
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 12 January 2016
Sec. Striated Muscle Physiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00422
This article is part of the Research TopicMitochondria in Skeletal Muscle Health, Aging and DiseasesView all 14 articles
 Vanina Romanello1*
Vanina Romanello1* Marco Sandri1,2,3,4*
Marco Sandri1,2,3,4*Loss of muscle mass and force occurs in many diseases such as disuse/inactivity, diabetes, cancer, renal, and cardiac failure and in aging-sarcopenia. In these catabolic conditions the mitochondrial content, morphology and function are greatly affected. The changes of mitochondrial network influence the production of reactive oxygen species (ROS) that play an important role in muscle function. Moreover, dysfunctional mitochondria trigger catabolic signaling pathways which feed-forward to the nucleus to promote the activation of muscle atrophy. Exercise, on the other hand, improves mitochondrial function by activating mitochondrial biogenesis and mitophagy, possibly playing an important part in the beneficial effects of physical activity in several diseases. Optimized mitochondrial function is strictly maintained by the coordinated activation of different mitochondrial quality control pathways. In this review we outline the current knowledge linking mitochondria-dependent signaling pathways to muscle homeostasis in aging and disease and the resulting implications for the development of novel therapeutic approaches to prevent muscle loss.
Nearly 40–50% of total body mass in non-obese mammals is composed by skeletal muscles. Striated muscles are plastic tissues that can undergo adaptive changes in their structure or functional properties to meet new challenges. For example, muscle mass can either increase or decrease in response to metabolic demands like exercise or inactivity. Moreover, muscle strength or resistance to fatigue can be modulated by training or detraining. The regulation of muscle size, due to its limited proliferative capacity, is determined by the coordinated balance between protein synthesis and protein degradation. Mechanical overload or anabolic hormonal stimulation shifts the balance toward protein synthesis with consequent increases in fiber size, a process called hypertrophy. Conversely, in catabolic conditions protein degradation exceeds protein synthesis leading to muscle weakness and muscle atrophy. The decrease in cell size is mainly due to loss of organelles, cytoplasm and proteins. Skeletal muscle atrophy occurs in several pathological conditions like disuse, denervation, immobilization, sepsis, burn injury, cancer, AIDS, diabetes, heart and renal failure and during aging. Importantly, the decrease of muscle mass increases morbity, impairs the efficacy of many therapeutic treatments and contributes to mortality. Muscle mass is mainly controlled by two major signaling pathways: TGFβ/Smads and IGF1-AKT-mTOR-FoxO. TGFβ superfamily of ligands regulate the size of muscles via the Smad transcription factors (Sandri, 2013). Smad2/3 control catabolic genes while Smad1/5/8 regulate anabolic genes (Sartori et al., 2013, 2014). The IGF1-AKT- mTOR axis increases protein synthesis by stimulating the translational machinery while simultaneously blocking FoxOs transcription factors and protein degradation pathways (Sandri, 2013). Two main ATP-dependent proteolytic systems are activated during muscle atrophy in order to contribute to muscle loss. The ubiquitin-proteasome system degrades predominantly myofibrillar proteins, whereas the autophagy-lysosome system removes dysfunctional organelles, protein aggregates as well as unfolded and toxic proteins. Muscle atrophy requires the activation of gene transcription programs that regulate the expression of a subset of genes that are named atrophy-related genes or atrogenes (Bodine et al., 2001; Gomes et al., 2001; Lecker et al., 2004; Sandri et al., 2004; Sacheck et al., 2007). These atrogenes belong to several fundamental biological processes such as the ubiquitin-proteasome and autophagy-lysosome systems, protein syntesis, ROS detoxification, DNA repair, unfolding protein response (UPR), mitochondria function and energy balance. FoxO family of transcription factors (FoxO1, FoxO3, and FoxO4), which are targets of AKT, are key mediators of the catabolic response during atrophy (Sandri et al., 2004; Mammucari et al., 2007; Milan et al., 2015). Indeed, specific inhibition of FoxOs in muscle protects from cancer cachexia-, fasting- or denervation-induced atrophy (Judge et al., 2014; Milan et al., 2015) as they are critical for the regulation of several atrogenes. Moreover, at least half of the atrogenes require FoxOs for their up or downregulation (Milan et al., 2015). FoxO-dependent atrogenes include the E3 Ubiquitin ligases Atrogin-1, MuRF-1, MUSA1, SMART, and several autophagy-related genes such as LC3, GABARAPL1, BNIP3, CATHEPSIN L (Bodine et al., 2001; Sandri et al., 2004; Mammucari et al., 2007; Milan et al., 2015). Therefore, FoxOs coordinate both major proteolytic systems of the cell, the autophagy-lysosome and the ubiquitin-proteasome.
Interestingly, in addition to genes linked with proteolytic pathways, more than 10% of the atrophy-related genes are directly involved in energy production. Furthermore, several genes coding for enzymes important in glycolysis and oxidative phosphorylation are coordinately suppressed in atrophying muscles (Lecker et al., 2004). These interesting findings suggest that alterations in mitochondria and the mitochondrial network morphology can have potential deleterious consequences for the maintenance of muscle mass and function. Recent data shows that the mitochondrial network communicates with myonuclei to adapt muscle function to the physiological or pathological demands. Here we will review the principal pathways that control mitochondrial quality and the importance of mitochondrial network in the regulation of muscle mass and metabolism.
Mitochondria are continuously challenged by reactive oxygen species (ROS), an inexorable by-product of oxidative phosphorylation. In order to prevent an excessive production of ROS but also the release of dangerous factors such as cytochrome c, AIF or endonuclease G, mammalian cells contain several systems that maintain mitochondrial integrity and function. This mitochondrial quality control includes pathways related to protein folding and degradation as well as systems involved in organelle shape, movement and turnover (Figure 1). The activation of each specific quality control system depends on the degree of mitochondrial damage.
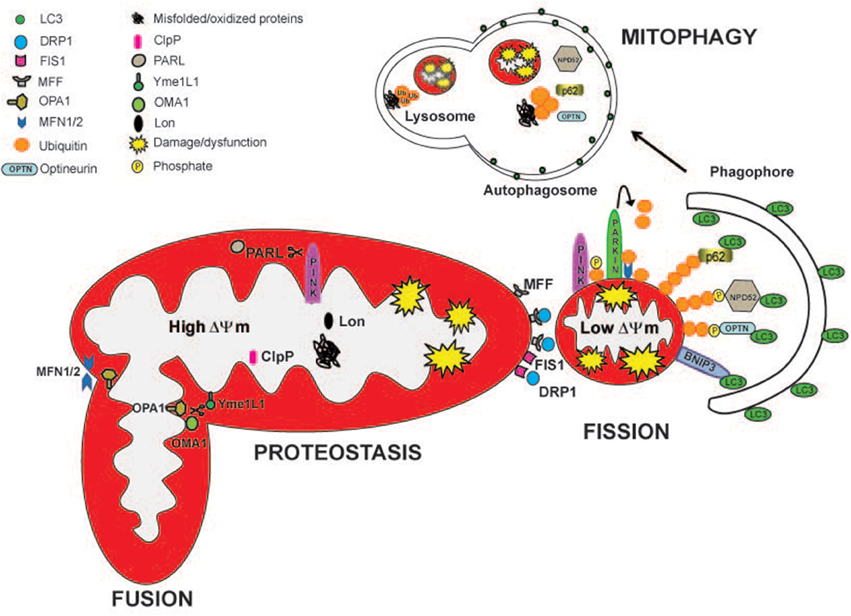
Figure 1. Mitochondrial quality control pathways are depicted. The actions of specific mitoproteases, Lon, ClpP, Oma1, Yme1L1, and PARL, maintain mitochondrial proteostasis and regulate mitochondrial function. PARL, Oma1, and Yme1L1 control mitochondrial dynamics by proteollytically processing Opa1 protein, which is important for mitochondrial fusion and cristae remodeling. PARL degrades PINK1, regulating mitophagy. Drp1, Fis, and Mff are the major proteins involved in mitochondrial fission. Partially damaged mitochondrial network divides into two fragments with different ΔΨm. The fragment with higher ΔΨm rejoins the functional mitochondrial network through mitochondrial fusion. Depolarized organelles will be removed by mitophagy. Bnip3 acts as a mitophagic receptor, binding to LC3 to tether mitochondria to the autophagosome. PINK1 accumulates on the surface of depolarized mitochondria where it phosphorylates ubiquitinated OMM proteins and the Parkin UBL domain. Parkin will further promote the ubiquitination of the outer mitochondrial membrane proteins, like MFN1/2. Then, the ubiquitinated proteins can be recognized by the adaptors p62/SQSTM1, Optineurin (OPTN), and NPD52 to initiate mitophagy.
Proteomic studies identified in human mitochondria approximately 1200 proteins (Calvo et al., 2015). However, only 1% of these proteins is encoded by mitochondrial DNA (mtDNA) (13 proteins in humans). Therefore, the remaining 99% is encoded by nuclear DNA (nDNA) genes, which are synthesized by ribosomes in cytoplasm and are imported into mitochondria (Harbauer et al., 2014). The incorporation of these precursors into specific mitochondria subcompartments such as the outer mitochondrial membrane (OMM), the intermembrane space (IMS), the inner mitochondrial membrane (IMM) and the mitochondrial matrix requires a specific and highly regulated import machineries (Harbauer et al., 2014). To maintain the proteostasis in each mitochondrial compartment, specific proteases (mitoproteases) will rapidly degrade misfolded or oxidized proteins. Mitoproteases-mediated quality control is the first line of defense against a mild mitochondrial damage. In the mitochondrial matrix, protein turnover is controlled by 3 AAA proteases: the soluble Lon and ClpP and the membrane-bound m-AAA. Protein degradation in the IMS is controlled by the membrane-bound i-AAA Yme1L1, the soluble HtrA2/Omi, the metallopeptidases OMA1 and the rhomboid protease PARL. Different reports showed that these mitoproteases do not only monitor mitochondrial protein quality but they can also decide mitochondrial fate. For example, m -AAA, Yme1L1, HtrA2, OMA1, and PARL l cleave the profusion protein Opa1 affecting mitochondrial morphology and function (Alavi and Fuhrmann, 2013). In addition, PARL modulates mitophagy by degrading the mitophagy protein PINK1 (Jin et al., 2010). For a review on the roles and the pathological relevance of these mitoproteases please refer to Quiros et al. (2015). Beside subcompartment-specific proteases, mitochondrial protein turnover is regulated also by the cytosolic ubiquitin-proteasome system (UPS). The UPS marks proteins for proteasomal degradation by the covalent linkage of a chain of ubiquitin proteins. A proteomic study in mouse cardiac muscle identified numerous OMM as well as IMS, IMM, and matrix proteins to be ubiquitinated (Jeon et al., 2007). A still unanswered question is how the cytosolic proteasome can degrade integral mitochondrial membrane proteins. During ER stress, misfolded proteins accumulate into the ER lumen and need to be retrotranslocated into the cytosol where they are flagged with ubiquitin and degraded by the proteasome in a process called ER-associated protein degradation (ERAD). Mitochondria have an ERAD-like mechanism, the mitochondria-associated degradation (MAD) pathway (Heo et al., 2010). ERAD and MAD pathways share some key components like the AAA ATPase p97 and the cofactor Npl4 (Ye et al., 2003; Heo et al., 2010). p97/Npl4 complex regulates the retrotranslocation of proteins from both the ER (Ye et al., 2001, 2003) and OMM (Heo et al., 2010; Tanaka et al., 2010; Xu et al., 2011). In mitochondria, p97 provides the driving force to extract Mfn1, Mfn2 and the anti-apoptotic protein Mcl1 from the OMM and chaperoned them for their degradation by the proteasome in the cytosol (Tanaka et al., 2010; Xu et al., 2011). Several components of the UPS are recruited to mitochondria such as the E3 ligases Parkin, March V/Mitol, Huwe, and Mulan (Livnat-Levanon and Glickman, 2011). These mitochondrial E3 ligases are a good example of the synergistic crosstalk between different mitochondria quality control systems. For instance, the activity of these ubiquitin ligases impacts on mitochondrial morphology and half-life. Indeed, Parkin-mediated ubiquitination of Mfn1, Mfn2, and VDAC1 blocks/reduces mitochondrial fusion and promotes mitophagy (Narendra et al., 2008; Gegg et al., 2010; Geisler et al., 2010; Tanaka et al., 2010). On the other hand, in physiological conditions Parkin avoid mitochondrial fragmentation by repressing Drp1 levels (Wang et al., 2011). Finally, MarchV/Mitol ubiquitinates Drp1, Fis1, and mitofusins 1 and 2 (Nakamura et al., 2006; Yonashiro et al., 2006; Karbowski et al., 2007). One intriguing issue is whether intramitochondrial proteins that do not face the cytosol can be substrates of the MAD pathway. Even though a retrotranslocation machinery has not been identified, some evidence indicates that the UPS controls IMM proteins. In fact, as a consequence of proximity to the respiratory chain, IMM proteins are exposed to ROS generated by mitochondrial respiration and therefore, oxidized. For example, the mitochondrial uncoupling protein 2 (UCP2) (Azzu and Brand, 2010; Bragoszewski et al., 2013) and the subunit of the OXPHOS complex V OSCP have been found to be retrotranslocated to the OMM for ubiquitination and proteasome degradation (Margineantu et al., 2007). Indeed, proteasome inhibition leads to accumulation of the IMM proteins UCP2, COXI, III, IV, OSCP (Margineantu et al., 2007; Azzu and Brand, 2010). Although the role of the UPS in different cellular processes has been well characterized, studies concerning the interaction with mitochondria are only at the beginning. Several critical issues related to how substrates are identified and how they are transported out for degradation need to be solved in the future.
Under stress conditions, when the degradation pathways are not sufficient to blunt the damage and restore a normal mitochondrial function, a retrograde signal is activated which coordinates nuclear gene expression. This mitochondria-to-nucleus response is named mitochondrial unfolding protein response (UPRmt) (Zhao et al., 2002). The ultimate purpose of UPRmt is to maintain proteostasis by promoting the expression of chaperones to improve protein folding, inhibit protein synthesis to alleviate ER stress and to induce expression of m-AAA proteases like ClpP and Yme1 to remove damaged proteins. For reviews on this topic see (Jovaisaite and Auwerx, 2015; Schulz and Haynes, 2015).
Mitochondrial dynamics is defined by the capacity of the organelle to rapidly change its size, shape and distribution by continuously alternating fusion and fission events. Fusion leads to elongated mitochondria with increased interconnectivity into a tubular network. On the contrary, fission fragments the network into unconnected shorter organelles. In physiological conditions, most mammalian cells show a continuous filamentous mitochondrial network with the exception of cardiomyocytes that display fragmented mitochondria that do not form a network (Song and Dorn, 2015). However, the mitochondrial pool rapidly changes its morphology according to the cellular needs. Increased mitochondrial fusion facilitates the redistribution of metabolites, proteins and mtDNA within the network. Moreover, fusion between healthy and damaged organelles allows to dilute the damaged material into the healthy network, avoids the accumulation of dysfunctional mitochondria and maintains their overall fitness (function) (Twig et al., 2008). On the other hand, mitochondrial fragmentation is a mechanism that segregates dysfunctional or damaged components of the mitochondrial network, allowing their removal via mitophagy (Otera and Mihara, 2011).
However, excessive fission generates isolated mitochondria which are less efficient in ATP production and are dysfunctional because they consume ATP to maintain their membrane potential (Benard et al., 2007).
Mitochondrial structure is optimized to ensure respiration and ATP production with a minimal release of ROS. This architecture must be preserved during fusion and fission events. Therefore, when mitochondria fuse they follow a scheme which starts with mitochondrial tethering, continues with fusion of OMM and concludes with fusion of IMM. In mammals, the membrane-bound GTPases mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy protein 1 (Opa1) are required for fusion of the OMM and IMM respectively.
