 Tobias Weinberger
Tobias Weinberger Christian Schulz
Christian Schulz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 07 April 2015
Sec. Striated Muscle Physiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00107
This article is part of the Research Topic Cardiac Remodeling: New insights in physiological and pathological adaptations View all 11 articles
Ischemic heart disease is a common condition and a leading cause of mortality and morbidity. Macrophages, besides their role in host defense and tissue homeostasis, are critical players in the pathophysiological processes induced by myocardial infarction. In this article we will summarize the current understanding of the role of monocytes and macrophages in myocardial damage and cardiac remodeling in relation to their origin and developmental paths. Furthermore, we describe their potential implications in therapeutic strategies to modulate myocardial healing and regeneration.
Cardiac diseases, especially acute myocardial infarction and congestive heart failure, are among the most frequent causes of death in the western world and are on the uprise in developing countries. Even though the survival after myocardial infarction has significantly improved by early revascularization therapy and drug treatment, a significant number of patients develop heart failure. A broad spectrum of local and systemic mechanisms are initiated in myocardial infarction and contribute to cardiac remodeling. If uncontrolled, they may lead to negative changes in the geometry, structure and function of the ventricle and thus may have deleterious effects on cardiac function in the long term. Therefore, great interest lies in the discovery of new therapeutic strategies to treat the conditions, which precede heart failure.
In myocardial infarction, occlusion of a coronary vessel (i.e., after coronary artery plaque rupture) results in acutely diminished oxygen supply to the myocardium, which leads to myocyte necrosis and acute inflammation. As the human heart has only limited regeneration capacity (Bergmann et al., 2009), infarct healing is–in addition to restoration of oxygen supply–very much dependent on the inflammatory response. Macrophages are an integral part of the innate immune response. They are equipped with an array of pathogen recognition receptors which can activate phagocytosis of pathogens and the secretion of cytokines and chemokines. They present antigens on their cell surface by constitutively expressed major histocompatibility complex II (MHC II) and thereby interact closely with the adaptive immune system.
In vertebrates, macrophages are found in all organs, where they adopt distinct phenotypes and functions that are shaped by the environment of the organ of residence (Gosselin et al., 2014; Lavin et al., 2014). Macrophages are also found in larger numbers in the mammalian heart (Jung et al., 2000; Epelman et al., 2014a; Heidt et al., 2014; Molawi et al., 2014).
Macrophages have long been thought to be of importance in cardiac remodeling after myocardial infarction in humans (Mallory et al., 1939). In autopsy reports macrophage numbers associated closely with different stages of myocardial infarction (Chang et al., 2013). Likewise, macrophages play a critical role in experimental infarction models including rodents (Yang et al., 2002) and swine (Vilahur et al., 2011). In this review we will summarize the functions of monocytes and macrophages in myocardial infarction and outline potential therapeutic strategies to improve infarct healing and outcome by manipulating distinct subsets of mononuclear phagocytes.
The immune response to myocardial ischemia can be divided into distinct but overlapping inflammatory phases, in a simplified view an early (3–4 days) and a late (7–10 days) phase. The kinetics are dependent on the pathological conditions and respective disease models (e.g., species analyzed, chronic ligation vs. ischemia and reperfusion model). Within 30 min after induction of infarction through ligation of the left anterior descending artery (LAD), monocytes are recruited to the site of injury as shown by in vivo microscopy (Jung et al., 2013; Nahrendorf and Swirski, 2013). However, the applied reporter mice did not permit distinguishing between the monocyte subsets involved. In mice, blood monocytes are divided into two principal subsets: Ly6Chi CCR2+ CX3CR1lo “inflammatory” monocytes (CD14+ in humans) and a less frequent subset of Ly6Clo CCR2− CX3CR1hi monocytes (CD14dim CD16+ in humans). Ly6Clo monocyte “patrol” the luminal side of the endothelium of small blood vessels under homeostatic and inflammatory conditions (Auffray et al., 2007). Initial work suggested that the early phase after infarction is dominated by inflammatory (Ly6Chi) monocytes (Nahrendorf et al., 2007). They phagocytose toxic molecules and cell debris, have proteolytic activity and produce inflammatory cytokines like interleukin (IL)-1α, IL-6 and TNFα (Nahrendorf et al., 2007; Troidl et al., 2009; Woollard and Geissmann, 2010; Leuschner et al., 2012). Thus, by exercising inflammatory functions they are potentially harmful in this early phase and promote myocardial damage (Kaikita et al., 2004). In contrast to Ly6Chi monocytes, non-classical Ly6Clo monocytes were thought to play a role in the later stages after infarction (Nahrendorf et al., 2007). Through secretion of anti-inflammatory cytokines and growth factors, like VEGF and TGF-ß, they could promote angiogenesis, extracellular matrix synthesis and myocardial healing (Nahrendorf et al., 2007; Troidl et al., 2009). However, the clear categorization of monocyte subsets into distinct phases and functions after infarction has recently been challenged. Ly6Clo monocytes are critically dependent on the transcription factor Nr4a1 for their development and survival (Hanna et al., 2011). Interestingly, absence of Ly6Clo monocytes in Nr4a1-deficient animals does not abrogate the bi-phasic inflammatory response. These findings might be explained by the concept that Ly6Clo cells derive from Ly6Chi monocytes, which display high plasticity and develop into either proinflammatory or anti-inflammatory monocytes/macrophages (Hilgendorf et al., 2014; Ismahil et al., 2014). The transcription factor Nr4a1 thereby seems to serve as a master switch that controls the polarity of monocytes and could represent a potential target to modulate the local inflammatory response, which drives adverse cardiac remodeling. The findings also indicate that inflammatory Ly6Chi monocytes are the predominant monocyte subset to be recruited to the infarct area not only in the first days of infarction but throughout the course of post-infarct remodeling (Nahrendorf et al., 2010; Hilgendorf et al., 2014; Ismahil et al., 2014). This is supported by data in humans demonstrating that peak levels of the corresponding subset of inflammatory (CD14+) monocytes correlate negatively with the recovery of left ventricular function after acute myocardial infarction (Tsujioka et al., 2009). In mice, inhibition of Ly6Chi inflammatory monocyte recruitment into the infarcted myocardium by genetic ablation of C-C chemokine receptor type 2 (CCR2) reduced adverse cardiac remodeling (Kaikita et al., 2004).
