 Rosalba Putti
Rosalba Putti Vincenzo Migliaccio
Vincenzo Migliaccio Lillà Lionetti
Lillà Lionetti
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 20 January 2016
Sec. Striated Muscle Physiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00426
This article is part of the Research Topic Mitochondria in Skeletal Muscle Health, Aging and Diseases View all 14 articles
It has been suggested that skeletal muscle mitochondria play a key role in high fat (HF) diet induced insulin resistance (IR). Two opposite views are debated on mechanisms by which mitochondrial function could be involved in skeletal muscle IR. In one theory, mitochondrial dysfunction is suggested to cause intramyocellular lipid accumulation leading to IR. In the second theory, excess fuel within mitochondria in the absence of increased energy demand stimulates mitochondrial oxidant production and emission, ultimately leading to the development of IR. Noteworthy, mitochondrial bioenergetics is strictly associated with the maintenance of normal mitochondrial morphology by maintaining the balance between the fusion and fission processes. A shift toward mitochondrial fission with reduction of fusion protein, mainly mitofusin 2, has been associated with reduced insulin sensitivity and inflammation in obesity and IR development. However, dietary fat source during chronic overfeeding differently affects mitochondrial morphology. Saturated fatty acids induce skeletal muscle IR and inflammation associated with fission phenotype, whereas ω-3 polyunsaturated fatty acids improve skeletal muscle insulin sensitivity and inflammation, associated with a shift toward mitochondrial fusion phenotype. The present minireview focuses on mitochondrial bioenergetics and morphology in skeletal muscle IR, with particular attention to the effect of different dietary fat sources on skeletal muscle mitochondria morphology and fusion/fission balance.
Skeletal muscle seems to play a central role in whole body insulin resistance (IR) and metabolic syndrome associated with high fat (HF) feeding, obesity and aging (see Corpeleijn et al., 2009; DeFronzo and Tripathy, 2009; Lark et al., 2012).
Some evidence suggested that cytosolic ectopic accumulation of fatty acids (FA) metabolites, such as diacylglycerols (DAG) and/or Ceramides, (Yu et al., 2002; Adams et al., 2004), underlies IR development in skeletal muscle (lipotoxicity theory) (reviewed in Lark et al., 2012). Numerous evidence has also suggested a link between elevated systemic and tissue inflammation with IR (inflammatory theory) (see Shenk et al., 2008; Lark et al., 2012). The effectors of IR in HF diet-induced inflammation are suggested to involve hyperactivation of stress-sensitive Ser/Thr kinases, such as JNK and IKKβ, which in turn inhibits insulin receptor/IRS1 axis. Several mechanisms were proposed to explain the link between inflammation and IR: endoplasmic reticulum (ER) stress (Ozcan et al., 2004; Lionetti et al., 2009; Mollica et al., 2011), oxidative stress (Lark et al., 2012), signaling through inflammation-associated receptors, such as TLR4 signaling (Uysal et al., 1997; Shi et al., 2006), and partitioning/activation of c-SRC (a key mediator of JNK activation) by saturated FA (Holzer et al., 2011; Figure 1A).
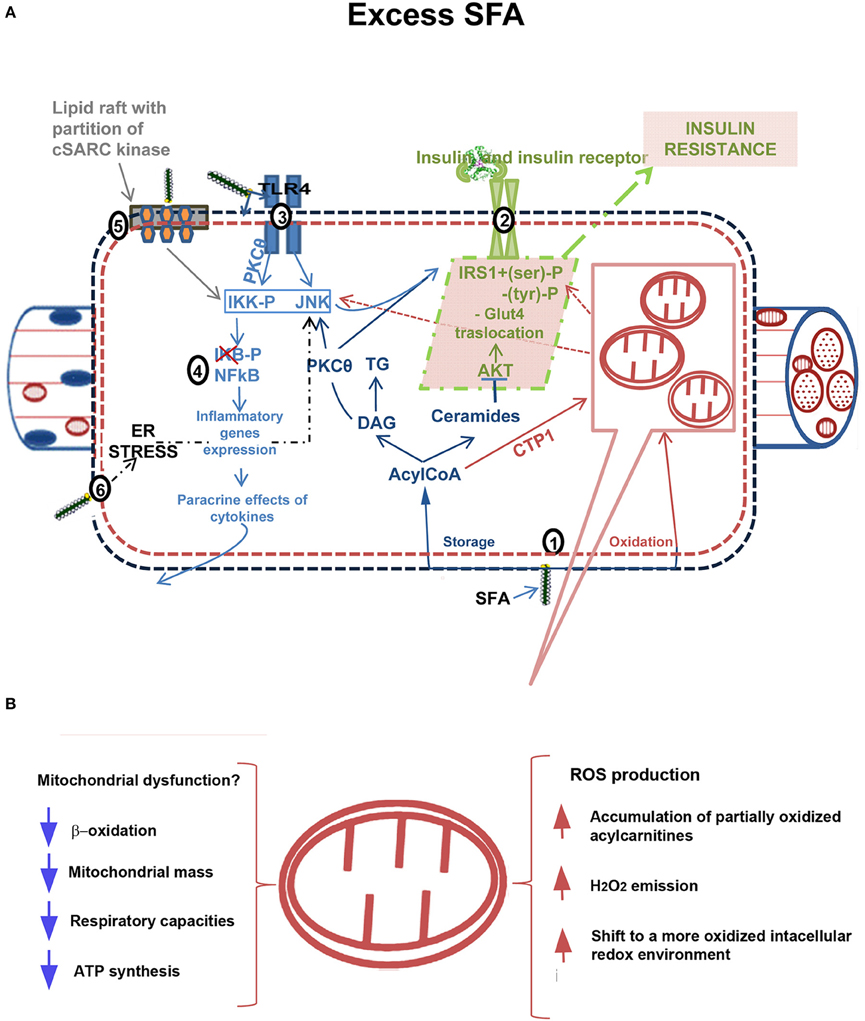
Figure 1. Mechanism linking excess fatty acids to insulin resistance in skeletal muscle. (A): (1) Excess free fatty acids (FFAs) are esterified in AcylCoAs, substrates involved in both synthetic and oxidative pathways. In the synthetic pathway, they are either stored as Triacylglycerols (TG) in lipid droplets or accumulated in metabolites (DAGs, Ceramides) that act as signaling molecules and may interfere with normal cellular signaling. DAGs are associated with membrane translocation and activation of Protein kinase C theta (PKC-θ), increased IRS1 serine/threonine phosphorylation and decreased insulin-stimulated IRS1 tyrosine phosphorylation, whereas Ceramides impair insulin action by inhibiting protein kinase PKB/AKT (dark blue). In the oxidative pathway, AcylCoAs are imported into the mitochondria by carnitine palmitoyltransferase-1 (CPT-1) shuttle and degraded via β-oxidation (in purple). (2) Insulin signaling pathway impaired by excess FFA (in green). Among FFAs, saturated FAs (SFAs) stimulate the activation of several inflammatory pathways. (3) Receptor-mediated mechanisms, as those of Toll like receptors 2/4 (TLR 2/4), activate serine kinases inhibitor kappaB kinase (IKK) and c-JUN NH2-terminal kinase (JNK). The activation of PKCθ also