 Brian L. Jones
Brian L. Jones Stephen M. Smith
Stephen M. Smith
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 30 March 2016
Sec. Integrative Physiology
Volume 7 - 2016 | https://doi.org/10.3389/fphys.2016.00116
This article is part of the Research Topic Physiology and pathophysiology of the extracellular calcium-sensing receptor View all 17 articles
Though both clinicians and scientists have long recognized the influence of extracellular calcium on the function of muscle and nervous tissue, recent insights reveal that the mechanisms allowing changes in extracellular calcium to alter cellular excitability have been incompletely understood. For many years the effects of calcium on neuronal signaling were explained only in terms of calcium entry through voltage-gated calcium channels and biophysical charge screening. More recently however, it has been recognized that the calcium-sensing receptor is prevalent in the nervous system and regulates synaptic transmission and neuronal activity via multiple signaling pathways. Here we review the multiplicity of mechanisms by which changes in extracellular calcium alter neuronal signaling and propose that multiple mechanisms are required to describe the full range of experimental observations.
The importance of extracellular calcium in regulating the behavior of excitable tissues was first recognized by Sydney Ringer when he became aware that a very effective physiological saline he developed was contaminated with calcium (Ringer, 1883). Upon this discovery, Ringer quickly determined that calcium at a concentration of approximately 1 mM was essential to maintain the viability and function of isolated frog hearts and solutions derived from Ringer's work have been employed by physiologists studying the heart and many other organ systems ever since (Miller, 2004). It was some time before the importance of calcium on neuronal excitability was recognized, but despite more than 100 years of inquiry, the mechanisms by which calcium alters the excitability of neurons remain incompletely elucidated. In this mini-review we will examine some of the mechanisms by which extracellular calcium influences neuronal signaling by altering both intrinsic excitability and synaptic transmission.
The distribution of calcium in the brain is characterized by steep transmembrane electrochemical gradients that are transiently attenuated as a result of large activity-dependent changes in both the intracellular and extracellular calcium concentration. The blood brain barrier defends extracellular calcium in the brain from changes in serum calcium (Jones and Keep, 1988) and at rest brain extracellular calcium is maintained at 1.1 mM (Hansen, 1985; Zhang et al., 1990; Nilsson et al., 1993, 1996). At the same time, neuronal intracellular calcium is orders of magnitude lower ranging between 50 and 100 nM, but rising rapidly to 10–100 μM in microdomains near open voltage-gated calcium channels when action potentials invade presynaptic terminals (Zucker, 1996). In contrast, during neuronal activity extracellular calcium can fall sharply as calcium is displaced to the intracellular compartment. These transient drops in calcium are facilitated by the small extracellular volume of the brain (only 12–20% of total volume; Rusakov et al., 1998) and restricted diffusion (up to five-fold slower than in free solution; Kullmann et al., 1999). The small volume and limited accessibility of the synaptic cleft led to the prediction that pre- and postsynaptic calcium influx during neurotransmission will significantly reduce calcium in the cleft following an action potential, possibly to as low as 0.3 mM (Smith, 1992; Vassilev et al., 1997b; Egelman and Montague, 1998, 1999; Rusakov et al., 1998). At the surface of the brain, ion-selective electrodes have shown that extracellular calciumfalls to 0.8 mM for tens of seconds following focal stimulation at rates of 20 Hz (Nicholson et al., 1978) and decreases to 0.1 mM have been recorded as a result of focal brain trauma (Nilsson et al., 1996). Importantly, it has been shown that short trains of action potentials can reduce extracellular calcium and impact synaptic transmission (Rusakov and Fine, 2003). The observed fall in extracellular calcium in the cortex at times of high activity (Nicholson et al., 1978) along with the extreme sensitivity of synaptic mechanism to extracellular calcium (Dodge and Rahamimoff, 1967) prompts us to ask how neurons respond to this change.
Even before calcium was identified as the trigger for exocytosis, reductions in extracellular calcium were known to increase the likelihood of action potential initiation (Frankenhaeuser and Hodgkin, 1957) by altering the properties of the neuronal ion channels. These effects on ion-channel activity can be divided into two major categories: those mediated by direct activity of calcium on ion-channel biophysics and those mediated indirectly by second messenger systems that are coupled to extracellular calcium concentrations by specific receptors. While many types of ion channels are involved in fine tuning neuronal excitability, voltage-gated sodium channels are central. Early work showed changes in excitability were mediated by shifts in the activation properties of the sodium conductance (Frankenhaeuser and Hodgkin, 1957) though subsequently extracellular calcium was found to alter the activity of other types of ion channels (Hablitz et al., 1986; Immke and McCleskey, 2001; Ma et al., 2012).
The most widely recognized model for the impact of calcium on sodium channel activity is surface charge screening (aka, surface potential theory), whereby interactions between multivalent cations like calcium and negatively charged phospholipids in the neuronal membrane serve to alter the intramembrane electric field that regulates the activity of voltage-gated ion channels in a concentration dependent manner (Hille, 2001). The idea is that membrane bound negative charges influence the local potential (Figure 1A, solid blue curve) and thereby reduce the intramembranous electric field (broken blue line) established by the transmembrane electrochemical gradient (black broken line). Extracellular divalents are adsorbed to the membranous negative charges and attenuate their impact on the existing voltage field (red broken line). Hence voltage-dependent channels within the intramembranous electric field have altered activity. In the case of a high extracellular calcium concentration, the intramembranous field is increased and the probability of channels being activated is decreased resulting in a reduction in excitability.