Future studies are needed to clearly define the role and impact of monocyte subsets in the course of infarction. In any case, preclinical (Panizzi et al., 2010) and clinical data (Tsujioka et al., 2009; Aoki et al., 2011) indicate that monocyte/macrophage recruitment needs to be tightly controlled as dysregulated infiltration results in infarct expansion, left ventricular dilation, and heart failure. In detail, monocytosis disturbs resolution of inflammation in murine infarcts and consequently enhances left ventricular remodeling (Panizzi et al., 2010). This is associated with the extent of myocardial salvage and the recovery of left ventricular function after acute infarction (Tsujioka et al., 2009).
As a side note, neutrophils accumulate in larger numbers during the early phase of ischemic injury, but disappear within approximately 24 h whereas the monocyte/macrophage response is sustained (Jung et al., 2013; Nahrendorf and Swirski, 2013). In addition to the recruitment of blood derived monocytes, tissue-resident macrophages in the heart represent another source of innate immune cells, which likely contribute to a local inflammatory process in the ischemic myocardium (Epelman et al., 2014a).
The formerly believed rational, that blood-derived monocytes are the only source of macrophages in the heart, has been challenged by recent work establishing the yolk sac as a common source of macrophages in adult tissues (Ginhoux et al., 2010; Schulz et al., 2012; Gomez Perdiguero et al., 2015). In line with these findings, also cardiac macrophages have their origin, at least to some extent, in the yolk sac (YS). Tamoxifen-induced pulse-labeling of Csf1r+ YS macrophages between embryonic days E8.5 and E9.5, when definitive hematopoiesis is not yet present, labeled a proportion of resident macrophages in the adult mouse heart (Epelman et al., 2014a; Molawi et al., 2014) (Figure 1A). These initial fate-mapping strategies were useful to assess the qualitative contribution of YS progenitors to adult tissue macrophages (Schulz et al., 2012; Epelman et al., 2014b). More recent work assessed the quantitative potential of YS progenitors for tissue resident macrophages in vivo, and established the YS as the key source of macrophages in adult tissues (Gomez Perdiguero et al., 2015). While YS-derived macrophages initially display a common signature (Schulz et al., 2012), their distinct phenotype and function is later shaped by local environmental cues specific for the respective tissue of residence (Gosselin et al., 2014; Lavin et al., 2014). Tissue macrophages perform specific duties, such as homeostatic functions and immune surveillance in other tissues (Davies et al., 2013). Nevertheless, their role in inflammatory processes in the heart needs to be determined.
