 Stephen J. Pandol
Stephen J. Pandol Mouad Edderkaoui
Mouad Edderkaoui- 1Departments of Medicine and Biological Sciences, Cedars-Sinai Medical Center, Los Angeles, CA, USA
- 2Veterans Affairs Greater Los Angeles Healthcare System and University of California, Los Angeles, Los Angeles, CA, USA
Pancreatic ductal adenocarcinoma is a devastating disease characterized by a dense desmoplastic stroma. Chemo- and radio-therapeutic strategies based on targeting cancer cells have failed in improving the outcome of this cancer suggesting important roles for stroma in therapy resistance. Cells in the tumor stroma have been shown to regulate proliferation, resistance to apoptosis and treatments, epithelial to mesenchymal transition (EMT) and stemness of cancer cells. Stellate cells in their activated state have been thought over the past decade to only have tumor promoting roles. However, recent findings suggest that stellate cells may have protective roles as well. The present review highlights the latest findings on the role of two major components of tumor stroma, pancreatic stellate cells and macrophages, in promoting or inhibiting pancreatic cancer, focused on their effects on EMT and cancer stemness.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States. The 5-year survival rate remains at 5% making it one of the worst outcomes among cancers (Siegel et al., 2012).
PDAC is unique among solid tumors because of the extremely dense desmoplastic stroma that surrounds the cancer cell glands of this tumor. In fact, the majority of the tumor volume is made of stroma compared to a much smaller volume occupied by the cancer cells. Understanding of the populations of the cells present in the stroma is needed to better target the correct sub-populations.
Stellate Cells in the Context of Pancreatic Cancer
Pancreatic stellate cells (PSCs) were first isolated in 1998 by two groups- one from Australia and one Germany (Apte et al., 1998; Bachem et al., 1998). Since then extensive work has been done to understand the role of PSC in promoting pancreatic fibrosis and cancer. There has been general agreement for over a decade that the PSCs promote pancreatic cancer growth, metastasis and resistance to treatment, with observed association between strong stroma and worse outcome (Erkan et al., 2008; Beatty et al., 2011). Indeed, stellate cells promote the growth of cancer cells in sub-cutaneous mouse models of pancreatic cancer (Bachem et al., 2005). One mechanism through which PSCs mediate their pro-cancer effect is through secretion of large amounts of extracellular matrix proteins, responsible in large part for the fibrotic reaction characterizing human PDAC (Apte et al., 2004) as well as for inducing resistance to apoptosis in the cancer cells (Edderkaoui et al., 2005). Activated PSCs secrete growth factors, metallo-proteases, and cytokines shown to stimulate cancer cell proliferation, migration and metastasis of the pancreatic cancer cells (Wehr et al., 2011).
Stellate Cells Regulate Epithelial-Mesenchymal Transition (EMT) and Stemness of the Cancer Cells
A major cause of the poor outcome for patients with pancreatic cancer is the ability of the cancer cells to metastasize and to develop resistance to treatments mainly through development of cancer stemness characteristics. EMT is a process by which epithelial cells lose their cell polarity and cell-cell adhesion while gaining migratory and invasive properties to become mesenchymal stem cells. The importance of the process of EMT is that it is responsible for both metastasis and therapy resistance through development of cancer stemness (Mani et al., 2008; Arumugam et al., 2009). Cancer stem cells markers such as ALDH1A1, ABCG2, and nestin are highly expressed in metastatic cancer cells compared to non-metastatic cancer cells in animal models of pancreatic cancer. The metastatic cells show strong expression of EMT markers indicating the association between EMT, cancer stemness and metastasis (Matsuda et al., 2014).
