 Lin Cai
Lin Cai Guangpu Fan
Guangpu Fan Fang Wang1
Fang Wang1 Si Liu
Si Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 30 May 2017
Sec. Lipid and Fatty Acid Research
Volume 8 - 2017 | https://doi.org/10.3389/fphys.2017.00356
Background: We previously reported that lysophosphatidic acid (LPA) promoted cardiomyocyte hypertrophy in vitro via one of its G protein-coupled receptor subtypes, LPA3. In this study, we examined the role of LPA3 in cardiac hypertrophy induced by isoproterenol (ISO) and myocardial infarction.
Methods: In vitro, neonatal rat cardiomyocytes (NRCMs) were subjected to LPA3 knocked-down, or pretreated with a β-adrenergic receptor (β-AR) antagonist (propranolol) before LPA/ISO treatment. Cardiomyocyte size and hypertrophic gene (ANP, BNP) mRNA levels were determined. In vivo, and wild-type mice were implanted subcutaneously with an osmotic mini-pump containing ISO or vehicle for 2 weeks; echocardiography was performed to determine the heart weight/body weight ratio, cardiomyocyte cross-sectional area, and level of ANP mRNA expression. and wild-type mice were subjected to permanent coronary artery ligation or sham surgery for 4 weeks; cardiac function, including the degree of hypertrophy and infarction size, was determined.
Results: In vitro, we found that knocked-down LPA3 in NRCMs did not attenuate ISO-induced hypertrophy, and propranolol was unable to abolish LPA-induced hypertrophy. In vivo, chronic ISO infusion caused cardiac hypertrophy in wild-type mice, while hypertrophic responses to ISO infusion were not attenuated in mice. However, in a myocardial infarction (MI) model, mice exhibited reduced cardiac hypertrophy compared to wild-type mice at 4 weeks post-MI, which was associated with reduced cardiac function and increased infarct size.
Conclusions: Our data show that LPA3 appears to play a protective role in myocardial hypertrophy post-MI, but does not appear to be involved in the hypertrophy that occurs in response to β-AR stimulation in vivo and in vitro. These results implicate LPA-LPA3 lipid signaling in cardiac hypertrophy occurring after pathological insults like MI, which presents a new variable in β-AR-independent hypertrophy. Thus, modulation of LPA3 signaling might represent a new strategy for preventing the stressed myocardium from ischemia injury.
Pathological cardiac hypertrophy is a powerful and independent risk factor for heart failure and mortality (Haider et al., 1998; Okin et al., 2006). β-adrenergic receptor (β-AR) stimulation and the renin-angiotensin-aldosterone system (RAAS) are both thought to be involved in this condition (Gupta et al., 2007). However, although β-AR blockade and angiotensin-converting enzyme inhibition are associated with an improvement in left ventricular performance and the reversal of left ventricular remodeling, left ventricular systolic function can still deteriorate, resulting in a high incidence of heart failure that is a huge burden on healthcare resources (Ma et al., 2010; Dinicolantonio et al., 2015; Mozaffarian et al., 2016). New mechanisms need to be identified in order to better understand and treat both cardiac hypertrophy and the associated heart failure.
Lysophosphatidic acid (LPA) is a small glycerophospholipid, which is involved in multiple biological actions including proliferation, survival, migration, apoptosis, and differentiation via its six, seven-transmemberane G protein-coupled receptors (GPCRs), LPA1–LPA6 (Choi et al., 2010; Kihara et al., 2014; Yung et al., 2014). Our previous study found that cardiomyocyte hypertrophy could be induced by LPA, via Akt and ERK-NFκB signaling pathways (Chen et al., 2008). Moreover, we have recently identified pro-hypertrophic effects of LPA on neonatal rat cardiomyocytes (NRCMs) are mediated through the LPA3 receptor (encoded by Lpar3) (Yang et al., 2013b). However, whether hypertrophic signaling induced by LPA-LPA3 is the same or different from other well-established pro-hypertrophic pathways, and whether the role of LPA3 in ventricular remodeling is good or bad, are currently unknown. In the present study, we investigated whether LPA-LPA3 is involved in hypertrophy induced by β-AR activation in vitro and in vivo, and assessed possible roles for LPA3 signaling in the heart following myocardial infarction (MI) in mice.
The mice used in this study were described previously (Yang et al., 2002; Ye et al., 2005; Choi et al., 2008). All mice were bred from a BALBc strain, and backcrossed for more than 10 generations. This study was carried out in accordance with the “Regulation to the Care and Use of Experimental Animals” of the Beijing Council on Animal Care study (1996). The protocol was approved by the Fuwai Hospital Animal Care and Use Committee.
The culture of NRCMs isolated from 1- to 3-day-old Sprague-Dawley rats was carried out as previously described (Chen et al., 2002). DMEM containing 10% fetal bovine serum, penicillin/streptomycin (1,000 U/ml each) with 100 mM 5-Bromo-2-deoxyUridine was added to inhibit the growth of cardiac fibroblasts. After 24 h of culture, the cells were washed and starved overnight in serum-free medium prior to use within experiments.
Small interfering RNA (siRNA) (Invitrogen, Carlsbad, CA, USA) for LPA3 was transfected into cardiomyocytes using Lipofectamine™ RNAiMAX according to the manufacturer's protocol, as previously described (Yang et al., 2013b). Stealth siRNA duplex target sequences were 5′-UACACCACCACCAUGAUGAAGAAGG-3′ and 5′-CCUUCUUCAUCAUGGUGGUGGUGUA-3′. The sequence for the stealth siRNA low-GC duplex was used as a negative control. The cells were transfected with the stealth siRNA at 20 nM.
