 Jem D. Lane
Jem D. Lane Andrew Tinker
Andrew Tinker- 1William Harvey Heart Centre, Barts and The London School of Medicine and Dentistry, London, United Kingdom
- 2Department of Cardiac Electrophysiology, Barts Heart Centre, St Bartholomew's Hospital, London, United Kingdom
Anti-arrhythmic drugs are a mainstay in the management of symptoms related to arrhythmias, and are adjuncts in prevention and treatment of life-threatening ventricular arrhythmias. However, they also have the potential for pro-arrhythmia and thus the prediction of arrhythmia predisposition and drug response are critical issues. Clinical trials are the latter stages in the safety testing and efficacy process prior to market release, and as such serve as a critical safeguard. In this review, we look at some of the lessons to be learned from approaches to arrhythmia prediction in patients, clinical trials of drugs used in the treatment of arrhythmias, and the implications for the design of pre-clinical safety pharmacology testing.
Introduction
Cardiac arrhythmias range from the benign to the life-threatening. The former typically arise in patients with structurally and functionally normal hearts, while the latter more commonly arise in those with acquired or genetically-determined abnormalities in cardiac structure or cellular electrophysiology. The two modalities currently available to directly target arrhythmias with the aim of prevention and/or eradication are anti-arrhythmic drugs and catheter ablation. Pharmacotherapy has been around for over 100 years, with quinine one of the first to be used (Sneader, 2005), and ironically, one of the first to be associated with inducing arrhythmia (Schwartz et al., 2016). For the majority of agents in use today, efficacy was based on clinical observation rather than a priori understanding of molecular mechanisms. Anti-arrhythmic drugs have retained a key role in the therapy of heart rhythm disorders, despite the advent of ablation. However, their potential to cause harm through pro-arrhythmic effects has placed constraints on the use of many existing drugs, and restricted the release of new agents to the market. The “catch-22” facing such drugs is the requirement to alter cardiac electrophysiology enough, and under the right circumstances, so as to prevent or terminate arrhythmias. Yet at the same time, they must not do so too much or they risk triggering drug-induced arrhythmias. Thus, it would seem a fine balance has to be achieved. In fact, what is required is detailed knowledge of the mechanisms of the arrhythmia requiring treatment at the cellular, tissue and organ levels, and its vulnerable parameter(s) (Task Force of the Working Group on Arrhythmias of the European Society of Cardiology, 1991; Rosen and Janse, 2010). Even more problematic is that non-cardiac drugs sometimes developed for relatively benign conditions can lead to malignant ventricular arrhythmias (Bednar et al., 2002).
Estimates of the incidence of drug-induced arrhythmia require the patient to come to medical attention, and that the diagnosis be considered. The fact that many have concomitant structural heart disease makes disentangling drug-induced from endogenous arrhythmia difficult. Partly because of this, attention is focused on torsade de pointes (TdP), which outside the setting of long QT syndrome (LQTS) is rare. TdP is also characteristic of non-cardiac drug-induced long QT in patients with normal hearts. With these considerations in mind, estimates have been made (Sarganas et al., 2014).
Safety pharmacology seeks to exclude drugs with a significant risk of pro-arrhythmia. The challenge is to set the threshold at the correct level, so as to allow safe drugs to continue through development and on to the market, and this is dependent on the methods employed in risk assessment. These methods are in the process of being modified, in light of advances in our understanding of cellular electrophysiology, and the models available. In this review, we focus on anti-arrhythmic drugs, and the role of clinical data in informing our approaches to assessment of their risks of pro-arrhythmia. We adhere to the Vaughan-Williams classification system in referring to drugs by class, acknowledging its limitations.
Key Concepts in Cardiac Safety Pharmacology
Ion Channels and Cellular Electrophysiology
Cardiomyocyte electrophysiology serves as the basis for understanding arrhythmia mechanisms, pharmacological anti-arrhythmic and pro-arrhythmic effects. The currents responsible for generating these action potentials differ in atrial and ventricular cardiomyocytes, and sinoatrial/atrioventricular nodal tissue, as well as within different regions of each chamber (Nerbonne and Kass, 2005; Grant, 2009; Figure 1).
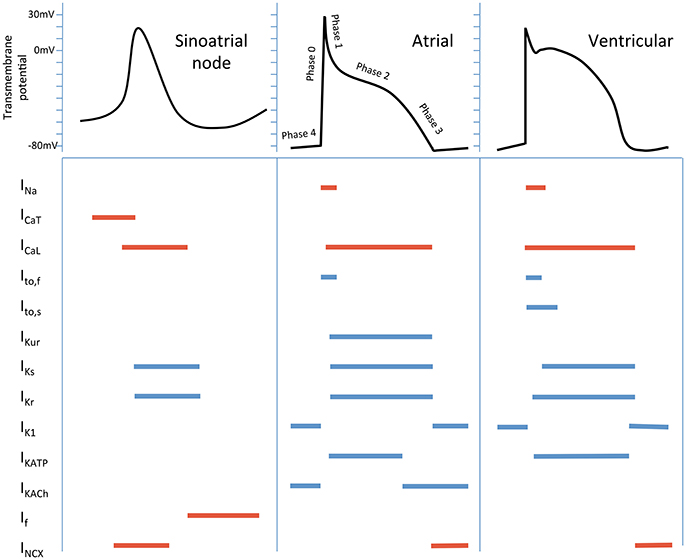
Figure 1. Schematic cardiac action potentials from different regions of the heart, with the currents that generate them. Colored lines indicate the phase of the action potential that the current participates in. Inward currents are in red, outward currents in blue. Currents - INa, inward Na+; ICaT, T-type Ca2+; ICaL, L- type Ca2+; Ito,f, fast transient outward; Ito,s, slow transient outward; IKur, ultra-rapid K+ delayed rectifier; IKs, slow K+ delayed rectifier; IKr, rapid K+ delayed rectifier; IK1, inward rectifier; IKATP, ADP-activated K+ channel; IKACh, muscarinic-gated K+ channel; If, “funny” current; INCX, Na+/Ca2+ exchange current.
Activation results in depolarization of the cellular membrane, which if of sufficient magnitude to attain threshold voltage, leads to generation of an action potential. This may then excite a neighboring cell via gap junctions. If the source current from one cell or group of cells is sufficient to depolarize the neighboring cells (the “sink”), propagation occurs. This cyclical process of transmembrane and intercellular ionic fluxes requires reversal of the activation process, and this is termed repolarization. Refractoriness is a distinct though closely linked concept to repolarization, and describes the state of a cell or tissue which is unexcitable, and unable to undergo depolarization.
Mechanisms of Ventricular Arrhythmias
Traditionally, at a cellular and tissue level these have been divided into disorders of impulse formation, disorders of conduction/propagation, or a combination of both (Zipes et al., 2005). With regards to tachyarrhythmias, the three most common mechanisms are abnormal automaticity, triggered activity and re-entry. The latter two are considered most relevant to ventricular arrhythmias. Triggered activity takes the form of either early (EADs) or delayed afterdepolarizations (DADs). EADs usually occur with delayed repolarization, which can cause “repolarization instability,” rendering cells more susceptible to premature depolarization (Shah et al., 2005). The postulated mechanisms relate either to arrest of repolarization due to diminished outward K+ currents, or abnormal Ca2+ influx, either through L-type calcium channels or the Na+/Ca2+ exchange pump (Pogwizd and Bers, 2004; Shah et al., 2005). They are best described as triggers for TdP in the setting of long QT syndrome (LQTS). DADs occur during phase 4 following completion of repolarization. They result from release of Ca2+ from the sarcoplasmic reticulum, which raises intracellular Ca2+ concentration ([Ca2+]i). The Na+/Ca2+ exchanger extrudes this, with resultant import of Na+ and a net inward current which causes premature depolarization (Nattel and Carlsson, 2006; Figure 2).