Recently, the role of PSCs in up-regulating EMT has become more evident. Co-culture of cancer cells with PSCs induces a fibroblast-like appearance of the cancer cells (Kikuta et al., 2010). The cancer cells assume fibroblast characteristics such as increased migration; and expression of mesenchymal markers Vimentin, Snail-1 and Zeb; and decrease expression of epithelial markers in the cancer cells (Kikuta et al., 2010; Mizuuchi et al., 2014). The mechanisms underlying the promotion of EMT are still under investigation and are not established. Because PSCs produce significant amounts of Transforming growth factor-beta (TGF-β) (Shek et al., 2002), there has been interest in the role of TGF-β in EMT. In one study, the PSC-induced EMT induction was not altered by treatment with anti-TGF-β-neutralizing antibody, suggesting no role of TGF-β in this process (Kikuta et al., 2010). Differently, another study showed that specific neutralizing anti-TGF-β prevented EMT, cancer stem cells phenotype, and tumorigenicity (Al-Assar et al., 2014).
There is accumulating evidence that small non-coding microRNAs (miRNAs) mediate the interaction between PSCs and cancer cells. Co-culture of pancreatic cancer cells with PSCs led to increased expression of miR-210. PSCs-induced miR-210 up-regulation was inhibited by inhibitors of ERK and PI3K/Akt pathways. Inhibition of miR-210 expression decreased the expression of Vimentin and Snail-1 as well as cell migration, and increased the membrane-associated expression of β-catenin in Panc-1 cells co-cultured with PSCs indicating its role in regulating EMT and migration of the pancreatic cancer cells (Takikawa et al., 2013).
The role of hypoxia, which is highly present in solid tumors, on tumor promotion was highly investigated in the last few years, Hypoxia increases the activity of PSCs in vitro leading to its migration, secretion of collagen I and VEGF (Masamune et al., 2008; Erkan et al., 2009). The fibrosis-related gene connective tissue growth factor (CTGF/CCN2) protects cells from hypoxia-mediated apoptosis, providing an in vivo selection for tumor cells that express high levels of CTGF/CCN2. Indeed, CTGF/CCN2 expression and secretion was increased in hypoxic pancreatic tumor cells in vitro, and co-localized with hypoxia in pancreatic tumor xenografts and clinical pancreatic adenocarcinomas (Bennewith et al., 2009).
PSCs enhance the cancer stem cell phenotype of pancreatic cancer cells by inducing the expression of genes such as ABCG2, Nestin and LIN28 leading to an enhanced radio-resistance of pancreatic cancer cells in in vitro and in vivo systems of pancreatic cancer (Hamada et al., 2012; Al-Assar et al., 2014). The expression of several EMT and cancer stem cell markers is associated with significant in vivo tumorigenicity.
Very importantly, PSCs show greater activity when isolated from patients after undergoing chemo-radiation therapy as measured by the ability of PSCs to migrate, expand and contract (Cabrera et al., 2014). This data together with the high level of resistance to treatments developed in pancreatic cancer patients suggest that PSC activity may contribute to the development of resistance to therapy in the cancer cells. Indeed, PSCs were found to promote in vitro sphere formation and invasiveness of pancreatic cancer stem cells in an activin/Alk4 receptor -dependent manner (Lonardo et al., 2012). They also stimulated metastasis and the ability to form colonies in mouse orthotopic model of pancreatic cancer (Hwang et al., 2008). In another study, PSCs mediated radioprotection of pancreatic cancer cells in a β1 integrin dependent manner (Mantoni et al., 2011).
Clinical treatment of pancreatic cancer patients with Nab-paclitaxel combined with gemcitabine significantly decreased tumor growth and collagen staining as well as decreased the level of activated stellate cells (Alvarez et al., 2013). There was no association between the level of the secreted protein acidic and rich in cysteine (SPARC) and metastasis or the effect of Nab-paclitaxel (Schneeweiss et al., 2014).
Controversial Role of Stellate Cells
More recently, emerging data is challenging the established PSC pro-cancer role and provides strong evidence for an anti-cancer role of PSCs.
