 Ramya Vajapey1
Ramya Vajapey1 David Rini
David Rini Peter Abadir
Peter Abadir
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 24 November 2014
Sec. Mitochondrial Research
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00439
This article is part of the Research Topic Mitochondria: Hubs of cellular signaling, energetics and redox balance View all 18 articles
Aging is associated with the accumulation of various deleterious changes in cells. According to the free radical and mitochondrial theory of aging, mitochondria initiate most of the deleterious changes in aging and govern life span. The failure of mitochondrial reduction-oxidation (redox) homeostasis and the formation of excessive free radicals are tightly linked to dysregulation in the Renin Angiotensin System (RAS). A main rate-controlling step in RAS is renin, an enzyme that hydrolyzes angiotensinogen to generate angiotensin I. Angiotensin I is further converted to Angiotensin II (Ang II) by angiotensin-converting enzyme (ACE). Ang II binds with equal affinity to two main angiotensin receptors—type 1 (AT1R) and type 2 (AT2R). The binding of Ang II to AT1R activates NADPH oxidase, which leads to increased generation of cytoplasmic reactive oxygen species (ROS). This Ang II-AT1R–NADPH-ROS signal triggers the opening of mitochondrial KATP channels and mitochondrial ROS production in a positive feedback loop. Furthermore, RAS has been implicated in the decrease of many of ROS scavenging enzymes, thereby leading to detrimental levels of free radicals in the cell. AT2R is less understood, but evidence supports an anti-oxidative and mitochondria-protective function for AT2R. The overlap between age related changes in RAS and mitochondria, and the consequences of this overlap on age-related diseases are quite complex. RAS dysregulation has been implicated in many pathological conditions due to its contribution to mitochondrial dysfunction. Decreased age-related, renal and cardiac mitochondrial dysfunction was seen in patients treated with angiotensin receptor blockers. The aim of this review is to: (a) report the most recent information elucidating the role of RAS in mitochondrial redox hemostasis and (b) discuss the effect of age-related activation of RAS on generation of free radicals.
It is well accepted that mitochondria are the major source of ATP, which is the fuel for many cellular processes. However, mitochondrial function extends well beyond bioenergetics (Kang and Pervaiz, 2012). Accumulating evidence suggests that mitochondria are also signaling organelles that interact with the rest of the cell through ROS and an array of other signaling complexes (Shigenaga et al., 1994; Turrens, 2003; Chandel, 2014). Earlier studies suggested that mitochondrial ROS were detrimental byproducts associated with several pathological conditions (Turrens, 2003; Chandel, 2014). However, more recent studies suggest that ROS may have fundamental cellular functions—acting as signaling molecules (Chandel, 2014), stabilizing hypoxia inducible factors (HIFs), and inducing gene expression to promote cellular adaptation to low oxygen levels (Chandel et al., 1998, 2000a). ROS also play a role in tumor necrosis factor (TNF) receptor signaling (Chandel et al., 2000b, 2001; Nemoto et al., 2000; Chandel, 2014) and defense against pathogens (Yu et al., 2012).
RAS is a key hormonal pathway that affects virtually every organ. While many of the endocrine (circulating), paracrine (cell-to-different cell), and autocrine (cell-to-same cell) effects of the RAS are believed to be mediated through the canonical extracellular RAS, an independent and differentially regulated intracellular RAS has also been proposed (Robertson and Khairallah, 1971; Inagami et al., 1986, 1988, 1990; Hunt et al., 1992; Sadoshima et al., 1993; Mercure et al., 1998; Vila-Porcile and Corvol, 1998; Cook et al., 2001; Sherrod et al., 2005; Kumar et al., 2007, 2008, 2012; Peters, 2008, 2012; Abadir et al., 2011, 2012). The major components of RAS are (1) angiotensinogen, derived from the liver; (2) renin, derived from the juxtaglomerular cells of afferent arterioles; (3) angiotensin converting enzyme (ACE), a dipeptidyl carboxypeptidase; (4) angiotensin II, and (5) angiotensin II receptors. Angiotensinogen is a globular protein that serves as a substrate for renin, a glycoproteolytic enzyme. The first step of the RAS pathway is conversion of angiotensinogen to angiotensin I by renin. Angiotensin I is then converted to angiotensin II via ACE enzyme (Griendling et al., 1993). At all levels -endocrine, paracrine, and autocrine—Ang II binds to two main receptor subtypes, AT1R and AT2R. Both belong to the G-protein-coupled receptor family but differ in terms of tissue distribution and cell signaling pathways. Ang II binds with equal affinity to AT1R and AT2R. These receptors, in turn, activate multiple signal transduction pathways that include signaling molecules like Nitric Oxide (NO·), Protein Tyrosine Phosphatases (PTP), and Mitogen-activated protein kinase (MAPK) (Abadir, 2011; Abadir et al., 2012). AT1R and AT2R stimulation generally leads to opposing actions (Abadir, 2011; Abadir et al., 2012) summarized in Table 1.

