 Daniela Guarino1,2,3
Daniela Guarino1,2,3 Giorgio Iervasi
Giorgio Iervasi Rosa Maria Bruno
Rosa Maria Bruno
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 14 September 2017
Sec. Autonomic Neuroscience
Volume 8 - 2017 | https://doi.org/10.3389/fphys.2017.00665
This article is part of the Research TopicNeurocardiovascular Diseases: New Aspects of the Old IssuesView all 16 articles
Obesity is reaching epidemic proportions globally and represents a major cause of comorbidities, mostly related to cardiovascular disease. The autonomic nervous system (ANS) dysfunction has a two-way relationship with obesity. Indeed, alterations of the ANS might be involved in the pathogenesis of obesity, acting on different pathways. On the other hand, the excess weight induces ANS dysfunction, which may be involved in the haemodynamic and metabolic alterations that increase the cardiovascular risk of obese individuals, i.e., hypertension, insulin resistance and dyslipidemia. This article will review current evidence about the role of the ANS in short-term and long-term regulation of energy homeostasis. Furthermore, an increased sympathetic activity has been demonstrated in obese patients, particularly in the muscle vasculature and in the kidneys, possibily contributing to increased cardiovascular risk. Selective leptin resistance, obstructive sleep apnea syndrome, hyperinsulinemia and low ghrelin levels are possible mechanisms underlying sympathetic activation in obesity. Weight loss is able to reverse metabolic and autonomic alterations associated with obesity. Given the crucial role of autonomic dysfunction in the pathophysiology of obesity and its cardiovascular complications, vagal nerve modulation and sympathetic inhibition may serve as therapeutic targets in this condition.
Obesity is a challenge for global public health. The worldwide prevalence of obesity has nearly doubled in the past decades (World Health Organization). Obesity may induce the onset of other conditions leading to overt cardiovascular disease, such as glucose intolerance, dyslipidemia, impaired glucose tolerance and type 2 diabetes, hypertension, and kidney failure (Martin-Rodriguez et al., 2015; Soares et al., 2015).
In this framework, there is a strong need to reach a deeper understanding of the basic mechanisms coupling energy balance with glucose homeostasis (Flier, 2001; Obici and Rossetti, 2003), in order to develop new treatments able to counteract obesity and thus decrease the risk of cardiovascular disease. The autonomic nervous system (ANS) plays a major role in the integrated regulation of food intake, involving satiety signals and energy expenditure: thus ANS dysregulation might favor body weight gain. Conversely, obesity might trigger alterations in the sympathetic regulation of cardiovascular function, thus favoring the development of cardiovascular complications and events. This article is aimed at reviewing the role of ANS in the pathophysiology of obesity, and thus to identify possible new therapeutic targets for the treatment of obesity and its complications.
Body weight is regulated by a complex homeostatic system, whose main components are the modulation of appetite and satiety and the modulation of energy expenditure and energy storage in the adipose tissue. This homeostatic system is aimed at maintaining a stable body weight and requires the existence of a network of signals conveying information from the periphery to the central nervous system (CNS), where these signals are integrated and contribute to long-term and short-term regulation of body weight (Cummings and Schwartz, 2003). Peripheral signals involved in energy homeostasis can be classified as short-acting signals, such as gastric distension and gut hormone release, which are acutely affected by ingested nutrients and modulating satiety, and long-acting signals, such as leptin and insulin, which regulate overall body weight and adiposity.
It is clear that any dysfunction in the pathways involved in maintaining body weight homeostasis may lead to weight gain and obesity. The ANS plays a central role in the communication between the CNS and the gastrointestinal system either in short-term or in long-term regulation of body weight (Figure 1). Going into detail, vagal afferents to the brain are crucial for information transfer from gut hormones and CNS and as a mediator of sense of satiety after gastric distension.
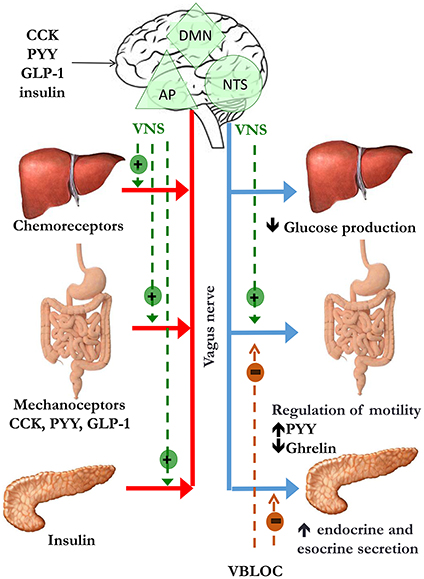
Figure 1. Peripheral signals of satiety and gastric emptying reach the nucleus of the solitary tract/area postrema complex (NST/AP) via afferent vagal nerves (red line). The NTS projects to the dorsal motor nucleus (DMN). This pathway modulates intestinal motility and secretion, glucose production and pancreatic secretion via efferent vagal nerves (blue line). The suggested site of action of vagal nerve stimulation (VNS) is indicated by the dotted green lines, while mechanism of weight loss hypothesized vagal nerve blockade includes decrease in gastric emptying, increase in gut hormones release and inhibition of pancreatic esocrine secretion (dotted orange lines).
The main mediators of short-term regulation of body weight through the sensation of satiety are:
- Gastric distension (mediated by vagal afferents) (Figure 1);
- Gut hormones release. Indeed the gastrointestinal tract, in addition to its primary role in digestion and adsorption of nutrients, regulates food ingestion by gut hormones. Interestingly, part of their action is mediated by vagal afferents. The action of gut hormones on vagal afferent neurons is now recognized to be an early step in controlling nutrient delivery to the intestine by regulating food intake and gastric emptying. Therefore, gut hormones and vagal afferent neurons have been considered playing an important role in the pathogenesis of obesity (Dockray, 2014).
Satiety is a result of neuro-humoral stimuli generated during food intake, leading to control of meal size and termination (Woods et al., 1998): thus it is not surprising that an altered sense of satiety has been involved in the pathogenesis of obesity. The main hypothalamic areas involved in the control of both hunger and satiety are the arcuate nucleus (ARC), the paraventricular nucleus, the dorsomedial and ventromedial hypothalamus, and the lateral hypothalamic area. These areas are influenced by different peripheral signals coming from the liver and gut, the endocrine pancreas and the adipocytes, which could act directly on neurons in the CNS or through afferent neurons. Indeed, the afferent vagal pathways are probably the most important link between the gut and the brain for satiety signal modulation (Berthoud, 2008a). Vagal afferent neurons receive post-ingestive information from the gastrointestinal tract by mechanoreceptor stimulation (Ikramuddin et al., 2014) in response to gastric distension, by gut hormone release in response to nutritional composition of food consumed, and by direct action of some nutrients, such as short chain fatty acids (Baskin et al., 1999; Obici et al., 2002; Brown et al., 2006; Capasso and Izzo, 2008; Shin et al., 2009; Scherer et al., 2011; Iwasaki et al., 2013). Finally, vagal afferents receive metabolic information by chemoreceptors located in the hepatoportal system (Yi et al., 2010; Figure 1). Signals from peripheral receptors reach via vagal afferents the nucleus of the solitary tract/area postrema (NTS/AP) complex in the brain stem, which integrates sensory information from the gastrointestinal tract and abdominal viscera and taste information from the oral cavity (Travers et al., 1987). NTS projects back to the gut via vago-vagal autonomic reflexes through the dorsal motor nucleus. The stimulation of this pathway leads to gut responses, including control of intestinal transit time and motility (i.e., delayed gastric emptying) (Forster et al., 1990), absorption rate and exposure of enteroendocrine cells (EECs) to nutrients, with changes in gastrointestinal hormones and pancreatic secretion, involved in satiety (Li and Owyang, 1994; Berthoud, 2008b).
Cholecystokinin (CCK) is an anorectic hormone secreted by different tissues, including the I-cells of the small intestine (Buffa et al., 1976), with the main effect of reducing meal size and duration (Kissileff et al., 2003). Its release pattern suggests that CCK plays a role in meal termination and early phase satiety (Burton-Freeman et al., 2002).
CCK binds A-type receptors, found either in the periphery or in the brain, and B-type receptors, found only in the brain (Fink et al., 1998). CCK may act directly on the CNS (Blessing, 1997) and/or peripherally via vagal afferent fibers (Corp et al., 1993; Burdyga et al., 2003). Some authors reported that the main mechanism trough which CCK regulates food intake is the inhibition of gastric emptying (Moran and Kinzig, 2004). Furthermore, Wank (1995) and Granger et al. (1980) CCK induces gastrointestinal vasodilation acting on CCK-A receptors placed on abdominal vagal afferents projecting to NTS. This pathway involves also caudal and rostral ventrolateral medulla neurons, thus leading to suppression of sympathetic vascular tone (Sartor and Verberne, 2002, 2006, 2008). The role of alteration of CCK secretion in obesity is uncertain: indeed, obese patients exhibit higher CCK plasmatic levels that lean individuals, either in fasting conditions or after a high-fat meal (Little et al., 2005).
Peptide YY (PYY) is released by the L-cells of the gastrointestinal tract, in response to a meal in proportion to calories, and to luminal content of fatty acids, fibers and bile acid (Adrian et al., 1985; Onaga et al., 2002). Its actions in the brainstem and in the gut are mediated by Y1 and Y2 receptors (Yang, 2002). PYY acts mainly via the Y2 receptor (Dumont et al., 1995), identified on both intestinal vagal afferents and within the ARC: both pathways may thus be involved in the anorectic effects of Y2 receptor activation (Fetissov et al., 2004; Koda et al., 2005). Central and peripheral specific binding sites of PYY have been identified in NTS/AP and in dorsal motor nucleus (Parker and Herzog, 1999), as well as in in enterocytes, myenteric and submucosal neurons (Cox, 2007a,b). PYY release in the post-prandial period seems to be induced also by the indirect stimulation of endocrine L-cells through vagal neural pathways (Fu-Cheng et al., 1997; Lin and Taylor, 2004). In animal models, PYY release was blocked by atropine, a nicotinic ganglionic blocker (Lin and Taylor, 2004), while intravenous administration of bethanechol (a muscarinic cholinergic agonist) stimulated PYY release (Dumoulin et al., 1995). PYY acts also as a counterregulatory hormone for ghrelin release via growth hormone secretagogue receptor, expressed in the nodose ganglion of vagal nerves (Neary et al., 2003) and in the ARC. PYY plasma concentrations are lower in obese in comparison to lean individuals either in the fasting period (Batterham et al., 2003) or in the post-prandial period (le Roux et al., 2006). The latter phenomenon could be responsible of impaired satiety signal in obesity, since PYY infusion reduces caloric intake both in obese and lean individuals (Batterham et al., 2003). Experimental data suggest that electrical vagal stimulation may increase PYY secretion from the isolated ileum in pigs (Sheikh et al., 1989).
Pancreatic Polypeptide (PP) is secreted by cells located at the periphery of the pancreatic islets, in the esocrine pancreas and distal gut (Track, 1980; Ekblad and Sundler, 2002) in response to food intake. PP has inhibitory effects on gastric emptying, and delays the post-prandial rise in insulin (Schmidt et al., 2005). The vagal nerve controls both PP basal and post-prandial release. Surgical or pharmacological vagal blockade causes a marked reduction in meal-induced PP release in dogs (Niebel et al., 1987) and humans (Meguro et al., 1995).
The role of PP in obesity pathogenesis is controversial. Some authors reported a blunted post-prandial PP increase in obese individuals (Lassmann et al., 1980; Glaser et al., 1988), and no differences have been reported in circulating PP between obese subjects and lean individuals (Jorde and Burhol, 1984). However, since plasma PP concentrations are almost exclusively under vagal control, they can be used as an indicator of vagal activity in a number of experimental settings (Schwartz, 1983; Arosio et al., 2004).
Glucagon-like peptide-1 (GLP-1) is an anorectic hormone, member of the incretin family. It is cleaved from preproglucagon within the intestine, where it is released by endocrine L-cells of the distal gut (Wettergren et al., 1997). GLP-1 levels rises post-prandially in response to a meal and fall in the fasting state. GLP-1 release is proportional to the calories ingested (Kreymann et al., 1987; Orskov et al., 1994) and it is particularly responsive to carbohydrates (Lavin et al., 1998) and fats (Frost et al., 2003). Some authors have suggested that circulating GLP-1 levels are reduced in obesity and normalized with weight loss (Verdich et al., 2001). GLP-1 mediates glucose-dependent insulinotropic effects in a number of species, including humans (Holst et al., 1987; Mojsov et al., 1987). Furthermore, it inhibits gastric acid secretion and gastric emptying (Imeryuz et al., 1997; Edvell and Lindstrom, 1999; Sheikh, 2013). The effects of GLP-1 on appetite regulation are mediated by the GLP-1 receptor. GLP-1 receptors are found not only in peripheral tissues (Bullock et al., 1996) but also in CNS areas (Kastin et al., 2002) involved in the regulation of satiety and induction of taste aversion, such as NTS/AP and ARC (Turton et al., 1996). In animal models GLP-1 actions on CNS seem to be mediated by afferent vagal fibers (Ronveaux et al., 2015). Indeed, vagotomy attenuates the satiating effect of GLP-1 (Nakabayashi et al., 1996; Abbott et al., 2005). Recent data showed that an intact vagal nerve is necessary for the inhibition of food intake by intravenous GLP-1 in human patients undergoing vagotomy and pyloroplasty (Plamboeck et al., 2013). Furthermore, some evidence suggest that GLP-1 crosses the blood brain barrier to act directly on CNS receptors (Kastin et al., 2002).
Ghrelin is an orexigenic hormone, primarily secreted by endocrine cells in the oxyntic mucosa of the stomach. Ghrelin stimulates eating behavior and is involved in meal initiation; ghrelin suppression after a meal is crucial to provide a feedback signaling to brain and stop food intake (Kojima et al., 1999; Cummings et al., 2001; Tschop et al., 2001). Thus it is not surprising that obese individuals, though exhibiting lower fasting ghrelin levels than lean individuals, lack the physiological ghrelin suppression in the post-prandial phase: this phenomenon could lead to increased food consumption and, finally, obesity (English et al., 2002).