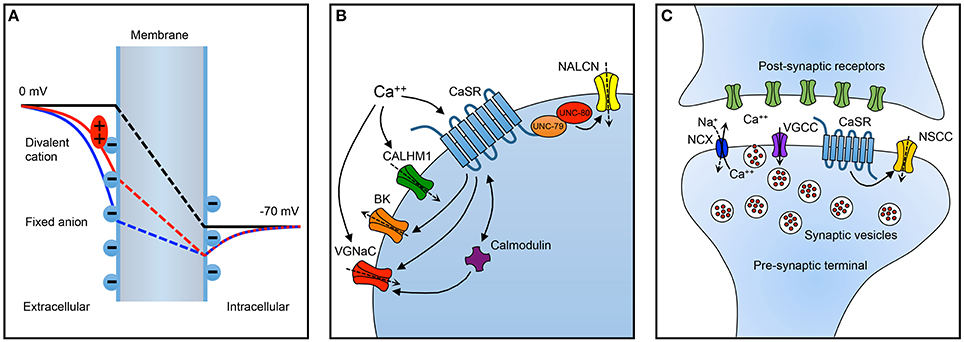
Figure 1. Summary of important neuronal targets of extracellular calcium. (A) Impact on resting membrane electric field of surface charge screening. The transmembrane potential is illustrated in three scenarios: without fixed anions (black line), with fixed anions (blue line), and with fixed anions and divalent cations (red line). The electric field produced by the transmembrane electrochemical gradient alone (black line) is attenuated by membrane associated negative charges in the absence of divalent cations (blue line). When divalent cations interact with (screen/adsorp to) the fixed anions, the influence of the fixed charges on intramembranous electric field is reduced (red line). Consequently, the activity of ion channels within the membrane and sensitive to the electric field may be altered by changes in the concentration of divalent cations. (B) Summary of targets of extracellular calcium on neuronal excitability and (C) synaptic transmission. BK, calcium-activated potassium channel; CALHM1, calcium homeostasis modulator 1; CaSR, calcium-sensing receptor; NALCN, Na-leak channel non-selective; NCX, sodium/calcium exchanger; NSCC, non-specific cation channel; VGCC, voltage-gated calcium channel; VGNaC, voltage-gated sodium channel. Dashed arrows reflect direction of current under typical conditions. Inwards arrows are depolarizing/excitatory. During high activity levels, the NCX replenishes extracellular calcium in the synaptic cleft.
However, surface charge screening does not account for all of the calcium-dependent gating phenomena exhibited by voltage-gated channels. At its simplest the surface potential theory predicts a uniform action on all types of voltage-dependent channels. While sodium channel activation is enhanced by reductions in extracellular calcium other types of voltage-gate ion-channels exhibit different dependence on extracellular calcium, ranging from sensitive to indifferent (Han et al., 2015). Beyond charge screening, other work has identified at least two other distinct biophysical mechanisms through which changes in extracellular calcium can alter the activity of voltage-gated sodium channels (Armstrong and Cota, 1991). Voltage-gated sodium channels have a number of extracellular moieties that also interact directly with calcium and so alter channel kinetics through changes in conformation or stability (Armstrong and Cota, 1990). Also, calcium ions are able to directly block sodium channels likely through interactions with specific amino acid residues lining the channel's pore (Santarelli et al., 2007).
Direct activation of non-selective cation channels following reductions in extracellular calcium, also depolarizes neurons and increases excitability (Hablitz et al., 1986; Xiong et al., 1997; Immke and McCleskey, 2001). Calcium homeostasis modulator 1 (CALHM1) is another non-selective cation channel that is both voltage- and calcium-dependent (Ma et al., 2012) positioning it to be a strong mediator of calcium-dependent excitability. In fact, neurons deficient in CALHM1 lost all calcium-dependent excitability (Ma et al., 2012) implying that in some neurons surface charge screening may not contribute to calcium-dependent excitability.