Ozdemir et al. (2014) showed that deletion of activated stellate cells in transgenic mice led to invasive and undifferentiated tumors, enhanced EMT, cancer stemness and decreased survival. The deletion of stellate cells resulted in decreases in both non-invasive precursor pancreatic intraepithelial neoplasia (PanIN) and the PDAC stage of the disease. Importantly, the authors observed an association between fewer activated stellate cells in the tumors with reduced survival in PDAC patients confirming their animal observations (Ozdemir et al., 2014). Finally, depletion of PSCs induced suppression of immune surveillance with increased CD4+Foxp3+Tregs in mouse tumors (Ozdemir et al., 2014). Of note, CD4+Foxp3+Tregs are cells of the immune system that suppress immune responses allowing the cancer cells to survive the immune checkpoint.
Simultaneously with this study, another study confirmed this data showing that depletion of stromal cells from pancreatic tumors, through genetic or pharmacological targeting of the Sonic hedgehog (SH) pathway, results in a reduced stromal content as expected, but led to a poorly differentiated histology, increased vascularity and proliferation, and reduced survival in an animal model of pancreatic cancer. Of note, SH drives the formation of a fibroblast-rich desmoplastic stroma in the tumor (Rhim et al., 2014).
Very importantly, the secretory protein periostin that has been suggested to function as a cell adhesion molecule and promote the invasiveness or growth rate of tumors is highly expressed by PSCs compared to cancer cells in humans (Erkan et al., 2007; Kanno et al., 2008). In addition, Recombinant periostin increased activation of the PSCs. These effects were reversed by silencing periostin expression and secretion by small interfering RNA transfection. Periostin stimulated cancer cell growth and induced their chemoresistance as well as resistance to starvation and hypoxia (Erkan et al., 2007).
This data suggest that PSC activation is a response by the body to prevent cancer development. The mechanism mediating this effect is not well known.
However, data from Kanno et al. (2008) showed that high concentration of recombinant periostin promoted cell migration of the cancer cells mediated by Akt kinase activation. They also found that lower doses of periostin had opposite effect causing expression of epithelial phenotype markers and reduction in mesenchymal markers resulting in reduced cell migration. The findings suggest that periostin has concentration-dependent dual effects on the development of pancreatic cancer (Kanno et al., 2008), especially on EMT and migration of the cancer cells. In the Erkan et al. study, periostin induced activation of the PSCs but decreased their invasiveness (Erkan et al., 2007). Periostin is highly expressed by PSC and its level may be a key regulator of the pro-EMT/cancer and the anti-EMT/cancer effects of PSC. That is, high expression of periostin by PSC will induce EMT, migration metastasis and stemness of the cancer cells; whereas, low doses of periostin will do the opposite.
The findings listed above indicate that there are potentially divergent roles for stellate cells in pancreatic cancer. That is, there may be pro-cancer, neutral, and anti-cancer effects of this stromal component. One possibility is that there are sub-populations and/or sub-phenotypes of PSCs that play different roles in the promotion or prevention of PDAC. The level of periostin expressed by PSC could be a good marker to start with. The different sub-phenotypes of PSCs may be affected by the other tumor microenvironment components. Targeting all of the PSCs together may not necessarily lead to an anti-cancer effect. Further studies are needed to better understand the potential divergent roles of PSCs before defining a treatment strategy based on targeting PSCs.
Role of Macrophages in Promoting Pancreatic Cancer
In addition to the PSCs, macrophages are a major component of the pancreatic tumor microenvironment. The link between inflammation and pancreatic cancer pathogenesis is well established (Yadav and Lowenfels, 2013).
Recently published studies suggest that the macrophages infiltrating the pancreas drive the acinar to ductal metaplasia, a key early process in pancreas carcinogenesis. Pancreatic acinar cells have the capacity to undergo metaplasia to a ductal cell phenotype in the setting of acute or chronic inflammation, representing an important link to PDAC. The process involves inflammatory cytokines such as RANTES and tumor necrosis factor-alpha (TNF-α) and inflammatory intracellular signals such as nuclear factor-κB (NF-κB) as well as growth factors such as TGF-α and epidermal growth factor-receptor (EGFR) (De Lisle and Logsdon, 1990; Sandgren et al., 1990; Song et al., 1999; Means et al., 2003). Genetically engineered mouse models using mutant Kras support the concept that acinar to ductal metaplasia precedes PanIN and PDAC development (Song et al., 1999; Means et al., 2005). Importantly, induction of KrasG12D mutation in acinar cells in mice leads to their transformation to PanIN lesions even in the absence of pancreas injury (Habbe et al., 2008).