contributes to IKK and JNK activation. Altogether, these kinases catalyze IRS1 serine phosphorylation and lead to a reduction of insulin-induced IRS1 tyrosine phosphorylation, impairing insulin action. Moreover, IKK/NFkB axis (4) triggers expression of inflammatory genes with cytokines production (e.g., Tumor necrosis factor, TNFα), which in turn activate intracellular pathways promoting insulin resistance development (in light blue). (5) SFAs enter the cellular membranes and incorporate into them reducing membrane fluidity and creating or expanding subdomains rich in cholesterol and sphingolipids (lipid raft). They induce clustering and activation of cytosolic cSRC. cSRC activity is required for JNK1 activation and inhibition of insulin signaling (in grey). (6) Endoplasmic reticulum stress (ER stress), induced by lipotoxicity, contributes to activate inflammatory pathways and impair insulin signaling. (B): Putative role of mitochondria in development of IR. Mitochondrial dysfunction in presence of excess FFAs leads to intramyocellular lipid accumulation due to impaired β-oxidation. Decreased mitochondrial mass, respiratory capacities and ATP synthesis have been found in obesity and diabetes. Alternatively, excess FFA within mitochondria in the absence of increased energy demand stimulates oxidative stress with high rates of ROS production and H2O2 emission and a shift to a more oxidized intracellular redox environment, ultimately leading to the development of IR.
In recent years, different reviews focused on mechanism(s) by which mitochondrial bioenergetics (Fisher-Wellman and Neufer, 2012; Lark et al., 2012; Muoio and Neufer, 2012; Holloszy, 2013) and morphology (Liesa and Shirihai, 2013; Montgomery and Turner, 2015) may be linked to the etiology of IR in skeletal muscle. In the present review, the challenging debate on the involvement of mitochondrial dysfunction in IR will be briefly reviewed. Then, the main aim of the review will be to underline the importance of including mitochondrial morphology/dynamics and dietary fat source in the debate on skeletal muscle mitochondria involvement in IR etiology and to highlight the need of further research studies to clarify the involved mechanism(s).
Two leading theories on mechanisms underlying skeletal muscle IR onset focus on mitochondria, although with opposite views (Figure 1B). In one theory, mitochondrial dysfunction is suggested to cause intramyocellular lipid accumulation leading to IR (Kelley et al., 1999; Lowell and Shulman, 2005; reviewed in Civitarese and Ravussin, 2008; Montgomery and Turner, 2015). In this case, the strategies to accelerate flux through β-oxidation should improve insulin sensitivity. In the second theory, excess fuel within mitochondria in the absence of increased energy demand stimulates mitochondrial oxidant production and emission, ultimately leading to the development of IR (Fisher-Wellman and Neufer, 2012; Figure 1B). In this case, elevated flux through β-oxidation without added energy demand is viewed as an underlying cause of IR disease (Muoio and Neufer, 2012).
Several studies have revealed a reduction in skeletal mitochondrial mass in obesity and type 2 diabetes (Kelley et al., 2002; Morino et al., 2005; Ritov et al., 2005), decreased ATP synthesis in insulin resistant offspring of patients with type 2 diabetes (Petersen et al., 2004, 2005) and decreased maximal respiration rates in skeletal muscle isolated mitochondria from type 2 diabetics (Mogensen et al., 2007). Moreover, with the limitation that gene expression is not a direct assessment of function itself, HF diet has been shown to coordinately down-regulate genes required for mitochondrial oxidative phosphorylation in human and rodents skeletal muscle (Sparks et al., 2005). Interestingly, in skeletal muscle from obese or diabetic patients, decreased activity of electron transport chain and reduced number of mitochondria have been mainly reported in skeletal muscle mitochondria located beneath the sarcolemmal membrane (SS mitochondria) (Ritov et al., 2005). SS mitochondria also displayed lower respiratory capacities in presence of succinate as substrates in adult rats exhibiting HF diet-induced IR (Lionetti et al., 2007). The two mitochondrial subpopulations (SS and intermyiofibrillar, IMF) are differentiated not only by the different localization but also by the different functions (Cogswell et al., 1993; Mollica et al., 2006): SS mitochondria could be more affected by the impairing effect of saturated FA due to their localization beneath the sarcolemmal membrane.