The excitability of neurons is also influenced indirectly by complex second messenger systems coupled to membrane receptors (Figure 1B). The calcium-sensing receptor (CaSR), a widely expressed G-protein coupled receptor, exhibits a punctate staining pattern in the cortex and cerebellum consistent with localization to nerve terminals (Ruat et al., 1995). Direct recordings from neocortical terminals, demonstrated that extracellular calcium regulated a membrane receptor and indirectly modulated a non-selective cation channel (Smith et al., 2004). Using a combination of pharmacological probes and a mutant mouse the terminal extracellular calcium receptor was identified as CaSR (Chen et al., 2010). In hippocampal neurons reductions in extracellular calcium increased neuronal excitability via another indirect mechanism. A non-selective cation channel (NSCC) NALCN (Na-Leak Channel Non-selective), was activated by decreases in extracellular calcium and mediated the vast majority of calcium-dependent excitability (Lu et al., 2010). This signaling pathway required two intracellular proteins, UNC-79 and UNC-80, and an unidentified membranous receptor (Lu et al., 2010). The authors went on to hypothesize that CaSR may be the receptor that detected and transduced changes in extracellular calcium into changes in neuronal excitability. In this model, low extracellular calcium was transduced into activation of a depolarizing current mediated by NALCN and increased neuronal excitability, controversially minimizing the contribution of surface charge screening (Lu et al., 2010). Other consequences of extracellular calcium signaling are suggested by work showing that CaSR may inhibit some neuronal potassium channels (Vysotskaya et al., 2014). Interestingly, CaSR activation was also proposed to activate other types of neuronal potassium channels (Vassilev et al., 1997a). Similarly, an unusually non-selective channel in neuronal soma was reported to be activated by CaSR agonists (Ye et al., 1996). The impact of decreased extracellular calcium on CaSR modulation seems to favor channel activation but the overall effect will depend on the balance of channel activation and block.
Intracellular changes in calcium, as a result of changes in extracellular calcium, may modulate channel activity and neuronal excitability. Calmodulin, a calcium sensitive signaling protein that is modulated by calcium entry, regulated sodium channel activity (Kim et al., 2004). Specifically, calmodulin interacted with an intracellular domain of voltage-gated sodium channels and so modified their gating behaviors (Sarhan et al., 2012). Notably, calmodulin has also been shown to regulate the cell surface expression and signaling from the CaSR providing a potential mechanism for cross-talk between these distinct calcium signaling pathways in the modulation of neuronal excitability (Huang et al., 2010). Thus, there are multiple direct and indirect mechanisms by which extracellular calcium can impact the intrinsic excitability of neurons and while surface charge theory provides a common mechanism across all neurons it would be surprising if these other mechanisms did not operate in parallel and mediate variability in calcium dependent excitability between neuronal types.
Calcium is an important signal on both the pre- and post-synaptic sides of the synapse where it triggers exocytosis (Douglas, 1968; Katz, 1969), plasticity (Lynch et al., 1983; Malenka et al., 1988; Bliss and Collingridge, 1993) and alters gene expression (Greenberg et al., 1992). The early and reproducible observation that synaptic efficacy is dependent on the fourth power of extracellular calcium highlights the importance of calcium in the exocytotic process and has been confirmed in a number of preparations (Dodge and Rahamimoff, 1967; Dudel, 1981; Augustine and Charlton, 1986; Zucker et al., 1991; Bollmann et al., 2000; Schneggenburger and Neher, 2000). Calcium activates the exocytotic machinery after entry through N-, P/Q-, and R- type voltage-activated calcium channels (Wheeler et al., 1994; Jun et al., 1999; Wu et al., 1999; Rozov et al., 2001). Numerous forms of synaptic plasticity have been described with varied rates of onset and durations lasting from milliseconds to hours (Katz and Miledi, 1969; Lynch et al., 1983; Malenka et al., 1988; Bliss and Collingridge, 1993; Zucker, 1993; Fisher et al., 1997; DeMaria et al., 2001; Kreitzer and Regehr, 2001; Rozov et al., 2001), all of which are affected by cleft calcium emphasizing its important regulatory role on synaptic function.
The broad dynamic range of extracellular calcium along with the exceedingly steep dependence of synaptic release probability on extracellular calcium (Dodge and Rahamimoff, 1967) leads to the hypothesis that even modest falls in cleft calcium will render the synapse much less effective at conducting signals. Indeed, a widely observed fourth-order proportionality implies that a reduction of the cleft calcium by one third could reduce synaptic efficacy by up to 80%. Accordingly, maneuvers reducing cleft calcium reduce synaptic efficacy (Borst and Sakmann, 1999a). Nevertheless, sustained phasic synaptic transmission has been observed at rates of up to 800 Hz (Taschenberger and von Gersdorff, 2000), indicating that either falls in cleft calcium do not occur at all synapses or there are compensatory mechanisms to reduce the effect of the fall of extracellular calcium at the synaptic cleft. The mechanism by which reductions in extracellular calciumreduce release probability and potential compensatory mechanisms remain incompletely understood, but similar to the impact of extracellular calcium on synaptic transmission can be divided into direct biophysical mechanisms and indirect mechanisms mediated by second messenger systems.
Dissociation of calcium from negative charged macromolecules, release from synaptic vesicles, and extracellular cation exchangers have been proposed to attenuate the fall in cleft calcium during episodes of high activity (Grohovaz et al., 1996; Borst and Sakmann, 1999a; Hartig et al., 2001), but the functional impact is uncertain. Similar to its impact on overall neuronal excitability, at the terminal reduced calcium is predicted to left-shift the voltage-dependence of sodium and calcium channels increasing the probability of release. Another putative, but incompletely understood compensatory mechanism observed at the calyx of Held and hippocampal nerve terminals is the broadening of presynaptic action potentials with repeated stimulation (Borst and Sakmann, 1999b; Geiger and Jonas, 2000). As calcium entry occurs during the repolarization phase of an action potential, spike broadening is a highly effective way of increasing calcium entry by prolonging depolarization (Sabatini and Regehr, 1997). Ion exchangers may also provide a mechanism to sustain synaptic transmission during periods of high activity. In parallel fiber-to-Purkinje neuron synapses, transient reversal of the sodium/calcium exchanger promotes calcium influx and enhanced glutamatergic transmission (Roome et al., 2013).