Expression of mutant Kras in pancreatic acinar cells expedites their transformation to a duct-like phenotype by inducing local inflammation. Specifically, KrasG12D induces the expression of intercellular adhesion molecule-1 (ICAM-1), which serves as chemo-attractant for macrophages.
Infiltrating macrophages amplify the formation of KrasG12D-caused abnormal pancreatic structures by re-modeling the extracellular matrix and providing cytokines such as TNF-α. Depletion of macrophages or treatment with a neutralizing antibody for ICAM-1 in mice expressing oncogenic Kras under an acinar cell-specific promoter both resulted in a decreased formation of abnormal structures and decreased progression of acinar to ductal metaplasia to PanIN lesions (Liou et al., 2014). Of note, we have shown before that ICAM-1 is also expressed in acinar cells during pancreatitis (Zaninovic et al., 2000).
Both M1 and M2 macrophage phenotypes were found in the pancreatic tumor microenvironment. M1 macrophages are characterized by high expression of inducible nitric oxide synthase (iNOS), IL-10, major histocompatibility complex II (MHC-II), and low IL-12 among others. M2 macrophages express low level or no iNOS, IL-10, and MHC-II, and high levels of Arginase1, CD206, and C-C chemokine receptor 3 (CCR3). Greater levels of tumor-infiltrating M2 macrophages are significantly associated with shorter survival, whereas M1high/M2low correlated significantly with longer survival and suggested using the M1/M2 ratio as independent prognosticator (Ino et al., 2013). Analysis of pancreatic cancer tissues from 36 patients showed that the number of infiltrating macrophages in tumor tissue was significantly greater in patients with metastases to lymph nodes compared to tumor tissue without metastasis (Gardian et al., 2012).
Co-culture with macrophages or with tumor associated macrophage-conditioned media significantly reduced apoptosis and activation of the caspase-3 pathway during gemcitabine treatment of pancreatic cancer cells pointing to a survival effect that the macrophages provide to cancer cells (Weizman et al., 2014). Also, reducing macrophage recruitment and activation in PDAC models in mice demonstrated improved response to gemcitabine compared with controls (Weizman et al., 2014). The data by Weizman et al. showed that decreasing macrophage recruitment augmented the response of PDAC to chemotherapy and that tumor associated macrophages induced up-regulation of cytidine deaminase (CDA), the enzyme that metabolizes gemcitabine following its transport into the cell (Weizman et al., 2014).
Liu et al. showed recently that co-culture with M2-polarized tumor associated macrophages up-regulated the expression of mesenchymal markers Vimentin and Snail and decreased the epithelial marker E-cadherin in pancreatic cancer cells (Liu et al., 2013). Furthermore, co-culture of tumor associated macrophages with pancreatic cancer cells increased proliferation and migration of the cancer cells. They demonstrated that TLR4 and IL-10 mediate the cross talk between tumor associated macrophages and cancer cells (Liu et al., 2013).
All these data indicate that a bidirectional interaction between cancer cells and macrophages determines the fate of their phenotypes, more or less metastatic and resistant to treatment for cancer cells, and pro-inflammatory or pro-cancer for macrophages.
Interaction between Macrophages and Stellate Cells
When quiescent pancreatic stellate cells are co-cultured with macrophage cell lines, the stellate cells showed morphological changes consistent with myofibroblast morphology (Shi et al., 2014). In the presence of Heparin-binding EGF (HB-EGF) many of the stellate cells co-cultured with macrophages were positive for their activation marker αSMA (Shi et al., 2014).
In the same study they found that PSCs have the potential to regulate macrophage function, inducing the production of multiple cytokines, partially through PSC production of IL-6 (Shi et al., 2014). However, caution should be taken as these studies were performed in cell lines and not in cells freshly isolated from animals or humans (Shi et al., 2014).