Ghrelin suppression after meals, which is crucial to reduce caloric intake, is induced by several factors include changes in plasma insulin, intestinal osmolarity, and enteric neural signaling, but a key role for vagal signaling has been also hypothesized (Date et al., 2002; Lee et al., 2002). Indeed in healthy humans vagal stimulation, achieved by modified sham feeding technique (in which nutrients are chewed and tasted but not swallowed) has an inhibitory effect on ghrelin release comparable to real feeding (Arosio et al., 2004; Heath et al., 2004).
Ghrelin plays also a role in long-term body weight regulation, acting as an adiposity signal, communicating the state of energy stores to the brain. Thus fasting ghrelin levels are reduced in obese individuals, and increase after weight loss (Cummings, 2006). However, gastric bypass is associated with markedly suppressed ghrelin levels: this phenomenon possibly favor a greater weight loss after this surgical procedure (Cummings et al., 2002).
Insulin, beyond its established role in glucose (Obici et al., 2002) and lipid metabolism (Scherer et al., 2011), is also involved in satiety pathway acting on CNS. Chronic or acute intracerebroventricular administration of insulin reduces food intake and body weight in a variety of species. Insulin receptors are expressed in the CNS neurons, especially in the ARC (Plum et al., 2005), and participate in the food intake control (Baskin et al., 1999; Brown et al., 2006). On the other hand, insulin could act on its peripheral receptors located in the nodose ganglion (Iwasaki et al., 2013). Hyperphagia and obesity could be, at least in part, caused by impaired response to insulin of nodose ganglion neurons (Iwasaki et al., 2013).
Chronic hyperinsulinemia is a feature of obesity, aimed at restoring energy balance and limiting weight gain in a compensatory fashion. However, it may act as a maladaptive mechanism, inducing sympathetic overactivity (Landsberg, 1986).
Leptin is a hormone released by the white adipose tissue (WAT), whose main actions are to suppress appetite and to regulate glucose metabolism (Elmquist et al., 1998; Elias et al., 2000). However, leptin pathways are involved also in energy expenditure control, as reviewed below. Leptin plasma levels decrease during fasting and increase after overfeeding, whereas leptin administration decreases food intake in animals and humans (Campfield et al., 1995; Heymsfield et al., 1999). The ARC is the most important site involved in leptin-related food intake (Satoh et al., 1997; Haynes, 2000). Within the ARC, two antagonistically acting neuronal populations, the neuropeptide Y (NPY) and proopiomelanocortinergic (POMC) neurons, were identified as immediate downstream targets of leptin. Even though leptin receptors are expressed on both neuronal populations, leptin stimulation of NPY neurons decreases their firing and attenuates food intake, whereas its actions on POMC neurons are opposite (Pandit et al., 2017).
While genetic syndromes characterized by leptin deficiency present hyperfagia and obesity (Zhang et al., 1994), most obese individuals rather have hyperleptinemia (Schwartz et al., 1997), due to desensitization of its own receptor (Considine et al., 1996).
SNS is involved in regulation of secretory function of WAT, especially for leptin secretion. Indeed, acute treatment with catecholamines in in vitro experimental human studies reduces circulating leptin through β1 and β2 receptors (Scriba et al., 2000). Furthermore, sympathetic activation induced by cold exposure induces not only increased metabolic rate and mobilization of free fatty acids, but also a rapid decrease in leptin gene expression and plasma leptin levels (Trayhurn et al., 1995).
The ANS seems to play a role, though not entirely clear, in energy expenditure and storage. In humans, the energy is stored mainly in the WAT under the action of insulin, from where can be mobilized mainly by activation of SNS. Furthermore, SNS might increase energy expenditure by acting either on brown adipose tissue (BAT) thermogenesis or on the cardiovascular system: this neuronal pathway is modulated by leptin (Pandit et al., 2017)
It is well known that lipolysis in the WAT is regulated by SNS and insulin, the principal initiator of lipolysis and a potent inhibitor of lipolysis respectively (Goodridge and Ball, 1965; Prigge and Grande, 1971). Indeed, sympathetic nerve stimulation results in fatty acid release (Rosell, 1966), while sympathetic or ganglionic blockade inhibits lipid mobilization (Gilgen et al., 1962). On the other hand, adrenal medullary catecholamines have no effects on lipid mobilization (Takahashi and Shimazu, 1981), confirming that lipolysis is induced by increased SNS outflow directed to WAT (Rebuffe-Scrive, 1991). Kreier et al. (2002) hypothesized also a parasympathetic innervation of WAT in animal models, possibly modulating insulin-mediated glucose uptake and free fatty acid metabolism in an anabolic way, thus promoting lipid accumulation. According to this hypothesis, lipid accumulation in obesity could be due either to a decrease in SNS activity or by an increase in parasympathetic activity (Bartness, 2002). However, other studies failed to demostrate parasympathetic innervation in WAT (Giordano et al., 2006).
Total energy expenditure is composed of resting metabolic rate (including cardiorespiratory work and the maintenance of transmembrane ion gradients at rest), physical activity and thermogenesis (shivering and non-shivering), and the termic effect of food. SNS activation induces total energy expenditure, either increasing cardiorespiratory work or increasing thermogenesis.
It is well known that the SNS plays a pivotal role in both blood pressure and metabolic homeostatic control by regulating cardiac output, peripheral vascular resistance, and heat production, which account for a large fraction of resting metabolic rate (Goran, 2000). Indeed, pharmacological adrenergic blockade is able to reduce resting energy expenditure (Welle et al., 1991; Monroe et al., 2001; Shibao et al., 2007).
At variance to what was previously thought, BAT is not present only in children, but also in lean and obese adult humans (Virtanen et al., 2009). Its main function is to increase energy expenditure by inducing cold- or diet-stimulated heat production (van der Lans et al., 2013), and by uncoupling oxidative phosphorylation from ATP synthesis through the uncoupling protein-1 in BAT mitochondria (Cannon and Nedergaard, 2004; Saito, 2013). Functional BAT in adults is detectable after exposure to mild cold (Saito et al., 2009) and its activity is inversely related to body mass index and body fat percentage (van Marken Lichtenbelt et al., 2009). Lean subjects increase energy expenditure in response to mild cold, whereas obese subjects have a blunted cold-induced thermogenesis (Wijers et al., 2010).
BAT thermogenesis is regulated by sympathetic nerves. As previously stated, sympathetic activation results in mobilization from WAT of fatty acids, which are then used by BAT to dissipate energy as heat (Figure 2). As far as sympathetic control is concerned, patients with surgical unilateral sympathectomy show a detectable uptake of 18F-f luorodeoxyglucose (18F-FDG) in BAT by positron emission tomography on the unaffected side, but not on the side of surgical sympathectomy (Lebron et al., 2010). Administration of β-adrenergic receptor blockade reduces BAT 18F-FDG uptake (Soderlund et al., 2007) in patients with known or suspected cancer as well as in a patient with paraganglioma, a condition characterized by a massively increased metabolic BAT activity, induced by excess circulating catecholamines (Cheng et al., 2012). The role of α-receptors and α-blockade is less clear. In a patient with catecholamine-secreting paraganglioma, BAT 18F-FDG uptake was suppressed after α-blockade (Sondergaard et al., 2015). The sympathomimetic drug ephedrine activates BAT in lean but not in obese subjects, though the degree of activation is substantially lower than observed after cold exposure (Carey et al., 2013). Conversely, the effect of parasympathetic nervous system on BAT appears to be indirect. Indeed, in animal models, the suppression of NE release in BAT, induced by ghrelin infusion, is abolished after vagotomy (Mano-Otagiri et al., 2009). The authors hypothesized that the vagal nerve mediates the peripheral action of ghrelin, thus inhibiting sympathetic traffic directed to BAT. The interaction between vagal and BAT activity was confirmed in patients undergoing vagal nerve stimulation (VNS) for refractory epilepsy: VNS induced a BAT-mediated increase in energy expenditure (Vijgen et al., 2013).
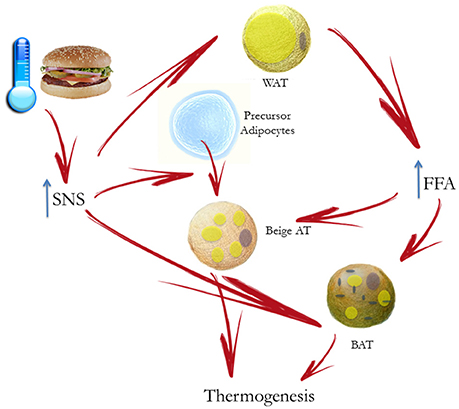
Figure 2. Cold- or diet-stimulated sympathetic activation results in mobilization of free fatty acids (FFA) by white adipose tissue (WAT) and regulation of brown adipose tissue (BAT) thermogenesis. The principal substrate for BAT is constituted by fatty acids to increase energy expenditure inducing heat production. Chronic sympathetic nervous system (SNS) activation also induces the conversion of “beige” adipose tissue in WAT, which also contribute to adaptive thermogenesis.
Furthermore, chronic sympathetic activation produces a remarkable induction of uncoupling protein1-positive brown-like adipocytes in white fat pads, called “beige” adipose tissue, which also contribute to adaptive thermogenesis and body fat reduction (Cousin et al., 1992; Inokuma et al., 2006; Figure 2). In humans it has been suggested that BAT is mostly composed of beige cells and is inducible in response to appropriate sympathetic stimulation. In healthy human participants, with undetectable or low BAT activity, daily 2-h cold exposure at 17°C for 6 weeks resulted in increased BAT activity. Changes in BAT activity and body fat content were negatively correlated (Yoneshiro et al., 2013).
It is important to note that leptin has a crucial role in regulation of energy expenditure through SNS. Indeed, leptin has been shown to increase energy expenditure acting both on the cardiovascular system and BAT thermogenesis via the hypothalamus (Pandit et al., 2017). The ARC represents the main site of action of leptin on SNS. In particular, CNS leptin administration does not affect sympathetic nerve activity after ARC destruction (Haynes, 2000). However, Fischer showed that leptin may increase energy expenditure by inducing a pyrexic increase in body temperature by reducing heat loss, rather than affecting BAT thermogenesis (Fischer et al., 2016).
On the other hand, in animal studies leptin administration in different CNS areas increases sympathetic outflow to the kidneys, the adipose tissue, the skeletal muscle vasculature and adrenal glands (Dunbar et al., 1997; Elmquist et al., 1997; Haynes et al., 1997), thus causing an increase in energy expenditure (Woods and Stock, 1996) and in sympathetic vasomotor activity (Marsh et al., 2003). The latter mechanism is involved in pathogenesis of obesity –induced hypertension, as explained later (see Section Sympathetic Overactivity in Obesity).
Taken together, these results suggest that BAT thermogenesis is an appealing target in obesity treatment. However, while promising evidence in experimental animals demonstrate that it is possible to impair BAT thermogenesis (i.e., by beta-adrenergic blockade), no intervention has so far been able to increase it (Tupone et al., 2014).
An increased SNS activity has been demonstrated in obese patients, particularly in the muscle vasculature and in the kidneys, possibily contributing to increased cardiovascular risk. Though SNS activation is similar in hypertensive and normotensive obese individuals, sympathetic contribution to blood pressure via vasoconstriction is greater in the hypertensive ones, confirming a role for sympathetic activation in the pathogenesis of obesity-related hypertension. Conversely, sympathetic overactivity is not effective in favoring energy expenditure and thus weight loss. Selective leptin resistance, obstructive sleep apnea syndrome, hyperinsulinemia and low ghrelin levels are possible mechanisms underlying sympathetic activation in obesity. Weight loss is able to reverse metabolic and SNS alterations associated with obesity.
It is well known that excess weight is associated with ANS dysfunction, and particularly with increased sympathetic traffic. Landsberg was the first researcher speculating that increased SNS activity in response to weight gain is an adaptive mechanism to increase resting energy expenditure and promote restoration of the antecedent weight (Landsberg, 1986, 2001), while other authors suggested that prolonged sympathetic overactivity might induce weight gain, due to reduced capacity to dissipate excessive calories, mediated by downregulation of β adrenoceptors (van Baak, 2001; Feldstein and Julius, 2009; Figure 3). On the other hand, some authors suggested that a reduced sympathetic activity is rather implied in obesity pathogenesis, inducing a lower rate of thermogenesis and a positive energy balance (Bray, 1991). However, several studies conducted with sophisticated techniques supported the Landsberg's hypothesis of SNS overactivity in obese individuals, with or without hypertension (Landsberg, 1986).
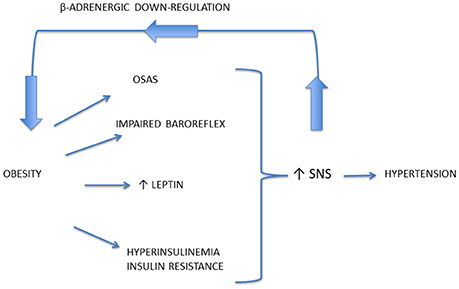
Figure 3. Mechanisms responsible for the occurrence of sympathetic activation in obesity-related hypertension. Prolonged sympathetic nervous system (SNS) overactivity might induce weight gain, due to downregulation of beta-adrenoceptors, thus reducing the capacity to dissipate excessive calories.