There is considerable evidence that the CaSR is intimately involved with regulating synaptic transmission Figure 1C. The CaSR is present in 80–90% of nerve terminals in the cerebral cortex (Smith et al., 2004; Chen et al., 2010) and its impact on synaptic transmission is complex indicating that it may be mediated by several mechanisms. In acutely isolated neocortical nerve terminals, decreases in extracellular calcium activated voltage-dependent NSCC currents indirectly via the CaSR (Smith et al., 2004; Phillips et al., 2008; Chen et al., 2010). Theoretically, NSCC activation at the nerve terminal following decreased CaSR activation (Smith et al., 2004) may depolarize the local membrane potential, inactivate voltage-dependent calcium channels, and thereby reduce the probability of evoked release. However, the voltage-dependence of the terminal NSCC means very few of the NSCCs would be activated at negative potentials making this unlikely to be a major effect. Another possibility is that NSCC activity following reduced CaSR activation could lead to action potential broadening which might prolong the duration of calcium entry and facilitate synaptic transmission. The absence of delay of activation of NSCC currents following rapid depolarizations (sub millisecond) and the ability of action potential waveforms to trigger these currents supported this hypothesis (Smith et al., 2004). Consistent with this idea, CaSR activation reduced excitatory transmission between pairs of neocortical neurons (Phillips et al., 2008). Furthermore, deletion of CaSR substantially increased excitatory synaptic transmission in neocortical neurons, and variance-mean analysis indicated this was due to an increase in release probability (Phillips et al., 2008). Thus, CaSR-NSCC signaling in nerve terminals would seem ideally placed to serve to increase release probability in situations where extracellular calcium was low thereby maintaining the fidelity of synaptic transmission during periods of high activity. However, although the NSCC currents were rapidly activated and likely to influence action potential shape the CaSR is a GPCR and unlikely to respond rapidly. Indeed in isolated terminals the pathway took a few seconds to respond to changes in extracellular calcium. These relatively slow kinetics indicate the CaSR-NSCC signaling pathway in terminals is more likely to detect and respond to sustained changes in calcium that persist for a few seconds and not those that develop over a few milliseconds (Smith et al., 2004; Chen et al., 2010). Endogenous modulators of CaSR in the periphery include magnesium, L-amino acids, polyamines, and γ-glutamyl peptides besides calcium (Leach et al., 2015). It remains unclear how much these agents modulate signaling in neurons but identification of central actions may reveal other physiological roles for CaSR in neurons as suggested for beta-amyloid (Conley et al., 2009).
Increasing attention has turned to spontaneous release of neurotransmitters with the recognition that action potential-evoked and spontaneous release mechanisms are distinct (Kavalali, 2015). Interestingly, CaSR activation by direct and allosteric agonists stimulate release of glutamate independent of intracellular calcium (Vyleta and Smith, 2011). In addition, deletion of CaSR substantially reduced spontaneous glutamate release. In other words, CaSR activation had opposite effects on evoked and spontaneous release of the major excitatory neurotransmitter (Phillips et al., 2008; Vyleta and Smith, 2011). It is as yet unclear how CaSR could have opposite effects on exocytosis of these apparently distinct populations of vesicles that reside in the same nerve terminals. However, we recognize that these apparently opposite actions mechanistically mirror the actions that CaSR stimulation has on release of parathyroid hormone and calcitonin (Garrett et al., 1995). The importance of CaSR signaling at nerve terminals has also been emphasized by the finding that spontaneous release of GABA, the major inhibitory neurotransmitter, is also strongly enhanced by CaSR activation (Smith et al., 2012).
Given the apparent abundance of CaSR it seems surprising that a role for CaSR was not suggested sooner. However, CaSR signaling may have been difficult to detect because “physiological” experiments frequently employed supraphysiological levels of calcium and magnesium concentrations (Smith et al., 2004; Chen et al., 2010). This approach ensured CaSR was at near-saturation attenuated our ability to detect changes in CaSR signaling. Another confounder is that in studies of the effects of decreased extracellular calcium, magnesium concentrations were often increased with the presumption that magnesium would only obviate the effects of surface charge screening. Since CaSR and spontaneous release are stimulated by magnesium (Vyleta and Smith, 2011; Smith et al., 2012) this experimental approach minimized the contribution of CaSR. The importance of employing physiological concentrations of divalent ions was emphasized by comparing neuronal activity and synaptic transmission in vivo and in acute brain slices (Sanchez-Vives and McCormick, 2000; Lorteije et al., 2009).
