 Sebastián Dubó1
Sebastián Dubó1 David Gallegos2
David Gallegos2 Lissette Cabrera2,3
Lissette Cabrera2,3 Luis Sobrevia4,5,6
Luis Sobrevia4,5,6 Leandro Zúñiga7
Leandro Zúñiga7 Marcelo González2,8*
Marcelo González2,8*- 1Department of Kinesiology, Faculty of Medicine, Universidad de Concepción, Concepción, Chile
- 2Vascular Physiology Laboratory, Department of Physiology, Faculty of Biological Sciences, Universidad de Concepción, Concepción, Chile
- 3Department of Morphophysiology, Faculty of Medicine, Universidad Diego Portales, Santiago, Chile
- 4Cellular and Molecular Physiology Laboratory (CMPL), Division of Obstetrics and Gynecology, Faculty of Medicine, School of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
- 5Department of Physiology, Faculty of Pharmacy, Universidad de Sevilla, Seville, Spain
- 6Faculty of Medicine and Biomedical Sciences, University of Queensland Centre for Clinical Research (UQCCR), University of Queensland, Herston, QLD, Queensland, Australia
- 7Centro de Investigaciones Médicas, Escuela de Medicina, Universidad de Talca, Talca, Chile
- 8Group of Research and Innovation in Vascular Health (GRIVAS-Health), Chillán, Chile
Impairment of insulin signaling on diabetes mellitus has been related to cardiovascular dysfunction, heart failure, and sudden death. In human endothelium, cationic amino acid transporter 1 (hCAT-1) is related to the synthesis of nitric oxide (NO) and insulin has a vascular effect in endothelial cells through a signaling pathway that involves increases in hCAT-1 expression and L-arginine transport. This mechanism is disrupted in diabetes, a phenomenon potentiated by excessive accumulation of reactive oxygen species (ROS), which contribute to lower availability of NO and endothelial dysfunction. On the other hand, electrical remodeling in cardiomyocytes is considered a key factor in heart failure progression associated to diabetes mellitus. This generates a challenge to understand the specific role of insulin and the pathways involved in cardiac function. Studies on isolated mammalian cardiomyocytes have shown prolongated action potential in ventricular repolarization phase that produces a long QT interval, which is well explained by attenuation in the repolarizing potassium currents in cardiac ventricles. Impaired insulin signaling causes specific changes in these currents, such a decrease amplitude of the transient outward K+ (Ito) and the ultra-rapid delayed rectifier (IKur) currents where, together, a reduction of mRNA and protein expression levels of α-subunits (Ito, fast; Kv 4.2 and IKs; Kv 1.5) or β-subunits (KChIP2 and MiRP) of K+ channels involved in these currents in a MAPK mediated pathway process have been described. These results support the hypothesis that lack of insulin signaling can produce an abnormal repolarization in cardiomyocytes. Furthermore, the arrhythmogenic potential due to reduced Ito current can contribute to an increase in the incidence of sudden death in heart failure. This review aims to show, based on pathophysiological models, the regulatory function that would have insulin in vascular system and in cardiac electrophysiology.
General Overview of Cardiovascular Regulation by Insulin in Health and Disease
Insulin is a peptide hormone produced by the β cells, of the islets of Langerhans. Functionally, is the most potent anabolic hormone, promoting the synthesis and storage of carbohydrates, lipids and proteins, while inhibiting their degradation and release into circulation. This hormone stimulates the uptake of glucose, amino acids, and fatty acids; and increases the expression of enzymes involved in the glycogen, lipid, and protein synthesis. At the same time, it reduces the activity of the enzymes that catalyse degradation of same molecules (Saltiel and Kahn, 2001). Cellular effects of insulin are mediated by the activation of insulin receptors (IRs A/B), and a signaling pathway involved in the regulation of nutrients metabolism, mitochondrial biogenesis, cellular grown, proliferation, differentiation, and migration (Nystrom and Quon, 1999). IRs are glycoproteins consisting of an extracellular α-subunit (135 kDa) and a transmembrane β-subunit (95 kDa). These receptors are allosteric enzymes in which the α-subunit regulates tyrosine kinase activity of the β-subunits. The binding of insulin to the α-subunit results in dimerization, forming the α2β2 complex in the plasma membrane. The complex α2β2 generates the autophosphorylation of β-subunit at tyrosine (Tyr) 1158 (Tyr1158), Tyr1162, and Tyr1163, the first step in the activation cascade induced by insulin (Patti and Kahn, 1998). Phosphorylation of IR induces the tyrosine kinase activity toward insulin receptor substrate (IRS) 1 (IRS-1) or 2 (IRS-2), generating binding sites for Src homology 2 (SH2) domain proteins, including phosphatidylinositol 3-kinase (PI3K), RAS guanine nucleotide exchange factor complex known as growth factor receptor-bound protein 2/son of sevenless (GRB2/SOS), and SH2 domain-containing protein tyrosine phosphatase-2 (SHP2) and other SH2 proteins (Copps and White, 2012). These proteins provide specific docking sites for the recruitment of downstream signaling proteins, leading to activation of mitogen-activated protein kinase (MAPK), protein kinase B (PKB/Akt) and protein kinase C (PKC) signaling cascades (White, 2003; Schultze et al., 2012).
Since the early nineties, the cardiovascular actions of insulin have been studied in humans, and the main information was collected in several works published by Alain Baron and Helmut Steinberg. In 1990, in a study made in obese and lean subjects determined a correlation between insulin resistance (hyperglycaemia and hyperinsulinemia), and lack of vasodilatory effects of insulin in leg blood flow (LBF). These obese subjects showed a lower capacity for uptake D-glucose and changes in cardiac function, such as reduction in cardiac index [cardiac output (CO)/body surface area] and stroke volume (SV) without change in heart rate (HR) (Baron et al., 1990). Authors concluded that insulin resistance could be a result of insulin's actions both in glucose uptake and hemodynamic regulation, highlighting the in vivo role of insulin as an endocrine regulator of cardiovascular physiology. Later, healthy subjects under a protocol of euglycemic hyperinsulinemia showed increases of CO, SV, HR, and LBF; and decreases of mean arterial pressure (MAP), systemic vascular resistance (SVR), and leg vascular resistance (LVR). Correlation analysis showed that the insulin-mediated glucose uptake is inversely proportional to MAP or SVR, supporting the hemodynamic basis for insulin resistance (Baron et al., 1993). The effects of insulin are part of physiological regulation of cardiovascular system, with a delicate interaction with other signals, like norepinephrine (NE) and acetylcholine (Ach). Obese insulin-resistant subjects show more sensitivity to contractile effects of NE, and lower clearance of NE in response to insulin (Baron et al., 1994). More recently, a reduction was shown in Ach-dependent forearm blood flow in obese patients, but this effect disappears in the group of obese subjects without metabolic syndrome (Schinzari et al., 2015). Even though it is not possible to discard a deleterious effect of obesity per se on the cardiovascular system, mechanisms that we discuss in this review show that insulin resistance could be a primary cause of cardiovascular disease in metabolic disorders, leading to a deterioration of endothelium-dependent vasodilatation and higher sensibility to vasoconstrictors, like NE or other locally released molecules. The accumulated evidence shown by the literature makes it possible to establish that insulin-dependent blood flow increments are blunted in obese, and type 2 diabetes mellitus (T2DM) patients, conditions associated with insulin resistance. Physiological regulation of blood flow by insulin depends on a mechanism that involves the activation of PI3K, PKB, and nitric oxide (NO) synthesis (Baron, 2002; Steinberg and Baron, 2002). Due to the increasing prevalence of diabetes and insulin resistance, a main objective of this article is to make a critical review of physiological mechanisms regulated by insulin both in vasculature and in the heart. Thus, we can propose a hypothesis for pathological mechanisms of cardiovascular disease in diabetes or insulin resistance.
Diabetes mellitus (DM) is characterized by chronic hyperglycaemia which results from defects in insulin secretion (type 1 DM), insulin action (T2DM), or both (WHO, 1998). Diabetes is a common disease that affects ~382 million people worldwide, and the costs to society are high and escalating. By the end of 2013, diabetes caused 5.1 million deaths and cost USD 548 billion in healthcare spending (IFD, 2013). Although treatment has been improved over the last decades, diabetes is related with several complications like acute hyperglycaemic or hypoglycaemic events, kidney disease, eye disease, peripheral vascular disease, coronary artery disease, cerebrovascular disease, and congestive heart failure (Bethel et al., 2007).
Cardiovascular diseases (CVD) represent the leading causes of morbidity and mortality in patients with diabetes (Gregg et al., 2014), with cardiac and vascular dysfunctions as key steps of the pathophysiological mechanism. Vascular dysfunction is primarily caused by endothelial dysfunction, a phenomenon related with deterioration in the capacity of these cells to synthesize NO. NO is synthesized from the semi-essential cationic amino acid L-arginine and molecular oxygen (O2) by endothelial NO synthase (eNOS), leading to the formation of NO and the neutral amino acid L-citrulline (Palmer et al., 1988). The enzyme depends on several cofactors, including Ca2+/calmodulin complex, tetrahydrobiopterin (BH4), nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), and flavin mononucleotide (FMN) (Knowles and Moncada, 1994; Sessa, 1994). In absence of NO signaling there is a disturbance in vascular homeostasis, triggering a series of events leading to pathologies like as hypertension, hypercholesterolemia, renal vascular insufficiency, and chronic heart failure (Vanhoutte, 2003; Wierzbicki et al., 2004; Yang et al., 2007). As mentioned above, several studies in diabetic patients have demonstrated deficiency in endothelium-dependent vasodilation, a disturbance that generates an imbalance in vascular tone, which ultimately leads to the development of endothelial dysfunction (Williams et al., 1996; Rask-Madsen and King, 2007). Mechanisms underlying the decrease in NO-dependent dilatation in diabetes include decreased bioavailability of tetrahydrobiopterin (BH4) cofactor and uncoupling of eNOS (Cai et al., 2005), increased activity of arginase (an enzyme that competes with the eNOS for L-arginine; Vanhoutte and Tang, 2008), high levels of asymmetric dimethylarginine (ADMA, endogenous inhibitor of eNOS; Xiong et al., 2003); increased production of superoxide anion () and peroxynitrite (ONOO−) synthesis (Cosentino et al., 2003; Quijano et al., 2007), inactivation of NO by advanced glycation end products (Gao et al., 2008) and abnormal responses of vascular smooth muscle cells (VSMC) (Lesniewski et al., 2008; Shi and Vanhoutte, 2008).
On the other hand, a main cardiac disorder is diabetic cardiomyopathy (DCM), a pathology associated with alterations in molecular metabolism and structure of cardiomyocytes (Fang et al., 2004). These changes include, among others, a ventricular electric remodeling characterized by long-term alterations in ion channel activity that could be a compensatory or adaptive mechanism (Casis and Echevarria, 2004). Electrical remodeling has been related with contractile failure due to abnormal increase of intracellular Ca2+ concentration or to arrhythmias that lead to sudden death (Straus et al., 2006). Most electrical disturbances are related to the abnormal repolarization of cardiac action potential (Shimoni et al., 2004). Electrophysiological studies have clearly identified the different types of voltage-dependent currents (inward or outward currents), which contribute to the repolarization phase in the mammalian myocardium (Nerbonne and Kass, 2005). Outward currents are clearly differentiated and cardiac myocyte express voltage-dependent K+ channels (Kv) that contribute to the development of action potential (Nerbonne, 2000). Changes in the functional properties of the Kv channels can generate dramatic effects in myocardial action potential, and also in the normal cardiac rhythm generation (Lengyel et al., 2007). Cardiovascular complications in diabetes have been associated to a series of electrocardiographic disturbances, such as QT interval prolongation, even in short-term diabetes (Veglio et al., 2002a,b; Zhang et al., 2011). Experimental evidence suggests that specific remodeling of some K+ channels in the ventricle is related with these alterations (Casis and Echevarria, 2004).
Insulin Regulation of Vascular System
Relevance of L-Arginine Transport in Vascular Physiology
In endothelial cells, uptake of the substrate L-arginine for NO synthesis is mediated by transport systems called y+, y+L, b0, +, and B0, +. The main transport system involved in this process is the system y+, responsible for ~85% of L-arginine transport in physiological state (Devés and Boyd, 1998). This system includes the family of proteins known as cationic amino acids transporters (CATs), formed by CAT-1, CAT-2A, CAT-2B, CAT-3, and CAT-4 (Verrey et al., 2004; Casanello et al., 2007). Members of the CATs family are encoded by genes SLC7A 1, 2, 3, and 4 (Verrey et al., 2004). CAT-1, encoded by SLC7A1 gene, is ubiquitously expressed, while CAT-2A and CAT-3 are constitutively expressed in liver and brain, respectively. CAT-2B is induced under inflammatory conditions in a variety of cells including T cells and macrophages. The CAT-4 gene sequence is 41–42% identical to the other members of the family CATs, but its transport activity has not been described. The function of CATs was suggested because of their structural similarity to Saccharomyces cerevisiae, permease for L-histidine and L-arginine. Its function was verified by expression in Xenopus laevis oocytes for kinetic transport studies (Closs et al., 1997). CAT-1, CAT-2B, and CAT-3 are Na+ independent transporters, presenting medium affinity for its substrate (Km ~50–250 μM), whereas CAT-2A has low affinity for cationic amino acids (Km ~2–5 mM) (Devés and Boyd, 1998; Palacín et al., 1998; Mann et al., 2003).
