
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 18 November 2015
Sec. Striated Muscle Physiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00336
This article is part of the Research Topic Mitochondria in Skeletal Muscle Health, Aging and Diseases View all 14 articles
 Terence E. Ryan1,2
Terence E. Ryan1,2 Cameron A. Schmidt1,2
Cameron A. Schmidt1,2 Tom D. Green1,2
Tom D. Green1,2 David A. Brown1,2
David A. Brown1,2 P. Darrell Neufer1,2
P. Darrell Neufer1,2 Joseph M. McClung1,2*
Joseph M. McClung1,2*Critical limb ischemia (CLI) is the most severe clinical presentation of peripheral arterial disease and manifests as chronic limb pain at rest and/or tissue necrosis. Current clinical interventions are largely ineffective and therapeutic angiogenesis based trials have shown little efficacy, highlighting the dire need for new ideas and novel therapeutic approaches. Despite a decade of research related to skeletal muscle as a determinant of morbidity and mortality outcomes in CLI, very little progress has been made toward an effective therapy aimed directly at the muscle myopathies of this disease. Within the muscle cell, mitochondria are well positioned to modulate the ischemic cellular response, as they are the principal sites of cellular energy production and the major regulators of cellular redox charge and cell death. In this mini review, we update the crucial importance of skeletal muscle to CLI pathology and examine the evolving influence of muscle and endothelial cell mitochondria in the complex ischemic microenvironment. Finally, we discuss the novelty of muscle mitochondria as a therapeutic target for ischemic pathology in the context of the complex co-morbidities often associated with CLI.
Peripheral artery disease (PAD) presents as either symptom-free, intermittent claudication (IC, pain with exertion that is relieved with rest) or critical limb ischemia (CLI, pain at rest with or without tissue necrosis or gangrene). CLI carries alarmingly high morbidity and mortality rates and patients have a risk of major amputation or death that approaches 40% in 1 year (Dormandy et al., 1999; Hirsch et al., 2001; Taylor et al., 2009). A common misconception is that CLI represents the natural progression of IC in patients; however the same degree of stenosis can present as symptom-free, IC, or CLI, implying that factors other than limb blood flow contribute to pathology. Despite recent advances in stem cell biology and genetics (Matzke and Lepantalo, 2001; Chalothorn et al., 2007; Dokun et al., 2008; Chalothorn and Faber, 2010; Wang et al., 2010, 2012; Katwal and Dokun, 2011), surprisingly little progress has been made toward effective therapeutic options for CLI, warranting the consideration of alternative and novel treatment approaches. Limb skeletal muscle is uniquely positioned to alter the clinical course of CLI due to its inherent plasticity, role as a paracrine signaling organ, and reservoir of endogenous pluripotent progenitor cells (Seale et al., 2001; Chargé and Rudnicki, 2004; Abou-Khalil et al., 2010). Currently, PAD research is overwhelmingly focused on limb collateral vessel development and nascent conduit promotion and survival (Annex, 2013), while potential alternative therapies directed at limb muscle in CLI have been slow to develop. In this mini-review we highlight the importance of skeletal muscle in the manifestation of CLI and discuss the potential influence of muscle and endothelial cell mitochondria on the ischemic limb.
Variations in the clinical course of CLI raise the intriguing possibility that disease manifestation is in part dependent on genetic determinants of susceptibility to ischemia (Matzke and Lepantalo, 2001; Chalothorn et al., 2007; Dokun et al., 2008; Chalothorn and Faber, 2010; Wang et al., 2010, 2012; Katwal and Dokun, 2011). The genetics of PAD are not well understood (Gudmundsson et al., 2002; Knowles et al., 2007; Messina, 2008; Katwal and Dokun, 2011; Leeper et al., 2012; Murabito et al., 2012) but present a complicated paradigm whereby differential determinants could direct the ischemic responses of multiple cell types (endothelial, muscle, fibroblast, etc.) in the affected limb. In this regard, inbred mouse strains have dramatically different responses to a murine model of PAD, analogous to the range of responses seen in humans. For example, limb perfusion recovers rapidly and without tissue loss in C57BL/6J (BL6) mice while BALB/cJ mice display significant tissue necrosis and poor perfusion recovery (Chalothorn et al., 2007; Dokun et al., 2008; Chalothorn and Faber, 2010; Wang et al., 2010). Inherent genetic differences in muscle regeneration are known to occur in BALB/cJ mice (Grounds, 1987; Grounds and McGeachie, 1989; McGeachie and Grounds, 1995; Mitchell et al., 1995; Roberts et al., 1997; Lagrota-Candido et al., 2010), and includes temporal alterations in the expression of traditional vascular growth factors and their receptors (McClung et al., 2012) that coincide with the strain-dependent segregation of limb blood flow. Differentiating muscle cells secrete traditional vascular growth factors that act as both autocrine and paracrine factors to stimulate maturation in both endothelial and muscle cells (McClung et al., 2012, 2015; Mofarrahi et al., 2015) and represent a unique source of regenerative signals that could potentially be harnessed to improve the local ischemic microenvironment. Because a large proportion of murine pre-clinical limb ischemia work is performed in mice on either a mixed or largely BL6 background, regeneration from ischemic muscle myopathy is often masked or ignored.
In a clinical CLI scenario, focusing on solely the vascular response is predicated on the idea that the ischemic muscle tissue is dispensable, at least short-term. Treatments that induce revascularization and/or nascent collateral vessel formation have proven ineffective to date (Annex, 2013; Hammer and Steiner, 2013; Cooke and Losordo, 2015) and indicate that a “restoration of flow approach” is not independently sufficient to rescue the limb. It is likely that myopathy and vasculopathy are interrelated components of a coordinated tissue response to CLI. Recent insights into the skeletal muscle response indicate that while the background genetics of an individual contributes to the density of pre-existing collateral vessels and the endogenous ability to generate nascent collateral vessels and capillaries, this simply isn't the sole determinant of pathology. The plasticity of the skeletal muscle facilitates temporal ischemic degeneration/regeneration in this environment, whereby genetically pre-determined deficits in muscle regenerative processes would result in cellular apoptosis and tissue necrosis that could negatively impact both endogenous neovascularization and/or the survival of a vessel graft. Simply put, limb muscle tissue that is already necrotic or beyond repair by endogenous regenerative mechanisms is representative of a local ischemic environment that is unable to sustain or promote neovascularization (Figure 1). Intricate coordination of therapies targeting muscle plasticity may be required to allow tissue survival and facilitate recovery until blood flow can be fully restored by surgical intervention and/or collateral vessel formation.
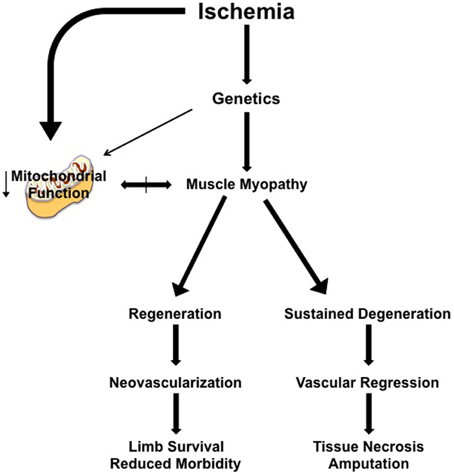
Figure 1. Simplified model of the proposed role of muscle myopathy in the progression of limb pathology in critical limb ischemia. Individual genetics play a role in determining the severity of the ischemic manifestation of limb pathology. Clinical interventions (endovascular or revascularization in nature) occur after the patient clinically presents with identifiable symptoms/manifestation of PAD, at a time when muscle myopathy is initiated or ongoing. Ischemic muscle myopathy involves muscle degeneration/regeneration cycles that: (1) function properly and result in a limb tissue microenvironment that is supportive of neovascularization and/or the clinical intervention, reducing morbidity and increasing the likelihood of limb survival, or (2) improperly function, resulting in a microenvironment that promotes continued degenerative myopathy and vascular regression that ultimately leads to tissue necrosis, morbidity, and secondary amputation. The role of mitochondrial function in ischemic limb muscle myopathy is not currently understood, but represents an exciting area for therapeutic exploration.