In a different study, culture of peripheral blood mononuclear cells (PBMC) with PSCs supernatants or IL-6 promoted PBMC differentiation into Myeloid-derived suppressor cells MDSC (CD11b+CD33+) phenotype and a subpopulation of polymorphonuclear CD11b+CD33+CD15+ cells (Mace et al., 2013). IL-6 was an important mediator as its neutralization inhibited PSC supernatant-mediated STAT3 phosphorylation and MDSC differentiation (Mace et al., 2013). Importantly, the level of cytokines produced by different PSCs was different suggesting different phenotypes of the PSCs and suggesting using these cytokines as markers for different sub-populations of PSCs.
Accumulating evidence indicate that targeting PSCs and/or macrophages needs to be focused on targeting specific sub-populations of these cells. Even though M2 macrophage populations are a good target, further studies are needed to determine markers of the pro-cancer PSCs (Figure 1).
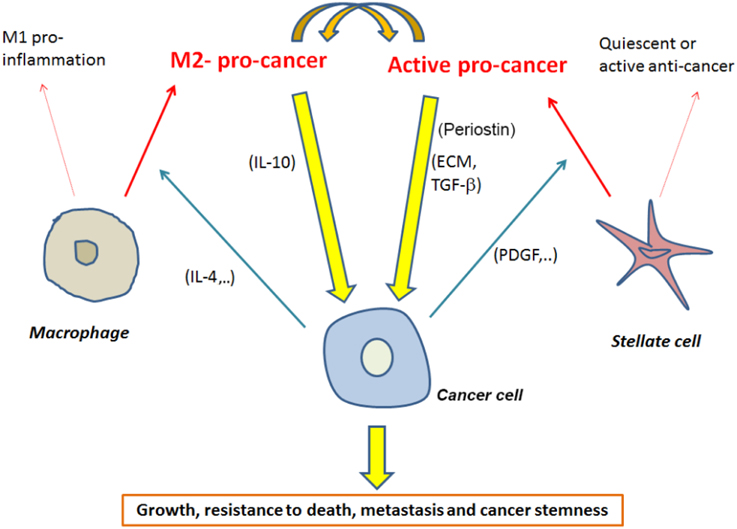
Figure 1. Interactions between macrophages and stellate cells mediating promotion of pancreatic cancer with a short list of possible mediators of these interactions.
Multiple studies have tried targeting pathways rather than targeting the whole cell populations. Therapies against molecules or pathways such as vascular endothelial growth factor (VEGF) (Taeger et al., 2011), sonic hedgehog (Olive et al., 2009), and hyaluronic acid (Provenzano et al., 2012) are now tested. Other studies including our work suggest that the IL-6 and STAT3 pathway is a relevant target due to its constitutive activation in the pancreatic cancer cells, immunosuppressive cells, and in PSC within the stroma (Corcoran et al., 2011; Lesina et al., 2011; Huang et al., 2012).
In summary, we have gained a lot of knowledge about the interactions between cancer cells and cells present in the tumor microenvironment, especially the interactions between cancer cells, macrophages, and PSCs. Ample work is needed to understand how cells in the tumor microenvironment regulate PSCs phenotype. Periostin could be is a good candidate to differentiate between different populations of PSCs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by K01AA019996, P01CA163200 and Department of Veterans Affairs.
