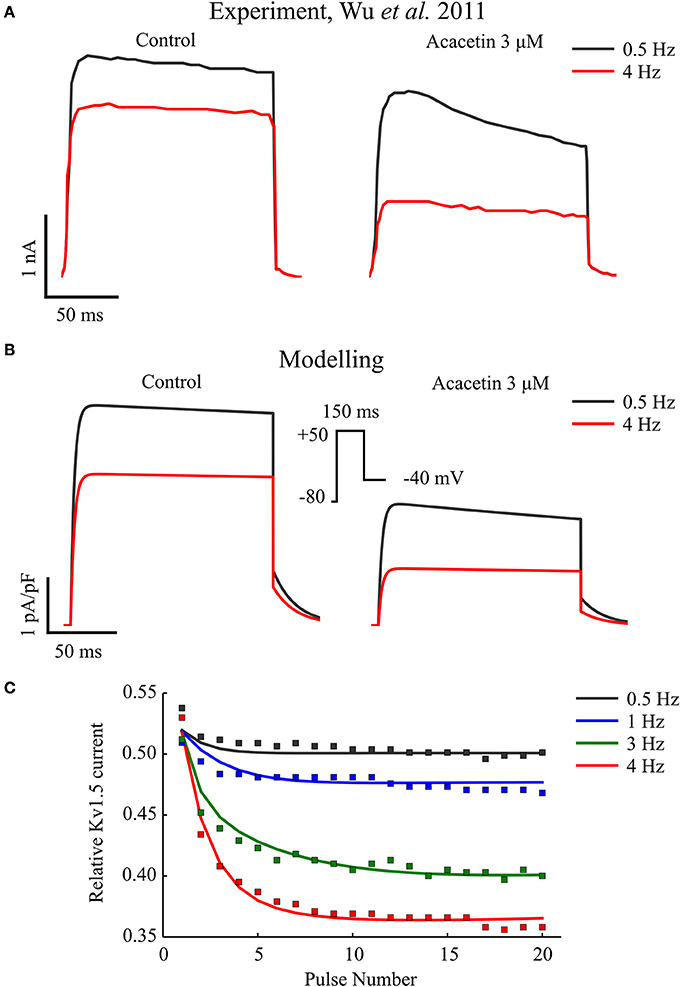
Figure 1. Frequency-dependent inhibition of IKur by acacetin. (A) Experimental and (B) simulated traces of KV1.5 channel current elicited from the 20th voltage step repeating at 0.5 and 4 Hz in control (left) and after exposure to 3 μM of acacetin (right). (C) Relative remaining IKur following application of acacetin at various frequencies plotted against the pulse number of the voltage step. The simulated data (lines) were compared with experimental values (squares). The relative fraction was obtained by normalizing the end-step current measured from each pulse following application of acacetin to that of control. Experimental data were digitalized from Wu et al. (2011).
Modeling Effect of Acacetin on Ito, IKr, and IKs
In addition to inhibiting IKur in the atria, acacetin potently blocks both Ito and IK,ACh, and also modulates IKr and IKs, exhibiting multiple K+-current block. The parameters of Hill equations describing use-dependent inhibitions of these channels by acacetin are shown in Table 1. In the simulations, the effects of acacetin on these channels were modeled using a simple pore block model (Yuan et al., 2015). In the present study, we did not simulate the effects of acacetin on IK,ACh inhibitions as the role of autonomic regulation on AF is beyond the scope of the study.
Simulations of the Effects of Acacetin on Human Ventricle
The effects of acacetin on human ventricular APs are unknown, although experimental data demonstrated that acacetin at 30 μM did not affect the heart rate and QT interval in isolated rabbit hearts (Li et al., 2008). In the present study, it was assumed that similar effects on the K+ currents (Ito, IKr, IKs) in atrial myocytes could be extrapolated to the ventricular myocytes. We acknowledge that IKur is negligible in ventricles (Ravens and Wettwer, 2011), therefore in simulations of blocking IKur alone, the ventricular electrophysiology was not affected.
Tissue Models
The effects of acacetin and INa blockers on atrial and ventricular electrophysiology were further evaluated using tissue models. The monodomain equation (Clayton et al., 2011) was employed to simulate the excitation wave propagation in the myocardium. 1D models of human atrial strands were used to quantify the effects of channel blockers on atrial conduction velocity and APD restitution properties. Changes in ventricular depolarization and repolarization in response to these drugs were evaluated using a 1D model representing a transmural strand of ventricular tissue. In order to evaluate the antiarrhythmic effects of the channel blockers on re-entrant excitations in atria in the setting of cAF-induced remodeling, both idealized 2D models representing an isotropic slab of atrial tissue and an anatomically accurate 3D model of the human atria (Aslanidi et al., 2011; Colman et al., 2013, 2017; Whittaker et al., 2017) were employed to simulate the behavior of re-entrant excitations in atrial tissue. Pseudo-ECGs (pECGs) (Gima and Rudy, 2002; Baher et al., 2006) were computed as a measure of the excitation rates of in-tissue with sprial excitation waves. Detailed descriptions of these tissue models and pECGs are given in Online Supplementary Material 1.2, 1.3.
Results
The updated Colman et al. human atrial model was first used to simulate effects cAF-induced remodeling on the action potential (AP) and calcium transient (CaT). Details are presented in Online Supplementary Material 2.1. The resultant changes in APD, APD restitution and CaT following cAF-induced remodeling as compared to those under the normal condition showed good agreement with previous experimental (Bosch et al., 1999; Osaka et al., 2000; Workman et al., 2001; Dobrev and Ravens, 2003; Voigt et al., 2012) and simulation studies (Zhang et al., 2005; Grandi et al., 2011; Colman et al., 2013, 2017; Wilhelms et al., 2013).
Effects of Application of Acacetin on Human Atrial Cells
To reveal the roles of inhibition of individual channels by acacetin in modulating cellular AP by acacetin, both the individual and combined block of Ito, IKr, IKur, and IKs by acacetin (3.2 μM) in simulated SR at cycle length 1,000 ms without (normal) or with cAF-related electrical remodeling were simulated. Figures 2A,B illustrates the effects of individual and combined K+-channel block by acacetin on AP waveform. The alterations to APD relative to the control are summarized in Figures 2C,D.
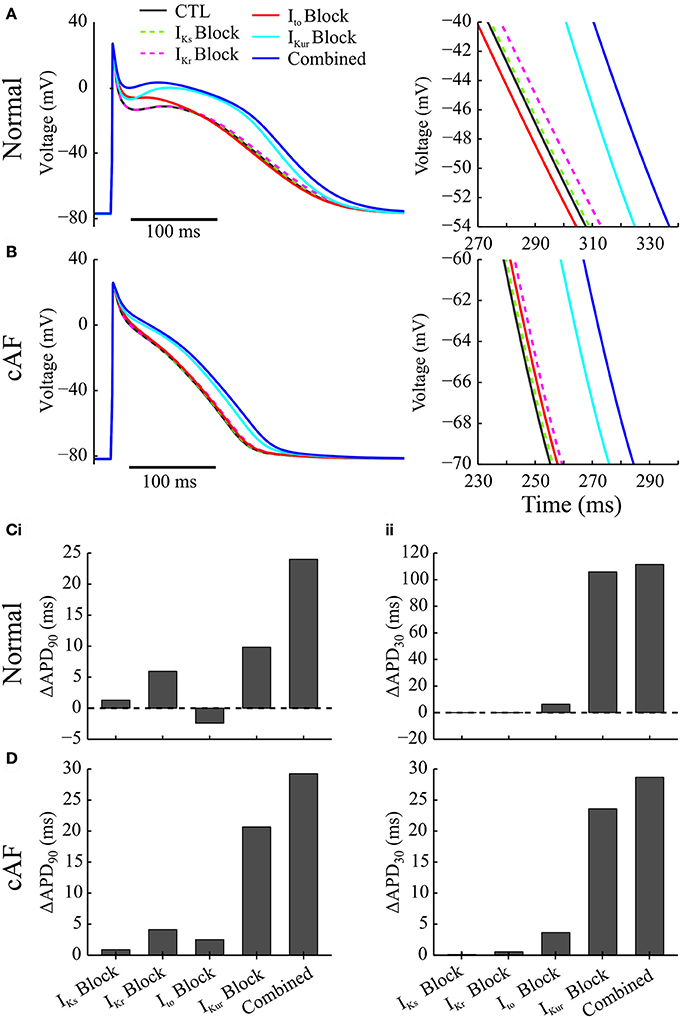
Figure 2. Effects of individual vs. combined block of K+-currents by acacetin (3.2 μM) on the human atrial AP in normal and cAF-remodeled myocytes paced at 1 Hz. (A,B) Effects on the atrial AP in (A) normal and (B) cAF-remodeled myocytes; a zoomed-in view for the traces of AP during phase-3 is plotted to the right. (C,D) Alterations in the (i) APD90 and (ii) APD30 by the simulated block obtained from atrial cells in (C) normal and (D) cAF-remodeled myocytes.
In the absence of electrical remodeling, in normal myocytes at a cycle length of 1,000 ms, simulated IKs or IKr block by acacetin (3.2 μM) presented no significant alterations to the atrial AP: although the atrial repolarization was delayed by 1.3 and 5.9 ms, respectively, the plateau phase was not affected, which is consistent with the minimal potency of acacetin on these channels (Table 1). Similar effects were also obtained from our simulated IKr and IKs block by the compound in the cAF-remodeled atrial cells.
Selective block of Ito (alone) by acacetin elevated the atrial plateau potential in both normal and cAF-remodeled myocytes paced at 1 Hz, and this led to modest prolongations in APD30 (by 6.4 and 3.6 ms for normal and AF-remodeling myocytes, respectively). The changes in APD90 due to Ito block varied between the two conditions: under normal conditions the atrial APD90 was shortened by 2.4 ms, whereas it was prolonged by 3.7 ms following cAF-remodeling at a stimulus rate of 1 Hz.
In contrast, blocking IKur alone by acacetin resulted in a pronounced alteration to the shape and duration of the AP in both normal and cAF-remodeled myocytes. The inhibition in IKur significantly elevated the plateau potential of atrial AP (by 7.1 and 5.7 mV in normal and cAF-remodeled myocytes, respectively), and this was accompanied by marked prolongations in APD30 (by 105.9 and 23.6 ms in normal and cAF-remodeled myocytes, respectively). The prolongation in APD90 induced by the IKur block was 9.8 ms for normal atrial cells, and was more pronounced (23.6 ms) in cAF-remodeled myocytes, despite that IKur was down-regulated by cAF-remodeling.
We note that combined effects of acacetin (3.2 μM) on multiple K+-currents produced greater alterations to the AP than those of any individual blocking effect. We have quantified effects produced by the combined block and compared it with the sum of the changes seen in each individual block. Synergistic effects were observed in the changes in APD90, represented by a further prolongation of 9.3 and 1.1 ms in APD90 in normal and cAF-remodeled myocytes, respectively. Additionally, the effects of IKur block dominated the AP-modulation by the compound, which is consistent with the high potency of acacetin on the channel (Table 1).
Effects of Sodium Blocker and Acacetin on cAF-Remodeled Atrial Myocytes and Ventricular Cells
Effects on Single Myocyte AP and INa
Individual and combined effects of Na+-block (indicated by Bl·INa) and K+-block by acacetin (3.2 μM) on human atrial electrophysiology after cAF-remodeling were simulated to assess any anti-AF benefits. Effects of acacetin (representing K+-block) were simulated in different settings: (i) IKur block alone (denoted by Bl·IKur) and (ii) combined block of all K+-currents in Table 1 (denoted by Comb·Bl·IX). In addition, the effects of Na+- and K+- block on human ventricular myocytes were also studied to assess the atrial-selectivity of the block. The results are shown in Figures 3A–C and quantitative measurements are shown in Figures 4A–C.