However, although correlative studies seem to implicate mitochondrial dysfunction and impaired β-oxidation as predisposing risk factors for IR, still uncertain is whether diminished fat oxidation reflects a cause or a late stage consequence of the disease process (reviewed in Muoio, 2010). In fact, obese/diabetic humans never use their mitochondrial capacity for lipid oxidation; therefore, a marginal decline in oxidative potential has little relevance in causing lipotoxicity and IR in sedentary individuals. Moreover, it has been suggested that early stages of obesity and IR are accompanied by increased, rather than reduced β-oxidation (Muoio, 2010). These findings question the concept that mitochondrial dysfunction is a primary cause of IR (Hoeks et al., 2010, 2011), as also underscored by the study of Bonnard et al. (2008), showing mitochondrial dysfunction in skeletal muscle after 16 weeks, but not after 4 weeks HF feeding, while muscle IR was observed after both 4 and 16 weeks of HF feeding. In addition, mitochondrial deficiency, severe enough to impair fat oxidation in resting muscle, cause an increase, not a decrease, in insulin action (reviewed in Holloszy, 2013). Altogether, these studies suggest that deficiency of mitochondria in muscle does not cause IR (reviewed in Holloszy, 2013).
An alternative mechanism to explain the connection between mitochondria and IR focused on reactive oxygen species (ROS) production (reviewed in Muoio and Neufer, 2012; Holloszy, 2013). Lefort et al. (2010) showed normal oxidative capacity, decreased mitochondrial mass and high rates of ROS production in mitochondria isolated from skeletal muscle of obese insulin resistant individuals. Moreover, FA overload within the mitochondria results in the accumulation of partially oxidized acyl-carnitines, increased mitochondrial hydrogen peroxide (H2O2) emission and a shift to a more oxidized intracellular redox environment (Anderson et al., 2009; reviewed in Lark et al., 2012). H2O2 emission may induce IR by directly targeting protein involved in the glucose uptake process. On the other hand, given the sensitivity of cellular phosphatase to redox state, it has been suggested that elevated mitochondrial H2O2 production may decrease phosphatase tone in cells, increasing the inhibition state of insulin signaling proteins by stress-sensitive kinases (reviewed in Lark et al., 2012).
Although the primary role of skeletal muscle mitochondrial dysfunction in the pathogenesis of IR and type 2 diabetes is under debate (Hoeks et al., 2010, 2011; Holloszy, 2013), it is generally accepted that in this disease a mitochondrial defect occurs, possibly secondary to a fat intake increase.
It is well known that mitochondrial morphology is highly variable, ranging between long tubular mitochondria and short circular ones and it is maintained through a dynamic balance between fusion and fission processes (Westermann, 2010, 2012), which allow mitochondria to redistribute in a cell, exchange contents and repair damaged mitochondria. These two opposing processes are finely regulated by mitochondrial fusion proteins mitofusins 1 and 2 (Mfn1 and Mfn2), and optic atrophy gene 1 (OPA1) (Cipolat et al., 2004; Palmer et al., 2011) and by mitochondrial fission protein dynamin-related protein 1 (DRP1) and fission protein 1 (Fis1) (Nunnari et al., 2002; Liesa et al., 2009).
Several pieces of evidence suggested that mitochondrial dynamic behavior play a key role in mitochondrial health, bioenergetics function, quality control, and cell viability. Notably, disruption of mitochondrial dynamics has been found in IR and type 2 diabetes (Bach et al., 2003, 2005; Yu et al., 2006; Liesa and Shirihai, 2013).
The group of Zorzano showed that decreased expression of Mfn2 and altered expression of OPA1 participated in obesity and type 2 diabetes development in both patients and rodent models (Bach et al., 2003; Zorzano et al., 2009a,b, 2010; Hernández-Alvarez et al., 2010; Quirós et al., 2012). In obese Zucker rats, skeletal muscle mitochondrial network was reduced by 25% associated with a repression of Mfn2 (Bach et al., 2003). In addition, skeletal muscle of obese subjects and type 2 diabetic patients also showed a reduced expression of Mfn2 mRNA and Mfn2 protein compared to lean subjects (Bach et al., 2003, 2005). Mfn2 repression was detected in the skeletal muscles of both obese and non-obese type 2 diabetic patients (Bach et al., 2005). Notably, the expression of one of the mitochondrial proteases involved in OPA1 processing, presenilin-associated rhomboid-like (PARL), was also reduced in diabetic animals. In humans, a positive linear correlation between PARL mRNA levels and insulin sensitivity has been reported (Walder et al., 2005). These data suggest multiple alterations in mitochondrial fusion in IR. However, reduction of Mfn2 expression together with decreased mitochondrial size in skeletal muscle in obesity and type 2 diabetes states allow proposing a relevant role for Mfn2 in IR (Civitarese and Ravussin, 2008; Zorzano et al., 2009a,b). In agreement with this suggestion, a positive correlation between Mfn2 expression in skeletal muscle and insulin sensitivity has been reported (Bach et al., 2005). It is of interest that the involvement of Mfns in diet-induced obesity via the regulation of leptin resistance and systemic energy metabolism was also revealed (Dietrich et al., 2013; Schneeberger et al., 2013; reviewed in Putti et al., 2015). Moreover, it has been suggested that there is an association between increased mitochondrial fission, mitochondrial bioenergetics and fat induced-IR in skeletal muscle (Jheng et al., 2012). Indeed, in differentiated C2C12 muscle cells mitochondrial fragmentation and increased mitochondrion associated-DRP1 and Fis1 was induced by excess palmitate and this fission phenotype was associated with increased oxidative stress, loss of ATP production and reduced insulin-stimulated glucose uptake. These authors also found smaller and shorter mitochondria and increased mitochondrial fission machinery in the skeletal muscle of mice with genetic or diet-induced obesity. Furthermore, inhibition of mitochondrial fission improved muscle insulin signaling and systemic insulin sensitivity in obese mice (Jheng et al., 2012).