In cytoplasm, L-arginine is used for synthesis of proteins, NO, urea, creatine, agmatine, polyamines, and other molecules. Intracellular concentration of L-arginine is near to 1 mM, but the Km of eNOS by L-arginine is ~3 μM and, as mentioned, the Km of hCAT-1/2B for L-arginine is ~50–250 μM (Shin et al., 2011). These inconsistencies between cytoplasmic concentrations of L-arginine and the affinity for substrate of L-arginine/NO system, constitutes the “L-arginine paradox.” This paradox is under discussion among researchers, especially given the growing relevance of L-arginine transport from extracellular space and the concept of compartmentalization of different pools of L-arginine (Karbach et al., 2011; Simon et al., 2013). In a simple but very elegant experiment, Shin et al. demonstrated that L-arginine transport mediated by a facilitated diffusion system is absolutely necessary for NO synthesis: when EA.hy926 endothelial cells are incubated with modified L-arginine, which has the ability to enter cells by passive diffusion, eNOS is not able to synthesize NO. Additionally, in knockdown cells for hCAT-1 (induced by siRNA), L-citrulline synthesis is significantly reduced, showing the relevance of this specific transporter (Shin et al., 2011). This evidence is supported by hCAT-1 localization studies. In porcine aortic endothelial cells (PAEC) CAT-1 colocalize with eNOS and caveolin-1 (Cav-1, structural protein of caveolae; McDonald et al., 1997). The subcellular localization through overexpression of CAT-1 fused to green fluorescent protein (GFP) has been studied in different cell lines (Closs et al., 2006). In hamster kidney baby cells (HKB) it has been noted that CAT-1-GFP is localized predominantly in caveolin associated domains in plasma membrane (Lu and Silver, 2000). In another study, CAT-1-EGFP was mainly found in intracellular vesicles of U373MG, human glioblastoma cells, and to a lesser extent a lesser extent in the plasma membrane (Wolf et al., 2002). In Madin-Darby canine kidney epithelial cells (MDCK) and human embryonic kidney 293 cells (HEK293), the expression of CAT-1-GFP was detected, predominantly in the basolateral plasma membrane (Cariappa et al., 2002; Kizhatil and Albritton, 2002). Meanwhile, more research into HEK293 showed that mCAT-1-GFP is located in filopodia, and near to Golgi (Masuda et al., 1999). Recently, Guo et al. showed that CAT-1-GFP it expressed in plasma membrane in colocalization with VE-caherin and the authors propose that CAT-1 have a role as cellular adhesion molecule (CAM) and that the incubation with extracellular L-arginine augments L-arginine transport via promoting the CAT-1 shift from endothelial cells junctions to the free surface of ECs (Guo et al., 2015). Therefore, the L-arginine paradox has a potential explanation in the characteristics of CAT-1 cell surface expression, generating functional clusters for ensure an adequate supply of L-arginine for eNOS activity. Due to their physiological relevance, we focus on determining changes induced by insulin and/or diabetes on activity and expression of CAT-1.
Regulation of hCAT-1 by Insulin in Normoglycaemic or Hyperglycaemic Environment
Insulin increases the mRNA expression levels of hCAT-1 and hCAT-2B in human umbilical vein endothelial cells (HUVEC), correlates with high rate of L-arginine transport and NO synthesis (González et al., 2004) in a mechanism that involves high abundance of the protein in the plasma membrane (González et al., 2011). Insulin exerts a vasodilatory effect (González et al., 2011; Guzmán-Gutiérrez et al., 2012) through a signaling pathway involving PI3K, PKB/Akt, and MAPK activity that activates hCAT-1 and eNOS in endothelial cells (González et al., 2004). It has been described that this stimulation by insulin is caused by the activation of the promoter of the SLC7A1, gene encoding hCAT-1, via a mechanism that involves multiple binding sequences for the transcriptional factor specificity protein 1 (Sp1), located between 117 and 105 upstream base pairs from the transcription start site (TSS) (González et al., 2011). The SLC7A1 promoter belongs to the TATA-less group, so binding sites for Sp1 located near the TSS would be responsible for both basal expression and regulation of gene expression to stimulation by growth factors (Sobrevia and González, 2009). These effects have been observed with physiologic concentrations of insulin (0.1–10 nM), so we propose that the expression of SLC7A1 and hCAT-1 activity would be under a tonic regulation by physiological levels of plasma insulin. Moreover, it is reported that similar concentrations of insulin (1 μM) induces an acute (5 min) increase of L-arginine transport by a mechanism dependent on PI3K activity and PKC in human aortic endothelial cells (HAEC) (Kohlhaas et al., 2011)
As was mentioned above, diabetes is related with hyperglycaemia and insulin resistance, which are conditions that stimulate the oxidative stress in vasculature. Furthermore, exposure of HUVEC to high extracellular concentrations of D-glucose increases the synthesis of dependent of NAD(P)H oxidase, which reacts with the NO to generate ONOO−, contributing to endothelial dysfunction (Sobrevia and González, 2009; González et al., 2015). Long-term incubation (24 h) of HUVEC with 25 mM D-glucose increases the transport of L-arginine and cGMP accumulation in a similar magnitude to that observed in HUVEC from pregnancies with gestational diabetes mellitus (Sobrevia et al., 1996, 1997). Increased transport of L-arginine in both chronic incubation with D-glucose and gestational diabetes has been linked to increased mRNA levels for hCAT-1 and eNOS activity (Vásquez et al., 2004, 2007). In HAEC, prolonged incubation (7 days) with 25 mM D-glucose induces decreased eNOS activity (determined by nitrite content) and lower abundance of the protein and mRNA level (Furfine et al., 1994). This effect is associated with decreased activity of eNOS promoter (Srinivasan et al., 2004). In HUVEC, the hyperglycaemic environment induces an increase in the abundance of the eNOS protein (Vásquez et al., 2007), an effect that would be associated with the activity of a signaling pathway involving the PI3K and Akt. It has also been shown that in BAEC, there is a reduced production of insulin-dependent NO when cells were incubated with high extracellular concentration of D-glucose, an effect that seems to depend on a signaling pathway involving the type 1 insulin receptor (IR-1), PI3K and the inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) (Kim et al., 2006). Furthermore, increased production of cGMP induced by D-glucose in HUVEC is blocked by incubating the cells with 1 nM insulin (Sobrevia et al., 1997); and the incubation with 1 nM insulin (8 h) is sufficient to block the effect of D-glucose on the decrease of adenosine transport (Muñoz et al., 2006), an important vasoactive nucleoside (San Martín and Sobrevia, 2006). More recently, González et al. (2015) demonstrated that high extracellular concentration of D-glucose increases the expression of hCAT-1 and L-arginine transport, inducing high synthesis of NO associated with NADPH oxidase-dependent synthesis in HUVEC. On other hand, insulin (in normoglycaemic environment) increases L-arginine transport and NO synthesis without changes in levels, showing a physiological response that results in relaxation of umbilical vein (González et al., 2011). Interestingly, the effect of high D-glucose is associated with high contractile response to U46619 (thromboxane A2 analog) and hydrogen peroxide (H2O2). Co-incubation with insulin recovers the adequate response to vasoconstrictors and decreases the NADPH oxidase-dependent ROS (González et al., 2015). These mechanisms induced by high D-glucose could be responsible for endothelial dysfunction in diabetes, a scenario in which the L-arginine and NO convert from “good guys” to “bad guys” in company with high levels of ROS. In these conditions, insulin reduces the oxidative stress to improve bioavailability of NO, as long as the endothelial cell retains its capacity to respond appropriately to the hormone (Figure 1). In fact, in diabetic patients there is a significant reduction of forearm blood flow in response to insulin infusion, and an increase in L-arginine clearance (L-arginine incorporation to the tissue) during insulin infusion was 41% less in diabetic subjects compared to controls after adjusting for covariates age, BMI, MAP, and HDL cholesterol (Rajapakse et al., 2013). The data discussed in this section confirm the potential association between T2DM (and gestational diabetes), insulin resistance and endothelial dysfunction. Hence, further research must be focused on detailed mechanisms that regulate the cardiovascular actions of insulin in other states, such as hypertension and heart failure. Following this principle, in the second part of this article we review the effects of disturbances in cardiac insulin signaling.
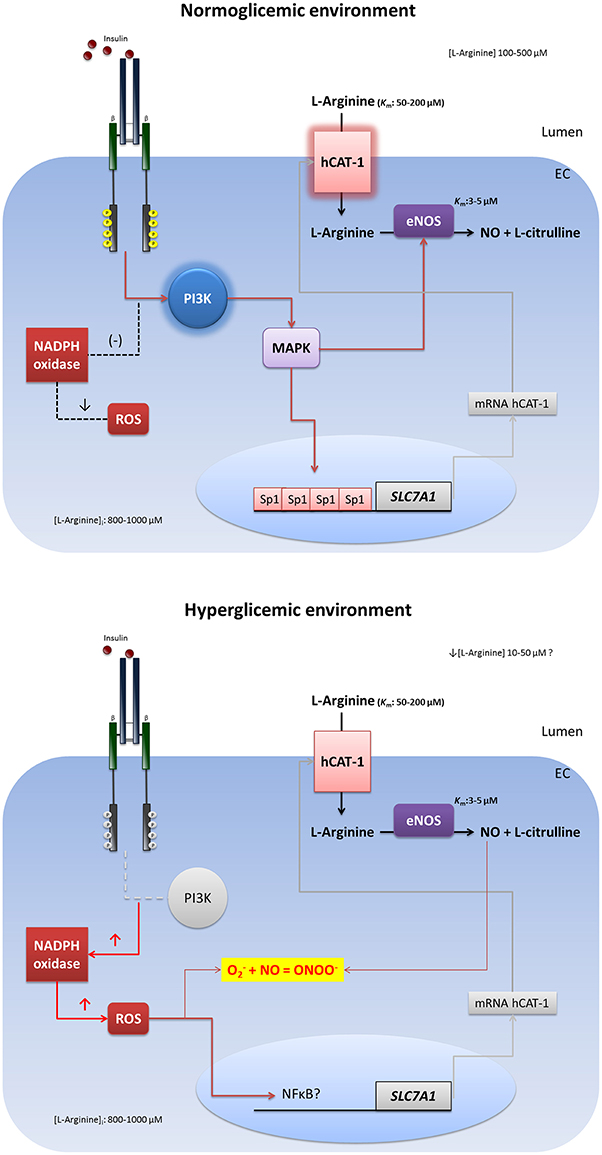
Figure 1. Impairment insulin signaling in endothelial hyperglycemic environment. In physiological state, insulin induces a signaling pathway that involves the activation of phosphatidylinositol 3-kinase (PI3K), mitogen activated protein kinase (MAPK), and specificity protein 1 (Sp1). This pathway induces the transcriptional activity of the SLC7A1 gene and the plasma membrane expression of hCAT-1 for increased the L-arginine transport and nitric oxide (NO) synthesis in endothelial cells. In hyperglycaemia, insulin resistance causes deterioration of intracellular signaling that reduces the transcriptional activity of SLC7A1, diminishing the L-arginine transport and NO synthesis. Also the insulin resistance abolishes the antioxidant capacity of the hormone, increasing the reactive oxygen species (ROS) and lowering the bioavailability of NO through the reaction with superoxide () to form peroxynitrite (ONOO−).
Insulin Regulation of Cardiac Function
Overview About Action Potential of Cardiomyocytes
Normal mechanical function of the heart in mammals depends on proper electrical functioning, which is reflected in the sequential activation of pacemaker cells and subsequent electrical propagation through the ventricles. Coordinated electrical functioning of the heart is recorded by a surface electrocardiogram (ECG). Propagation activity and electromechanical coordination of ventricles also depends on coupling among cells, mediated by gap junctions (Kanno and Saffitz, 2001). Generation of action potential (AP) in myocardium results from sequential activation and inactivation of ion channels, which can lead input currents (Na+ and Ca2+) and an output repolarization currents (K+) (Figure 2) (Antzelevitch and Dumaine, 2002; Nerbonne and Kass, 2005).
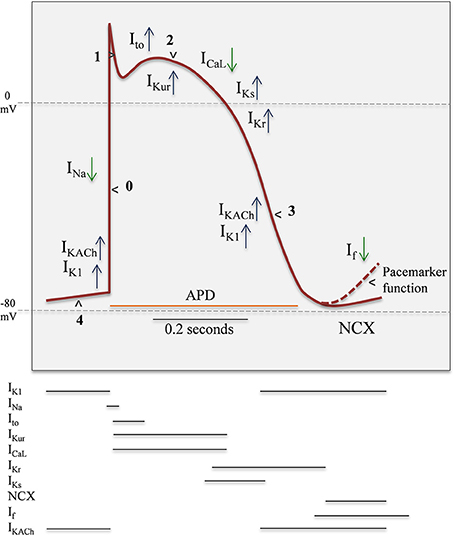
Figure 2. Representation of ventricular Action Potential (AP) and the temporal ionic current contribution. Blue arrow up represents outward currents, green arrow down represents inward currents. AP have five well-differenced states: 4, Resting; 0, Upstroke; 1, Early repolarization; 2, Plateau; 3, Final repolarization. The time- and voltage-dependent properties of the voltage-gated inward Na+ (Nav) and Ca2+ (Cav) currents expressed in human atrial and ventricular myocytes are similar. In contrast, there are multiple types of K+ currents, particularly Kv currents, contributing to ventricular action potential repolarization. The properties of the various Kv currents are distinct, and in contrast to the inward currents, there are multiple Kv currents expressed in individual myocytes throughout the myocardium. IK1, inward rectifier current; Ito, transient outward current; IK, delayed rectification currents: Ikur, ultra-rapid; Ikr, rapid; Iks, slow; ICaL, L-type Ca2+ current; Ina, sodium current; NCX, Na+/Ca2+ exchanger; If, funny current; IKACh, G-protein-activated K+ current.
AP differ in distinct regions of the heart, and contribute to the spreading of excitation way through the myocardium and the subsequent generation of normal heart beat (Kléber and Rudy, 2004). Changes in functional properties, or in the expression of ion channels in the myocardium, as a result of mutations (Antzelevitch, 2003) or myocardial diseases (Cesario et al., 2006), can lead to changes in the AP, synchronization or spread, including the heart's predisposition to potentially life-threatening arrhythmias (Akar et al., 2000; Casis et al., 2000; Casis and Echevarria, 2004). Given the clinical relevance of heart rhythm, it is of considerable interest to analyse the cellular and molecular mechanisms that contribute to the generation and maintenance of normal heart rhythm. Animal non-human studies suggests that small changes in time- and voltage-dependence properties of ion channels, in the myocardial sarcolemma, may have profound effects in both the action potentials time as well in the refractory period and rhythmicity (Delmar, 1992; Delisle et al., 2004).
In atrial myocytes, ventricular myocytes and Purkinje fibers, the onset of AP is fast (Phase 0) and given principally by voltage-dependent Na+ (Nav) channels activity (Fozzard, 2002). In contrast, in pacemaker cells of sino-atrial (SA) node (SAN) and atrio-ventricular (AV) node (AVN), this phase 0 is markedly slower. Phase 0 in Purkinje fibers and myocytes is followed by a transient repolarization (phase 1), reflecting Nav channel inactivation and the activation of the fast transient voltage-gated outward K+ current (Ito, f) (Niwa and Nerbonne, 2010). This transient repolarization, which can be quite prominent in Purkinje and ventricular cells, influences the shape and the time of the action potential plateau (phase 2). Plasma membrane depolarization also activates voltage-gated Ca2+ (Cav) currents, and the subsequent influx of Ca2+ through L-type Cav channels, during the phase 2 (plateau), triggering the excitation-contraction coupling in myocardium (Bers and Perez-Reyes, 1999). The driving force for K+ efflux is high during the plateau phase, and when Cav channels are inactivated, the outward K+ currents predominate, resulting in repolarization (phase 3). As a result, the voltage of plasma membrane returns to the resting potential. In contrast to Nav and Cav currents, there are multiple types of voltage-gated K+ (Kv) currents, as well as non-voltage-gated, inwardly rectifying K+ (Kir) currents (Figure 2 and Table 1). At least two types of transient outward currents, Itof and Itos, and several delayed rectifiers including IKr (IK rapid), IKs (IK slow), and IKur (IK ultra-rapid) have been distinguished (Brunet et al., 2004). The time- and voltage-dependent properties of Kv currents identified in myocytes from different species and/or from different regions of the heart present a remarkable similarity, suggesting that the same (or very similar) molecular entities contribute to the generation of each of the various types of Kv channels (Table 1) in different cells/species.