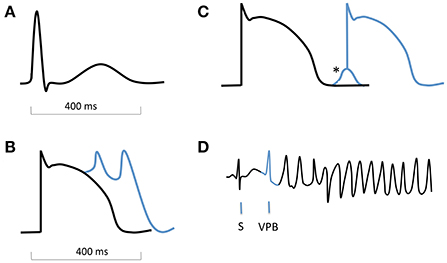
Figure 2. Schematic ECG, action potentials with afterdepolarizations, and onset of torsade de pointes. (A) normal QRS complex and T wave on an ECG. (B) action potential with early afterdepolarizations (EADs). (C) action potential with delayed afterdepolarization (DAD, *). (D) ECG showing onset of TdP, with a sinus beat (S) followed by a ventricular premature beat (VPB, blue) which is triggered by an EAD.
Re-entry refers to a circus movement of wavefront propagation, and wavelength is defined as the product of conduction velocity and effective refractory period (ERP), and as such, it represents the length (or volume) of tissue that is refractory to new impulses. For re-entry to occur, wavelength must be shorter than the re-entrant circuit path length. The difference between these is known as the “excitable gap”—the zone of non-refractory tissue between the wavefront and wavetail. In theory therefore, prolonging wavelength beyond path length should be antiarrhythmic. Indeed, this is the mechanism of “Class III” anti-arrhythmics, though ironically, the discipline of safety pharmacology in relation to anti-arrhythmic drugs has arisen largely as a result of this effect. More complex iterations of re-entry have been proposed, incorporating functional refractoriness. In particular, the “leading circle model”, and rotors are considered important in our attempts to understand complex arrhythmias such as torsade de pointes and ventricular fibrillation (Figure 3). “Substrate” is the term used to refer to abnormal myocardium that either produces triggered activity, or by virtue of fibrosis and/or altered cellular electrophysiology, aids the creation of a path suitable for re-entry, or fosters wave break and rotor formation.
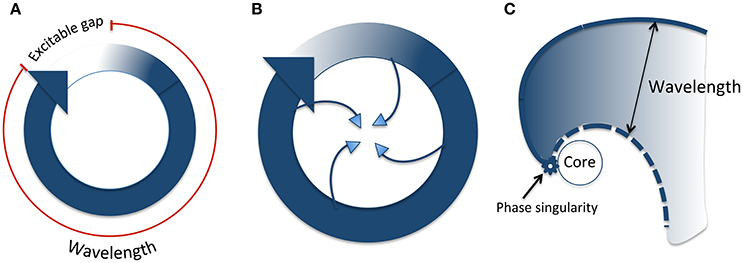
Figure 3. Types of re-entry. (A) Classical anatomical re-entry. The wavelength is the product of conduction velocity and refractory period (shown in red). The excitable gap is the section of the circuit which is unexcited, ahead of the wavefront. (B) Leading circle re-entry. The wavefront impinges on the wavetail such that there is no excitable gap. In addition, centripetal invasion creates a central region of functional refractoriness. (C) Rotor re-entry. The wavefront and wavetail meet at a phase singularity, which rotates around an unexcited core. The wavelength (distance between the wavefront and tail) varies according to distance from the phase singularity. Modified from Pandit and Jalife (2013).
QTc
The QT interval on the electrocardiogram (ECG) reflects the time between depolarization and repolarization of the ventricles. This interval varies with heart rate, so that a correction must be made (QTc). Measurement of the QT interval and adjustment for heart rate (utilizing the R-R interval—the time between successive QRS complexes) are deemed two of the major challenges of electrocardiography (Rautaharju et al., 2009). Various formulae are available, and while based on measurements from only 39 subjects, Bazett's correction is most commonly used (QTc = QT/√(R-R). The QT interval is relied upon as an easily accessed biomarker, reflecting repolarization. Its relevance is borne out by its prolongation in the LQTS, and its presaging TdP. Nevertheless, it has a number of shortcomings (Rautaharju et al., 2009; Sager et al., 2014).
Repolarization Reserve and Risk Modifiers
As with most rare but serious occurrences, a single factor is rarely sufficient on its own to lead to a ventricular arrhythmia. In the case of TdP in particular, this is due to repolarization reserve. This describes a degree of redundancy among repolarizing currents, such that if one is reduced, others may compensate to a degree, maintaining action potential duration, and preventing EADs (Roden and Abraham, 2011). Nevertheless, reserve only protects up to a point, and when several factors act in concert, protection may be lost and arrhythmia may ensue. Ion channel polymorphisms with subclinical effects, impaired clearance of an ion channel-blocking drug, concurrent use of more than one drug, female sex (Makkar et al., 1993; Gaborit et al., 2010) and hormonal derangement (Lane et al., 2012) are just a few of the factors that may modify risk (Figure 4).
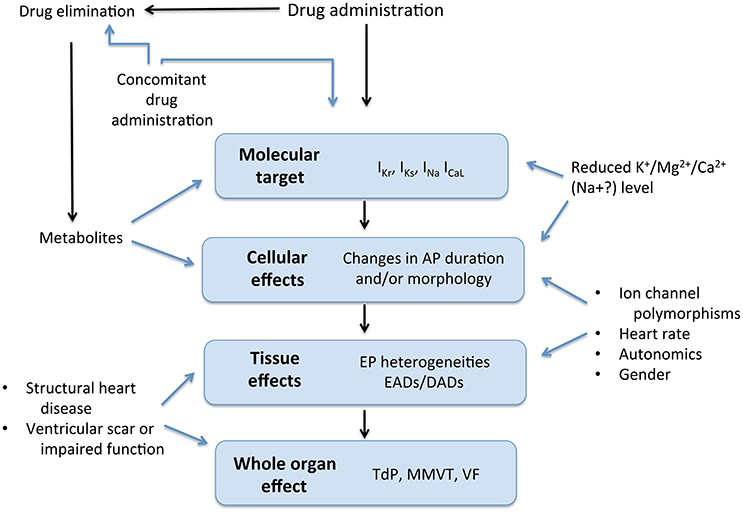
Figure 4. Drug effects and interactions. AP, Action potential; EP, electrophysiological; EAD, early afterdepolarization; DAD, delayed afterdepolarization; TdP, torsade de pointes; MMVT, monomorphic ventricular tachycardia; VF, ventricular fibrillation. Modified from Roden (2004).
Drug-induced Arrhythmias
Whilst the focus of safety pharmacology for anti-arrhythmics is the potential to induce ventricular tachyarrhythmias, it is worth considering other arrhythmias that may result. For example, a number of drugs have been shown to trigger atrial fibrillation (AF) (Strickberger et al., 1997; van der Hooft et al., 2004; Kaakeh et al., 2012). For example, there are good data for adenosine, dobutamine, theophyllines and acute alcohol excess precipitating AF (Strickberger et al., 1997; van der Hooft et al., 2004; Kaakeh et al., 2012). In the setting of reduced clearance or concomitant administration, atrioventricular (AV) nodal-blocking drugs such as beta-blockers and calcium channel antagonists may induce heart block.
Ventricular arrhythmias may occur as a result of therapy with Class I agents (Falk, 1989; The Cardiac Arrhythmia Suppression Trial (CAST) Investigators, 1989; Tisdale and Miller, 2010), though this is exceedingly rarely seen in practice, likely as a result of the restriction of use of these drugs to patients without evidence of QRS or QT prolongation on the ECG, and structurally normal hearts. Closer attention has been paid to drugs that prolong the QT interval due to the risk of precipitating TdP. Probably this largely stems from the fact there are more drugs that affect repolarizing K+ currents than INa, so the incidence of arrhythmias is higher due to more widespread use. It may be that in addition, repolarizing currents have less reserve than does INa, and phase 3 of the action potential is as a result, more vulnerable.