A shift toward fission was also found in skeletal muscle of HF diet (HFD)-induced obese mice by Liu et al. (2014). Notably, these authors also faced the question of whether mitochondrial dynamics exists in skeletal muscle in vivo. It should be considered that mitochondria in skeletal muscle are rigidly located between bundles of myofilaments in a highly regular “crystal like” pattern (Vendelin et al., 2005) and therefore, their motility and dynamics may be very restricted. Liu et al. (2014) confirmed that mitochondria are dynamic organelles in skeletal muscle in vivo, by demonstrating that they exchange contents within the whole mitochondrial population through nanotunneling-mediated mitochondrial fusion. In this way, mitochondria can bypass the restriction of myofilament and exchange mitochondrial matrix contents even if they are distant. This dynamic communication among mitochondria in skeletal muscle may protect from injury by preventing the accumulation of detrimental metabolites. Liu et al. (2014) also showed that this dynamic behavior was inhibited in skeletal muscle of HFD–induced obese mice associated with decreased Mfn2 and increased Fis1 and DRP1 expression compared to normal diet fed mice. This impaired mitochondrial fusion in skeletal muscle of HFD-induced obese mice was accompanied with damaged mitochondrial respiratory function and decreased ATP content. Therefore, the authors suggest that mitochondrial dynamics play an important role in regulating mitochondrial function, including respiration rate and ATP production (Liu et al., 2014).
It has been suggested that diverse dietary fat sources have different effects on obesity and associated diseases development. Saturated FA are well known to induce both obesity and related disease, whereas omega 3 polyunsaturated FA (ω-3 PUFA) from fish oil have been shown in many studies to protect against these metabolic diseases (Xin et al., 2008; Gonzalez-Periz et al., 2009; Abete et al., 2011). The effect of ω-3 PUFA on metabolic disease has been extensively studied during the past three decades since the first studies such as the one by Storlien et al. (1987) showing that fish oil prevents IR induced by high-fat feeding in rats. Further studies confirmed that ω-3 PUFA had an anti-obesity effect and enhanced insulin sensitivity and glucose homeostasis in rodent models of IR: the replacement of a small proportion of the diet with ω-3 PUFAs from fish oil completely prevents the development of skeletal muscle IR (Storlien et al., 1991; Fryer et al., 1995). Recent studies hypothesized that ω-3 PUFAs protect glucose tolerance, in part by preventing the accumulation of bioactive lipid mediators that interfere with the insulin signaling pathway (Lanza et al., 2013). Lanza et al. (2013) evaluated the influence of dietary ω-3 PUFAs on mitochondrial physiology and muscle lipid metabolites in the context of 10 weeks high-fat feeding in mice. They found a lower content of long-chain Acyl-CoAs and Ceramides in the presence of fish oil, whereas mitochondrial oxidative capacity was similarly increased with or without fish oil. Several studies have also indicated that ω-3 PUFAs possess anti-inflammatory properties that prevent and reverse the development of IR in mice which are fed a high-fat diet in an adiponectin-dependent manner (Kalupahana et al., 2010, 2011). On the other hand, unsaturated FA prevent c-Src membrane partitioning and activation and block JNK activation with consequent beneficial effects on insulin sensitivity (Holzer et al., 2011). Considering the anti-inflammatory properties of ω-3 PUFAs, in a recent study, we compared the effects of chronic high-fish oil and high-lard diets on obesity-related inflammation by evaluating serum and tissue adipokine levels and histological features in insulin-sensitive tissues (white adipose tissue, liver and skeletal muscle) (Lionetti et al., 2014b). We showed that the replacement of lard (saturated FA) with fish oil (ω-3 PUFAs) in chronic high-fat feeding attenuated the development of systemic and tissue inflammation. Indeed, compared with a high-lard diet, a high-fish oil diet resulted in a lower degree of systemic inflammation and IR that were associated with a lower ectopic lipid depot, inflammation degree and IR in the skeletal muscle (Lionetti et al., 2014b). In a further study on the same experimental design, we confirmed that the replacement of lard with fish oil in HF diet had preventive effects on obesity and systemic inflammation and IR development as well as we showed a fusion mitochondrial phenotype in association with the improvement of IR in skeletal muscle (Lionetti et al., 2013). As for preventive effects on obesity, body weight gain after 6 weeks of HF diet was lower in fish oil fed rats compared to lard fed rats. As for skeletal muscle IR, we showed that high-lard diet induced a defect in the skeletal muscle insulin signaling pathway with a lower immune-reactivity to IRS1 and pIRS (Tyr632), in agreement with other authors (Yaspelkis et al., 2009; Yuzefovych et al., 2013). On the other hand, a high fish oil diet elicited IRS1 and pIRS (Tyr632) immune-reactivity similar to a control diet, in agreement with ameliorated systemic insulin sensitivity (Lionetti et al., 2013). We cannot exclude the possibility that the fish oil protective effect was due to indirect effects of differences on adiposity. We also showed that the beneficial effects of fish oil feeding on skeletal muscle IR development was associated with changes in protein involved in mitochondrial dynamic behavior, with a greater number of immunoreactive fibers for Mfn2 and OPA1 proteins, as well as a weaker immunostaining for DRP1 and Fis1 compared to high lard feeding. Skeletal muscle electron microscopy observations also suggested a prominent presence of fission events in high-lard diet fed rats, and fusion events in high-fish oil diet fed rats (Lionetti et al., 2013).