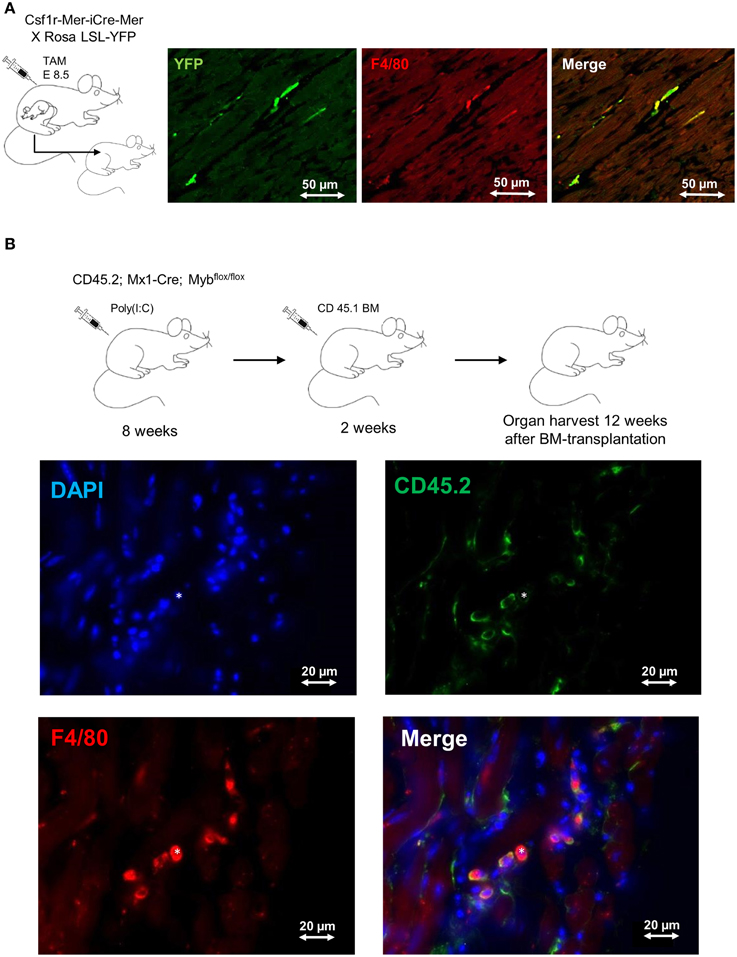
Figure 1. Fate mapping analysis of cardiac macrophages. (A) Frozen tissue sections of 1 year old mouse. FVB strain Csf1rMeriCreMer females mated with BL6 ROSA LSL-YFP/LSL-YFP male were pulse labeled by injection of 75μg/g hydroxytamoxifen on embryonic day 8.5 (E8.5). This labeling regime allowed the specific labeling of Csf1r+ cells derived from the yolk sac, as definitive hematopoiesis is not present at that time point. 1 year old mice were stained for F4/80 (red) and YFP (green); double-positive cells represent yolk-sac derived macrophages. Bars 50 μm (Schulz et al., 2012; Epelman et al., 2014a). (B) Frozen tissue sections of 6 month old Myb-deficient BM chimera (CD45.1/CD45.2). BM ablation was obtained through deletion of Myb by injection of poly(I:C) into CD45.2; Mx1-Cre; Mybflox/flox mice. Whole BM (107 cells) from CD 45.1 mice was transplanted after BM-ablation as described earlier (Schulz et al., 2012). The heart was examined 12 weeks after BM transplantation and sections were stained with DAPI (blue), F4/80 (red) and CD45.2 (green); double-positive (F4/80 positive, CD45.2 positive) cells represent host-derived macrophages; (*) represents a donor-derived macrophage (F4/80 positive, CD45.2 negative). Bars 20 μm.
It remains unknown to date, for how long YS-derived macrophages reside in adult tissues. In many organs, such as the brain and the liver, these cells are not replaced at steady state (Gomez Perdiguero et al., 2015) and can persist independently of HSCs (Schulz et al., 2012) (Figure 1B). However, a specific environment (e.g., intestine) or bacterial infections may result in replacement of tissue macrophages by bone marrow-derived monocytes (Bain et al., 2014; Bleriot et al., 2015). In the intestine, YS-derived macrophages are replaced by bone marrow monocytes within the first weeks of life due to substantial changes of the environment. Interestingly, the number of YS-derived resident macrophages in the mouse heart also seems to be impermanent and declines with age. As mice grow older the proliferation rate of cardiac macrophages decreases and is probably insufficient to maintain the resident macrophage pool (Molawi et al., 2014). Likewise, resident macrophages in the heart are lost as a consequence of myocardial infarction. They need to be substituted either through monocyte intermediates, which are recruited from the circulation, or by local proliferation and expansion of resident cells. Further experimental evidence is needed to define the quantitative contribution of circulation monocytes to the cardiac macrophage pool during aging and in the setting of myocardial injury (Heidt et al., 2014; Lavine et al., 2014; Molawi et al., 2014). Finally, it is still unknown to what extent monocyte-derived macrophages are distinct from resident macrophages with respect to their effector functions in tissues (Schulz and Massberg, 2014). Undoubtedly, the findings will have implications for our understanding of cardiac homeostasis and disease.
Recently, it has become clear that cardiac macrophages consist of several different subpopulations (Epelman et al., 2014a; Heidt et al., 2014; Lavine et al., 2014; Molawi et al., 2014). Elaborate multicolor flow cytometry studies allowed a comprehensive characterization of different macrophage subsets by cell surface markers including CD11b, F4/80, Cx3Cr1, major histocompatibility complex II (MHC II), CCR2 and Ly6C.