Mfn1 and Mfn2 are integrated into the OMM and have two cytosolic heptad repeat domains (HR1 and HR2) which interact to form a dimeric anti parallel coiled-coil structure. Both mitofusins can consequently interact homotypically (Mfn1-Mfn1, Mfn2-Mfn2) or heterotypically (Mfn1-Mfn2) to promote the tethering of different adjacent mitochondria and fusion of the OMMs (Koshiba et al., 2004). These proteins have a high degree of homology but they do not have the same functions. Mfn1 has higher GTPase and organelle fusion activity while Mfn2 has greater affinity for GTP (Ishihara et al., 2004). Aditionally, Mfn2 and not Mfn1 is expressed on the mitochondria-associated endoplasmic reticulum membranes (MAM) and to a lesser extent to the endoplasmic/sarcoplasmic reticulum (ER/SR) (de Brito and Scorrano, 2009). This unique distribution allows for the close communication between the two organelles. Indeed, Mfn2 bridges mitochondria to ER/SR facilitating important processes linked to ER-mitochondria interactions like calcium homeostasis and signaling (de Brito and Scorrano, 2009; Ngoh et al., 2012; Sebastian et al., 2012; Munoz et al., 2013; Ainbinder et al., 2015). Recent data indicates Mfn2 as a modulator of the UPR during ER stress. In fact, Mfn2 deficiency leads to fragmented ER network and ER stress (Ngoh et al., 2012; Sebastian et al., 2012; Munoz et al., 2013; Bhandari et al., 2015) by chronic activation of PERK (Munoz et al., 2013). Mfn2, like the other mitochondria-shaping proteins is not free from post-translational modifications. For instance, when Mfn2 is phosphorylated by PINK1, it becomes a receptor for the ubiquitin ligase Parkin (Chen and Dorn, 2013). In addition, the E3 ligases HUWE1 and Mul1 also promote Mfn2 ubiquitination and proteasomal degradation (Leboucher et al., 2012; Lokireddy et al., 2012). Conversely, the formation of a Lys63- polyubiquitin chain by the E3 ligase MARCHV/Mitol does not induce Mfn2 degradation but increases its activity as well as MAM formation and function (Sugiura et al., 2013). Altogether, Mfn2 promotes mitochondrial fusion, enhances ER-mitochondria and activates mitophagy via PINK1/Parkin pathway.
The profusion protein Opa1 is regulated by proteolytic processing. There are several splicing variants of Opa1 (8 in humans and 4 in mice) which are expressed in a tissue specific manner. Some of them are cleaved to generate soluble short Opa1 (Opa1S) from long Opa1 isoforms (Opa1L) (Ishihara et al., 2006). Opa1L is anchored to the IMM by a transmembrane domain at the N-terminus and can be further cleaved in exon 5 (site S1) by OMA1, a process that is mitochondrial membrane potential (ΔΨm)-dependent (Ehses et al., 2009; Head et al., 2009). The mitoprotease Yme1 cut the site S2 in exon 5b of a subset of Opa1L belonging to the splicing variants 4, 6, 7, and 8 (Anand et al., 2014). As aforementioned, other mitoproteases such as m -AAA, HtrA2, and PARL l are also involved in Opa1 processing (Alavi and Fuhrmann, 2013). Furthermore, Opa1 is cleaved in its C-terminal region by an unknown cysteine protease in the post-prandial liver, in a process dependent on Mfn2 and independent of OMA1 (Sood et al., 2014). Opa1-dependent mitochondrial fusion needs Mfn1 (Cipolat et al., 2004) as well as Opa1S and Opa1L forms (Frezza et al., 2006; Song et al., 2007). However, under stress conditions fusion can rely only on Opa1L while Opa1S forms are dispensable (Tondera et al., 2009; Anand et al., 2014). In addition, oligomerization of soluble and membrane-bound OPA1 isoforms controls cristae remodeling and the assembly of respiratory chain complexes into supercomplexes, a structure that enhances mitochondrial respiration (Cogliati et al., 2013). On the contrary, complete conversion of Opa1L into Opa1S inhibits fusion and increases mitochondrial fission (Ishihara et al., 2006; Anand et al., 2014).
Mitochondrial fission depends on the cytosolic GTPase dynamin-related protein 1 (Drp1). Drp1 is recruited to the OMM where it assembles into multimeric ring complexes that form active GTP-dependent mitochondrial fission sites (Smirnova et al., 2001). Drp1 lacks hydrophobic membrane-binding domains and for this reason its recruitment on OMM is dependent on mitochondrial membrane proteins that act as receptors. Accordingly, Drp1 interacts with the integral OMM proteins: Fis1, Mff, mitochondrial elongation factor 2/mitochondrial dynamics protein 49 (MIEF2/MiD49) and MIEF1/MiD51. Fis1 is the major Drp1 receptor in yeasts (Karren et al., 2005). However, increasing evidence suggests that in mammals Fis1 is not the only OMM-anchor receptor of Drp1. For example, downregulation of Fis1 leads to mitochondrial elongation, even if the recruitment of Drp1 to mitochondria is not reduced (Lee et al., 2004). Furthermore, conditional deletion of Fis1 in a carcinoma model did not lead to defects in mitochondrial fission, indicating that Fis1 is not essential for fission (Otera et al., 2010). Recently, new components of the mammalian fission machinery were identified (Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011). Mff, the OMM-anchored mitochondrial fission factor, colocalizes with Drp1 on mammalian mitochondria. Its overexpression promotes mitochondrial fission with increased Drp1 recruitment to mitochondria. On the contrary, silencing Mff generates elongated mitochondria and Drp1 cytosolic distribution. Moreover, Drp1 affinity for Mff is higher than for Fis, suggesting that Mff preferentially functions as Drp1 receptor (Otera et al., 2010). MIEF1/MiD51 and the variant MIEF2/MiD49 recruit Drp1 to mitochondria independently of Fis1or Mff. Moreover, overexpression of MiD51 does not induce mitochondrial fragmentation but instead prevents mitochondrial fission by sequestering and inhibiting Drp1 (Palmer et al., 2011, 2013; Zhao et al., 2011; Loson et al., 2013). MIEF1/MiD51 preferentially binds Fis1 but can also interact with DRP1 and consequently, depending from the levels of Fis1, two different protein complexes are generated. When the level of Fis1 is high, it sequesters MIEF1/MiD51 that cannot anymore interact and inhibit DRP1 allowing mitochondrial fission. In contrast, the downregulation of Fis1 releases MIEF1/MiD51 that can now block Drp1 resulting in mitochondrial fusion (Zhao et al., 2011). Finally, in yeast Drp1 has been suggested to act as a regulatory factor not only of fission but also of fusion (Huang et al., 2011). In fact, it has been reported that mitochondria-localized Drp1 can interact with the HR1 domain of MFN2 to promote mitochondrial fusion (Huang et al., 2011).
The complexity of Drp1 regulation has been recently reviewed (Elgass et al., 2013; Otera et al., 2013), here we will briefly summarize some critical steps. Drp1-dependent mitochondrial fission is regulated by post-translational modifications like ubiquitination, phosphorylation and SUMOylation to ensure a rapid adaptation to cellular needs (Otera et al., 2013). For example, Drp1 is targeted for proteasomal degradation by Parkin-mediated ubiquitination (Wang et al., 2011) while the ubiquitination mediated by MARCHV/Mitol is controversial with different outcomes depending on cell context (Nakamura et al., 2006; Karbowski et al., 2007). Sumoylation of Drp1 by the SUMO E3 ligase mitochondria-anchored protein ligase (MAPL) stimulates mitochondrial fission (Braschi et al., 2009). Phosphorylation of different residues of DRP1 cause opposing effects. During mitosis, when organelles are inherited by daughter cells, mitochondrial fission activity is promoted by Cdk1-cyclin B-dependent Drp1 phosphorylation at Ser616 (in humans) in the GTPase effector domain (GED) (Taguchi et al., 2007). However, Drp1 activity is also induced by the posphorylation of Ser637 in the GED by the calcium/calmodulin-dependent protein kinase I alpha (CaMK1 alpha). As a result, this phosphorylation increases mitochondrial translocation of Drp1 because it facilitates the interaction with Fis1 (Han et al., 2008). However, phosphorylation of the same Ser637 by the protein kinase A (PKA) has an opposite effect causing DRP1 retention in the cytosol (Cribbs and Strack, 2007). This inhibition is counteracted by the calcium dependent phosphatase, calcineurin (Cribbs and Strack, 2007; Cereghetti et al., 2008). An interesting model of how post-translational modulation of fission can impinge on both, fusion and autophagy is what happens during early stages of starvation. Under this condition, mitochondrial fission is impaired due to the inhibition of Drp1. Two different reports indicate that starvation in cells leads to increased phosphorylation of Ser637 due to PKA activity as well as to decreased phosphorylation of Ser616. As a consequence, Drp1 is retained in the cytosol and mitochondria are elongated and spared from autophagic degradation. The resulting tubular mitochondrial network displays an increased number and density of cristae and presents more dimers of the ATP synthase (Gomes et al., 2011; Rambold et al., 2011). In conclusion, the shaping machinery adapts mitochondrial morphology to the bioenergetic requirements of the cell determining the homeostasis and the fate of the cell. Even though, our knowledge of the mechanisms controlling mitochondria dynamics is growing, many aspects of this process are still unclear.
Depending from the type of injury, mitochondria can adopt two different routes for delivering the damaged components to lysosome for degradation. When mitochondria deterioration is mild without global mitochondrial depolarization, local removal of mitochondrial content can be done by the generation of vesicles that bud-off from mitochondria and contain matrix components (Soubannier et al., 2012a,b). These mitochondria-derived vescicles (MDV) are 70–150 nm in size and do not require the fission machinery for their biogenesis. MDVs carry oxidized proteins coming from different mitochondrial compartments and are directed to lysosomes (Soubannier et al., 2012b). Therefore, mitochondrial oxidative stress induces MDVs via Pink-Parkin system (McLelland et al., 2014) independently of the autophagy-related proteins Atg5, Rab9, or Beclin (McLelland et al., 2014). Since Drp1 fission activity is not required for MDV budding (Soubannier et al., 2012a,b), the identification of the machinery necessary for MDV biogenesis is still unknown. This system works in conjunction with other mitochondria quality control pathways like mitochondrial proteolysis and dynamics and precedes mitophagy. In fact, when mitochondria are irreversibly damaged, they are excised from the mitochondrial network by the fission machinery and sequestered into autophagic vesicles for their degradation in the lysosome. This selective mitochondrial degradation via autophagy is called mitophagy. Apart from removal of dysfunctional mitochondria, mitophagy is essential for mitochondria turnover in the basal state and during the development of specialized cells such as red blood cells (Sandoval et al., 2008). The loss of mitochondrial membrane potential is a major trigger for mitophagy (Elmore et al., 2001). The selectivity of mitophagy is controlled by mitochondria dynamics and by the proteins PINK1, Parkin, Bnip3L/Nix, and Bnip3.
Recessively inherited forms of Parkinson's disease are associated with loss-of-function mutations of the PTEN-induced kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin. Under basal conditions Pink1 is imported into the IMM where it is cleaved by PARL in a voltage dependent manner. The resulting fragments will be further degraded by the cytosolic UPS (Jin et al., 2010). Therefore, healthy mitochondria have undetectable levels of PINK1. However, when ΔΨm is lost, full length PINK1 is not further imported and accumulates on OMM. Here, PINK1 phosphorylates ubiquitin at Ser65 of ubiquitinated OMM proteins and the ubiquitin-like domain of Parkin (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014). Once phosphorylated, Parkin enhances the mitophagy signal by generating more ubiquitin chains on OMM proteins that can be further substrates for PINK1 (Lazarou et al., 2015). Subsequently, the autophagic adaptors optineurin, NDP52 (Lazarou et al., 2015) bind the phosphorylated ubiquitins tagging the OMM proteins with LC3 to initiate mitophagy. The contribution of the autophagy adaptor p62/SQSTM1 has been found to be dispensable for mitophagy but important for perinuclear clustering of depolarized mitochondria (Narendra et al., 2010; Lazarou et al., 2015). However, these data were obtained in vitro and thus the physiological relevance in vivo needs to be validated. For instance, while p62 and NBR1 are well expressed in adult muscles, optineurin and NDP52 proteins are barely detectable (Nicot et al., 2014; Lazarou et al., 2015). Bnip3 and Nix (also called Bnip3L) belong to the BH3-only proteins and are implicated in both apoptosis and mitophagy. These proteins translocate to mitochondria, form homodimers and disrupt mitochondrial membrane potential (Sandoval et al., 2008). They contain two evolutionary conserved LIR (LC3 Interacting Region) domains to bind LC3 and GABARAP. Nix preferentially interacts with GABARAP whereas Bnip3 binds LC3 (Novak et al., 2010; Hanna et al., 2012). These results indicate that Bnip3 and Nix have an important role in mitochondrial turnover, acting as autophagy receptors which tether the mitochondria to the autophagosome. Moreover, the induction of autophagy by the overexpression of Bnip3 in cardiomyocytes requires Drp1 translocation to mitochondria to mediate mitochondrial fission (Lee et al., 2011).
Energy requirements during contraction in skeletal muscles increase instantaneously up to 100-fold the consumption of ATP (Blei et al., 1993; Gaitanos et al., 1993). Therefore, to support this high demand of ATP, skeletal and cardiac muscles rely on oxidative phosphorylation for energy production. Indeed these tissues contain the greatest amount of mitochondria. Several reports identified within the myofibers two distinct mitochondrial populations that differ in their subcellular localization, morphology as well as biochemical and functional properties (Howald et al., 1985; Hoppeler et al., 1987; Romanello and Sandri, 2013). Subsarcolemmal (SS) mitochondria are located just underneath the sarcolemma and have a large, lamellar shape. In contrast, the intermyofibrillar (IMF) mitochondria are smaller, more compact, and located between the contractile filaments. However, the existence of two separated mitochondrial pools was challenged by recent findings on high-resolution, three-dimensional electron microscopy (FIB-SEM). Dahl and collaborators showed that SS and IMF are physically connected in human muscles (Dahl et al., 2015). Moreover, Ballaban's lab identified 4 different mitochondrial morphologies: the paravascular mitochondria (PVM), I-band mitochondria (IBM), fiber parallel mitochondria (FPM), and cross fiber connection mitochondria (CFCM) (Glancy et al., 2015). All these mitochondria are highly interconnected forming a conductive pathway that can rapidly distribute energy in the form of electrical conduction throughout myofibers, enabling muscles to respond immediately to changes in energy requirements.
There is compelling evidence that regular exercise has many beneficial effects for human health and life span (Neufer et al., 2015). The effects of exercise are sensed by most organs including cardiovascular, neuroendocrine, respiratory and musculoskeletal systems. Importantly, some forms of exercise training counteract skeletal muscle loss (Hourde et al., 2013; Cohen et al., 2015). On the other hand, physical inactivity has been identified as the fourth risk factor of morbidity/disability and mortality (Neufer et al., 2015). Beneficial adaptations to endurance exercise training (moderate intensity with prolonged duration) in skeletal muscle include the activation of transcriptional dependent pathways that impinge on mitochondrial biogenesis (Little et al., 2011), mitochondrial quality and mitophagy (Grumati et al., 2011; He et al., 2012a; Lo Verso et al., 2014).