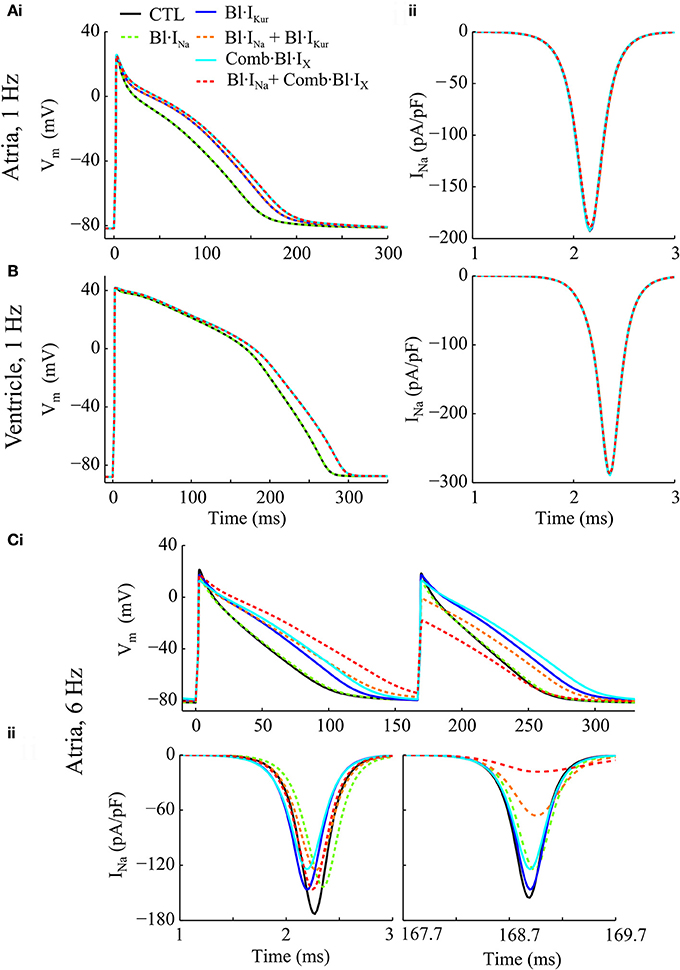
Figure 3. Simulated AP and INa traces of cAF-remodeled atrial myocytes and ventricular cells in response to Na+- and K+- block regimens. (Ai) APs from cAF-remodeled atrial myocytes paced at 1 Hz; (Aii) Time courses of corresponding INa during the upstroke phase. (B) Simulated time courses of AP and INa of a ventricular cell paced at 1 Hz. (C) Illustration of (i) APs and (ii) the corresponding time courses of INa of a cAF-remodeled myocyte paced at 6 Hz. In these simulations, rate constants for INa blocker were: KA = 100 ms−1· M−1, KI = 100 ms−1· M−1, IA = 1 ms−1, II = 0.01 ms−1.
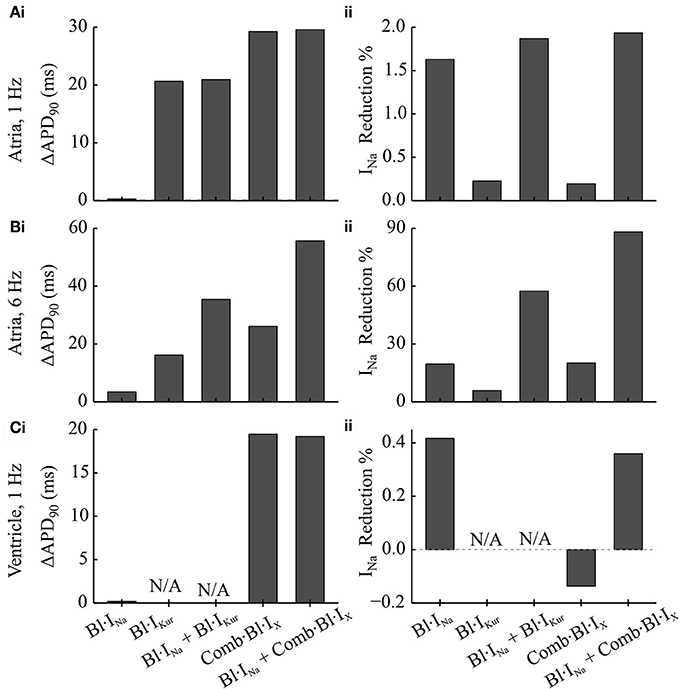
Figure 4. Simulated changes in the APD and peak INa following the applications of Na+- and K+- block in comparison to the drug-free condition in cAF-remodeled atrial myocytes or ventricular cells. (A,B) Changes in (i) APD and (ii) peak INa measured from a cAF-remodeled atrial myocyte paced at (A) 1 Hz and (B) 6 Hz. In the presence of alternans, the changes in APD were quantified by comparing the corresponding longer APs. The fractional reductions in peak INa were calculated from the INa of the corresponding shorter APs. (C) Changes in (i) APD and (ii) peak INa measured from an in silico ventricular myocyte paced at 1 Hz.
For atrial myocytes paced at 1 Hz, Bl·IKur, and Comb·Bl·IX prolonged atrial APD (Figure 4Ai and as presented in Effects of Application of Acacetin on Human Atrial Cells) whilst their effects on peak INa were minimal (reducing peak INa by less than 0.3%). Bl·INa alone slightly reduced the peak INa by 1.63% without affecting the APD. The fractional inhibition in INa by INa-block was slightly increased by the addition of Bl·IKur or Comb·Bl·IX (Figure 4Aii). In the ventricles, the simulated application of acacetin induced a prolongation of 19.5 ms in APD90 compared with that in control (drug-free) condition (Figures 3B, 4Ci). Bl·INa alone showed a negligible inhibitory effect on the ventricular INa (by 0.42%), which was also not affected by combining Bl·INa and Comb·Bl·IX (Figure 4Cii).
In atrial myocytes paced at 6 Hz, AP alternans were observed under the drug-free condition (Figure 3Ci): the APD varied between 100.1 and 88.1 ms. In the presence of AP alternans, the changes in APD by the Na+- and K+- blockers were quantified by comparing the corresponding big APs at baseline and after drug actions. These values were selected based on the characteristics of AP (APD prolongations seen in the long AP, and reduced INa for the short AP) that may be anti-arrhythmic. The fractional reductions in peak INa were calculated from the INa associated with the shorter APs. The results showed that applying Bl·IKur or Comb·Bl·IX alone both abolished the AP alternans while prolonging the APD to 116.3 and 126.3 ms and reducing the peak INa by 5.9 and 20.1%, respectively. The application of Bl·INa alone produced a minor APD prolongation (3.5 ms) and a reduction of 16.2% in peak INa. Combining block of INa with Bl·IKur or Comb·Bl·IX promoted the genesis of AP alternans, resulting in substantial prolongations in the APD of the big APs (by 35.4 ms for Bl·IKur + Bl·INa, and 55.6 ms for Comb·Bl·IX + Bl·INa) and dramatic decreases in the peak INa (by 57.5% for Bl·IKur + Bl·INa and 88.2% for Comb·Bl·IX + Bl·INa) in the corresponding small APs. These results suggest that the combined block of Bl·INa and Comb·Bl·IX/Bl·IKur exhibited synergistic antiarrhythmic effects manifested by prolongation in APD and reduction in peak INa. However, an increased susceptibility to AP alternans was observed at a fast pacing rate of 6 Hz, which may be potentially proarrhythmic at fast heart rates.
Effects on Steady-State Restitutions of APD and Conduction Velocity
Steady-state APD restitutions of cAF-remodeled human atria were simulated at both the cellular and tissue levels. In single myocyte simulations (Figure S5 in Online Supplementary Material 2.2), APD was prolonged over the entire range of simulated basic cycle lengths (BCL) for Bl·IKur and Comb·Bl·IX as compared to the control (drug-free) conditions. The reduction in peak INa in Bl·INa was rate-dependent and significantly greater at fast pacing rates. K+-block alone (Bl·IKur or Comb·Bl·IX) slightly shifted the rate-dependence of peak INa to larger BCLs. In comparison to the effects of individual current block scenarios, synergistic reductions in peak INa were observed following combined blocks of Bl·INa with Bl·IKur or Comb·Bl·IX over a wide range of BCLs. As compared to the drug-free conditions, AP alternans were observed at greater BCLs after K+-block, and this was further increased by combined Na+- and K+-block (Figure S5 in Online Supplementary Material 2.2).
Using a 1D model of atrial strands, the atrial APD and conduction velocity (CV) restitutions, as well as the rate-adaptation of in-tissue upstroke velocity (Vmax), were evaluated (Figure 5). The atrial activation-recovery interval (ARI) was not affected by Bl·INa alone, whereas it was substantially lengthened by K+-block as compared to control (Figure 5A). Applying K+-block alone also shifted the CV and Vmax restitution curves rightwards (i.e., to higher BCLs) (Figures 5B,C). These rate-adaptations of Vmax and CV were progressively enhanced by Bl·INa and the combined block over a wide range of BCLs. Synergistically enhanced rate-dependent reductions in Vmax and CV were observed in response to the combined blocks. Furthermore, K+-block increased the critical BCLs for conduction block as compared to the drug-free condition (Figure 5C).
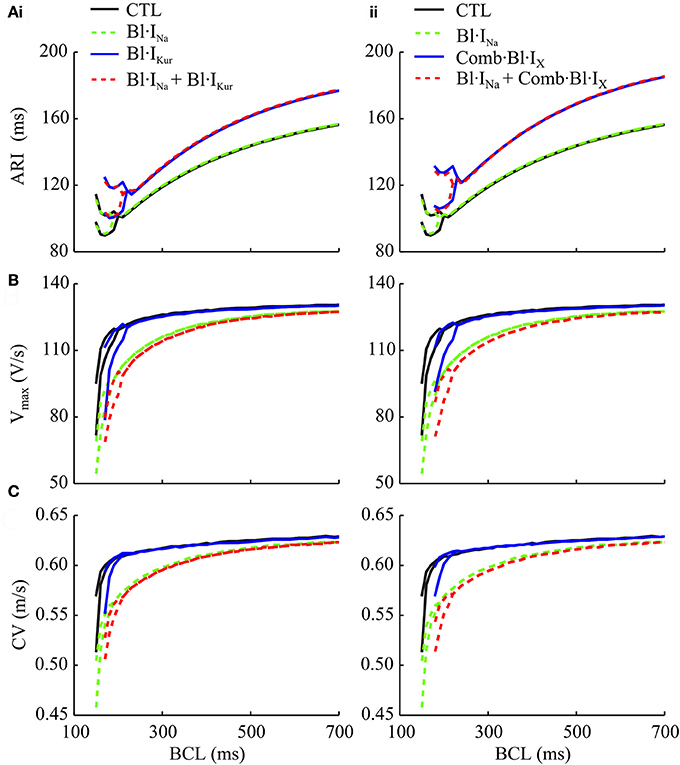
Figure 5. Simulated activation-recovery intervals (ARI) (A), Vmax (B), and CV (conduction velocity) (C) measured in 1D cAF-remodeled atrial strand models as a function of BCL for control (drug-free), individual and combined Na+- and (i) IKur block and (ii) action of acacetin. ARI values were measured as the interval between the time at which the AP depolarizes to −20 mV and the time it reaches a 90% repolarization.