At present, clinical practice relies largely on drug indication, ECG markers, indices of cardiac contractility, electrolyte levels and concurrent use of other medication with QT prolonging effects (Drew et al., 2010) to guide risk assessment. More accurate evidence-based scoring systems have been developed (discussed below). Of the ECG biomarkers available, QRS duration, QT interval, T wave morphology and non-sustained or sustained VT are the most easily assessed and clinically useful (Wellens et al., 2014). For example, there has been interesting work done looking at periodic oscillations in repolarization as measured using the T-wave. The authors found a low frequency oscillation <0.1 Hz associated with sympathetic activity but not heart rate variability or respiratory ventilation. It correlated strongly with outcomes after myocardial infarction (Rizas et al., 2014, 2017). However, it is complex to measure. In the absence of a more readily available and accessible measure of cardiac repolarization, the QT interval has retained its role as an important biomarker despite its many shortcomings (Hondeghem, 2006). However, even without the difficulties in measurement, reliance on this oversimplifies the assessment of drug-induced repolarization disturbance (Hondeghem, 2006).
Pre-clinical Approaches to Screening
A major issue for industry is identifying cardiac risk for non-cardiac drugs: there seems to be little interest in developing new antiarrhythmic agents for ventricular arrhythmia for the reasons discussed below. However, it is also important not to exaggerate potential toxicity and discard potentially useful agents. Until recently, screening for pro-arrhythmia was based on the International Conference on Harmonization non-clinical and clinical evaluation guidelines, S7B and E14, respectively (International Conference on Harmonsation of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2005a,b). Essentially, these focused on measurement of the human ether-a-go-go related gene (hERG) channel current IKr, and the ECG parameter QTc, as means of identifying drugs with the potential to cause TdP. Heterologous expression systems and animal models have been central to pre-clinical screening, with guinea pig, rabbit, dog and monkey being the most utilized species (Friedrichs et al., 2005; Champeroux et al., 2015). And non-rodent models have demonstrated good correlation of in vivo QT measurements with those in humans (Vargas et al., 2015).
Whilst effective at excluding torsadogenic compounds from market release, proposals for a new screening paradigm have come about due to concerns about oversensitivity and low specificity for detecting pro-arrhythmic potential with the S7B/E14 guidelines, as well as a drive to reduce the number of animals involved in experiments (Lu et al., 2008; Sager et al., 2014; NC3Rs). In addition, improvements in understanding of ion channel physiology, species differences in both cardiac electrophysiology and pharmacokinetics (Haushalter et al., 2008), developments in computer modeling, and the advent of stem cell technology, have reached a stage where it is advantageous to try to incorporate them in the process. A new paradigm known as the Comprehensive in vitro Proarrhythmia Assay (CiPA) has therefore been proposed, and is supported by a number of national and international government and commercial bodies (CiPA project, 2000). CiPA recommends a move toward human-based approaches, with screening of multiple ion channels and computer modeling central to this. There is also the aspiration to use human induced pluripotent stem cell (iPSC) models. Overall there is a move away from the emphasis on IKr and the QT interval, due to recognition of the co-dependence and interplay of ionic currents, multichannel effects of drugs (Li et al., 2017), and the shortcomings of the QT interval and importance of other ECG parameters such as the PR and QRS intervals (Sager et al., 2014).
CiPA is still in the process of being validated (Cavero et al., 2016; Colatsky et al., 2016), and has not yet been accepted to supersede the S7B/E14 guidelines. The hope that iPSC derived cardiomyocytes can assume a confirmatory role within the framework, is ambitious and perhaps the least certain of CiPA's four components, given their relative novelty. There is a growing acceptance that these cells are immature compared to native adult myocytes and are more fetal in terms of their electrophysiology and other properties (Veerman et al., 2015; Rodriguez et al., 2016). The latter may be circumvented by use of human cardiac tissue, for example from organ donors (Page et al., 2016), though this is not without its own difficulties, chiefly the lack of availability in many countries. It may be worthwhile to calibrate findings in iPSC cardiomyocytes with those from human myocytes to validate measurements. Nevertheless, a reappraisal of existing guidelines' strengths and weaknesses, and attempts to enhance the accuracy of cardiac safety testing by making use of new techniques and improved understanding, is commendable. And importantly, the new paradigm is being systematically validated prior to implementation.
Clinical Trials on Antiarrhythmic Drugs
Clinical trials have been of paramount importance in the field of safety pharmacology for anti-arrhythmics. Unexpected findings have brought about the widespread use of beta-blockers in heart failure, and the restricted use of many other drugs such as flecainide and sotalol. They enable assessment of hard endpoints rather than surrogates, and provide opportunities to test repolarization and activation reserve in vivo. The main stages in this development process are illustrated in Figure 5. An overview of two of the most important trials is provided, prior to looking at the evidence relating to a number of anti-arrhythmic drugs. Rather than provide an exhaustive account of clinical trials involving anti-arrhythmic drugs, we try to focus on randomized trials that have been instructive in terms of safety, or changed practice.
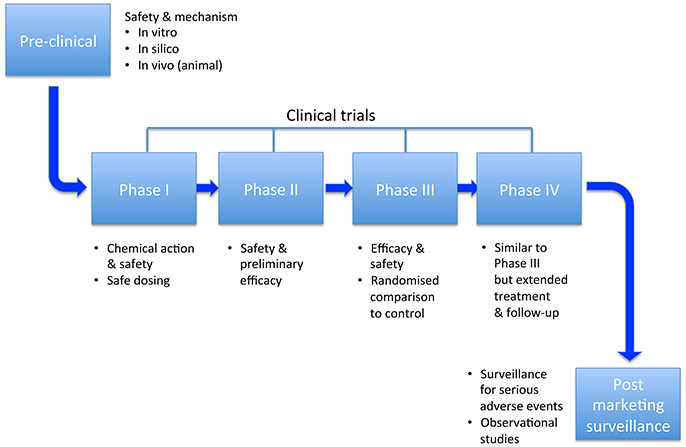
Figure 5. Milestones of research and development.
Cardiac Arrhythmia Suppression Trial (CAST)
This landmark randomized controlled trial (RCT) was both disappointing and hugely influential. To investigate whether suppression of ventricular premature beats (VPBs) in patients following myocardial infarction (MI) reduced their risk of sudden death, patients were assigned to the Na+ channel/INa blockers, encainide, flecainide, moricizine, or placebo (flecainide also has some hERG/IKr-blocking effects, but INa blockade is pharmacodynamically more important). A preliminary report of the drug titration phase in 1989 revealed that despite their apparent suppression of VPBs, there was an excess of arrhythmic deaths in patients assigned to encainide or flecainide (The Cardiac Arrhythmia Suppression Trial (CAST) Investigators, 1989). This was confirmed in the full report of 1498 patients assigned to these two drugs or placebo. An excess of both arrhythmic and non-arrhythmic cardiac deaths were seen (Echt et al., 1991). A few points are noteworthy regarding the study. Firstly, beta-blocker use was low by contemporary standards: between 20 and 30% across all groups. Calcium channel blocker use, primarily diltiazem, was high (47–53%), as was digitalis (16–24%). Secondly, mean baseline left ventricular ejection fraction (LVEF) was 39–40%. The second part of the study comparing moricizine to placebo is less widely discussed, but found similar results (The Cardiac Arrhythmia Suppression Trial II Investigators, 1992). The drug was withdrawn in 2007 (Structural Bioinformatics Group at Charité, 2000). The fallout has resulted in avoidance of flecainide (and other “Class IC” drugs) in patients with “structural heart disease”—particularly ischaemic heart disease with a history of MI, but extrapolated to essentially anyone with any abnormality in ventricular structure and function. The rationale for this has been questioned (Kramer and Josephson, 2010). In terms of possible mechanisms for the observed pro-arrhythmia, slowing of conduction velocity with resultant facilitation of re-entry has been posited (Ruskin, 1989), though late development of ischaemia and accumulation of high drug levels may also have contributed (Aliot et al., 2011).