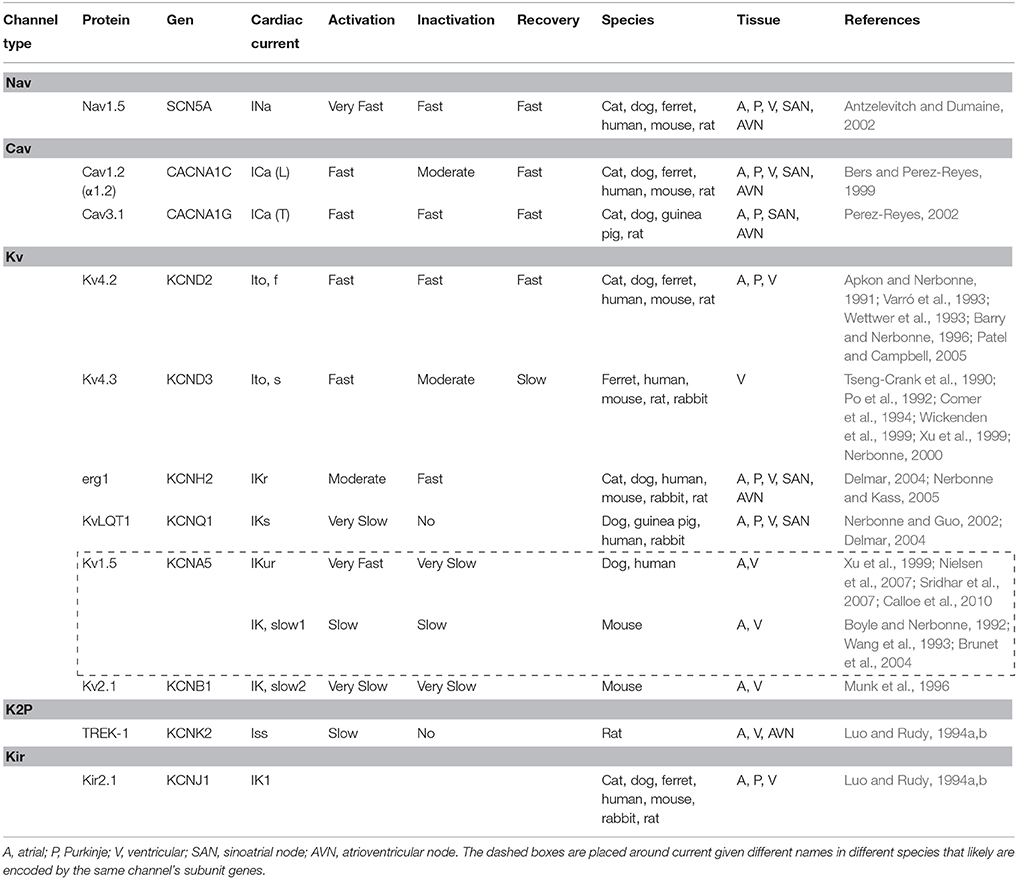
Table 1. Cardiac currents contributing to action potential repolarization.
Voltage-Dependent Potassium Channels
Based on biophysical differences and pharmacological sensitivities there are two main classes of Kv currents: transient outward currents (Ito) and delayed rectifier (IK) currents (Barry and Nerbonne, 1996; Nerbonne and Guo, 2002). The Ito currents are activated and rapidly inactivated on a depolarization stimuli (~−30 mV) and underlie the early phase (phase 1) of repolarization in ventricular and atrial cells in rat (Apkon and Nerbonne, 1991), mouse (Benndorf and Nilius, 1998), cat (Furukawa et al., 1990), rabbit (Hiraoka and Kawano, 1989), dog (Litovsky and Antzelevitch, 1998), and humans (Varró et al., 1993; Wettwer et al., 1993). The Ito currents influence Cav channel activation and the balance between inward and outward currents during phase 2. Cardiac IK currents are activated at similar plasma membrane potentials and present variable kinetics. These currents are determinant for the latter phase of repolarization that determine the diastolic potential (Barry and Nerbonne, 1996).
The two components of the Ito currents identified in the heart cell types show different kinetics of recovery (Xu et al., 1999). The Ito rapid current component (Ito fast, Itof) is recovered rapidly in the range of 60–100 ms (Patel and Campbell, 2005). In contrast, the slow component (Ito slow, Itos) recovers slowly with time constants in the order of seconds (Xu et al., 1999). Both are differentially expressed and contribute to heterogeneity of the action potential shape (Brunet et al., 2004).
Structurally, Kv channels are comprised of four α subunits that form the channel pore (MacKinnon, 1991), each subunit with six transmembrane segments (denoted S1–S6) including the voltage sensor S4 (Snyders, 1999), a single pore region between S5 and S6, and C- and N-terminal intracellular domains. The pore region works as a K+ selectivity filter (Doyle et al., 1998). In Kv1 to Kv4, the amino acids sequence at the N-terminal domain, which precedes S1, plays a critical role in the assembly Kvα subunit (Figure 3) (McKeown et al., 2008). Evidence suggests that Kv α-subunits of the Kv4 subfamily are responsible for a rapidly activating, inactivating, and recovering cardiac transient outward Kv channels referred to as Itof (Table 1). In rat and mouse ventricular myocytes exposed to antisense oligodeoxynucleotides targeted against Kv4.2 or Kv4.3, Itof density is reduced by < 50% (Fiset et al., 1997; Guo et al., 2002). Significant reductions in rat ventricular Itof density are also seen in cells exposed to an adenoviral construct encoding a truncated Kv4.2 subunit (Kv4.2ST) that functions as a negative dominant (Johns et al., 1997). In addition, it has been reported that Itof is eliminated in ventricular myocytes isolated from transgenic mice, expressing a pore mutant of Kv4.2, W362F (Barry et al., 1998). Taken together, these results demonstrate that members of the Kv4 subfamily underlie Itof in mouse and rat ventricles. Biochemical studies have also shown that Kv4.2 and Kv4.3 are associated in adult mouse ventricles, suggesting that functional ventricular Itof reflect the heteromeric assembly of the Kv4.2 and Kv4.3 α-subunits (Guo et al., 2002). In dog and human myocardium, however, Kv4.2 appears not to be expressed (Kong et al., 1998), suggesting that only Kv4.3 contributes to Itof, in larger mammals. Moreover, the genetic deletion of Kv4.2 abolishes Itof current, revealing a critical role of Kv4.2 channels in generating these currents in rodents (Guo et al., 2005).
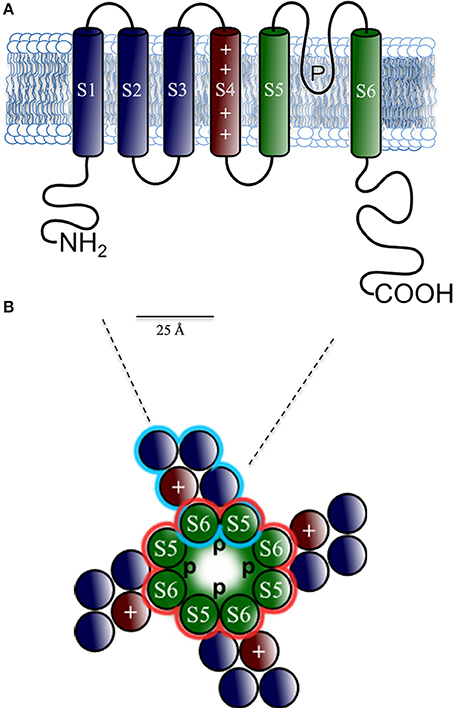
Figure 3. Predicted protein topology of cardiac K+−channel α subunits. Schematic representations of (A) the six-transmembrane (S1–S6) one P-loop (6Tm-1P) voltage-dependent K+−channel (Kv) α-subunits topology that conduct IKur, Ito, Iks, IKr, and If in the heart; in red voltage sensor (S4); in green the pore region formed by S5 and S6 segments. Note the amino (NH2) and carboxyl (COOH) terminal tails located intracellular, which serves as assembly sites for β-subunits as KCHiP2. (B) Structure of the tetrameric assembly of the Kv Channel viewed from the extracellular side. The S5-P-loop-S6 region assembles with three other pore domains (central P) functions as a K+ selectivity filter.
Biophysical properties of slow transient outward myocardial Kv currents, referred as Itos, are different. Considerable evidence supports that myocardial Itos currents are encoded by the Kv1 α-subunit, Kv1.4. Heterologous expression of Kv1.4 yields slowly recovering (seconds), 4-aminopyridine (4-AP, Kv inhibitor) sensitive transient Kv currents (Tseng-Crank et al., 1990; Po et al., 1992; Comer et al., 1994; Wickenden et al., 1999) that closely resemble Itos (Fermini et al., 1992; Brahmajothi et al., 1999; Wang et al., 1999; Xu et al., 1999). In addition, expression of Kv1.4 has been demonstrated at the transcript level in rat (Tseng-Crank et al., 1990; Roberds and Tamkun, 1991; Wickenden et al., 1999), ferret (Brahmajothi et al., 1999), canine (Dixon et al., 1996), and human (Tamkun et al., 1991) hearts, and at the protein level in mouse, rat and ferret ventricles (London et al., 1998; Brahmajothi et al., 1999; Wickenden et al., 1999) and rabbit atrium (Wang et al., 1999). Direct support for this hypothesis was complemented on myocytes isolated from (Kv1.4−∕−) mice with a targeted disruption in the kcna4 (Kv1.4) locus (London et al., 1998). Waveforms of outward currents in cells isolated from Kv1.4−∕− ventricles are indistinguishable from those recorded in wild-type ventricular cells (Guo et al., 1999). In cells isolated from the interventricular septum from Kv1.4−∕−, however, Itos is undetectable, thereby demonstrating that Kv1.4 underlies Itos (Guo et al., 1999).
Some studies suggest that additional subunits are required for the generation of cardiac Ito currents. Indeed, a number of molecularly diverse accessory subunits can modulate biophysical properties and cell surface expression of heterologously-expressed Kv4 channels (Guo et al., 2000; Abbott et al., 2007). Of these accessory subunits, only the cytoplasmic KChIP2 protein has been shown directly to be an essential component of myocardial Itof (Grubb et al., 2012). Co-expression of KChIP subunits dramatically increases the cell surface expression of Kv4 channels, Itos inactivation and markedly accelerates recovery (An et al., 2000). Also, KChIP2 co-immunoprecipitates with Kv 4.2 (and Kv4.3) α subunits in adult mouse ventricles (Guo et al., 2002) and the targeted deletion of the KChIP2 locus (kcnip2) abrogates ventricular Itof (Kuo et al., 2001). Interestingly, KChIP2 protein expression is dramatically reduced in Kv4.2−∕− ventricles (Guo et al., 2005), suggesting that expression of the Kv4 and KChIP2 proteins are reciprocally regulated. Other regulatory subunit that colocalize with Kv4.2 is the MinK-related peptide (MiRP) (McCrossan and Abbott, 2004; Roepke et al., 2008). Although MiRP does not affect the densities of expressed Kv4.2-KChIP2 currents (Liu et al., 2008), targeted deletion of the gene encoding MiRP result in reduced (< 25%) ventricular Itof densities without changes in total or surface Kv4.2 expression (Roepke et al., 2008).
The ultra-rapid potassium current IKur is well expressed in the atria, where it contributes to repolarization (Amos et al., 1996). The molecular constituent of IKur is the Kv1.5 potassium channel (Boyle and Nerbonne, 1992; Wang et al., 1993). Direct experimental support for this hypothesis was provided with the demonstration that exposure to antisense oligonucleotide targeted against Kv1.5 selectively attenuates IKur in isolated adult human (Snyders et al., 1993; Feng et al., 1997) and rat atrial myocytes (Bou-Abboud and Nerbonne, 1999). The important physiological role for Kv1.5 in human atria is suggested by the finding that IKur densities and Kv1.5 protein expression are reduced markedly in atria of patients with chronic atrial fibrillation (Van Wagoner et al., 1997). Although, IKur has predominantly been reported in atria, it has also been suggested to play a role in canine and human ventricles (Nielsen et al., 2007; Sridhar et al., 2007; Calloe et al., 2010).
Insulin Regulation of Cardiac Electrical Function
Insulin has a direct effect in cardiomyocytes, in glucose transport and oxidation, glycolysis, glycogen and protein synthesis (Abel, 2004; Bertrand et al., 2008). In vivo, many of insulin's effects on cardiac function and metabolism are related to systemic responses such as an increment in peripheral and coronary vasodilatation, and sodium and water intake by the kidney (Muniyappa et al., 2007).
Insulin is a key regulator of cardiac growth as reported in studies carried out in cardiac insulin receptor knockout (CIRKO) mice. In this study, the cardiac index was reduced by 22–28%, possibly due to a decrease in myocyte size rather than a reduction in cell number (Belke et al., 2002). The role of insulin in cell size regulation is supported by studies in Drosophila with disrupted homolog of vertebrate insulin signaling, were the knock-out model for IRS, had less than half the size of wild-type flies, owing to fewer and smaller cells (Böhni et al., 1999). In experimental mice with targeted deletion of IRS-1 or IRS-2, there has been evidence of a reduction in postnatal cardiac size (Araki et al., 1994), which is indicative of conserved roles for insulin signaling in the determination of cardiac growth. Inhibition of PI3K function in mouse heart using a negative dominant transgene, results in a similar phenotype generated in CIRKO mice (Shioi et al., 2000). Negative dominant PI3K transgenic mice showed 16% reduction in cardiac index and myocyte surface area reduction of 18% (Belke et al., 2002). Thus, impaired PI3K activity represents a mechanism that explains the cardiac size phenotype in CIRKO mice.
In addition, insulin could increase cardiac contractility, affecting cardiac output, and mediates the cellular hypertrophy and generates an antiapoptotic effect on cardiomyocytes by activating other intermediary intracellular signaling pathways (Belke et al., 2002). Changes in these pathways and in the pathologic processes linked to status of insulin deficiency or insulin resistance could contribute to an increased risk of cardiac hypertrophy, pathological remodeling of the left ventricle and heart failure, among others (Cesario et al., 2006).
Diverse evidences suggests that the stimulatory effect of insulin involves K+ channel protein synthesis, which is responsible for maintaining both Ito and IKur currents; although it is proposed that the hormone regulates differentially these two currents (Shimoni et al., 1998; Rozanski and Xu, 2002; Lengyel et al., 2007; Torres-Jacome et al., 2013). In a recent review, Ballou et al. collected and summarized several studies that shows that PI3K signaling regulates many aspects of proteins involved in AP in the heart, including protein expression, trafficking and gating Nav1.5, Kv11.1, Kv7.1, and Cav1.2 in cardiomyocytes. This evidence comes from studies where drugs that inhibit PI3K signaling cause QT prolongation, but the role of PI3K in transcriptional regulation of genes in cardiomyocytes is relatively unexplored (Ballou et al., 2015).