Mitochondrial biogenesis requires a coordinated action on nuclear and mitochondrial genomes that is regulated by the coactivators PGC-1α and PGC-1β (peroxisome proliferator-activated receptor-γ coactivator-1α and β) PGC-1 family members are preferentially expressed in tissues enriched in mitochondria like heart, adipose tissue and slow-twitch skeletal muscle. Exercise, fasting and cold exposure highly regulate their expression. Since PGC-1α and PGC-1β are coactivators that lack DNA binding domains, they elicit their function by modulating the activity of several transcription factors including PPARs, nuclear respiratory factors (NRFs), myocyte enhancing factors (MEFs), estrogen-related receptor (ERR), forkhead box (FoxOs), and yin-yang (YY1) (Lin et al., 2005; Olesen et al., 2010; Zechner et al., 2010). Exercise greatly stimulates PGC-1α via a transcription-dependent upregulation and by several post-translational modifications like phosphorylation and deacetylation (Wright et al., 2007; Cantó et al., 2009; Egan et al., 2010). Moreover, acute endurance exercise promotes the translocation of PGC-1α from the cytosol to the nucleus (Wright et al., 2007) and mitochondria (Safdar et al., 2011). This subcellular relocalization helps nuclear and mitochondrial crosstalk to promote mitochondrial biogenesis (Safdar et al., 2011). Indeed, both Tfam and nuclear gene products are imported into mitochondria where they regulate the expression of mitochondrial proteins required for ATP synthesis. Interestingly, deletion of PGC-1α or PGC-1β in mice showed mild phenotypes (Leone et al., 2005; Lelliott et al., 2006; Vianna et al., 2006; Sonoda et al., 2007). Although, exercise capacity was found reduced in PGC-1α knockout mice (Leone et al., 2005; Handschin et al., 2007; Wende et al., 2007), exercise-induced mitochondrial adaptations in PGC-1α knockout mice were similar to wild-type animals (Leick et al., 2008; Geng et al., 2010). These confilicting results can be probably explained because these two coactivators are redundant and partially compensate each other. In fact, when PGC-1β is deleted on a generalized PGC-1α-deficient background (Zechner et al., 2010) it was shown that these factors share a subset of target genes and display overlapping functions. Accordingly, exercise performance, in this model, is greatly decreased compared to single PGC-1α or PGC-1β knockout animals (Zechner et al., 2010). PGC-1α and PGC-1β are necessary for the maintenance of mitochondrial function (Zechner et al., 2010). Therefore, the deletion of both, PGC-1α and PGC-1β induce severe mitochondrial dysfunction that leads to rapid depletion of glycogen stores and to premature fatigue during exercise (Zechner et al., 2010). Moreover, PGC-1α and PGC-1β control mitochondrial dynamics by stimulating Mfn1 and Mfn2 gene expression in a ERRα-dependent manner (Soriano et al., 2006; Russell et al., 2013). Accordingly, Mfn1, Mfn2, and Drp1 are severely downregulated in PGC-1α and PGC-1β muscle-specific knockout mice (Zechner et al., 2010). Moreover, PGC-1α fine tunes autophagy regulation during some forms of disuse atrophy (Vainshtein et al., 2015). Therefore, PGC-1 coactivators not only increase mitochondrial content but also mitochondrial quality by modulating fusion/fission processes and mitophagy. Surprisingly, PGC-1α and PGC-1β muscle-specific knockout mice do not exhibit defects in muscle fiber type formation in terms of myosin content (Zechner et al., 2010). This is in contrast with a previous study that showed an increase of type I fibers in transgenic mice that specifically express PGC-1α in fast glycolytic muscles (Lin et al., 2002). PGC-1α mRNA levels drop in different atrophying conditions like denervation (Sandri et al., 2006), hind limb suspension (Cannavino et al., 2015) aging (Chabi et al., 2008) or type 2 diabetic patients (Patti et al., 2003). Importantly, maintaining PGC-1α levels high during catabolic conditions, either by the use of transgenic mice or by transfecting adult muscle fibers, spares muscle mass during hind limb suspension, sarcopenia, cardiac cachexia, denervation, fasting, or expression of constitutively active FoxO3 (Sandri et al., 2006; Wenz et al., 2009; Geng et al., 2011; Cannavino et al., 2015). Similar beneficial effects have been recently obtained by overexpression of PGC-1β, an homolog of PGC-1α (Brault et al., 2010). The positive action on muscle mass of these cofactors is caused by the inhibition of the autophagic-lysosomal and the ubiquitin proteasome degradation pathways. PGC-1α and β reduce protein breakdown by inhibiting the transcriptional activity of FoxO3 and NF-κB, but they do not affect protein synthesis (Brault et al., 2010). Thus, these cofactors can prevent the excessive activation of proteolytic systems by inhibiting the action of the pro-atrophy transcription factors without perturbing the translational machinery.
A new splicing variant transcript from PGC-1α gene, PGC-1α4, was identified and shown to be involved in the regulation of muscle mass (Ruas et al., 2012). PGC-1α4 expression is induced with resistance exercise protocols (fewer repetitions at high muscle loads aimed at increasing muscle mass) and not with endurance exercise. Skeletal muscle-specific transgenic overexpression of PGC-1α4 results into hypertrophic and stronger mice (Ruas et al., 2012). PGC-1α4 controls muscle mass by inducing IGF1 and repressing the myostatin pathway. Importantly, PGC-1α4 overexpression counteract muscle loss induced by hindlimb suspension and cancer-cachexia (Ruas et al., 2012). However, the results obtained by Ruas and collegues are in contrast with different reports analyzing exercise-mediated adaptations in human muscles (Ydfors et al., 2013; Lundberg et al., 2014; Silvennoinen et al., 2015). In humans, PGC-1α4 is regulated transiently with exercise regardless of mode (resistance or endurance) (Ydfors et al., 2013; Lundberg et al., 2014; Silvennoinen et al., 2015). Moreover, chronic training adaptations, such as increases in muscle mass and force do not correlate with changes in the expression of PGC-1α4 after resistance exercise or after the combination of aerobic and resistance exercise (Lundberg et al., 2014).
The beneficial effects of exercise can be also mediated by contraction-induced changes in calcium homeostasis, ATP consumption and ROS production. The alteration in calcium levels activates calcium-sensitive signaling such as calcium/calmodulin-dependent protein kinases (CaMK) and calcineurin/NFAT systems that are important for fiber type regulation and muscle plasticity (Wu et al., 2002; Blaauw et al., 2013).
ER-mitochondria crosstalk is essential for excitation-calcium coupling during muscle contraction. Indeed a great amount of ATP produced by mitochondria is consumed by SERCA pumps. Moreover, the positioning of intermyofibrillar mitochondria nearby to SR makes them affected by the calcium wave of contraction. In this context, the role of mitochondrial calcium uptake in the maintenance of skeletal muscle mass was recently investigated (Mammucari et al., 2015). In vivo gain- and loss-of-function experiments in skeletal muscle showed that modulation of mitochondrial calcium uniporter (MCU) controls mitochondrial volume. Overexpression of MCU in muscles leads to increased protein synthesis with a resulting hypertrophic phenotype which impinge on IGF1/AKT and PGC-1α4 pathways. Moreover, MCU overexpression protects from denervation-induced atrophy. Supporting the importance of MCU in skeletal muscle is the identification of mutations of MICU1, an MCU regulator, in patients with proximal muscle weakness, learning difficulties and extrapiramidal motor disorder (Logan et al., 2014). In addition, different reports linked calcium homeostasis with the control of muscle mass (Blaauw et al., 2013). Therefore, the investigation of calcium entry pathways in skeletal muscle mitochondria open the possibility to better understand the role of calcium in ER-mitochondria communication and its impacts on muscle mass regulation.
On the other side ATP depletion modifies the AMP/ATP ratio, activating the energy sensor AMP-activated protein kinase (AMPK). The activation of this signal transducer is crucial for the metabolic adaptations in response to energy stress. AMPK inhibition of anabolic pathways together with stimulation of catabolic pathways are aimed at conserving/restoring ATP (Ha et al., 2015). In skeletal muscle AMPK activation is sufficient to increase glucose uptake (by increasing GLUT4 translocation), fatty acid oxidation and mitochondrial biogenesis. In fact, AMPK-dependent phosphorylation of PGC-1α is required for the induction of PGC-1α promoter and for the transcription of many AMPK target genes (Jäger et al., 2007). Summarizing, both calcium-dependent pathways and AMPK modulate the activity and the expression of PGC-1α (Wu et al., 2002; Jäger et al., 2007). Therefore, many of the beneficial effects of exercise converge on the activation of PGC-1 signaling which is central for the mitochondrial and metabolic adaptation to exercise.
Different reports demonstrated that acute as well chronic exercise stimulates autophagy in different tissues (Grumati et al., 2011; He et al., 2012a,b; Lo Verso et al., 2014). Experiments conducted in inducible Atg7-deficient mice unraveled the mechanisms behind autophagy induction following exercise (Lo Verso et al., 2014). Interestingly, autophagy impairment does not affect exercise performance suggesting that autophagy is not required to sustain muscle contraction during exercise training (Lira et al., 2013; Lo Verso et al., 2014). However, autophagy is critical for muscle homeostasis after eccentric contractions. In fact, autophagy is necessary to remove dysfunctional mitochondria that have been altered by exercise in order to prevent excessive ROS production (Lo Verso et al., 2014). It is well known that exercise training and muscle contraction cause an increase in ROS production and a transient oxidative stress (Davies et al., 1982; Powers and Jackson, 2008). Indeed, oxidative stress has an important role in muscle signaling (Finkel, 1998; Rhee et al., 2000; Sena and Chandel, 2012), but can also induce damage to contractile proteins and organelles (Carnio et al., 2014). Although counterintuitive, antioxidant treatment leads to exercise intolerance in wild-type animals and do not ameliorate the physical performance of autophagy-deficient mice (Lo Verso et al., 2014). Moreover, control animals treated with antioxidants display mitochondrial dysfunction and autophagy impairment. Different reports support a crucial role of oxidative stress signaling during exercise in the maintenance of mitochondrial fitness (Gomez-Cabrera et al., 2005, 2008) and in the induction of molecular regulators of insulin sensitivity and antioxidant defense (Ristow et al., 2009). All together these results are consistent with the mitohormesis concept (Ristow and Schmeisser, 2014). Mitohormesis, the non-linear response to mitochondrial ROS formation, exhibits a dual dose-response (Ristow and Schmeisser, 2014). Accordingly, high ROS levels are detrimental due to the induction of oxidative stress with cellular damage. On the contrary, low ROS levels are essential for maintenance of cellular function as well as for improving oxidative stress for the promotion of health and lifespan (Ristow and Schmeisser, 2011; Owusu-Ansah et al., 2013). Therefore, understanding how mitohormesis is regulated could promise a potential application in the promotion of cellular homeostasis and longevity.
Although both endurance as well as resistance exercise can promote significant health benefits, the molecular response signature will be different according to the frequency, intensity, duration and the type of exercise (resistance or endurance). The objectives of resistance exercise are an increase in muscle mass, strength and power. The resistance exercise-induced adaptive hypertrophic response is mediated by the anabolic factor insulin growth factor 1 (IGF1) via the IGF1/PI3K/AKT/mTOR pathway. Indeed, increased IGF1 levels precedes muscle hypertrophy achieved by functional overload (Adams and Haddad, 1996). Moreover, IGF1 stimulates fatty acid uptake and insuling sensitivity (Clemmons, 2012) explaining in part some of the beneficial effects of exercise. ATP citrate lyase (ACL) is a cytosolic enzyme that catalyzes the conversion of mitochondria-derived citrate into oxaloacetate and Acetyl-CoA important for lipid synthesis and acetylation. ACL activity is controlled by a post-translational modification via the IGF1/PI3K/AKT pathway. Overexpression of ACL in muscles improved mitochondrial function via stabilization of respiratory chain supercomplexes formation due to increases in cardiolipin synthesis (Das et al., 2015). Thus, IGF1 treatment of myotubes significantly increases mitochondrial respiration due to IGF1-dependent ACL activation suggesting that the activation of the IGF1-ACL pathway increases protein synthesis as well as stimulating the formation of the ATP necessary for this anabolic process (Das et al., 2015). The correlation of mitochondrial fitness to muscle homeostasis is confirmed by the fact that ACL overexpression in human myotubes is sufficient to induce hypertrophy while ACL activity is downregulated in sarcopenic and in dexamethasone-induced atrophic muscles (Das et al., 2015). All together, these different models offer a useful platform to further investigate the molecular mechanisms explaining how improvements in mitochondrial function can be translated into an increase in protein synthesis and therefore in hypertrophy.
Muscle atrophy is an active process, controlled by specific signaling pathways and transcriptional programs (Lecker et al., 2004; Sandri, 2013). Of note, the muscle wasting transcriptome signature is characterized by the coordinated reduction of genes encoding key enzymes for ATP production and glycolysis (Lecker et al., 2004). Accordingly, alterations in mitochondrial morphology and function are frequently associated with atrophying muscles in aging (see Section “The Role of Disrupted Mitochondria Quality Control Pathways in Aging Sarcopenia”) as well as in several wasting conditions such as burn injury (Porter et al., 2013), intensive care unit-acquired weakness (Friedrich et al., 2015), insulin resistance (Crescenzo et al., 2014), chronic obstructive pulmonary disease (COPD) (Mathur et al., 2014), cancer cachexia (Antunes et al., 2014), and in different neuromuscular disorders (Katsetos et al., 2013). Over the last years, several in vivo genetic perturbation models indicate a key role of mitochondrial morphology and mitochondrial dysfunction signaling in the activation of nuclear programs controlling muscle loss. The maintenance of a functional mitochondrial network is indeed particularly important for tissues that are highly structured and metabolically active such as neurons, cardiac and skeletal muscles. These tissues are constituted by post-mitotic cells that do not divide and consequently, cannot dilute damaged/dysfunctional mitochondria through cellular division. Therefore, post-mitotic tissues depend on the activation of mitochondria quality control pathways to preserve or restore mitochondrial function.
Mitochondrial damage is attenuated with different defensive strategies including the activation of mitochondrial proteolytic systems, mitochondrial dynamics and the lysosomal degradation of mitochondrial cargo. For this reason, a failure in any of these systems predisposes to tissue dysfunction and degeneration (Figure 2). The importance of the actions of mitochondrial proteases, which remove misfolded or damaged mitochondrial proteins, in the control of muscle mass was confirmed by data obtained with several knockout animal models. Loss in the activities of the proteases PARL (Cipolat et al., 2006), Omi/HtrA2 (Jones et al., 2003; Martins et al., 2004), and Hax1 (Chao et al., 2008) lead to a similar phenotype, characterized by mitochondrial dysfunction, increased apoptosis, neurodegeneration and atrophy. A fine equilibrium of mitochondrial fusion/fission processes is needed to preserve muscle mass and prevent muscle wasting. This balance is regulated according to the physiological needs of cells.
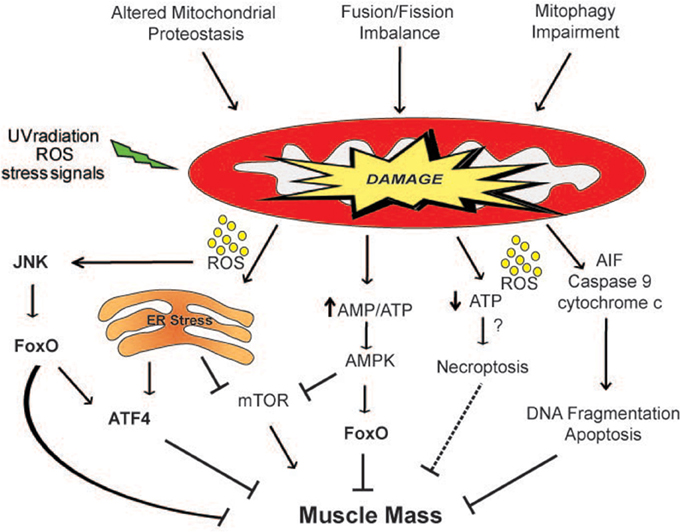
Figure 2. Mitochondria-derived signaling pathways controlling muscle mass. Mitochondrial network accumulates damage when the mitochondrial quality control mechanisms are impaired. ROS produced by the defective organelles induces muscle atrophy through the activation of the JNK-FoxO signaling pathway and of ER stress. Moreover, the release of dangerous factors such as AIF, cytochrome c and ROS can induce apoptosis and/or necroptosis. The activation of the energy sensor AMPK by an increase in the AMP/ATP ratio inhibits mTOR and directly phosphorylates FoxO3 (Ser413 and Ser588) increasing its transcriptional activity, affecting muscle mass. The dashed line indicates a mechanism that needs more studies.