NRCMs were placed on coverslips in six-well culture plates and treated with LPA (5 μM) or ISO (1 μM) for 24 h, or pretreated with propranolol (1 μM) for half an hour before LPA/ISO stimulation. In the transfection experiments, cells were transfected with siRNA of LPA3 or the negative control for 24 h, then exposed to LPA or ISO for 24 h. The proportion of cells staining positively for α-actinin was measured to determine the degree of cardiomyocyte hypertrophy. For α-actinin staining, cells were fixed with 4% paraformaldehyde for 30 min, and subsequently permeabilized with 0.1% Triton X-100 for 10 min and 1% BSA for 30 min at room temperature. After washing, cells were incubated with mouse monoclonal antibody against α-actinin (1:250, Abcam, Cambridge, MA, USA) at 37°C for 2 h and a fluorescein-conjugated goat-anti-mouse IgG (1:300, Zhongshan Jinqiao Biotechnology, Beijing, China) at 37°C for 1 h. DAPI staining marked the nuclei. The cells were visualized under a fluorescence microscope (Zeiss, Oberkochen, Germany) and laser confocal microscopy (Leica, Wetzlar, Germany). At least 100 cardiomyocytes in 20–30 fields were examined in each group.
Micro-osmotic pumps (model 1002, Alzet) releasing ISO (Sigma, St. Louis, MO, USA; 60 mg/kg/day in PBS) or vehicle (PBS) were implanted subcutaneously into age-matched (8–10 weeks old) male mice and wild-type littermates after being anesthetized by intraperitoneal administration of tribromoethanol (400 mg/kg). Hypertrophy was assessed by echocardiography using a VisualSonics Vevo 2100 ultrasound system (VisualSonics, Inc., Toronto, ON, Canada). Ventricular measurements in M-mode were taken before and 13 days after implantation. The following day mice were sacrificed for histological and molecular analysis.
Myocardial infarction (MI) experiments were performed on 8–10 week-old and wild-type mice. The MI model was characterized as previously described (Fan et al., 2015). Mice were anesthetized by intraperitoneal administration of tribromoethanol (400 mg/kg) and ventilated with a rodent respirator, then the left anterior descending coronary artery was permanently occluded using a 7-0 polypropylene suture, and the occlusion was confirmed by blanching of the anterior wall of the left ventricle. For non-infarcted controls, mice underwent sham operation where the ligature around the left anterior descending coronary artery was not tied. Animals were recovered from anesthesia under warm conditions with normal ventilation. Four weeks after surgery, cardiac function was assessed by echocardiography using a VisualSonics Vevo 2100 ultrasound system (VisualSonics, Inc.), then animals were sacrificed and hearts were excised for further analysis.
Hearts were fixed in 10% formalin overnight at room temperature and embedded in paraffin. Subsequently, the hearts were cut serially from the apex to the base. Each short-axis section (5 μm), collected with an interval of 200 μm, was stained with Picrosirius red for morphometric analysis. The experiments were executed following standard procedures. The infarct size was calculated as a percentage of the total left ventricle (LV) wall circumference from each of the three LV sections. Scar circumstance was calculated using Image Analysis Software.
WGA staining was carried out to measure the cross-sectional area of cardiac myocytes. Following antigen retrieval using citric acid buffer (10 mM, pH 6.0), the sections were blocked with 1% bovine serum albumin for 30 min at room temperature and then incubated with Alexa Fluor®594-WGA (Invitrogen) for 1 h at room temperature. The slides were washed three times in PBS, mounted using an anti-fade mounting media containing DAPI (Zhongshan Jinqiao Biotechnology), and imaged was performed using a Leica DM6000B microscope.
Total RNA was extracted from mice ventricular tissue and cultured cardiomyocytes using Trizol, and was then quantified using a Nanodrop 2000 spectrophotometer. cDNA was generated from total RNA (1 μg) using M-MLV reverse transcriptase and oligo(dT)15 primer. qRT-PCR was performed using a SYBR Green PCR Master Mix and an Applied Biosystems 7500 (ABI, Foster City, CA, USA). The primer pairs used in this study are listed in Table 1.

Table 1. Primers for quantitative real-time PCR detection.
All data are presented as mean ± SEM. Differences among groups were assessed by a one-way analysis of variance (ANOVA) followed by a post-hoc Tukey's test. Comparisons between two groups were performed using a Student's t-test. All statistical analyses were performed with GraphPad Prism 6.0. A value of p < 0.05 was considered to indicate a statistically significant difference.
To test whether LPA3 is involved in an agonist of β-AR, ISO-induced hypertrophy, we used RNAi technology to knock down LPA3 expression in neonatal rat cardiomyocytes (Figure 1A). Cardiomyocyte size and the expression of atrial natriuretic factor (ANP) and brain natriuretic factor (BNP) mRNA were significantly increased after LPA or ISO treatment (Figures 1B–D). LPA3 interference resulted in a significant reduction in LPA-induced cardiomyocyte hypertrophy, but had no effect on ISO-induced ANP and BNP mRNA expression (Figure 1B) or cardiomyocyte size (Figures 1C,D). These results suggest that LPA3 is not involved in ISO-induced cardiomyocyte hypertrophy in vitro.
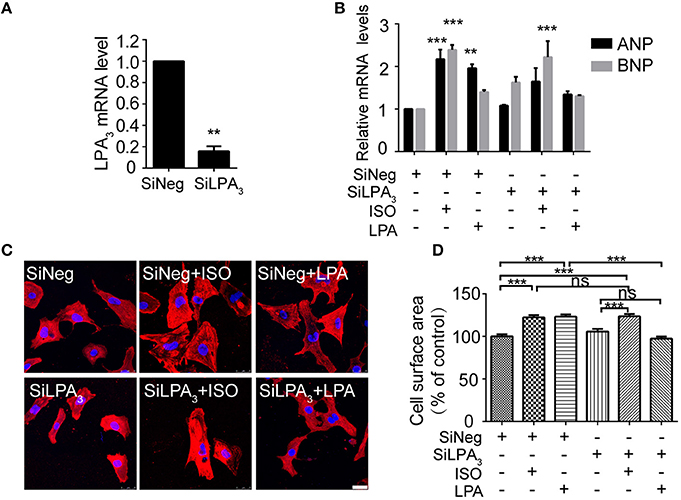
Figure 1. LPA3 silencing does not abolish ISO-induced cardiomyocyte hypertrophy. Rat neonatal cardiomyocytes were transfected with siRNA of LPA3 for 24 h. The level of LPA3 mRNA was measured by qPCR (A). After knockdown of LPA3, NRCMs were treated with ISO or LPA for 24h. The mRNA levels for pro-hypertrophic genes (ANP and BNP) were compared in the indicated groups (B). NRCMs were stained using an α-actinin antibody and DAPI to measure the average cell surface area (C,D, scale bar, 100 μm. Over 100 individual cells were analyzed per group). **p < 0.01, ***p < 0.001, ns p > 0.05 vs. SiNeg.