The finding on the effects of different dietary FA on skeletal muscle mitochondrial fusion/fission proteins may be associated with effects on inflammatory processes involved in IR development. Indeed, Bach et al. (2005) suggested that TNF-α inhibits Mfn2 gene expression in cells in culture, suggesting that inflammatory parameters may play a regulatory role on Mfn2. In agreement, we showed that pro-inflammatory dietary saturated FA reduced Mfn2 expression and induced fission phenotype in skeletal muscle (Lionetti et al., 2013). On the other hand, the anti-inflammatory effect of dietary ω-3 PUFAs was associated with no reduction in skeletal muscle Mfn2 content and a tendency to mitochondrial fusion. This shift toward fusion may be an adaptive mechanism to counteract cellular stress induced by chronic HF diet, by allowing functional mitochondria to complement dysfunctional mitochondria. In accordance, myocytes cultured with docosahexaenoic acid exhibited a higher mitochondrial mass with a higher proportion of large and elongated mitochondria with downregulation of fission genes DRP1 and Fis1 (Casanova et al., 2014).
The pro-fusion effect of ω-3 PUFAs dietary fat on skeletal muscle mitochondria is in agreement with the results found in liver mitochondria, where a shift toward mitochondrial fusion phenotype was also suggested (Zhang et al., 2011; Lionetti et al., 2014a).
The mechanism underlying fish oil/ω-3 PUFAs mitochondrial fusion stimulation may involve receptor-mediated signaling and/or lipid membrane composition, among other factors. Indeed, ω-3 PUFAs are incorporated into cellular membranes and may affect lipid-protein interactions as well as membrane fluidity, and therefore the function of embedded proteins. Recently, it has been suggested a role for ω-3 PUFAs in reorganizing the composition of the mitochondrial membrane, while promoting improvements in ADP sensitivity, determined as mitochondrial responses during ADP titration (Herbst et al., 2014). Moreover, it is well known that saturated FA incorporation reduces membrane fluidity, whereas PUFA do not have such effect (Clamp et al., 1997; Stulnig et al., 2001; Holzer et al., 2011). Further studies are needed to elucidate these mechanisms.
Recent evidence highlighted an association between mitochondrial morphology and IR development in skeletal muscle. Few works on different dietary fat source started to underline the different effect of saturated and ω-3 PUFAs on skeletal muscle IR and mitochondrial protein involved in dynamics behavior, suggesting an association between beneficial protective effect of ω-3 PUFAs toward IR development and mitochondrial fusion phenotype (Figure 2). However, it is important to underline that most of the data present in literature on skeletal muscle mitochondrial morphology and IR are from associational studies. Therefore, there is an urgent requirement for in vivo mechanistic studies to confirm the associational relationship between mitochondrial morphology/dynamics and IR development.
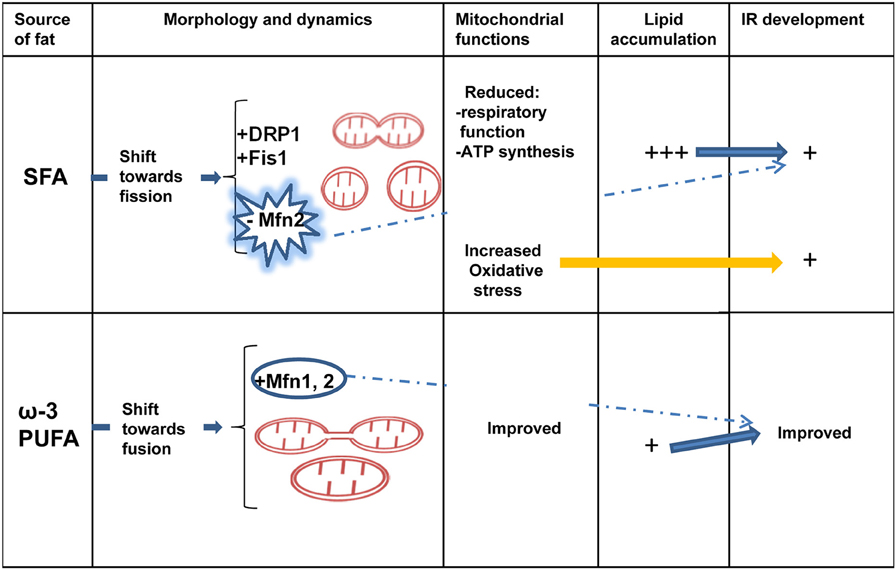
Figure 2. Dietary fat source, mitochondrial dynamics and skeletal muscle IR. Diverse dietary fat sources induce different effects on mitochondrial morphology/dynamics and IR development in skeletal muscle. Saturated fatty acids (SFA) elicit a shift toward fission processes (decreased Mfn2 and increased Fis1 and DRP1 expression) associated with reduced mitochondrial function, increased oxidative stress, lipid accumulation and IR development. Ω-3 polyunsaturated fatty acids (PUFAs) induce Mfn2 expression and fusion processes associated with less lipid accumulation and improved IR. IR, insulin resistance; Mfn2, mitofusin 2; DRP1, dynamin-related protein 1; Fis1, fission protein 1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abete, I., Goyenechea, E., Zulet, M. A., and Martínez, J. A. (2011). Obesity and metabolic syndrome: potential benefit from specific nutritional components. Nutr. Metab. Cardiovasc. Dis. 21, B1–B15. doi: 10.1016/j.numecd.2011.05.001
Adams, J. M. II, Pratipanawatr, T., Berria, R., Wang, E., DeFronzo, R. A., Sullards, M. C., et al. (2004). Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53, 25–31. doi: 10.2337/diabetes.53.1.25
Anderson, E. J., Lustig, M. E., Boyle, K. E., Woodlief, T. L., Kane, D. A., Lin, C. T., et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581. doi: 10.1172/JCI37048
Bach, D., Naon, D., Pich, S., Soriano, F. X., Vega, N., Rieusset, J., et al. (2005). Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 54, 2685–2693. doi: 10.2337/diabetes.54.9.2685
Bach, D., Pich, S., Soriano, F. X., Vega, N., Baumgartner, B., Oriola, J., et al. (2003). Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 278, 17190–17197. doi: 10.1074/jbc.M212754200
Bonnard, C., Durand, A., Peyrol, S., Chanseaume, E., Chauvin, M. A., Morio, B., et al. (2008). Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Invest. 118, 789–800. doi: 10.1172/jci32601
Casanova, E., Baselga-Escudero, L., Ribas-Latre, A., Arola-Arnal, A., Bladé, C., Arola, L., et al. (2014). Epigallocatechin gallate counteracts oxidative stress in docosahexaenoxic acid-treated myocytes. Biochim. Biophys. Acta 1837, 783–791. doi: 10.1016/j.bbabio.2014.01.014
Cipolat, S., Martins de Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 15927–15932. doi: 10.1073/pnas.0407043101
Civitarese, A. E., and Ravussin, E. (2008). Mitochondrial energetics and insulin resistance. Endocrinology 149, 950–954. doi: 10.1210/en.2007-1444
Clamp, A. G., Ladha, S., Clark, D. C., Grimble, R. F., and Lund, E. K. (1997). The influence of dietary lipids on the composition and membrane fluidity of rat hepatocyte plasma membrane. Lipids 32, 179–184. doi: 10.1007/s11745-997-0022-3
Cogswell, A. M., Stevens, R. J., and Hood, D. A. (1993). Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am. J. Physiol. 264, C383–C389.