Survival with Oral d-Sotalol (SWORD) Trial
This RCT recruited a similar patient demographic to CAST: those with LVEF <40% and a history of MI (Waldo et al., 1996). The objective was to evaluate whether a phase 3 K+ channel (hERG/IKr) blocker, d-sotalol, reduced all-cause mortality compared to placebo. The trial was stopped prematurely due to an increased risk of death in the d-sotalol group (5.0 vs. 3.1%, relative risk 1.65, p = 0.006) (Waldo et al., 1996). This was presumed to be primarily due to arrhythmias; unfortunately beyond the fact that the risk of death was higher in women, justification for this assumption could be challenged. In terms of possible mechanisms for arrhythmic death, beta-blocker use was again low pre-randomization (32–33%), and digoxin use was high (48–50%). Importantly though, patients were initiated on 100 mg twice daily of d-sotalol, and if tolerated with a QTc < 520 ms, the dose was increased to 200 mg twice daily. Then, if this dose was tolerated with a QTc < 560 ms, patients remained on this dose for the study's duration. Such QT prolongation is well-established as a risk for TdP (Makkar et al., 1993; Drew et al., 2010), and would be inconceivable in a modern trial.
Amiodarone
Amiodarone interacts with multiple ion channels, resulting in reduced INa, IKr, IKs, ICaL, as well as antagonizing α- and β-adrenoceptors and acetylcholine receptors (Zimetbaum, 2012; Darbar, 2014). It has been studied in a large number of randomized trials in the setting of AF or ventricular arrhythmias (Doval et al., 1994; Julian et al., 1997; Roy et al., 2000; Bardy et al., 2005; Singh et al., 2005; Connolly et al., 2006; Le Heuzey et al., 2010). Paradoxically it often prolongs the QT interval, yet has long been known to have a low incidence of TdP, possibly due to its actions on inward currents (Lazzara, 1989; Vorperian et al., 1997; Roden, 2004). There is a higher risk of bradycardic events nevertheless (Vorperian et al., 1997). More recently, a meta-analysis of over 8,000 patients in RCTs comparing amiodarone to placebo or control found amiodarone was associated with a reduction in sudden cardiac death, though not a significant reduction in overall mortality (Piccini et al., 2009). Notably, in the GESICA trial it was found to confer improved survival in the setting of heart failure (Doval et al., 1994). And in the European Myocardial Infarct Amiodarone Trial (EMIAT), it was demonstrated to reduce arrhythmic deaths by 35% in those with LVEF ≤ 40%, though had no effect on all-cause or cardiac mortality (Julian et al., 1997). The lack of benefit on overall mortality was supported by SCD-HeFT (Bardy et al., 2005). Thus, it is one of the few drugs considered safe for use in patients with a history of MI and reduced LV function.
Beta-Adrenoceptor Antagonists
This class of drugs exerts effects via antagonism of β1 and/or β2-adrenoceptor signaling. β1-adrenoceptors signal via the stimulatory G protein, Gs, and the cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) cascade. PKA increases ICaL, IKs and possibly INa (Brodde and Michel, 1999; Grant, 2009). β2-adrenoceptor signaling is more complex: it also couples to Gs, but can also be induced to couple to Gi, the inhibitory isoform (Xiao et al., 1995). This increases IKACh, and may negatively regulate ICaL (Nagata et al., 2000; Zuberi et al., 2010). β-adrenoceptor antagonists are the most studied anti-arrhythmics, due to their use for both supraventricular and ventricular arrhythmias, as well as heart failure and hypertension (Packer et al., 1996, 2002; MERIT-HF Study Group, 1999; The Cardiac Insufficiency Bisoprolol Study II, 1999; Pedersen et al., 2014; Katritsis et al., 2017). They have been demonstrated to reduce mortality in heart failure, including the risk of sudden cardiac death (Hjalmarson, 1997; MERIT-HF Study Group, 1999; The Cardiac Insufficiency Bisoprolol Study II, 1999; Packer et al., 2002). Risk of pro-arrhythmia is essentially limited to the small risk of AV conduction block, which in the absence of overdose, severe renal dysfunction or concomitant AV nodal-blocking drug use, occurs extremely rarely.
Dofetilide
A “pure” IKr blocker, dofetilide was investigated in patients with severe LV impairment and heart failure as a treatment for AF in the DIAMOND-CHF study (Torp-Pedersen et al., 1999). It performed better than placebo in converting patients with AF to sinus rhythm, though the rate of conversion by 1 month was low (12% vs. 1%). Maintenance of sinus rhythm was also higher for the dofetilide group. It was shown to be associated with a reduced rate of hospitalization for worsening heart failure. However, there was a 3.3% rate of TdP in those treated with the drug. A subsequent trial (DIAMOND-MI) investigated use of the drug in patients with recent MI and LV dysfunction (Køber et al., 2000). Again, there was no effect on all-cause or cardiac mortality, nor on arrhythmic deaths. It showed some efficacy in restoring sinus rhythm in those with AF, but there was a TdP event rate of approximately 1%. Further trials, predominantly in AF and atrial flutter have confirmed its anti-arrhythmic efficacy, but also its pro-arrhythmic potential (Bianconi et al., 2000; Singh et al., 2000). Thus, dofetilide exhibits reasonable anti-arrhythmic efficacy, and does not appear to increase mortality, yet there is a significant risk of TdP such that its use requires close monitoring (Abraham et al., 2015; Schwartz et al., 2016). Therefore, whilst current guidelines indicate it can be used to treat atrial flutter acutely (Katritsis et al., 2017), alternative drug therapy and catheter ablation have rendered this largely obsolete, in Europe at least.
Dronedarone
This multichannel blocking drug is similar to amiodarone but with reduced extra-cardiac effects (Tadros et al., 2016). Despite a promising start in trials such as EURIDIS/ADONIS and ATHENA (Singh et al., 2007; Hohnloser et al., 2009), subsequent trials in patients with permanent AF and heart failure did not support its anti-arrhythmic potency, and moreover, it was associated with worsening of heart failure and increased mortality (Køber et al., 2008; Connolly et al., 2011). Nevertheless, there does not appear to be a significant pro-arrhythmic tendency, reinforcing the notion that drugs with multichannel effects and complex actions can still be safe, in this regard at least.
Alternative and Evolving Clinical Approaches
The preceding discussion has shown that there is room for improvement in prediction of pro-arrhythmia. At the clinical level, strategies can broadly be divided into those focusing on the drugs, and those focusing on patient factors. Haverkamp et al addressed both in 2001 (Haverkamp et al., 2001). They identified many of the clinical risk factors still in use today, and came up with what is to our knowledge the first attempt to stratify drugs according to propensity to induce TdP. The list of drugs was limited, and the classification was not developed. Around the same time, the Georgetown University Center for Education and Research on Therapeutics (GUCERT) was awarded money to investigate the potential of drugs to induce TdP. Subsequently based in Arizona and renamed, AZCERT, a not-for-profit organization published lists of drugs known to be associated with, and causative of QT prolongation and TdP at www.qtdrugs.org. Currently the lists are available at www.crediblemeds.org. Brugadadrugs.org is a similar website set up by the University of Amsterdam Academic Medical Center, providing advice on drugs to avoid, and drugs with possible therapeutic use for patients with Brugada syndrome (University of Amsterdam Academic Medical Center, 2017).
Clinical risk factors for ventricular fibrillation (VF) were evaluated by Da Costa et al in 91 patients with pause-dependent TdP in the setting of QT prolongation. LVEF, presence of structural heart disease, and an index of QT dispersion were found to be significant predictors (Da Costa et al., 2000). And clinical scoring systems based on patient factors have been developed. For example, Tisdale et al utilized data from 900 patients to develop a scoring system to predict QTc prolongation, and then validated this in 300 additional patients (Tisdale et al., 2013). Female sex, diagnosis of MI, sepsis, LV dysfunction, administration of QT-prolonging drugs, use of loop diuretics, serum K+ < 3.5 mEq/L and QT interval on admission >450 ms were identified as independent risk factors. The system had reasonable sensitivity and specificity. Although useful, it utilized a biomarker rather than a patient outcome as an endpoint. Such risk scores are likely to gain importance as electronic prescribing becomes more widespread, with automated alert systems also becoming more feasible (Haugaa et al., 2013).