To further explore whether the β-AR participates in LPA-induced cardiomyocyte hypertrophy, a β-AR antagonist, propranolol, was used. After propranolol (1 μM) pretreatment for 30 min, ISO failed to increase cardiomyocyte size and ANP and BNP mRNA levels. However, LPA-induced cardiomyocyte hypertrophy was not significantly inhibited (Figures 2A–C). Taken together, our results indicate that ISO and LPA promote cardiomyocyte hypertrophy in vitro by independent mechanisms.
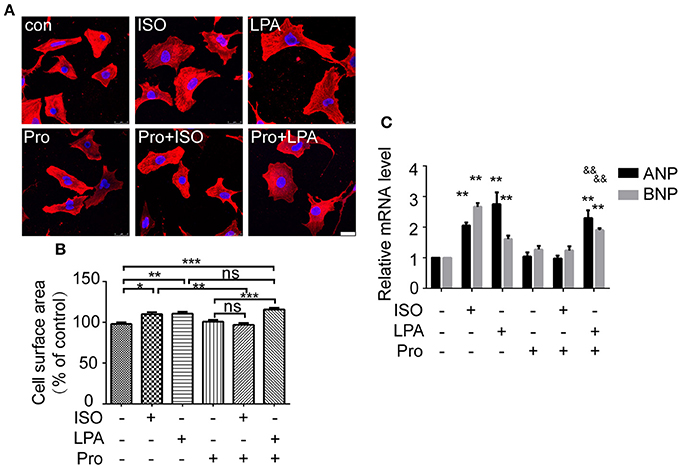
Figure 2. LPA-induced cardiomyocyte hypertrophy is not affected by a β-adrenergic receptor antagonist. Serum-starved NRCMs were treated with propranolol (Pro) for 0.5 h before LPA or ISO stimulation for 24 h. NRCMs were stained with an α-actinin antibody and DAPI to measure the average cell surface area (A,B, scale bar, 100 μm, over 100 individual cells were analyzed per group). The mRNA levels for pro-hypertrophic genes (ANP and BNP) were compared in the indicated groups (C). *p < 0.05, **p < 0.01, ***p < 0.001, ns p > 0.05 vs. control. &&p < 0.01 vs. Pro.
We further assessed whether the absence of LPA3 attenuates ISO-induced cardiac hypertrophy in vivo. and wild-type mice were subcutaneously implanted with an osmotic pump delivering ISO (60 mg/kg/day) for 2 weeks. Echocardiology data showed that left ventricular anterior and posterior wall thickness, and the left ventricular weight/body weight ratio were significantly increased in wild-type mice after ISO infusion (Figures 3A,C–E). mice showed similar responses to ISO infusion. Furthermore, heart rate was significantly increased in both wild-type and mice after ISO infusion (Table 2, Figure 3B).
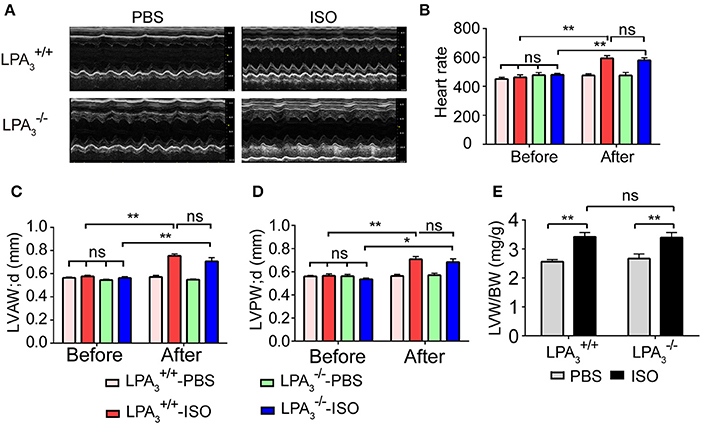
Figure 3. Loss of LPA3 does not affect echocardiographic parameters associated with ISO-induced cardiac hypertrophy. After ISO (60 mg/kg/d in PBS) or vehicle (PBS) infusion for 2 weeks, M-mode echocardiograms of hearts from wild-type or LPA3 knockout mice were recorded. Representative examples are shown in (A). Echocardiographic parameters including heartrate (B), LV diastolic anterior wall (LVAWd) (C), LV diastolic posterior wall (LVPWd) (D), and the left ventricular weight/body weight ratio (E) were measured. N = 8–11 mice per group. *p < 0.05, **p < 0.01, ns p > 0.05.
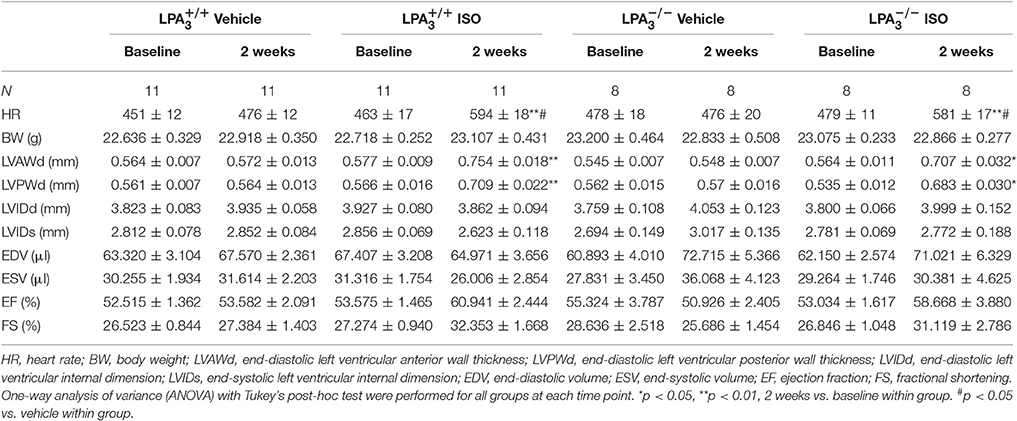
Table 2. Echocardiographic characteristics of the different study groups at baseline and 2 weeks post-ISO infusion.