Table 1. Opposing functions of AT1R and AT2R.
Evidence supporting an important role for RAS in mitochondrial function/dysfunction comes from many sources (Cook and Re, 2012; Ellis et al., 2012; Garcia et al., 2012; Gwathmey et al., 2012; Li et al., 2012; Singh et al., 2012; Wangler et al., 2012; Yu et al., 2012; Zaobornyj and Ghafourifar, 2012; Ferder et al., 2013; Sovari et al., 2013). The main function of mitochondria is to generate ATP via the electron transport chain. Electrons are transferred through complex I to complex IV with oxygen as the final electron acceptor. Damaged ETC complexes may no longer accept electrons, leading to generation of ROS (Kregel and Zhang, 2007). AT1R knock-out mice exhibit a notable phenotype with increased mitochondrial numbers and average lifespan extension exceeding 25% (Benigni et al., 2009). Other clues of the influence of RAS on mitochondria may be gleaned from prior work, which demonstrated that Ang II infusion in rodents induced cardiomyopathy by increasing mitochondrial ROS generation. In these animals, overexpression of catalase specific for mitochondria, but not peroxisomes, protected them against cardiac hypertrophy, fibrosis, and diastolic dysfunction (Dai et al., 2009; Dikalov and Nazarewicz, 2013). The recent identification of a functional intra-mitochondrial angiotensin system (MAS) provided additional insight into the RAS interface with mitochondria (Eto et al., 2002; Kumar et al., 2008; Abadir et al., 2011, 2012). Recently, changes in MAS and a novel role for AT1R on mitochondrial respiration in diabetes were reported (Persson et al., in press). In mitochondria from renal tubular cells, expression of AT2R decreases while AT1R increases with age. Chronic administration of losartan, an angiotensin receptor blocker, prevented age-related decrease of mitochondrial AT2R. Other parts of the system including renin and ACE have also been localized intracellularly, with evidence suggesting their presence in the nucleus and mitochondria (Vidotti et al., 2004; Abadir et al., 2012). Interestingly, this intracellular system is independent of circulating RAS; ACE inhibitors fail to block intracellular ACE (Cristovam et al., 2008).
Several groups demonstrated a tight link between RAS, mitochondria, and a host of age-related pathologic conditions (Inagami, 2011; Carey, 2012; Conti et al., 2012; Cook and Re, 2012; Dai et al., 2012; Ellis et al., 2012; Gao et al., 2012; Garcia et al., 2012; Gwathmey et al., 2012; Horan et al., 2012; Li et al., 2012; Singh et al., 2012; Wangler et al., 2012; Yu et al., 2012; Zaobornyj and Ghafourifar, 2012). In addition, RAS dysregulation aggravates several acute and chronic diseases, many of which have been linked to mitochondrial dysfunction [atherosclerosis (Warnholtz et al., 1999), kidney disease (Ma et al., 1998), myocardial damage after infarction (Kuno et al., 2002), cerebral infarct size after ischemia (Panahpour and Dehghani, 2012)]. Locally activated RAS in heart tissue has been implicated in cardiac hypertrophy and fibrosis (Kumar et al., 2008). The mechanism by which intracellular angiotensin II (iAng II) affects cardiac tissue has been a topic of debate for years. One possible mechanism is that iAng II interacts with intracellular AT1R or AT1-like receptors to bring about the observed changes. Another mechanism involves iAng II binding directly to chromatin to promote the transcription of growth factors like insulin, PDGF, and FGF-2 (Baker et al., 2004). iAng II may also may also affect Ca+2 fluxes and activate phospholipase C and PKC by binding to AT1R on sarcolemma, mitochondria, or internalized receptors (Eto et al., 2002; Baker et al., 2004).