Effects of Combined Na+- and K+- Block on the AF-Selectivity of INa block
AF-selectivity of Na+ blockers in cAF-remodeled hearts was examined by varying the drug binding and unbinding constants over wide parameters spaces to provide information concerning drug-Na+-channel interactions for various drug candidates. This was done by independently changing LA and LI for fixed {KA, KI} (Online Supplementary Material 2.3); and then varying KA and KI for fixed {LA, LI}. In this way we obtained the maximum AF-selectivity over the parameter space of drug binding and unbinding kinetics. Simulations with varied KA and KI were repeated for Bl·INa + Bl·IKur and Bl·INa + Comb·Bl·IX.
Figures 6A–D illustrates the block efficacy (defined in Equation 4), rate-selectivity, atrial-selectivity and the resultant AF-selectivity for Bl·INa alone and the combined block as a function of KA and KI. For Bl·INa alone, the block efficacy increased with increase of KI, whereas the rate-selectivity was reduced by increasing KA or KI. The AF-selectivity reached a maximum value of 9 at KA = 1 and KI=500 ms-1·M-1. The combined blocks achieved significantly greater AF-selectivity than Bl·INa alone: the maximum attainable AF-selectivity was increased by nearly 6-fold for Bl·IKur + Bl·INa and more than 14-fold for Comb·Bl·IX + Bl·INa as compared to Bl·INa alone (Figure 6C). These dramatic increases were attributed to the significantly greater values in all metrics contributing to the AF-selectivity. The maximal block efficacy achieved by Bl·INa alone was 0.77, and was increased to 0.81 and 0.94 for Bl·IKur + Bl·INa and Comb·Bl·IX + Bl·INa, respectively. A more appreciable increase in the maximal rate-selectivity was observed by the combined blocks as compared to Bl·INa alone (8-fold for Bl·IKur + Bl·INa and nearly 10-fold for Comb·Bl·IX + Bl·INa). Additionally, the atrial-selectivity was also increased by the combined block, although to a lesser extent. Bl·IKur + Bl·INa exhibited a greater atrial-selectivity than that of Comb·Bl·IX + Bl·INa since Bl·IKur was assumed to have no effect on the ventricles.
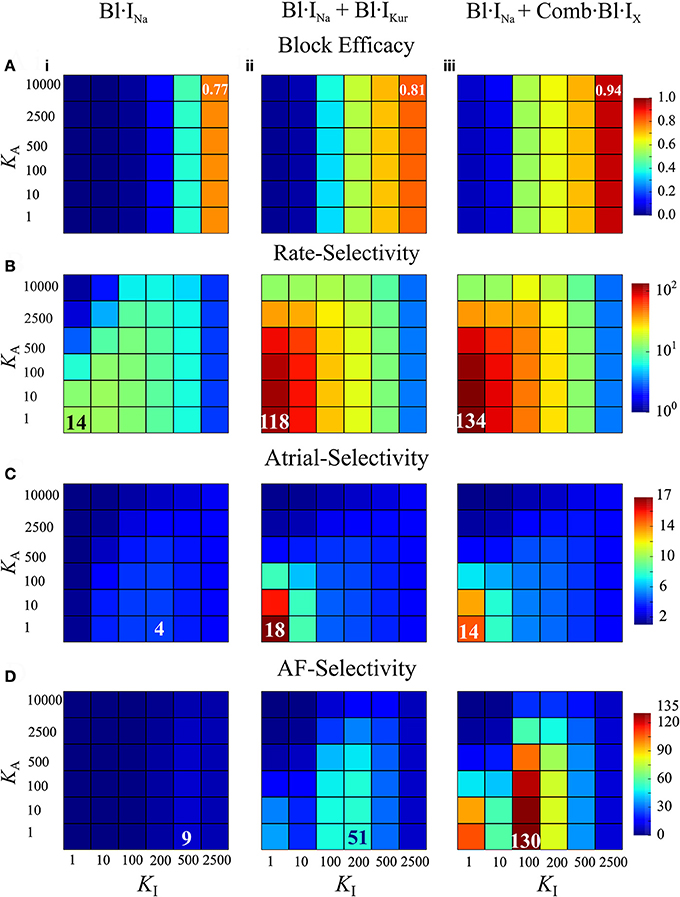
Figure 6. Block efficacy, and the rate-, atrial-, and AF-selectivity as a function of the open- and inactivated- state binding rates (KA, KI) for Na+-block or combined Na+- and K+-block computed from cAF-remodeled atrial myocytes or ventricular myocytes. (A) Block efficacy; (B) rate-selectivity; (C) atrial-selectivity, and (D) AF-selectivity. In each column shown are data from Bl·INa (i), combined Bl·INa and Bl·IKur (ii), and combined Bl·INa and Comb·Bl·IX (iii). LA=1 ms-1,LI=0.01 ms-1. Unit of KA, KI: ms−1·M−1.
Furthermore, these simulations revealed that the block efficacy, rate-selectivity and atrial-selectivity were strongly dependent on the inactivation state binding rate KI. These measures were also dependent on the open-state binding kinetics KA, but to a much lesser extent. The block efficacy was mainly determined by KI: an increase in KI led to a significant increase in the block efficacy. In combined block, the maximal rate- and atrial-selectivity were observed for KA=KI=1 ms-1·M-1 and increases in KI resulted in substantial reductions in the rate- and atrial-selectivity. In Bl·INa alone, the parameter set KA=1 ms-1·M-1, KI=200 ms-1·M-1 produced a maximal value in atrial-selectivity. Collectively, the optimal KI that maximized AF-selectivity was 200 ms−1·M−1 for Bl·INa and Bl·IKur + Bl·INa, and smaller (100 ms−1·M−1) for Comb·Bl·IX + Bl·INa. The optimal KA=1 ms-1·M-1 was seen for all conditions. Increasing KA consistently resulted in a smaller rate- and atrial-selectivity and therefore reduced AF-selectivity. These results suggest that the inactivation-state binding rate might be a more favorable targeting parameter than the open-state binding kinetics in optimizing AF-selectivity of Na+-blockers.
Effects of INa and IKur Block on Spiral Excitation Events in cAF-Remodeled Atria
Two-Dimensional Simulations
Using the cross-shock protocol, spiral waves were initiated in a 2D model representing a tissue slab of cAF-remodeled human atria. For each condition, a 10-s episode of electrical activity was simulated. Representative snapshots of the re-entrant waves in control (drug-free) and following application of drugs are presented in Figure 7A. The trajectories of the tips of re-entrant rotors under these conditions were traced and are shown in Figure 7B. The number of rotors during the time course of wave evolution was also measured (Figure 7C). The simulated pseudo-ECGs, membrane potential traces extracted from a representative myocyte and the corresponding fractional block of INa and IKur are detailed in Online Supplementary Material 2.4.
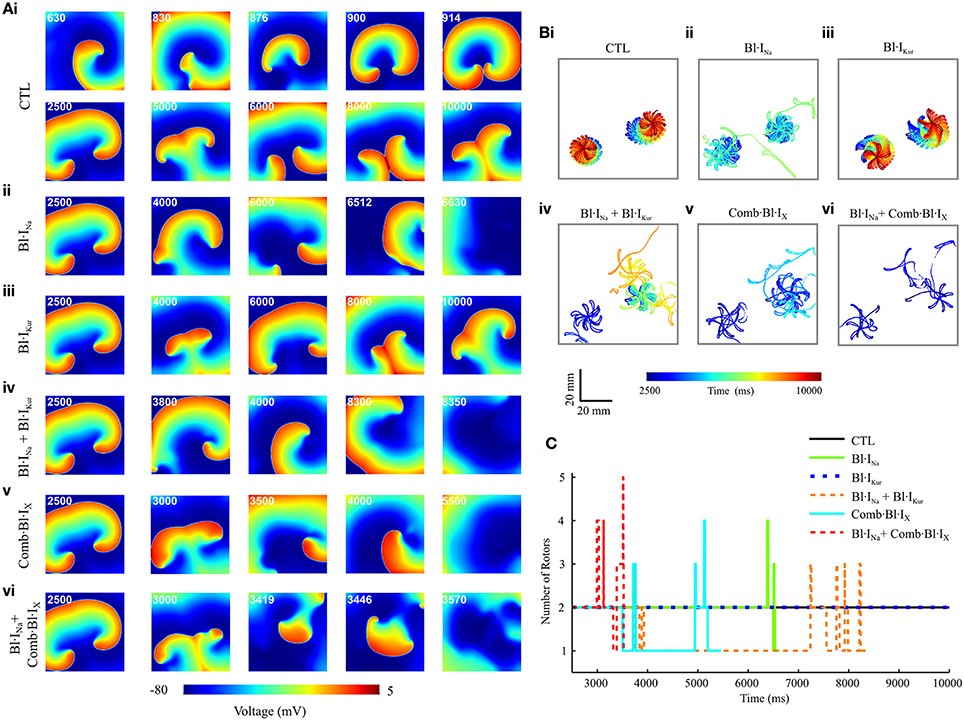
Figure 7. Snapshots of simulated re-entrant excitation events, tip trajectories of re-entrant waves and the number of rotor in a 2D model of cAF-remodeled atrial tissue slab in drug-free condition or after applying the drugs. (A) Snapshots of simulated re-entrant excitation events; The time sequence (ms) is indicated at the top left corner of each screenshot. (B) Tip trajectories of re-entrant waves. (C) Temporal evolution of total number of rotors represented by the number of spiral wave tips in tissue. In both (A,B), (i) Drug-free (CTL) condition, (ii) Bl·INa alone, (iii) Bl·IKur alone, (iv) Combined Bl·INa and Bl·IKur, (v) Applying Comb·Bl·IX alone, and (vi) Combined Bl·INa and Comb·Bl·IX. Rate constants for INa block: KA=1 ms-1·M-1,KI=100 ms-1·M-1,LA=1 ms-1, LI=0.01 ms-1.
Under the control (drug-free) condition, a single rotor was formed at approximately t = 630 ms; this broke into two spiral waves at t = 830 ms. These two rotors were stably anchored with star-shaped tip trajectories at the bottom half of the slab and persisted throughout the rest of the simulated 10-s episode (Figures 7Ai,Bi,C). In simulating drug actions, each drug was applied at t = 2,500 ms. For Bl·INa the dual rotors progressively became unstable, and the tips of the spiral waves meandered out of the tissue at approximately t = 6,000 ms, leading to self-termination of the re-entrant waves (Figures 7Aii,Bii,C). The dual rotors persisted throughout the period of the simulation after applying Bl·IKur alone (Figures 7Aiii,Biii,C). For Bl·INa + Bl·IKur, the rotor at the bottom left corner of the slab became unstable and meandered out of the tissue at approximately t = 3,800 ms, whereas the trajectory of the second rotor was confined to a small tissue area until t = 7,000 ms and then gradually became chaotic, forming up to 3 transient rotors that self-terminated at t = 8,334 ms (Figures 7Aiv,Biv,C). A similar but more marked effect was seen in the simulations that addressed the aggregate effects of acacetin: the bottom left rotor quickly meandered out of the tissue at t = 3,510 ms whilst the tip trajectory of the other rotor became chaotic and terminated at t = 5,439 ms (Figures 7Av,Bv,C). We note that the combined Bl·INa and Comb·Bl·IX exerted the strongest potency in terminating re-entrant excitations in these simulations: the two rotors transiently turned unstable and chaotic and self-terminated at t = 3,555 ms, with up to 5 rotors during the excitation in the slab (Figures 7Avi,Bvi,C).