Morphological data show that ISO infusion resulted in a significant increase in heart size (Figure 4A), heart to body ratio (Figure 4B), and cardiomyocyte cross-sectional area in wild-type mice (Figures 4C,D). However, there was no attenuation of these hypertrophic responses in mice (Figures 4A–D). Furthermore, we assessed the mRNA levels of genes associated with hypertrophy in the four treatment groups. We observed a similar alteration to ANP expression in both and hearts after ISO treatment (Figure 4E). These findings support the hypothesis that LPA3 deficiency does not affect ISO-induced cardiac hypertrophy in vivo.
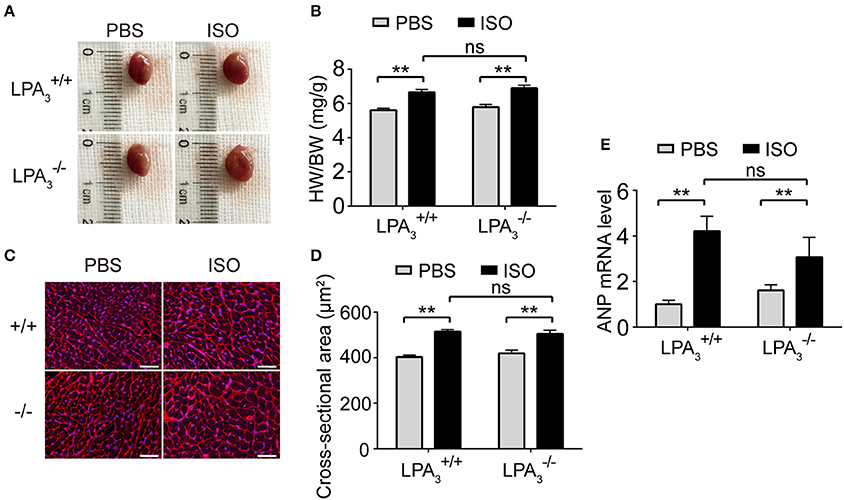
Figure 4. Loss of LPA3 does not affect ISO-induced cardiac hypertrophy. After ISO (60 mg/kg/day in PBS) infusion for 2 weeks in wild-type and LPA3 knockout mice, heart size (A) and the heart weight/body weight ratio (B, n = 8–11 mice per group) were measured. The cross-sectional area of cardiomyocytes was determined by wheat germ agglutinin staining (C,D, scale bar, 50 μm, n = 5 mice per group). The level of ANP mRNA expression was compared between the indicated groups (E, n = 6–8 mice). **p < 0.01, ns p > 0.05.
Since it is known that LPA-induced hypertrophy is different from the pathological hypertrophy induced by the β-AR, we decided to investigate further the role of LPA3-induced hypertrophy in pathological ventricular remodeling. At 4 weeks after MI, animals showed marked increases in the heart weight/body weight ratio, levels of ANP mRNA expression, and cardiomyocyte cross-sectional area (Figures 5A–D). Compared with infarcted mice, there was a tendency for a reduced heart weight/body weight ratio (Figure 5A), a notable decrease in ANP mRNA levels (Figure 5B), and a significant decrease in cardiomyocyte cross-sectional area (Figures 5C,D) in mice post-MI. These results indicate that LPA3 is involved in cardiac hypertrophy after MI.
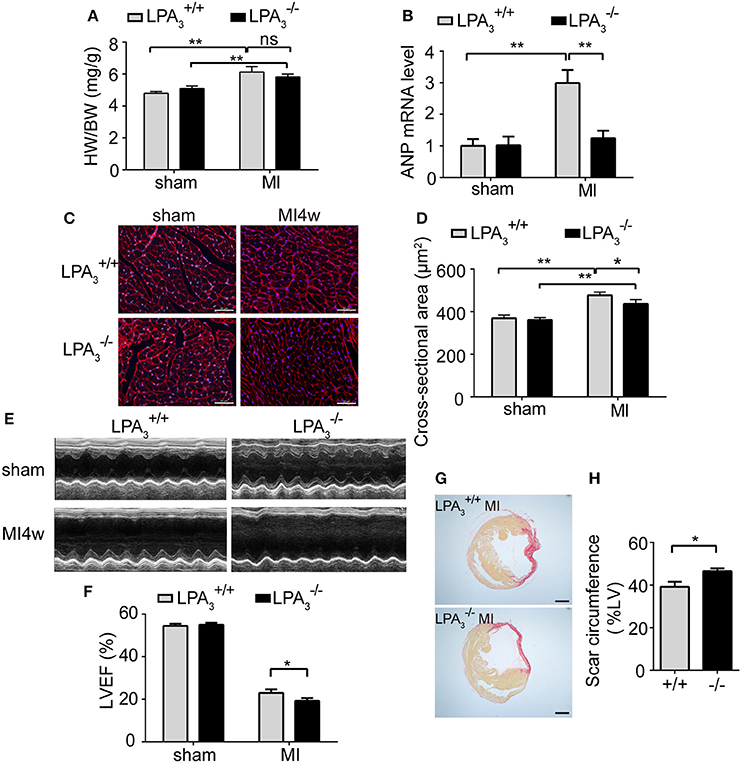
Figure 5. Loss of LPA3 attenuates cardiac hypertrophy, but exacerbates infarct size and loss of heart function following an MI. Four weeks after Ml, the heart weight/body weight ratio (A, n = 13–14 mice per group) and ANP mRNA levels (B, n = 4 mice per group) were compared in and wild-type mice. Cardiomyocyte cross-sectional area was determined by wheat germ agglutinin staining. (C,D, scale bar, 50 μm, n = 5 mice per group). (E) Representative echocardiography images and LVEF measurement (F, n = 13–14 mice per group) in the indicated groups. Scar circumference was measured and expressed as a percentage of the total area of the LV myocardium. Representative images (G) and measurement of the scar circumference (H, scale bar, 1,000 μm, n = 5 mice per group) were determined by Sirius red staining. *p < 0.05, **p < 0.01, ns p > 0.05.