However, a study of isolated cardiomyocytes from T1DM mice induced by STZ and treated with wortmannin (inhibitor of PI3K) prior to incubation with insulin, showed that insulin is still able to restore the densities of Ito currents without effect on other potassium currents (Ui et al., 1995). This indicates that this hormone could not induce its effects through PI3K pathway in T1DM (Shimoni et al., 1998). Insulin stimulates another important signaling pathway, MAPK/ERK, which can lead to cell proliferation and protein synthesis (Guo, 2014); furthermore, its activation can be blocked by PD98059, a specific inhibitor of the phosphorylation of ERK1/2 (Lazar et al., 1995). In isolated cardiomyocytes from T1DM rats treated with PD98059 (30 min) prior to the addition of insulin, blockage is perceived in the insulin-dependent return of the densities of potassium Ito currents (Shimoni et al., 1998). These results suggest that the effects on the maintenance of these currents and their respective channels occur in a MAP-kinase dependent process (Shimoni et al., 1998).
Cardiac Disease in Diabetes Mellitus
Studies have found that diabetic patients have an increased risk of CVD (Gregg et al., 2014). Common complications are acute myocardial infarction (181.5 events/1000 people/year) and stroke (126.2 events/1000 people/year; Bethel et al., 2007). In a cohort of geriatric American adults with T2DM between 2004 and 2010, cardiovascular complications have the highest incidence, followed by eye disease and hypoglycaemic events (Huang et al., 2014). The presence of diabetes is associated with a high risk of cardiac failure (Aneja et al., 2008), and is present in the 75% of patients with idiopathic dilated cardiomyopathy (Bertoni et al., 2003). The “Framingham Heart” Study (Kannel and McGee, 1979) showed that the incidence of heart failure is 2 and 5 times higher for diabetic men and women, respectively. Diabetic patients with macrovascular complications showed a strong association with cardiomyopathy, and its association is well correlated with the duration and severity of hyperglycaemia (Poornima et al., 2006).
Diabetic Cardiomyopathy
DM affects cardiac structure and function, which under certain conditions can lead to a condition called DCM. Rubler et al. described this condition first in 1972 and defined it as “ventricular dysfunction that occurs independently of coronary artery disease and hypertension” (Rubler et al., 1972). In addition, DCM is characterized by diastolic dysfunction, which becomes more apparent in presence of hypertension or myocardial ischemia (Fang et al., 2004). In humans, the DCM is also characterized by diastolic dysfunctions, which precede the development of systolic dysfunction (Liu et al., 2001). These findings also could be found in patients with T2DM, where diastolic dysfunction affected 30% of the patients (Boudina and Abel, 2007).
The effects of DCM lead to cardiomegaly, left ventricular dysfunction, ventricular electrical remodeling and symptoms of congestive heart failure (Casis et al., 2000). The resulting electrophysiological changes contribute to an increase in the incidence of cardiac arrhythmias and sudden death (Casis et al., 2000). Electrical changes have been associated with the surface ECG abnormalities, including T and QT wave prolongation (Casis et al., 2000; Casis and Echevarria, 2004).
Insulin resistance in the heart is a factor that contributes significantly to DCM (Mandavia et al., 2013). Therefore, it is important to distinguish between the secondary effects to environmental disturbances in states of insulin resistance and specific changes that occur in the signaling pathways that are intrinsic to heart tissue. These effects determine, among others, changes in cardiomyocyte AP as might be the extension of repolarization phase (Lengyel et al., 2007), and thus infer the pathophysiological mechanism that would play insulin on heart function.
Morphological Changes in the Ventricular Action Potential
The morphology of AP is clearly altered in T1DM and T2DM, showing a prolonged QT in myocytes isolated from STZ models in either, mice (Magyar et al., 1992; Casis et al., 2000) or rabbits (Torres-Jacome et al., 2013). AP duration in cells stimulated at 1 Hz increases in isolated cells from diabetic mice (Casis et al., 2000). These cells shown, phase 1 (early repolarization) less pronounced and a longer phase 2 (plateau). Otherwise, the resting membrane potential and the AP amplitude were unchanged (Casis et al., 2000). Moreover, T2DM models (db/db mice, a leptin receptor mutant that develops obesity, diabetes, hyperinsulinemia and increased levels of triglycerides and fatty acids; Buchanan et al., 2005) shown similar prolongation of action potentials (Figure 4) (Shimoni, 2001).
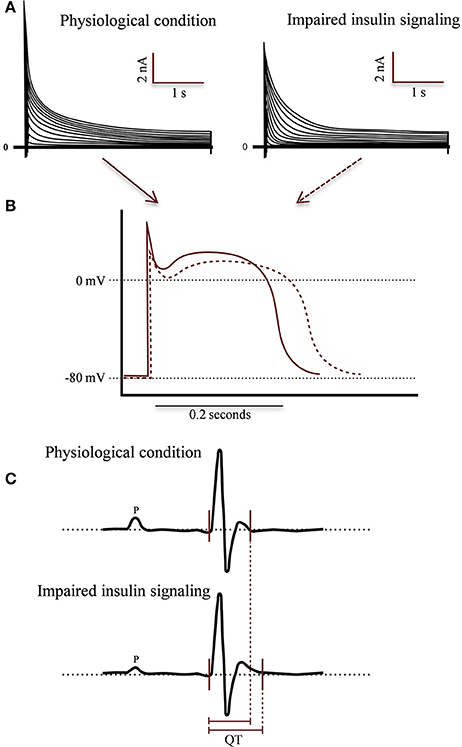
Figure 4. Morphological changes associated to impaired insulin in different experimental models: T1DM, T2DM, and CIRKO. (A) Representative whole cell voltage-gated outward K currents recorded from Wild-type (WT) animal (left) and impaired insulin signaling condition (right). Outward K+ currents are reduced in ventricle of experimental models. (B) Representative action potentials waveforms recorded from left ventricular myocytes isolated from WT (solid red line) and experimental mice (dashed red line). Insulin signaling alterations produce a prolonged action potential duration (APD) in experimental ventricular cells. Effect of absent or impaired cardiac insulin signaling at the level of the whole organism by measuring surface ECG activity in anesthetized mice. In (C) is illustrate the comparison of ECG recordings from WT and experimental mice. Analyses of the ECG parameters confirmed a significant prolongation mainly in QT intervals in experimental relative to control littermates. Relative differences in QT intervals are indicated below the records by red solid lines.
Prolongation of action potentials is due to, at less in part, by a decrease in the output repolarizing currents (Ito and IK) (Shimoni et al., 1999, 2004; Qin et al., 2001). The amplitude of the currents (Ito and IK) are reduced in myocytes isolated from T1DM murine models (Torres-Jacome et al., 2013). In addition, myocytes isolated from db/db mice (12th week of age), as a T2DM model, exhibit a sustained attenuation of the output currents compared to the db/+ mouse (Shimoni, 2001; Shimoni et al., 2004). Therefore, despite the dramatically different levels of insulin circulating in the STZ diabetic rat and db/db mouse, the changes of action potential and ventricular output K+ currents are similar, suggesting that these effects could be generated by an impaired ligand-receptor coupling or absence of insulin signaling pathway (Shimoni et al., 2004).
Despite the total output currents of K+ are impaired (Figure 4), is necessary to determine which one is most relevant electrical change. In dogs treated with alloxan (a toxic glucose analog that destroys the β-pancreatic cells) to induce T1DM, after 8 weeks, were evaluated the different currents contribution to the AP through voltage-clamp experiments. In this case, no changes were found in neither amplitude nor voltage-dependent IKr and ICa activation currents. Conversely, the amplitude of the Ito and IKur currents significantly decreased by 69.3 and 54.6% in myocyte cells isolated from diabetic models, respectively. In an effort to clarify the effects of insulin, after a week from developed diabetes, dogs were treated with insulin (0.5–1.0 IU/kg day-1s.c), and an increase in current density was evident only in the Ito and IKur currents (Lengyel et al., 2007). This result is consistent with findings obtained from myocytes isolated from hearts of diabetic mice (STZ model) incubated with insulin, in which the amplitudes of these currents were restored (Magyar et al., 1992; Xu et al., 1996, 2002; Shimoni et al., 1998).
Several animal models have been used to characterize the effects of diabetes on the heart (Shimoni et al., 2004; Boudina and Abel, 2010) and include a variety of systemic effects (Bugger and Abel, 2009). Diminished glucose oxidation rates in cardiomyocytes occur as early as 48 h after the induction of diabetes by STZ treatment, which is reversed by insulin treatment (Chen et al., 1984). Insulin-stimulated cardiac glucose uptake and their utilization are also impaired in type 2 diabetic hearts (Mazumder et al., 2004). Furthermore, in hyperinsulinemic animal models of T2DM, such as the Zucker diabetic rat, the impaired insulin signal transduction in cardiac muscle (Kolter et al., 1997) is also associated with diminished glucose utilization and increased fatty acid utilization in the heart (Chatham, 2001). In all of these models it is difficult to separate the relative contribution of intrinsic defects in cardiomyocyte insulin signaling from the conditions generated by the altered systemic metabolism, such as hyperglycaemia and hyperlipidaemia.
The cardiac-specific insulin receptor knockout (CIRKO) mice model, development by cre/loxP recombination to specifically inactivate insulin signaling in cardiomyocytes in vivo, preserve insulin signaling in other cells such as endothelial cells, VSMC, and liver and skeletal muscle cells (Brüning et al., 1998; Abel et al., 1999) and let to investigate the specific role of impaired cardiac insulin signaling without the additional conditions associated to systemic metabolic perturbations (Belke et al., 2002). CIRKO mice develop contractile dysfunction but not heart failure, associated with decreased glucose and fatty acid oxidation (Belke et al., 2002). Also develop mitochondrial dysfunction, with an increased mitochondrial superoxide production, and display fatty acid-induced mitochondrial uncoupling (Boudina et al., 2009). Likewise, CIRKO mice represent a useful model to dissect the mechanisms and the effects of insulin in the heart, in the absence of systemic effects of insulin deficiency, or the additional conditions associated to systemic effects of insulin treatment as occur in many models of diabetes.
Effects of Diabetes on Gene Expression of Kv Channels
Decreased of outward currents induced by diabetes conditions may be attributed to deficient protein expression (Howarth et al., 2009), changes in biophysical properties, or both (Lopez-Izquierdo et al., 2014). Until now, there is no difference in voltage dependence of inactivation, inactivation kinetics, or recovery from inactivation state. It is suggested that these effects are explained by an abnormal protein expression (Lopez-Izquierdo et al., 2014).
The study of the cardiac gene expression by RT-PCR considering the genes: kcnd2 and kcnd3 encoding the Ito α-subunit Kv4.2 and Kv4.3, respectively (Frank-Hansen et al., 2005); kcne2 encoding to MiRP1, Ito and IKs β-subunit (Roepke et al., 2008), on tissue extracted from left ventricles of mice with STZ-induced diabetes showed significant reductions of ~80% in the expression of kcnd2 and kcne2, whereas no significant differences were shown for kcnd3 (Howarth et al., 2009). These results were replicated in CIRKO mice, where KChIP2, Kv4.2 accessory subunit had about 50% reduction. Therefore, Kv4.2 and its assembly with specific accessory subunits such KChIP2 and MiRP1, would be relevant in maintaining transient K+ current, mainly in its fast component (Itof) (Lopez-Izquierdo et al., 2014).
Studies in animal models of diabetes also reveal a reduction in the mRNA and protein levels of the components of Itof (Qin et al., 2001). Based on the analysis of protein levels of ion channels evaluated by western blot obtained for CIRKO mice left ventricle, show a reduction of ~50% in the abundance of Kv4.2 and Kv1.5 (Lopez-Izquierdo et al., 2014). In contrast, other studies using a T1DM rat or CIRKO mice model, report a reduction in Kv4.2 protein expression, but not for the Kv4.3 channel, were neither the messenger nor protein expression were down-regulated (Kääb et al., 1998; Nishiyama et al., 2001). KChIP decreased by 40% when compared to the control group (Lopez-Izquierdo et al., 2014). Furthermore, immunofluorescence analysis shows a reduction of immunoreactivity for Kv4.2, Kv4.3, and Kv1.5 protein, in isolated ventricular cardiomyocytes from T1DM models vs. healthy controls, and no changes of Kv2.1 (IKr determining protein) were found (Torres-Jacome et al., 2013). Moreover, in extracted tissue from ventricles of diabetic animals evaluated by immunohistochemistry, a reduction has been shown in the number of cardiomyocytes with immunopositive reactivity to Kv4.2 and MiRP1 (IKur β-subunit), consistent with the reduced expression genes that encode them (Howarth et al., 2009). Therefore, a reduction in Kv4.2, Kv1.5 messenger and protein provides an explanation for the reduced Ito and IKur currents, observed in impaired insulin signaling conditions, respectively. At the same time, interaction with accessory subunits, such as KChiP2 and MiRP-1, would be relevant to the maintenance of these currents (Grubb et al., 2012).
In addition, the results previously mentioned suggest a regulatory role of insulin in the expression of different potassium channels in diabetic cardiomyocytes. Incubation of isolated myocytes with insulin can recover the protein expression of Kv4.2 and Kv1.5 channels, and also restores the amplitude of potassium currents when tested after 3, 4, 5, and 6 h incubation (Shimoni et al., 1998). In the same way, the use of protein synthesis blockers, such as cycloheximide, blocks these currents' recovery (Rozanski et al., 1998). Furthermore, the effect of insulin depends on the integrity of the cytoskeleton, hence protein traffic inhibitors, as cytochalasin D, are able to attenuate this effect (Shimoni et al., 1999; Yang et al., 2002). Presumably, it is necessary that newly synthesized channels can effectively translocate to the cell membrane to maintain these specific repolarizing currents.
Clinical Implications
Insulin is a hormone with pleiotropic physiological functions, so it is normal to conclude that deterioration of insulin signaling also has pleiotropic dysfunctions. In this review, we discuss two main mechanism that govern the cardiovascular physiology, one mechanism in endothelial cell related with L-arginine transport and NO bioavailability, and a second main mechanism related with the regulation of Kv channels on cardiomyocytes.