Similar to extracellular RAS, intracellular RAS has been implicated in many pathological conditions as well. Intracellularly, RAS is highly active in producing increased amounts of iAng II in mice with advanced heart failure. Increased ventricular hypertrophy or fibrosis was also observed (De Mello and Gerena, 2008). The mechanism by which Ang II is produced has not been defined yet. Pressure overload and mechanical stretch of cardiomyocytes after myocardial infarction may cause secretion of local Ang II. After examination of levels of expression of angiotensinogen, renin, ACE and AT1 genes, stretched cardiac myocytes were observed to have higher levels of mRNA and RAS enzymes than un-stretched myocytes (Malhotra et al., 1999). This increased Ang II seems to work through a mineralocorticoid receptor because the administration of an aldosterone receptor antagonist mitigated the effect of Ang II on inward Ca+2 current in failing hearts (De Mello and Gerena, 2008). Ang II further caused cell swelling in failing cardiac myocytes via activation of ionic channels and decrease in gap junction permeability. These molecular changes may lead to altered gene expression, which could be contributing to cardiac remodeling. In addition, changes in ionic and gap junction permeability can decrease action potential duration and conduction velocity. All of these factors may lead to cardiac arrhythmias by causing electrical uncoupling, mechanical de-synchronization, and cardiac remodeling (De Mello and Frohlich, 2011). Furthermore, diabetic patients seem to have upregulated intracellular RAS activity because high glucose in rat mesangial cells resulted in ~30-fold increase in intracellular renin (iRenin) activity and increased iAng II concentrations localized mostly to the nucleus. Localization of iAng II to the nucleus suggests that the mechanism of action of iAng II is both cytoplasmic and nuclear. Renin and chymase (an alternative Ang II-generating enzyme), were implicated in the glucose-induced increase in Ang II rather than ACE; this confirms that the increased Ang II is not due to increased uptake of circulating Ang II but rather due to localized tissue synthesis (Re et al., 1984; Vidotti et al., 2004; Kumar et al., 2007).
Mitochondria play a critical role in redox chemistry. Mitochondrial redox balance is the process by which, under physiological conditions, mitochondria maintain a dynamic balance between ROS generation, their transport, and an array of antioxidant systems (glutathione, glutathione peroxidase, glutathione reductase, MnSOD, catalase, and thioredoxin system) in response to fluctuations in cellular energy demand (Aon et al., 2010; Cortassa et al., 2014).
ROS are generated from various sources including NADPH oxidase (NOX2 and NOX4), uncoupled nitric oxide synthase (NOS), xanthine oxidase (XO), and mitochondria. Of these, mitochondria are the main source of ROS (Nickel et al., 2014).
ROS are generated in the respiratory chain, mainly at the level of complex I and III (Murphy, 2009; Kembro et al., 2014) although recent evidence also involves complex II (Drose, 2013).
Electron transfer between the respiratory complexes in the respiratory chain generates a proton motive force composed of proton and electrical gradients that then drives ATP synthesis at the level of ATP synthase. NADH and FADH2, generated in the Tricarboxylic Acid (TCA) cycle act as electron donors for the electron transport chain (ETC) (Rich and Marechal, 2010). Oxygen is the final acceptor of four electrons transferred from the ETC, and converted to H2O (Mitchell, 1961; Liu et al., 2002). If less than four electrons are transferred to oxygen, ROS are produced (Kregel and Zhang, 2007). Damaged ETC complexes may no longer accept electrons. Excess ROS can damage respiratory complexes and initiate a vicious cycle of ROS overflow thus highlighting the fact that mitochondria can be both source and victim of oxidative stress (Daiber, 2010).
ADP and Ca+2, can modulate the rate of ATP generation according to energy demand. ADP stimulates ATP synthesis via F1F0 ATPase, driven mainly by the electrical component of the proton motive force (Wood, 2006). The dissipation of the mitochondrial membrane potential generates a “pull” of electrons from NADH at the level of complex I, or FADH2 from succinate at the level of complex II thus increasing O2 consumption.