The anti-arrhythmic benefits of combined Na+- and K+- block were clearly revealed by additional simulations assuming the use of a different Na+-blocker (KA=1 ms-1·M-1,KI=200 ms-1·M-1,LA=1 ms-1,LI=0.01 ms-1) and at a reduced dose (75%) of both Na+-blocker and acacetin. The life span of re-entrant excitations was measured and shown in Figure 8A. Also, the pECG was computed and the segment from t = 3,000 ms to 500 ms before the termination of re-entries (or the end of the simulation if the rotor sustained) was analyzed using the Fast Fourier Transform to obtain the dominant frequency (DF) of the re-entrant excitations, which is illustrated in Figure 8B.
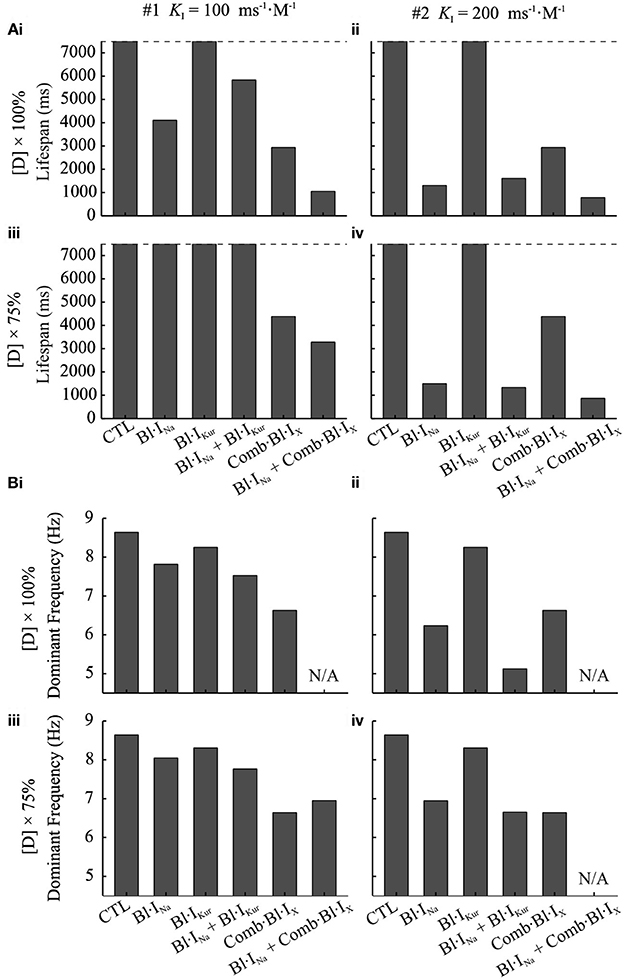
Figure 8. Dominant frequency and lifespan of re-entrant waves in 2D simulations using the cAF-remodeled atrial tissue model in the drug-free condition or after applying drugs. (A) Lifespan of the spiral waves. (B) DF of spiral waves. The results of K+-block alone are shown in both left and right panels for the purpose of comparison. In the left column (i,iii) of (A,B), Bl·INa was simulated with KI=100 ms-1·M-1; in the right column (ii,iv), KI=200 ms-1·M-1. In the top panels (i,ii) of (A,B), [DK+]=3.2μM, [DNa+]=60μM; in the bottom panels (iii,iv), [DK+]= 2.4μM, [DNa+]= 45 μM. In all panels, KA=1 ms-1·M-1, LA=1 ms-1, LI=0.01 ms-1.
At the simulated doses of acacetin, applying Bl·IKur alone did not lead to termination of re-entrant waves within the duration of the simulation (7,500 ms after TDrug), whereas the rotors were terminated in the simulations for Comb·Bl·IX at both doses (Figure 8Ai–iv), thus demonstrating enhanced anti-AF benefits of combined K+-channel block. For the simulated Bl·INa alone, the Na+-blocker with KI=100 ms-1·M-1 led to termination of AF at the control dose (lifespan of 4,102 ms) but not at the reduced dose; increasing KI of the Na+-blocker to 200 ms−1·M−1 resulted in a reduced lifespan (1,305 and 1,459 ms for [DNa+]=60 and 45 μM, respectively). The lifespan for the combined Bl·INa + Comb·Bl·IX was consistently shorter than that of any individual applications of Bl·INa or Comb·Bl·IX alone in all cases. A similar augmented anti-arrhythmic effect (shown as shortened lifespan of re-entry) was also observed for the combined Bl·INa + Bl·IKur for [DNa+] = 45 μM and KI=200 ms-1·M-1 (Figure 8Aiv) but not for the rest of the cases.
A consistent decrease in the DF was observed in the drug-modulated re-entrant excitations as compared to those in the drug-free condition (Figure 8Bi–iv). In the drug-free condition, the DF extracted from the pECG was 8.63 Hz, which is within the range of similar clinical data (Jarman et al., 2012). Applying Na+- or K+- block individually resulted in slowing of the rate of the rotors, and this was dependent on the concentrations and parameters of the blockers. For Bl·IKur the DF was 8.25 Hz with the control dose and 8.30 Hz for the reduced dose. In the simulations with Comb·Bl·IX, the DF was 6.63 Hz and was not affected by the 25% reduction in the dose of the compound. For Bl·INa alone the DF was 7.81 Hz and substantially smaller (6.24 Hz) for Na+-blockers of KI=100 ms-1·M-1 and KI=200 ms-1·M-1, respectively. An enhanced deceleration of the rotors was observed for Bl·INa + Bl·IKur in all cases. The DF for simulations with Comb·Bl·IX + Bl·INa was not computed due to the short lifespan in these events.
3D Simulations
The antiarrhythmic effects of acacetin and Na+-block on the re-entrant waves in the cAF-remodeled atria were also evaluated using our 3D anatomical model of the human atria (Colman et al., 2013, 2017). A 10-s episode of sustained re-entrant excitation was first initiated in the cAF-remodeled atria in the drug-free condition; this produced the initial conditions of the 3D model for additional 10-s episode simulations. Next, the behavior of electrical waves of another 10-s episode simulation in the drug-free condition and after applying the selected blockers was analyzed and compared. Figure 9A shows snapshots of excitation wave evolution following the 10-s episode of re-entrant excitation events. pECGs computed from the excitation waves are shown in Figure 9Bi–iv. Figures 9C,D illustrates power spectrum analyses of the pECGs and a comparison of the lifespan of the electrical waves in the drug-free condition and after applying the drugs/blockers. In the drug-free condition, stable re-entrant waves around the pulmonary veins and left atrium were observed; the power spectrum density (PSD) manifests a single-focused peak around 8.16 Hz. Following applying Bl·INa, the re-entrant waves became less organized and also decelerated. This was characterized by a smaller dominant frequency (6.58 Hz) and less focused PSD distribution; the excitation was not terminated. Following the application of Comb·Bl·IX, the re-entrant wave soon became unstable and eventually disappeared after t = 3,279 ms. Note that the peak PSD amplitude was much smaller and its distribution was much broader as compared to the drug-free condition. The combined drugs further destabilized the re-entrant waves and reduced the lifespan to approximately 1,120 ms.
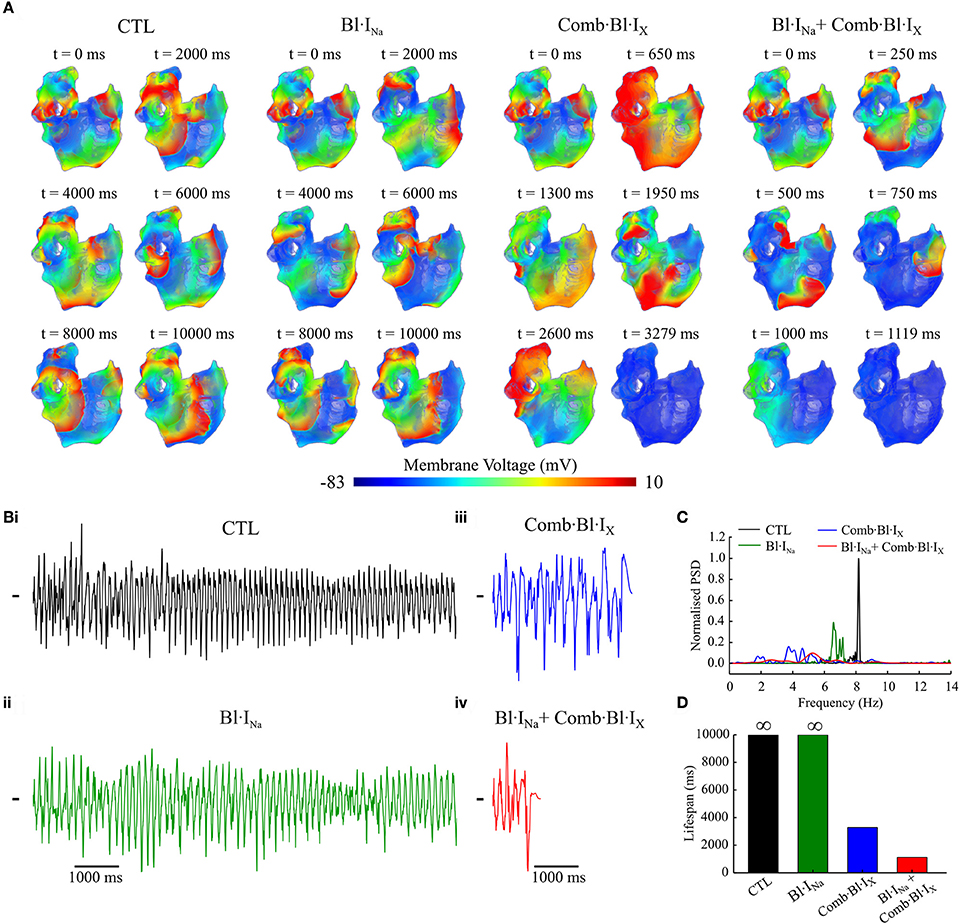
Figure 9. Simulated re-entrant excitations computed using the 3D anatomical model of the cAF-remodeled human atria in response to application of channel blockers as compared to that of the drug-free (CTL) condition. (A) Snapshots of electrical excitation waves. Drugs were applied at t = 0 ms. (B) Simulated pECGs. (C) Power spectrum density (PSD) obtained from the pECGs. PSDs were normalized to the maximum value of that in control. (D) Lifespan of re-entrant excitations in these simulated atria for various conditions. The symbol ∞ indicates the spiral waves were sustained throughout the 10-s episode of simulation. KA=1 ms-1·M-1, KI=100 ms-1·M-1,LA=1ms-1, LI=0.01 ms-1; [DK+]=3.2μM, [DNa+]=60μM.
Simulated Effects of Acacetin and Na+-Current Blocker on the QT Interval
Further simulations were performed using a 1D ventricular transmural strand model to evaluate the changes in the waveform of electrograms in consequence of applying the drugs. A comparison of the computed electrogram waveforms are illustrated in Figures 10A,B, and the QT intervals are quantified in Figure 10C. Blocking small fractions of IKr, IKs and Ito in the human ventricles by acacetin slightly increased the QT interval by 21 ms. The electrograms were not noticeably affected by applications of the Na+-blocker with the simulated parameters. Results from both cases did not show dramatic QT prolongation, indicating no dramatic effects affecting ventricular repolarization process which might promote ventricular arrhythmias.