The importance of striated muscle to ischemic outcomes is readily accepted in cardiac ischemia/reperfusion, and there are numerous clinical trials involving therapeutic targeting of the cardiomyocyte (clinicaltrials.gov: NCT01502774, NCT01374321, NCT01172171, NCT00966563, NCT01572909). Therapies aimed at the skeletal muscle represent an untapped arena with great potential to advance the field of CLI research. Table 1 highlights the clinical work verifying limb skeletal muscle's role in PAD mortality over the last 10-years. Documented histochemical evidence of skeletal muscle myopathies and necrosis in PAD patients exists (Rissanen et al., 2002; Pipinos et al., 2007, 2008a); however the majority of the field operates under the assumption that myopathy is not important (Sealock et al., 2014). In stark contrast, muscle biologists studying neuromuscular diseases, dystrophy, or myofibrillar myopathies have embraced the contributions of the vasculature to the pathologic manifestations of their respective diseases. Abnormal skeletal muscle perfusion and resultant ischemia are believed to contribute to the pathology of Duchenne and Becker muscular dystrophies, and have spawned the “two hit (ischemia-metabolic stress) hypothesis” for muscle injury in these diseases (Asai et al., 2007). This hypothesis has driven approaches to treat these muscular dystrophies with phosphodiesterase-5 inhibitors like tadalafil, which improves blood flow, in an attempt to circumvent the ischemic component and improve muscle bioenergetics (Martin et al., 2012). Given the similarities between dystrophic myopathies and those identified with CLI, therapies aimed at skeletal muscle could be effective treatments for tissue degeneration and dysfunction during ischemia while also providing benefits to the vascular compartment of the affected limb. Interestingly, a common pathology linking neuromuscular disorders involving degeneration/regeneration is mitochondrial dysfunction (Katsetos et al., 2013). Mitochondria have recently garnered attention in the PAD literature (Brass, 2013; Hiatt et al., 2015) and may represent an evolutionarily conserved “starting point” for investigation into CLI myopathy.
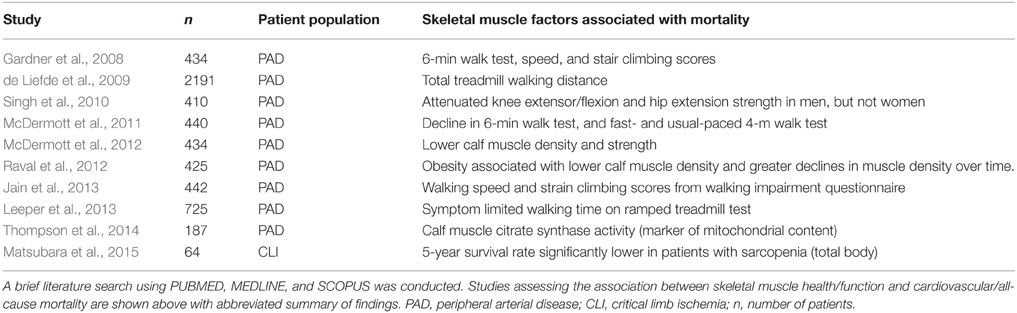
Table 1. Clinical studies implicating skeletal muscle function with mortality.
Mitochondria have numerous roles in the muscle cell, including the generation and maintenance of energy and redox charge, gatekeeping the mortality of ischemic cells (Karch and Molkentin, 2015; Shirihai et al., 2015), and the production of reactive oxygen species (ROS). Mitochondria also communicate with the rest of the cell through “signals” such as metabolites, cytochrome c release, and via redox-dependent cascades. Decreased muscle metabolism, impaired mitochondrial respiration, decreased expression of mitochondrial enzymes, increased oxidative stress, and somatic mutations in mitochondrial genes have been reported in limb muscle of patients' with PAD (Keller et al., 1985; Hands et al., 1986; Zatina et al., 1986; Bhat et al., 1999; Brass and Hiatt, 2000; Pipinos et al., 2000a, 2006, 2008b; Brass et al., 2004; Isbell et al., 2006; Schocke et al., 2008; Wurdeman et al., 2012; Weiss et al., 2013; Koutakis et al., 2014). Using non-invasive magnetic resonance spectroscopy, several labs have demonstrated that limb muscle from PAD patients' displays slower phosphocreatine (PCr) recovery, indicative of a lower muscle/mitochondrial oxidative capacity (Keller et al., 1985; Hands et al., 1986; Pipinos et al., 2000a,b; Isbell et al., 2006; Schocke et al., 2008). Interpretation of these data can be complicated by the influence of an intact but poorly functioning vascular system, perpetuating the idea that the reduced PCr recovery rates are more related to poor perfusion during the recovery period. Gastrocnemius muscle biopsies from PAD patients, however, also demonstrate reduced mitochondrial content and enzyme activity ex vivo (where oxygen delivery is not a limitation; Pipinos et al., 2003, 2006, 2007), and pre-clinical studies have recapitulated these findings (Pipinos et al., 2008b; Lejay et al., 2015). It is not currently known whether alterations in mitochondrial content or function cause ischemic muscle myopathy, but a recent report linked muscle mitochondrial content (reported as citrate synthase protein abundance) to PAD mortality (Thompson et al., 2014).
A lack of oxygen delivery to limb muscle tissue induces a progressive accumulation of ischemic injury that manifests as declining muscle function (Pipinos et al., 2007, 2008a; McDermott et al., 2012; Cluff et al., 2013; Weiss et al., 2013; Koutakis et al., 2014). A potential source for this tissue injury may be mitochondrial-derived ROS and the resulting oxidative stress with chronically elevated ROS. Pipinos et al. reported the first indirect evidence for skeletal muscle “oxidative stress” in patients with PAD (Pipinos et al., 2006). Recent work from this group suggests that these same indirect markers of oxidative stress may be related to disease severity (Fontaine Stage and ABI;Weiss et al., 2013). The potential also exists for repeated ischemia-reperfusion events in skeletal muscle from CLI patients (Lejay et al., 2014). When blood flow and pressure is low, arterial blockages may result in low oxygen tensions in muscle tissue that could be severe enough to inhibit mitochondrial complex IV (cytochrome c oxidase) and consequently electron flow in the electron transport system. This would result in the accumulation of metabolites and reducing equivalents (NADH and FADH2) that, upon re-oxygenation by surgical intervention or endogenous collateral flow with activity or mechanical loading, would be rapidly metabolized. These ischemia-reperfusion events have been well documented to produce large amounts of ROS in cardiac, brain, liver and renal tissues (Chouchani et al., 2014) and could be intermittently triggered by small amounts of physical activity or mechanical loading. For additional details on oxidative stress with PAD, we would recommend other excellent reviews (Brass, 1996; Pipinos et al., 2007, 2008a).