Corpeleijn, E., Saris, W. H., and Blaak, E. E. (2009). Metabolic flexibility in the development of insulin resistance and type 2 diabetes: effects of lifestyle. Obes. Rev. 10, 178–193. doi: 10.1111/j.1467-789X.2008.00544.x
DeFronzo, R. A., and Tripathy, D. (2009). Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32, S157–S163. doi: 10.2337/dc09-S302
Dietrich, M. O., Liu, Z. W., and Horvath, T. L. (2013). Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 155, 188–199. doi: 10.1016/j.cell.2013.09.004
Fisher-Wellman, K. H., and Neufer, P. D. (2012). Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 23, 142–153. doi: 10.1016/j.tem.2011.12.008
Fryer, L. G. D., Orfali, K. A., Holness, M. J., Saggerson, E. D., and Sugden, M. C. (1995). The long-term regulation of skeletal muscle pyruvate dehydrogenase kinase by dietary lipid is dependent on fatty acid composition. Eur. J. Biochem. 229, 741–748. doi: 10.1111/j.1432-1033.1995.tb20522.x
Gonzalez-Periz, A., Horrillo, R., Ferre, N., Gronert, K., Dong, B., Morán-Salvador, E., et al. (2009). Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 23, 1946–1957. doi: 10.1096/fj.08-125674
Herbst, E. A. F., Paglialunga, S., Gerling, C., Whitfield, J., Mukai, K., Chabowski, A., et al. (2014). Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 592, 1341–1352. doi: 10.1113/jphysiol.2013.267336
Hernández-Alvarez, M. I., Thabit, H., Burns, N., Shah, S., Brema, I., Hatunic, M., et al. (2010). Subjects with early-onset type 2 diabetes show defective activation of the skeletal muscle PGC-1{alpha}/Mitofusin-2 regulatory pathway in response to physical activity. Diabetes Care 33, 645–651. doi: 10.2337/dc09-1305
Hoeks, J., van Herpen, N. A., Mensink, M., Moonen-Kornips, E., van Beurden, D., Hesselink, M. K., et al. (2010). Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes 59, 2117–2125. doi: 10.2337/db10-0519
Hoeks, J., Wilde, J. D., Hulshof, M. F., Berg, S. A., Schaart, G., Dijk, K. W., et al. (2011). High fat diet-induced changes in mouse muscle mitochondrial phospholipids do not impair mitochondrial respiration despite insulin resistance. PLoS ONE 6:e27274. doi: 10.1371/journal.pone.0027274
Holloszy, J. O. (2013). “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes 62, 1036–1040. doi: 10.2337/db12-1107
Holzer, R. G., Park, E. J., Li, N., Tran, H., Chen, M., Choi, C., et al. (2011). Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 147, 173–184. doi: 10.1016/j.cell.2011.08.034
Jheng, H. F., Tsai, P. J., Guo, S. M., Kuo, L. H., Chang, C. S., Su, I. J., et al. (2012). Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 32, 309–319. doi: 10.1128/MCB.05603-11
Kalupahana, N. S., Claycombe, K. J., and Moustaid-Moussa, N. (2011). (n-3) Fatty acids alleviate adipose tissue inflammation and insulin resistance: mechanistic insights. Adv. Nutr. 2, 304–316. doi: 10.3945/an.111.000505
Kalupahana, N. S., Claycombe, K., Newman, S., Stewart, T., Siriwardhana, N., Matthan, N., et al. (2010). Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation. J. Nutr. 140, 1915–1922. doi: 10.3945/jn.110.125732
Kelley, D. E., Goodpaster, B., Wing, R. R., and Simoneau, J. A. (1999). Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity and weight loss. Am. J. Physiol. 277, E1130–E1141.