It is important to underline that obesity causes a selective and differentiated increase in sympathetic activity rather than generalized SNS activation. This crucial issue has been investigated by techniques such as microneurography, which allows recording directly spontaneous efferent activity of post-ganglionic SNS fibers controlling muscle vascular tone (Vallbo et al., 2004), and regional NE spillover, which is crucial in order to investigate organs like heart and kidney, whose efferent nerve traffic is not directly recordable in humans. Several studies highlighted that obesity is characterized by SNS overactivity directed to the muscle vasculature by means of microneurography (Grassi et al., 1995, 2004; Alvarez et al., 2004). In obese individuals, increased MSNA is obtained by recruitment of additional nervous fibers, as demonstrated by single fiber recordings, at variance to the increased firing frequency observed in essential hypertension (Lambert et al., 2007). MSNA values, although increased in both central and peripheral obesity, are greater in individuals with an abdominal or central distribution of body fat (Grassi et al., 2004), particularly with visceral obesity (Alvarez et al., 2004). Several reflex abnormalities were shown in obesity, such as impaired baroreflex sensitivity (Grassi et al., 1995), central chemoreflex hypersensitivity (Narkiewicz et al., 1999a) and blunted muscle metaboreflex (Negrao et al., 2001); conversely, MSNA responses to mental stress and cold pressure test were similar in obese and in lean subjects (Kuniyoshi et al., 2003).
Furthermore, an increased adrenergic tone in the renal district was also demonstrated, while the sympathetic outflow to the heart is not elevated or even reduced, as demonstrated by cardiac norepinephrine spillover (Esler et al., 2006). It has been hypothesized that cardiac sympathetic tone is reduced in human obesity in response to volume overload (Messerli et al., 1983), in part induced also by sodium retention mediated by high renal SNS activity (DiBona, 1992). An altered autonomic modulation of heart rate has been also demonstrated by the technique of spectral analysis of heart rate variability (Hirsch et al., 1991; Tonhajzerova et al., 2008), with conflicting findings (Matsumoto et al., 1999; Antelmi et al., 2004).
An impaired autonomic regulation in the post-prandial phase has also been suggested. As mentioned above, SNS inhibition is the physiological response to fasting, in order to limit weight loss during starvation (Young and Landsberg, 1977), while food ingestion, particularly of carbohydrate-rich food, induces an increase in SNS activity (Young and Landsberg, 1977; Welle, 1995). This physiological response is blunted in obese individuals in comparison to lean individuals, though energy expenditure was similar and no correlation between SNS activity and the thermic effect of the food has been demonstrated (Tentolouris et al., 2003). The blunted post-prandial increase in sympathetic tone, demonstrated also in adult obese individuals (Xu et al., 2014) may thus represent a mechanism of inhibition of post-prandial thermogenesis, thus favoring weight gain, though conflicting data exist (Emdin et al., 2001). However, these results do not allow drawing firm conclusions, since only autonomic modulation of heart rate has been explored, which may not represent sympathetic traffic directed to the adipose tissue.
Finally, it is important to note that sympathetic overactivity characterizing obesity has deleterious cardiovascular consequences, including the development of hypertension, but it is not effective in increasing energy expenditure and favoring weight loss as expected (see Section Role the ANS in Energy Homeostasis). Indeed, acute ganglionic blockade (Shibao et al., 2007), did not change energy expenditure in individuals with central obesity, supporting the Landsberg's hypothesis of sympathetic activation in obesity as a compensatory but ineffective strategy induced by weight gain. However, preliminary data suggest that contribution of SNS after gastric bypass might be very small: this fact might make more difficult to maintain weight loss after surgery (Curry et al., 2013).
Adrenergic activation plays an important role in pathophysiological mechanisms underlying the development, maintenance, and progression of essential hypertension (Grassi et al., 2015) and is suspected to contribute in particular to the development of hypertension in obese humans (Hall et al., 2012). Julius et al. first proposed that increased sympathetic activity in hypertension was the primary defect leading to insulin resistance and weight gain in obese adults (Julius et al., 2000). In young overweight individuals, SNS activity is directly related to the degree of cardiac, renal, and vascular dysfunction, suggesting that sympathetic neural drive may be a major player in CV risk development (Lambert et al., 2010).
Mechanisms underlying obesity-related hypertension are not fully understood. Indeed, a great importance has been given to activation of renal sympathetic nerves, causing sodium retention, increased renin secretion, and impaired renal-pressure natriuresis (Hall et al., 2012). Though renal NE spillover is similar in normotensive and hypertensive obese individuals, an exaggerated effect of SNS activation has been reported. Indeed Shibao and coauthors demonstrated that after ganglionic blockade with trimethaphan, hypertensive obese patients exhibited a greater BP fall than the normotensive ones (Shibao et al., 2007). Central mechanisms may be relevant in obesity-related hypertension and include activation of leptin and POMC pathway, and obstructive sleep apnea syndrome, with activation of chemoreceptor-mediated reflexes related to intermittent hypoxia (Figure 3). Furthermore, among peripheral mechanisms of sympathetic activation, hyperinsulinemia might play a role.
As already mentioned, leptin has central sympathoexcitatory effects, demonstrated in a number of experimental studies (Haynes et al., 1999; Lim et al., 2013). Indeed, obese mice with leptin or leptin-receptor deficiency showed no increase in arterial pressure (Mark et al., 1999). The sympathoexcitatory and hypertensive effect of leptin seems to be mediated by melanocortin-4 receptor (MC4R) (Tallam et al., 2005). These findings were confirmed also in MC4R deficient humans, who show a low prevalence of hypertension, despite the presence of severe obesity (Greenfield et al., 2009).
Based on this piece of evidence, Mark et al. suggested that some forms of obesity may be characterized by a “selective leptin resistance,” limited to its favorable metabolic effects (satiety and weight loss), while its sympathoexcitatory effects on the cardiovascular system are maintained (Correia et al., 2002; Mark et al., 2002; Rahmouni et al., 2005). In humans, a number of studies confirmed the association between leptin and hypertension. Human leptin deficiency was associated with early-onset morbid obesity and metabolic syndrome without SNS activation or hypertension (Ozata et al., 1999). Conversely, higher leptin levels in obese hypertensive in comparison to obese normotensive individuals have been reported (Kunz et al., 2000; Golan et al., 2002). Furthermore, in the Copenhagen City Heart Study increased plasma leptin levels predicted the risk of developing hypertension (Asferg et al., 2010). However, acute of chronic administration of leptin in humans failed to induce a sustained BP or SNS activity increase, thus the role of leptin in causing sympathetic activation in obesity still need to be fully clarified (Mark, 2013).
OSAS is a condition characterized by repetitive episodes of upper airway narrowing or occlusion, causing chronic intermittent hypoxia and sleep fragmentation (Dempsey et al., 2010). Obesity is a major risk factor for OSAS, which in turn may induce BP increase not only during nighttime but also during daytime (Brooks et al., 1997). The role of OSAS as a determinant of sympathetic overactivity has been reported not only in obese (Somers et al., 1995; Narkiewicz et al., 1998) but also in lean subjects (Grassi et al., 2005). Interestingly, some authors suggest that obesity per se is not associated to increased sympathetic traffic to the muscle vasculature, but this alteration is present only when obesity is accompanied by OSAS (Narkiewicz et al., 1998). Mechanisms of hypertension development during OSAS include sympathetic activation due to chemoreflex activation, secondary to repetitive hypoxic episodes at nighttime, but also alterations in vascular function and structure caused by oxidative stress and inflammation (Bruno et al., 2013). A sustained reduction in MSNA was demonstrated in normotensive patients with OSAS after both 6 and 12 months of continuous positive airway pressure therapy (Narkiewicz et al., 1999b).
Some authors suggest that chronic hyperinsulinemia may act as a maladaptive mechanism, inducing SNS overactivity in obesity (Landsberg, 1986, 2001). However, this hypothesis has not been supported by later studies. Indeed, insulin administration has a direct vasodilatory effect during acute euglycemic hyperinsulinemic clamp: thus the increase in MSNA and norepinephrine levels reported in healthy individuals and hypertensive patients may be a consequence of baroreflex activation (Rowe et al., 1981; Anderson et al., 1991, 1992). However, a modest increase in BP was observed in healthy individuals when supraphysiological insulin concentrations are obtained (Rowe et al., 1981). Interestingly, in elderly subjects with normal BP, acute elevations of plasma insulin during hyperinsulinemic/euglycemic clamp caused vasoconstriction, accompanied by a blunted increase in norepinephrine and heart rate, as compared to young individuals, while no changes in BP were observed in either group. The authors suggested that the insulin-induced vasoconstriction is not due to exaggerated insulin-induced sympathetic activation but rather to a reduction in the vasodilator action of insulin (Hausberg et al., 1997). Despite hyperinsulinemia, intracerebroventricular administration of insulin antagonists did not affect renal sympathetic nerve activity in experimental animals, adding to the evidence that insulin does not promote obesity hypertension by chronically stimulating the SNS (Lim et al., 2013).
Beyond its established role in appetite regulation, ghrelin has beneficial effects on blood pressure (BP) and cardiovascular function (Virdis et al., 2016), possibly modulating ANS activity. In experimental animals, intracerebral infusion of ghrelin reduced BP; however, it is still not clear whether this effect was mediated by modulation of sympathetic traffic (Matsumura et al., 2002; Prior et al., 2014). Lambert et al. investigated the effects of supraphysiological doses of intravenous ghrelin in lean and obese individuals. Ghrelin did not influence SNS activity controlling resting calf vascular tone; however, ghrelin infusion blunted BP and muscle sympathetic nerve activity (MSNA) responses to acute mental stress after short-term ghrelin infusion either in lean or obese individuals (Lambert et al., 2011).
Several studies have shown that sympathetic activation reported in obese subjects is reversed by weight loss (Muscelli et al., 1998; Nault et al., 2007; Perugini et al., 2010). This topic is extensively reviewed elsewhere (Lambert et al., 2015). Straznicky reported a marked sympathoinhibition secondary to diet-induced weight loss, evaluated by MSNA and whole-body plasma norepinephrine spillover rate (Straznicky et al., 2005). However, bariatric surgery is the most effective treatment for obesity, allowing to achieve up to 70% of excess weight loss (Buchwald et al., 2004). It is also well known that bariatric surgery improves the main defects responsible for obesity-associated hyperglycaemia, namely insulin resistance and beta-cell dysfunction (Ferrannini, 1998; Nannipieri et al., 2011). Few data explored the role of bariatric surgery in reduction of SNS activity. Pontiroli et al. showed a restoration of sympathovagal balance evaluated by heart rate variability in 24 subjects with severe obesity 6 months after gastric banding (Pontiroli et al., 2013), while Lips et al. showed an improvement in heart rate variability, although explored only in the time domain, after 3 months very low-calorie diet or gastric bypass (Lips et al., 2013). However, these two studies, using spectral analysis of RR interval, did not provide a measure of sympathetic activity. In 23 severely obese, non-diabetic, individuals, MSNA was measured before and after 10% weight loss induced by laparoscopic adjustable gastric band. Noteworthy, a significant reduction in BP, MSNA, fasting insulin and creatinine clearance was found, whereas cardiac and sympathetic baroreflex sensitivity were improved (Lambert et al., 2014). Seravalle et al. evaluated the effect of weight loss secondary to sleeve gastrectomy or caloric-restricted diet on the ANS. Six months after surgery, waist circumference, leptin levels and MSNA were reduced in the surgery group, which persisted 12 months after surgery (Seravalle et al., 2014). Conversely, insulin sensitivity, evaluated by Homeostatic Model Assessment (HOMA) index, was reduced after 6 months, but returned to pre-surgery values after 12 months, suggesting that sympathetic deactivation induced by weight loss might not influence insulin sensitivity (Seravalle et al., 2014). However, this conclusion is limited by the fact that HOMA index is a rough index of insulin sensitivity; furthermore, since it is derived from fasting insulin and glucose levels, it is related to hepatic insulin sensitivity rather than peripheral insulin sensitivity, which is conceivably more influenced by changes in sympathetic tone.
SNS activity after gastric bypass surgery seem to be lower than those of obese individuals and thus might blunt energy expenditure, with negative consequences for weight maintenance (Curry et al., 2013). We do not know whether different interventions, i.e., sleeve gastrectomy might lead to the same phenomenon.
Finally, it is important to note that the surgical procedure per se might have a direct impact on the autonomic innervation of the gastrointestinal tract. During surgery, sleeve gastrectomy and Roux-en-Y gastric bypass (RYGB) may damage the gastric branches of the vagal nerve in a different manner. Infact in the sleeve gastrectomy the stomach is cut longitudinally, damaging the very distal branches of the gastric vagal nerve, while in the RYGB the stomach is cut transversely, resulting in a damage of the gastric vagal branches very close to their origin from the esophageal plexus (Ballsmider et al., 2015). Thus, it is conceivable that the effects of bariatric surgery on brain-gut axys may be influenced by the surgically-induced anatomical alterations, which may affect the integrity of vagal innervation between the hindbrain feeding centers and the gastrointestinal tract.
Based on the physiopatological background above described, it is clear the modulation of ANS may induce weight loss and/or reduce cardiovascular risk in obese patients. VNS, achieved by implantable or transcutaneous devices, has been associated with a significant weight loss in small, non-randomized pilot studies. Vagal nerve blockade yelded either neutral or positive effects in term of weight loss in small sham-controlled studies, but even in this case further evidence is needed. Sympathetic inhibition accompanied weight loss achieved by diet or surgery. Interventions targeting SNS are able to improve cardiometabolic profile in obese individuals.