The ultimate aim of precision medicine is to tailor treatment to the specific patient and the genetic make-up is likely to play a major role in determining this. There have been substantial efforts to understand the genomic architecture of the heritability of the QT interval in the general population. A variety of loci have been identified from genome wide association study (GWAS) findings (Arking et al., 2014). It was shown that one of the signals in the nitric oxide synthase 1 adaptor protein predicted predisposition to drug-induced long QT syndrome (Jamshidi et al., 2012). Furthermore, Strauss and colleagues created a “genetic QT” score, and investigated its ability to predict drug-induced QTc prolongation, and TdP (Strauss et al., 2017). While it was a significant predictor of both, the predictive power was modest, leaving much of the variability unaccounted for.
Implications for Safety Pharmacology
The literature on the use of antiarrhythmic drugs illustrates a number of important points.
There Is No Universal Biomarker Predicting Risk
The QTc remains an important biomarker, though it is far from the only clinical marker of a drug's pro-arrhythmic risk. Clinical trials have demonstrated that amiodarone and beta-blockers remain two of the safest agents in terms of pro-arrhythmia. Their mechanisms are different, yet they both exert anti-arrhythmic effects, and cardiac contra-indications are few. Importantly, amiodarone confounds the predictive power of IKr and QTc screening, by virtue of its APD and QT-prolonging effects, with minimal associated pro-arrhythmic risk. It highlights the oversensitivity of the S7B and E14 guidelines: had it not been in use already, one of the most effective and safe (in arrhythmia terms) drugs may have been excluded from the market. Amiodarone's cardiac safety, together with flecainide's and sotalol's pro-arrhythmogenicity, serve as the strongest reminders of the current importance of clinical trials and post-marketing surveillance in bringing to light unexpected, unpredicted and counterintuitive findings; of how predictions based on theory may not be borne out in practice. But where this is the case, there is an opportunity to learn.
Underlying Patient Pathology Is Important
The presence of pre-existing cardiac conditions such as LV impairment and ischaemic heart disease modulate risk of pro-arrhythmia, such that use of certain drugs, deemed safe in those with structurally normal hearts, is given careful consideration in patients with a history of these conditions. They, and other risk modifiers, such as female sex, diuretic use, hypokalaemia, and concomitant use of other QT-prolonging drugs, identified in clinical reports and risk models (Drew et al., 2010), must be included in in silico models (Wiśniowska and Polak, 2016) if computer modeling and prediction is to fully realize its potential. Identification of drugs with significant risk of arrhythmia may enable us to gain insight into the reasons for this. For example, the list of drugs available at www.crediblemeds.org may have arisen due to similarities in the behavior of the drug molecules in their interaction with ion channels, or alternatively their pharmacokinetics. Ultimately, feedback such as this to pre-clinical models, and clinical trials' validation of these models will hopefully lead, via an iterative process, to greater confidence in the predictive powers of computer, animal and stem cell models (Carusi et al., 2012), with a greater burden of safety testing and prediction occurring in these, rather than in trials in humans. Those models that cannot predict with reasonable accuracy must be honed, or discarded. Increased pre-clinical predictive accuracy should allow more compounds to reach the clinical trial stage. This, together with post-marketing surveillance, will retain a key role in highlighting unexpected findings, due to our inability to completely account for human physiology, pathophysiology, pharmacokinetics, and pharmacodynamics, as well as inter-individual variability in these factors, in any model.
Ion Channels Remodel in Disease and Disease Specific Models May Be Necessary
Many of the experimental and computational approaches rely on the assessment of a compound against parameters or cells derived from healthy normal individuals. However, it is clear that the expression of ion channels significantly remodels in pathological states and this may account for proarrhythmia under such conditions. For example, in atrial fibrillation the expression of L-type calcium currents in the atria is reduced and this in itself generates a substrate for further atrial fibrillation (Wijffels et al., 1995; Gaspo et al., 1997; Yue et al., 1999). It is clear that ion channel remodeling also occurs in many other cardiac pathologies although is not so well defined (Nattel et al., 2007). On this background individual genetic differences are likely to modify the response (Munroe and Tinker, 2015). Thus, we may see the development of disease specific computational models and/or engineered cellular assay systems. In this regard the development of computational approaches to explore models with large numbers of varying parameters is likely to be valuable (Britton et al., 2013, 2017).
Author Contributions
Both authors have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the British Heart Foundation (FS/12/11/29289 and RG/15/15/31742 and facilitated by the NIHR Biomedical Research Centre at Barts.
References
Abraham, J. M., Saliba, W. I., Vekstein, C., Lawrence, D., Bhargava, M., Bassiouny, M., et al. (2015). Safety of oral dofetilide for rhythm control of atrial fibrillation and atrial flutter. Circ. Arrhythm. Electrophysiol. 8, 772–776. doi: 10.1161/CIRCEP.114.002339
Aliot, E., Capucci, A., Crijns, H. J., Goette, A., and Tamargo, J. (2011). Twenty-five years in the making: flecainide is safe and effective for the management of atrial fibrillation. Europace 13, 161–173. doi: 10.1093/europace/euq382
Arking, D. E., Pulit, S. L., Crotti, L., van der Harst, P., Munroe, P. B., Koopmann, T. T., et al. (2014). Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat. Genet. 46, 826–836. doi: 10.1038/ng.3014
Bardy, G., Lee, K., Mark, D., and Poole, J. (2005). Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N. Engl. J. Med. 352, 225–237. doi: 10.1056/NEJMoa043399
Bednar, M. M., Harrigan, E. P., and Ruskin, J. N. (2002). Torsades de pointes associated with nonantiarrhythmic drugs and observations on gender and QTc. Am. J. Cardiol. 89, 1316–1319. doi: 10.1016/S0002-9149(02)02337-8
Bianconi, L., Castro, A., Dinelli, M., Alboni, P., Pappalardo, A., Richiardi, E., et al. (2000). Comparison of intravenously administered dofetilide versus amiodarone in the acute termination of atrial fibrillation and flutter. a multicentre, randomized, double-blind, placebo-controlled study. Eur. Heart J. 21, 1265–1273. doi: 10.1053/euhj.1999.2039
Britton, O. J., Abi-Gerges, N., Page, G., Ghetti, A., Miller, P. E., and Rodriguez, B. (2017). Quantitative comparison of effects of dofetilide, sotalol, quinidine, and verapamil between human ex vivo trabeculae and in silico ventricular models incorporating inter-individual action potential variability. Front. Physiol. 8:597. doi: 10.3389/fphys.2017.00597
Britton, O. J., Bueno-Orovio, A., Van Ammel, K., Lu, H. R., Towart, R., Gallacher, D. J., et al. (2013). Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc. Natl. Acad. Sci. U.S.A. 110, E2098–E2105. doi: 10.1073/pnas.1304382110
Brodde, O. E., and Michel, M. C. (1999). Adrenergic and muscarinic receptors in the human heart. Pharmacol. Rev. 51, 651–690.