Fused mitochondria are preferred when optimal mitochondrial function is needed. Indeed intermixing of mitochondrial content by mitochondrial fusion increases the bioenergetic capacity of the cell and is essential for the excitacion-contraction coupling in skeletal muscles (Eisner et al., 2014). Moreover, under conditions of high energy demand, mitochondrial fusion has a survival function, by protecting mitochondria against mitophagy, optimizing ATP production and leading to a sustained cell viability during nutrient deprivation (Gomes et al., 2011; Rambold et al., 2011). The biological importance of mitochondrial fusion in early development is revealed by the embryonic lethality of mouse knockouts for Opa1, Mfn1, and Mfn2 (Chen et al., 2003; Davies et al., 2007). However, few conditional tissue-specific knockout mice have been generated (Chen et al., 2011; Zhang et al., 2011) and some of them show a lethal phenotype therefore, limiting the studies of the role of these factors in mitochondrial function in adulthood. Therefore, little is known about the contribution of fusion machinery to mitochondrial function and tissue homeostasis in most tissues in vivo, including skeletal muscle fibers. The neurodegenerative disorders dominant optic atrophy (DOA) (Amati-Bonneau et al., 2008) and Charcot-Marie-Tooth type 2A (CMT2A) (Zuchner et al., 2004), are associated with loss of function mutations of Opa1 and Mfn2 genes, respectively. Noteworthy, patients affected with both DOA and CMT2A develop myopathies (Yu-Wai-Man et al., 2010; Feely et al., 2011). Likewise, neuromuscular defects related to precocious age-dependent axonal and myelin degeneration are present in mouse models resembling the Opa1 mutations most frequently associated with DOA patients (Alavi et al., 2009; Sarzi et al., 2012). Skeletal muscle from these models have alterations at the ultrastructural level with increased smaller mitochondria. In addition, mitochondria display disorganized cristae and accumulate lipid droplets (Sarzi et al., 2012).
Further support in the role of Opa1 in the homeostatic control of muscle mass comes from the observations obtained with a genetic model of controlled Opa1overexpression (Civiletto et al., 2015; Varanita et al., 2015). Opa1 transgenic mice are protected from acute muscle loss induced by denervation (Varanita et al., 2015) as well as from chronic muscle loss in a model of myopathy caused by muscle-specific deletion of the mitochondrial subunit COX15 (Civiletto et al., 2015). The analysis of the expression levels of the major atrogenes belonging to the ubiquitin-proteasome and of the autophagy-lysosome systems revealed a reduction of MuRF1 and LC3 expression levels in denervated Opa1 transgenic muscles. Furthermore, increased levels of Opa1 during denervation were sufficient to blunt denervation-induced mitochondrial dysfunction (Varanita et al., 2015).
In line with the relevance of mitochondrial fusion proteins in the normal physiology of skeletal muscles, Mfn2 downregulation induces changes in ER-mitochondria communication that alters mitochondrial calcium buffering (Sebastian et al., 2012; Ainbinder et al., 2015). Moreover, Mfn2 protein levels are reduced in several catabolic conditions (Bach et al., 2005; Hernandez-Alvarez et al., 2010; Lokireddy et al., 2012). Mul1 is a FoxO-dependent ubiquitin-ligase involved in the ubiquitination and further degradation of Mfn2 during muscle atrophy. Accordingly, the maintenance of high Mfn2 proteins levels in starved muscles achieved by the downregulation of Mul1 partially protected from starvation-induced muscle loss (Lokireddy et al., 2012). On the contrary, the ablation of Mfn1 and Mfn2 in muscle-specific knockout mice induces a profound muscle atrophy. These models display mitochondrial dysfunction and compensatory mitochondrial proliferation, reduction of mtDNA as well as accumulation of point mutations and deletions of the mitochondrial genome (Chen et al., 2010). Mechanistically, Mfn2 deficiency in muscles induces a remodeling of the mitochondrial network leading to mitochondrial dysfunction and ROS production (Sebastian et al., 2012). Oxidative stress signaling is sufficient to activate an UPR that blunts insulin signaling via JNK and leads to insulin resistance (Sebastian et al., 2012). Mitochondrial fission is an essential component of the mitochondrial quality control mechanism. When mitochondria divide, two organelles are generated, one with high membrane potential that might undergo fusion and another fragment with low mitochondrial potential and reduced levels of OPA1 that will not rejoin the mitochondrial network and will be likely removed by autophagy (Twig et al., 2008).
Impairment of fission leads to the disruption of the selective mitochondrial autophagic degradation followed by accumulation of dysfunctional organelles (Twig et al., 2008; Dagda et al., 2009; Kageyama et al., 2014; Ikeda et al., 2015; Song et al., 2015). In fact, mutations in mitochondrial fission components can be lethal. An infant girl born with a dominant-negative mutation of Drp1 presented alterations in brain development and in metabolism that cause neonatal lethality (Waterham et al., 2007). Moreover, genetic perturbations leading to the ablation of Drp1 in heart or brain are lethal (Ishihara et al., 2009; Wakabayashi et al., 2009; Kageyama et al., 2014; Ikeda et al., 2015; Song et al., 2015). Three different reports investigate Drp1 role in heart physiology. Disrupted mitochondrial fission, obtained with cardiac-specific ablation of Drp1, induces accumulation of defective mitochondria due to impaired autophagy/mitophagy, that over-time promotes cardiomyocyte death (Kageyama et al., 2014; Ikeda et al., 2015; Song et al., 2015). In addition, abnormalities in mitochondrial fission has also been implicated in neurodegenerative disorders such as Alzheimer's, Huntington, and Parkinson (Chaturvedi and Flint Beal, 2013). In skeletal muscle, atrophying conditions such as fasting, denervation or the overexpression of a constitutively active form of FoxO3 are characterized by alterations of the mitochondrial network (Romanello et al., 2010). Furthermore, the disruption of the mitochondrial network is a crucial amplificatory loop for the muscle atrophy program. The acute overexpression in muscles of the fission machinery per se promotes changes of mitochondrial morphology which are followed by mitochondrial dysfunction, energy stress and AMPK activation. As a consequence, AMPK modulates FoxO3 transcriptional activity in order to induce muscle atrophy via the activation of ubiquitin-proteasome and autophagy pathways (Romanello et al., 2010). Accordingly, inhibition of fission machineries or downregulation of AMPK is sufficient to protect against muscle loss in atrophying conditions (Romanello et al., 2010). These findings pave the way to further investigate the causal link between changes in mitochondria morphology and alterations in muscle homeostasis. A recent report investigates the consequences of mitochondrial network remodeling on muscle growth. The constitutive muscle-specific overexpression of Drp1 (Touvier et al., 2015) leads to muscle mass loss and decreased exercise performance (Touvier et al., 2015). In this mouse model, stress-induced mitochondria-dependent signals activate both, the UPRmt and the eIF2α-ATF4-Fgf21 axis causing a reduction in protein synthesis and a blockade of growth hormones actions that prevent muscle growth (Touvier et al., 2015).
Mitophagy is a housekeeping mechanism important to keep under control the turnover of mitochondria in both, the basal state as well as under stress conditions. A finely tuned autophagic system is required for muscle maintenance. Notably, genetic manipulations to activate or inhibit autophagy results into muscle atrophy (Mammucari et al., 2007; Masiero et al., 2009; Romanello et al., 2010). Therefore, autophagy needs to be properly regulated, otherwise it can become detrimental instead of being protective. Several lines of evidence demonstrate that constitutive basal autophagy, which preserves mitochondrial function, is crucial for skeletal muscle homeostasis. Indeed, dysregulation of autophagic flux is detrimental for myofiber health and is a common feature of a group of muscle disorders with alterations of lysosomal proteins such as Danon or Pompe Disease as well as in the Vici Syndrome (Sandri et al., 2013). Moreover, defective autophagy plays a role in congenital muscular dystrophies caused by defects in collagen VI production (Grumati et al., 2010), laminin A/C (Ramos et al., 2012), or dystrophin (De Palma et al., 2012). These dystrophic models have in common a hyperactivation of Akt/mTOR signaling pathway that inhibits autophagy. Dystrophic muscles present accumulation of structurally altered mitochondria together with myofiber degeneration. Noteworthy, reactivation of autophagy flux by dietary or pharmacological tools, like rapamycin, cyclosporine A, or AICAR rescues the dystrophic phenotype by clearing the abnormal mitochondria (Grumati et al., 2010; De Palma et al., 2012; Pauly et al., 2012; Ramos et al., 2012). In agreement, suppression of autophagy by muscle-specific ablation of Atg5 and Atg7, the E3 ubiquitin ligases necessary for autophagosome formation, exacerbates fasting- and denervation- induced atrophy (Masiero et al., 2009). Moreover, impairment of autophagy induces accumulation of abnormal mitochondria, induction of oxidative stress, apoptosis, muscle atrophy, weakness and several features of myopathy (Raben et al., 2008; Masiero et al., 2009). Likewise, reduction of mitophagy caused by ablation of Pink and Parkin induces mitochondrial dysfunction and increased sensitivity to oxidative stress followed by muscle degeneration (Clark et al., 2006; Park et al., 2006; Billia et al., 2011). On the contrary, autophagic flux is increased in muscles in both exercise (Grumati et al., 2011; Lo Verso et al., 2014) and catabolic conditions (Sandri, 2013). For example, in skeletal muscle, mitochondrial dysfunction caused by the transient overexpression of FoxO3, Mul1 (Lokireddy et al., 2012), Bnip3, or Nix (Romanello et al., 2010) triggers autophagy and induces muscle atrophy (Mammucari et al., 2007; Romanello et al., 2010). Naf-1, the nutrient-deprivation autophagy- factor 1, is a ER-Bcl2 interacting protein identified as a novel autophagy regulator. Its downregulation is accompanied by the presence of abnormal mitochondria and increased autophagy that leads to muscle weakness and atrophy (Chang et al., 2012).
Oxidative stress is increased when autophagy is dysregulated. ROS has detrimental effects both within and outside mitochondria. Indeed, autophagy deficient muscle show an increase of myosin and actin oxidation that reduces their sliding properties and causes muscle weakness. Anti-oxidant treatment restores a normal force generation and sliding properties even if it does not rescue muscle atrophy (Carnio et al., 2014). Mutations of the Superoxide Dismutase (SOD1) gene is behind 20% of cases of inherited amyotrophic lateral sclerosis (ALS). ALS is a neurodegenerative disease characterized by motoneuron degeneration and muscle atrophy. Aberrant ROS production affects mitochondrial function producing a feedback loop that in turn exacerbates mitochondrial damage. Transgenic muscle-specific mice expressing the SOD1 G93A mutation, an ALS-associated human SOD1 mutation display muscle atrophy which is mainly through autophagy activation (Dobrowolny et al., 2008).
Skeletal muscle plays a significant role in the metabolic control of glucose, lipids, and energy. Importantly, these processes are coordinated with changes in mitochondrial content, shape, and/or function. Therefore, the question now arises as to how modulation of the mitochondrial quality pathways and specifically of mitophagy can affect muscle metabolism. Despite conflicting results, two different studies investigate the connection between autophagy/mitophagy and metabolic regulation in muscles. The first one, uses transgenic mice with knock-in mutations on Bcl2 phosphorylation sites that cannot trigger autophagy by fasting or exercise. These mice show decreased exercise endurance, impaired exercise-induced glucose uptake and are more sensitive to high fat diet-induced glucose intolerance (He et al., 2012a). Since the transgene is expressed in all cell types, it cannot be ruled out whether the observed effects are resulting from the combination of the actions exerted by different metabolically active tissues (liver, heart, muscle, brain). In the second study, muscle-specific autophagy deficient mice (Atg7 knockout) are protected from high-fat diet induced obesity and insulin resistance. The protective effects depend on dysfunctional mitochondrial signals sufficient to activate an UPR via ATF4, which induces Fgf21 expression (Kim et al., 2013). Therefore, it is not clear how autophagy can fine-tune the metabolic pathways in response to different cellular cues. An important future goal is to establish the metabolic impact of mitochondrial quality control pathways in skeletal muscle.
As already discussed, ROS act in a hormetic fashion (Ristow and Schmeisser, 2014). The beneficial or detrimental action will depend on the level and persistance of ROS flow, as well as on the antioxidant capacity of target cells (Barbieri and Sestili, 2012). While low levels of ROS are associated with positive effects on muscle physiology, on the other hand, excessive production of free radicals accelerates muscle proteolysis (Powers et al., 2012; Reid et al., 2014). Accordingly, the exposure of oxidants to myotubes results in muscle atrophy (McClung et al., 2009; Gilliam et al., 2012). In addition, several models of disuse atrophy like mechanical ventilation, limb immobilization, hindlimb unloading or bed rest are associated with increased ROS production (Pellegrino et al., 2011). Recent evidence indicates mitochondrial ROS production as a required signaling step to induce mitochondrial dysfunction and muscle disuse atrophy (Min et al., 2011; Powers et al., 2011; Talbert et al., 2013b). Mechanistically, oxidative stress can impinge on proteolytic pathways in multiple ways; First, ROS can regulate NF-kB and FoxO transcriptional factors (Dodd et al., 2010) activating the autophagy-lysosome and the ubiquitin-proteasome systems (Taillandier et al., 1996; Levine et al., 2008, 2011; Andrianjafiniony et al., 2010; Hussain et al., 2010; Brocca et al., 2012). Second, the muscle proteases calpain (Taillandier et al., 1996; McClung et al., 2009; Andrianjafiniony et al., 2010; Nelson et al., 2012; Talbert et al., 2013a) and caspase-3 (Levine et al., 2008; Nelson et al., 2012; Talbert et al., 2013a) are activated during disuse atrophy. Third, oxidized proteins are more prone to proteolysis because: (a) their susceptibility to be degraded by caspase-3 and calpain is enhanced, in part because of unfolding of the proteins (Smuder et al., 2010), (b) they can be directly degraded by the proteasome without being ubiquitinated (Grune et al., 2003), and (c) ubiquitin conjugation activity is increased by oxidative stress (Shang et al., 1997).
Summarizing, all these data strongly support the involvement of mitochondrial morphology and/or function in the activation of signaling pathways that control muscle mass.
Sarcopenia is defined as the progressive age-related decline in muscle mass and force. It is considered a primary risk for developing major human pathologies that can result into disability and increased vulnerability to death. It has been estimated that roughly, 1.1 and 1.9/kg of muscle mass are lost per decade in women and men, respectively (Janssen et al., 2000). Of note, muscle strength precedes muscle loss, declining three-times faster than muscle loss (Goodpaster et al., 2006). Age-associated muscle loss begins at a slow rate around 30 years of age, then it accelerates and starts to be noticeable after 45 years of age (Janssen et al., 2000). Beyond the sixth decade of life more severe alterations such as spinal motor neurons alterations can be observed (Booth et al., 1994) while the most affected age is over 80 years old (Cruz-Jentoft et al., 2010). The average life expectancy of human beings is rapidly increasing. According to the National Institute on Aging (NIH), in 2010 8% of the world's population were people over 65 years old and this number is expected to double by 2050. Although the resulting clinical, economical and social relevance of sarcopenia, the mechanisms that drive the age-related loss of muscle mass and function are not completely understood. Sarcopenia is a complex multifactorial process. Recently, nine primary hallmarks of aging were specified (Lopez-Otin et al., 2013). Among these, mitochondrial dysfunction has been long suggested as one of the mechanisms of aging sarcopenia (Trounce et al., 1989; Melov et al., 2007; Gouspillou et al., 2010, 2014a,b; Picard et al., 2011; Carnio et al., 2014).