Since vagal afferents convey to the CNS the gastric distension signal and satiety signals evoked by gut hormones, it is not surprising that vagal stimulation has been proposed as a weight loss intervention. Several studies, carried out in obese animals, showed that VNS suppressed food intake and weight gain. Bugajski et al. suggested that VNS, achieved by implantable electronic devices, mimics activation of gastric mechanoceptors and jejunal chemoceptors, thus resulting in decreased food intake and weight loss in obese rats (Bugajski et al., 2007; Figure 1). The limitations of this study are the monolateral VNS and the use of constant voltage stimulation (Bugajski et al., 2007). Bilateral VNS with constant current stimulation induced stable weight loss in obese minipigs (Val-Laillet et al., 2010). Furthermore, patients treated with vagal stimulation for severe depression experienced a relevant weight loss (Pardo et al., 2007) (Table 1). However, this approach is limited by its high cost and invasiveness, potential need for reintervention for mechanical failure and/or battery re-placement, and side effects (Ventureyra, 2000). More recently, transcutaneous auricular VNS (taVNS) has been proposed to treat disorders such as epilepsy (Miro et al., 2015) and depression, drawing inspiration from auricular acupuncture of traditional chinese medicine (Rong et al., 2016). The rationale for using taVNS is that anatomical studies showed that the ear is the only place on the surface of the human body where afferent vagal nerve distribution is present (Wang et al., 2014). Indeed, a branch of the vagal nerve provides sensory innervation of the “cymba conchae” of the external ear (Peuker and Filler, 2002). Thus, the direct stimulation of the afferent vagal nerve fibers on the ear may produce similar effects as classic VNS without the burden of surgical intervention (Henry, 2002). Indeed, cymba conchae stimulation of auricolar vagal branch activated the NTS and other vagal projections within the brainstem and forebrain in healthy adults (Frangos et al., 2015). Furthermore, in a pilot randomized clinical trial, Huang et al reported an improvement of in the 2-h glucose tolerance and systolic BP in after a 12-week treatment with taVNS in comparison with sham technique (Huang et al., 2014) (Table 1). Finally, taVNS is able to acutely reduce MSNA and shift cardiac autonomic function toward parasympathetic predominance in healthy volunteers (Clancy et al., 2014). These promising findings suggest that in obese and glucose-intolerant individuals, taVNS may not only restore insulin resistance and secretion, but also counteract obesity-related autonomic dysfunction (Lambert et al., 2010; Seravalle et al., 2014) and thus play a role in reducing its cardiovascular burden.
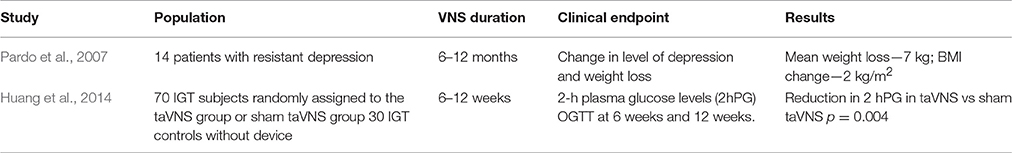
Table 1. Human studies investigating the role of VNS in weight loss and glucose control.
On the other hand, gastric emptying is under the control of vagal efferent fibers. Vagotomy, in experimental animals (Smith et al., 1983) as well as in humans (Kral, 1978) is able to delay gastric emptying and impair gastric accommodation to food, thus inducing weight loss. Since pancreatic secretion is under vagal control, interruption of vagal efferent fibers induces malabsorption (Camilleri et al., 2008). Furthermore, vagotomy in rats prevents the physiological ghrelin increase in fasting conditions (Williams et al., 2003). Thus, intermittent electric stimulation of vagal fibers, inducing blockade of the neural transmission, has been tested as a novel weight-loss intervention (Table 2).
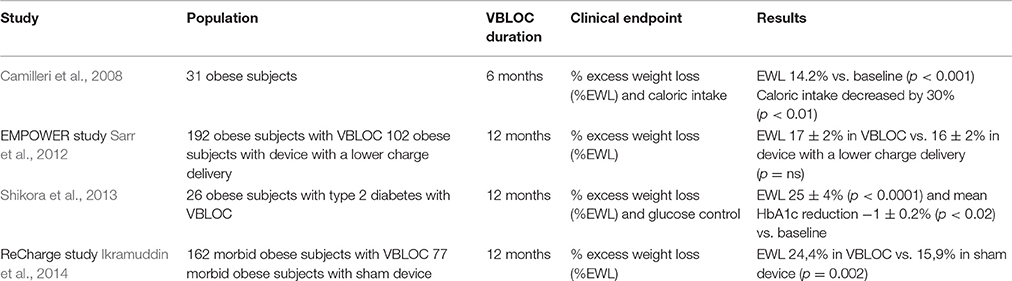
Table 2. Human studies investigating the role of vagal nerve blockade (VBLOC) in weight loss, glucose control and caloric intake.
The EMPOWER study evaluated the effects of intermittent, bilateral blockade of bilateral subdiaphragmatic vagal nerves to stop both ascending and descending neural traffic, speculating its involvement in satiety, reduced food intake and weight loss in morbid obese individuals (Figure 1). However, despite the solid scientific background linking vagal activity and obesity, extensively described in the previous sections, the EMPOWER study yelded negative results: vagal blockade induced a similar weight loss than the control group, which had the same device with a lower charge delivery; interestingly, weight loss was related to device use time in both groups, suggesting that what was supposed to be a sham therapy was active as well (Sarr et al., 2012). This hypothesis is confirmed by the ReCharge study, in which vagal nerve blockade was obtained by using a device that delivered at least 12 h of therapy per day and was compared a sham control device that had no possibility of delivering therapy. Individuals undergoing vagal blockade therapy achieved a greater weight loss than the sham control group, although the pre-established efficacy outcomes were not achieved (Ikramuddin et al., 2014) (Table 2).
Given the above-described role of SNS in the pathophysiology of obesity and its cardiovascular consequences, SNS inhibition is considered a potential therapeutic target in obesity. As reviewed above, it is important to underline that interventions aimed at inducing weight loss by diet or surgery are able to achieve a significant reduction in SNS tone, in particular in the muscle vasculature (Lambert et al., 2015).
Indeed, a number of mechanistic studies demonstrated that acute pharmacologic ganglionic blockade by trimetaphan is able to reduce blood pressure (Shibao et al., 2007), to improve insulin sensitivity (Gamboa et al., 2014) and to reverse endothelial function (Gamboa et al., 2016) in obesity, in particular if associated with hypertension. However, ganglionic blockers cannot be used chronically, given their unfavorable profile in terms of adverse effects.
A significant antihypertensive effect of a combined α and ß-blockade has been reported in dietary mediated obesity in dogs consuming high fat diets (Hall et al., 2001) and in obese individuals in which a greater reduction in BP in comparison to lean subjects was reported after 1 month of treatment (Wofford et al., 2001). Adrenergic blockade produced a significantly greater decrease in BP in obese than in lean patients with hypertension (Wofford et al., 2001), in line to the results reported with ganglionic blockade (Shibao et al., 2007). A study suggested also that the use of a BP-lowering central sympatholytic drug, moxonidine, might induce a small but significant weight loss, together with a reduction in blood pressure, triglycerides and fasting blood glucose (Chazova and Schlaich, 2013), though another study failed to demonstrate any impact on insulin sensitivity (Masajtis-Zagajewska et al., 2010). In contrast, β-blockers may exert negative or neutral effects on body weight and lipid and glucose profile (Lambert et al., 2015). However, some authors suggest that β-blockers may be first-choice drug in the treatment of hypertension in young adults, which is mainly linked to sympathetic overactivity due to overweight and obesity (Cruickshank, 2017).
In the past decade, great interest has been placed in device-based therapies targeting SNS for the treatment of refractory hypertension, such as renal denervation and baroceptor activating therapy (Bruno et al., 2013). Given the presence of sympathetic activation in obesity and its possible role in pathogenesis of obesity-associated hypertension, as described above, it may be expected that sympathetic inhibition might have a relevant impact in obese patients. Indeed, renal denervation seems able to restore insulin sensitivity in obese dogs (Iyer et al., 2016) but not in obese hypertensive mice (Asirvatham-Jeyaraj et al., 2016). Bilateral renal denervation greatly attenuated sodium retention and hypertension in obese dogs fed a high-fat diet (Kassab et al., 1995).
Glucose tolerance and glycemic control was significantly improved 3 and 6 months after renal denervation in 10 patients with resistant hypertension and OSAS: in this study, BP, but not BMI, was significantly reduced (Witkowski et al., 2011). This finding was confirmed in a larger cohort of resistant hypertensive patients, in whom renal denervation induced a reduction in blood fasting glucose, insulin, and HOMA-IR after 3 months (Mahfoud et al., 2011). However, the BP-lowering effect of such procedures has been recently questioned; furthermore, obese patients seem to benefit less of renal denervation in terms of BP reduction (Id et al., 2016).
In conclusion, obesity is accompanied by increased morbidity and mortality, mostly related to cardiovascular disease, and represents a major issue for global healthcare. Thus, the study of mechanisms underlying its pathogenesis is crucial to identify novel targets for its treatment. The ANS plays a major role in the integrated short-term regulation of weight, modulating the satiety signal and energy expenditure. The afferent vagal pathways are probably the most important link between the gut and the brain and interact in a complex way with gut hormones. SNS has the physiological function of increasing lipolysis and energy expenditure, through sympathetic innervation in white and brown adipose tissue; thus it is abnormally activated in obesity in a compensatory but ineffective fashion. Sympathetic activation may favor the development of hypertension and organ damage in obesity and lead to overt cardiovascular disease. Though preliminary clinical trials exploring autonomic modulation as a treatment for obesity yelded contrasting results, mechanistic and physiopathological studies strongly support this therapeutic strategy as an appealing and promising approach for obesity treatment.
DG drafted the manuscript. RMB designed and reviewed critically the article. MN, GI, and ST reviewed critically the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbott, C. R., Monteiro, M., Small, C. J., Sajedi, A., Smith, K. L., Parkinson, J. R., et al. (2005). The inhibitory effects of peripheral administration of peptide YY(3-36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res. 1044, 127–131. doi: 10.1016/j.brainres.2005.03.011
Adrian, T. E., Ferri, G. L., Bacarese-Hamilton, A. J., Fuessl, H. S., Polak, J. M., and Bloom, S. R. (1985). Human distribution and release of a putative new gut hormone, peptide YY. Gastroenterology 89, 1070–1077. doi: 10.1016/0016-5085(85)90211-2
Alvarez, G. E., Ballard, T. P., Beske, S. D., and Davy, K. P. (2004). Subcutaneous obesity is not associated with sympathetic neural activation. Am. J. Physiol. Heart Circ. Physiol. 287, H414–H418. doi: 10.1152/ajpheart.01046.2003
Anderson, E. A., Balon, T. W., Hoffman, R. P., Sinkey, C. A., and Mark, A. L. (1992). Insulin increases sympathetic activity but not blood pressure in borderline hypertensive humans. Hypertension 19(6 Pt 2), 621–627. doi: 10.1161/01.HYP.19.6.621
Anderson, E. A., Hoffman, R. P., Balon, T. W., Sinkey, C. A., and Mark, A. L. (1991). Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J. Clin. Invest. 87, 2246–2252. doi: 10.1172/JCI115260
Antelmi, I., de Paula, R. S., Shinzato, A. R., Peres, C. A., Mansur, A. J., and Grupi, C. J. (2004). Influence of age, gender, body mass index, and functional capacity on heart rate variability in a cohort of subjects without heart disease. Am. J. Cardiol. 93, 381–385. doi: 10.1016/j.amjcard.2003.09.065
Arosio, M., Ronchi, C. L., Beck-Peccoz, P., Gebbia, C., Giavoli, C., Cappiello, V., et al. (2004). Effects of modified sham feeding on ghrelin levels in healthy human subjects. J. Clin. Endocrinol. Metab. 89, 5101–5104. doi: 10.1210/jc.2003-032222
Asferg, C., Mogelvang, R., Flyvbjerg, A., Frystyk, J., Jensen, J. S., Marott, J. L., et al. (2010). Leptin, not adiponectin, predicts hypertension in the Copenhagen City Heart Study. Am. J. Hypertens. 23, 327–333. doi: 10.1038/ajh.2009.244
Asirvatham-Jeyaraj, N., Fiege, J. K., Han, R., Foss, J., Banek, C. T., Burbach, B. J., et al. (2016). Renal denervation normalizes arterial pressure with no effect on glucose metabolism or renal inflammation in obese hypertensive mice. Hypertension 68, 929–936. doi: 10.1161/HYPERTENSIONAHA.116.07993
Ballsmider, L. A., Vaughn, A. C., David, M., Hajnal, A., Di Lorenzo, P. M., and Czaja, K. (2015). Sleeve gastrectomy and Roux-en-Y gastric bypass alter the gut-brain communication. Neural Plast. 2015:601985. doi: 10.1155/2015/601985
Bartness, T. J. (2002). Dual innervation of white adipose tissue: some evidence for parasympathetic nervous system involvement. J. Clin. Invest. 110, 1235–1237. doi: 10.1172/JCI0217047
Baskin, D. G., Figlewicz Lattemann, D., Seeley, R. J., Woods, S. C., Porte, D. Jr., and Schwartz, M. W. (1999). Insulin and leptin: dual adiposity signals to the brain for the regulation of food intake and body weight. Brain Res. 848, 114–123. doi: 10.1016/S0006-8993(99)01974-5
Batterham, R. L., Cohen, M. A., Ellis, S. M., Le Roux, C. W., Withers, D. J., Frost, G. S., et al. (2003). Inhibition of food intake in obese subjects by peptide YY3-36. N. Engl. J. Med. 349, 941–948. doi: 10.1056/NEJMoa030204
Berthoud, H. R. (2008a). Vagal and hormonal gut-brain communication: from satiation to satisfaction. Neurogastroenterol. Motil. 20(Suppl. 1), 64–72. doi: 10.1111/j.1365-2982.2008.01104.x
Berthoud, H. R. (2008b). The vagus nerve, food intake and obesity. Regul. Pept. 149, 15–25. doi: 10.1016/j.regpep.2007.08.024
Blessing, W. W. (1997). Inadequate frameworks for understanding bodily homeostasis. Trends Neurosci. 20, 235–239. doi: 10.1016/S0166-2236(96)01029-6
Bray, G. A. (1991). Obesity, a disorder of nutrient partitioning: the MONA LISA hypothesis. J. Nutr. 121, 1146–1162.