References
Al-Assar, O., Demiciorglu, F., Lunardi, S., Gaspar-Carvalho, M. M. I., McKenna, W. G. I., Muschel, R. M., et al. (2014). Contextual regulation of pancreatic cancer stem cell phenotype and radioresistance by pancreatic stellate cells. Radiother. Oncol. 111, 243–251. doi: 10.1016/j.radonc.2014.03.014
Alvarez, R., Musteanu, M., Garcia-Garcia, E., Lopez-Casas, P. P., Megias, D., Guerra, C., et al. (2013). Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 109, 926–933. doi: 10.1038/bjc.2013.415
Apte, M. V., Haber, P. S., Applegate, T. L., Norton, I. D., Korsten, M. A., Pirola, R. C., et al. (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43, 128–133. doi: 10.1136/gut.43.1.128
Apte, M. V., Park, S., Phillips, P. A., Santucci, N., Goldstein, D., Kumar, R. K., et al. (2004). Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 29, 179–187. doi: 10.1097/00006676-200410000-00002
Arumugam, T., Ramachandran, V., Fournier, K. F., Wang, H., Marquis, L., Abbruzzese, J. L., et al. (2009). Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 69, 5820–5828. doi: 10.1158/0008-5472.CAN-08-2819
Bachem, M. G., Schneider, E., Gross, H., Weidenbach, H., Schmid, R. M., Menke, A., et al. (1998). Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115, 421–432. doi: 10.1016/S0016-5085(98)70209-4
Bachem, M. G., Schunemann, M., Ramadani, M., Siech, M., Beger, H., Buck, A., et al. (2005). Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921. doi: 10.1053/j.gastro.2004.12.036
Beatty, G. L., Chiorean, E. G., Fishman, M. P., Saboury, B., Teitelbaum, U. R., Sun, W., et al. (2011). CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616. doi: 10.1126/science.1198443
Bennewith, K. L., Huang, X., Ham, C. M., Graves, E. E., Erler, J. T., Kambham, N., et al. (2009). The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 69, 775–784. doi: 10.1158/0008-5472.CAN-08-0987
Cabrera, M. C., Tilahun, E., Nakles, R., Diaz-Cruz, E. S., Charabaty, A., Suy, S., et al. (2014). Human pancreatic cancer-associated stellate cells remain activated after in vivo chemoradiation. Front. Oncol. 4:102. doi: 10.3389/fonc.2014.00102
Corcoran, R. B., Contino, G., Deshpande, V., Tzatsos, A., Conrad, C., Benes, C. H., et al. (2011). STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 71, 5020–5029. doi: 10.1158/0008-5472.CAN-11-0908
De Lisle, R. C., and Logsdon, C. D. (1990). Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur. J. Cell Biol. 51, 64–75.
Edderkaoui, M., Hong, P., Vaquero, E. C., Lee, J. K., Fischer, L., Friess, H., et al. (2005). Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G1137–G1147. doi: 10.1152/ajpgi.00197.2005
Erkan, M., Kleeff, J., Gorbachevski, A., Reiser, C., Mitkus, T., Esposito, I., et al. (2007). Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 132, 1447–1464. doi: 10.1053/j.gastro.2007.01.031
Erkan, M., Michalski, C. W., Rieder, S., Reiser-Erkan, C., Abiatari, I., Kolb, A., et al. (2008). The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 6, 1155–1161. doi: 10.1016/j.cgh.2008.05.006
Erkan, M., Reiser-Erkan, C., Michalski, C. W., Deucker, S., Sauliunaite, D., Streit, S., et al. (2009). Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 11, 497–508. doi: 10.1593/neo.81618
Gardian, K., Janczewska, S., Olszewski, W. L., and Durlik, M. (2012). Analysis of pancreatic cancer microenvironment: role of macrophage infiltrates and growth factors expression. J. Cancer 3, 285–291. doi: 10.7150/jca.4537
Habbe, N., Shi, G., Meguid, R. A., Fendrich, V., Esni, F., Chen, H., et al. (2008). Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. U.S.A. 105, 18913–18918. doi: 10.1073/pnas.0810097105
Hamada, S., Masamune, A., Takikawa, T., Suzuki, N., Kikuta, K., Hirota, M., et al. (2012). Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 421, 349–354. doi: 10.1016/j.bbrc.2012.04.014
Huang, C., Huang, R., Chang, W., Jiang, T., Huang, K., Cao, J., et al. (2012). The expression and clinical significance of pSTAT3, VEGF and VEGF-C in pancreatic adenocarcinoma. Neoplasma 59, 52–61. doi: 10.4149/neo_2012_007
Hwang, R. F., Moore, T., Arumugam, T., Ramachandran, V., Amos, K. D., Rivera, A., et al. (2008). Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926. doi: 10.1158/0008-5472.CAN-07-5714