Over the past decade a number of reports have underlined the potential of CaSR as a therapeutic target in diseases of the nervous system. Familial idiopathic epilepsy was linked to dominantly inherited CaSR mutations across three generations (Kapoor et al., 2008). The signaling pathways by which changes in CaSR activity might relate to epilepsy are not known, but the evidence implicating the CaSR in neuronal excitability and maintenance of high-frequency synaptic transmission suggests a plausible mechanism by which changes in CaSR activity could underpin a disorder of neuronal activity. In parallel, CaSR levels have been found to be increased in animal models following induction of seizures as well as traumatic brain injury (Mudo et al., 2009; Kim et al., 2011) hinting at a potential role for CaSR in the development of epilepsy following status epilepticus or traumatic brain injury. Intriguingly, CaSR antagonists were shown to reduce CaSR expression levels, brain tissue loss and neurological deficits, in animal models of traumatic brain injury and cerebral ischemia (Kim et al., 2013, 2014). Furthermore, links between beta amyloid and CaSR signaling may be important in the development of Alzheimer's disease and hypoxic brain injury (Bai et al., 2015; Dal Pra et al., 2015).
Extracellular calcium ions are recognized, like intracellular calcium ions, as important regulators of neuronal function in the central and peripheral nervous systems. The action of extracellular calcium is complex and its actions via CaSR and surface charge screening affect numerous ion channels impacting neuronal excitability and many forms of synaptic transmission. An important goal for the field is to determine the relative contributions of these signaling pathways to neuronal function to facilitate our understanding behind the role of CaSR signaling in pathogenesis of acute neurological diseases like stroke, traumatic brain injury, and epilepsy.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by Merit Review Award (BX002547) from U.S. Department of Veterans Affairs and NIGMS (R01 GM097433).
Armstrong, C. M., and Cota, G. (1990). Modification of sodium channel gating by lanthanum. Some effects that cannot be explained by surface charge theory. J. Gen. Physiol. 96, 1129–1140. doi: 10.1085/jgp.96.6.1129
Armstrong, C. M., and Cota, G. (1991). Calcium ion as a cofactor in Na channel gating. Proc. Natl. Acad. Sci. U.S.A. 88, 6528–6531. doi: 10.1073/pnas.88.15.6528
Augustine, G. J., and Charlton, M. P. (1986). Calcium dependence of presynaptic calcium current and post-synaptic response at the squid giant synapse. J. Physiol. 381, 619–640. doi: 10.1113/jphysiol.1986.sp016347
Bai, S., Mao, M., Tian, L., Yu, Y., Zeng, J., Ouyang, K., et al. (2015). Calcium sensing receptor mediated the excessive generation of beta-amyloid peptide induced by hypoxia in vivo and in vitro. Biochem. Biophys. Res. Commun. 459, 568–573. doi: 10.1016/j.bbrc.2015.02.141
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Bollmann, J. H., Sakmann, B., and Borst, J. G. (2000). Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289, 953–957. doi: 10.1126/science.289.5481.953
Borst, J. G., and Sakmann, B. (1999a). Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J. Physiol. 521(Pt 1), 123–133. doi: 10.1111/j.1469-7793.1999.00123.x
Borst, J. G., and Sakmann, B. (1999b). Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 347–355. doi: 10.1098/rstb.1999.0386
Chen, W., Bergsman, J. B., Wang, X., Gilkey, G., Pierpoint, C. R., Daniel, E. A., et al. (2010). Presynaptic external calcium signaling involves the calcium-sensing receptor in neocortical nerve terminals PLoS ONE 5:e8563. doi: 10.1371/journal.pone.0008563
Conley, Y. P., Mukherjee, A., Kammerer, C., DeKosky, S. T., Kamboh, M. I., Finegold, D. N., et al. (2009). Evidence supporting a role for the calcium-sensing receptor in Alzheimer disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150b, 703–709. doi: 10.1002/ajmg.b.30896
Dal Pra, I., Chiarini, A., and Armato, U. (2015). Antagonizing amyloid-beta/calcium-sensing receptor signaling in human astrocytes and neurons: a key to halt Alzheimer's disease progression? Neural Regen. Res. 10, 213–218. doi: 10.4103/1673-5374.152373
DeMaria, C. D., Soong, T. W., Alseikhan, B. A., Alvania, R. S., and Yue, D. T. (2001). Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411, 484–489. doi: 10.1038/35078091
Dodge, F. Jr., and Rahamimoff, R. (1967). Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193, 419–432. doi: 10.1113/jphysiol.1967.sp008367
Douglas, W. W. (1968). Stimulus-secretion coupling: the concept and clues from chromaffin and other cells. Br. J. Pharmacol. 34, 453–474. doi: 10.1111/j.1476-5381.1968.tb08474.x
Dudel, J. (1981). The effect of reduced calcium on quantal unit current and release at the crayfish neuromuscular junction. Pflug. Arch. 391, 35–40. doi: 10.1007/BF00580691
Egelman, D. M., and Montague, P. R. (1998). Computational properties of peri-dendritic calcium fluctuations. J. Neurosci. 18, 8580–8589.