Meanwhile, we further investigated the effect of LPA3 deficiency on cardiac function post-MI. mice manifested no pathological abnormalities with respect to cardiac function at baseline. After MI challenge, the ejection fraction percentage (EF%) was lower in both and mice, but mice exhibited more pronounced LV dilatation and more severe contractile dysfunction (Table 3, Figures 5E,F). Sirius red staining at 4 weeks after MI further displayed a significantly enlarged infarct size in mice compared with wild-type control animals (P < 0.05; Figures 5G,H). These findings suggest that LPA3 might play a protective role against MI.
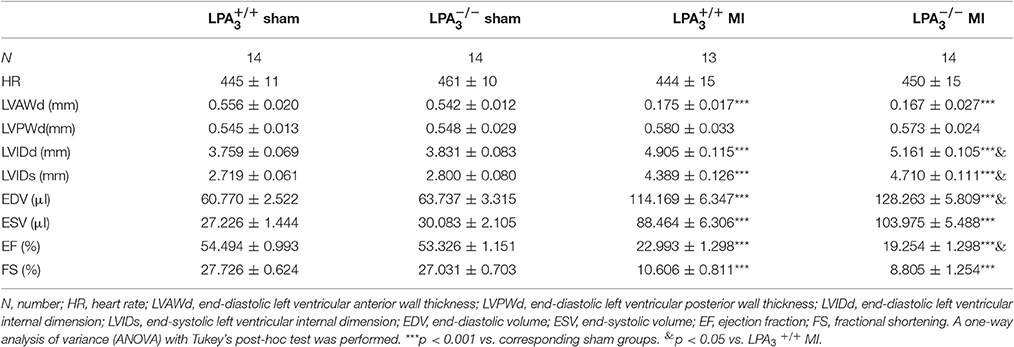
Table 3. Echocardiographic characteristics of the different study groups 4 weeks after a myocardial infarction.
In the present study, we first demonstrated that LPA3 signaling is not involved in ISO-induced cardiac hypertrophy in vitro and in vivo. However, LPA3 deficiency attenuated cardiac hypertrophy while aggravating cardiac dysfunction after MI, suggesting a protective role of LPA3 in cardiac function. These results implicate LPA-LPA3 lipid signaling as a new variable in cardiac hypertrophy, independent of the β-AR system, which can also affect cardiac function.
Our previous study showed that LPA stimulates cardiomyocyte hypertrophy through LPA3 (Yang et al., 2013b). Here we find that hypertrophy induced by LPA-LPA3 signaling is independent of ISO-β-AR system. ISO, a catecholamine, is a synthetic β- adrenergic agonist that induces cardiomyocyte hypertrophy in experimental animals (Oudit et al., 2003; Hohimer et al., 2005; Cha et al., 2009). β-AR is one of the most important GPCRs and plays a central role in sympathetic regulation of cardiac function. With sustained ISO stimulation, β-AR binds to Gs protein to elevate cystolic cAMP. cAMP activates PKA to cause abnormal diastolic sarcoplasmic reticulum (SR) Ca2+ leak, or directly activate Epac thus inducing NFAT dependent fetal phenotype gene elevation by a CaMKIIδ-dependent manner, which eventually both give rise to hypertrophy and heart failure (Tada and Kirchberger, 1975; Antos et al., 2001; Morel et al., 2005; Metrich et al., 2008; Grimm and Brown, 2010; Ruiz-Hurtado et al., 2012; Pereira et al., 2013). As for LPA-LPA3 lipid signaling system, LPA3 is also one member of GPCRs. LPA induces cardiomyocyte hypertrophy through a mechanism involving both Gi and the small G protein Rho (Hilal-Dandan et al., 2004), which is different from G proteins that ISO-β-AR couples to. After binding to Gi, LPA3 activates Akt and ERK-NFκB, and then promotes the expression of fetal phenotype gene markers ANP and BNP (Chen et al., 2008; Yang et al., 2013b). These data suggest that β-AR and LPA3 mediate distinct effectors and, in support of this, the present study provides experimental evidence for LPA-LPA3 signaling being distinct from ISO-β-AR-mediated myocardial hypertrophy. It is worth noting that another important GPCR in the cardiovascular system, AT1aR, interacts with the β-AR system, since AT1aR−/− mice showed remarkable repression of cardiac hypertrophy and oxidative stress in response to ISO stimulation (Zhang et al., 2007), in contrast to LPA3 signaling. Whether LPA3 contributes to myocardial hypertrophy via the related RAAS or represents a new system, awaits further study.
We found that LPA3 deficiency attenuated cardiac hypertrophy but aggravated cardiac dysfunction after MI. Cardiac hypertrophy involved in myocardial remodeling includes compensated hypertrophy and decompensated hypertrophy, which are associated with different signaling pathways (Tham et al., 2015). Decompensated hypertrophy results in pathological hypertrophy and heart failure, which involves the activation of NFAT signaling, β-AR-CaMKII signaling, cGMP-PKG signaling, and PKC-MAPKs signaling (Bernardo et al., 2010; Shimizu and Minamino, 2016). Overactivation of β-AR correlates with cardiotoxic outcomes such as hypertrophy and heart failure (Osadchii, 2007; Pleger et al., 2007; El-Armouche and Eschenhagen, 2009). While blockade of β-AR remarkably reduces cardiac hypertrophy and promotes heart function in mouse model (Ni et al., 2011). As Gs-cAMP-PKA and cAMP-CaMKII-Epac-NFAT are central mechanisms of β-AR-induced hypertrophy, gain- and loss- of function studies show that they are all involved in pathological hypertrophy. For example, transgenic over-expression studies indicate that Gsα (Gaudin et al., 1995), PKA (Antos et al., 2001), or NFAT(Wilkins et al., 2004) are all sufficient for inducing pathological hypertrophy and heart failure in vivo. Gain of CaMKII function leads to cardiac hypertrophy, while inhibition of CaMKII ameliorates myocardial hypertrophy and improves heart function (Kirchhefer et al., 1999; Ai et al., 2005; Bossuyt et al., 2008). Although, the boundary between compensated and decompensated hypertrophy is not clearly elucidated, some signaling pathways are considered to be beneficial (Tham et al., 2015; Shimizu and Minamino, 2016). For example, the insulin-like growth factor 1 (IGF1) -phosphoinositide-3-kinase (p110α)—protein kinase B (Akt) signaling pathway appears to be involved in mediating physiological heart growth such as postnatal and exercise-induced hypertrophy, but not pathological hypertrophy like that associated with pressure overload (e.g., by constricting the ascending aorta) (McMullen et al., 2003, 2004; Luo et al., 2005; Debosch et al., 2006; Kim et al., 2008). Among the mitogen-activated protein kinases (MAPKs) superfamily, ERK1/2 has been reported to mediate both adaptive (Gαq mediated) (Bueno et al., 2000) and maladaptive (via Gβγ) (Lorenz et al., 2009) processes through different phosphorylation sites within the heart. As the underlying mechanisms of LPA-induced cardiomyocyte hypertrophy involve activation of Akt and ERK-NFκB (Chen et al., 2008; Yang et al., 2013b), with Gαq-dependent signaling by LPA3 reported in other cell systems like vascular smooth muscle (Kim et al., 2006), we speculate that in contrast with detrimental effects of ISO-β-AR activation, LPA-LPA3 signaling might represent another upstream stimulus for adaptive hypertrophy.