As a matter of fact, an increase in energy demand, e.g., higher cardiac workload under exercising conditions, will increase both ADP and Ca2+ uptake by the mitochondria to increase ATP supply to match the demand (Cortassa et al., 2006; Murphy, 2009). Energized mitochondria will exhibit higher levels of ATP and NAD(P)H and lower electron flow thus increasing the probability of O·−2generation in the respiratory chain. Consequently, mitochondrial ROS production is highly dependent on the energetic and redox status of mitochondria (Kang and Pervaiz, 2012; Cortassa et al., 2014).
As mentioned before, angiotensin II can bind to two major receptors: AT1R and AT2R. Ang II binding to AT1R in the plasma membrane has been implicated in increased ROS production (Figure 1). Ang II-AT1R can activate NADPH oxidase, leading to increased generation of cytoplasmic ROS. This Ang II-AT1R–NADPH-ROS signal triggers the opening of mitochondrial KATP (mtKATP) channels (Figure 2) that in turn activates mitochondrial ROS production in a positive feedback loop (Daiber, 2010). Opening of mtKATP channels decreases mitochondrial membrane potential. This triggers the opening of mitochondrial permeability transition (MPT) channel. The loss of membrane potential due to opening of the inner membrane anion channel (IMAC) (Aon et al., 2003, 2007) or the permeability transition pore (PTP) (Zorov et al., 2000) can produce a burst of mitochondrial ROS leading to ROS-induced ROS release (Zorov et al., 2000; Aon et al., 2003; Zhang et al., 2007).
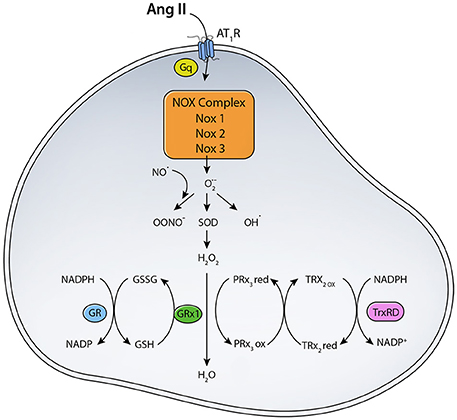
Figure 1. Scheme for RAS induced ROS generation. The binding of Ang II to AT1R activates NADPH oxidase that transfers an electron from NADPH to O2 generating O·−2. Location and expression level of different NOX enzymes determine their function. (SOD, Superoxide dismutase; TrxRD, thioredoxin reductase; GR, Glutathione Reductase; GPX, Glutathione peroxidase; PRx3, peroxiredoxin 3; TRx2, thioredoxin).
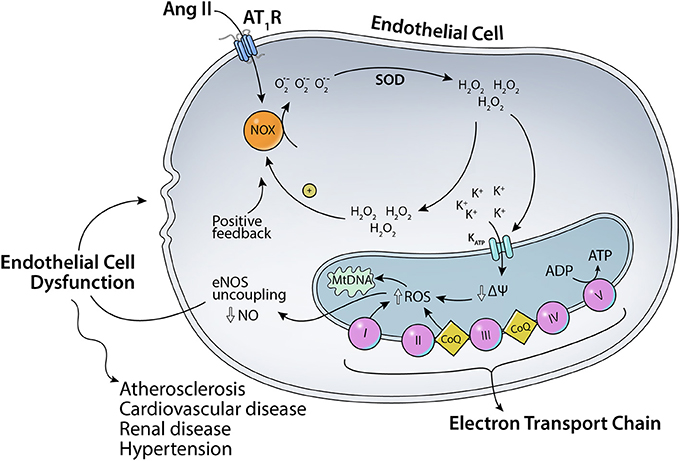
Figure 2. Scheme for the effects of Ang II on mitochondrial KATP channels. Ang II-AT1R–NADPH-ROS signal triggers the opening of mitochondrial KATP channels and mitochondrial ROS production in a positive feedback loop.
(i) Complex I or NADH: ubiquinone oxidoreductase is considered one of the major source of superoxide production in the ETC complexes (Brand et al., 2004). Electrons from succinate can flow in reverse to complex I to reduce NAD+ to NADH. This reverse electron transfer (RET) increases ROS production at the level of complex I. When treated with ADP or malonate, both of which block RET, mitochondrial ROS emission decreases (Liu et al., 2002). Which component of complex I—FMN, Fe-S clusters, or ubiquinone—is producing superoxide is not clear (Brand et al., 2004). Ang II has been shown to damage complex I, thus contributing to ROS generation. Specifically, Ang II is able to down-regulate ND5, a component of complex I, via oxidation of mtDNA thus decreasing electron flow and the likelihood of ROS generation (Ricci et al., 2005).