It has been shown that muscle mitochondrial content declines with age (Conley et al., 2000; Short et al., 2005; Chabi et al., 2008), however, there is no clear consensus (Mathieu-Costello et al., 2005; Callahan et al., 2014; Gouspillou et al., 2014b). Mitochondrial biogenesis is important for determining mitochondrial content and function. It depends on the transcription, translation and import of new proteins into pre-existing organelles. In agreement to the observed age-associated mitochondrial content reduction and dysfunction, PGC1-α mRNA and protein levels are reduced in aged muscles (Short et al., 2005; Chabi et al., 2008; Safdar et al., 2010; Ghosh et al., 2011; Joseph et al., 2012; Ibebunjo et al., 2013). On the other hand, muscle-specific PGC1-α transgenic mice are protected from the reduction in mitochondrial function and content observed during aging (Wenz et al., 2009). Moreover, overexpression of PGC1-α is sufficient to prevent age-related muscle loss (Wenz et al., 2009). Blunted mitochondrial biogenesis in aging is supported also by the fact that nascent mitochondrial precursors are more prone to be degraded by cytosolic factors (Huang et al., 2010).
Several mechanisms necessary for mitochondrial homeostasis are dysregulated in sarcopenia. In fact, during aging, ROS production increases over a certain threshold, overcoming cellular antioxidant defense. Therefore, the initial homeostatic response of ROS signaling is subverted and accelerates the age-associated damage (Lopez-Otin et al., 2013). The resulting oxidative stress, induces post-translational modifications which compromise protein function (Baraibar et al., 2013). The target proteins modified by oxidative stress (the oxy-proteome components) in aged muscles have been identified recently, by the resolution of carbonylated proteins (an indicator of oxidized damaged proteins) by two-dimensional gel electrophoresis (Lourenco Dos Santos et al., 2015). Many of them are mitochondrial proteins which affect mitochondrial proteostasis (Lourenco Dos Santos et al., 2015; Ross et al., 2015).
Despite certain discrepancy between reports, the balance between fusion and fission is altered in sarcopenic muscles. Some studies reported that muscle mitochondria in old rats are smaller than young controls (Ljubicic et al., 2009; Iqbal et al., 2013). On the other hand, giant mitochondria were observed in aged muscles of houseflies (Rockstein and Bhatnagar, 1965), muscle cells of humans (Beregi et al., 1988), mice (Beregi et al., 1988), and rats (Navratil et al., 2008). Analysis of the longitudinal vs. transversal mitochondrial orientation with transmission electron microscopy reveals that muscle mitochondria from aged mice are much more enlarged and branched than young muscles (Leduc-Gaudet et al., 2015). Even though the authors did not find any difference in the expression of the dynamic machineries between young and old mice, they suggest that the alteration of mitochondrial morphology in old muscles is the result of a fusion/fission imbalance toward mitochondrial fusion (Bori et al., 2012; Leduc-Gaudet et al., 2015). In contrast, other groups find a downregulation only of Opa1 (Navratil et al., 2008; Joseph et al., 2012) or an upregulation of the fission machinery (Iqbal et al., 2013). Interestingly, a transcriptomic and proteomic study found that reduced expression of all the components of the mitochondria dynamic machineries correlates with age-related muscle loss in rats (Ibebunjo et al., 2013). The employment of new tools, like FIB-SEM, would be useful to correlate mitochondrial dynamics alterations with mitochondrial function in sarcopenic muscles.
Several evidences indicate an alteration in mitochondrial turnover during sarcopenia. Considering the decline of mitochondrial biogenesis together with the progressive accumulation of macromolecules and dysfunctional organelles it seems to create a likely basis for impaired mitochondrial function and removal during aging. In agreement, some autophagic regulators like LC3 lipidation, Atg7, p62, beclin1, Bnip3, Parkin, and LAMP2 decrease with age (Russ et al., 2012; Joseph et al., 2013; Carnio et al., 2014; Gouspillou et al., 2014b). Moreover, a genetic muscle-specific model of autophagy deletion (Atg7 knockout mice) displays precocious aging characterized by increased oxidative stress, mitochondrial dysfunction, muscle loss and weakness and degeneration of neuromuscular junctions. All together, these results put mitophagy central in age-related mitochondrial dysfunction and critical to prevent age-related denervation and weakness. In contrast, others found increased levels of lipidated LC3 (Wenz et al., 2009), Atg7, beclin1 (Fry et al., 2013), parkin, and p62 (O'Leary et al., 2013). The divergence in these results highlight the necessity of complementing the analysis of the expression of single autophagic regulators with assays to evaluate the complete autophagic process by flux measurements for the correct interpretation of the results (Klionsky et al., 2012).
Even though, aging sarcopenia is an unavoidable consequence of getting old, it can be reduced with regular exercise and with nutritional interventions. Notably, the benefits of both strategies are mediated by activation of autophagy. Exercise training increases mitochondrial content and turnover by activating both, mitochondrial biogenesis (Little et al., 2011) and autophagy (Vainshtein et al., 2014). As mitochondrial turnover and function are both altered in sarcopenic muscles, exercise seems to be a good countermeasure to improve mitochondrial function (Jubrias et al., 2001; Short et al., 2003; Menshikova et al., 2006; Melov et al., 2007) and therefore, to reduce the sarcopenic process (Jubrias et al., 2001; Melov et al., 2007; Kern et al., 2014; Zampieri et al., 2015). Interestingly, Melov et al. (2007) defined the aging transcriptome signature. The sarcopenic muscle profile was enriched with genes associated with mitochondrial dysfunction compared with young muscles (Melov et al., 2007). Genes important for energy production and mitochondrial function decreased with age. Examples of these genes are the genes encoding the ubiquinol-cytochrome C reductase hinge protein (UQCRH), the β subunit of succinyl-CoA synthase (SUCLA2), and the C subunit of succinate dehydrogenase (SDH). Six months of resistance training were sufficient to revert the transcriptome expression profile of the genes associated with mitochondrial metabolism and electron transport to the expression levels characteristic of young people. However, at the functional/phenotypical levels resistance exercise partially improved muscle strength (Melov et al., 2007). Importantly, the benefits of exercise in reverting the sarcopenic phenotype do not have the same effect on advanced age (over 80 years of age) which have limited muscle plasticity (Slivka et al., 2008; Raue et al., 2009), with the oldest ones being the most affected by the consequences of aging muscle atrophy (Cruz-Jentoft et al., 2010). Caloric restriction (CR) is a powerful nutritional intervention that has been reported to increase lifespan, to maintain metabolism at a more youthful-like state and to prevent chronic diseases (Fontana and Partridge, 2015). The investigation of the effects of lifelong CR in rats muscles demonstrated that CR increases the expression of some autophagic proteins like LC3, Atg7, and 9 and LAMP2 attenuating the age-dependent decline in autophagy. Therefore, the chronic activation of autophagy decreases oxidative damage as well as apopoptotic DNA fragmentation and is sufficient to reduce age-related myocyte degeneration maintaining skeletal muscle homeostasis (Wohlgemuth et al., 2010). Chronic moderate CR adaptation in muscles from humans (30% lower energy intake) and rats is mediated by three major pathways: IGF1/insulin/FoxO, mitochondrial biogenesis, and inflammation (Mercken et al., 2013). CR-mediated responses lead to reduction of IGF1/AKT pathway with the consequent increase of FoxO3 and 4 transcript as well as an upregulation of FoxO-dependent antioxidant system and autophagy-related genes (Mercken et al., 2013). Moreover, CR induces a transcriptional reprogramming profile of molecular pathways that become more similar to the profile of young individuals (Mercken et al., 2013). The involvement of age-dependent mitochondrial decline in sarcopenia as well as the physiological significance of this decline clearly needs more studies. In years to come it will be exciting to see if manipulations aimed at enhancing mitochondria quality control pathways will be sufficient to promote a healthy aging of muscles.
In the last years our understanding of the different mechanisms controlling mitochondrial morphology/function and the impact of these pathways in the homeostatic control of muscle mass has experienced an unprecedented advance. However, there are still many challenges that should be addressed. First, since the components of the mitochondria quality control system are closely interconnected, the alteration in one system can impinge on the activation/inhibition of the other repair mechanism. Therefore, this interplay, which is of fundamental importance for the integration of mitochondrial function within the network, should be considered in future studies. Second, mitochondria are social organelles that establish direct or indirect (via mitochondria-derived vesicles) contacts with other cellular components. Indeed, mitochondrial communication with the endoplasmic reticulum, peroxisomes, and lysosomes/vacuoles are hot topics of study at the moment. Then, it will be of great interest to investigate the functional consequences of these interactions in the physiology of muscle in the future. Finally, mitochondria-derived metabolites can shuttle within the cell and within the whole organism. Thus, exploring the interrelation between mitochondria quality control pathways and the metabolic regulation of muscle mass as well as the muscle-interorgans crosstalk should give additional insights to understand the way these relationships can be manipulated to treat human diseases.
VR and MS wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to colleagues whose studies were not cited owing to space limitations. We thank Bert Blaauw for the critical reading. Our work is supported from ERC (282310-MyoPHAGY), Italian Ministry of Education (MiUR) (PRIN 2010/2011), Foundation Leducq, STINT program and CARIPARO to MS.
Adams, G. R., and Haddad, F. (1996). The relationships among IGF-1, DNA content, and protein accumulation during skeletal muscle hypertrophy. J. Appl. Physiol. (1985) 81, 2509–2516.
Ainbinder, A., Boncompagni, S., Protasi, F., and Dirksen, R. T. (2015). Role of Mitofusin-2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 57, 14–24. doi: 10.1016/j.ceca.2014.11.002
Alavi, M. V., and Fuhrmann, N. (2013). Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics. Mol. Neurodegener. 8:32. doi: 10.1186/1750-1326-8-32
Alavi, M. V., Fuhrmann, N., Nguyen, H. P., Yu-Wai-Man, P., Heiduschka, P., Chinnery, P. F., et al. (2009). Subtle neurological and metabolic abnormalities in an Opa1 mouse model of autosomal dominant optic atrophy. Exp. Neurol. 220, 404–409. doi: 10.1016/j.expneurol.2009.09.026
Amati-Bonneau, P., Valentino, M. L., Reynier, P., Gallardo, M. E., Bornstein, B., Boissiere, A., et al. (2008). OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131, 338–351. doi: 10.1093/brain/awm298
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. doi: 10.1083/jcb.201308006
Andrianjafiniony, T., Dupré-Aucouturier, S., Letexier, D., Couchoux, H., and Desplanches, D. (2010). Oxidative stress, apoptosis, and proteolysis in skeletal muscle repair after unloading. Am. J. Physiol. Cell Physiol. 299, C307–C315. doi: 10.1152/ajpcell.00069.2010
Antunes, D., Padrão, A. I., Maciel, E., Santinha, D., Oliveira, P., Vitorino, R., et al. (2014). Molecular insights into mitochondrial dysfunction in cancer-related muscle wasting. Biochim. Biophys. Acta. 1841, 896–905. doi: 10.1016/j.bbalip.2014.03.004
Azzu, V., and Brand, M. D. (2010). Degradation of an intramitochondrial protein by the cytosolic proteasome. J. Cell Sci. 123, 578–585. doi: 10.1242/jcs.060004
Bach, D., Naon, D., Pich, S., Soriano, F. X., Vega, N., Rieusset, J., et al. (2005). Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes 54, 2685–2693. doi: 10.2337/diabetes.54.9.2685
Baraibar, M. A., Gueugneau, M., Duguez, S., Butler-Browne, G., Bechet, D., and Friguet, B. (2013). Expression and modification proteomics during skeletal muscle ageing. Biogerontology 14, 339–352. doi: 10.1007/s10522-013-9426-7
Barbieri, E., and Sestili, P. (2012). Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 982794. doi: 10.1155/2012/982794
Benard, G., Bellance, N., James, D., Parrone, P., Fernandez, H., Letellier, T., et al. (2007). Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 120, 838–848. doi: 10.1242/jcs.03381
Beregi, E., Regius, O., Hüttl, T., and Göbl, Z. (1988). Age-related changes in the skeletal muscle cells. Z. Gerontol. 21, 83–86.
Bhandari, P., Song, M., and Dorn, G. W. II. (2015). Dissociation of mitochondrial from sarcoplasmic reticular stress in Drosophila cardiomyopathy induced by molecularly distinct mitochondrial fusion defects. J. Mol. Cell. Cardiol. 80, 71–80. doi: 10.1016/j.yjmcc.2014.12.018
Billia, F., Hauck, L., Konecny, F., Rao, V., Shen, J., and Mak, T. W. (2011). PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. U.S.A. 108, 9572–9577. doi: 10.1073/pnas.1106291108
Blaauw, B., Schiaffino, S., and Reggiani, C. (2013). Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 3, 1645–1687. doi: 10.1002/cphy.c130009
Blei, M. L., Conley, K. E., and Kushmerick, M. J. (1993). Separate measures of ATP utilization and recovery in human skeletal muscle. J. Physiol. 465, 203–222. doi: 10.1113/jphysiol.1993.sp019673
Bodine, S. C., Latres, E., Baumhueter, S., Lai, V. K., Nunez, L., Clarke, B. A., et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708. doi: 10.1126/science.1065874
Booth, F. W., Weeden, S. H., and Tseng, B. S. (1994). Effect of aging on human skeletal muscle and motor function. Med. Sci. Sports Exerc. 26, 556–560. doi: 10.1249/00005768-199405000-00006
Bori, Z., Zhao, Z., Koltai, E., Fatouros, I. G., Jamurtas, A. Z., Douroudos, I. I., et al. (2012). The effects of aging, physical training, and a single bout of exercise on mitochondrial protein expression in human skeletal muscle. Exp. Gerontol. 47, 417–424. doi: 10.1016/j.exger.2012.03.004
Bragoszewski, P., Gornicka, A., Sztolsztener, M. E., and Chacinska, A. (2013). The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol. Cell. Biol. 33, 2136–2148. doi: 10.1128/MCB.01579-12
Braschi, E., Zunino, R., and McBride, H. M. (2009). MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 10, 748–754. doi: 10.1038/embor.2009.86
Brault, J. J., Jespersen, J. G., and Goldberg, A. L. (2010). Peroxisome proliferator-activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J. Biol. Chem. 285, 19460–19471. doi: 10.1074/jbc.M110.113092
Brocca, L., Cannavino, J., Coletto, L., Biolo, G., Sandri, M., Bottinelli, R., et al. (2012). The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J. Physiol. 590, 5211–5230. doi: 10.1113/jphysiol.2012.240267
Callahan, D. M., Bedrin, N. G., Subramanian, M., Berking, J., Ades, P. A., Toth, M. J., et al. (2014). Age-related structural alterations in human skeletal muscle fibers and mitochondria are sex specific: relationship to single-fiber function. J. Appl. Physiol. (1985). 116, 1582–1592. doi: 10.1152/japplphysiol.01362.2013
Calvo, S. E., Clauser, K. R., and Mootha, V. K. (2015). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. doi: 10.1093/nar/gkv1003. [Epub ahead of print].