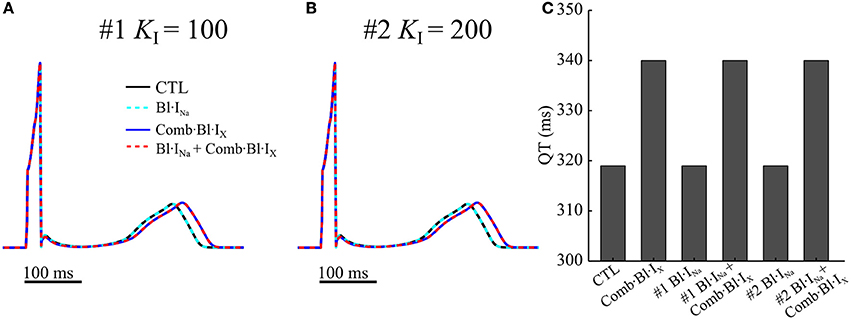
Figure 10. Computed pECGs from a 1D strand model for human ventricular transmural strands in drug-free (CTL) condition and in response to anti-arrhythmic drugs. In (A,B) two sets of parameters (#1 KI = 100 ms−1·M−1, #2 KI = 200 ms−1·M−1) for INa block were simulated. (C) QT intervals measured from the pECGs.
Discussion
Even decades after goal-directed work, successful development of effective and safe antiarrhythmic drugs for treating AF has not been accomplished and remains a major unmet clinical need. In a recent study on the canine heart, enhanced anti-arrhythmic effects and AF-selectivity of INa blockade by additional IKur block was demonstrated (Aguilar et al., 2015). Whether similar effects could be obtained in the human atria, especially following cAF-induced electrical remodeling which reduces IKur, remained unclear. How the combined Na+ and K+-block modulates the QT interval also remained incompletely understood. In this study, the effects of IKur (combined with a modest block in Ito, IKr, and IKs as presented by acacetin, a compound shown to be effective in anti-AF treatment) and INa block (two potentially effective atrial-selective block on human atrial electrophysiology) were investigated in silico using multiscale models of the human atria and state-dependent block scheme. The simulation results demonstrate that both Na+-block and K+-block exhibited anti-arrhythmic effects in the atria following cAF-remodeling, despite reduced IKur by the remodeling. The present study highlighted that in addition to combined Na+- and K+-block, combined multi-K+-channels also exerted beneficial synergistic antiarrhythmic effects when compared with single channel block whilst having modest impact on ventricular repolarization (QT interval). This study suggests that multi-channel block (either combined Na+-K+-block, or combined multi-K+-block) may be a favorable strategy for the development of novel pharmaceutical therapies for AF.
Effects of INa block
An atrial-ventricular difference in the properties of INa, especially in the voltage dependence of steady-state inactivation, has been reported (Li et al., 2002; Burashnikov et al., 2007; Chen et al., 2016; Fan et al., 2016; Caves et al., 2017). In these studies, the voltage dependent steady-state inactivation curves for INa were found to be negatively shifted (by 5–16 mV) in atrium as compared to the ventricular parameters. This difference gave rise to an on-going interest in developing an atrial-selective blocker of INa as a strategy in terminating AF (Burashnikov et al., 2007; Antzelevitch and Burashnikov, 2009; Zygmunt et al., 2011; Morotti et al., 2016; Caves et al., 2017). As was done in previous studies (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015), in this study, the kinetic parameters in drug actions of Na+-blockers were varied over wide parameter spaces to reveal AF-selectivity of Na+-blockers in the ventricles and fibrillating atria. Our results demonstrated that in the presence of AF-remodeling, an atrial-selective block of INa could produce different effects between atrial and ventricular cells (Figures 3, 4) and that the AF-selectivity could be maximized by optimizing the binding and unbinding rates of the Na+-blocker (Figure 6). Note also that, the fractional inhibition of INa by the Na+-blocker exhibited a substantial dependence on the rate of pacing (Figures 3, 4 and Online Supplementary Material 2.2), which was quantified using the rate-selectivity (Figure 6).
At the cellular level, Na+-block resulted in a significant inhibition in INa at fast pacing rates, but minor effects within the range of normal heart rates in the atria (Figures 5, 6). The antiarrhythmic effects of these changes were demonstrated in simulations of multicellular atrial tissue. In a 1D atrial strand model, applying Na+-block progressively enhanced the rate-adaptations of Vmax and CV over a larger range of BCLs, whereas the atrial APD was not affected. At fast pacing rates, Vmax and CV were decreased significantly, suggesting reduced excitabilities of atrial myocytes (Figure 5). These results are in concordance with the recent study (Aguilar et al., 2015) where similar effects of Na+-block on the canine atria were demonstrated in silico and experimentally in coronary-perfused hearts.
In 2D tissue simulations, this Na+-block shortened the lifespan and caused slowing in the excitation rate of the spiral waves (Figures 7, 8). In the 3D anatomical model, applying Na+-block alone produced antiarrhythmic effects by slowing the re-entrant excitations (Figure 9). Furthermore, in our 1D model of transmural ventricular strand, the simulations suggested that the Na+-block had minimal impact on ventricular repolarization, as judged by modest QT interval prolongation. These results demonstrated that Na+-block could be beneficial in suppressing re-entrant activities in the cAF-remodeled atria, with modest impact on ventricular repolarization.
Effects of K+-Current Block
K+-current blockers delay the repolarization phase of the AP and thus prolong atrial APD and refractory period. This can cause disruptions and eventually termination of the re-entrant circuits (Hancox et al., 2016). However, K+-channel blockers such as dofetilide and sotalol (which potently inhibit IKr) have a substantial risk of prolonging QT interval and promoting Torsades de pointes arrhythmias (Hondeghem and Snyders, 1990; Yap and Camm, 2003). In principle, blocking atrial-specific K+-channels may exert antiarrhythmic effects in the atria while minimizing potential risks of adverse effects in the ventricles. IKur is believed to be such an atrial-selective substrate for drug interventions, and effects of IKur block have been extensively studied (Burashnikov and Antzelevitch, 2008; Li et al., 2008; Tsujimae et al., 2008; Almquist et al., 2010; Pavri et al., 2012; Scholz et al., 2013; Loose et al., 2014; Ford et al., 2016). Interestingly, many existing IKur blockers potently block other K+-channels including Ito and IK,ACh (Gögelein et al., 2004; Wirth et al., 2007; Burashnikov and Antzelevitch, 2008; Li et al., 2008). The additional blockades of these channels may contribute to the antiarrhythmic effects of those drugs, which warrant further investigations.
In this study, acacetin, a compound initially isolated from the traditional Chinese medicine Xuelianhua, was selected as a representative IKur blocker. The effects of acacetin on atrial electrophysiology were evaluated in two ways: (a) the effects of acacetin blocking IKur only; and (b) the full actions of acacetin on the targeting channels (Ito, IKur, IKr, and IKs) (Li et al., 2008). This approach allowed for investigations into the effects of IKur block alone as well as the potential benefits of additional-but-modest inhibition of other K+-currents in the human atria.
Selective IKur Block
Blocking IKur with 3.2 μM acacetin exerted APD prolongation (9.8 ms) under the baseline/normal conditions (Figure 2). Experimental data show that dependent on the baseline AP waveform the effect of IKur block on human atrial APD70−90 under normal (SR) conditions can manifest as prolongation or shortening in the APD, (Workman et al., 2001; Wettwer et al., 2004; Schotten et al., 2007; Burashnikov and Antzelevitch, 2008; Loose et al., 2014). Additionally, the prolongation in APD by IKur block observed in the present study is similar to our previous paper (Colman et al., 2017) concerning the effects of genetically down-regulated IKur. Moreover, inhibiting IKur under normal conditions elevated the AP plateau potential and prolonged APD30 (Figure 2). Both effects matched well with experimental studies (Workman et al., 2001; Wettwer et al., 2004; Schotten et al., 2007; Burashnikov and Antzelevitch, 2008; Loose et al., 2014) and our simulation study (Colman et al., 2017).
We note that in the cAF-remodeling cells, a more pronounced prolongation in APD (by 23.6 ms for 1 Hz and 16.2 ms at 6 Hz, Figure 4) was observed in the presence of 3.2 μM acacetin, despite that this current was down-regulated by cAF-remodeling (Wagoner et al., 1997; Brandt et al., 2000; Christ et al., 2008). These results are in accordance with previous experimental results of blocking IKur with MK-0448 (Pavri et al., 2012; Loose et al., 2014). In addition, IKur block exhibited enhanced rate-dependent adaptations in APD both at the cellular (Online Supplementary Material 2.2) and 1D strand models (Figure 5). Importantly, the CV restitution curve shifted toward higher BCLs, indicating that this tissue is less capable of conduction of atrial excitation waves at high rate while maintaining conduction of slow waves (Figure 5). In 2D tissue simulations, applying IKur block alone (3.2 μM acacetin) destabilized the cores of rotors (i.e., potential organizing centers for AF), and slightly slowed their excitation rates, but failed to terminate them (Figure 7), suggesting a limited efficacy of terminating AF by IKur block alone. Similarly, a recent modeling study by Aguilar et al. (2017) suggested that the antiarrhythmic efficacy of IKur block was substantially decreased in the presence of AF-induced electrical remodeling. Also, the experimental study (Burashnikov and Antzelevitch, 2008) showed that block of IKur by 4-AP of small doses had limited efficacy in suppressing AF in canine atria. This may represent the fact that IKur is reduced at high frequencies (as discussed/suggested in Feng et al., 1998a; Burashnikov and Antzelevitch, 2008; Wu et al., 2011) and shown in Figure 1) as well as by cAF-induced remodeling (Wagoner et al., 1997; Brandt et al., 2000; Christ et al., 2008). In addition, IKur is primarily active during phase 2 of AP, and hence pure IKur block exerted a relatively greater prolongation in APD30 than APD90 (Figure 3), in contrast to other K+-block including dofetilide which mediates anti-AF effects by prolonging the terminal phase of the AP (Roukoz and Saliba, 2007).
In this study, IKur block was simulated using a state-dependent block model, which successfully reproduced the use- and rate-dependent inhibition of acacetin (Figure 1). The rate-dependent block of IKur exerted a higher fractional inhibition in the current at faster pacing rates, which likely produces greater anti-AF effects in the presence of high-frequency excitations as seen during AF. Along with the previous modeling studies on investigating effects of IKur block (Almquist et al., 2010; Scholz et al., 2013; Ellinwood et al., 2017), this study demonstrated the importance of explicitly considering the kinetic properties of the block in computational efforts of understanding the consequences and underlying mechanisms of IKur block.
Effects of Combined K+-Current Block
The combined K+-block (as exhibited by acacetin and many other IKur blockers) resulted in synergistic APD prolongation as well as an increased efficacy in terminating re-entry in tissue as compared to the pure IKur block.
Note that at the single myocyte level, the combined actions of acacetin produced greater prolongation in atrial APD than the sum of changes due to drug-induced block of individual channel in normal and cAF-remodeled myocytes (Figure 2). Additionally, the combined K+-block increased the rate-additivity of APD as compared to the pure IKur block (Online Supplementary Material 2.2). This was also consistently observed in the 1D simulation (Figure 5). In the setting of pure IKur block, the elevated and prolonged plateau phase of the AP could promote the activation of IKr/IKs, which in return may accelerate the repolarization of AP-phase 3 (Colman et al., 2017). Therefore, additional inhibition in IKr by an identical fraction is expected to result in a greater APD prolongation than a pure IKur or IKr/IKs block.