(ii) Cytochrome bc1 complex or complex III is another important source of O·−2generation. Coenzyme Q, or ubiquinol (QH2), is the electron donor for complex III. Reduced coenzyme Q (QH2) diffuses to the inner side of mitochondrial membrane to the Qo site, where it transfers one electron to cytochrome c bound to complex III. Cytochrome c then transfers electrons to complex IV. Another QH2 transfers an electron to the oxidized coenzyme Q (Q), reducing it to QH2 near the matrix side of the membrane (Qi site) (Quinlan et al., 2011). After losing an electron to cytochrome c and prior to transferring an electron to Q, ubiquinol may form the intermediate ubisemiquinone (Q.). Electrons can leak (Turrens, 2003) from Q· to oxygen, forming O·−2. Antimycin, a Qi site inhibitor, elicits large amounts of O·−2 when O2 reacts with ubisemiquinone bound to the Qo site (Murphy, 2009). Under normal conditions, O·−2generation from complex I and III varies according to tissue conditions…. For example, complex III appears to be the primary source of O·−2 in heart and lung mitochondria whereas complex I is the major source in the brain (Turrens and Boveris, 1980; Turrens et al., 1982; Barja and Herrero, 1998; Turrens, 2003). In general, acute and chronic infusion of Ang II has been shown to decrease expression of electron transport chain proteins (Larkin et al., 2004). Excessive ROS production due to Ang II can impair complex I and III activities, increasing electron leakage (Prathapan et al., 2014). More information is needed to delineate the role of RAS in the modulation of complex III.
(iii) NADPH oxidase (NOX) family proteins: NOX is a family of transmembrane proteins that may be source of ROS. NADPH oxidase works as a nonspecific host defense system by releasing large amounts of ROS during infections (Thrasher et al., 1994). When cytoplasmic NADPH oxidase is activated, it moves toward membrane-associated cytochrome b558 to form a complex. Cytochrome b558 regulates the enzymatic activity of NADPH oxidase by transferring one electron to molecular oxygen, which gets reduced to O·−2 (Bayraktutan et al., 1998). While neutrophil NADPH oxidase produces ROS in bursts, vascular NADPH oxidases produce low levels of O·−2 continuously (Cai et al., 2003). NADPH-derived cytoplasmic ROS can mediate mtKATP opening enabling K+ influx that produces membrane depolarization and alkalinization of the matrix (Di et al., 2007). This matrix alkalization has been shown to increase H2O2 in the presence of an mtKATP opener (Pain et al., 2000; Heinzel et al., 2005; Andrukhiv et al., 2006; Daiber, 2010).
Location and expression level of the different NOX enzymes determine their function. NOX1 is abundant in colon epithelium and has been reported to play a role in host defense in intestinal crypts and on luminal surface (Szanto et al., 2005). NOX2 is expressed in granulocytes and monocyte/macrophages. ROS generation through NOX2 activation has been shown to play a role in killing microbes and inactivating microbial virulent factors. The exact localization and level of expression of the different NOX enzymes are still not completely sorted out yet. NOX3 has been generally located in the cochlear and vestibular sensory epithelia, and spiral ganglion. NOX4 has been shown to be expressed in the kidney and suggested that it plays a role in protection of the vasculature during inflammatory stress. ROS generation through NOX4 has also been shown to induce HIF-1α transcription factors, which in turn leads to activation of erythropoietin gene in the kidney (Bedard and Krause, 2007; Jung et al., 2008; Schroder et al., 2012). NOX5 is expressed in various tissues including spleen, lymph nodes, and vascular smooth muscle cells (Bedard and Krause, 2007). NOX5 increases Jak2 phosphorylation, which increases vascular smooth muscle cell (VSMC) proliferation in human coronary arteries, aorta, and splenic vessels (Fulton, 2009).