Cannavino, J., Brocca, L., Sandri, M., Grassi, B., Bottinelli, R., and Pellegrino, M. A. (2015). The role of alterations in mitochondrial dynamics and PGC-1alpha over-expression in fast muscle atrophy following hindlimb unloading. J. Physiol. 593, 1981–1995. doi: 10.1113/jphysiol.2014.286740
Cantó, C., Gerhart-Hines, Z., Feige, J. N., Lagouge, M., Noriega, L., Milne, J. C., et al. (2009). AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060. doi: 10.1038/nature07813
Carnio, S., LoVerso, F., Baraibar, M. A., Longa, E., Khan, M. M., Maffei, M., et al. (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 8, 1509–1521. doi: 10.1016/j.celrep.2014.07.061
Cereghetti, G. M., Stangherlin, A., Martins de Brito, O., Chang, C. R., Blackstone, C., Bernardi, P., et al. (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808. doi: 10.1073/pnas.0808249105
Chabi, B., Ljubicic, V., Menzies, K. J., Huang, J. H., Saleem, A., and Hood, D. A. (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7, 2–12. doi: 10.1111/j.1474-9726.2007.00347.x
Chang, N. C., Nguyen, M., Bourdon, J., Risse, P. A., Martin, J., Danialou, G., et al. (2012). Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 21, 2277–2287. doi: 10.1093/hmg/dds048
Chao, J. R., Parganas, E., Boyd, K., Hong, C. Y., Opferman, J. T., and Ihle, J. N. (2008). Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 452, 98–102. doi: 10.1038/nature06604
Chaturvedi, R. K., and Flint Beal, M. (2013). Mitochondrial diseases of the brain. Free Radic. Biol. Med. 63, 1–29. doi: 10.1016/j.freeradbiomed.2013.03.018
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, H., Vermulst, M., Wang, Y. E., Chomyn, A., Prolla, T. A., McCaffery, J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. doi: 10.1016/j.cell.2010.02.026
Chen, Y., and Dorn, G. W. II. (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Chen, Y., Liu, Y., and Dorn, G. W. II. (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331. doi: 10.1161/CIRCRESAHA.111.258723
Cipolat, S., Martins de Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U.S.A. 101, 15927–15932. doi: 10.1073/pnas.0407043101
Cipolat, S., Rudka, T., Hartmann, D., Costa, V., Serneels, L., Craessaerts, K., et al. (2006). Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175. doi: 10.1016/j.cell.2006.06.021
Civiletto, G., Varanita, T., Cerutti, R., Gorletta, T., Barbaro, S., Marchet, S., et al. (2015). Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 21, 845–854. doi: 10.1016/j.cmet.2015.04.016
Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., et al. (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166. doi: 10.1038/nature04779
Clemmons, D. R. (2012). Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol. Metab. Clin. North Am. 41, 425–443, vii–viii. doi: 10.1016/j.ecl.2012.04.017
Cogliati, S., Frezza, C., Soriano, M. E., Varanita, T., Quintana-Cabrera, R., Corrado, M., et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. doi: 10.1016/j.cell.2013.08.032
Cohen, S., Nathan, J. A., and Goldberg, A. L. (2015). Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 14, 58–74. doi: 10.1038/nrd4467
Conley, K. E., Jubrias, S. A., and Esselman, P. C. (2000). Oxidative capacity and ageing in human muscle. J. Physiol. 526(Pt 1), 203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x
Crescenzo, R., Bianco, F., Mazzoli, A., Giacco, A., Liverini, G., and Iossa, S. (2014). Mitochondrial efficiency and insulin resistance. Front. Physiol. 5:512. doi: 10.3389/fphys.2014.00512
Cribbs, J. T., and Strack, S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944. doi: 10.1038/sj.embor.7401062
Cruz-Jentoft, A. J., Landi, F., Topinková, E., and Michel, J. P. (2010). Understanding sarcopenia as a geriatric syndrome. Curr. Opin. Clin. Nutr. Metab. Care 13, 1–7. doi: 10.1097/MCO.0b013e328333c1c1
Dagda, R. K., Cherra, S. J. III, Kulich, S. M., Tandon, A., Park, D., and Chu, C. T. (2009). Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855. doi: 10.1074/jbc.M808515200
Dahl, R., Larsen, S., Dohlmann, T. L., Qvortrup, K., Helge, J. W., Dela, F., et al. (2015). Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol. (Oxf). 213, 145–155. doi: 10.1111/apha.12289
Das, S., Morvan, F., Jourde, B., Meier, V., Kahle, P., Brebbia, P., et al. (2015). ATP citrate lyase improves mitochondrial function in skeletal muscle. Cell Metab. 21, 868–876. doi: 10.1016/j.cmet.2015.05.006
Davies, K. J., Quintanilha, A. T., Brooks, G. A., and Packer, L. (1982). Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 107, 1198–1205. doi: 10.1016/S0006-291X(82)80124-1
Davies, V. J., Hollins, A. J., Piechota, M. J., Yip, W., Davies, J. R., White, K. E., et al. (2007). Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 16, 1307–1318. doi: 10.1093/hmg/ddm079
de Brito, O. M., and Scorrano, L. (2009). Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9, 222–226. doi: 10.1016/j.mito.2009.02.005
De Palma, C., Morisi, F., Cheli, S., Pambianco, S., Cappello, V., Vezzoli, M., et al. (2012). Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 3:e418. doi: 10.1038/cddis.2012.159
Dobrowolny, G., Aucello, M., Rizzuto, E., Beccafico, S., Mammucari, C., Boncompagni, S., et al. (2008). Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436. doi: 10.1016/j.cmet.2008.09.002
Dodd, S. L., Gagnon, B. J., Senf, S. M., Hain, B. A., and Judge, A. R. (2010). Ros-mediated activation of NF-kappaB and Foxo during muscle disuse. Muscle Nerve. 41, 110–113. doi: 10.1002/mus.21526
Egan, B., Carson, B. P., Garcia-Roves, P. M., Chibalin, A. V., Sarsfield, F. M., Barron, N., et al. (2010). Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J. Physiol. 588, 1779–1790. doi: 10.1113/jphysiol.2010.188011
Ehses, S., Raschke, I., Mancuso, G., Bernacchia, A., Geimer, S., Tondera, D., et al. (2009). Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 187, 1023–1036. doi: 10.1083/jcb.200906084
Eisner, V., Lenaers, G., and Hajnóczky, G. (2014). Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J. Cell Biol. 205, 179–195. doi: 10.1083/jcb.201312066
Elgass, K., Pakay, J., Ryan, M. T., and Palmer, C. S. (2013). Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 1833, 150–161. doi: 10.1016/j.bbamcr.2012.05.002
Elmore, S. P., Qian, T., Grissom, S. F., and Lemasters, J. J. (2001). The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 15, 2286–2287. doi: 10.1096/fj.01-0206fje
Feely, S. M., Laura, M., Siskind, C. E., Sottile, S., Davis, M., Gibbons, V. S., et al. (2011). MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 76, 1690–1696. doi: 10.1212/WNL.0b013e31821a441e
Finkel, T. (1998). Oxygen radicals and signaling. Curr. Opin. Cell Biol. 10, 248–253. doi: 10.1016/S0955-0674(98)80147-6
Fontana, L., and Partridge, L. (2015). Promoting health and longevity through diet: from model organisms to humans. Cell 161, 106–118. doi: 10.1016/j.cell.2015.02.020
Frezza, C., Cipolat, S., Martins de Brito, O., Micaroni, M., Beznoussenko, G. V., Rudka, T., et al. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189. doi: 10.1016/j.cell.2006.06.025
Friedrich, O., Reid, M. B., Van den Berghe, G., Vanhorebeek, I., Hermans, G., Rich, M. M., et al. (2015). The sick and the weak: neuropathies/myopathies in the critically ill. Physiol. Rev. 95, 1025–1109. doi: 10.1152/physrev.00028.2014
Fry, C. S., Drummond, M. J., Glynn, E. L., Dickinson, J. M., Gundermann, D. M., Timmerman, K. L., et al. (2013). Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J. Gerontol. A Biol. Sci. Med. Sci. 68, 599–607. doi: 10.1093/gerona/gls209
Gaitanos, G. C., Williams, C., Boobis, L. H., and Brooks, S. (1993). Human muscle metabolism during intermittent maximal exercise. J. Appl. Physiol. (1985) 75, 712–719.
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H., and Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 19, 4861–4870. doi: 10.1093/hmg/ddq419
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Geng, T., Li, P., Okutsu, M., Yin, X., Kwek, J., Zhang, M., et al. (2010). PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 298, C572–C579. doi: 10.1152/ajpcell.00481.2009
Geng, T., Li, P., Yin, X., and Yan, Z. (2011). PGC-1alpha promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am. J. Pathol. 178, 1738–1748. doi: 10.1016/j.ajpath.2011.01.005
Ghosh, S., Lertwattanarak, R., Lefort, N., Molina-Carrion, M., Joya-Galeana, J., Bowen, B. P., et al. (2011). Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 60, 2051–2060. doi: 10.2337/db11-0121
Gilliam, L. A., Moylan, J. S., Patterson, E. W., Smith, J. D., Wilson, A. S., Rabbani, Z., et al. (2012). Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 302, C195–C202. doi: 10.1152/ajpcell.00217.2011
Glancy, B., Hartnell, L. M., Malide, D., Yu, Z. X., Combs, C. A., Connelly, P. S., et al. (2015). Mitochondrial reticulum for cellular energy distribution in muscle. Nature 523, 617–620. doi: 10.1038/nature14614
Gomes, L. C., Di Benedetto, G., and Scorrano, L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598. doi: 10.1038/ncb2220
Gomes, M. D., Lecker, S. H., Jagoe, R. T., Navon, A., and Goldberg, A. L. (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U.S.A. 98, 14440–14445. doi: 10.1073/pnas.251541198
Gomez-Cabrera, M. C., Borrás, C., Pallardó, F. V., Sastre, J., Ji, L. L., and Vina, J. (2005). Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 567, 113–120. doi: 10.1113/jphysiol.2004.080564
Gomez-Cabrera, M. C., Domenech, E., Romagnoli, M., Arduini, A., Borras, C., Pallardo, F. V., et al. (2008). Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 87, 142–149.
Goodpaster, B. H., Park, S. W., Harris, T. B., Kritchevsky, S. B., Nevitt, M., Schwartz, A. V., et al. (2006). The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J. Gerontol. A Biol. Sci. Med. Sci. 61, 1059–1064. doi: 10.1093/gerona/61.10.1059
Gouspillou, G., Bourdel-Marchasson, I., Rouland, R., Calmettes, G., Biran, M., Deschodt-Arsac, V., et al. (2014a). Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell. 13, 39–48. doi: 10.1111/acel.12147
Gouspillou, G., Bourdel-Marchasson, I., Rouland, R., Calmettes, G., Franconi, J. M., Deschodt-Arsac, V., et al. (2010). Alteration of mitochondrial oxidative phosphorylation in aged skeletal muscle involves modification of adenine nucleotide translocator. Biochim. Biophys. Acta 1797, 143–151. doi: 10.1016/j.bbabio.2009.09.004
Gouspillou, G., Sgarioto, N., Kapchinsky, S., Purves-Smith, F., Norris, B., Pion, C. H., et al. (2014b). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 28, 1621–1633. doi: 10.1096/fj.13-242750
Grumati, P., Coletto, L., Sabatelli, P., Cescon, M., Angelin, A., Bertaggia, E., et al. (2010). Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 16, 1313–1320. doi: 10.1038/nm.2247
Grumati, P., Coletto, L., Schiavinato, A., Castagnaro, S., Bertaggia, E., Sandri, M., et al. (2011). Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7, 1415–1423. doi: 10.4161/auto.7.12.17877
Grune, T., Merker, K., Sandig, G., and Davies, K. J. (2003). Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 305, 709–718. doi: 10.1016/S0006-291X(03)00809-X
Ha, J., Guan, K. L., and Kim, J. (2015). AMPK and autophagy in glucose/glycogen metabolism. Mol. Aspects Med. doi: 10.1016/j.mam.2015.08.002. [Epub ahead of print].
Han, X. J., Lu, Y. F., Li, S. A., Kaitsuka, T., Sato, Y., Tomizawa, K., et al. (2008). CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 182, 573–585. doi: 10.1083/jcb.200802164
Handschin, C., Chin, S., Li, P., Liu, F., Maratos-Flier, E., Lebrasseur, N. K., et al. (2007). Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Biol. Chem. 282, 30014–30021. doi: 10.1074/jbc.M704817200
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., and Gustafsson, Å. B. (2012). Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104. doi: 10.1074/jbc.M111.322933
Harbauer, A. B., Zahedi, R. P., Sickmann, A., Pfanner, N., and Meisinger, C. (2014). The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 19, 357–372. doi: 10.1016/j.cmet.2014.01.010
He, C., Bassik, M. C., Moresi, V., Sun, K., Wei, Y., Zou, Z., et al. (2012a). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. doi: 10.1038/nature10758
He, C., Sumpter, R. Jr., and Levine, B. (2012b). Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 8, 1548–1551. doi: 10.4161/auto.21327
Head, B., Griparic, L., Amiri, M., Gandre-Babbe, S., and van der Bliek, A. M. (2009). Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 187, 959–966. doi: 10.1083/jcb.200906083
Heo, J. M., Livnat-Levanon, N., Taylor, E. B., Jones, K. T., Dephoure, N., Ring, J., et al. (2010). A stress-responsive system for mitochondrial protein degradation. Mol. Cell. 40, 465–480. doi: 10.1016/j.molcel.2010.10.021
Hernandez-Alvarez, M. I., Thabit, H., Burns, N., Shah, S., Brema, I., Hatunic, M., et al. (2010). Subjects with early-onset type 2 diabetes show defective activation of the skeletal muscle PGC-1{alpha}/Mitofusin-2 regulatory pathway in response to physical activity. Diabetes Care 33, 645–651. doi: 10.2337/dc09-1305
Hoppeler, H., Hudlicka, O., and Uhlmann, E. (1987). Relationship between mitochondria and oxygen consumption in isolated cat muscles. J. Physiol. 385, 661–675. doi: 10.1113/jphysiol.1987.sp016513
Hourdé, C., Joanne, P., Medja, F., Mougenot, N., Jacquet, A., Mouisel, E., et al. (2013). Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. Am. J. Pathol. 182, 1509–1518. doi: 10.1016/j.ajpath.2013.01.020
Howald, H., Hoppeler, H., Claassen, H., Mathieu, O., and Straub, R. (1985). Influences of endurance training on the ultrastructural composition of the different muscle fiber types in humans. Pflugers Arch. 403, 369–376. doi: 10.1007/BF00589248
Huang, J. H., Joseph, A. M., Ljubicic, V., Iqbal, S., and Hood, D. A. (2010). Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 65, 138–146. doi: 10.1093/gerona/glp201
Huang, P., Galloway, C. A., and Yoon, Y. (2011). Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS ONE 6:e20655. doi: 10.1371/journal.pone.0020655
Hussain, S. N., Mofarrahi, M., Sigala, I., Kim, H. C., Vassilakopoulos, T., Maltais, F., et al. (2010). Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am. J. Respir. Crit. Care Med. 182, 1377–1386. doi: 10.1164/rccm.201002-0234OC
Ibebunjo, C., Chick, J. M., Kendall, T., Eash, J. K., Li, C., Zhang, Y., et al. (2013). Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 33, 194–212.
Ikeda, Y., Shirakabe, A., Maejima, Y., Zhai, P., Sciarretta, S., Toli, J., et al. (2015). Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116, 264–278. doi: 10.1161/CIRCRESAHA.116.303356
Iqbal, S., Ostojic, O., Singh, K., Joseph, A. M., and Hood, D. A. (2013). Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve. 48, 963–970. doi: 10.1002/mus.23838
Ishihara, N., Eura, Y., and Mihara, K. (2004). Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 117, 6535–6546. doi: 10.1242/jcs.01565
Ishihara, N., Fujita, Y., Oka, T., and Mihara, K. (2006). Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 25, 2966–2977. doi: 10.1038/sj.emboj.7601184
Ishihara, N., Nomura, M., Jofuku, A., Kato, H., Suzuki, S. O., Masuda, K., et al. (2009). Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966. doi: 10.1038/ncb1907
Jäger, S., Handschin, C., St-Pierre, J., and Spiegelman, B. M. (2007). AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U.S.A. 104, 12017–12022. doi: 10.1073/pnas.0705070104
Janssen, I., Heymsfield, S. B., Wang, Z. M., and Ross, R. (2000). Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J. Appl. Physiol. (1985) 89, 81–88.