Kelley, D. E., He, J., Menshikova, E. V., and Ritov, V. B. (2002). Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950. doi: 10.2337/diabetes.51.10.2944
Lanza, I. R., Blachnio-Zabielska, A., Johson, M. L., Schimke, J. M., Jakaitis, D. R., Lebrasseur, N. K., et al. (2013). Influence of fish oil on skeletal muscle mitochondrial energetics and lipid metabolites during high-fat diet. Am. J. Physiol. Endocrinol. Metab. 304, E1391–E1403. doi: 10.1152/ajpendo.00584.2012
Lark, D. S., Fisher-Wellman, K. H., and Neufer, P. D. (2012). High-fat load: mechanism(s) of insulin resistance in skeletal muscle. Int. J. Obes. 2, S31–S36. doi: 10.1038/ijosup.2012.20
Lefort, N., Glancy, B., Bowen, B., Willis, W. T., Bailowitz, Z., De Filippis, E. A., et al. (2010). Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes 59, 2444–2452. doi: 10.2337/db10-0174
Liesa, M., Palacín, M., and Zorzano, A. (2009). Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 89, 799–845. doi: 10.1152/physrev.00030.2008
Liesa, M., and Shirihai, O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. doi: 10.1016/j.cmet.2013.03.002
Lionetti, L., Mollica, M. P., Crescenzo, R., D'Andrea, E., Ferraro, M., Bianco, F., et al. (2007). Skeletal muscle subsarcolemmal mitochondrial dysfunction in high-fat fed rats exhibiting impaired glucose homeostasis. Int. J. Obes. 31, 1596–1604. doi: 10.1038/sj.ijo.0803636
Lionetti, L., Mollica, M. P., Donizzetti, I., Gifuni, G., Sica, R., Pignalosa, A., et al. (2014a). High-lard and high-fish-oil diets differ in their effects on function and dynamic behavior of rat hepatic mitochondria. PLoS ONE 9:e92753. doi: 10.1371/journal.pone.0092753
Lionetti, L., Mollica, M. P., Lombardi, A., Cavaliere, G., Gifuni, G., and Barletta, A. (2009). From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 19, 146–152. doi: 10.1016/j.numecd.2008.10.010
Lionetti, L., Mollica, M. P., Sica, R., Donizzetti, I., Gifuni, G., and Pignalosa, A. (2014b). Differential effects of high-fish oil and high-lard diets on cells and cytokines involved in the inflammatory process in rat insulin-sensitive tissues. Int. J. Mol. Sci. 15, 3040–3063. doi: 10.3390/ijms15023040
Lionetti, L., Sica, R., Mollica, M. P., and Putti, R. (2013). High-lard and high-fish oil diets differ in their effects on insulin resistance development, mitochondrial morphology and dynamic behavior in rat skeletal muscle. Food Nutr. Sci. 4, 105–112. doi: 10.4236/fns.2013.49A1017
Liu, R., Jin, P., Yu, L., Wang, Y., Han, L., Shi, T., et al. (2014). Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS ONE 9:e92810. doi: 10.1371/journal.pone.0092810
Lowell, B. B., and Shulman, G. I. (2005). Mitochondrial dysfunction and type 2 diabetes. Science 307, 384–387. doi: 10.1126/science.1104343
Mogensen, M., Sahlin, K., Fernström, M., Glintborg, D., Vind, B. F., Beck-Nielsen, H., et al. (2007). Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56, 1592–1599. doi: 10.2337/db06-0981
Mollica, M. P., Lionetti, L., Crescenzo, R., D'Andrea, E., Ferraro, M., Liverini, G., et al. (2006). Heterogeneous bioenergetic behavior of subsarcolemmal and intermyofibrillar mitochondria in fed and fasted rats. Cell. Mol. Life Sci. 63, 358–366. doi: 10.1007/s00018-005-5443-2
Mollica, M. P., Lionetti, L., Putti, R., Cavaliere, G., Gaita, M., and Barletta, A. (2011). From chronic overfeeding to hepatic injury: role of endoplasmic reticulum stress and inflammation. Nutr. Metab. Cardiovasc. Dis. 21, 222–230. doi: 10.1016/j.numecd.2010.10.012
Montgomery, M. K., and Turner, N. (2015). Mitochondrial dysfunction and insulin resistance: an update. Endocr. Connect. 4, R1–R15. doi: 10.1530/EC-14-0092
Morino, K., Petersen, K. F., Dufour, S., Befroy, D., Frattini, J., Shatzkes, N., et al. (2005). Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Invest. 115, 3587–3593. doi: 10.1172/JCI25151
Muoio, D. M. (2010). Intramuscolar triacylglycerol and insulin resistance: guilty as charged or wrongly accused? Biochim. Biophys. Acta 1801, 281–288. doi: 10.1016/j.bbalip.2009.11.007
Muoio, D. M., and Neufer, P. D. (2012). Lipid-induced mitochondrial stress and insulin resistance. Cell Met. 15, 595–605. doi: 10.1016/j.cmet.2012.04.010
Nunnari, J., Wong, E. D., Meeusen, S., and Wagner, J. A. (2002). Studying the behavior of mitochondria. Methods Enzymol. 351, 381–393. doi: 10.1016/S0076-6879(02)51859-0
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi, N. N., Ozdelen, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–456. doi: 10.1126/science.1103160
Palmer, C. S., Osellame, L. D., Stojanovski, D., and Ryan, M. T. (2011). The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Sign. 23, 1534–1545. doi: 10.1016/j.cellsig.2011.05.021
Petersen, K. F., Dufour, S., Befroy, D., Garcia, R., and Shulman, G. I. (2004). Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 350, 664–671. doi: 10.1056/NEJMoa031314
Petersen, K. F., Dufour, S., and Shulman, G. I. (2005). Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2:e233. doi: 10.1371/journal.pmed.0020233
Putti, R., Sica, R., Migliaccio, V., and Lionetti, L. (2015). Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol. 6:109. doi: 10.3389/fphys.2015.00109