The physiological role of NO is well known in human vasculature, so the largest contribution of this review is the establishment that insulin resistance is closely related with potential reduction of hCAT-1 expression and/or activity in endothelial cells. Reduction of L-arginine transport is a primarily cause of endothelial dysfunction and induces a reduction of NO synthesis due to restriction of substrate availability for eNOS. Cardiovascular pathologies associated to insulin resistance are well documented, and we propose that the significant reduction of L-arginine transport mediated by lower expression and/or alteration of plasma membrane localization of the transporter could be a link between the impairment of insulin signaling and vascular disease. Clinical evidence shows that the forearm L-arginine uptake is reduced in subjects with essential hypertension or with family history of hypertension. In peripheral blood mononuclear cells (PBMCs) isolated from the same patients, a reduction was seen of Vmax of L-arginine transport and lower sensitivity to Ach-dependent vasodilatation, parameter that was reverted by co-infusion of L-arginine (Schlaich et al., 2004). Also a reduction in agonist-induced blood flow was observed in response to insulin in patients with T2DM, an alteration that is associated to lower capacity of transport of L-arginine and NO synthesize (Rajapakse et al., 2013). Additionally, in CHF patients it is determined that there is a reduced capacity for transport of L-arginine from plasma to the tissues with reduction of transcardiac fractional extraction of L-arginine and lower expression of hCAT-1 in myocardial tissue, without changes in hCAT-2B expression. Also the transcardiac L-citrulline flux, as a parameter of NO synthesis in heart, is decreased in CHF patients (Kaye et al., 2002). These data indicate the presence of an abnormal L-arginine transport in CHF, suggesting a shared mechanism between endothelial dysfunction and heart disease, related with hCAT-1 down-regulation and impairment of NO synthesis.
Altogether, this information demonstrates that there is a positive feedback mechanism between insulin secretion, insulin signaling, L-arginine/NO pathway, and cardiac function that is disrupted in pathophysiological conditions such as metabolic syndrome, insulin resistance and diabetes.
In regards to the physiological role of insulin in the regulation of Kv channels and their association with cardiac electrical function specifically, this relationship would be given by an altered intracellular signaling in a similar way to the pathological processes involved in diabetes mellitus (Casis and Echevarria, 2004). Diabetic patients show electrocardiographic changes associated with a high incidence of cardiac arrhythmias (Veglio et al., 1999, 2002a,b, 2004). Specifically, long QT syndromes (duration of ventricular depolarization and repolarization of the myocardium) have been reported as a result of an increase in the time of the ventricular action potential. This phenomenon has been linked to a three-fold increased risk of sudden cardiac death (Straus et al., 2006). Even more, studies have shown a significantly increase in relative death risk for subjects with QTc (QTc, heart rate-corrected QT interval) longer than 0.43–0.44 s (Brown et al., 2001). This relationship has been shown for all cause mortality, cardiac mortality, and sudden death (Festa et al., 2000).
Prevalence of QTc prolongation has been evaluated in different cohorts of diabetic subjects (Veglio et al., 1999, 2002a,b) and in the National Health and Nutrition Examination Survey III (NHANES III) population (Brown et al., 2001; Zhang et al., 2011). In T1DM patients, the prevalence of an abnormally prolonged, corrected QT was 16% in the whole population, 11% in males and 21% in females. QTc in insulin-dependent diabetic female patients is longer than in male patients, even in the absence of diabetic complications known to increase the risk of corrected QT prolongation (Veglio et al., 1999). While in T2DM patients, prevalence of increased QTc duration was 25.8%, with no sex differences, showing a considerably high prevalence of increased QTc and their association with Coronary Heart Disease (Veglio et al., 2002a,b). Finally, in the NHANES III survey, the prevalence of QTc prolongation in diabetic subjects was nearly twice that of non-diabetic subjects (Brown et al., 2001; Zhang et al., 2011).
Different studies explain the action potential prolongation as a response to a reduction of repolarizing K+ currents, in cardiomyocytes isolated from type 1 to 2 diabetic models (Magyar et al., 1992; Shimoni et al., 1994; Casis et al., 2000; Shimoni, 2001; Torres-Jacome et al., 2013). To examine the effects of impaired insulin signaling on these electrophysiological changes, recent studies have measured the electrocardiographic activity in anesthetized CIRKO mice, in order to assess the effect of the lack of cardiomyocyte specific insulin receptor (Lopez-Izquierdo et al., 2014). When was compared the electrocardiographic recordings from CIRKO mice vs. control mice, it has been demonstrated a significant prolongation of the QRS, QT, and QTc intervals (Lopez-Izquierdo et al., 2014).
The specific contribution of impaired insulin signaling in gene expression for cardiac Kv channels, lead to a QT interval prolongation in experimental models (Figure 5) (Howarth et al., 2009). This is similar to the findings that were reported for diabetic patients (Veglio et al., 2002a,b). These results are independent of the systemic metabolic changes that join the disease.
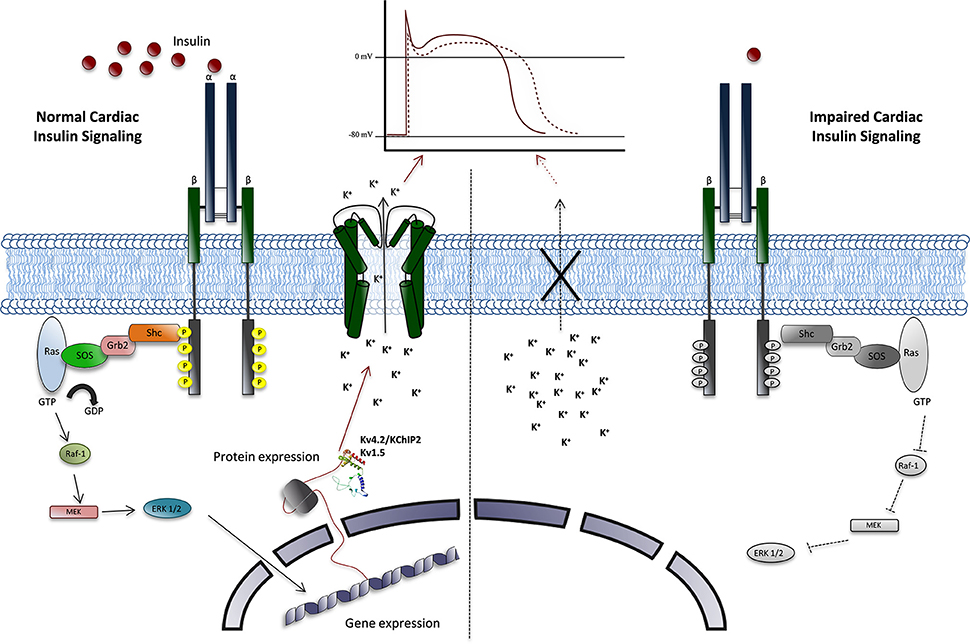
Figure 5. Proposal model of Insulin regulation of cardiac Kv channels. Normal cardiac Insulin signaling (right side) plays an important role in maintaining the repolarizing currents, in a Ras/MAP Kinase signaling pathway-mediated process. This regulation is achieved through gene expression and protein, which, specifically, determine the Kv4.2 and Kv1.5 α subunits, were need translocate to cellular membrane to generate Ito and IKur currents, which determine the appropriate action potential in ventricular cells. Furthermore, in disease, impaired insulin signaling (left side) and the reduce Ito and IKur currents amplitude, leads to an increase in the length of AP, manly in phase 2, due to the reduced availability of Kv channels with an increased risk of cardiac arrhythmias associated.
Concluding Remarks
In summary, insulin has general effects on all tissues and a strong relationship with cardiovascular function. Specifically, through experimental models of impaired intracellular signaling, which occurs in pathological conditions such as diabetes, it is possible to elucidate the regulatory effects that tie this particular ligand to the vascular and cardiac function.
Diabetes involves a series of prevalent cardiovascular complications such as DCM, a disease that appears as relevant for studying the effects of insulin resistance. This is because it includes a number of pathophysiological events, which include the ventricular electric remodeling.
Insulin plays an important role in maintaining the current repolarizing, specifically the Ito and IKur components, which determine the appropriate action potential in ventricular cells. This regulation is achieved through genes and proteins expression, which determine the Kv4.2 and Kv1.5 α subunits, β subunits and KChIP2 and MiRP1 composition, in a Ras/MAP Kinase signaling pathway-mediated process (Figure 5).
Outside of metabolic function, it is also evidenced that insulin, through the lack of its signaling pathways, plays a regulatory role in the cardiac electrical activity through voltage-dependent potassium channels and can produce electrocardiographic changes, such as QT prolongation, and increased risk of sudden death in diabetic patients.
Author Contributions
SD and MG participated in bibliographic research, writing, edition, and figures design; DG and LC participated in bibliographic research in vascular effects; LS and LZ participate as vascular function and channels experts, respectively.
Funding
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT 11100192, 1150377) and Vicerrectoría de Investigación y Desarrollo, Universidad de Concepción (VRID-Asociativo 213.A84.014-1.0), Chile.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all research staff at the Vascular Physiology Laboratory of Universidad de Concepción for their technical support and the Department of Kinesiology of Universidad de Concepción for administrative support. Part of this work was the thesis of Sebastián Dubó to obtain the degree of Masters in Human Physiology (Faculty of Biological Sciences, Universidad de Concepción, Chile).
Abbreviations
Ach, acetylcholine; AP, action potential; AsODNs, antisense oligonucleotides; BH4, tetrahydrobiopterin; Cav, voltage-gated calcium channel; CIRKO, cardiac-specific insulin receptor knock-out; DCM, Diabetic Cardiomyopathy; DM, Diabetes Mellitus; DOCK1, dedicator of cytokinesis protein 1; EC, electrocardiogram; eNOS, endothelial NO synthase; GAB1, Grb-2 associated protein; HAEC, human aortic endothelial cells; hCAT-1, cationic aminoacid transporter; HEKC, human embryonic kidney cells; HUVEC, human umbilical vein endothelial cells; IK, delayed rectifier potassium channel; IKr, rapidly activating delayed rectifier potassium channel; IKs, slowly activating delayed rectifier potassium channel; IKur, ultra-rapid potassium current; IR, Insulin receptor; IRS, insulin receptor substrate; Ito, transient outward potassium current; KChIP2, voltage-gated potassium (Kv) channel-interacting proteins; Kir, inwardly rectifying potassium channels; Kv, voltage-gated potassium channel; MAPK, Mitogen-activated protein kinases; MiRP, MinK-related peptide 1; Nav, voltage-gated sodium channel; NE, norepinephrine; NO, nitric oxide; PI3K, phosphatidylinositide-3-kinase; PKC, protein kinase C; QTc, heart rate-corrected QT interval; SHC, Src homology 2-domain containing protein; STZ, streptozotocin; T1DM, Type 1 Diabetes Mellitus; T2DM, Type 2 Diabetes Mellitus; Tyr, tyrosine amino acid; WHO, World Health Organization.
References
Abbott, G. W., Xu, X., and Roepke, T. K. (2007). Impact of ancillary subunits on ventricular repolarization. J. Electrocardiol. 40, S42–S46. doi: 10.1016/j.jelectrocard.2007.05.021
Abel, E. D., Kaulbach, H. C., Tian, R., Hopkins, J. C., Duffy, J., Doetschman, T., et al. (1999). Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J. Clin. Invest. 104, 1703–1714. doi: 10.1172/JCI7605
Akar, F. G., Laurita, K. R., and Rosenbaum, D. S. (2000). Cellular basis for dispersion of repolarization underlying reentrant arrhythmias. J. Electrocardiol. 33, 23–31. doi: 10.1054/jelc.2000.20313
Amos, G. J., Wettwer, E., Metzger, F., Li, Q., Himmel, H. M., and Ravens, U. (1996). Differences between outward currents of human atrial and subepicardial ventricular myocytes. J. Physiol. 491, 31–50. doi: 10.1113/jphysiol.1996.sp021194
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., and Mendoza, G. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. doi: 10.1038/35000592
Aneja, A., Tang, W. H., Bansilal, S., Garcia, M. J., and Farkouh, M. E. (2008). Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am. J. Med. 121, 748–757. doi: 10.1016/j.amjmed.2008.03.046
Antzelevitch, C. (2003). Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. J. Cardiovasc. Electrophysiol. 14, 1259–1272. doi: 10.1046/j.1540-8167.2003.03316.x
Antzelevitch, C., and Dumaine, R. (2002). “Section 2: Electrical heterogeneity in the heart: physiological, pharmacological and clinical implications,” in Handbook of Physiology. The Cardiovascular System. The Heart. Vol. I, ed P. F. Solaro (Bethesda, MD: Am. Physiol. Soc.), 654–692.
Apkon, M., and Nerbonne, J. M. (1991). Characterization of two distinct depolarization-activated K+ currents in isolated adult rat ventricular myocytes. J. Gen. Physiol. 97, 973–1011. doi: 10.1085/jgp.97.5.973
Araki, E., Lipes, M. A., Patti, M. E., Brüning, J. C., Haag, B., Johnson, R. S., et al. (1994). Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372, 186–190. doi: 10.1038/372186a0
Ballou, L. M., Lin, R. Z., and Cohen, I. S. (2015). Control of cardiac repolarization by phosphoinositide 3-kinase signaling to ion channels. Circ. Res. 116, 127–137. doi: 10.1161/CIRCRESAHA.116.303975
Baron, A. D. (2002). Insulin resistance and vascular function. J. Diabetes Complications 16, 92–102. doi: 10.1016/S1056-8727(01)00209-4
Baron, A. D., Brechtel, G., Johnson, A., Fineberg, N., Henry, D. P., and Steinberg, H. O. (1994). Interactions between insulin and norepinephrine on blood pressure and insulin sensitivity. Studies in lean and obese men. J. Clin. Invest. 93, 2453–2462. doi: 10.1172/JCI117254
Baron, A. D., Brechtel-Hook, G., Johnson, A., and Hardin, D. (1993). Skeletal muscle blood flow. A possible link between insulin resistance and blood pressure. Hypertension 21, 129–135. doi: 10.1161/01.HYP.21.2.129
Baron, A. D., Laakso, M., Brechtel, G., Hoit, B., Watt, C., and Edelman, S. V. (1990). Reduced postprandial skeletal muscle blood flow contributes to glucose intolerance in human obesity. J. Clin. Endocrinol. Metab. 70, 1525–1533. doi: 10.1210/jcem-70-6-1525
Barry, D. M., and Nerbonne, J. M. (1996). Myocardial potassium channels: electrophysiological and molecular diversity. Annu. Rev. Physiol. 58, 363–394. doi: 10.1146/annurev.ph.58.030196.002051
Barry, D. M., Xu, H., Schuessler, R. B., and Nerbonne, J. M. (1998). Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 α subunit. Circ. Res. 83, 560–567. doi: 10.1161/01.RES.83.5.560
Belke, D., Betuing, S., Tuttle, M., Graveleau, C., Young, M., Abel, E., et al. (2002). Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Invest. 109, 629–639. doi: 10.1172/JCI0213946
Benndorf, K., and Nilius, B. (1998). Properties of an early outward current in single cells of the mouse ventricle. Gen. Physiol. Biophys. 7, 449–466.