Egelman, D. M., and Montague, P. R. (1999). Calcium dynamics in the extracellular space of mammalian neural tissue. Biophys. J. 76, 1856–1867. doi: 10.1016/S0006-3495(99)77345-5
Fisher, S. A., Fischer, T. M., and Carew, T. J. (1997). Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 20, 170–177. doi: 10.1016/S0166-2236(96)01001-6
Frankenhaeuser, B., and Hodgkin, A. L. (1957). The action of calcium on the electrical properties of squid axons. J. Physiol. 137, 218–244. doi: 10.1113/jphysiol.1957.sp005808
Garrett, J. E., Tamir, H., Kifor, O., Simin, R. T., Rogers, K. V., Mithal, A., et al. (1995). Calcitonin-secreting cells of the thyroid express an extracellular calcium receptor gene. Endocrinology 136, 5202–5211.
Geiger, J. R., and Jonas, P. (2000). Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron 28, 927–939. doi: 10.1016/S0896-6273(00)00164-1
Greenberg, M. E., Thompson, M. A., and Sheng, M. (1992). Calcium regulation of immediate early gene transcription. J. Physiol. 86, 99–108. doi: 10.1016/s0928-4257(05)80013-0
Grohovaz, F., Bossi, M., Pezzati, R., Meldolesi, J., and Tarelli, F. T. (1996). High resolution ultrastructural mapping of total calcium: electron spectroscopic imaging/electron energy loss spectroscopy analysis of a physically/chemically processed nerve-muscle preparation. Proc. Natl. Acad. Sci. U.S.A. 93, 4799–4803. doi: 10.1073/pnas.93.10.4799
Hablitz, J. J., Heinemann, U., and Lux, H. D. (1986). Step reductions in extracellular Ca2+ activate a transient inward current in chick dorsal root ganglion cells. Biophys. J. 50, 753–757. doi: 10.1016/S0006-3495(86)83515-9
Han, P., Trinidad, B. J., and Shi, J. (2015). Hypocalcemia-induced seizure: demystifying the calcium paradox. ASN Neuro. 7, 1–9. doi: 10.1177/1759091415578050
Hartig, W., Singer, A., Grosche, J., Brauer, K., Ottersen, O. P., and Bruckner, G. (2001). Perineuronal nets in the rat medial nucleus of the trapezoid body surround neurons immunoreactive for various amino acids, calcium- binding proteins and the potassium channel subunit Kv3.1b. Brain Res. 899, 123–133. doi: 10.1016/S0006-8993(01)02211-9
Huang, Y., Zhou, Y., Wong, H. C., Castiblanco, A., Chen, Y., Brown, E. M., et al. (2010). Calmodulin regulates Ca2+-sensing receptor-mediated Ca2+ signaling and its cell surface expression. J. Biol. Chem. 285, 35919–35931. doi: 10.1074/jbc.M110.147918
Immke, D. C., and McCleskey, E. W. (2001). Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat. Neurosci. 4, 869–870. doi: 10.1038/nn0901-869
Jones, H. C., and Keep, R. F. (1988). Brain fluid calcium concentration and response to acute hypercalcaemia during development in the rat. J. Physiol. 402, 579–593. doi: 10.1113/jphysiol.1988.sp017223
Jun, K., Piedras-Renteria, E. S., Smith, S. M., Wheeler, D. B., Lee, S. B., Lee, T. G., et al. (1999). Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)- subunit. Proc. Natl. Acad. Sci. U.S.A. 96, 15245–15250. doi: 10.1073/pnas.96.26.15245
Kapoor, A., Satishchandra, P., Ratnapriya, R., Reddy, R., Kadandale, J., Shankar, S. K., et al. (2008). An idiopathic epilepsy syndrome linked to 3q13.3-q21 and missense mutations in the extracellular calcium sensing receptor gene. Ann. Neurol. 64, 158–167. doi: 10.1002/ana.21428
Katz, B. (1969). The Release of Neural Transmitter Substances. Liverpool: Liverpool University Press.
Katz, B., and Miledi, R. (1969). Spontaneous and evoked activity of motor nerve endings in calcium Ringer. J. Physiol. 203, 689–706. doi: 10.1113/jphysiol.1969.sp008887
Kavalali, E. T. (2015). The mechanisms and functions of spontaneous neurotransmitter release. Nat. Rev. Neurosci. 16, 5–16. doi: 10.1038/nrn3875
Kim, J., Ghosh, S., Liu, H., Tateyama, M., Kass, R. S., and Pitt, G. S. (2004). Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 279, 45004–45012. doi: 10.1074/jbc.M407286200
Kim, J. Y., Ho, H., Kim, N., Liu, J., Tu, C. L., Yenari, M. A., et al. (2014). Calcium-sensing receptor (CaSR) as a novel target for ischemic neuroprotection. Ann. Clin. Transl. Neurol 1, 851–866. doi: 10.1002/acn3.118
Kim, J. Y., Kim, N., Yenari, M. A., and Chang, W. (2011). Mild Hypothermia Suppresses Calcium-Sensing Receptor (CaSR) Induction Following Forebrain Ischemia While Increasing GABA-B Receptor 1 (GABA-B-R1) expression. Transl. Stroke Res. 2, 195–201. doi: 10.1007/s12975-011-0082-4