Because mitochondria are a major source of both reductive power (e.g., NADPH) and oxidants (superoxide anion and hydrogen peroxide), they serve as a metabolic rheostat controlling cellular redox homeostasis. Flux through both the reductive and oxidative arms contributes to redox signaling through redox modifications to cysteine residues that regulate the structure/function of target proteins (Go and Jones, 2013). Post-translational modifications such as S-nitrosylation, glutathionylation, sulfenylation, and disulfide bond formation are also considered mechanisms of redox signaling. Although the redox signaling field is at an early stage, recent studies suggest regulation of several cellular pathways relevant to the ischemic microenvironment including: muscle autophagy (Rahman et al., 2014), contractile dysfunction (reviewed in Powers et al., 2011), atrophy (Lawler et al., 2003), mitochondrial fission and fusion (reviewed in Willems et al., 2015), vascular growth and remodeling (reviewed in Bir et al., 2012), gene stability (Mikhed et al., 2015), and cellular proliferation and death (Wang et al., 2013; L'honoré et al., 2014). An oxidative shift with elevated ROS production in one cell type may have a direct and/or indirect effect on other resident cell types. Although it is difficult to imagine that charged, highly reactive oxygen/nitrogen species arising within subcellular organelles (e.g., mitochondria) or from cytosolic enzymes (e.g., xanthine oxidase) could escape the oxidant buffering systems (e.g., glutathione peroxidases, peroxiredoxins, superoxide dismutase, catalase) and travel to neighboring cells, ROS species, particularly those not carrying a charge (e.g., H2O2), produced by membrane bound enzymes (e.g., NADPH oxidase) may be capable of directly affecting nearby cells. It is likely that altered redox homeostasis in one cell would dramatically alter the local microenvironment through paracrine signaling. For example, skeletal muscle redox alterations have been shown to decrease endothelial cell angiogenic properties via the HIF-1α signaling cascade (Dromparis et al., 2014). Further, HIF-1α is a known transcriptional regulator for vascular endothelial growth factor (VEGF), which plays a vital role in angiogenesis (Rhoads et al., 2009).
Early research on the cellular bioenergetics of endothelial cells (ECs) suggested a heavy cellular reliance on glycolytic metabolism for the energy requirements of normal processes (Dobrina and Rossi, 1983; Leighton et al., 1987; Krützfeldt et al., 1990; Laing et al., 1992). These studies reported high activities of key enzymes in glycolytic metabolism (phosphofructokinase, hexokinase), high rates of lactate production in aerobic conditions, and low rates of glucose oxidation especially when high levels of glucose are present (Crabtree Effect). Additionally, ECs have a relatively low mitochondrial content, [less than 5% of the cell volume vs. 5–20% in skeletal muscle (Hoppeler et al., 1981; Groschner et al., 2012; Dahl et al., 2015; Jacobs et al., 2015)]. Some studies suggest mitochondrial ATP production is dispensable in ECs (Quintero et al., 2006) and there appears to be supportive evidence in that limb ECs are resistant to ischemic insult in CLI patients (Mertens et al., 1990; Noll et al., 1990). There is also a distinct body of research, however, indicating that mitochondria are critical organelles to the viability and function of ECs (Quintero et al., 2006; Goveia et al., 2014; Eelen et al., 2015). Mesenchymal stem cells form tunneling nanotubes that transfer mitochondria to ECs to rescue cellular aerobic respiration and stave off apoptosis induced by ischemia/reperfusion (Liu et al., 2014), a response that could be particularly important in stroke patients (Chan, 2005; Li et al., 2012; Lejay et al., 2014; Mishiro et al., 2014). Capillary EC mitochondrial cytopathies decrease angiogenesis and precede myofiber injury in early infants with mitochondrial diseases, a finding termed “mitochondrial angiopathy” (Sarnat et al., 2012). Overexpression of mitochondrial Thioredoxin-2 (Trx2) improves EC proliferation and arteriogenesis in the ischemic limb (Dai et al., 2009) and cancer researchers now utilize mitochondrial uncouplers in attempts to reduce tumor size due to their effects on neovascularization (Coutelle et al., 2014). These recent findings support an integral role for the mitochondria in the regulation of EC function and indicate this organelle's potential as a therapeutic target for CLI.
Mitochondria are dynamic organelles that rely on complex signals orchestrating dynamic fission and fusion events believed to be responsible for regulating mitochondrial quality control. Fission and fusion are involved in the elimination of damaged/dysfunctional mitochondria (Song et al., 2015) which may serve as major sources of ROS. A cell's decision to remove dysfunctional mitochondria plays a vital role in limiting cellular damage/apoptosis while maintaining cell function. Damaged and depolarized mitochondria are targeted by PTEN-induced putative kinase 1 (PINK1), which drives Parkin-mediated mitophagic engulfment by autophagosomes (termed “mitophagy,” for detailed reviews see Dorn and Kitsis, 2015; Shirihai et al., 2015). Recent preclinical evidence suggests that defects in mitophagy exacerbate cardiomyocyte injury and decrease survival following ischemia/reperfusion (Song et al., 2014), indicating an increased cellular sensitivity to ischemic stress (Kubli et al., 2013). Mitophagy is critically important to the plasticity of skeletal muscle (Liesa and Shirihai, 2013) and is a unique process that could be similarly important to the health of the vasculature in the ischemic limb. Ischemia/reperfusion-induced impairments in EC mitochondrial respiratory capacity have been intricately linked to accelerated fission caused by excessive oxidative and nitrosative stress (Giedt et al., 2012). Moreover, siRNA-knockdown of mitofusin-1 or mitofusin-2 impairs EC angiogenic function in vitro and increases markers of apoptosis under stress (serum-deprivation; Lugus et al., 2011). There are no current investigations into the potential role of mitophagy in limb muscle pathology with CLI, although mitochondrial dynamics provide an attractive candidate for exploration. The accumulation of damaged mitochondria is likely to lead to increased ROS, an oxidative shift in the redox environment, and impaired energy production; all factors contributing to a pathologic ischemic microenvironment.
There are numerous risk factors linked to the CLI manifestation in PAD patients (Fowkes et al., 2013; Nehler et al., 2014). The two strongest risk factors for CLI, smoking and diabetes, are particularly provocative in terms of the subject matter of this review due to the ability of both to impair mitochondrial function in multiple cellular compartments of the ischemic limb.
From a physiologic perspective, smoking impairs microvascular reactivity (Ijzerman et al., 2003), increases intima-media carotid wall thickness (Howard et al., 1994), decreases flow-mediated dilation in the brachial artery (Langham et al., 2015) and increases the likelihood of atherosclerotic lesion formation (Yanbaeva et al., 2007). Pre-clinically, chronic cigarette smoke exposure severely alters vascular structure and function, including facilitating oxidative and nitrosative stress (Talukder et al., 2011). ECs exposed to cigarette smoke extract in vitro have lower mitochondrial integrity, rapid loss of mitochondrial membrane potential, and arrest of cell cycle progression (Henderson et al., 2008). Interference of the respiratory chain by either hydroquinone or carbon monoxide is believed to be a key component of smoking induced mitochondrial dysfunction in skeletal muscles (Degens et al., 2015), as well as impaired oxygen delivery due to carbon monoxide binding with hemoglobin/myoglobin. Interestingly, the combination of high-fat diet and nicotine results in increased oxidative stress and substantial lipid accumulation adjacent to swollen intramyofibrillar mitochondria in peripheral skeletal muscle (Sinha-Hikim et al., 2014). Taken together, the global cellular response to smoking demonstrates the potential for smoking to alter not only physiologic vessel function and the time-course of atherosclerotic lesion formation, but also the health of peripheral muscle cells. Interestingly, only a small series of research studies examine the muscle regenerative aspect, several of which are linked with healing rates after orthopedic surgery (Karim et al., 2006; Lundgreen et al., 2014; Mall et al., 2014). There are, however, a number of studies demonstrating ultrastructural and functional alterations in cardiomyocyte mitochondria after exposure to cigarette smoke (Yamada et al., 2009; Hu et al., 2013; Tippetts et al., 2014). Further work is necessitated to directly examine the effects of smoking on the mitochondria of the ischemic limb muscle, but this area represents an exciting arena with the potential to result in singular therapies for multiple co-morbidities associated with smoking and cardiovascular disease.