Carusi, A., Burrage, K., and Rodríguez, B. (2012). Bridging experiments, models and simulations: an integrative approach to validation in computational cardiac electrophysiology. AJP Hear. Circ. Physiol. 303, H144–H155. doi: 10.1152/ajpheart.01151.2011
Cavero, I., Guillon, J. M., Ballet, V., Clements, M., Gerbeau, J. F., and Holzgrefe, H. (2016). Comprehensive in vitro Proarrhythmia Assay (CiPA): pending issues for successful validation and implementation. J. Pharmacol. Toxicol. Methods 81, 21–36. doi: 10.1016/j.vascn.2016.05.012
Champeroux, P., Thireau, J., Judé, S., Laigot-Barbé, C., Maurin, A., Sola, M. L., et al. (2015). Short-term variability in QT interval and ventricular arrhythmias induced by dofetilide are dependent on high-frequency autonomic oscillations. Br. J. Pharmacol. 172, 2878–2891. doi: 10.1111/bph.13093
CiPA project (2000). Available online at: http://cipaproject.org
Colatsky, T., Fermini, B., Gintant, G., Pierson, J. B., Sager, P., Sekino, Y., et al. (2016). The comprehensive in Vitro proarrhythmia assay (CiPA) initiative - update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20. doi: 10.1016/j.vascn.2016.06.002
Connolly, S. J., Camm, A. J., Halperin, J. L., Joyner, C., Alings, M., Amerena, J., et al. (2011). Dronedarone in high-risk permanent atrial fibrillation. N. Engl. J. Med. 365, 2268–2276. doi: 10.1056/NEJMoa1109867
Connolly, S. J., Dorian, P., Roberts, R. S., Gent, M., Bailin, S., Fain, E. S., et al. (2006). Comparison of β-blockers, amiodarone plus β-blockers, or Sotalol for prevention of shocks from implantable cardioverter defibrillators. the OPTIC study: a randomized trial. JAMA 295, 165–171. doi: 10.1001/jama.295.2.165
Da Costa, A., Chalvidan, T., Belounas, A., Messier, M., Viallet, M., Mansour, H., et al. (2000). Predictive factors of ventricular fibrillation triggered by pause-dependent torsades de pointes associated with acquired long QT interval: role of QT dispersion and left ventricular function. J. Cardiovasc. Electrophysiol. 11, 990–997. doi: 10.1111/j.1540-8167.2000.tb00171.x
Darbar, D. (2014). Cardiac Electrophysiology from Cell to Bedside. Edited by D. Zipes and J. Jalife. Philadelphia, PA: Elsevier Saunders.
Doval, H. C., Nul, D. R., Grancelli, H. O., Perrone, S. V., Bortman, G. R., and Curiel, R. (1994). Randomised trial of low-dose amiodarone in severe congestive heart failure. Grupo de Estudio de la Sobrevida en la insuficiencia Cardiaca en Argentina (GESICA). Lancet 344, 493–498. doi: 10.1016/S0140-6736(94)91895-3
Drew, B. J., Ackerman, M. J., Funk, M., Gibler, W. B., Kligfield, P., Menon, V., et al. (2010). Prevention of torsade de pointes in hospital settings. J. Am. Coll. Cardiol. 55, 934–947. doi: 10.1016/j.jacc.2010.01.001
Echt, D. S., Liebson, P. R., Mitchell, L. B., Peters, R. W., Obias-Manno, D., Barker, A. H., et al. (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. the cardiac arrhythmia suppression trial. N. Engl. J. Med. 324, 781–788. doi: 10.1056/NEJM199103213241201
Falk, R. H. (1989). Flecainide-induced ventricular tachycardia and fibrillation in patients treated for atrial fibrillation. Ann. Intern. Med. 111, 107–111. doi: 10.7326/0003-4819-111-2-107
Friedrichs, G. S., Patmore, L., and Bass, A. (2005). Non-clinical evaluation of ventricular repolarization (ICH S7B): results of an interim survey of international pharmaceutical companies. J. Pharmacol. Toxicol. Methods 52, 6–11. doi: 10.1016/j.vascn.2005.05.001
Gaborit, N., Varro, A., Le Bouter, S., Szuts, V., Escande, D., Nattel, S., et al. (2010). Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J. Mol. Cell. Cardiol. 49, 639–646. doi: 10.1016/j.yjmcc.2010.06.005
Gaspo, R., Bosch, R. F., Talajic, M., and Nattel, S. (1997). Functional mechanisms underlying tachycardia-induced sustained atrial fibrillation in a chronic dog model. Circulation 96, 4027–4035. doi: 10.1161/01.CIR.96.11.4027
Grant, A. O. (2009). Cardiac ion channels. Circ. Arrhythm. Electrophysiol. 2, 185–194. doi: 10.1161/CIRCEP.108.789081
Haugaa, K. H., Bos, J. M., Tarrell, R. F., Morlan, B. W., Caraballo, P. J., and Ackerman, M. J. (2013). Institution-wide QT alert system identifies patients with a high risk of mortality. Mayo Clin. Proc. 88, 315–325. doi: 10.1016/j.mayocp.2013.01.013
Haushalter, T. M., Friedrichs, G. S., Reynolds, D. L., Barecki-Roach, M., Pastino, G., Hayes, R., et al. (2008). The cardiovascular and pharmacokinetic profile of dofetilide in conscious telemetered beagle dogs and cynomolgus monkeys. Br. J. Pharmacol. 154, 1457–1464. doi: 10.1038/bjp.2008.275
Haverkamp, W., Eckardt, L., Monnig, G., Schulze-Bah, E., Wedekin, H., Kirchhof, P., et al. (2001). Clinical aspects of ventricular arrhythmias associated with QT prolongation. Eur. Hear. J. Suppl. 3, 81–88. doi: 10.1016/S1520-765X(01)90010-0
Hjalmarson, A. (1997). Effects of beta blockade on sudden cardiac death during acute myocardial infarction and the postinfarction period. Am. J. Cardiol. 80, 35J–39J.
Hohnloser, S. H., Crijns, H. J., van Eickels, M., Gaudin, C., Page, R. L., Torp-Pedersen, C., et al. (2009). Effect of dronedarone on cardiovascular events in atrial fibrillation. N. Engl. J. Med. 360, 668–678. doi: 10.1056/NEJMoa0803778
Hondeghem, L. M. (2006). Thorough QT/QTc not so thorough: removes torsadogenic predictors from the T-wave, incriminates safe drugs, and misses profibrillatory drugs. J. Cardiovasc. Electrophysiol. 17, 337–340. doi: 10.1111/j.1540-8167.2006.00347.x
International Conference on Harmonsation of Technical Requirements for Registration of Pharmaceuticals for Human Use (2005a). The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. Available online at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073153.pdf
International Conference on Harmonsation of Technical Requirements for Registration of Pharmaceuticals for Human Use (2005b). The Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals. Available online at: http://www.ich.org/products/guidelines/safety/article/safety-guidelines.html
Jamshidi, Y., Nolte, I. M., Dalageorgou, C., Zheng, D., Johnson, T., Bastiaenen, R., et al. (2012). Common variation in the NOS1AP gene is associated with drug-induced QT prolongation and ventricular arrhythmia. J. Am. Coll. Cardiol. 60, 841–850. doi: 10.1016/j.jacc.2012.03.031
Julian, D. G., Camm, A. J., Frangin, G., Janse, M. J., Munoz, A., Schwartz, P. J., et al. (1997). Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet 349, 667–674. doi: 10.1016/S0140-6736(96)09145-3
Kaakeh, Y., Overholser, B. R., Lopshire, J. C., and Tisdale, J. E. (2012). Drug-induced atrial fibrillation. Drugs 72, 1617–1630. doi: 10.2165/11633140-000000000-00000
Katritsis, D. G., Boriani, G., Cosio, F. G., Hindricks, G., Jais, P., Josephson, M. E., et al. (2017). European Heart Rhythm Association (EHRA) consensus document on the management of supraventricular arrhythmias, endorsed by Heart Rhythm Society (HRS), Asia-Pacific Heart Rhythm Society (APHRS), and sociedad latinoamericana de estimulacion cardiaca y elect. Europace 19, 465–511. doi: 10.1093/europace/euw301
Køber, L., Bloch Thomsen, P. E., Møller, M., Torp-Pedersen, C., Carlsen, J., Sandøe, E., et al. (2000). Dofetilide in patients with recent myocardial infarction and left-ventricular dysfunction: a randomised trial. Lancet 356, 2052–2058. doi: 10.1016/S0140-6736(00)03402-4
Køber, L., Torp-Pedersen, C., McMurray, J. J., Gøtzsche, O., Lévy, S., Crijns, H., et al. (2008). Increased mortality after dronedarone therapy for severe heart failure. N. Engl. J. Med. 358, 2678–2687. doi: 10.1056/NEJMoa0800456
Kramer, D. B., and Josephson, M. E. (2010). Three questions for evidence-based cardiac electrophysiology. Circ. Cardiovasc. Qual. Outcomes 3, 704–709. doi: 10.1161/CIRCOUTCOMES.110.957381
Lane, J. D., Keenan, N. G., Bouloux, P., and Rogers, D. (2012). A heart without hormones. Lancet 379:1922. doi: 10.1016/s0140-6736(12)60314-6
Lazzara, R. (1989). Amiodarone and torsade de pointes. Ann. Intern. Med. 111, 549–551. doi: 10.7326/0003-4819-111-7-549
Le Heuzey, J. Y., De Ferrari, G. M., Radzik, D., Santini, M., Zhu, J., and Davy, J. M. (2010). A short-term, randomized, double-blind, parallel-group study to evaluate the efficacy and safety of dronedarone versus amiodarone in patients with persistent atrial fibrillation: the DIONYSOS study. J. Card. Electrophysiol. 21, 597–605. doi: 10.1111/j.1540-8167.2010.01764.x
Li, Z., Dutta, S., Sheng, J., Tran, P. N., Wu, W., Chang, K., et al. (2017). Improving the in silico assessment of proarrhythmia risk by combining hERG (Human Ether-à-go-go-Related Gene) channel-drug binding kinetics and multichannel pharmacology. Circ. Arrhythm. Electrophysiol. 10:e004628. doi: 10.1161/CIRCEP.116.004628
Lu, H. R., Vlaminckx, E., Hermans, A. N., Rohrbacher, J., Van Ammel, K., Towart, R., et al. (2008). Predicting drug-induced changes in QT interval and arrhythmias: QT-shortening drugs point to gaps in the ICHS7B Guidelines. Br. J. Pharmacol. 154, 1427–1438. doi: 10.1038/bjp.2008.191
Makkar, R. R., Fromm, B. S., Steinman, R. T., Meissner, M. D., and Lehmann, M. H. (1993). Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 270, 2590–2597. doi: 10.1001/jama.1993.03510210076031
MERIT-HF Study Group (1999). Effect of metoprolol CR/XL in chronic heart failure : metoprolol CR/XL randomised intervention trial in congestive heart failure (MERIT-HF). Lancet 353, 2001–2007.