Brooks, D., Horner, R. L., Kozar, L. F., Render-Teixeira, C. L., and Phillipson, E. A. (1997). Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J. Clin. Invest. 99, 106–109. doi: 10.1172/JCI119120
Brown, L. M., Clegg, D. J., Benoit, S. C., and Woods, S. C. (2006). Intraventricular insulin and leptin reduce food intake and body weight in C57BL/6J mice. Physiol. Behav. 89, 687–691. doi: 10.1016/j.physbeh.2006.08.008
Bruno, R. M., Rossi, L., Fabbrini, M., Duranti, E., Di Coscio, E., Maestri, M., et al. (2013). Renal vasodilating capacity and endothelial function are impaired in patients with obstructive sleep apnea syndrome and no traditional cardiovascular risk factors. J. Hypertens 31, 1456–1464. discussion: 1464. doi: 10.1097/HJH.0b013e328360f773
Buchwald, H., Avidor, Y., Braunwald, E., Jensen, M. D., Pories, W., Fahrbach, K., et al. (2004). Bariatric surgery: a systematic review and meta-analysis. JAMA 292, 1724–1737. doi: 10.1001/jama.292.14.1724
Buffa, R., Solcia, E., and Go, V. L. (1976). Immunohistochemical identification of the cholecystokinin cell in the intestinal mucosa. Gastroenterology 70, 528–532.
Bugajski, A. J., Gil, K., Ziomber, A., Zurowski, D., Zaraska, W., and Thor, P. J. (2007). Effect of long-term vagal stimulation on food intake and body weight during diet induced obesity in rats. J. Physiol. Pharmacol. 58(Suppl.1), 5–12.
Bullock, B. P., Heller, R. S., and Habener, J. F. (1996). Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 137, 2968–2978. doi: 10.1210/endo.137.7.8770921
Burdyga, G., Lal, S., Spiller, D., Jiang, W., Thompson, D., Attwood, S., et al. (2003). Localization of orexin-1 receptors to vagal afferent neurons in the rat and humans. Gastroenterology 124, 129–139. doi: 10.1053/gast.2003.50020
Burton-Freeman, B., Davis, P. A., and Schneeman, B. O. (2002). Plasma cholecystokinin is associated with subjective measures of satiety in women. Am. J. Clin. Nutr. 76, 659–667.
Camilleri, M., Toouli, J., Herrera, M. F., Kulseng, B., Kow, L., Pantoja, J. P., et al. (2008). Intra-abdominal vagal blocking (VBLOC therapy): clinical results with a new implantable medical device. Surgery 143, 723–731. doi: 10.1016/j.surg.2008.03.015
Campfield, L. A., Smith, F. J., Guisez, Y., Devos, R., and Burn, P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546–549. doi: 10.1126/science.7624778
Cannon, B., and Nedergaard, J. (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359. doi: 10.1152/physrev.00015.2003
Capasso, R., and Izzo, A. A. (2008). Gastrointestinal regulation of food intake: general aspects and focus on anandamide and oleoylethanolamide. J. Neuroendocrinol. 20(Suppl. 1), 39–46. doi: 10.1111/j.1365-2826.2008.01686.x
Carey, A. L., Formosa, M. F., Van Every, B., Bertovic, D., Eikelis, N., Lambert, G. W., et al. (2013). Ephedrine activates brown adipose tissue in lean but not obese humans. Diabetologia 56, 147–155. doi: 10.1007/s00125-012-2748-1
Chazova, I., and Schlaich, M. P. (2013). Improved hypertension control with the imidazoline agonist moxonidine in a multinational metabolic syndrome population: principal results of the MERSY study. Int. J. Hypertens. 2013:541689. doi: 10.1155/2013/541689
Cheng, W., Zhu, Z., Jin, X., Chen, L., Zhuang, H., and Li, F. (2012). Intense FDG activity in the brown adipose tissue in omental and mesenteric regions in a patient with malignant pheochromocytoma. Clin. Nucl. Med. 37, 514–515. doi: 10.1097/RLU.0b013e31824d2121
Clancy, J. A., Mary, D. A., Witte, K. K., Greenwood, J. P., Deuchars, S. A., and Deuchars, J. (2014). Non-invasive vagus nerve stimulation in healthy humans reduces sympathetic nerve activity. Brain Stimul. 7, 871–877. doi: 10.1016/j.brs.2014.07.031
Considine, R. V., Considine, E. L., Williams, C. J., Hyde, T. M., and Caro, J. F. (1996). The hypothalamic leptin receptor in humans: identification of incidental sequence polymorphisms and absence of the db/db mouse and fa/fa rat mutations. Diabetes 45, 992–994. doi: 10.2337/diab.45.7.992
Corp, E. S., McQuade, J., Moran, T. H., and Smith, G. P. (1993). Characterization of type A and type B CCK receptor binding sites in rat vagus nerve. Brain Res. 623, 161–166. doi: 10.1016/0006-8993(93)90024-H
Correia, M. L., Haynes, W. G., Rahmouni, K., Morgan, D. A., Sivitz, W. I., and Mark, A. L. (2002). The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes 51, 439–442. doi: 10.2337/diabetes.51.2.439
Cousin, B., Cinti, S., Morroni, M., Raimbault, S., Ricquier, D., Penicaud, L., et al. (1992). Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J. Cell Sci. 103(Pt 4), 931–942.
Cox, H. M. (2007a). Neuropeptide Y receptors; antisecretory control of intestinal epithelial function. Auton. Neurosci. 133, 76–85. doi: 10.1016/j.autneu.2006.10.005
Cox, H. M. (2007b). Peptide YY: a neuroendocrine neighbor of note. Peptides 28, 345–351. doi: 10.1016/j.peptides.2006.07.023
Cruickshank, J. M. (2017). The role of beta-blockers in the treatment of hypertension. Adv. Exp. Med. Biol. 956, 149–166. doi: 10.1007/5584_2016_36
Cummings, D. E. (2006). Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol. Behav. 89, 71–84. doi: 10.1016/j.physbeh.2006.05.022
Cummings, D. E., Purnell, J. Q., Frayo, R. S., Schmidova, K., Wisse, B. E., and Weigle, D. S. (2001). A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50, 1714–1719. doi: 10.2337/diabetes.50.8.1714
Cummings, D. E., and Schwartz, M. W. (2003). Genetics and pathophysiology of human obesity. Annu. Rev. Med. 54, 453–471. doi: 10.1146/annurev.med.54.101601.152403
Cummings, D. E., Weigle, D. S., Frayo, R. S., Breen, P. A., Ma, M. K., Dellinger, E. P., et al. (2002). Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N. Engl. J. Med. 346, 1623–1630. doi: 10.1056/NEJMoa012908
Curry, T. B., Somaraju, M., Hines, C. N., Groenewald, C. B., Miles, J. M., Joyner, M. J., et al. (2013). Sympathetic support of energy expenditure and sympathetic nervous system activity after gastric bypass surgery. Obesity 21, 480–485. doi: 10.1002/oby.20106
Date, Y., Murakami, N., Toshinai, K., Matsukura, S., Niijima, A., Matsuo, H., et al. (2002). The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123, 1120–1128. doi: 10.1053/gast.2002.35954
Dempsey, J. A., Veasey, S. C., Morgan, B. J., and O'Donnell, C. P. (2010). Pathophysiology of sleep apnea. Physiol. Rev. 90, 47–112. doi: 10.1152/physrev.00043.2008
DiBona, G. F. (1992). Sympathetic neural control of the kidney in hypertension. Hypertension 19, I28–I35. doi: 10.1161/01.HYP.19.1_Suppl.I28
Dockray, G. J. (2014). Gastrointestinal hormones and the dialogue between gut and brain. J. Physiol. 592, 2927–2941. doi: 10.1113/jphysiol.2014.270850
Dumont, Y., Fournier, A., St-Pierre, S., and Quirion, R. (1995). Characterization of neuropeptide Y binding sites in rat brain membrane preparations using [125I][Leu31,Pro34]peptide YY and [125I]peptide YY3-36 as selective Y1 and Y2 radioligands. J. Pharmacol. Exp. Ther. 272, 673–680.
Dumoulin, V., Dakka, T., Plaisancie, P., Chayvialle, J. A., and Cuber, J. C. (1995). Regulation of glucagon-like peptide-1-(7-36) amide, peptide YY, and neurotensin secretion by neurotransmitters and gut hormones in the isolated vascularly perfused rat ileum. Endocrinology 136, 5182–5188. doi: 10.1210/endo.136.11.7588257
Dunbar, J. C., Hu, Y., and Lu, H. (1997). Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes 46, 2040–2043. doi: 10.2337/diab.46.12.2040
Edvell, A., and Lindstrom, P. (1999). Initiation of increased pancreatic islet growth in young normoglycemic mice (Umea +/?). Endocrinology 140, 778–783. doi: 10.1210/endo.140.2.6514
Ekblad, E., and Sundler, F. (2002). Distribution of pancreatic polypeptide and peptide YY. Peptides 23, 251–261. doi: 10.1016/S0196-9781(01)00601-5
Elias, C. F., Kelly, J. F., Lee, C. E., Ahima, R. S., Drucker, D. J., Saper, C. B., et al. (2000). Chemical characterization of leptin-activated neurons in the rat brain. J. Comp. Neurol. 423, 261–281. doi: 10.1002/1096-9861(20000724)423:2<261::AID-CNE6>3.0.CO;2-6
Elmquist, J. K., Ahima, R. S., Maratos-Flier, E., Flier, J. S., and Saper, C. B. (1997). Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology 138, 839–842. doi: 10.1210/endo.138.2.5033
Elmquist, J. K., Maratos-Flier, E., Saper, C. B., and Flier, J. S. (1998). Unraveling the central nervous system pathways underlying responses to leptin. Nat. Neurosci. 1, 445–450. doi: 10.1038/2164
Emdin, M., Gastaldelli, A., Muscelli, E., Macerata, A., Natali, A., Camastra, S., et al. (2001). Hyperinsulinemia and autonomic nervous system dysfunction in obesity: effects of weight loss. Circulation 103, 513–519. doi: 10.1161/01.CIR.103.4.513
English, P. J., Ghatei, M. A., Malik, I. A., Bloom, S. R., and Wilding, J. P. (2002). Food fails to suppress ghrelin levels in obese humans. J. Clin. Endocrinol. Metab. 87:2984. doi: 10.1210/jcem.87.6.8738
Esler, M., Straznicky, N., Eikelis, N., Masuo, K., Lambert, G., and Lambert, E. (2006). Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 48, 787–796. doi: 10.1161/01.HYP.0000242642.42177.49
Feldstein, C., and Julius, S. (2009). The complex interaction between overweight, hypertension, and sympathetic overactivity. J. Am. Soc. Hypertens. 3, 353–365. doi: 10.1016/j.jash.2009.10.001
Ferrannini, E. (1998). Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: problems and prospects. Endocr. Rev. 19, 477–490. doi: 10.1210/edrv.19.4.0336
Fetissov, S. O., Kopp, J., and Hokfelt, T. (2004). Distribution of NPY receptors in the hypothalamus. Neuropeptides 38, 175–188. doi: 10.1016/j.npep.2004.05.009
Fink, H., Rex, A., Voits, M., and Voigt, J. P. (1998). Major biological actions of CCK–a critical evaluation of research findings. Exp. Brain Res. 123, 77–83. doi: 10.1007/s002210050546
Fischer, A. W., Hoefig, C. S., Abreu-Vieira, G., de Jong, J. M., Petrovic, N., Mittag, J., et al. (2016). Leptin raises defended body temperature without activating thermogenesis. Cell Rep. 14, 1621–1631. doi: 10.1016/j.celrep.2016.01.041
Flier, J. S. (2001). Diabetes. The missing link with obesity? Nature 409, 292–293. doi: 10.1038/35053251
Forster, E. R., Green, T., Elliot, M., Bremner, A., and Dockray, G. J. (1990). Gastric emptying in rats: role of afferent neurons and cholecystokinin. Am. J. Physiol. 258(4 Pt 1), G552–G556.
Frangos, E., Ellrich, J., and Komisaruk, B. R. (2015). Non-invasive access to the vagus nerve central projections via electrical stimulation of the external ear: fMRI evidence in humans. Brain Stimul. 8, 624–636. doi: 10.1016/j.brs.2014.11.018
Frost, G. S., Brynes, A. E., Dhillo, W. S., Bloom, S. R., and McBurney, M. I. (2003). The effects of fiber enrichment of pasta and fat content on gastric emptying, GLP-1, glucose, and insulin responses to a meal. Eur. J. Clin. Nutr. 57, 293–298. doi: 10.1038/sj.ejcn.1601520
Fu-Cheng, X., Anini, Y., Chariot, J., Castex, N., Galmiche, J. P., and Roze, C. (1997). Mechanisms of peptide YY release induced by an intraduodenal meal in rats: neural regulation by proximal gut. Pflugers Arch. 433, 571–579. doi: 10.1007/s004240050316
Gamboa, A., Figueroa, R., Paranjape, S. Y., Farley, G., Diedrich, A., and Biaggioni, I. (2016). Autonomic blockade reverses endothelial dysfunction in obesity-associated hypertension. Hypertension 68, 1004–1010. doi: 10.1161/HYPERTENSIONAHA.116.07681
Gamboa, A., Okamoto, L. E., Arnold, A. C., Figueroa, R. A., Diedrich, A., Raj, S. R., et al. (2014). Autonomic blockade improves insulin sensitivity in obese subjects. Hypertension 64, 867–874. doi: 10.1161/HYPERTENSIONAHA.114.03738
Gilgen, A., Maickel, R. P., Nikodijevic, O., and Brodie, B. B. (1962). Essential role of catecholamines in the mobilization of free fatty acids and glucose after exposure to cold. Life Sci. 1, 709–715. doi: 10.1016/0024-3205(62)90138-8
Giordano, A., Song, C. K., Bowers, R. R., Ehlen, J. C., Frontini, A., Cinti, S., et al. (2006). White adipose tissue lacks significant vagal innervation and immunohistochemical evidence of parasympathetic innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1243–R1255. doi: 10.1152/ajpregu.00679.2005
Glaser, B., Zoghlin, G., Pienta, K., and Vinik, A. I. (1988). Pancreatic polypeptide response to secretin in obesity: effects of glucose intolerance. Horm. Metab. Res. 20, 288–292. doi: 10.1055/s-2007-1010817
Golan, E., Tal, B., Dror, Y., Korzets, Z., Vered, Y., Weiss, E., et al. (2002). Reduction in resting metabolic rate and ratio of plasma leptin to urinary nitric oxide: influence on obesity-related hypertension. Isr. Med. Assoc. J. 4, 426–430.