In 2D simulations, the combined K+-block produced an enhanced efficacy in suppressing AF compared with the pure IKur block: promoting meandering of rotor tips (Figure 7B), shortening the lifespan of re-entries (Figure 8A) and slowing of spiral wave excitations (Figure 8B). Rotor meandering is one mechanism by which spiral waves may meet non-conducting boundaries to extinguish re-entry (Narayan et al., 2013; Pandit and Jalife, 2013; Rappel et al., 2015).
The effects of acacetin (3.2 μM) on the ventricular AP and QT interval was assessed in a single cell model and 1D transmural strand model by assuming similar blockade effects of the compound on the human ventricles and atria. It was shown that following applying acacetin, the ventricular repolarization and QT interval was both preserved with slight prolongations around 21 ms (Figure 10). Our results are close to the previous experimental study (Li et al., 2008) showing that QT intervals were not prolonged by acacetin in isolated rabbit hearts and anesthetised dogs.
The synergistic effects demonstrated by the combined K+-blocks have implications on developing novel pharmaceutical anti-AF therapies. Given that Ito, IKr and IKs contribute to the repolarizations of ventricular APs, inhibitions in these channels may promote risks of side effects in the ventricles. In this regard, combined block of atrial-specific K+ channels may be favorable. Recently, another two families of K+-channels that are dominantly expressed in the atria have been acknowledged: the small-conductance Ca2+-activated K+ (SK) channels (ISK) (Qi et al., 2014), and the two-pore K+ (K2P3.1) channel (ITASK−1) (Schmidt et al., 2015), further to the well-known constitutively active acetylcholine-activated K+ current (IK,ACh). Combined block of these atrial-specific channels may exert greater and safer antiarrhythmic effects in the atria, warranting future investigations.
Synergistic Effects of Combined Na+- and K+- Block
The present study reveals novel and significant synergistic effects of combined block of Na+- and K+-currents (INa and pure-IKur/multi-K+-block) and demonstrates the additional synergistic anti-arrhythmic effects derived from the multi-K+ channel block in cAF-remodeled atria.
In cAF-remodeled atria, combined Na+- and K+-block significantly increased the fractional INa inhibition and APD prolongation (Figures 3, 4) and promoted pronounced AP alternans at 6 Hz, with complex effects in human AF (Narayan et al., 2011). In the simulations varying the blockade kinetics of INa block, the combined block dramatically augmented the attainable maximal AF-selectivity in consequence of enhanced atrial-selectivity and rate-selectivity as compared to the pure Na+-block (Figure 6).
In the 1D model of an atrial strand, combined Na+ and K+-block produced synergistic reductions in Vmax and CV; the threshold of BCL allowing a 1:1 conduction was increased as compared to the control conditions (Figure 5). In simulated re-entrant waves in 2D and 3D atria, the combined Bl·INa + Comb·Bl·IX exhibited a greater efficacy in suppressing AF, with a decreased lifespan of rotors as compared to that by either individual block (Figures 7–9). Although the combined Bl·INa + Bl·IKur did not further reduce the lifespan of spiral waves as compared to the Bl·INa alone, the combination did lead to the extinction of one of the two rotors (Figures 7Aiv,Biv) and deceleration of re-entrant activations (Figure 8B). Follow-up simulations showed that consistent synergistic antiarrhythmic effects could be obtained with reduced doses of Na+- and K+- blockers.
The non-specific multi-channel blockade is increasingly recognized as a strategy for pharmaceutical therapy of AF both experimentally (Sicouri et al., 2010; Aguilar et al., 2015; Kirchhoff et al., 2015; Hartmann et al., 2016) and clinically (Koskinas et al., 2014; Reiffel et al., 2015). In a previous study (Aguilar et al., 2015), synergistic anti-arrhythmic effects were demonstrated both in silico and experimentally in healthy canine hearts. Additionally, the favorable synergistic antiarrhythmic effects have also been reported in combined block of ISK and INa in an experimental atrial-fibrillated guinea pig model (Kirchhoff et al., 2015). Also, the recent HARMONY trial (Reiffel et al., 2015) revealed synergistic AF-suppressing effects for combined use of ranolazine and dronedarone. While revealing the synergistic effects of combined Na+- and K+- block in cAF-remodeled human atria, this study supports and adds insights into the on-going efforts in developing multi-channel block as a strategy for the treatment of AF.
Limitations and Future Work
In the absence of the required detailed experimental data, when simulating effects of acacetin on Ito, IKs and IKr, the dose-dependence block of acacetin was assumed to be identical in both human atrial and ventricular cells. This assumption may warrant further investigations. In addition, the parameters of atrial Ito have been reported to be different from those of ventricular Ito in human (Amos et al., 1996). Previous studies reported that the IC50 of 4-AP block of atrial Ito was one-third of that of ventricular Ito (Amos et al., 1996; Nattel et al., 2000). If a similar atrial-vs.-ventricular difference in the IC50 of Ito and/or IKr/IKs could exist for acacetin, the effects of acacetin on the ventricular electrophysiology would be less significant than our simulations, which might result in to a smaller change in ventricular INa and APD for the combined block of Bl·INa and Comb·Bl·IX, and thus enhance the computed atrial-selectivity and AF-selectivity of the combined block. Given that applying acacetin in vivo did not prolong QT intervals in isolated rabbit hearts and anesthetised dogs (Li et al., 2008), any significant prolongation of the ventricular APD and QT interval is unlikely (Figures 3B, 4C, 10). Therefore, our assumption of no atrial-ventricular difference in the potency of acacetin on K+-currents may not affect our conclusions concerning the atrial-selectivity of combined Na+- and K+-block.
Additionally, in the absence of detailed experimental data for state-dependent block of Ito, IKr, and IKs by acacetin, the block of these channels was modeled using a single pore block model. The IC50 values (Table 1) were determined by fitting the concentration-response relation of the step current at 40 mV in previous experimental studies (Li et al., 2008; Wu et al., 2011). A recent study suggests that the IC50 values may be dependent on the voltage protocols applied, and this cannot be reflected by single pore models. In future studies, the pore block model for Ito, IKr, and IKs can be replaced by a state-dependent block model when such experimental data become available. Also, the present work did not attempt to model the effects of acacetin on IK,ACh, although the study shows the current is potently blocked by the compound. The 2D and 3D simulations of atrial tissue, while validated, may not fully capture the complexity of fibrosis-tissue interfaces which are seen in structurally remodeled atria and were not simulated in these monodomain experiments.
Thirdly, our simulation results showed a moderate QT prolongation of around 20 ms following applying both INa blocker and acacetin. While a QT prolongation of less than 5 ms does not raise a regulatory concern (Committee for Medicinal Products for Human Use, 2012), implications of QT prolongations between 5 and 20 ms remain inconclusive (Committee for Medicinal Products for Human Use, 2005). In the present study, though the extent of QT prolongation of 20 ms is far less than the threshold of discontinuation criteria of 60 ms as indicated in Committee for Medicinal Products for Human Use (2005), it would indeed raise a positive flag in thorough QT tests and necessitate extended safety assessment and intensive patient monitoring during late stages of trials (Committee for Medicinal Products for Human Use, 2012). On the other hand, the approach we used in accounting for the effects of acacetin on ventricular myocytes may result in upper bound of QT prolongation, since the potency of acacetin was assumed to be identical in atria and ventricles.
Fourthly, the threshold in BCL inducing AP alternans was increased by K+-block (Figure 5, Figure S5). However, AP alternans seen at slower pacing rates has been linked with occurrence of AF (Narayan et al., 2002, 2011). Therefore, the increased threshold in BCL developing AF by K+-block can be potentially proarrhythmic. The safety of K+-block and its proarrhythmic potential in the atria should be addressed in future studies.
Fifthly, there are limitations in the approaches used in simulating the INa blockers in single myocytes and tissue. Similar to previous studies (Aguilar-Shardonofsky et al., 2012; Aguilar et al., 2015), in our simulations, the drug action on INa was modeled through a state-dependent block assuming drugs binding to both activated and inactivated states of INa, and the gating variables of INa were modeled using an Hodgkin-Huxley scheme. The limitations in this approach outlined in Aguilar-Shardonofsky et al. (2012) therefore apply in the present study. The results of the use-dependent block may be affected by the models used (Aguilar-Shardonofsky et al., 2012). However, the previous study (Aguilar-Shardonofsky et al., 2012) compared this modeling scheme with simulations using a Markov model, showing qualitative agreement in major findings. Therefore, the major conclusions drawn from this study may not be affected by the selected modeling approach for INa and drug interactions. Furthermore, in optimizing the AF-selectivity of the INa and K+-current blockers, the concentrations of Na+ and K+ blockers were fixed at 60 uM. This may potentially impose limitations in discomposing the role of the binding parameters in the modulatory effects of the blockers because of the very slow kinetics of the drug binding to its targeted channel at this high concentration. It warrants further studies by varying the concentration of blockers to simulate the optimized effects of the AF-selectivity of INa blocker. In tissue simulations, effects of drugs were modeled by increasing their doses homogeneously, simultaneously and instantaneously. The realistic actions of INa blockers in tissue, however, may be different. Also, in tissue simulations a homogenous cell model was used. As previous study (Feng et al., 1998b) showed atria are electrically heterogeneous, future work is needed to assess how tissue heterogeneities affect the efficacy of atrial-selective pharmaceutical interventions. Furthermore, the current simulations did not take the cardiac autonomic regulation into account in order to take into considerations of acacetin on IKACh. Future studies on interactions of atrial-selective anti-arrhythmic drug actions and autonomic systems may also render valuable findings.
It is important to acknowledge that administration of class Ic agents for Na+-block can cause cardiac arrhythmia and increased mortality (Echt et al., 1991). Further investigations are therefore warranted to assess the safety of the simulated Na+-block in the heart, especially in the ventricles.
Conclusions
By using state-dependent drug block models and our mathematical models of the human atria, the antiarrhythmic effects of atrial selective Na+- and K+-blockers on the cAF-remodeled atria were evaluated. The combined block of multiple K+-currents as well as simultaneous block of Na+- and K+-currents produced synergistic antiarrhythmic effects. Our results suggest that developing multi-channel (multiple K+ currents and/or combined Na+- and K+-current) block is a potentially valuable strategy for the treatment of AF.
Author Contributions
HZ and HN conceived the study. HN designed experiments, developed and validated computational models, and performed numerical experiments. HN, DW, and WW analyzed data. HN, DW, WW, WG, SN, and HZ interpreted data and wrote the manuscript.