Jeon, H. B., Choi, E. S., Yoon, J. H., Hwang, J. H., Chang, J. W., Lee, E. K., et al. (2007). A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 357, 731–736. doi: 10.1016/j.bbrc.2007.04.015
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Jones, J. M., Datta, P., Srinivasula, S. M., Ji, W., Gupta, S., Zhang, Z., et al. (2003). Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 425, 721–727. doi: 10.1038/nature02052
Joseph, A. M., Adhihetty, P. J., Buford, T. W., Wohlgemuth, S. E., Lees, H. A., Nguyen, L. M., et al. (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell. 11, 801–809. doi: 10.1111/j.1474-9726.2012.00844.x
Joseph, A. M., Adhihetty, P. J., Wawrzyniak, N. R., Wohlgemuth, S. E., Picca, A., Kujoth, G. C., et al. (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 8:e69327. doi: 10.1371/journal.pone.0069327
Jovaisaite, V., and Auwerx, J. (2015). The mitochondrial unfolded protein response-synchronizing genomes. Curr. Opin. Cell Biol. 33, 74–81. doi: 10.1016/j.ceb.2014.12.003
Jubrias, S. A., Esselman, P. C., Price, L. B., Cress, M. E., and Conley, K. E. (2001). Large energetic adaptations of elderly muscle to resistance and endurance training. J. Appl. Physiol. (1985) 90, 1663–1670.
Judge, S. M., Wu, C. L., Beharry, A. W., Roberts, B. M., Ferreira, L. F., Kandarian, S. C., et al. (2014). Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer. 14:997. doi: 10.1186/1471-2407-14-997
Kageyama, Y., Hoshijima, M., Seo, K., Bedja, D., Sysa-Shah, P., Andrabi, S. A., et al. (2014). Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 33, 2798–2813. doi: 10.15252/embj.201488658
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Karbowski, M., Neutzner, A., and Youle, R. J. (2007). The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71–84. doi: 10.1083/jcb.200611064
Karren, M. A., Coonrod, E. M., Anderson, T. K., and Shaw, J. M. (2005). The role of Fis1p-Mdv1p interactions in mitochondrial fission complex assembly. J. Cell Biol. 171, 291–301. doi: 10.1083/jcb.200506158
Katsetos, C. D., Koutzaki, S., and Melvin, J. J. (2013). Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatr. Neurol. 20, 202–215. doi: 10.1016/j.spen.2013.10.010
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
Kern, H., Barberi, L., Lofler, S., Sbardella, S., Burggraf, S., Fruhmann, H., et al. (2014). Electrical stimulation counteracts muscle decline in seniors. Front. Aging Neurosci. 6:189. doi: 10.3389/fnagi.2014.00189
Kim, K. H., Jeong, Y. T., Oh, H., Kim, S. H., Cho, J. M., Kim, Y. N., et al. (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 19, 83–92. doi: 10.1038/nm.3014
Klionsky, D. J., Abdalla, F. C., Abeliovich, H., Abraham, R. T., Acevedo-Arozena, A., Adeli, K., et al. (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. doi: 10.4161/auto.19496
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Leboucher, G. P., Tsai, Y. C., Yang, M., Shaw, K. C., Zhou, M., Veenstra, T. D., et al. (2012). Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell. 47, 547–557. doi: 10.1016/j.molcel.2012.05.041
Lecker, S. H., Jagoe, R. T., Gilbert, A., Gomes, M., Baracos, V., Bailey, J., et al. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 18, 39–51. doi: 10.1096/fj.03-0610com
Leduc-Gaudet, J. P., Picard, M., St-Jean Pelletier, F., Sgarioto, N., Auger, M. J., Vallee, J., et al. (2015). Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923–17937. doi: 10.18632/oncotarget.4235
Lee, Y., Lee, H. Y., Hanna, R. A., and Gustafsson, A. B. (2011). Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931. doi: 10.1152/ajpheart.00368.2011
Lee, Y. J., Jeong, S. Y., Karbowski, M., Smith, C. L., and Youle, R. J. (2004). Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 15, 5001–5011. doi: 10.1091/mbc.E04-04-0294
Leick, L., Wojtaszewski, J. F., Johansen, S. T., Kiilerich, K., Comes, G., Hellsten, Y., et al. (2008). PGC-1alpha is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 294, E463–E474. doi: 10.1152/ajpendo.00666.2007
Lelliott, C. J., Medina-Gomez, G., Petrovic, N., Kis, A., Feldmann, H. M., Bjursell, M., et al. (2006). Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 4:e369. doi: 10.1371/journal.pbio.0040369
Leone, T. C., Lehman, J. J., Finck, B. N., Schaeffer, P. J., Wende, A. R., Boudina, S., et al. (2005). PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3:e101. doi: 10.1371/journal.pbio.0030101
Levine, S., Biswas, C., Dierov, J., Barsotti, R., Shrager, J. B., Nguyen, T., et al. (2011). Increased proteolysis, myosin depletion, and atrophic AKT-FOXO signaling in human diaphragm disuse. Am. J. Respir. Crit. Care Med. 183, 483–490. doi: 10.1164/rccm.200910-1487OC
Levine, S., Nguyen, T., Taylor, N., Friscia, M. E., Budak, M. T., Rothenberg, P., et al. (2008). Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N. Engl. J. Med. 358, 1327–1335. doi: 10.1056/NEJMoa070447
Lin, J., Handschin, C., and Spiegelman, B. M. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370. doi: 10.1016/j.cmet.2005.05.004
Lin, J., Wu, H., Tarr, P. T., Zhang, C. Y., Wu, Z., Boss, O., et al. (2002). Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418, 797–801. doi: 10.1038/nature00904
Lira, V. A., Okutsu, M., Zhang, M., Greene, N. P., Laker, R. C., Breen, D. S., et al. (2013). Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 27, 4184–4193. doi: 10.1096/fj.13-228486
Little, J. P., Safdar, A., Benton, C. R., and Wright, D. C. (2011). Skeletal muscle and beyond: the role of exercise as a mediator of systemic mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. 36, 598–607. doi: 10.1139/h11-076
Livnat-Levanon, N., and Glickman, M. H. (2011). Ubiquitin-proteasome system and mitochondria - reciprocity. Biochim. Biophys. Acta 1809, 80–87. doi: 10.1016/j.bbagrm.2010.07.005
Ljubicic, V., Joseph, A. M., Adhihetty, P. J., Huang, J. H., Saleem, A., Uguccioni, G., et al. (2009). Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging 1, 818–830.
Logan, C. V., Szabadkai, G., Sharpe, J. A., Parry, D. A., Torelli, S., Childs, A. M., et al. (2014). Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 46, 188–193. doi: 10.1038/ng.2851
Lokireddy, S., Wijesoma, I. W., Teng, S., Bonala, S., Gluckman, P. D., McFarlane, C., et al. (2012). The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 16, 613–624. doi: 10.1016/j.cmet.2012.10.005
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Loson, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell. 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lourenco Dos Santos, S., Baraibar, M. A., Lundberg, S., Eeg-Olofsson, O., Larsson, L., and Friguet, B. (2015). Oxidative proteome alterations during skeletal muscle ageing. Redox Biol. 5, 267–274. doi: 10.1016/j.redox.2015.05.006
Lo Verso, F., Carnio, S., Vainshtein, A., and Sandri, M. (2014). Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 10, 1883–1894. doi: 10.4161/auto.32154
Lundberg, T. R., Fernandez-Gonzalo, R., Norrbom, J., Fischer, H., Tesch, P. A., and Gustafsson, T. (2014). Truncated splice variant PGC-1alpha4 is not associated with exercise-induced human muscle hypertrophy. Acta Physiol. 212, 142–151. doi: 10.1111/apha.12310
Mammucari, C., Gherardi, G., Zamparo, I., Raffaello, A., Boncompagni, S., Chemello, F., et al. (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 10, 1269–1279. doi: 10.1016/j.celrep.2015.01.056
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458–471. doi: 10.1016/j.cmet.2007.11.001
Margineantu, D. H., Emerson, C. B., Diaz, D., and Hockenbery, D. M. (2007). Hsp90 inhibition decreases mitochondrial protein turnover. PLoS ONE 2:e1066. doi: 10.1371/journal.pone.0001066
Martins, L. M., Morrison, A., Klupsch, K., Fedele, V., Moisoi, N., Teismann, P., et al. (2004). Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 24, 9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004
Masiero, E., Agatea, L., Mammucari, C., Blaauw, B., Loro, E., Komatsu, M., et al. (2009). Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–515. doi: 10.1016/j.cmet.2009.10.008
Mathieu-Costello, O., Ju, Y., Trejo-Morales, M., and Cui, L. (2005). Greater capillary-fiber interface per fiber mitochondrial volume in skeletal muscles of old rats. J. Appl. Physiol. (1985) 99, 281–289. doi: 10.1152/japplphysiol.00750.2004
Mathur, S., Brooks, D., and Carvalho, C. R. (2014). Structural alterations of skeletal muscle in copd. Front. Physiol. 5:104. doi: 10.3389/fphys.2014.00104
McClung, J. M., Judge, A. R., Talbert, E. E., and Powers, S. K. (2009). Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. Am. J. Physiol. Cell Physiol. 296, C363–C371. doi: 10.1152/ajpcell.00497.2008
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. doi: 10.1002/embj.201385902
Melov, S., Tarnopolsky, M. A., Beckman, K., Felkey, K., and Hubbard, A. (2007). Resistance exercise reverses aging in human skeletal muscle. PLoS ONE 2:e465. doi: 10.1371/journal.pone.0000465
Menshikova, E. V., Ritov, V. B., Fairfull, L., Ferrell, R. E., Kelley, D. E., and Goodpaster, B. H. (2006). Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 61, 534–540. doi: 10.1093/gerona/61.6.534
Mercken, E. M., Crosby, S. D., Lamming, D. W., JeBailey, L., Krzysik-Walker, S., Villareal, D. T., et al. (2013). Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell. 12, 645–651. doi: 10.1111/acel.12088
Milan, G., Romanello, V., Pescatore, F., Armani, A., Paik, J. H., Frasson, L., et al. (2015). Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 6, 6670. doi: 10.1038/ncomms7670
Min, K., Smuder, A. J., Kwon, O. S., Kavazis, A. N., Szeto, H. H., and Powers, S. K. (2011). Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. (1985) 111, 1459–1466. doi: 10.1152/japplphysiol.00591.2011
Munoz, J. P., Ivanova, S., Sanchez-Wandelmer, J., Martinez-Cristobal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi: 10.1038/emboj.2013.168
Nakamura, N., Kimura, Y., Tokuda, M., Honda, S., and Hirose, S. (2006). MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 7, 1019–1022. doi: 10.1038/sj.embor.7400790
Narendra, D., Kane, L. A., Hauser, D. N., Fearnley, I. M., and Youle, R. J. (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106. doi: 10.4161/auto.6.8.13426
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803. doi: 10.1083/jcb.200809125
Navratil, M., Terman, A., and Arriaga, E. A. (2008). Giant mitochondria do not fuse and exchange their contents with normal mitochondria. Exp. Cell Res. 314, 164–172. doi: 10.1016/j.yexcr.2007.09.013
Nelson, W. B., Smuder, A. J., Hudson, M. B., Talbert, E. E., and Powers, S. K. (2012). Cross-talk between the calpain and caspase-3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit. Care Med. 40, 1857–1863. doi: 10.1097/CCM.0b013e318246bb5d
Neufer, P. D., Bamman, M. M., Muoio, D. M., Bouchard, C., Cooper, D. M., Goodpaster, B. H., et al. (2015). Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metab. 22, 4–11. doi: 10.1016/j.cmet.2015.05.011
Ngoh, G. A., Papanicolaou, K. N., and Walsh, K. (2012). Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 287, 20321–20332. doi: 10.1074/jbc.M112.359174
Nicot, A. S., Lo Verso, F., Ratti, F., Pilot-Storck, F., Streichenberger, N., Sandri, M., et al. (2014). Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 10, 1036–1053. doi: 10.4161/auto.28479
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
O'Leary, M. F., Vainshtein, A., Iqbal, S., Ostojic, O., and Hood, D. A. (2013). Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 304, C422–C430. doi: 10.1152/ajpcell.00240.2012
Olesen, J., Kiilerich, K., and Pilegaard, H. (2010). PGC-1alpha-mediated adaptations in skeletal muscle. Pflugers Arch. 460, 153–162. doi: 10.1007/s00424-010-0834-0
Otera, H., Ishihara, N., and Mihara, K. (2013). New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 1833, 1256–1268. doi: 10.1016/j.bbamcr.2013.02.002
Otera, H., and Mihara, K. (2011). Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 149, 241–251. doi: 10.1093/jb/mvr002
Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J., et al. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. doi: 10.1083/jcb.201007152
Owusu-Ansah, E., Song, W., and Perrimon, N. (2013). Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712. doi: 10.1016/j.cell.2013.09.021
Palmer, C. S., Elgass, K. D., Parton, R. G., Osellame, L. D., Stojanovski, D., and Ryan, M. T. (2013). Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 288, 27584–27593. doi: 10.1074/jbc.M113.479873
Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E., and Ryan, M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. doi: 10.1038/embor.2011.54
Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. doi: 10.1038/nature04788
Patti, M. E., Butte, A. J., Crunkhorn, S., Cusi, K., Berria, R., Kashyap, S., et al. (2003). Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. U.S.A. 100, 8466–8471. doi: 10.1073/pnas.1032913100
Pauly, M., Daussin, F., Burelle, Y., Li, T., Godin, R., Fauconnier, J., et al. (2012). AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 181, 583–592. doi: 10.1016/j.ajpath.2012.04.004
Pellegrino, M. A., Desaphy, J. F., Brocca, L., Pierno, S., Camerino, D. C., and Bottinelli, R. (2011). Redox homeostasis, oxidative stress and disuse muscle atrophy. J. Physiol. 589, 2147–2160. doi: 10.1113/jphysiol.2010.203232
Picard, M., Ritchie, D., Thomas, M. M., Wright, K. J., and Hepple, R. T. (2011). Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell. 10, 1047–1055. doi: 10.1111/j.1474-9726.2011.00745.x
Porter, C., Herndon, D. N., Sidossis, L. S., and Borsheim, E. (2013). The impact of severe burns on skeletal muscle mitochondrial function. Burns 39, 1039–1047. doi: 10.1016/j.burns.2013.03.018
Powers, S. K., Hudson, M. B., Nelson, W. B., Talbert, E. E., Min, K., Szeto, H. H., et al. (2011). Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 39, 1749–1759. doi: 10.1097/CCM.0b013e3182190b62
Powers, S. K., and Jackson, M. J. (2008). Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol. Rev. 88, 1243–1276. doi: 10.1152/physrev.00031.2007
Powers, S. K., Smuder, A. J., and Judge, A. R. (2012). Oxidative stress and disuse muscle atrophy: cause or consequence? Curr. Opin. Clin. Nutr. Metab. Care 15, 240–245. doi: 10.1097/mco.0b013e328352b4c2
Quiros, P. M., Langer, T., and Lopez-Otin, C. (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 16, 345–359. doi: 10.1038/nrm3984
Raben, N., Hill, V., Shea, L., Takikita, S., Baum, R., Mizushima, N., et al. (2008). Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 17, 3897–3908. doi: 10.1093/hmg/ddn292
Rambold, A. S., Kostelecky, B., Elia, N., and Lippincott-Schwartz, J. (2011). Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U.S.A. 108, 10190–10195. doi: 10.1073/pnas.1107402108