Goodridge, A. G., and Ball, E. G. (1965). Studies on the metabolism of adipose tissue. 18. In vitro effects of insulin, epinephrine and glucagon on lipolysis and glycolysis in pigeon adipose tissue. Comp. Biochem. Physiol. 16, 367–381. doi: 10.1016/0010-406X(65)90303-8
Goran, M. I. (2000). Energy metabolism and obesity. Med. Clin. North Am. 84, 347–362. doi: 10.1016/S0025-7125(05)70225-X
Granger, D. N., Richardson, P. D., Kvietys, P. R., and Mortillaro, N. A. (1980). Intestinal blood flow. Gastroenterology 78, 837–863.
Grassi, G., Dell'Oro, R., Facchini, A., Quarti Trevano, F., Bolla, G. B., and Mancia, G. (2004). Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J. Hypertens. 22, 2363–2369. doi: 10.1097/00004872-200412000-00019
Grassi, G., Facchini, A., Trevano, F. Q., Dell'Oro, R., Arenare, F., Tana, F., et al. (2005). Obstructive sleep apnea-dependent and -independent adrenergic activation in obesity. Hypertension 46, 321–325. doi: 10.1161/01.HYP.0000174243.39897.6c
Grassi, G., Mark, A., and Esler, M. (2015). The sympathetic nervous system alterations in human hypertension. Circ. Res. 116, 976–990. doi: 10.1161/CIRCRESAHA.116.303604
Grassi, G., Seravalle, G., Cattaneo, B. M., Bolla, G. B., Lanfranchi, A., Colombo, M., et al. (1995). Sympathetic activation in obese normotensive subjects. Hypertension 25(4 Pt 1), 560–563. doi: 10.1161/01.HYP.25.4.560
Greenfield, J. R., Miller, J. W., Keogh, J. M., Henning, E., Satterwhite, J. H., Cameron, G. S., et al. (2009). Modulation of blood pressure by central melanocortinergic pathways. N. Engl. J. Med. 360, 44–52. doi: 10.1056/NEJMoa0803085
Hall, J. E., Granger, J. P., do Carmo, J. M., da Silva, A. A., Dubinion, J., George, E., et al. (2012). Hypertension: physiology and pathophysiology. Compr. Physiol. 2, 2393–2442. doi: 10.1002/cphy.c110058
Hall, J. E., Hildebrandt, D. A., and Kuo, J. (2001). Obesity hypertension: role of leptin and sympathetic nervous system. Am. J. Hypertens. 14(6 Pt 2), 103S–115S. doi: 10.1016/S0895-7061(01)02077-5
Hausberg, M., Hoffman, R. P., Somers, V. K., Sinkey, C. A., Mark, A. L., and Anderson, E. A. (1997). Contrasting autonomic and hemodynamic effects of insulin in healthy elderly versus young subjects. Hypertension 29, 700–705. doi: 10.1161/01.HYP.29.3.700
Haynes, W. G. (2000). Interaction between leptin and sympathetic nervous system in hypertension. Curr. Hypertens. Rep. 2, 311–318. doi: 10.1007/s11906-000-0015-1
Haynes, W. G., Morgan, D. A., Djalali, A., Sivitz, W. I., and Mark, A. L. (1999). Interactions between the melanocortin system and leptin in control of sympathetic nerve traffic. Hypertension 33(1 Pt 2), 542–547. doi: 10.1161/01.HYP.33.1.542
Haynes, W. G., Sivitz, W. I., Morgan, D. A., Walsh, S. A., and Mark, A. L. (1997). Sympathetic and cardiorenal actions of leptin. Hypertension 30(3 Pt 2), 619–623. doi: 10.1161/01.HYP.30.3.619
Heath, R. B., Jones, R., Frayn, K. N., and Robertson, M. D. (2004). Vagal stimulation exaggerates the inhibitory ghrelin response to oral fat in humans. J. Endocrinol. 180, 273–281. doi: 10.1677/joe.0.1800273
Henry, T. R. (2002). Therapeutic mechanisms of vagus nerve stimulation. Neurology 59(6 Suppl. 4), S3–S14. doi: 10.1212/WNL.59.6_suppl_4.S3
Heymsfield, S. B., Greenberg, A. S., Fujioka, K., Dixon, R. M., Kushner, R., Hunt, T., et al. (1999). Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA 282, 1568–1575. doi: 10.1001/jama.282.16.1568
Hirsch, J., Leibel, R. L., Mackintosh, R., and Aguirre, A. (1991). Heart rate variability as a measure of autonomic function during weight change in humans. Am. J. Physiol. 261(6 Pt 2), R1418–R1423.
Holst, J. J., Orskov, C., Nielsen, O. V., and Schwartz, T. W. (1987). Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 211, 169–174. doi: 10.1016/0014-5793(87)81430-8
Huang, F., Dong, J., Kong, J., Wang, H., Meng, H., Spaeth, R. B., et al. (2014). Effect of transcutaneous auricular vagus nerve stimulation on impaired glucose tolerance: a pilot randomized study. BMC Complement. Altern. Med. 14:203. doi: 10.1186/1472-6882-14-203
Id, D., Bertog, S. C., Ziegler, A. K., Hornung, M., Hofmann, I., Vaskelyte, L., et al. (2016). Predictors of blood pressure response: obesity is associated with a less pronounced treatment response after renal denervation. Catheter. Cardiovasc. Interv. 87, E30–E38. doi: 10.1002/ccd.26068
Ikramuddin, S., Blackstone, R. P., Brancatisano, A., Toouli, J., Shah, S. N., Wolfe, B. M., et al. (2014). Effect of reversible intermittent intra-abdominal vagal nerve blockade on morbid obesity: the ReCharge randomized clinical trial. JAMA 312, 915–922. doi: 10.1001/jama.2014.10540
Imeryuz, N., Yegen, B. C., Bozkurt, A., Coskun, T., Villanueva-Penacarrillo, M. L., and Ulusoy, N. B. (1997). Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. Am. J. Physiol. 273(4 Pt 1), G920–G927.
Inokuma, K., Okamatsu-Ogura, Y., Omachi, A., Matsushita, Y., Kimura, K., Yamashita, H., et al. (2006). Indispensable role of mitochondrial UCP1 for antiobesity effect of beta3-adrenergic stimulation. Am. J. Physiol. Endocrinol. Metab. 290, E1014–E1021. doi: 10.1152/ajpendo.00105.2005
Iwasaki, Y., Shimomura, K., Kohno, D., Dezaki, K., Ayush, E. A., Nakabayashi, H., et al. (2013). Insulin activates vagal afferent neurons including those innervating pancreas via insulin cascade and Ca(2+)influx: its dysfunction in IRS2-KO mice with hyperphagic obesity. PLoS ONE 8:e67198. doi: 10.1371/journal.pone.0067198
Iyer, M. S., Bergman, R. N., Korman, J. E., Woolcott, O. O., Kabir, M., Victor, R. G., et al. (2016). Renal denervation reverses hepatic insulin resistance induced by high-fat diet. Diabetes 65, 3453–3463. doi: 10.2337/db16-0698
Jorde, R., and Burhol, P. G. (1984). Fasting and postprandial plasma pancreatic polypeptide (PP) levels in obesity. Int. J. Obes. 8, 393–397.
Julius, S., Valentini, M., and Palatini, P. (2000). Overweight and hypertension: a 2-way street? Hypertension 35, 807–813. doi: 10.1161/01.HYP.35.3.807
Kassab, S., Kato, T., Wilkins, F. C., Chen, R., Hall, J. E., and Granger, J. P. (1995). Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension 25(4 Pt 2), 893–897. doi: 10.1161/01.HYP.25.4.893
Kastin, A. J., Akerstrom, V., and Pan, W. (2002). Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J. Mol. Neurosci. 18, 7–14. doi: 10.1385/JMN:18:1-2:07
Kissileff, H. R., Carretta, J. C., Geliebter, A., and Pi-Sunyer, F. X. (2003). Cholecystokinin and stomach distension combine to reduce food intake in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R992–R998. doi: 10.1152/ajpregu.00272.2003
Koda, S., Date, Y., Murakami, N., Shimbara, T., Hanada, T., Toshinai, K., et al. (2005). The role of the vagal nerve in peripheral PYY3-36-induced feeding reduction in rats. Endocrinology 146, 2369–2375. doi: 10.1210/en.2004-1266
Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H., and Kangawa, K. (1999). Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660. doi: 10.1038/45230
Kral, J. G. (1978). Vagotomy for treatment of severe obesity. Lancet 1, 307–308. doi: 10.1016/S0140-6736(78)90074-0
Kreier, F., Fliers, E., Voshol, P. J., Van Eden, C. G., Havekes, L. M., Kalsbeek, A., et al. (2002). Selective parasympathetic innervation of subcutaneous and intra-abdominal fat–functional implications. J. Clin. Invest. 110, 1243–1250. doi: 10.1172/JCI0215736
Kreymann, B., Williams, G., Ghatei, M. A., and Bloom, S. R. (1987). Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 2, 1300–1304. doi: 10.1016/S0140-6736(87)91194-9
Kuniyoshi, F. H., Trombetta, I. C., Batalha, L. T., Rondon, M. U., Laterza, M. C., Gowdak, M. M., et al. (2003). Abnormal neurovascular control during sympathoexcitation in obesity. Obes. Res. 11, 1411–1419. doi: 10.1038/oby.2003.190
Kunz, I., Schorr, U., Klaus, S., and Sharma, A. M. (2000). Resting metabolic rate and substrate use in obesity hypertension. Hypertension 36, 26–32. doi: 10.1161/01.HYP.36.1.26
Lambert, E. A., Rice, T., Eikelis, N., Straznicky, N. E., Lambert, G. W., Head, G. A., et al. (2014). Sympathetic activity and markers of cardiovascular risk in nondiabetic severely obese patients: the effect of the initial 10% weight loss. Am. J. Hypertens. 27, 1308–1315. doi: 10.1093/ajh/hpu050
Lambert, E. A., Straznicky, N. E., Dixon, J. B., and Lambert, G. W. (2015). Should the sympathetic nervous system be a target to improve cardiometabolic risk in obesity? Am. J. Physiol. Heart Circ. Physiol. 309, H244–H258. doi: 10.1152/ajpheart.00096.2015
Lambert, E., Lambert, G., Ika-Sari, C., Dawood, T., Lee, K., Chopra, R., et al. (2011). Ghrelin modulates sympathetic nervous system activity and stress response in lean and overweight men. Hypertension 58, 43–50. doi: 10.1161/HYPERTENSIONAHA.111.171025
Lambert, E., Sari, C. I., Dawood, T., Nguyen, J., McGrane, M., Eikelis, N., et al. (2010). Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension 56, 351–358. doi: 10.1161/HYPERTENSIONAHA.110.155663
Lambert, E., Straznicky, N., Schlaich, M., Esler, M., Dawood, T., Hotchkin, E., et al. (2007). Differing pattern of sympathoexcitation in normal-weight and obesity-related hypertension. Hypertension 50, 862–868. doi: 10.1161/HYPERTENSIONAHA.107.094649
Landsberg, L. (1986). Diet, obesity and hypertension: an hypothesis involving insulin, the sympathetic nervous system, and adaptive thermogenesis. Q. J. Med. 61, 1081–1090.
Landsberg, L. (2001). Insulin-mediated sympathetic stimulation: role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J. Hypertens 19(3 Pt 2), 523–528. doi: 10.1097/00004872-200103001-00001
Lassmann, V., Vague, P., Vialettes, B., and Simon, M. C. (1980). Low plasma levels of pancreatic polypeptide in obesity. Diabetes 29, 428–430. doi: 10.2337/diab.29.6.428
Lavin, J. H., Wittert, G. A., Andrews, J., Yeap, B., Wishart, J. M., Morris, H. A., et al. (1998). Interaction of insulin, glucagon-like peptide 1, gastric inhibitory polypeptide, and appetite in response to intraduodenal carbohydrate. Am. J. Clin. Nutr. 68, 591–598.