Quirós, P. M., Ramsay, A. J., Sala, D., Fernández-Vizarra, E., Rodríguez, F., Peinado, J. R., et al. (2012). Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 31, 2117–2133. doi: 10.1038/emboj.2012.70
Ritov, V. B., Menshikova, E. V., He, J., Ferrell, R. E., Goodpaster, B. H., and Kelley, D. E. (2005). Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54, 8–14. doi: 10.2337/diabetes.54.1.8
Schneeberger, M., Dietrich, M. O., Sebástian, D., Imbernón, M., Castaño, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187. doi: 10.1016/j.cell.2013.09.003
Shenk, S., Saberi, M., and Olefsky, J. M. (2008). Insulin sensitiivyt: modulation by nutrients and inflammation. J. Clin. Invest. 118, 2992–3002. doi: 10.1172/JCI34260
Shi, H., Kokoeva, M. V., Inouye, K., Tzameli, I., Yin, H., and Flier, J. S. (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025. doi: 10.1172/JCI28898
Sparks, L. M., Xie, H., Koza, R. A., Mynatt, R., Hulver, M. W., Bray, G. A., et al. (2005). A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 54, 1926–1933. doi: 10.2337/diabetes.54.7.1926
Storlien, L. H., Jenkins, A. B., Chisholm, D. J., Pascoe, W. S., Khouri, S., and Kraegen, E. W. (1991). Influence of dietary fat composition on development of insulin resistance in rats: relationship to muscle triglyceride and ω-3 fatty acids in muscle phospholipid. Diabetes 40, 280–289. doi: 10.2337/diab.40.2.280
Storlien, L. H., Kraegen, E. W., Chisholm, D. J., Ford, G. L., Bruce, D. G., and Pascoe, W. S. (1987). Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science 237, 885–888. doi: 10.1126/science.3303333
Stulnig, T. M., Huber, J., Leitinger, N., Imre, E. M., Angelisova, P., Nowotny, P., et al. (2001). Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J. Biol. Chem. 276, 37335–37340. doi: 10.1074/jbc.M106193200
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., and Hotamisligil, G. S. (1997). Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614. doi: 10.1038/39335
Vendelin, M., Beraud, N., Guerrero, K., Andrienko, T., Kuznetsov, A. V., Olivares, J., et al. (2005). Mitochondrial regular arrangement in muscle cells: a “crystal-like” pattern. Am. J. Physiol. Cell Physiol. 288, C757–C767. doi: 10.1152/ajpcell.00281.2004
Walder, K., Kerr-Bayles, L., Civitarese, A., Jowett, J., Curran, J., Elliott, K., et al. (2005). The mitochondrial rhomboid protease PSARL is a new candidate gene for type 2 diabetes. Diabetologia 48, 459–468. doi: 10.1007/s00125-005-1675-9
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884. doi: 10.1038/nrm3013
Westermann, B. (2012). Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 1817, 1833–1838. doi: 10.1016/j.bbabio.2012.02.033
Xin, Y. N., Xuan, S. Y., Zhang, J. H., Zheng, M. H., and Guan, H. S. (2008). Omega-3 polyunsaturated fatty acids: a specific liver drug for non-alcoholic fatty liver disease (NAFLD). Med. Hypotheses. 71, 820–821. doi: 10.1016/j.mehy.2008.07.008
Yaspelkis, B. B. III, Kvasha, I. A., and Figuero, T. Y. (2009). High-fat feeding increases insulin receptor and IRS-1 coimmunoprecipitation with SOCS-3, Ikkα/B phos- phorylation and decreases PI-3 kinase activity in muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R1709–R1715. doi: 10.1152/ajpregu.00117.2009
Yu, C., Chen, Y., Cline, G. W., Zhang, D., Zong, H., Wang, Y., et al. (2002). Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50230–50236. doi: 10.1074/jbc.M200958200
Yu, T., Robotham, J. L., and Yoon, Y. (2006). Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U.S.A. 103, 2653–2658. doi: 10.1073/pnas.0511154103
Yuzefovych, L. V., Musiyenko, S. I., Wilson, G. L., and Rachek, L. I. (2013). Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS ONE 8:e54059. doi: 10.1371/journal.pone.0054059
Zhang, Y., Jiang, L., Hu, W., Zheng, Q., and Xiang, W. (2011). Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega-3 fatty acid-induced up-regulation of mitofusin 2. Metabolism 60, 767–775. doi: 10.1016/j.metabol.2010.07.026
Zorzano, A., Hernández-Alvarez, M. I., Palacín, M., and Mingrone, G. (2010). Alterations in the mitochondrial regulatory pathways constituted by the nuclear co-factors PGC-1alpha or PGC-1beta and mitofusin 2 in skeletal muscle in type 2 diabetes. Biochim. Biophys. Acta 1797, 1028–1033. doi: 10.1016/j.bbabio.2010.02.017
Zorzano, A., Liesa, M., and Palacín, M. (2009a). Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch. Physiol. Biochem. 115, 1–12. doi: 10.1080/13813450802676335
Keywords: mitochondrial fusion, mitochondrial fission, lard, fish oil, omega-3 fatty acids
Citation: Putti R, Migliaccio V, Sica R and Lionetti L (2016) Skeletal Muscle Mitochondrial Bioenergetics and Morphology in High Fat Diet Induced Obesity and Insulin Resistance: Focus on Dietary Fat Source. Front. Physiol. 6:426. doi: 10.3389/fphys.2015.00426
Received: 31 July 2015; Accepted: 27 December 2015;
Published: 20 January 2016.
Edited by:
Gilles Gouspillou, Université du Québec à Montréal, CanadaReviewed by:
Giovanni Solinas, University of Gothenburg, SwedenCopyright © 2016 Putti, Migliaccio, Sica and Lionetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lillà Lionetti, bGlsbGEubGlvbmV0dGlAdW5pbmEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.