Accumulating evidence suggests that the composition of macrophage subsets in the myocardium undergoes dynamic changes in the course of life. This needs to be kept in mind when discussing cardiac macrophage populations in the heart, especially as the macrophage subsets have different regenerative and inflammatory potential. In newborn mice cardiac macrophages display a MHC IIlo phenotype and have predominantly originated from the yolk sac. This subset of macrophages seems to have distinct, probably beneficial, effects on the myocardium under stress. As mice age, the macrophage phenotype diversifies into a MHC IIlo and a MHC IIhi phenotype. Fate mapping analysis in adult mice using pulse labeling of macrophages indicated that the contribution of embryo-derived cardiac macrophages to the MHC IIlo subset was higher than to MHC IIhi macrophages. Between birth and 30 weeks of age resident macrophages become increasingly replaced by monocyte-derived macrophages from the bone marrow (Lavine et al., 2014; Molawi et al., 2014).
Another surface marker to describe macrophage subsets is CCR2, the receptor for chemokine monocyte chemoattractant protein-1 (MCP-1/CCL2), which facilitates monocyte chemotaxis. This marker helps to identity macrophages, which recently derived from circulating monocytes (Epelman et al., 2014a; Lavine et al., 2014). Using CCR2 and MHC II, cardiac monocyte, and macrophage subsets of the neonatal and adult heart could be defined. In a nutshell, at least two subsets exist in the neonatal heart at steady state, namely a CCR2−/MHC IIlo subset derived predominantly from the yolk sac and a CCR2+/MHC IIlo subset derived from definitive hematopoiesis. In contrast, in the adult heart (6 weeks of age), the monocyte/macrophage population could be divided into 4 subsets. The CCR2− population had a diversified phenotype with MHC IIlo and MHC IIhi macrophage subsets and a mixed developmental origin. In contrast, the CCR2+ population derived exclusively from definitive hematopoiesis and could be subdivided into MHC IIhi and MHC IIlo populations (Lavine et al., 2014).
Besides the unknown influence of the origin of macrophages on myocardial repair, it has become clear that macrophages exhibit a distinct potential to facilitate regeneration during different stages of life. Subjecting neonatal mice to myocardial infarction on postnatal day 1 (P1), the ischemic myocardium has been shown to completely regenerate by proliferation of preexisting cardiomyocytes. This regeneration capacity is already lost during the first week after birth. Induction of myocardial infarction 14 days postpartum (P14) showed left ventricular scarring and cardiomyocyte hypertrophy comparable to the pathophysiological changes seen in adult animals after myocardial infarction (Porrello et al., 2013). Macrophages differed in number, localization, and gene expression profile in mice subjected to myocardial infarction on P1 and P14 after birth. Even more strikingly, the regenerative capacity of the neonatal heart after myocardial infarction was lost when macrophages were depleted using clodronate liposomes. The depletion of macrophages in neonates was accompanied by a loss of neovascularization (Aurora et al., 2014). These results stress the importance of macrophages in the regeneration potential of neonatal mice.
In fact, neonatal mice react to myocardial stress (e.g., after in vivo cardiomyocyte cell ablation) with an expansion of the MHC IIlo macrophage subpopulation. It could be shown that these macrophages possess proangiogenic and reparative properties which, at least partially, contribute to the regenerative capacity of the neonatal heart. Fate mapping analysis revealed that this macrophage subset is of yolk sac origin. In contrast, adult mice respond to myocardial injury with the recruitment of inflammatory MHC IIhi CCR2+ macrophages from the blood circulation and loss of potentially beneficial CCR2− (resident) macrophages. These mice developed pathological left ventricular remodeling including cardiomyocyte hypertrophy, fibrosis, and LV dilation resulting in a decreased LV function. Blocking the recruitment of CCR2+ monocytes into the injured myocardium preserved cardiac MHC IIlo macrophages in the adult heart and showed beneficial effects on coronary angiogenesis and attenuated inflammation (Lavine et al., 2014). This underscores a beneficial role of resident cardiac macrophages (Figure 2).