Bers, D. M., and Perez-Reyes, E. (1999). Ca2+ channels in cardiac myocytes: structure and function in Ca2+ influx and intracellular Ca2+ release. Cardiovasc. Res. 42, 339–360. doi: 10.1016/S0008-6363(99)00038-3
Bertoni, A. G., Tsai, A., Kasper, E. K., and Brancati, F. L. (2003). Diabetes and idiopathic cardiomyopathy: a nationwide case-control study. Diabetes Care 26, 2791–2795. doi: 10.2337/diacare.26.10.2791
Bertrand, L., Horman, S., Beauloye, C., and Vanoverschelde, J. L. (2008). Insulin signalling in the heart. Cardiovasc. Res. 79, 238–248. doi: 10.1093/cvr/cvn093
Bethel, M. A., Sloan, F. A., Belsky, D., and Feinglos, M. N. (2007). Longitudinal incidence and prevalence of adverse outcomes of diabetes mellitus in elderly patients. Arch. Intern. Med. 167, 921–927. doi: 10.1001/archinte.167.9.921
Böhni, R., Riesgo-Escovar, J., Oldham, S., Brogiolo, W., Stocker, H., Andruss, B. F., et al. (1999). Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 97, 865–875. doi: 10.1016/S0092-8674(00)80799-0
Bou-Abboud, E., and Nerbonne, J. M. (1999). Molecular correlates of the calcium-independent, depolarization-activated K+ currents in rat atrial myocytes. J. Physiol. 517, 407–420. doi: 10.1111/j.1469-7793.1999.0407t.x
Boudina, S., and Abel, E. D. (2007). Diabetic cardiomyopathy revisited. Circulation 115, 3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597
Boudina, S., and Abel, E. D. (2010). Diabetic Cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 11, 31–39. doi: 10.1007/s11154-010-9131-7
Boudina, S., Bugger, H., Sena, S., O'Neill, B. T., Zaha, V. G., Ilkun, O., et al. (2009). Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 19, 1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101
Boyle, W. A., and Nerbonne, J. M. (1992). Two functionally distinct 4-amino-pyridine-sensitive outward K+ currents in rat atrial myocytes. J. Gen. Physiol. 100, 1041–1067. doi: 10.1085/jgp.100.6.1041
Brahmajothi, M. V., Campbell, D. L., Rasmusson, R. L., Morales, M. J., Trimmer, J. S., and Nerbonne, J. M. (1999). Distinct transient outward potassium current (Ito) phenotypes and distribution of fast-inactivating potassium channel α-subunits in ferret left ventricular myocytes. J. Gen. Physiol. 113, 581–600. doi: 10.1085/jgp.113.4.581
Brown, D. W., Giles, W. H., Greenlund, K. J., Valdez, R., and Croft, J. B. (2001). Impaired fasting glucose, diabetes mellitus, and cardiovascular disease risk factors are associated with prolonged QTc duration. Results from the third national health and nutrition examination survey. J. Cardiovasc. Risk 8, 227–233. doi: 10.1177/174182670100800407
Brunet, S., Aimond, F., Li, H., Guo, W., Eldstrom, J., Nerbonne, J. M., et al. (2004). Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J. Physiol. 559, 103–120. doi: 10.1113/jphysiol.2004.063347
Brüning, J. C., Michael, M. D., Winnay, J. N., Hayashi, T., Hörsch, D., Accili, D., et al. (1998). A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569. doi: 10.1016/S1097-2765(00)80155-0
Buchanan, J., Mazumder, P., Hu, P., Chakrabarti, G., Roberts, M., Abel, E. D., et al. (2005). Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146, 5341–5349. doi: 10.1210/en.2005-0938
Bugger, H., and Abel, E. D. (2009). Rodent models of diabetic cardiomyopathy. Dis. Models Mech. 2, 454–466. doi: 10.1242/dmm.001941
Cai, S., Khoo, J., Mussa, S., Alp, N. J., and Channon, K. M. (2005). Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia 48, 1933–1940. doi: 10.1007/s00125-005-1857-5
Calloe, K., Soltysinska, E., Jespersen, T., Lundby, A., Antzelevitch, C., Olesen, S. P., et al. (2010). Differential effects of the transient outward K+ current activator NS5806 in the canine left ventricle. J. Mol. Cell. Cardiol. 48, 191–200. doi: 10.1016/j.yjmcc.2009.07.017
Cariappa, R., Heath-Monnig, E., Furesz, T. C., Kamath, S. G., and Smith, C. H. (2002). Stable polarized expression of hCAT-1 in an epithelial cell line. J. Membr. Biol. 186, 23–30. doi: 10.1007/s00232-001-0133-y
Casanello, P., Escudero, C., and Sobrevia, L. (2007). Equilibrative nucleoside (ENTs) and cationic amino acid (CATs) transporters: implications in foetal endothelial dysfunction in human pregnancy diseases. Curr. Vasc. Pharmacol. 5, 69–84. doi: 10.2174/157016107779317198
Casis, O., and Echevarria, E. (2004). Diabetic cardiomyopathy: electromechanical cellular alterations. Curr. Vasc. Pharmacol. 2, 237–248. doi: 10.2174/1570161043385655
Casis, O., Gallego, M., Iriarte, M., and Sánchez-Chapula, J. A. (2000). Effects of diabetic cardiomyopathy on regional electrophysiologic characteristics of rat ventricle. Diabetología 43, 101–109. doi: 10.1007/s001250050013
Cesario, D. A., Brar, R., and Shivkumar, K. (2006). Alterations in ion channel physiology in diabetic cardiomyopathy. Endocrinol. Metab. Clin. North. Am. 35, 601–610. doi: 10.1016/j.ecl.2006.05.002
Chatham, J. C. (2001). The impact of type 2 diabetes on the regulation of cardiac energy metabolism and function. J. Mol. Cell. Cardiol. 33, A153. doi: 10.1016/S0022-2828(01)90587-5
Chen, V., Ianuzzo, C. D., Fong, B. C., and Spitzer, J. J. (1984). The effects of acute and chronic diabetes on myocardial metabolism in rats. Diabetes 33, 1078–1084. doi: 10.2337/diab.33.11.1078
Closs, E. I., Boissel, J. P., Habermeier, A., and Rotmann, A. (2006). Structure and function of cationic amino acid transporters (CATs). J. Membr. Biol. 213, 67–77. doi: 10.1007/s00232-006-0875-7
Closs, E. I., Gräf, P., Habermeier, A., Cunningham, J. M., and Förstermann, U. (1997). Human cationic amino acid transporters hCAT-1, hCAT-2A, and hCAT-2B: three related carriers with distinct transport properties. Biochemistry 36, 6462–6468. doi: 10.1021/bi962829p
Comer, M. B., Campbell, D. L., Rasmusson, R. L., Lamson, D. R., Morales, M. J., and Zhang, Y. (1994). Cloning and characterization of an Ito-like potassium channel from ferret ventricle. Am. J. Physiol. 267, H1383–H1395.
Copps, K. D., and White, M. F. (2012). Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 55, 2565–2582. doi: 10.1007/s00125-012-2644-8
Cosentino, F., Eto, M., De Paolis, P., van der Loo, B., Bachschmid, M., Ullrich, V., et al. (2003). High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation 107, 1017–1023. doi: 10.1161/01.CIR.0000051367.92927.07
Delisle, B. P., Anson, B. D., Rajamani, S., and January, C. T. (2004). Biology of cardiac arrhythmias: ion channel protein trafficking. Circ. Res. 94, 1418–1428. doi: 10.1161/01.RES.0000128561.28701.ea
Delmar, M. (1992). Role of potassium currents on cell excitability in cardiac ventricular myocytes. J. Cardiovasc. Electrophysiol. 3, 474–486. doi: 10.1111/j.1540-8167.1992.tb00990.x
Delmar, M. (2004). Role of potassium currents on cell excitability in cardiac ventricular myocytes. J. Cardiovasc. Electrophysiol. 3, 474–486. doi: 10.1111/j.1540-8167.1992.tb00990.x
Devés, R., and Boyd, C. A. (1998). Transporters for cationic amino acids in animal cells: discovery, structure, and function. Physiol. Rev. 78, 487–545.
Dixon, J. E., Shi, W., Wang, H. S., McDonald, C., Yu, H., and Wymore, R. S. (1996). Role of the Kv4.3, K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ. Res. 79, 659–668. doi: 10.1161/01.RES.79.4.659
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77. doi: 10.1126/science.280.5360.69
Fang, Z. Y., Prins, J. B., and Marwick, T. H. (2004). Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr. Rev. 25, 543–567. doi: 10.1210/er.2003-0012
Feng, J., Wible, B., Li, G. R., Wang, Z., and Nattle, S. (1997). Antisense oligodeoxynucleotides directed against Kv1.5 mRNA specifically inhibit ultrarapid delayed rectifier K+ current in cultured adult human atrial myocytes. Circ. Res. 80, 572–579. doi: 10.1161/01.RES.80.4.572
Fermini, B., Wang, Z., Duan, D., and Nattel, S. (1992). Differences in rate dependence of transient outward current in rabbit and human atrium. Am. J. Physiol. 263, H1747–H1754.
Festa, A., D'Agostino, R. Jr., Rautaharju, P., Mykkänen, L., and Haffner, S. M. (2000). Relation of systemic blood pressure, left ventricular mass, insulin sensitivity, and coronary artery disease to QT interval duration in nondiabetic and type 2 diabetic subjects. Am. J. Cardiol. 86, 1117–1122. doi: 10.1016/S0002-9149(00)01170-X
Fiset, C., Clark, R. B., Shimoni, Y., and Giles, W. R. (1997). Shal-type channels contribute to the Ca2+-independent transient outward K+ current in rat ventricle. J. Physiol. 500, 51–64. doi: 10.1113/jphysiol.1997.sp021998
Fozzard, H. A. (2002). Cardiac sodium and calcium channels: a history of excitatory currents. Cardiovasc. Res. 55, 1–8. doi: 10.1016/S0008-6363(02)00407-8
Frank-Hansen, R., Larsen, L. A., Andersen, P., Jespersgaard, C., and Christiansen, M. (2005). Mutations in the genes KCND2 and KCND3 encoding the ion channels Kv4.2 and Kv4.3, conducting the cardiac fast transient outward current (Ito, f), are not a frequent cause of long QT syndrome. Clin. Chim. Acta 351, 95–100. doi: 10.1016/j.cccn.2004.08.017
Furfine, E. S., Harmon, M. F., Paith, J. E., Knowles, R. G., Salter, M., Kiff, R. J., et al. (1994). Potent and selective inhibition of human nitric oxide synthases. Selective inhibition of neuronal nitric oxide synthase by S-methyl-L-thiocitrulline and S-ethyl-L-thiocitrulline. J. Biol. Chem. 269, 26677–26683.
Furukawa, T., Myerburg, R. J., Furukawa, N., Bassett, A. L., and Kimura, S. (1990). Differences in transient outward currents of feline endocardial and epicardial myocytes. Circ. Res. 67, 1287–1291. doi: 10.1161/01.RES.67.5.1287
Gao, X., Zhang, H., Schmidt, A. M., and Zhang, C. (2008). AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 295, H491–H498. doi: 10.1152/ajpheart.00464.2008
González, M., Flores, C., Pearson, J. D., Casanello, P., and Sobrevia, L. (2004). Cell signalling-mediating insulin increase of mRNA expression for cationic amino acid transporters-1 and -2 and membrane hyperpolarization in human umbilical vein endothelial cells. Pflugers Arch. 448, 383–394. doi: 10.1007/s00424-004-1261-x
González, M., Gallardo, V., Rodríguez, N., Salomón, C., Westermeier, F., Guzmán-Gutiérrez, E., et al. (2011). Insulin-stimulated L-arginine transport requires SLC7A1 gene expression and is associated with human umbilical vein relaxation. J. Cell. Physiol. 226, 2916–2924. doi: 10.1002/jcp.22635
González, M., Rojas, S., Avila, P., Cabrera, L., Villalobos, R., Palma, C., et al. (2015). Insulin reverses D-glucose-increased nitric oxide and reactive oxygen species generation in human umbilical vein endothelial cells. PLoS ONE 10:e0122398. doi: 10.1371/journal.pone.0122398
Gregg, E., Li, Y., Wang, J., Rios, N., Ali, M., Rolka, D., et al. (2014). Changes in Diabetes-Related Complications in the United States, 1990–2010. N. Engl. J. Med. 370, 1514–1523. doi: 10.1056/NEJMoa1310799
Grubb, S., Calloe, K., and Thomsen, M. B. (2012). Impact of KChIP2 on cardiac electrophysiology and the progression of heart failure. Front. Physiol. 3:118. doi: 10.3389/fphys.2012.00118
Guo, L., Tian, S., Chen, Y., Mao, Y., Cui, S., Hu, A., et al. (2015). CAT-1 as a novel CAM stabilizes endothelial integrity and mediates the protective actions of l-Arg via a NO-independent mechanism. J. Mol. Cell. Cardiol. 87, 180–191. doi: 10.1016/j.yjmcc.2015.08.011
Guo, S. (2014). Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J. Endocrinol. 220, T1–T23. doi: 10.1530/JOE-13-0327
Guo, W., Jung, W. E., Marionneau, C., Aimond, F., Xu, H., Yamada, K. A., et al. (2005). Targeted deletion of Kv4.2 eliminates Ito,f and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ. Res. 97, 1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa
Guo, W., Li, H., Aimond, F., Johns, D. C., Rhodes, K. J., Nerbonne, J. M., et al. (2002). Role of heteromultimers in the generation of myocardial transient outward K+ currents. Circ. Res. 90, 586–593. doi: 10.1161/01.RES.0000012664.05949.E0
Guo, W., Li, H., London, B., and Nerbonne, J. M. (2000). Functional consequences of elimination of Ito,f and Ito,s: early afterdepolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 α subunit. Circ. Res. 87, 73–79. doi: 10.1161/01.RES.87.1.73
Guo, W., Xu, H., London, B., and Nerbonne, J. M. (1999). Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J. Physiol. 521, 587–599. doi: 10.1111/j.1469-7793.1999.00587.x
Guzmán-Gutiérrez, E., Westermeier, F., Salomón, C., González, M., Pardo, F., Leiva, A., et al. (2012). Insulin-increased L-arginine transport requires A(2A) adenosine receptors activation in human umbilical vein endothelium. PLoS ONE 7:e41705. doi: 10.1371/journal.pone.0041705
Hiraoka, M., and Kawano, S. (1989). Calcium-sensitive and insensitive transient outward current in rabbit ventricular myocytes. J. Physiol. 410, 187–212. doi: 10.1113/jphysiol.1989.sp017528
Howarth, F., Jacobson, M., Qureshi, M., Shafiullah, M., Hameed, R., Zilahi, E., et al. (2009). Altered gene expression may underlie prolonged duration of the QT interval and ventricular action potential in streptozotocin-induced diabetic rat heart. Mol. Cell. Biochem. 328, 57–65. doi: 10.1007/s11010-009-0074-9
Huang, E. S., Laiteerapong, N., Liu, J. Y., John, P. M., Moffet, H. H., and Karter, A. J. (2014). Rates of complications and mortality in older patients with diabetes mellitus: the diabetes and aging study. JAMA Intern. Med. 174, 251–258. doi: 10.1001/jamainternmed.2013.12956
International Diabetes Federation (IFD) (2013). Diabetes Atlas, 6th Edn. Available online at: www.idf.org/diabetesatlas
Johns, D. C., Nuss, H. B., and Marban, E. (1997). Suppression of neuronal and cardiac transient outward currents by viral gene transfer of dominant-negative Kv4.2 constructs. J. Biol. Chem. 272, 31598–31603. doi: 10.1074/jbc.272.50.31598
Kääb, S., Dixon, J., Duc, J., Ashen, D., Näbauer, M., Beuckelmann, D. J., et al. (1998). Molecular basis of transient outward potassium current downregulation in human heart failure. A decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98, 1383–1393. doi: 10.1161/01.CIR.98.14.1383
Kannel, W. B., and McGee, D. L. (1979). Diabetes and cardiovascular disease: the Framingham study. JAMA 241, 2035–2038. doi: 10.1001/jama.1979.03290450033020
Kanno, S., and Saffitz, J. E. (2001). The role of myocardial gap junctions in electrical conduction and arrhythmogenesis. Cardiovasc. Pathol. 10, 169–177. doi: 10.1016/S1054-8807(01)00078-3
Karbach, S., Simon, A., Slenzka, A., Jaenecke, I., Habermeier, A., Martiné, U., et al. (2011). Relative contribution of different l-arginine sources to the substrate supply of endothelial nitric oxide synthase. J. Mol. Cell. Cardiol. 51, 855–861. doi: 10.1016/j.yjmcc.2011.07.024
Kaye, D. M., Parnell, M. M., and Ahlers, B. A. (2002). Reduced myocardial and systemic L-arginine uptake in heart failure. Circ. Res. 91, 1198–1203. doi: 10.1161/01.RES.0000047506.52381.90
Kim, Y. J., Park, H. S., Lee, H. Y., Ha, E. H., Suh, S. H., and Oh, S. K. (2006). Reduced L-arginine level and decreased placental eNOS activity in preeclampsia. Placenta 27, 438–444. doi: 10.1016/j.placenta.2005.04.011
Kizhatil, K., and Albritton, L. M. (2002). System y+ localizes to different membrane subdomains in the basolateral plasma membrane of epithelial cells. Am. J. Physiol. Cell. Physiol. 283, C1784–C1794. doi: 10.1152/ajpcell.00061.2002
Kléber, A. G., and Rudy, Y. (2004). Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 84, 431–488. doi: 10.1152/physrev.00025.2003
Knowles, R. G., and Moncada, S. (1994). Nitric oxide synthases in mammals. Biochem. J. 298, 249–258. doi: 10.1042/bj2980249
Kohlhaas, C. F., Morrow, V. A., Jhakra, N., Patil, V., Connell, J. M., Petrie, J. R., et al. (2011). Insulin rapidly stimulates L-arginine transport in human aortic endothelial cells via Akt. Biochem. Biophys. Res. Commun. 412, 747–751. doi: 10.1016/j.bbrc.2011.08.048
Kolter, T., Uphues, I., and Eckel, J. (1997). Molecular analysis of insulin resistance in isolated ventricular cardiomyocytes of obese Zucker rats. Am. J. Physiol. 273, E59–E67.