NADPH oxidase is a major source of angiotensin II-induced ROS generation. NOX1 appears to have various roles in VSMCs, including Ang II-induced hypertrophy, serum-induced proliferation, and basic fibroblast-induced growth factor migration (Redmond and Cahill, 2012). It localizes in VSMCs from large arteries (Nguyen Dinh et al., 2013). NOX1 is needed under various physiologic conditions such as thrombin-induced migration, endothelial cell proliferation, cell growth, and vessel formation (Sunggip et al., 2013). Overproduction of O·−2 from NOX1 and NOX 2 can cause renin release, smooth muscle cell proliferation, and decreased NO·, all of which lead to endothelial dysfunction and increased sympathetic tone that in turn cause hypertension (Takac et al., 2012). Ang II can signal via phospholipase D and this pathway may cause VSMC proliferation and contractility through NADPH oxidase (Touyz and Berry, 2002). NOX1distributes in the plasma membrane, primarily generating O·−2 anions (Dikalova et al., 2005; Valente et al., 2012). Ang II activates NOX1 via NOX1-AT1R interaction. This is supported by a study showing that continuous infusion of Ang II was correlated with increased NOX1 mRNA expression (Valente et al., 2012). NOX1 functions are mostly growth-promoting being highly expressed in proliferating cells. It activates VEGF and matrix metalloproteinase, which promote angiogenesis (Lassegue et al., 2001; Wilkinson-Berka et al., 2014). NOX1 is also responsible for O·−2generation and is implicated in many pathological conditions including atherosclerosis, diabetes and hypertension (Chose et al., 2008; Briones et al., 2011).
NOX4 is found in subcellular compartments such as the nucleus, endoplasmic reticulum, and mitochondria (Wingler et al., 2001; Briones et al., 2011). Unlike NOX1, NOX4 typically generates H2O2 (Briones et al., 2011; Valente et al., 2012). NOX4 serves as an oxygen sensor and regulates erythropoietin synthesis in the kidneys (Geiszt et al., 2000). In VSMCs, NOX4 is found in focal adhesions and shown to maintain differentiation of cells whereas NOX 1 induces growth and proliferation. This phenomenon was seen in multiple tissues. For example, in coronary arteries, NOX4 expression was correlated with α-actin levels in osteoclasts, and over-expressed NOX4 was associated with increased osteoclast markers. NOX4 is important for maintaining physiologic function of many tissues (Clempus et al., 2007). NOX4 is not up- but instead down-regulated when treated with Ang II (Lassegue et al., 2001; Wingler et al., 2001). Both NOX2 and NOX4 are present in the aortic vascular smooth muscle. When infused with Ang II, NOX2 knock-out mice did not have significant hypertrophy. Therefore, NOX2 was deemed essential for Ang II-induced cardiac hypertrophy and NOX 4 did not seem to play a big role in Ang II-induced cardiac pathologies (Byrne et al., 2003). This argument is supported by many research groups that studied Ang II's effects on ROS generation. Interestingly, Ang II doubled the ratio of O·−2 to H2O2 ratio in quiescent cells and almost quadrupled it in proliferating cells. In addition, NOX4 is a source of H2O2in a variety of vascular cells including fibroblasts, endothelium, and smooth muscles. This constant generation of H2O2 might be essential to maintaining cell functions in angiogenesis during wound healing (Dikalov et al., 2008). Increased NOX4 expression is essential in tissue survival during ischemic stress, tissue remodeling, and regulation of glutathione. Interestingly, NOX4 has been implicated in cardiac hypertrophy, interstitial fibrosis, and apoptosis in cardiac muscle; however, cardiac function was still maintained (Zhang et al., 2010; Brewer et al., 2011; Schroder et al., 2012; Sunggip et al., 2013). Though some studies show NOX4 playing a role in pathophysiology, the great majority shows that the physiological role of this enzyme is more relevant.