It is well-known that GPCRs are much important drug targets. β-blockers and AngII receptor inhibitors have become widely used and effective cardiovascular medicines (Liao et al., 2004; Ouwerkerk et al., 2017). In the present study, we found that LPA3 deficiency led to reduced cardiac function and increased infarct size after MI. LPA3 also belongs to GPCR family, and our study hits that functionally enhances LPA3 might be a new strategy to protect heart from ischemia injury. The role of LPA signaling in cardiovascular diseases is broad. On one hand, LPA is reported to promote the progression of atherosclerotic vascular diseases (Siess et al., 1999; Kritikou et al., 2016). On the other hand, LPA has protective roles in cell survival. For example, LPA protects CD34+ cells from ischemia-induced apoptosis and the delivery of LPA-treated CD34+ cells into the infarcted heart improved cardiac function (Kostic et al., 2015). We also reported that LPA may protect cardiomyocytes from hypoxia/reperfusion-induced injury by activating LPA3 and suppressing mitochondrial apoptotic pathways in vitro (Yang et al., 2013a) and ex vivo (Chen et al., 2017). Here we found aggravated cardiac dysfunction causing by LPA3 deficiency appears to correlate with decreased hypertrophy, but there may be other, concomitant mechanisms, such as resistance to apoptosis. However, it needs further study to prove that anti-apoptosis action of LPA is responsible for myocardial protection of LPA-LPA3 during myocardial remodeling post-MI.
In conclusion, this study suggests that LPA3 participates in hypertrophy associated with myocardial remodeling post-MI, but does not appear to be involved in the induction of hypertrophy in response to β-AR stimulation in vitro and in vivo. LPA3 deficiency leads to dysfunction of the myocardium post-MI. Taken together, these results indicate that LPA-LPA3 lipid may be a novel regulator of cardiac hypertrophy, which in turn suggests that strategies aimed at LPA3 modulation could protect the myocardium against ischemic injury.
XC, XfC, FW, and LC conceived the studies. LC, GF, TL, and SL performed the experiments. LC and FW analyzed and interpreted data. JC provided the mice. LC wrote, and XC, FW, JC, and GF revised the manuscript. All authors have read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Ms. Qing Xu of Capital Medical University, Beijing, China for providing accurate echocardiography measurements. This work was supported by the National Natural Science Foundation of China (81470484), the National Natural Science Foundation of China (81300111), and the Doctoral Student's Innovation Fund of Chinese Academy of Medical Sciences and Peking Union Medical College (2014-0710-1020). JC was supported by the US National Institutes of Health (R01 HD050685 and R01 NS084398).
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Antos, C. L., Frey, N., Marx, S. O., Reiken, S., Gaburjakova, M., Richardson, J. A., et al. (2001). Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ. Res. 89, 997–1004. doi: 10.1161/hh2301.100003
Bernardo, B. C., Weeks, K. L., Pretorius, L., and McMullen, J. R. (2010). Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol. Ther. 128, 191–227. doi: 10.1016/j.pharmthera.2010.04.005
Bossuyt, J., Helmstadter, K., Wu, X., Clements-Jewery, H., Haworth, R. S., Avkiran, M., et al. (2008). Ca2+/calmodulin-dependent protein kinase IIδ and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ. Res. 102, 695–702. doi: 10.1161/CIRCRESAHA.107.169755
Bueno, O. F., De Windt, L. J., Tymitz, K. M., Witt, S. A., Kimball, T. R., Klevitsky, R., et al. (2000). The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350. doi: 10.1093/emboj/19.23.6341
Cha, H. N., Hong, G. R., Kim, Y. W., Kim, J. Y., Dan, J. M., and Park, S. Y. (2009). Deficiency of iNOS does not prevent isoproterenol-induced cardiac hypertrophy in mice. Korean J. Physiol. Pharmacol. 13, 153–159. doi: 10.4196/kjpp.2009.13.3.153
Chen, H., Liu, S., Liu, X., Yang, J., Wang, F., Cong, X., et al. (2017). Lysophosphatidic acid pretreatment attenuates myocardial ischemia/reperfusion injury in the immature hearts of rats. Front. Physiol. 8:153. doi: 10.3389/fphys.2017.00153
Chen, J., Chen, Y., Zhu, W., Han, Y., Han, B., Xu, R., et al. (2008). Specific LPA receptor subtype mediation of LPA-induced hypertrophy of cardiac myocytes and involvement of Akt and NFkappaB signal pathways. J. Cell. Biochem. 103, 1718–1731. doi: 10.1002/jcb.21564
Chen, X., Cui, Z., Zhang, F., Chang, W., and Chen, L. (2002). Angiotensin II and cAMP regulate AT(1)-mRNA expression in rat cardiomyocytes by transcriptional mechanism. Eur. J. Pharmacol. 448, 1–9. doi: 10.1016/S0014-2999(02)01900-3
Choi, J. W., Herr, D. R., Noguchi, K., Yung, Y. C., Lee, C. W., Mutoh, T., et al. (2010). LPA receptors: subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 50, 157–186. doi: 10.1146/annurev.pharmtox.010909.105753
Choi, J. W., Lee, C. W., and Chun, J. (2008). Biological roles of lysophospholipid receptors revealed by genetic null mice: an update. Biochim. Biophys. Acta 1781, 531–539. doi: 10.1016/j.bbalip.2008.03.004
Debosch, B., Treskov, I., Lupu, T. S., Weinheimer, C., Kovacs, A., Courtois, M., et al. (2006). Akt1 is required for physiological cardiac growth. Circulation 113, 2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231
Dinicolantonio, J. J., Fares, H., Niazi, A. K., Chatterjee, S., D'ascenzo, F., Cerrato, E., et al. (2015). beta-Blockers in hypertension, diabetes, heart failure and acute myocardial infarction: a review of the literature. Open Heart 2:e000230. doi: 10.1136/openhrt-2014-000230
El-Armouche, A., and Eschenhagen, T. (2009). β-Adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 14, 225–241. doi: 10.1007/s10741-008-9132-8
Fan, G. P., Wang, W., Zhao, H., Cai, L., Zhang, P. D., Yang, Z. H., et al. (2015). Pharmacological inhibition of focal adhesion kinase attenuates cardiac fibrosis in mice cardiac fibroblast and post-myocardial-infarction models. Cell Physiol. Biochem. 37, 515–526.