Lebron, L., Chou, A. J., and Carrasquillo, J. A. (2010). Interesting image. Unilateral F-18 FDG uptake in the neck, in patients with sympathetic denervation. Clin. Nucl. Med. 35, 899–901. doi: 10.1097/RLU.0b013e3181f49ff8
Lee, H. M., Wang, G., Englander, E. W., Kojima, M., and Greeley, G. H. Jr. (2002). Ghrelin, a new gastrointestinal endocrine peptide that stimulates insulin secretion: enteric distribution, ontogeny, influence of endocrine, and dietary manipulations. Endocrinology 143, 185–190. doi: 10.1210/endo.143.1.8602
le Roux, C. W., Batterham, R. L., Aylwin, S. J., Patterson, M., Borg, C. M., Wynne, K. J., et al. (2006). Attenuated peptide YY release in obese subjects is associated with reduced satiety. Endocrinology 147, 3–8. doi: 10.1210/en.2005-0972
Li, Y., and Owyang, C. (1994). Endogenous cholecystokinin stimulates pancreatic enzyme secretion via vagal afferent pathway in rats. Gastroenterology 107, 525–531. doi: 10.1016/0016-5085(94)90180-5
Lim, K., Burke, S. L., and Head, G. A. (2013). Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension 61, 628–634. doi: 10.1161/HYPERTENSIONAHA.111.00705
Lin, H. C., and Taylor, I. L. (2004). Release of peptide YY by fat in the proximal but not distal gut depends on an atropine-sensitive cholinergic pathway. Regul. Pept. 117, 73–76. doi: 10.1016/j.regpep.2003.10.008
Lips, M. A., de Groot, G. H., De Kam, M., Berends, F. J., Wiezer, R., Van Wagensveld, B. A., et al. (2013). Autonomic nervous system activity in diabetic and healthy obese female subjects and the effect of distinct weight loss strategies. Eur. J. Endocrinol. 169, 383–390. doi: 10.1530/EJE-13-0506
Little, T. J., Horowitz, M., and Feinle-Bisset, C. (2005). Role of cholecystokinin in appetite control and body weight regulation. Obes. Rev. 6, 297–306. doi: 10.1111/j.1467-789X.2005.00212.x
Mahfoud, F., Schlaich, M., Kindermann, I., Ukena, C., Cremers, B., Brandt, M. C., et al. (2011). Effect of renal sympathetic denervation on glucose metabolism in patients with resistant hypertension: a pilot study. Circulation 123, 1940–1946. doi: 10.1161/CIRCULATIONAHA.110.991869
Mano-Otagiri, A., Ohata, H., Iwasaki-Sekino, A., Nemoto, T., and Shibasaki, T. (2009). Ghrelin suppresses noradrenaline release in the brown adipose tissue of rats. J. Endocrinol. 201, 341–349. doi: 10.1677/JOE-08-0374
Mark, A. L. (2013). Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R566–R581. doi: 10.1152/ajpregu.00180.2013
Mark, A. L., Correia, M. L., Rahmouni, K., and Haynes, W. G. (2002). Selective leptin resistance: a new concept in leptin physiology with cardiovascular implications. J. Hypertens. 20, 1245–1250. doi: 10.1097/00004872-200207000-00001
Mark, A. L., Shaffer, R. A., Correia, M. L., Morgan, D. A., Sigmund, C. D., and Haynes, W. G. (1999). Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J. Hypertens 17(12 Pt 2), 1949–1953. doi: 10.1097/00004872-199917121-00026
Marsh, A. J., Fontes, M. A., Killinger, S., Pawlak, D. B., Polson, J. W., and Dampney, R. A. (2003). Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension 42, 488–493. doi: 10.1161/01.HYP.0000090097.22678.0A
Martin-Rodriguez, E., Guillen-Grima, F., Marti, A., and Brugos-Larumbe, A. (2015). Comorbidity associated with obesity in a large population: the APNA study. Obes. Res. Clin. Pract. 9, 435–447. doi: 10.1016/j.orcp.2015.04.003
Masajtis-Zagajewska, A., Majer, J., and Nowicki, M. (2010). Effect of moxonidine and amlodipine on serum YKL-40, plasma lipids and insulin sensitivity in insulin-resistant hypertensive patients-a randomized, crossover trial. Hypertens. Res. 33, 348–353. doi: 10.1038/hr.2010.6
Matsumoto, T., Miyawaki, T., Ue, H., Kanda, T., Zenji, C., and Moritani, T. (1999). Autonomic responsiveness to acute cold exposure in obese and non-obese young women. Int. J. Obes. Relat. Metab. Disord. 23, 793–800. doi: 10.1038/sj.ijo.0800928
Matsumura, K., Tsuchihashi, T., Fujii, K., Abe, I., and Iida, M. (2002). Central ghrelin modulates sympathetic activity in conscious rabbits. Hypertension 40, 694–699. doi: 10.1161/01.HYP.0000035395.51441.10
Meguro, T., Shimosegawa, T., Kikuchi, Y., Koizumi, M., and Toyota, T. (1995). Effects of cisapride on gallbladder emptying and pancreatic polypeptide and cholecystokinin release in humans. J. Gastroenterol. 30, 237–243. doi: 10.1007/BF02348671
Messerli, F. H., Sundgaard-Riise, K., Reisin, E., Dreslinski, G., Dunn, F. G., and Frohlich, E. (1983). Disparate cardiovascular effects of obesity and arterial hypertension. Am. J. Med. 74, 808–812. doi: 10.1016/0002-9343(83)91071-9
Miro, J., Jaraba, S., Mora, J., Puig, O., Castaner, S., Rodriguez-Bel, L., et al. (2015). Vagus nerve stimulation therapy is effective and safe for startle-induced seizures. J. Neurol. Sci. 354, 124–126. doi: 10.1016/j.jns.2015.04.048
Mojsov, S., Weir, G. C., and Habener, J. F. (1987). Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J. Clin. Invest. 79, 616–619. doi: 10.1172/JCI112855
Monroe, M. B., Seals, D. R., Shapiro, L. F., Bell, C., Johnson, D., and Parker Jones, P. (2001). Direct evidence for tonic sympathetic support of resting metabolic rate in healthy adult humans. Am. J. Physiol. Endocrinol. Metab. 280, E740–E744.
Moran, T. H., and Kinzig, K. P. (2004). Gastrointestinal satiety signals II. Cholecystokinin. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G183–G188. doi: 10.1152/ajpgi.00434.2003
Muscelli, E., Emdin, M., Natali, A., Pratali, L., Camastra, S., Gastaldelli, A., et al. (1998). Autonomic and hemodynamic responses to insulin in lean and obese humans. J. Clin. Endocrinol. Metab. 83, 2084–2090. doi: 10.1210/jc.83.6.2084
Nakabayashi, H., Nishizawa, M., Nakagawa, A., Takeda, R., and Niijima, A. (1996). Vagal hepatopancreatic reflex effect evoked by intraportal appearance of tGLP-1. Am. J. Physiol. 271(5 Pt 1), E808–E813.
Nannipieri, M., Mari, A., Anselmino, M., Baldi, S., Barsotti, E., Guarino, D., et al. (2011). The role of beta-cell function and insulin sensitivity in the remission of type 2 diabetes after gastric bypass surgery. J. Clin. Endocrinol. Metab. 96, E1372–E1379. doi: 10.1210/jc.2011-0446
Narkiewicz, K., Kato, M., Pesek, C. A., and Somers, V. K. (1999a). Human obesity is characterized by a selective potentiation of central chemoreflex sensitivity. Hypertension 33, 1153–1158. doi: 10.1161/01.HYP.33.5.1153
Narkiewicz, K., Kato, M., Phillips, B. G., Pesek, C. A., Davison, D. E., and Somers, V. K. (1999b). Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation 100, 2332–2335. doi: 10.1161/01.CIR.100.23.2332
Narkiewicz, K., van de Borne, P. J., Cooley, R. L., Dyken, M. E., and Somers, V. K. (1998). Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circulation 98, 772–776. doi: 10.1161/01.CIR.98.8.772
Nault, I., Nadreau, E., Paquet, C., Brassard, P., Marceau, P., Marceau, S., et al. (2007). Impact of bariatric surgery–induced weight loss on heart rate variability. Metab. Clin. Exp. 56, 1425–1430. doi: 10.1016/j.metabol.2007.06.006
Neary, N. M., Small, C. J., and Bloom, S. R. (2003). Gut and mind. Gut 52, 918–921. doi: 10.1136/gut.52.7.918
Negrao, C. E., Trombetta, I. C., Batalha, L. T., Ribeiro, M. M., Rondon, M. U., Tinucci, T., et al. (2001). Muscle metaboreflex control is diminished in normotensive obese women. Am. J. Physiol. Heart Circ. Physiol. 281, H469–H475.
Niebel, W., Eysselein, V. E., and Singer, M. V. (1987). Pancreatic polypeptide response to a meal before and after cutting the extrinsic nerves of the upper gastrointestinal tract and the pancreas in the dog. Dig. Dis. Sci. 32, 1004–1009. doi: 10.1007/BF01297191
Obici, S., and Rossetti, L. (2003). Minireview: nutrient sensing and the regulation of insulin action and energy balance. Endocrinology 144, 5172–5178. doi: 10.1210/en.2003-0999
Obici, S., Zhang, B. B., Karkanias, G., and Rossetti, L. (2002). Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 8, 1376–1382. doi: 10.1038/nm1202-798
Onaga, T., Zabielski, R., and Kato, S. (2002). Multiple regulation of peptide YY secretion in the digestive tract. Peptides 23, 279–290. doi: 10.1016/S0196-9781(01)00609-X
Orskov, C., Rabenhoj, L., Wettergren, A., Kofod, H., and Holst, J. J. (1994). Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 43, 535–539. doi: 10.2337/diab.43.4.535
Ozata, M., Ozdemir, I. C., and Licinio, J. (1999). Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J. Clin. Endocrinol. Metab. 84, 3686–3695. doi: 10.1210/jcem.84.10.5999
Pandit, R., Beerens, S., and Adan, R. A. H. (2017). Role of leptin in energy expenditure: the hypothalamic perspective. Am. J. Physiol. Regul. Integr. Comp. Physiol. 312, R938–R947. doi: 10.1152/ajpregu.00045.2016
Pardo, J. V., Sheikh, S. A., Kuskowski, M. A., Surerus-Johnson, C., Hagen, M. C., Lee, J. T., et al. (2007). Weight loss during chronic, cervical vagus nerve stimulation in depressed patients with obesity: an observation. Int. J. Obes. 31, 1756–1759. doi: 10.1038/sj.ijo.0803666
Parker, R. M., and Herzog, H. (1999). Regional distribution of Y-receptor subtype mRNAs in rat brain. Eur. J. Neurosci. 11, 1431–1448. doi: 10.1046/j.1460-9568.1999.00553.x
Perugini, R. A., Li, Y., Rosenthal, L., Gallagher-Dorval, K., Kelly, J. J., and Czerniach, D. R. (2010). Reduced heart rate variability correlates with insulin resistance but not with measures of obesity in population undergoing laparoscopic Roux-en-Y gastric bypass. Surg. Obes. Relat. Dis. 6, 237–241. doi: 10.1016/j.soard.2009.09.012
Peuker, E. T., and Filler, T. J. (2002). The nerve supply of the human auricle. Clin. Anat. 15, 35–37. doi: 10.1002/ca.1089
Plamboeck, A., Veedfald, S., Deacon, C. F., Hartmann, B., Wettergren, A., Svendsen, L. B., et al. (2013). The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G1117–G1127. doi: 10.1152/ajpgi.00035.2013
Plum, L., Schubert, M., and Bruning, J. C. (2005). The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 16, 59–65. doi: 10.1016/j.tem.2005.01.008
Pontiroli, A. E., Merlotti, C., Veronelli, A., and Lombardi, F. (2013). Effect of weight loss on sympatho-vagal balance in subjects with grade-3 obesity: restrictive surgery versus hypocaloric diet. Acta Diabetol. 50, 843–850. doi: 10.1007/s00592-013-0454-1
Prigge, W. F., and Grande, F. (1971). Effects of glucagon, epinephrine and insulin on in vitro lipolysis of adipose tissue from mammals and birds. Comp. Biochem. Physiol. B 39, 69–82. doi: 10.1016/0305-0491(71)90254-9
Prior, L. J., Davern, P. J., Burke, S. L., Lim, K., Armitage, J. A., and Head, G. A. (2014). Exposure to a high-fat diet during development alters leptin and ghrelin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 63, 338–345. doi: 10.1161/HYPERTENSIONAHA.113.02498
Rahmouni, K., Morgan, D. A., Morgan, G. M., Mark, A. L., and Haynes, W. G. (2005). Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes 54, 2012–2018. doi: 10.2337/diabetes.54.7.2012
Rebuffe-Scrive, M. (1991). Neuroregulation of adipose tissue: molecular and hormonal mechanisms. Int. J. Obes. 15(Suppl. 2), 83–86.
Rong, P., Liu, J., Wang, L., Liu, R., Fang, J., Zhao, J., et al. (2016). Effect of transcutaneous auricular vagus nerve stimulation on major depressive disorder: a nonrandomized controlled pilot study. J. Affect. Disord. 195, 172–179. doi: 10.1016/j.jad.2016.02.031
Ronveaux, C. C., Tome, D., and Raybould, H. E. (2015). Glucagon-like peptide 1 interacts with ghrelin and leptin to regulate glucose metabolism and food intake through vagal afferent neuron signaling. J. Nutr. 145, 672–680. doi: 10.3945/jn.114.206029
Rosell, S. (1966). Release of free fatty acids from subcutaneous adipose tissue in dogs following sympathetic nerve stimulation. Acta Physiol. Scand. 67, 343–351. doi: 10.1111/j.1748-1716.1966.tb03320.x
Rowe, J. W., Young, J. B., Minaker, K. L., Stevens, A. L., Pallotta, J., and Landsberg, L. (1981). Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes 30, 219–225. doi: 10.2337/diab.30.3.219
Saito, M. (2013). Brown adipose tissue as a regulator of energy expenditure and body fat in humans. Diabetes Metab. J. 37, 22–29. doi: 10.4093/dmj.2013.37.1.22
Saito, M., Okamatsu-Ogura, Y., Matsushita, M., Watanabe, K., Yoneshiro, T., Nio-Kobayashi, J., et al. (2009). High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58, 1526–1531. doi: 10.2337/db09-0530
Sarr, M. G., Billington, C. J., Brancatisano, R., Brancatisano, A., Toouli, J., Kow, L., et al. (2012). The EMPOWER study: randomized, prospective, double-blind, multicenter trial of vagal blockade to induce weight loss in morbid obesity. Obes. Surg. 22, 1771–1782. doi: 10.1007/s11695-012-0751-8
Sartor, D. M., and Verberne, A. J. (2002). Cholecystokinin selectively affects presympathetic vasomotor neurons and sympathetic vasomotor outflow. Am. J. Physiol. Regul. Integr. Comp. Physiol. 282, R1174–R1184. doi: 10.1152/ajpregu.00500.2001
Sartor, D. M., and Verberne, A. J. (2006). The sympathoinhibitory effects of systemic cholecystokinin are dependent on neurons in the caudal ventrolateral medulla in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1390–R1398. doi: 10.1152/ajpregu.00314.2006
Sartor, D. M., and Verberne, A. J. (2008). Abdominal vagal signalling: a novel role for cholecystokinin in circulatory control? Brain Res. Rev. 59, 140–154. doi: 10.1016/j.brainresrev.2008.07.002
Satoh, N., Ogawa, Y., Katsuura, G., Hayase, M., Tsuji, T., Imagawa, K., et al. (1997). The arcuate nucleus as a primary site of satiety effect of leptin in rats. Neurosci. Lett. 224, 149–152. doi: 10.1016/S0304-3940(97)00163-8
Scherer, T., O'Hare, J., Diggs-Andrews, K., Schweiger, M., Cheng, B., Lindtner, C., et al. (2011). Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 13, 183–194. doi: 10.1016/j.cmet.2011.01.008
Schmidt, P. T., Naslund, E., Gryback, P., Jacobsson, H., Holst, J. J., Hilsted, L., et al. (2005). A role for pancreatic polypeptide in the regulation of gastric emptying and short-term metabolic control. J. Clin. Endocrinol. Metab. 90, 5241–5246. doi: 10.1210/jc.2004-2089
Schwartz, M. W., Prigeon, R. L., Kahn, S. E., Nicolson, M., Moore, J., Morawiecki, A., et al. (1997). Evidence that plasma leptin and insulin levels are associated with body adiposity via different mechanisms. Diabetes Care 20, 1476–1481. doi: 10.2337/diacare.20.9.1476
Schwartz, T. W. (1983). Pancreatic polypeptide: a hormone under vagal control. Gastroenterology 85, 1411–1425.