Type II Diabetic patients with PAD are five times more likely to present clinically with CLI accompanied by tissue loss (Jude et al., 2001) and do not respond well to revascularization or endovascular interventions (Derubertis et al., 2008; Malmstedt et al., 2008). While diabetes may exacerbate the development of plaque blockages in the arteries, the impact of metabolic syndrome/diabetes on other tissue compartments has not been investigated in the context of CLI. As a common risk-factor, one explanation for the diabetic increase in PAD susceptibility could conceivably be exacerbated muscle damage originating from compromised mitochondrial function prior to the onset of ischemia. Diabetes both reduces skeletal muscle mitochondrial function (Kelley et al., 2002; Petersen et al., 2004; Bonnard et al., 2008) and increases mitochondrial fission, fragmentation, and ROS production in human venous ECs (Shenouda et al., 2011), indicating the potential for exacerbated ischemic responses in multiple cellular compartments of the ischemic limb. Chronic oxidative stress caused by nutrient oversupply to muscle mitochondria is implicated in reduced diabetic mitochondrial respiratory function (Bonnard et al., 2008; Anderson et al., 2009a,b), whereas mitochondrial-targeted antioxidants confer protection against diet-induced dysfunction (Hoehn et al., 2009; Anderson et al., 2009b; Lee et al., 2010). Taken together, these findings suggest the possibility that compromised muscle and endothelial mitochondrial function may be pre-conditioning the limb tissue to respond poorly to the ischemic insult in diabetic CLI patients, resulting in greater myopathy and sustained tissue degeneration regardless of genetic susceptibility.
A critical barrier to developing therapeutic strategies to PAD has been a lack of understanding of the mechanisms underlying the etiology and pathology of PAD. While the cause of PAD is unquestionably occlusive arterial disease, the limited success of surgical and angiogenic treatments suggest that factors other than blood flow may significantly contribute to patient outcomes. Physiologically, angiogenesis and neovascularization are directed by the metabolic demand of the resident tissue. Simply put, the return of blood flow will have little effect if the limb tissue is beyond repair. In this review, we have highlighted recent trends in CLI research that suggest limb musculature may be a viable and potentially parallel therapeutic option for both the myopathy and vasculopathy of CLI. Furthermore, limb muscle and EC mitochondria provide attractive specific targets for novel therapeutic intervention.
JM supported by NIH/NHLBI R00HL103797 and R01HL125695, and a Brody Brothers Endowment Award, DB supported by NIH/NHLBI R01 HL123647 and R15 HL122922, PN supported by NIH/NIDDK R01 DK096907.
David A. Brown has served as a consultant for Stealth BioTherapeutics, which is developing novel treatments for mitochondrial diseases. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abou-Khalil, R., Mounier, R., and Chazaud, B. (2010). Regulation of myogenic stem cell behavior by vessel cells: the “menage a trois” of satellite cells, periendothelial cells and endothelial cells. Cell Cycle 9, 892–896. doi: 10.4161/cc.9.5.10851
Anderson, E. J., Kypson, A. P., Rodriguez, E., Anderson, C. A., Lehr, E. J., and Neufer, P. D. (2009a). Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 54, 1891–1898. doi: 10.1016/j.jacc.2009.07.031
Anderson, E. J., Lustig, M. E., Boyle, K. E., Woodlief, T. L., Kane, D. A., Lin, C. T., et al. (2009b). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581. doi: 10.1172/JCI37048
Annex, B. H. (2013). Therapeutic angiogenesis for critical limb ischaemia. Nat. Rev. Cardiol. 10, 387–396. doi: 10.1038/nrcardio.2013.70
Asai, A., Sahani, N., Kaneki, M., Ouchi, Y., Martyn, J. A., and Yasuhara, S. E. (2007). Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE 2:e806. doi: 10.1371/journal.pone.0000806
Bhat, H. K., Hiatt, W. R., Hoppel, C. L., and Brass, E. P. (1999). Skeletal muscle mitochondrial DNA injury in patients with unilateral peripheral arterial disease. Circulation 99, 807–812. doi: 10.1161/01.CIR.99.6.807
Bir, S. C., Kolluru, G. K., Fang, K., and Kevil, C. G. (2012). Redox balance dynamically regulates vascular growth and remodeling. Semin. Cell Dev. Biol. 23, 745–757. doi: 10.1016/j.semcdb.2012.05.003
Bonnard, C., Durand, A., Peyrol, S., Chanseaume, E., Chauvin, M. A., Morio, B., et al. (2008). Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Invest. 118, 789–800. doi: 10.1172/jci32601
Brass, E. P. (1996). Skeletal muscle metabolism as a target for drug therapy in peripheral arterial disease. Vasc. Med. 1, 55–59.
Brass, E. P. (2013). Intermittent claudication: new targets for drug development. Drugs 73, 999–1014. doi: 10.1007/s40265-013-0078-3
Brass, E. P., and Hiatt, W. R. (2000). Acquired skeletal muscle metabolic myopathy in atherosclerotic peripheral arterial disease. Vasc. Med. 5, 55–59. doi: 10.1177/1358836X0000500109
Brass, E. P., Hiatt, W. R., and Green, S. (2004). Skeletal muscle metabolic changes in peripheral arterial disease contribute to exercise intolerance: a point-counterpoint discussion. Vasc. Med. 9, 293–301. doi: 10.1191/1358863x04vm572ra
Chalothorn, D., Clayton, J. A., Zhang, H., Pomp, D., and Faber, J. E. (2007). Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol. Genomics 30, 179–191. doi: 10.1152/physiolgenomics.00047.2007
Chalothorn, D., and Faber, J. E. (2010). Strain-dependent variation in collateral circulatory function in mouse hindlimb. Physiol. Genomics 42, 469–479. doi: 10.1152/physiolgenomics.00070.2010
Chan, P. H. (2005). Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann. N.Y. Acad. Sci. 1042, 203–209. doi: 10.1196/annals.1338.022
Chargé, S. B., and Rudnicki, M. A. (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238. doi: 10.1152/physrev.00019.2003
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijevic, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi: 10.1038/nature13909
Cluff, K., Miserlis, D., Naganathan, G. K., Pipinos, I. I., Koutakis, P., Samal, A., et al. (2013). Morphometric analysis of gastrocnemius muscle biopsies from patients with peripheral arterial disease: objective grading of muscle degeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R291–R299. doi: 10.1152/ajpregu.00525.2012
Cooke, J. P., and Losordo, D. W. (2015). Modulating the vascular response to limb ischemia: angiogenic and cell therapies. Circ. Res. 116, 1561–1578. doi: 10.1161/CIRCRESAHA.115.303565
Coutelle, O., Hornig-Do, H. T., Witt, A., Andree, M., Schiffmann, L. M., Piekarek, M., et al. (2014). Embelin inhibits endothelial mitochondrial respiration and impairs neoangiogenesis during tumor growth and wound healing. EMBO Mol. Med. 6, 624–639. doi: 10.1002/emmm.201303016
Dahl, R., Larsen, S., Dohlmann, T. L., Qvortrup, K., Helge, J. W., Dela, F., et al. (2015). Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol. 213, 145–155. doi: 10.1111/apha.12289
Dai, S., He, Y., Zhang, H., Yu, L., Wan, T., Xu, Z., et al. (2009). Endothelial-specific expression of mitochondrial thioredoxin promotes ischemia-mediated arteriogenesis and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 29, 495–502. doi: 10.1161/ATVBAHA.108.180349
Degens, H., Gayan-Ramirez, G., and Van Hees, H. W. (2015). Smoking-induced skeletal muscle dysfunction: from evidence to mechanisms. Am. J. Respir. Crit. Care Med. 191, 620–625. doi: 10.1164/rccm.201410-1830PP
de Liefde, I. I., Hoeks, S. S., van Gestel, Y. R., Klein, J., Bax, J. J., Verhagen, H. J., et al. (2009). The prognostic value of impaired walking distance on long-term outcome in patients with known or suspected peripheral arterial disease. Eur. J. Vasc. Endovasc. Surg. 38, 482–487. doi: 10.1016/j.ejvs.2009.02.022
Derubertis, B. G., Pierce, M., Ryer, E. J., Trocciola, S., Kent, K. C., and Faries, P. L. (2008). Reduced primary patency rate in diabetic patients after percutaneous intervention results from more frequent presentation with limb-threatening ischemia. J. Vasc. Surg. 47, 101–108. doi: 10.1016/j.jvs.2007.09.018
Dobrina, A., and Rossi, F. (1983). Metabolic properties of freshly isolated bovine endothelial cells. Biochim. Biophys. Acta 762, 295–301. doi: 10.1016/0167-4889(83)90084-8
Dokun, A. O., Keum, S., Hazarika, S., Li, Y., Lamonte, G. M., Wheeler, F., et al. (2008). A quantitative trait locus (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation 117, 1207–1215. doi: 10.1161/CIRCULATIONAHA.107.736447
Dormandy, J., Heeck, L., and Vig, S. (1999). The fate of patients with critical leg ischemia. Semin. Vasc. Surg. 12, 142–147.