Munroe, P. B., and Tinker, A. (2015). Genome-wide association studies and contribution to cardiovascular physiology. Physiol. Genomics 47, 365–375. doi: 10.1152/physiolgenomics.00004.2015
Nagata, K., Ye, C., Jain, M., Milstone, D. S., Liao, R., and Mortensen, R. M. (2000). Gαi2 but not Gαi3 is required for muscarinic inhibition of contractility and calcium currents in adult cardiomyocytes. Circ. Res. 87, 903–909. doi: 10.1161/01.RES.87.10.903
Nattel, S., and Carlsson, L. (2006). Innovative approaches to anti-arrhythmic drug therapy. Nat. Rev. Drug Discov. 5, 1034–1049. doi: 10.1038/nrd2112
Nattel, S., Maguy, A., Le Bouter, S., and Yeh, Y. H. (2007). Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 87, 425–456. doi: 10.1152/physrev.00014.2006
NC3Rs. National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs). Available online at: https://www.nc3rs.org.uk/the-3rs.
Nerbonne, J. M., and Kass, R. S. (2005). Molecular Physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. doi: 10.1152/physrev.00002.2005
Packer, M., Bristow, M. R., Cohn, J. N., Colucci, W. S., Fowler, M. B., Gilbert, E. M., et al. (1996). The effect of carvedilol on mortality and morbidity in patients with chronic heart failure. N. Engl. J. Med. 334, 1349–1355. doi: 10.1056/NEJM199605233342101
Packer, M., Fowler, M. B., Roecker, E. B., Coats, A. J., Katus, H. A., Krum, H., et al. (2002). Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 106, 2194–2199. doi: 10.1161/01.CIR.0000035653.72855.BF
Page, G., Ratchada, P., Miron, Y., Steiner, G., Ghetti, A., Miller, P. E., et al. (2016). Human ex vivo action potential model for pro-arrhythmia risk assessment. J. Pharmacol. Toxicol. Methods 81, 183–195. doi: 10.1016/j.vascn.2016.05.016
Pandit, S. V., and Jalife, J. (2013). Rotors and the dynamics of cardiac fibrillation. Circ. Res. 112, 849–862. doi: 10.1161/CIRCRESAHA.111.300158
Pedersen, C. T., Kay, G. N., Kalman, J., Borggrefe, M., Della-Bella, P., Dickfeld, T., et al. (2014). EHRA/HRS/APHRS expert consensus on ventricular arrhythmias. Heart Rhythm 11, e166–e196. doi: 10.1016/j.hrthm.2014.07.024
Piccini, J. P., Berger, J. S., and O'Connor, C. M. (2009). Amiodarone for the prevention of sudden cardiac death: a meta-analysis of randomized controlled trials. Eur. Heart J. 30, 1245–1253. doi: 10.1093/eurheartj/ehp100
Pogwizd, S. M., and Bers, D. M. (2004). Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 14, 61–66. doi: 10.1016/j.tcm.2003.12.002
Rautaharju, P. M., Surawicz, B., and Gettes, L. S. (2009). AHA/ACCF/HRS recommendations for the standardization and interpretation of the electrocardiogram. Part VI: the ST segment, T and U waves, and the QT interval. J. Am. Coll. Cardiol. 119, e241–e250. doi: 10.1161/CIRCULATIONAHA.108.191096
Rizas, K. D., McNitt, S., Hamm, W., Massberg, S., Kääb, S., Zareba, W., et al. (2017). Prediction of sudden and non-sudden cardiac death in post-infarction patients with reduced left ventricular ejection fraction by periodic repolarization dynamics: MADIT-II substudy. Eur. Heart J. 38, 2110–2118. doi: 10.1093/eurheartj/ehx161
Rizas, K. D., Nieminen, T., Barthel, P., Zürn, C. S., Kähönen, M., Viik, J., et al. (2014). Sympathetic activity-associated periodic repolarization dynamics predict mortality following myocardial infarction. J. Clin. Invest. 124, 1770–1780. doi: 10.1172/JCI70085
Roden, D. M. (2004). Drug-induced prolongation of the QT interval. N. Engl. J. Med. 350, 1013–1022. doi: 10.1056/NEJMra032426
Roden, D. M., and Abraham, R. L. (2011). Refining repolarization reserve. Hear. Rhythm 8, 1756–1757. doi: 10.1016/j.hrthm.2011.06.024
Rodriguez, B., Carusi, A., Abi-Gerges, N., Ariga, R., Britton, O., Bub, G., et al. (2016). Human-based approaches to pharmacology and cardiology: an interdisciplinary and intersectorial workshop. Europace 18, 1287–1298. doi: 10.1093/europace/euv320
Rosen, M. R., and Janse, M. J. (2010). Concept of the vulnerable parameter: the sicilian gambit revisited. J. Cardiovasc. Pharmacol. 55, 428–437. doi: 10.1097/FJC.0b013e3181bfaddc
Roy, D., Talajic, M., Dorian, P., Connolly, S., Eisenberg, M. J., Green, M., et al. (2000). Amiodarone to prevent recurrence of atrial fibrillation. canadian trial of atrial fibrillation investigators. N. Engl. J. Med. 342, 913–920. doi: 10.1056/NEJM200003303421302
Ruskin, J. N. (1989). Editorial: the cardiac arrhythmia suppression trial (CAST). N. Engl. J. Med. 321, 386–388. doi: 10.1056/NEJM198908103210608
Sager, P. T., Gintant, G., Turner, J. R., Pettit, S., and Stockbridge, N. (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the cardiac safety research consortium. Am. Heart J. 167, 292–300. doi: 10.1016/j.ahj.2013.11.004
Sarganas, G., Garbe, E., Klimpel, A., Hering, R. C., Bronder, E., and Haverkamp, W. (2014). Epidemiology of symptomatic drug-induced long QT syndrome and torsade de pointes in Germany. Europace 16, 101–108. doi: 10.1093/europace/eut214
Schwartz, P. J., Woosley, R. L., and Woosley, R. L. (2016). Predicting the unpredictable: drug-induced QT prolongation and torsades de pointes. J. Am. Coll. Cardiol. 67, 1639–1650. doi: 10.1016/j.jacc.2015.12.063
Shah, M., Akar, F. G., and Tomaselli, G. F. (2005). Molecular basis of arrhythmias. Circulation 112, 2517–2529. doi: 10.1161/CIRCULATIONAHA.104.494476
Singh, B. N., Connolly, S. J., Crijns, H. J. G. M., Roy, D., Kowey, P. R., Capucci, A., et al. (2007). Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N. Engl. J. Med. 357, 987–999. doi: 10.1056/NEJMoa054686
Singh, B. N., Singh, S. N., Reda, D. J., Tang, X. C., Lopez, B., Harris, C. L., et al. (2005). Amiodarone versus sotalol for atrial fibrillation. N. Engl. J. Med. 352, 1861–1872. doi: 10.1056/NEJMoa041705