Kong, W., Po, S., Yamagishi, T., Ashen, M. D., Stetten, G., and Tomaselli, G. F. (1998). Isolation and characterization of the human gene encoding Ito: further diversity by alternative mRNA splicing. Am. J. Physiol. 275, H1963–H1970.
Kuo, H. C., Cheng, C. F., Clark, R. B., Lin, J. J., Lin, J. L., Hoshijima, M., et al. (2001). A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of Ito and confers susceptibility to ventricular tachycardia. Cell 107, 801–813. doi: 10.1016/S0092-8674(01)00588-8
Lazar, D. F., Wiese, R. J., Brady, M. J., Mastick, C. C., Waters, S. B., Saltiel, A. R., et al. (1995). Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J. Biol. Chem. 270, 20801–20807. doi: 10.1074/jbc.270.35.20801
Lengyel, C., Virág, L., Bíró, T., Jost, N., Magyar, J., Biliczki, P., et al. (2007). Diabetes mellitus attenuates the repolarization reserve in mammalian heart. Cardiovasc. Res. 73, 512–520. doi: 10.1016/j.cardiores.2006.11.010
Lesniewski, L. A., Donato, A. J., Behnke, B. J., Woodman, C. R., Laughlin, M. H., Ray, C. A., et al. (2008). Decreased NO signaling leads to enhanced vasoconstrictor responsiveness in skeletal muscle arterioles of the ZDF rat prior to overt diabetes and hypertension. Am. J. Physiol. Heart Circ. Physiol. 294, H1840–H1850. doi: 10.1152/ajpheart.00692.2007
Litovsky, S. H., and Antzelevitch, C. (1998). Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ. Res. 62, 116–126. doi: 10.1161/01.RES.62.1.116
Liu, J. E., Palmieri, V., Roman, M. J., Bella, J. N., Fabsitz, R., Howard, B. V., et al. (2001). The impact of diabetes on left ventricular filling pattern in normotensive and hypertensive adults: the Strong Heart Study. J. Am. Coll. Cardiol. 37, 1943–1949. doi: 10.1016/S0735-1097(01)01230-X
Liu, W. J., Wang, H. T., Chen, W. W., Deng, J. X., Jiang, Y., and Liu, J. (2008). Co-expression of KCNE2 and KChIP2c modulates the electrophysiological properties of Kv4.2 current in COS-7 cells. Acta Pharmacol. Sin. 29, 653–660. doi: 10.1111/j.1745-7254.2008.00804.x
London, B., Wang, D. W., Hill, J. A., and Bennett, P. B. (1998). The transient outward current in mice lacking the potassium channel gene Kv1.4. J. Physiol. 509, 171–182. doi: 10.1111/j.1469-7793.1998.171bo.x
Lopez-Izquierdo, A., Pereira, R. O., Wende, A. R., Punske, B. B., Abel, E. D., and Tristani-Firouzi, M. (2014). The absence of insulin signaling in the heart induces changes in potassium channel expression and ventricular repolarization. Am. J. Physiol. Heart Circ. Physiol. 306, H747–H754. doi: 10.1152/ajpheart.00849.2013
Lu, X., and Silver, J. (2000). Ecotropic murine leukemia virus receptor is physically associated with caveolin and membrane rafts. Virology 276, 251–258. doi: 10.1006/viro.2000.0555
Luo, C. H., and Rudy, Y. (1994a). A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74, 1071–1096. doi: 10.1161/01.RES.74.6.1071
Luo, C. H., and Rudy, Y. (1994b). A dynamic model of the cardiac ventricular action potential. II. After depolarizations, triggered activity, and potentiation. Circ. Res. 74, 1097–1113. doi: 10.1161/01.RES.74.6.1097
MacKinnon, R. (1991). Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature 350, 232–235. doi: 10.1038/350232a0
Magyar, J., Rusznák, Z., Szentesi, P., Szûcs, G., and Kovács, L. (1992). Action potentials and potassium currents in rat ventricular muscle during experimental diabetes. J. Mol. Cell. Cardiol. 24, 841–853. doi: 10.1016/0022-2828(92)91098-P
Mandavia, C. H., Aroor, A. R., Demarco, V. G., and Sowers, J. R. (2013). Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci. 92, 601–608. doi: 10.1016/j.lfs.2012.10.028
Mann, G. E., Yudilevich, D. L., and Sobrevia, L. (2003). Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol. Rev. 83, 183–252. doi: 10.1152/physrev.00022.2002
Masuda, M., Kakushima, N., Wilt, S. G., Ruscetti, S. K., Hoffman, P. M., and Iwamoto, A. (1999). Analysis of receptor usage by ecotropic murine retroviruses, using green fluorescent protein-tagged cationic amino acid transporters. J. Virol. 73, 8623–8629.
Mazumder, P. K., O'Neill, B. T., Roberts, M. W., Buchanan, J., Boudina, S., Abel, E. D., et al. (2004). Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53, 2366–2374. doi: 10.2337/diabetes.53.9.2366
McCrossan, Z. A., and Abbott, G. W. (2004). The MinK-related peptides. Neuropharmacology 47, 787–821. doi: 10.1016/j.neuropharm.2004.06.018
McDonald, K. K., Zharikov, S., Block, E. R., and Kilberg, M. S. (1997). A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “arginine paradox”. J. Biol. Chem. 272, 31213–31216. doi: 10.1074/jbc.272.50.31213
McKeown, L., Swanton, L., Robinson, P., and Jones, O. T. (2008). Surface expression and distribution of voltage-gated potassium channels in neurons. Mol. Membr. Biol. 25, 332–343. doi: 10.1080/09687680801992470
Muniyappa, R., Montagnani, M., Koh, K. K., and Quon, M. J. (2007). Cardiovascular actions of insulin. Endocr. Rev. 28, 463–491. doi: 10.1210/er.2007-0006
Munk, A. A., Adjemian, R. A., Zhao, J., Ogbagherriel, A., and Shrier, A. (1996). Electrophysiological properties of morphologically distinct cells isolated from the rabbit atrio-ventricular node. J. Physiol. 493, 801–818. doi: 10.1113/jphysiol.1996.sp021424
Muñoz, G., San Martín, R., Farías, M., Cea, L., Vecchiola, A., Casanello, P., et al. (2006). Insulin restores glucose inhibition of adenosine transport by increasing the expression and activity of the equilibrative nucleoside transporter 2 in human umbilical vein endothelium. J. Cell. Physiol. 209, 826–835. doi: 10.1002/jcp.20769
Nerbonne, J. M. (2000). Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J. Physiol. 525, 285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x
Nerbonne, J. M., and Guo, W. (2002). Heterogeneous expression of voltage-gated potassium channels in the heart: roles in normal excitation and arrhythmias. J. Cardiovasc. Electrophysiol. 13, 406–409. doi: 10.1046/j.1540-8167.2002.00406.x
Nerbonne, J. M., and Kass, R. S. (2005). Physiology and molecular biology of ion channels contributing to ventricular repolarization. Physiol. Rev. 85, 1205–1253. doi: 10.1152/physrev.00002.2005
Nielsen, N. H., Winkel, B. G., Kanters, J. K., Schmitt, N., Hofman-Bang, J., Jensen, H. S., et al. (2007). Mutations in the Kv1.5 channel gene KCNA5 in cardiac arrest patients. Biochem. Biophys. Res. Commun. 354, 776–782. doi: 10.1016/j.bbrc.2007.01.048
Nishiyama, A., Ishii, D. N., Backx, P. H., Pulford, B. E., Birks, B. R., and Tamkun, M. M. (2001). Altered K+ channel gene expression in diabetic rat ventricle: isoform switching between Kv42 and Kv14. Am. J. Physiol. Heart Circ. Physiol. 281, H1800–H1807. Available online at: http://ajpheart.physiology.org/content/281/4/H1800
Niwa, N., and Nerbonne, J. M. (2010). Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J. Mol. Cell. Cardiol. 48, 12–25. doi: 10.1016/j.yjmcc.2009.07.013
Nystrom, F. H., and Quon, M. J. (1999). Insulin signalling: metabolic pathways and mechanisms for specificity. Cell Signal. 11, 563–574. doi: 10.1016/S0898-6568(99)00025-X
Palacín, M., Estévez, R., Bertran, J., and Zorzano, A. (1998). Molecular biology of mammalian plasma membrane amino acid transporters. Physiol. Rev. 78, 969–1054.
Palmer, R. M., Rees, D. D., Ashton, D. S., and Moncada, S. (1988). L-arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 153, 1251–1256. doi: 10.1016/S0006-291X(88)81362-7
Patel, S. P., and Campbell, D. L. (2005). Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J. Physiol. 569, 7–39. doi: 10.1113/jphysiol.2005.086223
Patti, M., and Kahn, C. (1998). The insulin receptor: a critical link in glucose homeostasis and insulin action. J. Basic Clin. Physiol. Pharmacol. 9, 89–109. doi: 10.1515/JBCPP.1998.9.2-4.89
Perez-Reyes, E. (2002). Molecular physiology of low voltage-activated T-type calcium channels. Physiol. Rev. 83, 117–161. doi: 10.1152/physrev.00018.2002
Po, S., Snyders, D. J., Baker, R., Tamkun, M. M., and Bennett, P. B. (1992). Functional expression of an inactivating potassium channel cloned from human heart. Circ. Res. 71, 732–736. doi: 10.1161/01.RES.71.3.732
Poornima, I. G., Parikh, P., and Shannon, R. P. (2006). Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ. Res. 98, 596–605. doi: 10.1161/01.RES.0000207406.94146.c2
Qin, D., Huang, B., Deng, L., El-Adawi, H., Ganguly, K., Sowers, J. R., et al. (2001). Downregulation of K+ channel genes expression in type I diabetic cardiomyopathy. Biochem. Biophys. Res. Commun. 283, 549–553. doi: 10.1006/bbrc.2001.4825
Quijano, C., Castro, L., Peluffo, G., Valez, V., and Radi, R. (2007). Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: direct measurements and formation of hydrogen peroxide and peroxynitrite. Am. J. Physiol. Heart Circ. Physiol. 293, H3404–H3414. doi: 10.1152/ajpheart.00761.2007
Rajapakse, N. W., Chong, A. L., Zhang, W. Z., and Kaye, D. M. (2013). Insulin-mediated activation of the L-arginine nitric oxide pathway in man, and its impairment in diabetes. PLoS ONE 8:e61840. doi: 10.1371/journal.pone.0061840
Rask-Madsen, C., and King, G. L. (2007). Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat. Clin. Pract. Endocrinol. Metab. 3, 46–56. doi: 10.1038/ncpendmet0366
Roberds, S. L., and Tamkun, M. M. (1991). Cloning and tissue-specific expression of five voltage-gated potassium channel cDNAs expressed in rat heart. Proc. Natl. Acad. Sci. U.S.A. 88, 1798–1802. doi: 10.1073/pnas.88.5.1798
Roepke, T. K., Kontogeorgis, A., Ovanez, C., Xu, X., Young, J. B., Purtell, K., et al. (2008). Targeted deletion of kcne2 impairs ventricular repolarization via disruption of IK, slow1 and Ito,f. FASEB J. 22, 3648–3660. doi: 10.1096/fj.08-110171
Rozanski, G. J., and Xu, Z. (2002). A metabolic mechanism for cardiac K+ channel remodelling. Clin. Exp. Pharmacol. Physiol. 29, 132–137. doi: 10.1046/j.1440-1681.2002.03618.x
Rozanski, G. J., Xu, Z., Zhang, K., and Patel, K. P. (1998). Altered K+ current of ventricular myocytes in rats with chronic myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 274, H259–H265.