Gaudin, C., Ishikawa, Y., Wight, D. C., Mahdavi, V., Nadal-Ginard, B., Wagner, T. E., et al. (1995). Overexpression of Gs alpha protein in the hearts of transgenic mice. J. Clin. Invest. 95, 1676–1683. doi: 10.1172/JCI117843
Grimm, M., and Brown, J. H. (2010). Beta-adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol. 48, 322–330. doi: 10.1016/j.yjmcc.2009.10.016
Gupta, S., Das, B., and Sen, S. (2007). Cardiac hypertrophy: mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 9, 623–652. doi: 10.1089/ars.2007.1474
Haider, A. W., Larson, M. G., Benjamin, E. J., and Levy, D. (1998). Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 32, 1454–1459. doi: 10.1016/S0735-1097(98)00407-0
Hilal-Dandan, R., Means, C. K., Gustafsson, B., Morissette, M. R., Adams, J. W., Brunton, L. L., et al. (2004). Lysophosphatidic acid induces hypertrophy of neonatal cardiac myocytes via activation of Gi and Rho. J. Mol. Cell. Cardiol. 36, 481–493. doi: 10.1016/j.yjmcc.2003.12.010
Hohimer, A. R., Davis, L. E., and Hatton, D. C. (2005). Repeated daily injections and osmotic pump infusion of isoproterenol cause similar increases in cardiac mass but have different effects on blood pressure. Can. J. Physiol. Pharmacol. 83, 191–197. doi: 10.1139/y04-137
Kihara, Y., Maceyka, M., Spiegel, S., and Chun, J. (2014). Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 171, 3575–3594. doi: 10.1111/bph.12678
Kim, J., Keys, J. R., and Eckhart, A. D. (2006). Vascular smooth muscle migration and proliferation in response to lysophosphatidic acid (LPA) is mediated by LPA receptors coupling to Gq. Cell. Signal. 18, 1695–1701. doi: 10.1016/j.cellsig.2006.01.009
Kim, J., Wende, A. R., Sena, S., Theobald, H. A., Soto, J., Sloan, C., et al. (2008). Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22, 2531–2543. doi: 10.1210/me.2008-0265
Kirchhefer, U., Schmitz, W., Scholz, H., and Neumann, J. (1999). Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 42, 254–261. doi: 10.1016/S0008-6363(98)00296-X
Kostic, I., Fidalgo-Carvalho, I., Aday, S., Vazao, H., Carvalheiro, T., Graos, M., et al. (2015). Lysophosphatidic acid enhances survival of human CD34+ cells in ischemic conditions. Sci. Rep. 5:16406. doi: 10.1038/srep16406
Kritikou, E., Van Puijvelde, G. H., Van Der Heijden, T., Van Santbrink, P. J., Swart, M., Schaftenaar, F. H., et al. (2016). Inhibition of lysophosphatidic acid receptors 1 and 3 attenuates atherosclerosis development in LDL-receptor deficient mice. Sci. Rep. 6:37585. doi: 10.1038/srep37585
Liao, Y., Asakura, M., Takashima, S., Ogai, A., Asano, Y., Shintani, Y., et al. (2004). Celiprolol, a vasodilatory β-blocker, inhibits pressure overload-induced cardiac hypertrophy and prevents the transition to heart failure via nitric oxide-dependent mechanisms in mice. Circulation 110, 692–699. doi: 10.1161/01.CIR.0000137831.08683.E1
Lorenz, K., Schmitt, J. P., Schmitteckert, E. M., and Lohse, M. J. (2009). A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 15, 75–83. doi: 10.1038/nm.1893
Luo, J., McMullen, J. R., Sobkiw, C. L., Zhang, L., Dorfman, A. L., Sherwood, M. C., et al. (2005). Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell. Biol. 25, 9491–9502. doi: 10.1128/MCB.25.21.9491-9502.2005
Ma, T. K., Kam, K. K., Yan, B. P., and Lam, Y. Y. (2010). Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br. J. Pharmacol. 160, 1273–1292. doi: 10.1111/j.1476-5381.2010.00750.x
McMullen, J. R., Shioi, T., Huang, W. Y., Zhang, L., Tarnavski, O., Bisping, E., et al. (2004). The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J. Biol. Chem. 279, 4782–4793. doi: 10.1074/jbc.M310405200
McMullen, J. R., Shioi, T., Zhang, L., Tarnavski, O., Sherwood, M. C., Kang, P. M., et al. (2003). Phosphoinositide 3-kinase(p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 100, 12355–12360. doi: 10.1073/pnas.1934654100
Metrich, M., Lucas, A., Gastineau, M., Samuel, J. L., Heymes, C., Morel, E., et al. (2008). Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 102, 959–965. doi: 10.1161/CIRCRESAHA.107.164947
Morel, E., Marcantoni, A., Gastineau, M., Birkedal, R., Rochais, F., Garnier, A., et al. (2005). cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 97, 1296–1304. doi: 10.1161/01.RES.0000194325.31359.86
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2016). Heart disease and stroke statistics-2016 update: a report from the american heart association. Circulation 133, e38–e360. doi: 10.1161/CIR.0000000000000366
Ni, L., Zhou, C., Duan, Q., Lv, J., Fu, X., Xia, Y., et al. (2011). beta-AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. PLoS ONE 6:e27294. doi: 10.1371/journal.pone.0027294