Funding
This work was supported by grants from the British Heart Foundation FS/14/5/30533, EPSRC (UK) (EP/J00958X/1; EP/I029826/1), MC-IRSES CORDIS3D (317766), NSFC (61179009), Shenzhen Science and Technology Innovation Committee (JCYJ20151029173639477; JSGG20160229125049615). SN is supported by grants from the National Institutes of Health (R01 Hl83359).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2017.00946/full#supplementary-material
References
Aguilar, M., Feng, J., Vigmond, E., Comtois, P., and Nattel, S. (2017). Rate-dependent role of IKur in human atrial repolarization and atrial fibrillation maintenance. Biophys. J. 112, 1997–2010. doi: 10.1016/j.bpj.2017.03.022
Aguilar, M., Xiong, F., Qi, X. Y., Comtois, P., and Nattel, S. (2015). Potassium channel blockade enhances atrial fibrillation-selective antiarrhythmic effects of optimized state-dependent sodium channel blockade. Circulation 132, 2203–2211. doi: 10.1161/CIRCULATIONAHA.115.018016
Aguilar-Shardonofsky, M., Vigmond, E. J., Nattel, S., and Comtois, P. (2012). In silico optimization of atrial fibrillation-selective sodium channel blocker pharmacodynamics. Biophys. J. 102, 951–960. doi: 10.1016/j.bpj.2012.01.032
Almquist, J., Wallman, M., Jacobson, I., and Jirstrand, M. (2010). Modeling the effect of Kv1.5 block on the canine action potential. Biophys. J. 99, 2726–2736. doi: 10.1016/j.bpj.2010.08.062
Amos, G. J., Wettwer, E., Metzger, F., Li, Q., Himmel, H. M., and Ravens, U. (1996). Differences between outward currents of human atrial and subepicardial ventricular myocytes. J. Physiol. 491, 31–50. doi: 10.1113/jphysiol.1996.sp021194
Antzelevitch, C., and Burashnikov, A. (2009). Atrial-selective sodium channel block as a novel strategy for the management of atrial fibrillation. J. Electrocardiol. 42, 543–548. doi: 10.1016/j.jelectrocard.2009.07.007
Aslanidi, O. V., Colman, M. A., Stott, J., Dobrzynski, H., Boyett, M. R., Holden, A. V., et al. (2011). 3D virtual human atria: a computational platform for studying clinical atrial fibrillation. Prog. Biophys. Mol. Biol. 107, 156–168. doi: 10.1016/j.pbiomolbio.2011.06.011
Baher, A., Qu, Z., Hayatdavoudi, A., Lamp, S. T., Yang, M.-J., Xie, F., et al. (2006). Short-term cardiac memory and mother rotor fibrillation. Am. J. Physiol. Heart Circ. Physiol. 292, H180–H189. doi: 10.1152/ajpheart.00944.2005
Bosch, R. F., Zeng, X., Grammer, J. B., Popovic, K., Mewis, C., and Kühlkamp, V. (1999). Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 44, 121–131. doi: 10.1016/S0008-6363(99)00178-9
Brandt, M. C., Priebe, L., Böhle, T., Südkamp, M., and Beuckelmann, D. J. (2000). The ultrarapid and the transient outward K+ current in human atrial fibrillation: their possible role in postoperative atrial fibrillation. J. Mol. Cell. Cardiol. 32, 1885–1896. doi: 10.1006/jmcc.2000.1221
Brennan, T., Fink, M., and Rodriguez, B. (2009). Multiscale modelling of drug-induced effects on cardiac electrophysiological activity. Eur. J. Pharm. Sci. 36, 62–77. doi: 10.1016/j.ejps.2008.09.013
Burashnikov, A., and Antzelevitch, C. (2008). Can inhibition of IKur promote atrial fibrillation? Heart Rhythm 5, 1304–1309. doi: 10.1016/j.hrthm.2008.05.020
Burashnikov, A., Diego, J. M. D., Zygmunt, A. C., Belardinelli, L., and Antzelevitch, C. (2007). Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation 116, 1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890
Caves, R. E., Cheng, H., Choisy, S. C., Gadeberg, H. C., Bryant, S. M., Hancox, J. C., et al. (2017). Atrial-ventricular differences in rabbit cardiac voltage-gated Na+ currents: basis for atrial-selective block by ranolazine. Heart Rhythm. 14, 1657–1664. doi: 10.1016/j.hrthm.2017.06.012
Chen, K.-H., Xu, X.-H., Sun, H.-Y., Du, X.-L., Liu, H., Yang, L., et al. (2016). Distinctive property and pharmacology of voltage-gated sodium current in rat atrial versus ventricular myocytes. Heart Rhythm. 13, 762–770. doi: 10.1016/j.hrthm.2015.11.022
Christ, T., Wettwer, E., Voigt, N., Hála, O., Radicke, S., Matschke, K., et al. (2008). Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br. J. Pharmacol. 154, 1619–1630. doi: 10.1038/bjp.2008.209
Clayton, R. H., Bernus, O., Cherry, E. M., Dierckx, H., Fenton, F. H., Mirabella, L., et al. (2011). Models of cardiac tissue electrophysiology: progress, challenges and open questions. Prog. Biophys. Mol. Biol. 104, 22–48. doi: 10.1016/j.pbiomolbio.2010.05.008
Colman, M. A., Aslanidi, O. V., Kharche, S., Boyett, M. R., Garratt, C., Hancox, J. C., et al. (2013). Pro-arrhythmogenic effects of atrial fibrillation-induced electrical remodelling: insights from the three-dimensional virtual human atria. J. Physiol. 591, 4249–4272. doi: 10.1113/jphysiol.2013.254987
Colman, M. A., Ni, H., Liang, B., Schmitt, N., and Zhang, H. (2017). In silico assessment of genetic variation in KCNA5 reveals multiple mechanisms of human atrial arrhythmogenesis. PLoS Comput. Biol. 13:e1005587. doi: 10.1371/journal.pcbi.1005587
Committee for Medicinal Products for Human Use (2005). ICH Note for Guidance on the Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-antiarrhythmic Drugs (ICH E14) (CHMP/ICH/2/04). London: EMEA. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002879.pdf
Committee for Medicinal Products for Human Use (2012). ICH Topic E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-antiarrhythmic Drugs Questions and Answers (EMA/CHMP/ICH/310133/2008). London: EMEA. Available online at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002878.pdf
Dobrev, D., Carlsson, L., and Nattel, S. (2012). Novel molecular targets for atrial fibrillation therapy. Nat. Rev. Drug Discov. 11, 275–291. doi: 10.1038/nrd3682
Dobrev, D., and Ravens, U. (2003). Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res. Cardiol. 98, 137–148. doi: 10.1007/s00395-003-0409-8
Echt, D. S., Liebson, P. R., Mitchell, L. B., Peters, R. W., Obias-Manno, D., Barker, A. H., et al. (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. N. Engl. J. Med. 324, 781–788. doi: 10.1056/NEJM199103213241201
Ellinwood, N., Dobrev, D., Morotti, S., and Grandi, E. (2017). Revealing kinetics and state-dependent binding properties of IKur-targeting drugs that maximize atrial fibrillation selectivity. Chaos Interdiscip. J. Nonlinear Sci. 27:093918. doi: 10.1063/1.5000226
Fan, X., Wang, C., Wang, N., Ou, X., Liu, H., Yang, Y., et al. (2016). Atrial-selective block of sodium channels by acehytisine in rabbit myocardium. J. Pharmacol. Sci. 132, 235–243. doi: 10.1016/j.jphs.2016.03.014
Feng, J., Xu, D., Wang, Z., and Nattel, S. (1998a). Ultrarapid delayed rectifier current inactivation in human atrial myocytes: properties and consequences. Am. J. Physiol. Heart Circ. Physiol. 275, H1717–H1725.
Feng, J., Yue, L., Wang, Z., and Nattel, S. (1998b). Ionic mechanisms of regional action potential heterogeneity in the canine right atrium. Circ. Res. 83, 541–551. doi: 10.1161/01.RES.83.5.541
Ford, J., Milnes, J., El Haou, S., Wettwer, E., Loose, S., Matschke, K., et al. (2016). The positive frequency-dependent electrophysiological effects of the Ikur inhibitor Xen-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm. 13, 555–564. doi: 10.1016/j.hrthm.2015.10.003
Gima, K., and Rudy, Y. (2002). Ionic current basis of electrocardiographic waveforms a model study. Circ. Res. 90, 889–896. doi: 10.1161/01.RES.0000016960.61087.86
Gögelein, H., Brendel, J., Steinmeyer, K., Strübing, C., Picard, N., Rampe, D., et al. (2004). Effects of the atrial antiarrhythmic drug AVE0118 on cardiac ion channels. Naunyn. Schmiedebergs Arch. Pharmacol. 370, 183–192. doi: 10.1007/s00210-004-0957-y
Grandi, E., Pandit, S. V., Voigt, N., Workman, A. J., Dobrev, D., Jalife, J., et al. (2011). Human atrial action potential and Ca2+ model sinus rhythm and chronic atrial fibrillation. Circ. Res. 109, 1055–1066. doi: 10.1161/CIRCRESAHA.111.253955
Haan, S., de Greiser, M., Harks, E., Blaauw, Y., Hunnik, A., van Verheule, S., et al. (2006). AVE0118, blocker of the transient outward current (Ito) and ultrarapid delayed rectifier current (IKur), fully restores atrial contractility after cardioversion of atrial fibrillation in the goat. Circulation 114, 1234–1242. doi: 10.1161/CIRCULATIONAHA.106.630905
Hancox, J. C., James, A. F., Marrion, N. V., Zhang, H., and Thomas, D. (2016). Novel ion channel targets in atrial fibrillation. Expert Opin. Ther. Targets 20, 947–958. doi: 10.1517/14728222.2016.1159300
Hartmann, N., Mason, F. E., Braun, I., Pabel, S., Voigt, N., Schotola, H., et al. (2016). The combined effects of ranolazine and dronedarone on human atrial and ventricular electrophysiology. J. Mol. Cell. Cardiol. 94, 95–106. doi: 10.1016/j.yjmcc.2016.03.012
Hondeghem, L. M., and Snyders, D. J. (1990). Class III antiarrhythmic agents have a lot of potential but a long way to go. Reduced effectiveness and dangers of reverse use dependence. Circulation 81, 686–690. doi: 10.1161/01.CIR.81.2.686
Jarman, J. W. E., Wong, T., Kojodjojo, P., Spohr, H., Davies, J. E., Roughton, M., et al. (2012). Spatiotemporal behavior of high dominant frequency during paroxysmal and persistent atrial fibrillation in the human left atrium. Circ. Arrhythm. Electrophysiol. 5, 650–658. doi: 10.1161/CIRCEP.111.967992
Kirchhoff, J. E., Goldin Diness, J., Sheykhzade, M., Grunnet, M., and Jespersen, T. (2015). Synergistic antiarrhythmic effect of combining inhibition of Ca2+-activated K+ (SK) channels and voltage-gated Na+ channels in an isolated heart model of atrial fibrillation. Heart Rhythm 12, 409–418. doi: 10.1016/j.hrthm.2014.12.010
Koskinas, K. C., Fragakis, N., Katritsis, D., Skeberis, V., and Vassilikos, V. (2014). Ranolazine enhances the efficacy of amiodarone for conversion of recent-onset atrial fibrillation. Europace 16, 973–979. doi: 10.1093/europace/eut407
Li, G.-R., Lau, C.-P., and Shrier, A. (2002). Heterogeneity of sodium current in atrial vs. epicardial ventricular myocytes of adult guinea pig hearts. J. Mol. Cell. Cardiol. 34, 1185–1194. doi: 10.1006/jmcc.2002.2053
Li, G.-R., Wang, H.-B., Qin, G.-W., Jin, M.-W., Tang, Q., Sun, H.-Y., et al. (2008). Acacetin, a natural flavone, selectively inhibits human atrial repolarization potassium currents and prevents atrial fibrillation in dogs. Circulation 117, 2449–2457. doi: 10.1161/CIRCULATIONAHA.108.769554