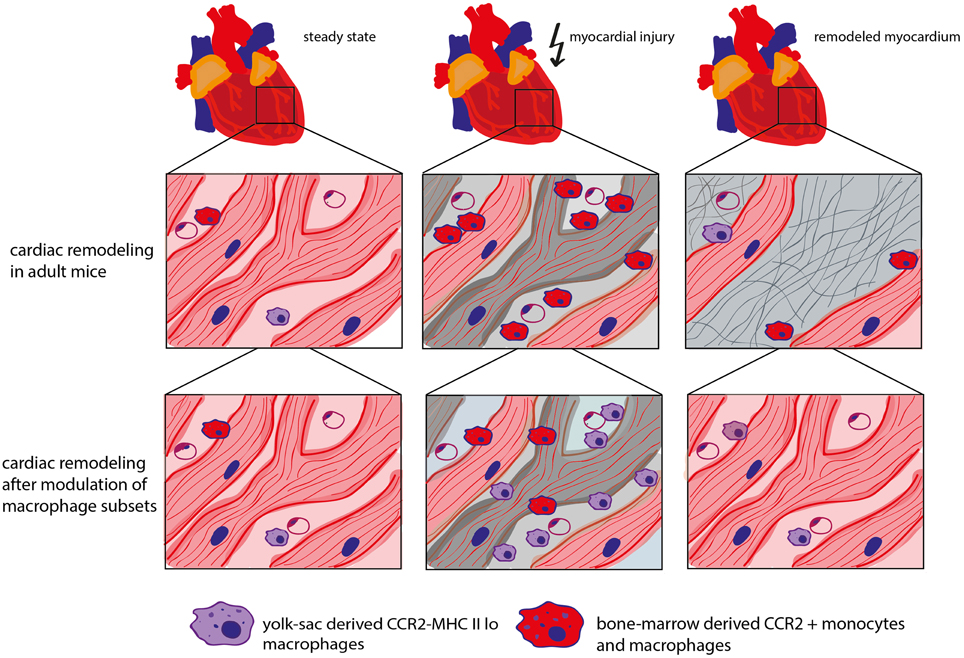
Figure 2. Role of different macrophage subtypes in cardiac remodeling. In steady state, the adult mouse heart contains various types of macrophage populations, which are of yolk sac (YS) or bone marrow (BM) origin dependent on the mouse age. CCR2− macrophages are of mixed origin but predominately derive from YS/embryonic precursors. CCR2+ monocytes and macrophages are derived from the bone marrow. In adult mice, myocardial stress triggers the depletion of resident cardiac macrophages followed by the recruitment of “inflammatory” CCR2+ monocytes. Inhibition of CCR2 leads to a decreased recruitment of CCR2+ monocytes into the myocardium and protects resident cardiac macrophages which has a beneficial impact on coronary angiogenesis, inflammation and cardiac remodeling (Kaikita et al., 2004; Lavine et al., 2014).
It is currently unknown whether the effects of the different macrophage populations on cardiac remodeling after in vivo cardiomyocyte cell ablation also apply to the myocardial infarction model. Intriguingly, myocardial infarction in adult mice is accompanied by the loss of resident cardiac macrophages. Consequently, blood-derived monocytes are recruited to the ischemic myocardium and repopulate the cardiac resident macrophage population (Heidt et al., 2014). Further studies are required to unravel the role of the different macrophage populations in infarct healing.
Monocytes and macrophages play a detrimental role in the pathophysiological processes triggered by infarction and therefore represent a potential therapeutic target to promote myocardial repair and functional regeneration. Yet we have only begun to understand the spatiotemporal relationships and functions of the different macrophage subsets in the course of infarct healing. It is likely that cell-type specific therapies as well as adequate timing of respective therapies will be able to achieve a more beneficial outcome in cardiovascular remodeling. In the following, we will highlight differential strategies targeting monocytes and macrophages, which have the potential to reduce the inflammatory burden in the post-infarct myocardium.
One approach that has been investigated for some time is blocking the recruitment of inflammatory monocytes. This can be achieved for example by interfering with chemokine gradients resulting in a reduction of circulating inflammatory monocytes (e.g., ablation of CCR2), or by inhibition of their recruitment into the ischemic myocardium (e.g., C-X-C chemokine receptor type 6 (CXCR6), macrophage migration inhibitory factor MIF). Reduction of inflammatory CCR2+ monocytes either using a knockout model of CCR2 or an antibody-mediated approach attenuated the inflammatory response after myocardial infarction and had beneficial effects on cardiac remodeling (Kaikita et al., 2004; Lavine et al., 2014). Likewise, disruption of the CXCL16-CXCR6 axis in an ischemia/reperfusion model, led to a decreased number of CD11b+ cells in the infarcted tissue resulting in improved cardiac function and attenuated cardiac remodeling through a diminished autophagic response (Zhao et al., 2013). Inhibition of MIF influences apoptotic pathways and other signaling cascades. Importantly, plasma MIF levels are associated with infarct size and extent of cardiac remodeling in humans. However, blockade of MIF is rather unspecific, and among other effects, also reduces the influx of neutrophils (Gao et al., 2011; Chan et al., 2013).
Another approach to modulate macrophage function is to modify their environment. As shown only recently, macrophage phenotypes and functions are shaped by the microenvironment of the organ of residence (Gosselin et al., 2014; Lavin et al., 2014). For example, transplantation of differentiated peritoneal macrophages into the lung environment induced reprogramming of the transcriptional landscape of these cells and their acquirement of new tissue specific functions. It is tempting to speculate that the same is true for the cardiac microenvironment.
Further, resident macrophages also interact with surrounding immune cells, including those of the adaptive immune system. Regulatory T cells have recently been shown to modulate monocyte/macrophage differentiation in the setting of myocardial infarction with a positive impact on wound healing and remodeling (Weirather et al., 2014). Whether enhancement of regulatory T-cells is beneficial for infarct repair is currently unknown. In addition, B lymphocytes interact with monocytes in the course of cardiac repair after myocardial infarction. In this setting, B cells facilitate mobilization and recruitment of Ly6Chi monocytes into the ischemic myocardium by secretion of CCL7. Genetic as well as antibody-induced depletion of B cells reduced circulatory monocytes and inflammatory Ly6Chi monocytes in the myocardium which improved cardiac function and attenuated myocardial injury (Zouggari et al., 2013). However, the precise mechanisms of the crosstalk between the adaptive immune system and monocytes/macrophages and its therapeutic potential in cardiac remodeling will require further experimental data.