Dorn, G. W. II, Kitsis, R. N. (2015). The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 116, 167–182. doi: 10.1161/CIRCRESAHA.116.303554
Dromparis, P., Sutendra, G., Paulin, R., Proctor, S., Michelakis, E. D., and McMurtry, M. S. (2014). Pioglitazone inhibits HIF-1alpha-dependent angiogenesis in rats by paracrine and direct effects on endothelial cells. J. Mol. Med. 92, 497–507. doi: 10.1007/s00109-013-1115-0
Eelen, G., De Zeeuw, P., Simons, M., and Carmeliet, P. (2015). Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 116, 1231–1244. doi: 10.1161/CIRCRESAHA.116.302855
Fowkes, F. G., Rudan, D., Rudan, I., Aboyans, V., Denenberg, J. O., McDermott, M. M., et al. (2013). Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet 382, 1329–1340. doi: 10.1016/S0140-6736(13)61249-0
Gardner, A. W., Montgomery, P. S., and Parker, D. E. (2008). Physical activity is a predictor of all-cause mortality in patients with intermittent claudication. J. Vasc. Surg. 47, 117–122. doi: 10.1016/j.jvs.2007.09.033
Giedt, R. J., Yang, C., Zweier, J. L., Matzavinos, A., and Alevriadou, B. R. (2012). Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free Radic. Biol. Med. 52, 348–356. doi: 10.1016/j.freeradbiomed.2011.10.491
Go, Y. M., and Jones, D. P. (2013). The redox proteome. J. Biol. Chem. 288, 26512–26520. doi: 10.1074/jbc.R113.464131
Goveia, J., Stapor, P., and Carmeliet, P. (2014). Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 6, 1105–1120. doi: 10.15252/emmm.201404156
Groschner, L. N., Waldeck-Weiermair, M., Malli, R., and Graier, W. F. (2012). Endothelial mitochondria–less respiration, more integration. Pflugers Arch. 464, 63–76. doi: 10.1007/s00424-012-1085-z
Grounds, M. D. (1987). Phagocytosis of necrotic muscle in muscle isografts is influenced by the strain, age, and sex of host mice. J. Pathol. 153, 71–82. doi: 10.1002/path.1711530110
Grounds, M. D., and McGeachie, J. K. (1989). A comparison of muscle precursor replication in crush-injured skeletal muscle of Swiss and BALBc mice. Cell Tissue Res. 255, 385–391. doi: 10.1007/BF00224122
Gudmundsson, G., Matthiasson, S. E., Arason, H., Johannsson, H., Runarsson, F., Bjarnason, H., et al. (2002). Localization of a gene for peripheral arterial occlusive disease to chromosome 1p31. Am. J. Hum. Genet. 70, 586–592. doi: 10.1086/339251
Hammer, A., and Steiner, S. (2013). Gene therapy for therapeutic angiogenesis in peripheral arterial disease - a systematic review and meta-analysis of randomized, controlled trials. VASA 42, 331–339. doi: 10.1024/0301-1526/a000298
Hands, L. J., Bore, P. J., Galloway, G., Morris, P. J., and Radda, G. K. (1986). Muscle metabolism in patients with peripheral vascular-disease investigated by P-31 nuclear-magnetic-resonance spectroscopy. Clin. Sci. 71, 283–290. doi: 10.1042/cs0710283
Henderson, B., Csordas, A., Backovic, A., Kind, M., Bernhard, D., and Wick, G. (2008). Cigarette smoke is an endothelial stressor and leads to cell cycle arrest. Atherosclerosis 201, 298–305. doi: 10.1016/j.atherosclerosis.2008.02.022
Hiatt, W. R., Armstrong, E. J., Larson, C. J., and Brass, E. P. (2015). Pathogenesis of the limb manifestations and exercise limitations in peripheral artery disease. Circ. Res. 116, 1527–1539. doi: 10.1161/CIRCRESAHA.116.303566
Hirsch, A. T., Criqui, M. H., Treat-Jacobson, D., Regensteiner, J. G., Creager, M. A., Olin, J. W., et al. (2001). Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA 286, 1317–1324. doi: 10.1001/jama.286.11.1317
Hoehn, K. L., Salmon, A. B., Hohnen-Behrens, C., Turner, N., Hoy, A. J., Maghzal, G. J., et al. (2009). Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 17787–17792. doi: 10.1073/pnas.0902380106
Hoppeler, H., Mathieu, O., Krauer, R., Claassen, H., Armstrong, R. B., and Weibel, E. R. (1981). Design of the mammalian respiratory system. VI Distribution of mitochondria and capillaries in various muscles. Respir. Physiol. 44, 87–111. doi: 10.1016/0034-5687(81)90078-5
Howard, G., Burke, G. L., Szklo, M., Tell, G. S., Eckfeldt, J., Evans, G., et al. (1994). Active and passive smoking are associated with increased carotid wall thickness. The atherosclerosis risk in communities study. Arch. Intern. Med. 154, 1277–1282. doi: 10.1001/archinte.1994.00420110125014
Hu, N., Han, X., Lane, E. K., Gao, F., Zhang, Y., and Ren, J. (2013). Cardiac-specific overexpression of metallothionein rescues against cigarette smoking exposure-induced myocardial contractile and mitochondrial damage. PLoS ONE 8:e57151. doi: 10.1371/journal.pone.0057151
Ijzerman, R. G., Serne, E. H., Van Weissenbruch, M. M., De Jongh, R. T., and Stehouwer, C. D. (2003). Cigarette smoking is associated with an acute impairment of microvascular function in humans. Clin. Sci. 104, 247–252. doi: 10.1042/cs1040247
Isbell, D. C., Berr, S. S., Toledano, A. Y., Epstein, F. H., Meyer, C. H., Rogers, W. J., et al. (2006). Delayed calf muscle phosphocreatine recovery after exercise identifies peripheral arterial disease. J. Am. Coll. Cardiol. 47, 2289–2295. doi: 10.1016/j.jacc.2005.12.069
Jacobs, R. A., Lundby, A. M., Fenk, S., Gehrig, S., Siebenmann, C., Fluck, D., et al. (2015). Twenty-eight days of exposure to 3,454 m increases mitochondrial volume density in human skeletal muscle. J. Physiol. doi: 10.1113/JP271118. [Epub ahead of print].