Ino, Y., Yamazaki-Itoh, R., Shimada, K., Iwasaki, M., Kosuge, T., Kanai, Y., et al. (2013). Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 108, 914–923. doi: 10.1038/bjc.2013.32
Kanno, A., Satoh, K., Masamune, A., Hirota, M., Kimura, K., Umino, J., et al. (2008). Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int. J. Cancer 122, 2707–2718. doi: 10.1002/ijc.23332
Kikuta, K., Masamune, A., Watanabe, T., Ariga, H., Itoh, H., Hamada, S., et al. (2010). Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 403, 380–384. doi: 10.1016/j.bbrc.2010.11.040
Lesina, M., Kurkowski, M. U., Ludes, K., Rose-John, S., Treiber, M., Klöppel, G., et al. (2011). Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19, 456–469. doi: 10.1016/j.ccr.2011.03.009
Liou, G. Y., Doppler, H., Necela, B., Edenfield, B., Zhang, L., Dawson, D. W., et al. (2014). Mutant Kras-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 5, 52–63. doi: 10.1158/2159-8290.CD-14-0474
Liu, C. Y., Xu, J. Y., Shi, X. Y., Huang, W., Ruan, T. Y., Xie, P., et al. (2013). M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 93, 844–854. doi: 10.1038/labinvest.2013.69
Lonardo, E., Frias-Aldeguer, J., Hermann, P. C., and Heeschen, C. (2012). Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 11, 1282–1290. doi: 10.4161/cc.19679
Mace, T. A., Ameen, Z., Collins, A., Wojcik, S., Mair, M., Young, G. S., et al. (2013). Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 73, 3007–3018. doi: 10.1158/0008-5472.CAN-12-4601
Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. doi: 10.1016/j.cell.2008.03.027
Mantoni, T. S., Lunardi, S., Al-Assar, O., Masamune, A., and Brunner, T. B. (2011). Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer Res. 71, 3453–3458. doi: 10.1158/0008-5472.CAN-10-1633
Masamune, A., Kikuta, K., Watanabe, T., Satoh, K., Hirota, M., and Shimosegawa, T. (2008). Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G709–G717. doi: 10.1152/ajpgi.90356.2008
Matsuda, Y., Yoshimura, H., Ueda, J., Naito, Z., Korc, M., and Ishiwata, T. (2014). Nestin delineates pancreatic cancer stem cells in metastatic foci of NOD/Shi-scid IL2Rgamma(null) (NOG) mice. Am. J. Pathol. 184, 674–685. doi: 10.1016/j.ajpath.2013.11.014
Means, A. L., Meszoely, I. M., Suzuki, K., Miyamoto, Y., Rustgi, A. K., Coffey, R. J. Jr., et al. (2005). Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132, 3767–3776. doi: 10.1242/dev.01925
Means, A. L., Ray, K. C., Singh, A. B., Washington, M. K., Whitehead, R. H., Harris, R. C. Jr., et al. (2003). Overexpression of heparin-binding EGF-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology 124, 1020–1036. doi: 10.1053/gast.2003.50150
Mizuuchi, Y., Aishima, S., Ohuchida, K., Shindo, K., Fujino, M., Hattori, M., et al. (2014). Anterior gradient 2 downregulation in a subset of pancreatic ductal adenocarcinoma is a prognostic factor indicative of epithelial-mesenchymal transition. Lab. Invest. 95, 193–206. doi: 10.1038/labinvest.2014.138
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., McIntyre, D., Honess, D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. doi: 10.1126/science.1171362
Ozdemir, B. C., Pentcheva-Hoang, T., Carstens, J. L., Zheng, X., Wu, C. C., Simpson, T. R., et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. doi: 10.1016/j.ccr.2014.04.005
Provenzano, P. P., Cuevas, C., Chang, A. E., Goel, V. K., Von Hoff, D. D., and Hingorani, S. R. (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. doi: 10.1016/j.ccr.2012.01.007
Rhim, A. D., Oberstein, P. E., Thomas, D. H., Mirek, E. T., Palermo, C. F., Sastra, S. A., et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. doi: 10.1016/j.ccr.2014.04.021
Sandgren, E. P., Luetteke, N. C., Palmiter, R. D., Brinster, R. L., and Lee, D. C. (1990). Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 61, 1121–1135. doi: 10.1016/0092-8674(90)90075-P
Schneeweiss, A., Seitz, J., Smetanay, K., Schuetz, F., Jaeger, D., Bachinger, A., et al. (2014). Efficacy of nab-paclitaxel does not seem to be associated with SPARC expression in metastatic breast cancer. Anticancer Res. 34, 6609–6615.