Loose, S., Mueller, J., Wettwer, E., Knaut, M., Ford, J., Milnes, J., et al. (2014). Effects of IKur blocker MK-0448 on human right atrial action potentials from patients in sinus rhythm and in permanent atrial fibrillation. Pharmacol. Ion Channels Channelopathies 5:26. doi: 10.3389/fphar.2014.00026
Luo, C. H., and Rudy, Y. (1994). A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74, 1071–1096. doi: 10.1161/01.RES.74.6.1071
Moreno, J. D., Zhu, Z. I., Yang, P.-C., Bankston, J. R., Jeng, M.-T., Kang, C., et al. (2011). A computational model to predict the effects of class i anti-arrhythmic drugs on ventricular rhythms. Sci. Transl. Med. 3:98ra83. doi: 10.1126/scitranslmed.3002588
Morotti, S., McCulloch, A. D., Bers, D. M., Edwards, A. G., and Grandi, E. (2016). Atrial-selective targeting of arrhythmogenic phase-3 early afterdepolarizations in human myocytes. J. Mol. Cell. Cardiol. 96, 63–71. doi: 10.1016/j.yjmcc.2015.07.030
Narayan, S. M., Bode, F., Karasik, P. L., and Franz, M. R. (2002). Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 106, 1968–1973. doi: 10.1161/01.CIR.0000037062.35762.B4
Narayan, S. M., Franz, M. R., Clopton, P., Pruvot, E. J., and Krummen, D. E. (2011). Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 123, 2922–2930. doi: 10.1161/CIRCULATIONAHA.110.977827
Narayan, S. M., Shivkumar, K., Krummen, D. E., Miller, J. M., and Rappel, W.-J. (2013). Panoramic electrophysiological mapping but not electrogram morphology identifies stable sources for human atrial fibrillation. Circ. Arrhythm. Electrophysiol. 6, 58–67. doi: 10.1161/CIRCEP.111.977264
Nattel, S., and Dobrev, D. (2017). Controversies about atrial fibrillation mechanisms: aiming for order in chaos and whether it matters. Circ. Res. 120, 1396–1398. doi: 10.1161/CIRCRESAHA.116.310489
Nattel, S., Matthews, C., Blasio, E. D., Han, W., Li, D., and Yue, L. (2000). Dose-dependence of 4-aminopyridine plasma concentrations and electrophysiological effects in dogs potential relevance to ionic mechanisms in vivo. Circulation 101, 1179–1184. doi: 10.1161/01.CIR.101.10.1179
O'Hara, T., Virág, L., Varr,ó, A., and Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7:e1002061. doi: 10.1371/journal.pcbi.1002061
Osaka, T., Itoh, A., and Kodama, I. (2000). Action potential remodeling in the human right atrium with chronic lone atrial fibrillation. Pacing Clin. Electrophysiol. 23, 960–965. doi: 10.1111/j.1540-8159.2000.tb00881.x
Pandit, S. V., and Jalife, J. (2013). Rotors and the dynamics of cardiac fibrillation. Circ. Res. 112, 849–862. doi: 10.1161/CIRCRESAHA.111.300158
Pavri, B. B., Greenberg, H. E., Kraft, W. K., Lazarus, N., Lynch, J. J., Salata, J. J., et al. (2012). Mk-0448, a specific Kv1.5 inhibitor safety, pharmacokinetics, and pharmacodynamic electrophysiology in experimental animal models and humans. Circ. Arrhythm. Electrophysiol. 5, 1193–1201. doi: 10.1161/CIRCEP.111.969782
Persson, F., Carlsson, L., Duker, G., and Jacobson, I. (2005). Blocking characteristics of hKv1.5 and hKv4.3/hKChIP2.2 after administration of the novel antiarrhythmic compound AZD7009. J. Cardiovasc. Pharmacol. 46, 7–17. doi: 10.1097/01.fjc.0000161405.37198.c1
Qi, X.-Y., Diness, J. G., Brundel, B. J. J. M., Zhou, X.-B., Naud, P., Wu, C.-T., et al. (2014). Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 129, 430–440. doi: 10.1161/CIRCULATIONAHA.113.003019
Rappel, W.-J., Zaman, J. A. B., and Narayan, S. M. (2015). Mechanisms for the termination of atrial fibrillation by localized ablation: computational and clinical studies. Circ. Arrhythm. Electrophysiol. 8, 1325–1333. doi: 10.1161/CIRCEP.115.002956
Ravens, U., and Wettwer, E. (2011). Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovasc. Res. 89, 776–785. doi: 10.1093/cvr/cvq398
Reiffel, J. A., Camm, A. J., Belardinelli, L., Zeng, D., Karwatowska-Prokopczuk, E., Olmsted, A., et al. (2015). The HARMONY trial: combined ranolazine and dronedarone in the management of paroxysmal atrial fibrillation: mechanistic and therapeutic synergism. Circ. Arrhythm. Electrophysiol. 8, 1048–1056. doi: 10.1161/CIRCEP.115.002856
Roukoz, H., and Saliba, W. (2007). Dofetilide: a new class III antiarrhythmic agent. Expert Rev. Cardiovasc. Ther. 5, 9–19. doi: 10.1586/14779072.5.1.9
Schmidt, C., Wiedmann, F., Voigt, N., Zhou, X.-B., Heijman, J., Lang, S., et al. (2015). Upregulation of K2P3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation 132, 82–92. doi: 10.1161/CIRCULATIONAHA.114.012657
Scholz, E. P., Carrillo-Bustamante, P., Fischer, F., Wilhelms, M., Zitron, E., Dössel, O., et al. (2013). Rotor termination is critically dependent on kinetic properties of IKur inhibitors in an in silico model of chronic atrial fibrillation. PLoS ONE 8:e83179. doi: 10.1371/journal.pone.0083179
Schotten, U., Haan, S., de Verheule, S., Harks, E. G. A., Frechen, D., Bodewig, E., et al. (2007). Blockade of atrial-specific K+-currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+-exchange. Cardiovasc. Res. 73, 37–47. doi: 10.1016/j.cardiores.2006.11.024
Sicouri, S., Burashnikov, A., Belardinelli, L., and Antzelevitch, C. (2010). Synergistic electrophysiologic and antiarrhythmic effects of the combination of ranolazine and chronic amiodarone in canine atria. Circ. Arrhythm. Electrophysiol. 3, 88–95. doi: 10.1161/CIRCEP.109.886275
Tamargo, J., Caballero, R., Gómez, R., and Delpón, E. (2009). IKur/Kv1.5 channel blockers for the treatment of atrial fibrillation. Expert Opin. Investig. Drugs 18, 399–416. doi: 10.1517/13543780902762850
Tsujimae, K., Murakami, S., and Kurachi, Y. (2008). In silico study on the effects of IKur block kinetics on prolongation of human action potential after atrial fibrillation-induced electrical remodeling. Am. J. Physiol. Heart Circ. Physiol. 294, H793–H800. doi: 10.1152/ajpheart.01229.2007
Voigt, N., and Dobrev, D. (2016). Atrial-selective potassium channel blockers. Card. Electrophysiol. Clin. 8, 411–421. doi: 10.1016/j.ccep.2016.02.005
Voigt, N., Li, N., Wang, Q., Wang, W., Trafford, A. W., Abu-Taha, I., et al. (2012). Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125, 2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306
Wagoner, D. R. V., Pond, A. L., McCarthy, P. M., Trimmer, J. S., and Nerbonne, J. M. (1997). Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 80, 772–781. doi: 10.1161/01.RES.80.6.772
Wettwer, E., Hála, O., Christ, T., Heubach, J. F., Dobrev, D., Knaut, M., et al. (2004). Role of ikur in controlling action potential shape and contractility in the human atrium influence of chronic atrial fibrillation. Circulation 110, 2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71
Wettwer, E., and Terlau, H. (2014). Pharmacology of voltage-gated potassium channel Kv1.5—impact on cardiac excitability. Curr. Opin. Pharmacol. 15, 115–121. doi: 10.1016/j.coph.2014.02.001
Whittaker, D. G., Ni, H., Harchi, A. E., Hancox, J. C., and Zhang, H. (2017). Atrial arrhythmogenicity of KCNJ2 mutations in short QT syndrome: insights from virtual human atria. PLoS Comput. Biol. 13:e1005593. doi: 10.1371/journal.pcbi.1005593
Wilhelms, M., Maleckar, M. M., Koivumäki, J. T., Dössel, O., and Seemann, G. (2013). Benchmarking electrophysiological models of human atrial myocytes. Front. Comput. Physiol. Med. 3:487. doi: 10.3389/fphys.2012.00487
Wirth, K. J., Brendel, J., Steinmeyer, K., Linz, D. K., Rütten, H., and Gögelein, H. (2007). In vitro and in vivo effects of the atrial selective antiarrhythmic compound AVE1231. J. Cardiovasc. Pharmacol. 49, 197–206. doi: 10.1097/FJC.0b013e318032002f
Woods, C. E., and Olgin, J. (2014). Atrial fibrillation therapy now and in the future drugs, biologicals, and ablation. Circ. Res. 114, 1532–1546. doi: 10.1161/CIRCRESAHA.114.302362
Workman, A. J., Kane, K. A., and Rankin, A. C. (2001). The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc. Res. 52, 226–235. doi: 10.1016/S0008-6363(01)00380-7
Wu, H.-J., Wu, W., Sun, H.-Y., Qin, G.-W., Wang, H.-B., Wang, P., et al. (2011). Acacetin causes a frequency- and use-dependent blockade of hKv1.5 channels by binding to the S6 domain. J. Mol. Cell. Cardiol. 51, 966–973. doi: 10.1016/j.yjmcc.2011.08.022
Yap, Y. G., and Camm, A. J. (2003). Drug induced QT prolongation and torsades de pointes. Heart 89, 1363–1372. doi: 10.1136/heart.89.11.1363
Yuan, Y., Bai, X., Luo, C., Wang, K., and Zhang, H. (2015). The virtual heart as a platform for screening drug cardiotoxicity. Br. J. Pharmacol. 172, 5531–5547. doi: 10.1111/bph.12996
Zhang, H., Garratt, C. J., Zhu, J., and Holden, A. V. (2005). Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc. Res. 66, 493–502. doi: 10.1016/j.cardiores.2005.01.020
Zhu, Y., Kyle, J. W., and Lee, P. J. (2006). Flecainide sensitivity of a Na channel long QT mutation shows an open-channel blocking mechanism for use-dependent block. Am. J. Physiol. Heart Circ. Physiol. 291, H29–H37. doi: 10.1152/ajpheart.01317.2005
Zygmunt, A. C., Nesterenko, V. V., Rajamani, S., Hu, D., Barajas-Martinez, H., Belardinelli, L., et al. (2011). Mechanisms of atrial-selective block of Na+ channels by ranolazine: I. Experimental analysis of the use-dependent block. Am. J. Physiol. Heart Circ. Physiol. 301, H1606–H1614. doi: 10.1152/ajpheart.00242.2011
Keywords: atrial-selective block, atrial fibrillation, sodium and potassium current block, multiscale simulation, synergistic antiarrhythmic effect
Citation: Ni H, Whittaker DG, Wang W, Giles WR, Narayan SM and Zhang H (2017) Synergistic Anti-arrhythmic Effects in Human Atria with Combined Use of Sodium Blockers and Acacetin. Front. Physiol. 8:946. doi: 10.3389/fphys.2017.00946
Received: 31 July 2017; Accepted: 08 November 2017;
Published: 23 November 2017.
Edited by:
Eleonora Grandi, University of California, Davis, United StatesReviewed by:
Christopher Huang, University of Cambridge, United KingdomAndrew G. Edwards, Simula Research Laboratory, Norway
Copyright © 2017 Ni, Whittaker, Wang, Giles, Narayan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henggui Zhang, henggui.zhang@manchester.ac.uk