Jain, A., Liu, K., Ferrucci, L., Criqui, M. H., Tian, L., Gurainik, J. M., et al. (2013). Declining walking impairment questionnaire scores are associated with subsequent increased mortality in peripheral arterial disease. J. Am. Coll. Cardiol. 61, 1820–1829. doi: 10.1016/j.jacc.2013.01.060
Jude, E. B., Oyibo, S. O., Chalmers, N., and Boulton, A. J. (2001). Peripheral arterial disease in diabetic and nondiabetic patients: a comparison of severity and outcome. Diabetes Care 24, 1433–1437. doi: 10.2337/diacare.24.8.1433
Karch, J., and Molkentin, J. D. (2015). Regulated necrotic cell death: the passive aggressive side of bax and bak. Circ. Res. 116, 1800–1809. doi: 10.1161/CIRCRESAHA.116.305421
Karim, A., Pandit, H., Murray, J., Wandless, F., and Thomas, N. P. (2006). Smoking and reconstruction of the anterior cruciate ligament. J. Bone Joint Surg. Br. 88, 1027–1031. doi: 10.1302/0301-620X.88B8.17189
Katsetos, C. D., Koutzaki, S., and Melvin, J. J. (2013). Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatr. Neurol. 20, 202–215. doi: 10.1016/j.spen.2013.10.010
Katwal, A. B., and Dokun, A. O. (2011). Peripheral arterial disease in diabetes: is there a role for genetics? Curr. Diab. Rep. 11, 218–225. doi: 10.1007/s11892-011-0188-9
Keller, U., Oberhansli, R., Huber, P., Widmer, L. K., Aue, W. P., Hassink, R. I., et al. (1985). Phosphocreatine content and intracellular pH of calf muscle measured by phosphorus NMR spectroscopy in occlusive arterial disease of the legs. Eur. J. Clin. Invest. 15, 382–388. doi: 10.1111/j.1365-2362.1985.tb00289.x
Kelley, D. E., He, J., Menshikova, E. V., and Ritov, V. B. (2002). Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950. doi: 10.2337/diabetes.51.10.2944
Knowles, J. W., Assimes, T. L., Li, J., Quertermous, T., and Cooke, J. P. (2007). Genetic susceptibility to peripheral arterial disease: a dark corner in vascular biology. Arterioscler. Thromb. Vasc. Biol. 27, 2068–2078. doi: 10.1161/01.ATV.0000282199.66398.8c
Koutakis, P., Weiss, D. J., Miserlis, D., Shostrom, V. K., Papoutsi, E., Ha, D. M., et al. (2014). Oxidative damage in the gastrocnemius of patients with peripheral artery disease is myofiber type selective. Redox Biol. 2, 921–928. doi: 10.1016/j.redox.2014.07.002
Krützfeldt, A., Spahr, R., Mertens, S., Siegmund, B., and Piper, H. M. (1990). Metabolism of exogenous substrates by coronary endothelial cells in culture. J. Mol. Cell. Cardiol. 22, 1393–1404. doi: 10.1016/0022-2828(90)90984-A
Kubli, D. A., Zhang, X., Lee, Y., Hanna, R. A., Quinsay, M. N., Nguyen, C. K., et al. (2013). Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 288, 915–926. doi: 10.1074/jbc.M112.411363
Lagrota-Candido, J., Canella, I., Pinheiro, D. F., Santos-Silva, L. P., Ferreira, R. S., Guimarães-Joca, F. J., et al. (2010). Characteristic pattern of skeletal muscle remodelling in different mouse strains. Int. J. Exp. Pathol. 91, 522–529. doi: 10.1111/j.1365-2613.2010.00737.x
Laing, R. A., Chiba, K., Tsubota, K., and Oak, S. S. (1992). Metabolic and morphologic changes in the corneal endothelium. The effects of potassium cyanide, iodoacetamide, and ouabain. Invest. Ophthalmol. Vis. Sci. 33, 3315–3324.
Langham, M. C., Zhou, Y., Chirico, E. N., Magland, J. F., Sehgal, C. M., Englund, E. K., et al. (2015). Effects of age and smoking on endothelial function assessed by quantitative cardiovascular magnetic resonance in the peripheral and central vasculature. J. Cardiovasc. Magn. Reson. 17, 19. doi: 10.1186/s12968-015-0110-8
Lawler, J. M., Song, W., and Demaree, S. R. (2003). Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic. Biol. Med. 35, 9–16. doi: 10.1016/S0891-5849(03)00186-2
Lee, H. Y., Choi, C. S., Birkenfeld, A. L., Alves, T. C., Jornayvaz, F. R., Jurczak, M. J., et al. (2010). Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 12, 668–674. doi: 10.1016/j.cmet.2010.11.004
Leeper, N. J., Kullo, I. J., and Cooke, J. P. (2012). Genetics of peripheral artery disease. Circulation 125, 3220–3228. doi: 10.1161/CIRCULATIONAHA.111.033878
Leeper, N. J., Myers, J., Zhou, M., Nead, K. T., Syed, A., Kojima, Y., et al. (2013). Exercise capacity is the strongest predictor of mortality in patients with peripheral arterial disease. J. Vasc. Surg. 57, 728–733. doi: 10.1016/j.jvs.2012.07.051
Leighton, B., Curi, R., Hussein, A., and Newsholme, E. A. (1987). Maximum activities of some key enzymes of glycolysis, glutaminolysis, Krebs cycle and fatty acid utilization in bovine pulmonary endothelial cells. FEBS Lett. 225, 93–96. doi: 10.1016/0014-5793(87)81137-7
Lejay, A., Choquet, P., Thaveau, F., Singh, F., Schlagowski, A., Charles, A. L., et al. (2015). A new murine model of sustainable and durable chronic critical limb ischemia fairly mimicking human pathology. Eur. J. Vasc. Endovasc. Surg. 49, 205–212. doi: 10.1016/j.ejvs.2014.12.010
Lejay, A., Meyer, A., Schlagowski, A. I., Charles, A. L., Singh, F., Bouitbir, J., et al. (2014). Mitochondria: mitochondrial participation in ischemia-reperfusion injury in skeletal muscle. Int. J. Biochem. Cell Biol. 50, 101–105. doi: 10.1016/j.biocel.2014.02.013
L'honoré, A., Commère, P. H., Ouimette, J. F., Montarras, D., Drouin, J., and Buckingham, M. (2014). Redox regulation by Pitx2 and Pitx3 is critical for fetal myogenesis. Dev. Cell 29, 392–405. doi: 10.1016/j.devcel.2014.04.006
Li, J., Ma, X., Yu, W., Lou, Z., Mu, D., Wang, Y., et al. (2012). Reperfusion promotes mitochondrial dysfunction following focal cerebral ischemia in rats. PLoS ONE 7:e46498. doi: 10.1371/journal.pone.0046498
Liesa, M., and Shirihai, O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. doi: 10.1016/j.cmet.2013.03.002
Liu, K., Ji, K., Guo, L., Wu, W., Lu, H., Shan, P., et al. (2014). Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 92, 10–18. doi: 10.1016/j.mvr.2014.01.008
Lugus, J. J., Ngoh, G. A., Bachschmid, M. M., and Walsh, K. (2011). Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J. Mol. Cell. Cardiol. 51, 885–893. doi: 10.1016/j.yjmcc.2011.07.023
Lundgreen, K., Lian, O. B., Scott, A., Nassab, P., Fearon, A., and Engebretsen, L. (2014). Rotator cuff tear degeneration and cell apoptosis in smokers versus nonsmokers. Arthroscopy 30, 936–941. doi: 10.1016/j.arthro.2014.03.027
Mall, N. A., Tanaka, M. J., Choi, L. S., and Paletta, G. A. Jr. (2014). Factors affecting rotator cuff healing. J. Bone Joint Surg. Am. 96, 778–788. doi: 10.2106/JBJS.M.00583
Malmstedt, J., Leander, K., Wahlberg, E., Karlström, L., Alfredsson, L., and Swedenborg, J. (2008). Outcome after leg bypass surgery for critical limb ischemia is poor in patients with diabetes: a population-based cohort study. Diabetes Care 31, 887–892. doi: 10.2337/dc07-2424
Martin, E. A., Barresi, R., Byrne, B. J., Tsimerinov, E. I., Scott, B. L., Walker, A. E., et al. (2012). Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci. Transl. Med. 4:162ra155. doi: 10.1126/scitranslmed.3004327
Matsubara, Y., Matsumoto, T., Aoyagi, Y., Tanaka, S., Okadome, J., Morisaki, K., et al. (2015). Sarcopenia is a prognostic factor for overall survival in patients with critical limb ischemia. J. Vasc. Surg. 61, 945–950. doi: 10.1016/j.jvs.2014.10.094
Matzke, S., and Lepantalo, M. (2001). Claudication does not always precede critical leg ischemia. Vasc. Med. 6, 77–80. doi: 10.1177/1358836X0100600202
McClung, J. M., Mccord, T. J., Keum, S., Johnson, S., Annex, B. H., Marchuk, D. A., et al. (2012). Skeletal muscle-specific genetic determinants contribute to the differential strain-dependent effects of hindlimb ischemia in mice. Am. J. Pathol. 180, 2156–2169. doi: 10.1016/j.ajpath.2012.01.032
McClung, J. M., Reinardy, J. L., Mueller, S. B., McCord, T. J., Kontos, C. D., Brown, D. A., et al. (2015). Muscle cell derived angiopoietin-1 contributes to both myogenesis and angiogenesis in the ischemic environment. Front. Physiol. 6:161. doi: 10.3389/fphys.2015.00161
McDermott, M. M., Liu, K., Ferrucci, L., Tian, L., Gurainik, J. M., Liao, Y., et al. (2011). Decline in functional performance predicts later increased mobility loss and mortality in peripheral arterial disease. J. Am. Coll. Cardiol. 57, 962–970. doi: 10.1016/j.jacc.2010.09.053
McDermott, M. M., Liu, K., Tian, L., Guralnik, J. M., Criqui, M. H., Liao, Y., et al. (2012). Calf muscle characteristics, strength measures, and mortality in peripheral arterial disease: a longitudinal study. J. Am. Coll. Cardiol. 59, 1159–1167. doi: 10.1016/j.jacc.2011.12.019
McGeachie, J. K., and Grounds, M. D. (1995). Retarded myogenic cell replication in regenerating skeletal muscles of old mice: an autoradiographic study in young and old BALBc and SJL/J mice. Cell Tissue Res. 280, 277–282. doi: 10.1007/BF00307799
Mertens, S., Noll, T., Spahr, R., Krützfeldt, A., and Piper, H. M. (1990). Energetic response of coronary endothelial cells to hypoxia. Am. J. Physiol. 258, H689–H694.