Shek, F. W., Benyon, R. C., Walker, F. M., McCrudden, P. R., Pender, S. L., Williams, E. J., et al. (2002). Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am. J. Pathol. 160, 1787–1798. doi: 10.1016/S0002-9440(10)61125-X
Shi, C., Washington, M. K., Chaturvedi, R., Drosos, Y., Revetta, F. L., Weaver, C. J., et al. (2014). Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab. Invest. 94, 409–421. doi: 10.1038/labinvest.2014.10
Siegel, R., Naishadham, D., and Jemal, A. (2012). Cancer statistics. CA Cancer J. Clin. 62, 10–29. doi: 10.3322/caac.20138
Song, S. Y., Gannon, M., Washington, M. K., Scoggins, C. R., Meszoely, I. M., Goldenring, J. R., et al. (1999). Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 117, 1416–1426. doi: 10.1016/S0016-5085(99)70292-1
Taeger, J., Moser, C., Hellerbrand, C., Mycielska, M. E., Glockzin, G., Schlitt, H. J., et al. (2011). Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol. Cancer Ther. 10, 2157–2167. doi: 10.1158/1535-7163.MCT-11-0312
Takikawa, T., Masamune, A., Hamada, S., Nakano, E., Yoshida, N., and Shimosegawa, T. (2013). miR-210 regulates the interaction between pancreatic cancer cells and stellate cells. Biochem. Biophys. Res. Commun. 437, 433–439. doi: 10.1016/j.bbrc.2013.06.097
Wehr, A. Y., Furth, E. E., Sangar, V., Blair, I. A., and Yu, K. H. (2011). Analysis of the human pancreatic stellate cell secreted proteome. Pancreas 40, 557–566. doi: 10.1097/MPA.0b013e318214efaf
Weizman, N., Krelin, Y., Shabtay-Orbach, A., Amit, M., Binenbaum, Y., Wong, R. J., et al. (2014). Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 33, 3812–3819. doi: 10.1038/onc.2013.357
Yadav, D., and Lowenfels, A. B. (2013). The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144, 1252–1261. doi: 10.1053/j.gastro.2013.01.068
Keywords: stellate cells, macrophages, pancreatic cancer, periostin, TGF-beta
Citation: Pandol SJ and Edderkaoui M (2015) What are the macrophages and stellate cells doing in pancreatic adenocarcinoma? Front. Physiol. 6:125. doi: 10.3389/fphys.2015.00125
Received: 18 February 2015; Accepted: 07 April 2015;
Published: 15 May 2015.
Edited by:
Atsushi Masamune, Tohoku University Graduate School of Medicine, JapanReviewed by:
Kyoko Shimizu, Tokyo Women's Medical University, JapanShin Hamada, Tohoku University Graduate School of Medicine, Japan
Mert Erkan, Klinikum rechts der Isar - Technische Universitaet Muenchen, Germany
Copyright © 2015 Pandol and Edderkaoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mouad Edderkaoui, Departments of Medicine, Cedars-Sinai Medical Center, Veterans Affairs-Los Angeles and University of California, Los Angeles, 8700 Beverly Boulevard, Davis Building, Suite 3100 Los Angeles, CA 90048, USA, mouad.edderkaoui@cshs.org