Kim, J. Y., Kim, N., Yenari, M. A., and Chang, W. (2013). Hypothermia and pharmacological regimens that prevent overexpression and overactivity of the extracellular calcium-sensing receptor protect neurons against traumatic brain injury. J. Neurotrauma 30, 1170–1176. doi: 10.1089/neu.2012.2691
Kreitzer, A. C., and Regehr, W. G. (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29, 717–727. doi: 10.1016/S0896-6273(01)00246-X
Kullmann, D. M., Min, M. Y., Asztely, F., and Rusakov, D. A. (1999). Extracellular glutamate diffusion determines the occupancy of glutamate receptors at CA1 synapses in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 395–402. doi: 10.1098/rstb.1999.0392
Leach, K., Conigrave, A. D., Sexton, P. M., and Christopoulos, A. (2015). Towards tissue-specific pharmacology: insights from the calcium-sensing receptor as a paradigm for GPCR (patho)physiological bias. Trends Pharmacol. Sci. 36, 215–225. doi: 10.1016/j.tips.2015.02.004
Lorteije, J. A., Rusu, S. I., Kushmerick, C., and Borst, J. G. (2009). Reliability and precision of the mouse calyx of Held synapse. J. Neurosci. 29, 13770–13784. doi: 10.1523/JNEUROSCI.3285-09.2009
Lu, B., Zhang, Q., Wang, H., Wang, Y., Nakayama, M., and Ren, D. (2010). Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron 68, 488–499. doi: 10.1016/j.neuron.2010.09.014
Lynch, G., Larson, J., Kelso, S., Barrionuevo, G., and Schottler, F. (1983). Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature 305, 719–721. doi: 10.1038/305719a0
Ma, Z., Siebert, A. P., Cheung, K. H., Lee, R. J., Johnson, B., Cohen, A. S., et al. (2012). Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 109, E1963–E1971. doi: 10.1073/pnas.1204023109
Malenka, R. C., Kauer, J. A., Zucker, R. S., and Nicoll, R. A. (1988). Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science 242, 81–84. doi: 10.1126/science.2845577
Miller, D. J. (2004). Sydney Ringer; physiological saline, calcium and the contraction of the heart. J. Physiol. 555, 585–587. doi: 10.1113/jphysiol.2004.060731
Mudo, G., Trovato-Salinaro, A., Barresi, V., Belluardo, N., and Condorelli, D. F. (2009). Identification of calcium sensing receptor (CaSR) mRNA-expressing cells in normal and injured rat brain. Brain Res. 1298, 24–36. doi: 10.1016/j.brainres.2009.08.074
Nicholson, C., ten Bruggencate, G., Stöckle, H., and Steinberg, R. (1978). Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J. Neurophysiol. 41, 1026–1039.
Nilsson, P., Hillered, L., Olsson, Y., Sheardown, M. J., and Hansen, A. J. (1993). Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats. J. Cereb. Blood Flow Metabol. 13, 183–192. doi: 10.1038/jcbfm.1993.22
Nilsson, P., Laursen, H., Hillered, L., and Hansen, A. J. (1996). Calcium movements in traumatic brain injury: the role of glutamate receptor-operated ion channels. J. Cereb. Blood Flow Metabol. 16, 262–270. doi: 10.1097/00004647-199603000-00011
Phillips, C. G., Harnett, M. T., Chen, W., and Smith, S. M. (2008). Calcium-sensing receptor activation depresses synaptic transmission. J. Neurosci. 28, 12062–12070. doi: 10.1523/JNEUROSCI.4134-08.2008
Ringer, S. (1883). A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J. Physiol. 4, 29–42.23. doi: 10.1113/jphysiol.1883.sp000120
Roome, C. J., Power, E. M., and Empson, R. M. (2013). Transient reversal of the sodium/calcium exchanger boosts presynaptic calcium and synaptic transmission at a cerebellar synapse. J. Neurophysiol. 109, 1669–1680. doi: 10.1152/jn.00854.2012
Rozov, A., Burnashev, N., Sakmann, B., and Neher, E. (2001). Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol. 531, 807–826. doi: 10.1111/j.1469-7793.2001.0807h.x
Ruat, M., Molliver, M. E., Snowman, A. M., and Snyder, S. H. (1995). Calcium sensing receptor: molecular cloning in rat and localization to nerve terminals. Proc. Natl. Acad. Sci. U.S.A. 92, 3161–3165. doi: 10.1073/pnas.92.8.3161
Rusakov, D. A., and Fine, A. (2003). Extracellular Ca2+ depletion contributes to fast activity-dependent modulation of synaptic transmission in the brain. Neuron 37, 287–297. doi: 10.1016/S0896-6273(03)00025-4
Rusakov, D. A., Harrison, E., and Stewart, M. G. (1998). Synapses in hippocampus occupy only 1-2% of cell membranes and are spaced less than half-micron apart: a quantitative ultrastructural analysis with discussion of physiological implications. Neuropharmacology 37, 513–521. doi: 10.1016/S0028-3908(98)00023-9
Sabatini, B. L., and Regehr, W. G. (1997). Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J. Neurosci. 17, 3425–3435.