Scriba, D., Aprath-Husmann, I., Blum, W. F., and Hauner, H. (2000). Catecholamines suppress leptin release from in vitro differentiated subcutaneous human adipocytes in primary culture via beta1- and beta2-adrenergic receptors. Eur. J. Endocrinol. 143, 439–445. doi: 10.1530/eje.0.1430439
Seravalle, G., Colombo, M., Perego, P., Giardini, V., Volpe, M., Dell'Oro, R., et al. (2014). Long-term sympathoinhibitory effects of surgically induced weight loss in severe obese patients. Hypertension 64, 431–437. doi: 10.1161/HYPERTENSIONAHA.113.02988
Sheikh, A. (2013). Direct cardiovascular effects of glucagon like peptide-1. Diabetol. Metab. Syndr. 5:47. doi: 10.1186/1758-5996-5-47
Sheikh, S. P., Holst, J. J., Orskov, C., Ekman, R., and Schwartz, T. W. (1989). Release of PYY from pig intestinal mucosa; luminal and neural regulation. Regul. Pept. 26, 253–266. doi: 10.1016/0167-0115(89)90193-6
Shibao, C., Gamboa, A., Diedrich, A., Ertl, A. C., Chen, K. Y., Byrne, D. W., et al. (2007). Autonomic contribution to blood pressure and metabolism in obesity. Hypertension 49, 27–33. doi: 10.1161/01.HYP.0000251679.87348.05
Shikora, S., Toouli, J., Herrera, M. F., Kulseng, B., Zulewski, H., Brancatisano, R., et al. (2013). Vagal blocking improves glycemic control and elevated blood pressure in obese subjects with type 2 diabetes mellitus. J. Obes. 2013:245683. doi: 10.1155/2013/245683
Shin, A. C., Zheng, H., and Berthoud, H. R. (2009). An expanded view of energy homeostasis: neural integration of metabolic, cognitive, and emotional drives to eat. Physiol. Behav. 97, 572–580. doi: 10.1016/j.physbeh.2009.02.010
Smith, D. K., Sarfeh, J., and Howard, L. (1983). Truncal vagotomy in hypothalamic obesity. Lancet 1, 1330–1331. doi: 10.1016/S0140-6736(83)92437-6
Soares, L. P., Fabbro, A. L., Silva, A. S., Sartorelli, D. S., Franco, L. F., Kuhn, P. C., et al. (2015). Prevalence of metabolic syndrome in the Brazilian Xavante indigenous population. Diabetol. Metab. Syndr. 7:105. doi: 10.1186/s13098-015-0100-x
Soderlund, V., Larsson, S. A., and Jacobsson, H. (2007). Reduction of FDG uptake in brown adipose tissue in clinical patients by a single dose of propranolol. Eur. J. Nucl. Med. Mol. Imaging 34, 1018–1022. doi: 10.1007/s00259-006-0318-9
Somers, V. K., Dyken, M. E., Clary, M. P., and Abboud, F. M. (1995). Sympathetic neural mechanisms in obstructive sleep apnea. J. Clin. Invest. 96, 1897–1904. doi: 10.1172/JCI118235
Sondergaard, E., Gormsen, L. C., Christensen, M. H., Pedersen, S. B., Christiansen, P., Nielsen, S., et al. (2015). Chronic adrenergic stimulation induces brown adipose tissue differentiation in visceral adipose tissue. Diabet. Med. 32, e4–e8. doi: 10.1111/dme.12595
Straznicky, N. E., Lambert, E. A., Lambert, G. W., Masuo, K., Esler, M. D., and Nestel, P. J. (2005). Effects of dietary weight loss on sympathetic activity and cardiac risk factors associated with the metabolic syndrome. J. Clin. Endocrinol. Metab. 90, 5998–6005. doi: 10.1210/jc.2005-0961
Takahashi, A., and Shimazu, T. (1981). Hypothalamic regulation of lipid metabolism in the rat: effect of hypothalamic stimulation on lipolysis. J. Auton. Nerv. Syst. 4, 195–205. doi: 10.1016/0165-1838(81)90044-8
Tallam, L. S., Stec, D. E., Willis, M. A., da Silva, A. A., and Hall, J. E. (2005). Melanocortin-4 receptor-deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension 46, 326–332. doi: 10.1161/01.HYP.0000175474.99326.bf
Tentolouris, N., Tsigos, C., Perea, D., Koukou, E., Kyriaki, D., Kitsou, E., et al. (2003). Differential effects of high-fat and high-carbohydrate isoenergetic meals on cardiac autonomic nervous system activity in lean and obese women. Metab. Clin. Exp. 52, 1426–1432. doi: 10.1016/S0026-0495(03)00322-6
Tonhajzerova, I., Javorka, M., Trunkvalterova, Z., Chroma, O., Javorkova, J., Lazarova, Z., et al. (2008). Cardio-respiratory interaction and autonomic dysfunction in obesity. J. Physiol. Pharmacol. 59(Suppl. 6), 709–718.
Travers, J. B., Travers, S. P., and Norgren, R. (1987). Gustatory neural processing in the hindbrain. Annu. Rev. Neurosci. 10, 595–632. doi: 10.1146/annurev.ne.10.030187.003115
Trayhurn, P., Duncan, J. S., and Rayner, D. V. (1995). Acute cold-induced suppression of ob (obese) gene expression in white adipose tissue of mice: mediation by the sympathetic system. Biochem. J. 311(Pt 3), 729–733. doi: 10.1042/bj3110729
Tschop, M., Wawarta, R., Riepl, R. L., Friedrich, S., Bidlingmaier, M., Landgraf, R., et al. (2001). Post-prandial decrease of circulating human ghrelin levels. J. Endocrinol. Invest. 24(6), Rc19–Rc 21. doi: 10.1007/BF03351037
Tupone, D., Madden, C. J., and Morrison, S. F. (2014). Autonomic regulation of brown adipose tissue thermogenesis in health and disease: potential clinical applications for altering BAT thermogenesis. Front. Neurosci. 8:14. doi: 10.3389/fnins.2014.00014
Turton, M. D., O'Shea, D., Gunn, I., Beak, S. A., Edwards, C. M., Meeran, K., et al. (1996). A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 379, 69–72. doi: 10.1038/379069a0
Val-Laillet, D., Biraben, A., Randuineau, G., and Malbert, C. H. (2010). Chronic vagus nerve stimulation decreased weight gain, food consumption and sweet craving in adult obese minipigs. Appetite 55, 245–252. doi: 10.1016/j.appet.2010.06.008
Vallbo, A. B., Hagbarth, K. E., and Wallin, B. G. (2004). Microneurography: how the technique developed and its role in the investigation of the sympathetic nervous system. J. Appl. Physiol. (1985) 96, 1262–1269. doi: 10.1152/japplphysiol.00470.2003
van Baak, M. A. (2001). The peripheral sympathetic nervous system in human obesity. Obes. Rev. 2, 3–14. doi: 10.1046/j.1467-789x.2001.00010.x
van der Lans, A. A., Hoeks, J., Brans, B., Vijgen, G. H., Visser, M. G., Vosselman, M. J., et al. (2013). Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Invest. 123, 3395–3403. doi: 10.1172/JCI68993
van Marken Lichtenbelt, W. D., Vanhommerig, J. W., Smulders, N. M., Drossaerts, J. M., Kemerink, G. J., Bouvy, N. D., et al. (2009). Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360, 1500–1508. doi: 10.1056/NEJMoa0808718
Ventureyra, E. C. (2000). Transcutaneous vagus nerve stimulation for partial onset seizure therapy. A new concept. Childs Nerv. Syst. 16, 101–102. doi: 10.1007/s003810050021
Verdich, C., Toubro, S., Buemann, B., Lysgard Madsen, J., Juul Holst, J., and Astrup, A. (2001). The role of postprandial releases of insulin and incretin hormones in meal-induced satiety–effect of obesity and weight reduction. Int. J. Obes. Relat. Metab. Disord. 25, 1206–1214. doi: 10.1038/sj.ijo.0801655
Vijgen, G. H., Bouvy, N. D., Leenen, L., Rijkers, K., Cornips, E., Majoie, M., et al. (2013). Vagus nerve stimulation increases energy expenditure: relation to brown adipose tissue activity. PLoS ONE 8:e77221. doi: 10.1371/journal.pone.0077221
Virdis, A., Lerman, L. O., Regoli, F., Ghiadoni, L., Lerman, A., and Taddei, S. (2016). Human ghrelin: a gastric hormone with cardiovascular properties. Curr. Pharm. Des. 22, 52–58. doi: 10.2174/1381612822666151119144458
Virtanen, K. A., Lidell, M. E., Orava, J., Heglind, M., Westergren, R., Niemi, T., et al. (2009). Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360, 1518–1525. doi: 10.1056/NEJMoa0808949
Wang, Z., Yu, L., Wang, S., Huang, B., Liao, K., Saren, G., et al. (2014). Chronic intermittent low-level transcutaneous electrical stimulation of auricular branch of vagus nerve improves left ventricular remodeling in conscious dogs with healed myocardial infarction. Circ. Heart Fail. 7, 1014–1021. doi: 10.1161/CIRCHEARTFAILURE.114.001564
Welle, S. (1995). Sympathetic nervous system response to intake. Am. J. Clin. Nutr. 62(5 Suppl.), 1118s–1122s.
Welle, S., Schwartz, R. G., and Statt, M. (1991). Reduced metabolic rate during beta-adrenergic blockade in humans. Metab. Clin. Exp. 40, 619–622. doi: 10.1016/0026-0495(91)90053-Y
Wettergren, A., Maina, P., Boesby, S., and Holst, J. J. (1997). Glucagon-like peptide-1 7-36 amide and peptide YY have additive inhibitory effect on gastric acid secretion in man. Scand. J. Gastroenterol. 32, 552–555. doi: 10.3109/00365529709025098
Wijers, S. L., Saris, W. H., and van Marken Lichtenbelt, W. D. (2010). Cold-induced adaptive thermogenesis in lean and obese. Obesity 18, 1092–1099. doi: 10.1038/oby.2010.74
Williams, D. L., Grill, H. J., Cummings, D. E., and Kaplan, J. M. (2003). Vagotomy dissociates short- and long-term controls of circulating ghrelin. Endocrinology 144, 5184–5187. doi: 10.1210/en.2003-1059
Witkowski, A., Prejbisz, A., Florczak, E., Kadziela, J., Sliwinski, P., Bielen, P., et al. (2011). Effects of renal sympathetic denervation on blood pressure, sleep apnea course, and glycemic control in patients with resistant hypertension and sleep apnea. Hypertension 58, 559–565. doi: 10.1161/HYPERTENSIONAHA.111.173799
Wofford, M. R., Anderson, D. C. Jr., Brown, C. A., Jones, D. W., Miller, M. E., and Hall, J. E. (2001). Antihypertensive effect of alpha- and beta-adrenergic blockade in obese and lean hypertensive subjects. Am. J. Hypertens 14(7 Pt 1), 694–698. doi: 10.1016/S0895-7061(01)01293-6
Woods, A. J., and Stock, M. J. (1996). Leptin activation in hypothalamus. Nature 381:745. doi: 10.1038/381745a0
Woods, S. C., Seeley, R. J., Porte, D. Jr., and Schwartz, M. W. (1998). Signals that regulate food intake and energy homeostasis. Science 280, 1378–1383. doi: 10.1126/science.280.5368.1378
Xu, X., Chen, D. D., Yin, J., and Chen, J. D. (2014). Altered postprandial responses in gastric myoelectrical activity and cardiac autonomic functions in healthy obese subjects. Obes. Surg. 24, 554–560. doi: 10.1007/s11695-013-1109-6
Yang, H. (2002). Central and peripheral regulation of gastric acid secretion by peptide YY. Peptides 23, 349–358. doi: 10.1016/S0196-9781(01)00611-8
Yi, C. X., la Fleur, S. E., Fliers, E., and Kalsbeek, A. (2010). The role of the autonomic nervous liver innervation in the control of energy metabolism. Biochim. Biophys. Acta 1802, 416–431. doi: 10.1016/j.bbadis.2010.01.006
Yoneshiro, T., Aita, S., Matsushita, M., Kayahara, T., Kameya, T., Kawai, Y., et al. (2013). Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Invest. 123, 3404–3408. doi: 10.1172/JCI67803
Young, J. B., and Landsberg, L. (1977). Stimulation of the sympathetic nervous system during sucrose feeding. Nature 269, 615–617. doi: 10.1038/269615a0
Keywords: autonomic nervous system, obesity, gut hormones, adipose tissue, energy expenditure, weight loss, vagal nerve stimulation, vagal nerve blockade
Citation: Guarino D, Nannipieri M, Iervasi G, Taddei S and Bruno RM (2017) The Role of the Autonomic Nervous System in the Pathophysiology of Obesity. Front. Physiol. 8:665. doi: 10.3389/fphys.2017.00665
Received: 02 February 2017; Accepted: 22 August 2017;
Published: 14 September 2017.
Edited by:
Tijana Bojić, INN Vinča University of Belgrade, SerbiaReviewed by:
Elisabeth Lambert, Swinburne University of Technology, AustraliaCopyright © 2017 Guarino, Nannipieri, Iervasi, Taddei and Bruno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Maria Bruno, cm9zYW1hcmlhLmJydW5vQHVuaXBpLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.