Rubler, S., Dlugash, J., Yuceoglu, Y. Z., Kumral, T., Branwood, A. W., and Grishman, A. (1972). New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 30, 595–602. doi: 10.1016/0002-9149(72)90595-4
Saltiel, A. R., and Kahn, C. R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806. doi: 10.1038/414799a
San Martín, R., and Sobrevia, L. (2006). Gestational diabetes and the adenosine/L-arginine/nitric oxide (ALANO) pathway in human umbilical vein endothelium. Placenta 27, 1–10. doi: 10.1016/j.placenta.2005.01.011
Schinzari, F., Iantorno, M., Campia, U., Mores, N., Rovella, V., Tesauro, M., et al. (2015). Vasodilator responses and endothelin-dependent vasoconstriction in metabolically healthy obesity and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 309, E787–E792. doi: 10.1152/ajpendo.00278.2015
Schlaich, M. P., Parnell, M. M., Ahlers, B. A., Finch, S., Marshall, T., Zhang, W. Z., et al. (2004). Impaired L-arginine transport and endothelial function in hypertensive and genetically predisposed normotensive subjects. Circulation 110, 3680–3686. doi: 10.1161/01.CIR.0000149748.79945.52
Schultze, S. M., Hemmings, B. A., Niessen, M., and Tschopp, O. (2012). PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert. Rev. Mol. Med. 14, e1. doi: 10.1017/S1462399411002109
Sessa, W. C. (1994). The nitric oxide synthase family of proteins. J. Vasc. Res. 31, 131–143. doi: 10.1159/000159039
Shi, Y., and Vanhoutte, P. M. (2008). Oxidative stress and COX cause hyper-responsiveness in vascular smooth muscle of the femoral artery from diabetic rats. Br. J. Pharmacol. 154, 639–651. doi: 10.1038/bjp.2008.110
Shimoni, Y. (2001). Inhibition of the formation or action of angiotensin II reverses attenuated K+ currents in type 1 and type 2 diabetes. J. Physiol. 537, 83–92. doi: 10.1111/j.1469-7793.2001.0083k.x
Shimoni, Y., Chuang, M., Abel, E. D., and Severson, D. L. (2004). Gender-dependent attenuation of cardiac potassium currents in type 2 diabetic db/db mice. J. Physiol. 555, 345–354. doi: 10.1113/jphysiol.2003.055590
Shimoni, Y., Ewart, H. S., and Severson, D. (1998). Type I and II models of diabetes produce different modifications of K+ currents in rat heart: role of insulin. J. Physiol. 507, 485–496. doi: 10.1111/j.1469-7793.1998.485bt.x
Shimoni, Y., Ewart, H. S., and Severson, D. (1999). Insulin stimulation of rat ventricular K+ currents depends on the integrity of the cytoskeleton. J. Physiol. 514, 735–745. doi: 10.1111/j.1469-7793.1999.735ad.x
Shimoni, Y., Firek, L., Severson, D., and Giles, W. (1994). Short-term diabetes alters K+ currents in rat ventricular myocytes. Circ. Res. 74, 620–628. doi: 10.1161/01.RES.74.4.620
Shin, S., Mohan, S., and Fung, H. L. (2011). Intracellular L-arginine concentration does not determine NO production in endothelial cells: implications on the “L-arginine paradox”. Biochem. Biophys. Res. Commun. 414, 660–663. doi: 10.1016/j.bbrc.2011.09.112
Shioi, T., Kang, P. M., Douglas, P. S., Hampe, J., Yballe, C. M., Lawitts, J., et al. (2000). The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548. doi: 10.1093/emboj/19.11.2537
Simon, A., Karbach, S., Habermeier, A., and Closs, E. I. (2013). Decoding the substrate supply to human neuronal nitric oxide synthase. PLoS ONE 8:e67707. doi: 10.1371/journal.pone.0067707
Snyders, D. J. (1999). Structure and function of cardiac potassium channels. Cardiovasc. Res. 42, 377–390. doi: 10.1016/S0008-6363(99)00071-1
Snyders, D. J., Tamkun, M. M., and Bennett, P. B. (1993). A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. J. Gen. Physiol. 101, 513–543. doi: 10.1085/jgp.101.4.513
Sobrevia, L., and González, M. (2009). A role for insulin on L-arginine transport in fetal endothelial dysfunction in hyperglycaemia. Curr. Vasc. Pharmacol. 7, 467–474. doi: 10.2174/157016109789043919
Sobrevia, L., Nadal, A., Yudilevich, D. L., and Mann, G. E. (1996). Activation of L-arginine transport (system y+) and nitric oxide synthase by elevated glucose and insulin in human endothelial cells. J. Physiol. 490, 775–781. doi: 10.1113/jphysiol.1996.sp021185
Sobrevia, L., Yudilevich, D. L., and Mann, G. E. (1997). Activation of A2-purinoceptors by adenosine stimulates L-arginine transport (system y+) and nitric oxide synthesis in human fetal endothelial cells. J. Physiol. 499, 135–140. doi: 10.1113/jphysiol.1997.sp021916
Sridhar, A., da Cunha, D. N., Lacombe, V. A., Zhou, Q., Fox, J. J., Hamlin, R. L., et al. (2007). The plateau outward current in canine ventricle, sensitive to 4-aminopyridine, is a constitutive contributor to ventricular repolarization. Br. J. Pharmacol. 152, 870–879. doi: 10.1038/sj.bjp.0707403
Srinivasan, S., Hatley, M. E., Bolick, D. T., Palmer, L. A., Edelstein, D., Brownlee, M., et al. (2004). Hyperglycaemia-induced superoxide production decreases eNOS expression via AP-1 activation in aortic endothelial cells. Diabetologia 47, 1727–1734. doi: 10.1007/s00125-004-1525-1
Steinberg, H. O., and Baron, A. D. (2002). Vascular function, insulin resistance and fatty acids. Diabetologia 45, 623–634. doi: 10.1007/s00125-002-0800-2
Straus, S. M., Kors, J. A., De Bruin, M. L., van der Hooft, C. S., Hofman, A., Heeringa, J., et al. (2006). Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J. Am. Coll. Cardiol. 47, 362–367. doi: 10.1016/j.jacc.2005.08.067
Tamkun, M. M., Knoth, K. M., Walbridge, J. A., Kroemer, H., Roden, D. M., and Glover, D. M. (1991). Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB J. 5, 331–337.
Torres-Jacome, J., Gallego, M., Rodríguez-Robledo, J. M., Sanchez-Chapula, J. A., and Casis, O. (2013). Improvement of the metabolic status recovers cardiac potassium channel synthesis in experimental diabetes. Acta Physiol. (Oxf.) 207, 447–459. doi: 10.1111/apha.12043
Tseng-Crank, J. C., Tseng, G. N., Schwartz, A., and Tanouye, M. A. (1990). Molecular cloning and functional expression of a potassium channel cDNA isolated from a rat cardiac library. FEBS Lett. 268, 63–68. doi: 10.1016/0014-5793(90)80973-M
Ui, M., Okada, T., Hazeki, K., and Hazeki, O. (1995). Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci. 20, 303–307. doi: 10.1016/S0968-0004(00)89056-8
Van Wagoner, D., Pond, A. L., McCarthy, P. M., Trimmer, J. S., and Nerbonne, J. M. (1997). Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 80, 772–781. doi: 10.1161/01.RES.80.6.772
Vanhoutte, P. M. (2003). Endothelial control of vasomotor function: from health to coronary disease. Circ. J. 67, 572–575. doi: 10.1253/circj.67.572
Vanhoutte, P. M., and Tang, E. H. (2008). Endothelium-dependent contractions: when a good guy turns bad! J. Physiol. 586, 5295–5304. doi: 10.1113/jphysiol.2008.161430
Varró, A., Nanasi, P. P., and Lathrop, D. A. (1993). Potassium currents in isolated human atrial and ventricular cardiocytes. Acta Physiol. Scand. 149, 133–142. doi: 10.1111/j.1748-1716.1993.tb09605.x
Vásquez, G., Sanhueza, F., Vásquez, R., González, M., San Martín, R., Casanello, P., et al. (2004). Role of adenosine transport in gestational diabetes-induced L-arginine transport and nitric oxide synthesis in human umbilical vein endothelium. J. Physiol. 560, 111–122. doi: 10.1113/jphysiol.2004.068288
Vásquez, R., Farías, M., Vega, J. L., Martin, R. S., Vecchiola, A., Casanello, P., et al. (2007). D-glucose stimulation of L-arginine transport and nitric oxide synthesis results from activation of mitogen-activated protein kinases p42/44 and Smad2 requiring functional type II TGF-beta receptors in human umbilical vein endothelium. J. Cell. Physiol. 212, 626–632. doi: 10.1002/jcp.21057
Veglio, M., Borra, M., Stevens, L. K., Fuller, J. H., and Perin, P. C. (1999). The relation between QTc interval prolongation and diabetic complications. The EURODIAB IDDM complication study group. Diabetologia 42, 68–75. doi: 10.1007/s001250051115
Veglio, M., Bruno, G., Borra, M., Macchia, G., Bargero, G., D'Errico, N., et al. (2002a). Prevalence of increased QT interval duration and dispersion in type 2 diabetic patients and its relationship with coronary heart disease: a population-based cohort. J. Intern. Med. 251, 317–324. doi: 10.1046/j.1365-2796.2002.00955.x
Veglio, M., Chinaglia, A., and Cavallo-Perin, P. (2004). QT interval, cardiovascular risk factors and risk of death in diabetes. J. Endocrinol. Invest. 27, 175–181. doi: 10.1007/BF03346265
Veglio, M., Giunti, S., Stevens, L. K., Fuller, J. H., and Perin, P. C. (2002b). Prevalence of Q-T interval dispersion in type 1 diabetes and its relation with cardiac ischemia: the EURODIAB IDDM complications study group. Diabetes Care 25, 702–707. doi: 10.2337/diacare.25.4.702
Verrey, F., Closs, E. I., Wagner, C. A., Palacin, M., Endou, H., and Kanai, Y. (2004). CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Arch. 447, 532–542. doi: 10.1007/s00424-003-1086-z
Wang, Z., Feng, J., Shi, H., Pond, A., Nerbonne, J. M., and Nattel, S. (1999). Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circ. Res. 84, 551–561. doi: 10.1161/01.RES.84.5.551
Wang, Z., Fermini, B., and Nattel, S. (1993). Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ. Res. 73, 1061–1076. doi: 10.1161/01.RES.73.6.1061
Wettwer, E., Amos, G., Gath, J., Zerkowski, H. R., Reidemeister, J. C., and Ravens, U. (1993). Transient outward current in human and rat ventricular myocytes. Cardiovasc. Res. 27, 1662–1669. doi: 10.1093/cvr/27.9.1662
White, M. F. (2003). Insulin signaling in health and disease. Science 302, 1710–1711. doi: 10.1126/science.1092952
Wickenden, A. D., Jegla, T. J., Kaprielian, R., and Backx, P. H. (1999). Regional contributions of Kv1.4, Kv4.2, and Kv4.3 to transient outward K+ current in rat ventricle. Am. J. Physiol. 276, H1599–H1607.
Wierzbicki, A. S., Chowienczyk, P. J., Cockcroft, J. R., Brett, S. E., Watts, G. F., Jenkins, B. S., et al. (2004). Cardiovascular risk factors and endothelial dysfunction. Clin. Sci. (Lond.) 107, 609–615. doi: 10.1042/CS20040078
Williams, S. B., Cusco, J. A., Roddy, M. A., Johnstone, M. T., and Creager, M. A. (1996). Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Am. Coll. Cardiol. 27, 567–574. doi: 10.1016/0735-1097(95)00522-6
Wolf, S., Janzen, A., Vékony, N., Martiné, U., Strand, D., and Closs, E. I. (2002). Expression of solute carrier 7A4 (SLC7A4) in the plasma membrane is not sufficient to mediate amino acid transport activity. Biochem. J. 364, 767–775. doi: 10.1042/bj20020084
World Health Organization (WHO) (1998). Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications. Part 1: Diagnosis and Classification of Diabetes Mellitus. Geneva.
Xiong, Y., Fu, Y. F., Fu, S. H., and Zhou, H. H. (2003). Elevated levels of the serum endogenous inhibitor of nitric oxide synthase and metabolic control in rats with streptozotocin-induced diabetes. J. Cardiovasc. Pharmacol. 42, 191–196. doi: 10.1097/00005344-200308000-00006
Xu, H., Guo, W., and Nerbonne, J. M. (1999). Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. J. Gen. Physiol. 113, 661–678. doi: 10.1085/jgp.113.5.661
Xu, Z., Patel, K. P., Lou, M. F., and Rozanski, G. J. (2002). Up-regulation of K(+) channels in diabetic rat ventricular myocytes by insulin and glutathione. Cardiovasc. Res. 53, 80–88. doi: 10.1016/S0008-6363(01)00446-1
Xu, Z., Patel, K. P., and Rozanski, J. G. (1996). Metabolic basis of decreased transient outward K+ current in ventricular myocytes from diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 271, H2190–H2196.
Yang, X., Salas, P. J., Pham, T. V., Wasserlauf, B. J., Smets, M. J., Myerburg, R. J., et al. (2002). Cytoskeletal actin microfilaments and the transient outward potassium current in hypertrophied rat ventriculocytes. J. Physiol. 541, 411–421. doi: 10.1113/jphysiol.2002.019562
Yang, Z., Venardos, K., Jones, E., Morris, B. J., Chin-Dusting, J., and Kaye, D. M. (2007). Identification of a novel polymorphism in the 3′UTR of the L-arginine transporter gene SLC7A1: contribution to hypertension and endothelial dysfunction. Circulation 115, 1269–1274. doi: 10.1161/circulationaha.106.665836
Keywords: insulin, L-arginine, nitric oxide, endothelium, cardiac potassium channels, ventricular repolarization, heart failure, insulin resistance
Citation: Dubó S, Gallegos D, Cabrera L, Sobrevia L, Zúñiga L and González M (2016) Cardiovascular Action of Insulin in Health and Disease: Endothelial L-Arginine Transport and Cardiac Voltage-Dependent Potassium Channels. Front. Physiol. 7:74. doi: 10.3389/fphys.2016.00074
Received: 18 December 2015; Accepted: 15 February 2016;
Published: 15 March 2016.
Edited by:
Agustín Guerrero-Hernández, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Carmen Valenzuela, Instituto de Investigaciones Biomédicas “Alberto Sols” CSIC-UAM, SpainCatherine Brenner Jan, INSERM - University Paris-Sud, France
Copyright © 2016 Dubó, Gallegos, Cabrera, Sobrevia, Zúñiga and González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo González, bWdvbnphbGV6b0B1ZGVjLmNs