Okin, P. M., Devereux, R. B., Nieminen, M. S., Jern, S., Oikarinen, L., Viitasalo, M., et al. (2006). Electrocardiographic strain pattern and prediction of new-onset congestive heart failure in hypertensive patients: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study. Circulation 113, 67–73. doi: 10.1161/CIRCULATIONAHA.105.569491
Osadchii, O. E. (2007). Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects. Heart Fail. Rev. 12, 66–86. doi: 10.1007/s10741-007-9007-4
Oudit, G. Y., Crackower, M. A., Eriksson, U., Sarao, R., Kozieradzki, I., Sasaki, T., et al. (2003). Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation 108, 2147–2152. doi: 10.1161/01.CIR.0000091403.62293.2B
Ouwerkerk, W., Voors, A. A., Anker, S. D., Cleland, J. G., Dickstein, K., Filippatos, G., et al. (2017). Determinants and clinical outcome of uptitration of ACE-inhibitors and beta-blockers in patients with heart failure: a prospective European study. Eur. Heart J. doi: 10.1093/eurheartj/ehx026. [Epub ahead of print].
Pereira, L., Cheng, H., Lao, D. H., Na, L., Van Oort, R. J., Brown, J. H., et al. (2013). Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 127, 913–922. doi: 10.1161/CIRCULATIONAHA.12.148619
Pleger, S. T., Boucher, M., Most, P., and Koch, W. J. (2007). Targeting myocardial β-adrenergic receptor signaling and calcium cycling for heart failure gene therapy. J. Card. Fail. 13, 401–414. doi: 10.1016/j.cardfail.2007.01.003
Ruiz-Hurtado, G., Dominguez-Rodriguez, A., Pereira, L., Fernandez-Velasco, M., Cassan, C., Lezoualc'h, F., et al. (2012). Sustained Epac activation induces calmodulin dependent positive inotropic effect in adult cardiomyocytes. J. Mol. Cell. Cardiol. 53, 617–625. doi: 10.1016/j.yjmcc.2012.08.004
Shimizu, I., and Minamino, T. (2016). Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 97, 245–262. doi: 10.1016/j.yjmcc.2016.06.001
Siess, W., Zangl, K. J., Essler, M., Bauer, M., Brandl, R., Corrinth, C., et al. (1999). Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. U.S.A. 96, 6931–6936. doi: 10.1073/pnas.96.12.6931
Tada, M., and Kirchberger, M. A. (1975). Regulation of calcium transport by cyclic AMP. A proposed mechanism for the beta-adrenergic control of myocardial contractility. Acta Cardiol. 30, 231–237.
Tham, Y. K., Bernardo, B. C., Ooi, J. Y., Weeks, K. L., and McMullen, J. R. (2015). Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch. Toxicol. 89, 1401–1438. doi: 10.1007/s00204-015-1477-x
Wilkins, B. J., Dai, Y. S., Bueno, O. F., Parsons, S. A., Xu, J., Plank, D. M., et al. (2004). Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 94, 110–118. doi: 10.1161/01.RES.0000109415.17511.18
Yang, A. H., Ishii, I., and Chun, J. (2002). In vivo roles of lysophospholipid receptors revealed by gene targeting studies in mice. Biochim. Biophys. Acta 1582, 197–203. doi: 10.1016/S1388-1981(02)00172-5
Yang, J., Chen, H., Xianyun, W., Xuesong, F., Xiangfeng, C., and Xi, C. (2013a). The protective role with its mechanism of lysophosphatidic acid on hypoxia/re-oxygenation induced neonatal rat's H9c2 cardiomyocyte injury. Chin. Circulat. J. 28, 222–225. doi: 10.3969/j.issn.1000-3614.2013.03.018
Yang, J., Nie, Y., Wang, F., Hou, J., Cong, X., Hu, S., et al. (2013b). Reciprocal regulation of miR-23a and lysophosphatidic acid receptor signaling in cardiomyocyte hypertrophy. Biochim. Biophys. Acta 1831, 1386–1394. doi: 10.1016/j.bbalip.2013.05.005
Ye, X., Hama, K., Contos, J. J., Anliker, B., Inoue, A., Skinner, M. K., et al. (2005). LPA3-mediated lysophosphatidic acid signalling in implantation and embryo spacing. Nature 435, 104–108. doi: 10.1038/nature03505
Yung, Y. C., Stoddard, N. C., and Chun, J. (2014). LPA receptor signaling: pharmacology, physiology, and pathophysiology. J. Lipid Res. 55, 1192–1214. doi: 10.1194/jlr.R046458
Keywords: lysophosphatidic acid, LPA3, hypertrophy, isoproterenol, MI
Citation: Cai L, Fan G, Wang F, Liu S, Li T, Cong X, Chun J and Chen X (2017) Protective Role for LPA3 in Cardiac Hypertrophy Induced by Myocardial Infarction but Not by Isoproterenol. Front. Physiol. 8:356. doi: 10.3389/fphys.2017.00356
Received: 09 February 2017; Accepted: 15 May 2017;
Published: 30 May 2017.
Edited by:
Naim Akhtar Khan, Université de Bourgogne, FranceReviewed by:
Monique Mulder, Erasmus University Rotterdam, NetherlandsCopyright © 2017 Cai, Fan, Wang, Liu, Li, Cong, Chun and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Chen, Y2hlbnhpZndAcHVtYy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.