Depletion of macrophages could represent another approach to modulate myocardial inflammation. However, this approach is likely to represent a double edged sword. Depletion of by injection of clodronate liposomes during the first week after myocardial injury led to reduced removal of necrotic cells, impaired neovascularization and increased scar formation. This resulted in a higher mortality of macrophage-depleted mice (van Amerongen et al., 2007; Ben-Mordechai et al., 2013; Frantz et al., 2013). Further, clodronate liposomes deplete most phagocytic cells, including dendritic cells (Leenen et al., 1998) and are therefore not specific. Still, phagocytic properties of mononuclear phagocytes may be harnessed to deliver drugs to their site of action, as reviewed elsewhere (Ben-Mordechai et al., 2015).
Finally, resident cardiac macrophages might be targeted through the alteration of intracellular signals, which for example induce proliferation or apoptosis. IL-4 has been shown to directly signal to tissue-resident macrophages and induce their proliferation above homeostatic levels (Jenkins et al., 2013). Targeting expression of the transcription factor MafB may inhibit macrophage apoptosis in tissues during inflammatory conditions (Hamada et al., 2014). Small-interfering RNA could thereby help achieve robust gene silencing. Delivery of respective particles in an encapsuled form revealed an efficient uptake by phagocytes (Aouadi et al., 2009). However, strategies targeting function of resident macrophages will have to account for their physiological role in tissue homeostasis (e.g., Nr4a1) in organs, including the heart (Schulz and Massberg, 2014).
In summary, modulation of macrophage functions represents an interesting therapeutic strategy. Its success, however, will depend on a deeper understanding of the heterogeneity of macrophage subsets and their developmental origin and molecular regulation. Further, the regulatory signals derived from the local microenvironment in the heart at steady state and in the course of infarction, which modulate the phenotype and functional specification of macrophages, require further investigation.
Despite their abundance in the myocardium, the function of the different cardiac macrophage populations at steady state and in response to stress is largely unknown. There is no doubt that future studies will reveal the precise contribution of monocyte/macrophage populations to myocardial remodeling in pathological conditions thereby integrating findings on their developmental origin and regulation. Of note, recent data in mouse models of infarction suggest that “time” may matter in various ways. First, monocyte/macrophage populations may have a distinct role at different stages of myocardial remodeling and thus, targeting these immune cells at specific time points could further improve wound healing and functional outcome. Second, it may also play a role at which age experimental models (e.g., myocardial infarction) are performed, as macrophage populations in the heart are –at least in mice– replaced over time (Molawi et al., 2014) and also the recovery potential abates with age (Lavine et al., 2014). These recent studies on the heterogeneity and kinetics of monocytes and macrophage subsets in the mammalian heart have paved the way for new therapeutical strategies to target infarct repair after myocardial infarction. Despite the recent advancement on our understanding of the pathomechanisms, specifically the kinetics and regulation of inflammatory processes and wound healing in the diseased heart, early reperfusion therapy for patients who have myocardial infarction remains the essential basis to preserve myocardial function and reduce mortality.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Anna Titova, Sue Chandraratne, Michael Lorenz and Sebastian Helmer for technical assistance. This work was supported by the DZHK (German Centre for Cardiovascular Research) and the BMBF (German Ministry of Education and Research).
Aoki, S., Nakagomi, A., Asai, K., Takano, H., Yasutake, M., Seino, Y., et al. (2011). Elevated peripheral blood mononuclear cell count is an independent predictor of left ventricular remodeling in patients with acute myocardial infarction. J. Cardiol. 57, 202–207. doi: 10.1016/j.jjcc.2010.10.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aouadi, M., Tesz, G. J., Nicoloro, S. M., Wang, M., Chouinard, M., Soto, E., et al. (2009). Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 458, 1180–1184. doi: 10.1038/nature07774
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Auffray, C., Fogg, D., Garfa, M., Elain, G., Join-Lambert, O., Kayal, S., et al. (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670. doi: 10.1126/science.1142883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aurora, A. B., Porrello, E. R., Tan, W., Mahmoud, A. I., Hill, J. A., Bassel-Duby, R., et al. (2014). Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382–1392. doi: 10.1172/JCI72181
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bain, C. C., Bravo-Blas, A., Scott, C. L., Gomez Perdiguero, E., Geissmann, F., Henri, S., et al. (2014). Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 15, 929–937. doi: 10.1038/ni.2967
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ben-Mordechai, T., Holbova, R., Landa-Rouben, N., Harel-Adar, T., Feinberg, M. S., Abd Elrahman, I., et al. (2013). Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J. Am. Coll. Cardiol. 62, 1890–1901. doi: 10.1016/j.jacc.2013.07.057