Messina, L. M. (2008). Elucidating the genetic basis of peripheral arterial disease: identification of a quantitative trait locus that determines the phenotypic response to experimental hindlimb ischemia. Circulation 117, 1127–1129. doi: 10.1161/CIRCULATIONAHA.107.752055
Mikhed, Y., Görlach, A., Knaus, U. G., and Daiber, A. (2015). Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 5, 275–289. doi: 10.1016/j.redox.2015.05.008
Mishiro, K., Imai, T., Sugitani, S., Kitashoji, A., Suzuki, Y., Takagi, T., et al. (2014). Diabetes mellitus aggravates hemorrhagic transformation after ischemic stroke via mitochondrial defects leading to endothelial apoptosis. PLoS ONE 9:e103818. doi: 10.1371/journal.pone.0103818
Mitchell, C. A., Grounds, M. D., and Papadimitriou, J. M. (1995). The genotype of bone marrow-derived inflammatory cells does not account for differences in skeletal muscle regeneration between SJL/J and BALB/c mice. Cell Tissue Res. 280, 407–413. doi: 10.1007/BF00307814
Mofarrahi, M., McClung, J. M., Kontos, C. D., Davis, E. C., Tappuni, B., Moroz, N., et al. (2015). Angiopoietin-1 enhances skeletal muscle regeneration in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R576–R589. doi: 10.1152/ajpregu.00267.2014
Murabito, J. M., White, C. C., Kavousi, M., Sun, Y. V., Feitosa, M. F., Nambi, V., et al. (2012). Association between chromosome 9p21 variants and the ankle-brachial index identified by a meta-analysis of 21 genome-wide association studies. Circ. Cardiovasc. Genet. 5, 100–112. doi: 10.1161/CIRCGENETICS.111.961292
Nehler, M. R., Duval, S., Diao, L., Annex, B. H., Hiatt, W. R., Rogers, K., et al. (2014). Epidemiology of peripheral arterial disease and critical limb ischemia in an insured national population. J. Vasc. Surg. 60, 686.e2–695.e2. doi: 10.1016/j.jvs.2014.03.290
Noll, T., Wissemann, P., Mertens, S., Krützfeldt, A., Spahr, R., and Piper, H. M. (1990). Hypoxia tolerance of coronary endothelial cells. Adv. Exp. Med. Biol. 277, 467–476. doi: 10.1007/978-1-4684-8181-5_52
Petersen, K. F., Dufour, S., Befroy, D., Garcia, R., and Shulman, G. I. (2004). Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 350, 664–671. doi: 10.1056/NEJMoa031314
Pipinos, I. I., Judge, A. R., Selsby, J. T., Zhu, Z., Swanson, S. A., Nella, A. A., et al. (2007). The myopathy of peripheral arterial occlusive disease: part 1. Functional and histomorphological changes and evidence for mitochondrial dysfunction. Vasc. Endovasc. Surg. 41, 481–489. doi: 10.1177/1538574407311106
Pipinos, I. I., Judge, A. R., Selsby, J. T., Zhu, Z., Swanson, S. A., Nella, A. A., et al. (2008a). The myopathy of peripheral arterial occlusive disease: Part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc. Endovasc. Surg. 42, 101–112. doi: 10.1177/1538574408315995
Pipinos, I. I., Judge, A. R., Zhu, Z., Selsby, J. T., Swanson, S. A., Johanning, J. M., et al. (2006). Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med. 41, 262–269. doi: 10.1016/j.freeradbiomed.2006.04.003
Pipinos, I. I., Sharov, V. G., Shepard, A. D., Anagnostopoulos, P. V., Katsamouris, A., Todor, A., et al. (2003). Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J. Vasc. Surg. 38, 827–832. doi: 10.1016/S0741-5214(03)00602-5
Pipinos, I. I., Shepard, A. D., Anagnostopoulos, P. V., Katsamouris, A., and Boska, M. D. (2000a). Phosphorus 31 nuclear magnetic resonance spectroscopy suggests a mitochondrial defect in claudicating skeletal muscle. J. Vasc. Surg. 31, 944–952. doi: 10.1067/mva.2000.106421
Pipinos, I. I., Shepard, A. D., Anagnostopoulos, P. V., Katsamouris, A., and Boska, M. D. (2000b). Phosphorus 31 nuclear magnetic resonance spectroscopy suggests a mitochondrial defect in claudicating skeletal muscle. J. Vasc. Surg. 31, 944–952. doi: 10.1067/mva.2000.106421
Pipinos, I. I., Swanson, S. A., Zhu, Z., Nella, A. A., Weiss, D. J., Gutti, T. L., et al. (2008b). Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R290–R296. doi: 10.1152/ajpregu.90374.2008
Powers, S. K., Ji, L. L., Kavazis, A. N., and Jackson, M. J. (2011). Reactive oxygen species: impact on skeletal muscle. Compr. Physiol. 1, 941–969. doi: 10.1002/cphy.c100054
Quintero, M., Colombo, S. L., Godfrey, A., and Moncada, S. (2006). Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. U.S.A. 103, 5379–5384. doi: 10.1073/pnas.0601026103
Rahman, M., Mofarrahi, M., Kristof, A. S., Nkengfac, B., Harel, S., and Hussain, S. N. (2014). Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxid. Redox Signal. 20, 443–459. doi: 10.1089/ars.2013.5410
Raval, Z., Liu, K., Tian, L., Ferrucci, L., Gurainik, J. M., Liao, Y., et al. (2012). Higher body mass index is associated with more adverse changes in calf muscle characteristics in peripheral arterial disease. J. Vasc. Surg. 55, 1015–1024. doi: 10.1016/j.jvs.2011.10.105
Rhoads, R. P., Johnson, R. M., Rathbone, C. R., Liu, X., Temm-Grove, C., Sheehan, S. M., et al. (2009). Satellite cell-mediated angiogenesis in vitro coincides with a functional hypoxia-inducible factor pathway. Am. J. Physiol. Cell Physiol. 296, C1321–C1328. doi: 10.1152/ajpcell.00391.2008
Rissanen, T. T., Vajanto, I., Hiltunen, M. O., Rutanen, J., Kettunen, M. I., Niemi, M., et al. (2002). Expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 (KDR/Flk-1) in ischemic skeletal muscle and its regeneration. Am. J. Pathol. 160, 1393–1403. doi: 10.1016/S0002-9440(10)62566-7
Roberts, P., McGeachie, J. K., and Grounds, M. D. (1997). The host environment determines strain-specific differences in the timing of skeletal muscle regeneration: cross-transplantation studies between SJL/J and BALB/c mice. J. Anat. 191 (Pt 4), 585–594. doi: 10.1046/j.1469-7580.1997.19140585.x