Ramos, F. J., Chen, S. C., Garelick, M. G., Dai, D. F., Liao, C. Y., Schreiber, K. H., et al. (2012). Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 4, 144ra103. doi: 10.1126/scitranslmed.3003802
Raue, U., Slivka, D., Minchev, K., and Trappe, S. (2009). Improvements in whole muscle and myocellular function are limited with high-intensity resistance training in octogenarian women. J. Appl. Physiol. (1985) 106, 1611–1617. doi: 10.1152/japplphysiol.91587.2008
Reid, M. B., Judge, A. R., and Bodine, S. C. (2014). CrossTalk opposing view: the dominant mechanism causing disuse muscle atrophy is proteolysis. J. Physiol. 592, 5345–5347. doi: 10.1113/jphysiol.2014.279406
Rhee, S. G., Bae, Y. S., Lee, S. R., and Kwon, J. (2000). Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE. 2000:pe1. doi: 10.1126/stke.2000.53.pe1
Ristow, M., and Schmeisser, K. (2014). Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 12, 288–341. doi: 10.2203/dose-response.13-035.Ristow
Ristow, M., and Schmeisser, S. (2011). Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 51, 327–336. doi: 10.1016/j.freeradbiomed.2011.05.010
Ristow, M., Zarse, K., Oberbach, A., Kloting, N., Birringer, M., Kiehntopf, M., et al. (2009). Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8665–8670. doi: 10.1073/pnas.0903485106
Rockstein, M., and Bhatnagar, P. L. (1965). Age Changes in Size and Number of the Giant Mitochondria in the Flight Muscle of the Common Housefly (Musca Domestica L.). J. Insect Physiol. 11, 481–491. doi: 10.1016/0022-1910(65)90053-3
Romanello, V., Guadagnin, E., Gomes, L., Roder, I., Sandri, C., Petersen, Y., et al. (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785. doi: 10.1038/emboj.2010.60
Romanello, V., and Sandri, M. (2013). Mitochondrial biogenesis and fragmentation as regulators of protein degradation in striated muscles. J. Mol. Cell. Cardiol. 55, 64–72. doi: 10.1016/j.yjmcc.2012.08.001
Ross, J. M., Olson, L., and Coppotelli, G. (2015). Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin? Int. J. Mol. Sci. 16, 19458–19476. doi: 10.3390/ijms160819458
Ruas, J. L., White, J. P., Rao, R. R., Kleiner, S., Brannan, K. T., Harrison, B. C., et al. (2012). A PGC-1alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331. doi: 10.1016/j.cell.2012.10.050
Russ, D. W., Krause, J., Wills, A., and Arreguin, R. (2012). “SR stress” in mixed hindlimb muscles of aging male rats. Biogerontology 13, 547–555. doi: 10.1007/s10522-012-9399-y
Russell, A. P., Wada, S., Vergani, L., Hock, M. B., Lamon, S., Leger, B., et al. (2013). Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol. Dis. 49, 107–117. doi: 10.1016/j.nbd.2012.08.015
Sacheck, J. M., Hyatt, J. P., Raffaello, A., Jagoe, R. T., Roy, R. R., Edgerton, V. R., et al. (2007). Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 21, 140–155. doi: 10.1096/fj.06-6604com
Safdar, A., Hamadeh, M. J., Kaczor, J. J., Raha, S., Debeer, J., and Tarnopolsky, M. A. (2010). Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS ONE 5:e10778. doi: 10.1371/journal.pone.0010778
Safdar, A., Little, J. P., Stokl, A. J., Hettinga, B. P., Akhtar, M., and Tarnopolsky, M. A. (2011). Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 286, 10605–10617. doi: 10.1074/jbc.M110.211466
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Sandri, M. (2013). Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 45, 2121–2129. doi: 10.1016/j.biocel.2013.04.023
Sandri, M., Coletto, L., Grumati, P., and Bonaldo, P. (2013). Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 126, 5325–5333. doi: 10.1242/jcs.114041
Sandri, M., Lin, J., Handschin, C., Yang, W., Arany, Z. P., Lecker, S. H., et al. (2006). PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. U.S.A. 103, 16260–16265. doi: 10.1073/pnas.0607795103
Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., et al. (2004). Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412. doi: 10.1016/S0092-8674(04)00400-3
Sartori, R., Gregorevic, P., and Sandri, M. (2014). TGFbeta and BMP signaling in skeletal muscle: potential significance for muscle-related disease. Trends Endocrinol. Metab. 25, 464–471. doi: 10.1016/j.tem.2014.06.002
Sartori, R., Schirwis, E., Blaauw, B., Bortolanza, S., Zhao, J., Enzo, E., et al. (2013). BMP signaling controls muscle mass. Nat. Genet. 45, 1309–1318. doi: 10.1038/ng.2772
Sarzi, E., Angebault, C., Seveno, M., Gueguen, N., Chaix, B., Bielicki, G., et al. (2012). The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain 135, 3599–3613. doi: 10.1093/brain/aws303
Schulz, A. M., and Haynes, C. M. (2015). UPR-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta. doi: 10.1016/j.bbabio.2015.03.008. [Epub ahead of print].
Sebastian, D., Hernandez-Alvarez, M. I., Segales, J., Sorianello, E., Munoz, J. P., Sala, D., et al. (2012). Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 109, 5523–5528. doi: 10.1073/pnas.1108220109
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. doi: 10.1016/j.molcel.2012.09.025
Shang, F., Gong, X., and Taylor, A. (1997). Activity of ubiquitin-dependent pathway in response to oxidative stress. Ubiquitin-activating enzyme is transiently up-regulated. J. Biol. Chem. 272, 23086–23093. doi: 10.1074/jbc.272.37.23086
Short, K. R., Bigelow, M. L., Kahl, J., Singh, R., Coenen-Schimke, J., Raghavakaimal, S., et al. (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U.S.A. 102, 5618–5623. doi: 10.1073/pnas.0501559102
Short, K. R., Vittone, J. L., Bigelow, M. L., Proctor, D. N., Rizza, R. A., Coenen-Schimke, J. M., et al. (2003). Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes 52, 1888–1896. doi: 10.2337/diabetes.52.8.1888
Silvennoinen, M., Ahtiainen, J. P., Hulmi, J. J., Pekkala, S., Taipale, R. S., Nindl, B. C., et al. (2015). PGC-1 isoforms and their target genes are expressed differently in human skeletal muscle following resistance and endurance exercise. Physiol. Rep. 3:e12563. doi: 10.14814/phy2.12563
Slivka, D., Raue, U., Hollon, C., Minchev, K., and Trappe, S. (2008). Single muscle fiber adaptations to resistance training in old (>80 yr) men: evidence for limited skeletal muscle plasticity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R273–R280. doi: 10.1152/ajpregu.00093.2008
Smirnova, E., Griparic, L., Shurland, D. L., and van der Bliek, A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 12, 2245–2256. doi: 10.1091/mbc.12.8.2245
Smuder, A. J., Kavazis, A. N., Hudson, M. B., Nelson, W. B., and Powers, S. K. (2010). Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic. Biol. Med. 49, 1152–1160. doi: 10.1016/j.freeradbiomed.2010.06.025
Song, M., and Dorn, G. W. II. (2015). Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 21, 195–205. doi: 10.1016/j.cmet.2014.12.019
Song, M., Mihara, K., Chen, Y., Scorrano, L., and Dorn, G. W. II. (2015). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–285. doi: 10.1016/j.cmet.2014.12.011
Song, Z., Chen, H., Fiket, M., Alexander, C., and Chan, D. C. (2007). OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178, 749–755. doi: 10.1083/jcb.200704110
Sonoda, J., Mehl, I. R., Chong, L. W., Nofsinger, R. R., and Evans, R. M. (2007). PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. U.S.A. 104, 5223–5228. doi: 10.1073/pnas.0611623104
Sood, A., Jeyaraju, D. V., Prudent, J., Caron, A., Lemieux, P., McBride, H. M., et al. (2014). A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. U.S.A. 111, 16017–16022. doi: 10.1073/pnas.1408061111
Soriano, F. X., Liesa, M., Bach, D., Chan, D. C., Palacin, M., and Zorzano, A. (2006). Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes 55, 1783–1791. doi: 10.2337/db05-0509
Soubannier, V., McLelland, G. L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012a). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Soubannier, V., Rippstein, P., Kaufman, B. A., Shoubridge, E. A., and McBride, H. M. (2012b). Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 7:e52830. doi: 10.1371/journal.pone.0052830
Sugiura, A., Nagashima, S., Tokuyama, T., Amo, T., Matsuki, Y., Ishido, S., et al. (2013). MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell. 51, 20–34. doi: 10.1016/j.molcel.2013.04.023
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.M607279200
Taillandier, D., Aurousseau, E., Meynial-Denis, D., Bechet, D., Ferrara, M., Cottin, P., et al. (1996). Coordinate activation of lysosomal, Ca2+-activated and ATP-ubiquitin-dependent proteinases in the unweighted rat soleus muscle. Biochem. J. 316(Pt 1), 65–72.
Talbert, E. E., Smuder, A. J., Min, K., Kwon, O. S., and Powers, S. K. (2013a). Calpain and caspase-3 play required roles in immobilization-induced limb muscle atrophy. J. Appl. Physiol. (1985) 114, 1482–1489. doi: 10.1152/japplphysiol.00925.2012
Talbert, E. E., Smuder, A. J., Min, K., Kwon, O. S., Szeto, H. H., and Powers, S. K. (2013b). Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J. Appl. Physiol. (1985). 115, 529–538. doi: 10.1152/japplphysiol.00471.2013
Tanaka, A., Cleland, M. M., Xu, S., Narendra, D. P., Suen, D. F., Karbowski, M., et al. (2010). Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380. doi: 10.1083/jcb.201007013
Tondera, D., Grandemange, S., Jourdain, A., Karbowski, M., Mattenberger, Y., Herzig, S., et al. (2009). SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600. doi: 10.1038/emboj.2009.89
Touvier, T., De Palma, C., Rigamonti, E., Scagliola, A., Incerti, E., Mazelin, L., et al. (2015). Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 6:e1663. doi: 10.1038/cddis.2014.595
Trounce, I., Byrne, E., and Marzuki, S. (1989). Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet 1, 637–639. doi: 10.1016/S0140-6736(89)92143-0
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Vainshtein, A., Desjardins, E. M., Armani, A., Sandri, M., and Hood, D. A. (2015). PGC-1alpha modulates denervation-induced mitophagy in skeletal muscle. Skelet. Muscle 5, 9. doi: 10.1186/s13395-015-0033-y
Vainshtein, A., Grumati, P., Sandri, M., and Bonaldo, P. (2014). Skeletal muscle, autophagy, and physical activity: the menage a trois of metabolic regulation in health and disease. J. Mol. Med. 92, 127–137. doi: 10.1007/s00109-013-1096-z
Varanita, T., Soriano, M. E., Romanello, V., Zaglia, T., Quintana-Cabrera, R., Semenzato, M., et al. (2015). The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 21, 834–844. doi: 10.1016/j.cmet.2015.05.007
Vianna, C. R., Huntgeburth, M., Coppari, R., Choi, C. S., Lin, J., Krauss, S., et al. (2006). Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 4, 453–464. doi: 10.1016/j.cmet.2006.11.003
Wakabayashi, J., Zhang, Z., Wakabayashi, N., Tamura, Y., Fukaya, M., Kensler, T. W., et al. (2009). The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 186, 805–816. doi: 10.1083/jcb.200903065
Wang, H., Song, P., Du, L., Tian, W., Yue, W., Liu, M., et al. (2011). Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 286, 11649–11658. doi: 10.1074/jbc.M110.144238
Waterham, H. R., Koster, J., van Roermund, C. W., Mooyer, P. A., Wanders, R. J., and Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741. doi: 10.1056/NEJMoa064436
Wende, A. R., Schaeffer, P. J., Parker, G. J., Zechner, C., Han, D. H., Chen, M. M., et al. (2007). A role for the transcriptional coactivator PGC-1alpha in muscle refueling. J. Biol. Chem. 282, 36642–36651. doi: 10.1074/jbc.M707006200
Wenz, T., Rossi, S. G., Rotundo, R. L., Spiegelman, B. M., and Moraes, C. T. (2009). Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. U.S.A. 106, 20405–20410. doi: 10.1073/pnas.0911570106
Wohlgemuth, S. E., Seo, A. Y., Marzetti, E., Lees, H. A., and Leeuwenburgh, C. (2010). Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp. Gerontol. 45, 138–148. doi: 10.1016/j.exger.2009.11.002
Wright, D. C., Han, D. H., Garcia-Roves, P. M., Geiger, P. C., Jones, T. E., and Holloszy, J. O. (2007). Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1alpha expression. J. Biol. Chem. 282, 194–199. doi: 10.1074/jbc.M606116200
Wu, H., Kanatous, S. B., Thurmond, F. A., Gallardo, T., Isotani, E., Bassel-Duby, R., et al. (2002). Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 296, 349–352. doi: 10.1126/science.1071163
Xu, S., Peng, G., Wang, Y., Fang, S., and Karbowski, M. (2011). The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell. 22, 291–300. doi: 10.1091/mbc.E10-09-0748
Ydfors, M., Fischer, H., Mascher, H., Blomstrand, E., Norrbom, J., and Gustafsson, T. (2013). The truncated splice variants, NT-PGC-1alpha and PGC-1alpha4, increase with both endurance and resistance exercise in human skeletal muscle. Physiol. Rep. 1:e00140. doi: 10.1002/phy2.140
Ye, Y., Meyer, H. H., and Rapoport, T. A. (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656. doi: 10.1038/414652a
Ye, Y., Meyer, H. H., and Rapoport, T. A. (2003). Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84. doi: 10.1083/jcb.200302169
Yonashiro, R., Ishido, S., Kyo, S., Fukuda, T., Goto, E., Matsuki, Y., et al. (2006). A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 25, 3618–3626. doi: 10.1038/sj.emboj.7601249
Yu-Wai-Man, P., Griffiths, P. G., Gorman, G. S., Lourenco, C. M., Wright, A. F., Auer-Grumbach, M., et al. (2010). Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771–786. doi: 10.1093/brain/awq007
Zampieri, S., Pietrangelo, L., Loefler, S., Fruhmann, H., Vogelauer, M., Burggraf, S., et al. (2015). Lifelong physical exercise delays age-associated skeletal muscle decline. J. Gerontol. A Biol. Sci. Med. Sci. 70, 163–173. doi: 10.1093/gerona/glu006
Zechner, C., Lai, L., Zechner, J. F., Geng, T., Yan, Z., Rumsey, J. W., et al. (2010). Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 12, 633–642. doi: 10.1016/j.cmet.2010.11.008
Zhang, Z., Wakabayashi, N., Wakabayashi, J., Tamura, Y., Song, W. J., Sereda, S., et al. (2011). The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell. 22, 2235–2245. doi: 10.1091/mbc.E10-12-0933
Zhao, J., Liu, T., Jin, S., Wang, X., Qu, M., Uhlen, P., et al. (2011). Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 30, 2762–2778. doi: 10.1038/emboj.2011.198
Zhao, Q., Wang, J., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T., and Hoogenraad, N. J. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419. doi: 10.1093/emboj/cdf445
Keywords: atrophy, mitochondria, fission, fusion, biogenesis, autophagy, muscle, sarcopenia
Citation: Romanello V and Sandri M (2016) Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 6:422. doi: 10.3389/fphys.2015.00422
Received: 13 November 2015; Accepted: 22 December 2015;
Published: 12 January 2016.
Edited by:
Gilles Gouspillou, Université du Québec à Montréal, CanadaReviewed by:
Scott Powers, University of Florida, USACopyright © 2016 Romanello and Sandri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vanina Romanello, dmFuaW5hLnJvbWFuZWxsb0B1bmlwZC5pdA==;
Marco Sandri, bWFyY28uc2FuZHJpQHVuaXBkLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.