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ben-Mordechai, T., Palevski, D., Glucksam-Galnoy, Y., Elron-Gross, I., Margalit, R., and Leor, J. (2015). Targeting Macrophage Subsets for Infarct Repair. J. Cardiovasc. Pharmacol. Ther. 20, 36–51. doi: 10.1177/1074248414534916
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bergmann, O., Bhardwaj, R. D., Bernard, S., Zdunek, S., Barnabe-Heider, F., Walsh, S., et al. (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98–102. doi: 10.1126/science.1164680
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bleriot, C., Dupuis, T., Jouvion, G., Eberl, G., Disson, O., and Lecuit, M. (2015). Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 42, 145–158. doi: 10.1016/j.immuni.2014.12.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chan, W., White, D. A., Wang, X. Y., Bai, R. F., Liu, Y., Yu, H. Y., et al. (2013). Macrophage migration inhibitory factor for the early prediction of infarct size. J. Am. Heart Assoc. 2:e000226. doi: 10.1161/JAHA.113.000226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chang, J., Nair, V., Luk, A., and Butany, J. (2013). Pathology of myocardial infarction. Diagn. Histopathol. 19, 7–12. doi: 10.1016/j.mpdhp.2012.11.001
Davies, L. C., Jenkins, S. J., Allen, J. E., and Taylor, P. R. (2013). Tissue-resident macrophages. Nat. Immunol. 14, 986–995. doi: 10.1038/ni.2705
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Epelman, S., Lavine, K. J., Beaudin, A. E., Sojka, D. K., Carrero, J. A., Calderon, B., et al. (2014a). Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104. doi: 10.1016/j.immuni.2013.11.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Epelman, S., Lavine, K. J., and Randolph, G. J. (2014b). Origin and functions of tissue macrophages. Immunity 41, 21–35. doi: 10.1016/j.immuni.2014.06.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frantz, S., Hofmann, U., Fraccarollo, D., Schafer, A., Kranepuhl, S., Hagedorn, I., et al. (2013). Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 27, 871–881. doi: 10.1096/fj.12-214049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, X. M., Liu, Y., White, D., Su, Y., Drew, B. G., Bruce, C. R., et al. (2011). Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: a predominant role of anti-inflammation. J. Mol. Cell. Cardiol. 50, 991–999. doi: 10.1016/j.yjmcc.2010.12.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L., et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. doi: 10.1038/nature13989
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gosselin, D., Link, V. M., Romanoski, C. E., Fonseca, G. J., Eichenfield, D. Z., Spann, N. J., et al. (2014). Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. doi: 10.1016/j.cell.2014.11.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hamada, M., Nakamura, M., Tran, M. T., Moriguchi, T., Hong, C., Ohsumi, T., et al. (2014). MafB promotes atherosclerosis by inhibiting foam-cell apoptosis. Nat. Commun. 5, 3147. doi: 10.1038/ncomms4147
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanna, R. N., Carlin, L. M., Hubbeling, H. G., Nackiewicz, D., Green, A. M., Punt, J. A., et al. (2011). The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat. Immunol. 12, 778–785. doi: 10.1038/ni.2063
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heidt, T., Courties, G., Dutta, P., Sager, H. B., Sebas, M., Iwamoto, Y., et al. (2014). Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295. doi: 10.1161/CIRCRESAHA.115.303567
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hilgendorf, I., Gerhardt, L. M., Tan, T. C., Winter, C., Holderried, T. A., Chousterman, B. G., et al. (2014). Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 114, 1611–1622. doi: 10.1161/CIRCRESAHA.114.303204
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ismahil, M. A., Hamid, T., Bansal, S. S., Patel, B., Kingery, J. R., and Prabhu, S. D. (2014). Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res. 114, 266–282. doi: 10.1161/CIRCRESAHA.113.301720
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jenkins, S. J., Ruckerl, D., Thomas, G. D., Hewitson, J. P., Duncan, S., Brombacher, F., et al. (2013). IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J. Exp. Med. 210, 2477–2491. doi: 10.1084/jem.20121999
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jung, K., Kim, P., Leuschner, F., Gorbatov, R., Kim, J. K., Ueno, T., et al. (2013). Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ. Res. 112, 891–899. doi: 10.1161/CIRCRESAHA.111.300484
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jung, S., Aliberti, J., Graemmel, P., Sunshine, M. J., Kreutzberg, G. W., Sher, A., et al. (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaikita, K., Hayasaki, T., Okuma, T., Kuziel, W. A., Ogawa, H., and Takeya, M. (2004). Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 165, 439–447. doi: 10.1016/S0002-9440(10)63309-3
Lavin, Y., Winter, D., Blecher-Gonen, R., David, E., Keren-Shaul, H., Merad, M., et al. (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. doi: 10.1016/j.cell.2014.11.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lavine, K. J., Epelman, S., Uchida, K., Weber, K. J., Nichols, C. G., Schilling, J. D., et al. (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. U.S.A. 111, 16029–16034. doi: 10.1073/pnas.1406508111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leenen, P. J., Radosevic, K., Voerman, J. S., Salomon, B., van Rooijen, N., Klatzmann, D., et al. (1998). Heterogeneity of mouse spleen dendritic cells: in vivo phagocytic activity, expression of macrophage markers, and subpopulation turnover. J. Immunol. 160, 2166–2173.