Sarnat, H. B., Flores-Sarnat, L., Casey, R., Scott, P., and Khan, A. (2012). Endothelial ultrastructural alterations of intramuscular capillaries in infantile mitochondrial cytopathies: “mitochondrial angiopathy” Neuropathology 32, 617–627. doi: 10.1111/j.1440-1789.2012.01308.x
Schocke, M., Esterhammer, R., and Greiner, A. (2008). High-energy phosphate metabolism in the exercising muscle of patients with peripheral arterial disease. VASA 37, 199–210. doi: 10.1024/0301-1526.37.3.199
Seale, P., Asakura, A., and Rudnicki, M. A. (2001). The potential of muscle stem cells. Dev. Cell 1, 333–342. doi: 10.1016/S1534-5807(01)00049-1
Sealock, R., Zhang, H., Lucitti, J. L., Moore, S. M., and Faber, J. E. (2014). Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ. Res. 114, 660–671. doi: 10.1161/CIRCRESAHA.114.302931
Shenouda, S. M., Widlansky, M. E., Chen, K., Xu, G., Holbrook, M., Tabit, C. E., et al. (2011). Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 124, 444–453. doi: 10.1161/CIRCULATIONAHA.110.014506
Shirihai, O. S., Song, M., and Dorn, G. W. II. (2015). How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 116, 1835–1849. doi: 10.1161/CIRCRESAHA.116.306374
Singh, N., Liu, K., Tian, L., Criqui, M. H., Gurainik, J. M., Ferucci, L., et al. (2010). Leg strength predicts mortality in men but not in women with peripheral arterial disease. J. Vasc. Surg. 52, 624–631. doi: 10.1016/j.jvs.2010.03.066
Sinha-Hikim, I., Friedman, T. C., Shin, C. S., Lee, D., Ivey, R., and Sinha-Hikim, A. P. (2014). Nicotine in combination with a high-fat diet causes intramyocellular mitochondrial abnormalities in male mice. Endocrinology 155, 865–872. doi: 10.1210/en.2013-1795
Song, M., Chen, Y., Gong, G., Murphy, E., Rabinovitch, P. S., and Dorn, G. W. II. (2014). Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 115, 348–353. doi: 10.1161/CIRCRESAHA.115.304384
Song, M., Mihara, K., Chen, Y., Scorrano, L., and Dorn, G. W. II. (2015). Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–285. doi: 10.1016/j.cmet.2014.12.011
Talukder, M. A., Johnson, W. M., Varadharaj, S., Lian, J., Kearns, P. N., El-Mahdy, M. A., et al. (2011). Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am. J. Physiol. Heart Circ. Physiol. 300, H388–H396. doi: 10.1152/ajpheart.00868.2010
Taylor, S. M., Cull, D. L., Kalbaugh, C. A., Senter, H. F., Langan, E. M. III, Carsten, C. G. III et al. (2009). Comparison of interventional outcomes according to preoperative indication: a single center analysis of 2,240 limb revascularizations. J. Am. Coll. Surg. 208, 770–778. discussion: 778–780. doi: 10.1016/j.jamcollsurg.2009.01.025
Thompson, J. R., Swanson, S. A., Haynatzki, G., Koutakis, P., Johanning, J. M., Reppert, P. R., et al. (2014). Protein concentration and mitochondrial content in the gastrocnemius predicts mortality rates in patients with peripheral arterial disease. Ann. Surg. 261, 605–610. doi: 10.1097/SLA.0000000000000643
Tippetts, T. S., Winden, D. R., Swensen, A. C., Nelson, M. B., Thatcher, M. O., Saito, R. R., et al. (2014). Cigarette smoke increases cardiomyocyte ceramide accumulation and inhibits mitochondrial respiration. BMC Cardiovasc. Disord. 14:165. doi: 10.1186/1471-2261-14-165
Wang, K., Zhang, T., Dong, Q., Nice, E. C., Huang, C., and Wei, Y. (2013). Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 4:e537. doi: 10.1038/cddis.2013.50
Wang, S., Zhang, H., Dai, X., Sealock, R., and Faber, J. E. (2010). Genetic architecture underlying variation in extent and remodeling of the collateral circulation. Circ. Res. 107, 558–568. doi: 10.1161/CIRCRESAHA.110.224634
Wang, S., Zhang, H., Wiltshire, T., Sealock, R., and Faber, J. E. (2012). Genetic dissection of the Canq1 locus governing variation in extent of the collateral circulation. PLoS ONE 7:e31910. doi: 10.1371/journal.pone.0031910
Weiss, D. J., Casale, G. P., Koutakis, P., Nella, A. A., Swanson, S. A., Zhu, Z., et al. (2013). Oxidative damage and myofiber degeneration in the gastrocnemius of patients with peripheral arterial disease. J. Transl. Med. 11:230. doi: 10.1186/1479-5876-11-230
Willems, P. H., Rossignol, R., Dieteren, C. E., Murphy, M. P., and Koopman, W. J. (2015). Redox homeostasis and mitochondrial dynamics. Cell Metab. 22, 207–218. doi: 10.1016/j.cmet.2015.06.006
Wurdeman, S. R., Myers, S. A., Johanning, J. M., Pipinos, I. I., and Stergiou, N. (2012). External work is deficient in both limbs of patients with unilateral PAD. Med. Eng. Phys. 34, 1421–1426. doi: 10.1016/j.medengphy.2012.01.004
Yamada, S., Zhang, X. Q., Kadono, T., Matsuoka, N., Rollins, D., Badger, T., et al. (2009). Direct toxic effects of aqueous extract of cigarette smoke on cardiac myocytes at clinically relevant concentrations. Toxicol. Appl. Pharmacol. 236, 71–77. doi: 10.1016/j.taap.2009.01.008
Yanbaeva, D. G., Dentener, M. A., Creutzberg, E. C., Wesseling, G., and Wouters, E. F. (2007). Systemic effects of smoking. Chest 131, 1557–1566. doi: 10.1378/chest.06-2179
Keywords: skeletal muscle, vascular diseases, mitochondria, ischemia, peripheral arterial disease, angiogenesis
Citation: Ryan TE, Schmidt CA, Green TD, Brown DA, Neufer PD and McClung JM (2015) Mitochondrial Regulation of the Muscle Microenvironment in Critical Limb Ischemia. Front. Physiol. 6:336. doi: 10.3389/fphys.2015.00336
Received: 05 August 2015; Accepted: 02 November 2015;
Published: 18 November 2015.
Edited by:
Russell T. Hepple, McGill University, CanadaReviewed by:
Scott Powers, University of Florida, USACopyright © 2015 Ryan, Schmidt, Green, Brown, Neufer and McClung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph M. McClung, bWNjbHVuZ2pAZWN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.