Sanchez-Vives, M. V., and McCormick, D. A. (2000). Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 3, 1027–1034. doi: 10.1038/79848
Santarelli, V. P., Eastwood, A. L., Dougherty, D. A., Ahern, C. A., and Horn, R. (2007). Calcium block of single sodium channels: role of a pore-lining aromatic residue. Biophys. J. 93, 2341–2349. doi: 10.1529/biophysj.107.106856
Sarhan, M. F., Tung, C. C., Van Petegem, F., and Ahern, C. A. (2012). Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 109, 3558–3563. doi: 10.1073/pnas.1114748109
Schneggenburger, R., and Neher, E. (2000). Intracellular calcium dependence of transmitter release rates at a fast central synapse [see comments]. Nature 406, 889–893. doi: 10.1038/35022702
Smith, S. J. (1992). Do astrocytes process neural information? Prog. Brain Res. 94, 119–136. doi: 10.1016/S0079-6123(08)61744-6
Smith, S. M., Bergsman, J. B., Harata, N. C., Scheller, R. H., and Tsien, R. W. (2004). Recordings from single neocortical nerve terminals reveal a nonselective cation channel activated by decreases in extracellular calcium. Neuron 41, 243–256. doi: 10.1016/S0896-6273(03)00837-7
Smith, S. M., Chen, W., Vyleta, N. P., Williams, C., Lee, C. H., Phillips, C., et al. (2012). Calcium regulation of spontaneous and asynchronous neurotransmitter release. Cell Calcium 52, 226–233. doi: 10.1016/j.ceca.2012.06.001
Taschenberger, H., and von Gersdorff, H. (2000). Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J. Neurosci. 20, 9162–9173.
Vassilev, P. M., Ho-Pao, C. L., Kanazirska, M. P., Ye, C., Hong, K., Seidman, C. E., et al. (1997a). Cao-sensing receptor (CaR)-mediated activation of K+ channels is blunted in CaR gene-deficient mouse neurons. Neuroreport 8, 1411–1416. doi: 10.1097/00001756-199704140-00018
Vassilev, P. M., Mitchel, J., Vassilev, M., Kanazirska, M., and Brown, E. M. (1997b). Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys. J. 72, 2103–2116. doi: 10.1016/S0006-3495(97)78853-2
Vyleta, N. P., and Smith, S. M. (2011). Spontaneous glutamate release is independent of calcium influx and tonically activated by the calcium-sensing receptor. J. Neurosci. 31, 4593–4606. doi: 10.1523/JNEUROSCI.6398-10.2011
Vysotskaya, Z. V., Moss, C. R. II., Gilbert, C. A., Gabriel, S. A., and Gu, Q. (2014). Modulation of BK channel activities by calcium-sensing receptor in rat bronchopulmonary sensory neurons. Respir. Physiol. Neurobiol. 203, 35–44. doi: 10.1016/j.resp.2014.08.014
Wheeler, D. B., Randall, A., and Tsien, R. W. (1994). Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111. doi: 10.1126/science.7832825
Wu, L. G., Westenbroek, R. E., Borst, J. G. G., Catterall, W. A., and Sakmann, B. (1999). Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J. Neurosci. 19, 726–736.
Xiong, Z., Lu, W., and MacDonald, J. F. (1997). Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 7012–7017. doi: 10.1073/pnas.94.13.7012
Ye, C., Kanazirska, M., Quinn, S., Brown, E. M., and Vassilev, P. M. (1996). Modulation by polycationic Ca(2+)-sensing receptor agonists of nonselective cation channels in rat hippocampal neurons. Biochem. Biophys. Res. Commun. 224, 271–280. doi: 10.1006/bbrc.1996.1019
Zhang, E. T., Hansen, A. J., Wieloch, T., and Lauritzen, M. (1990). Influence of MK-801 on brain extracellular calcium and potassium activities in severe hypoglycemia. J. Cereb. Blood Flow Metabol. 10, 136–139. doi: 10.1038/jcbfm.1990.18
Zucker, R. S. (1993). Calcium and transmitter release. J. Physiol. 87, 25–36. doi: 10.1016/0928-4257(93)90021-k
Zucker, R. S. (1996). Exocytosis: a molecular and physiological perspective. Neuron 17, 1049–1055. doi: 10.1016/S0896-6273(00)80238-X
Zucker, R. S., Delaney, K. R., Mulkey, R., and Tank, D. W. (1991). “Presynaptic calcium in transmitter release and posttetanic potentiation,” in Annals Of The New York Academy Of Sciences, Vol. 635. Calcium Entry And Action At The Presynaptic Terminal; Conference, Baltimore, Maryland, Usa, October 15-17, 1990. Ix+506p, eds E. F. Stanley, M. C. Nowycky and D. J. Triggle (New York, NY: New York Academy Of Sciences), 191–207.
Keywords: calcium sensing receptor, nervous system, synaptic transmission, action potentials, ion channels, calcium, excitability
Citation: Jones BL and Smith SM (2016) Calcium-Sensing Receptor: A Key Target for Extracellular Calcium Signaling in Neurons. Front. Physiol. 7:116. doi: 10.3389/fphys.2016.00116
Received: 02 February 2016; Accepted: 14 March 2016;
Published: 30 March 2016.
Edited by:
Enikö Kallay, Medical University of Vienna, AustriaReviewed by:
Ubaldo Armato, University of Verona Medical School, ItalyCopyright © 2016 Jones and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen M. Smith, c21pc3RlcGhAb2hzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.