Leuschner, F., Rauch, P. J., Ueno, T., Gorbatov, R., Marinelli, B., Lee, W. W., et al. (2012). Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 209, 123–137. doi: 10.1084/jem.20111009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mallory, G. K., White, P. D., and Salcedo-Salgar, J. (1939). The speed of healing of myocardial infarction. a study of the pathologic anatomy in 72 cases. Am. Heart J. 18, 647–671.
Molawi, K., Wolf, Y., Kandalla, P. K., Favret, J., Hagemeyer, N., Frenzel, K., et al. (2014). Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 211, 2151–2158. doi: 10.1084/jem.20140639
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nahrendorf, M., Pittet, M. J., and Swirski, F. K. (2010). Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121, 2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nahrendorf, M., and Swirski, F. K. (2013). Monocyte and macrophage heterogeneity in the heart. Circ. Res. 112, 1624–1633. doi: 10.1161/CIRCRESAHA.113.300890
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nahrendorf, M., Swirski, F. K., Aikawa, E., Stangenberg, L., Wurdinger, T., Figueiredo, J. L., et al. (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037–3047. doi: 10.1084/jem.20070885
Panizzi, P., Swirski, F. K., Figueiredo, J. L., Waterman, P., Sosnovik, D. E., Aikawa, E., et al. (2010). Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J. Am. Coll. Cardiol. 55, 1629–1638. doi: 10.1016/j.jacc.2009.08.089
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Porrello, E. R., Mahmoud, A. I., Simpson, E., Johnson, B. A., Grinsfelder, D., Canseco, D., et al. (2013). Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. U.S.A. 110, 187–192. doi: 10.1073/pnas.1208863110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schulz, C., Gomez Perdiguero, E., Chorro, L., Szabo-Rogers, H., Cagnard, N., Kierdorf, K., et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90. doi: 10.1126/science.1219179
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schulz, C., and Massberg, S. (2014). Atherosclerosis—multiple pathways to lesional macrophages. Sci. Transl. Med. 6, 239ps2. doi: 10.1126/scitranslmed.3008922
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Troidl, C., Mollmann, H., Nef, H., Masseli, F., Voss, S., Szardien, S., et al. (2009). Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J. Cell. Mol. Med. 13, 3485–3496. doi: 10.1111/j.1582-4934.2009.00707.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsujioka, H., Imanishi, T., Ikejima, H., Kuroi, A., Takarada, S., Tanimoto, T., et al. (2009). Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J. Am. Coll. Cardiol. 54, 130–138. doi: 10.1016/j.jacc.2009.04.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Amerongen, M. J., Harmsen, M. C., van Rooijen, N., Petersen, A. H., and van Luyn, M. J. (2007). Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 170, 818–829. doi: 10.2353/ajpath.2007.060547
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vilahur, G., Juan-Babot, O., Pena, E., Onate, B., Casani, L., and Badimon, L. (2011). Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J. Mol. Cell. Cardiol. 50, 522–533. doi: 10.1016/j.yjmcc.2010.12.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weirather, J., Hofmann, U. D., Beyersdorf, N., Ramos, G. C., Vogel, B., Frey, A., et al. (2014). Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115, 55–67. doi: 10.1161/CIRCRESAHA.115.303895
Woollard, K. J., and Geissmann, F. (2010). Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol. 7, 77–86. doi: 10.1038/nrcardio.2009.228
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, F., Liu, Y. H., Yang, X. P., Xu, J., Kapke, A., and Carretero, O. A. (2002). Myocardial infarction and cardiac remodelling in mice. Exp. Physiol. 87, 547–555. doi: 10.1113/eph8702385
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, G., Wang, S., Wang, Z., Sun, A., Yang, X., Qiu, Z., et al. (2013). CXCR6 deficiency ameliorated myocardial ischemia/reperfusion injury by inhibiting infiltration of monocytes and IFN-gamma-dependent autophagy. Int. J. Cardiol. 168, 853–862. doi: 10.1016/j.ijcard.2012.10.022
Keywords: heart, development, myocardial infarction, macrophages, yolk sac, hematopoiesis, inflammation
Citation: Weinberger T and Schulz C (2015) Myocardial infarction: a critical role of macrophages in cardiac remodeling. Front. Physiol. 6:107. doi: 10.3389/fphys.2015.00107
Received: 09 February 2015; Accepted: 17 March 2015;
Published: 07 April 2015.
Edited by:
Leonardo Roever, Federal University of Uberlândia, BrazilReviewed by:
Douglas L. Jones, The University of Western Ontario, CanadaCopyright © 2015 Weinberger and Schulz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Schulz, Medizinische Klinik und Poliklinik I, Klinikum der Universität, Ludwig Maximilians-Universität, Marchioninistrasse 15, 81377 Munich, GermanyY2hyaXN0aWFuLnNjaHVsekBtZWQudW5pLW11ZW5jaGVuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.