Singh, S., Zoble, R. G., Yellen, L., Brodsky, M. A., Feld, G. K., Berk, M., et al. (2000). Efficacy and safety of oral dofetilide in converting to and maintaining sinus rhythm in patients with chronic atrial fibrillation or atrial flutter: the symptomatic atrial fibrillation investigative research on dofetilide (SAFIRE-D) study. Circulation 102, 2385–2390. doi: 10.1161/01.CIR.102.19.2385
Strauss, D. G., Vicente, J., Johannesen, L., Blinova, K., Mason, J. W., Weeke, P., et al. (2017). A common genetic variant risk score is associated with drug-induced QT prolongation and torsade de pointes risk: a Pilot study. Circulation 135, 1300–1310. doi: 10.1161/CIRCULATIONAHA.116.023980
Strickberger, S. A., Man, K. C., Daoud, E. G., Goyal, R., Brinkman, K., Knight, B. P., et al. (1997). Adenosine-induced atrial arrhythmia: a prospective analysis. Ann. Intern. Med. 127, 417–422. doi: 10.7326/0003-4819-127-6-199709150-00001
Structural Bioinformatics Group at Charité (2000). WITHDRAWN: A Resource for Withdrawn and Discontinued Drugs. Available online at: http://cheminfo.charite.de/withdrawn/
Tadros, R., Nattel, S., and Andrade, J. G. (2016). Dronedarone: basic pharmacology and clinical use. Card. Electrophysiol. Clin. 8, 453–465. doi: 10.1016/j.ccep.2016.02.008
Task Force of the Working Group on Arrhythmias of the European Society of Cardiology (1991). The sicilian gambit. a new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Circulation 84, 1831–1851.
The Cardiac Arrhythmia Suppression Trial (CAST) Investigators (1989). Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N. Engl. J. Med. 321, 406–412.
The Cardiac Arrhythmia Suppression Trial II Investigators. (1992). Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N. Engl. J. Med. 327, 227–233.
The Cardiac Insufficiency Bisoprolol Study II. (1999). (CIBIS-II): a randomised trial. Lancet 353, 9–13.
Tisdale, J. E., Jaynes, H. A., Kingery, J. R., Mourad, N. A., Trujillo, T. N., Overholser, B. R., et al. (2013). Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ. Cardiovasc. Qual. Outcomes 6, 479–487. doi: 10.1161/CIRCOUTCOMES.113.000152
Tisdale, J. E., and Miller, D. A. (2010). Drug-Induced Diseases. Prevention, Detection and Management. Bethesda, MD: American Society of Health-System Pharmacists.
Torp-Pedersen, C., Møller, M., Bloch-Thomsen, P. E., Køber, L., Sandøe, E., Egstrup, K., et al. (1999). Dofetilide in patients with congestive heart failure and left ventricular dysfunction. N. Engl. J. Med. 341, 857–865. doi: 10.1056/NEJM199909163411201
University of Amsterdam Academic Medical Center (2017). Available online at: http://www.brugadadrugs.org
van der Hooft, C. S., Heeringa, J., van Herpen, G., Kors, J. A., Kingma, J. H., and Stricker, B. H. (2004). Drug-induced atrial fibrillation. J. Am. Coll. Cardiol. 44, 2117–2124. doi: 10.1016/j.jacc.2004.08.053
Vargas, H. M., Bass, A. S., Koerner, J., Matis-Mitchell, S., Pugsley, M. K., Skinner, M., et al. (2015). Evaluation of drug-induced QT interval prolongation in animal and human studies: a literature review of concordance. Br. J. Pharmacol. 172, 4002–4011. doi: 10.1111/bph.13207
Veerman, C. C., Kosmidis, G., Mummery, C. L., Casini, S., Verkerk, A. O., and Bellin, M. (2015). Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. 24, 1035–1052. doi: 10.1089/scd.2014.0533
Vorperian, V. R., Havighurst, T. C., Miller, S., and January, C. T. (1997). Adverse effects of low dose-amiodarone: a meta-analysis. J. Am. Coll. Cardiol. 30, 791–798. doi: 10.1016/S0735-1097(97)00220-9
Waldo, A. L., Camm, A. J., deRuyter, H., Friedman, P. L., MacNeil, D. J., Pauls, J. F., et al. (1996). Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet 348, 7–12. doi: 10.1016/S0140-6736(96)02149-6
Wellens, H. J., Schwartz, P. J., Lindemans, F. W., Buxton, A. E., Goldberger, J. J., Hohnloser, S. H., et al. (2014). Risk stratification for sudden cardiac death: Current status and challenges for the future. Eur. Heart J. 35, 1642–1651. doi: 10.1093/eurheartj/ehu176
Wijffels, M. C., Kirchhof, C. J., Dorland, R., and Allessie, M. A. (1995). Atrial fibrillation begets atrial fibrillation. a study in awake chronically instrumented goats. Circulation 92, 1954–1968. doi: 10.1161/01.CIR.92.7.1954
Wiśniowska, B., and Polak, S. (2016). The role of interaction model in simulation of drug interactions and QT prolongation. Curr. Pharmacol. Rep. 2, 339–344. doi: 10.1007/s40495-016-0075-9
Xiao, R. P., Ji, X., and Lakatta, E. G. (1995). Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol. Pharmacol. 47, 322–329.
Yue, L., Melnyk, P., Gaspo, R., Wang, Z., and Nattel, S. (1999). Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ. Res. 84, 776–784. doi: 10.1161/01.RES.84.7.776
Zimetbaum, P. (2012). Antiarrhythmic drug therapy for atrial fibrillation. Circulation 125, 381–389. doi: 10.1161/CIRCULATIONAHA.111.019927
Zipes, D., Libby, P., Bonow, R., and Braunwald, E. (2005). Braunwald's Heart Disease. A Textbook of Cardiovascular Medicine. Philadelphia, PA: Elsevier Saunders.
Keywords: arrhythmias, cardiac, torsades de pointes, anti-arrhythmia agents, cardiac ion channels, long QT
Citation: Lane JD and Tinker A (2017) Have the Findings from Clinical Risk Prediction and Trials Any Key Messages for Safety Pharmacology? Front. Physiol. 8:890. doi: 10.3389/fphys.2017.00890
Received: 26 June 2017; Accepted: 20 October 2017;
Published: 06 November 2017.
Edited by:
Stefano Morotti, University of California, Davis, United StatesReviewed by:
Hugo M. Vargas, Amgen, United StatesNajah Abi Gerges, AnaBios Corporation, Inc., United States
Copyright © 2017 Lane and Tinker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew Tinker, YS50aW5rZXJAcW11bC5hYy51aw==