To maintain ROS balance, the production of ROS has to be matched with a ROS scavenging mechanisms (Figure 1). MnSOD in the mitochondrial matrix converts O·−2 to H2O2, which can diffuse across membranes. O·−2dismutation to H2O2 is important for signaling and avoiding oxidative damage, in case of oxidative stress (Kang and Pervaiz, 2012). H2O2 is scavenged by glutathione peroxidase (GPX) and peroxiredoxin (PRX). NADPH generated from mitochondrial transhydrogenase (Hoek and Rydstrom, 1988; Jackson, 2003) is the main electron of the GSH and Trx systems (Nickel et al., 2014). Under normal function, GSH and Trx mitochondrial systems are mainly responsible for offsetting most of the ROS produced by the respiratory chain, especially under state 3 respiration when electron flow is maximal (Aon et al., 2012). Other antioxidants localized to cytoplasmic and intermembrane space include Copper-Zinc superoxide dismutase (Cu-ZnSOD) and catalase (Aon et al., 2012). ROS can mediate increased expression of antioxidant enzymes via activation of the Nrf2-antioxidant response element signaling pathway (Nguyen et al., 2009). ROS-activated Nrf2 increases ARE-mediated gene expression, which includes many antioxidants such as glutathione S-transferase, quinine reductase, and heme oxygenase 1 (Bergelson et al., 1994; Alam et al., 1999). Levels of antioxidants are often decreased in disease states or under oxidative stress. For example, diabetes and insulin resistance are correlated with decreased antioxidant capacity due to decreased SOD and glutathione reductase activities (Aon et al., 2012). Under normal conditions, however, antioxidant enzymes play a major role in scavenging ROS generated from various sources like NADPH oxidase, NOS, xanthine oxidase, and mitochondrial electron transport chain enzymes.
Angiotensin II has been implicated in decreasing the activity of scavenging enzymes, thereby leading to detrimental levels of ROS. Ethanol ingestion is associated with oxidative stress and decreased GSH levels via activation of the RAS. When ethanol-fed rats were treated with the AT1R blocker losartan, GSH levels were maintained (Bechara et al., 2005). SOD are also targets of Ang II. Mammals possess three SOD isoforms, Cu-Zn SOD (SOD1), Mn SOD (SOD2), and extracellular SOD (SOD3) located in the cytoplasm, mitochondria and the extracellular space, respectively (Rodriguez-Iturbe et al., 2007). SOD1 is a major defense system against Ang II-triggered ROS in the kidneys. SOD1 knockout mice showed 4-fold increase in afferent arteriolar O·−2 levels when treated with Ang II (Carlstrom et al., 2010). In addition, in coronary artery disease patients, SOD3 levels were severely reduced in the human arterial walls and this decrease was associated with oxidative stress and reduced NO· bioavailability; these conditions were shown to contribute to the pathophysiology of the disease (Landmesser et al., 2007). When treated with the AT1R blocker losartan, an increase in NO· bioavailability and SOD3 activity were seen. A more than 200% increase in SOD3 activity was associated with reduced oxidative stress and improved endothelial function (Hornig et al., 2001). Catalase activity was diminished in Ang II-stimulated cardiomyocytes whereas ROS-elicited by Ang II in mesangial cells implied reduced catalase transcription (Venkatesan et al., 2007; Murtaza et al., 2008; Tan et al., 2008). Reduced catalase mRNA expression and protein levels were noted with Ang II treatment of VSMCs (Xiong et al., 2010). There is also some evidence about the modulation of the Trx system by the RAS system; ACE inhibitors have been shown to improve myocarditis via a mechanism involving the Trx system (Tanito et al., 2004; Touyz, 2004). The RAS not only contributes directly to ROS generation, but also it affects indirectly the redox balance via modulation of various antioxidant enzymes. Treatment methods involving inhibition of RAS enzymes, specifically Ang II type 1 receptor blocker, may be crucial in impeding pathophysiological behavior.
Angiotensin II interacts with various tissues including vascular, renal, and neuronal to induce numerous physiologic signaling cascades and functions. Griendling et al. discussed the role of Ang II in modulating growth-related signaling pathways via ROS signaling (Griendling and Ushio-Fukai, 2000). ROS-mediated oxidation can alter gene expression thorough signaling cascades induction, or interaction with transcription factors. H2O2 can reversibly inhibit tyrosine phosphatase PTP1B, a regulator of the insulin signaling pathway (Apel and Hirt, 2004; Combs, 2010). Recent research focuses on PTP1B inhibitors as potential therapeutic treatment for type 2 diabetes and obesity (Thareja et al., 2012). ROS also play a role in tyrosine phosphorylation as shown in platelet-derived growth factor where a transient increase in ROS inactivated tyrosine phosphatase (Finkel, 1998). Nontoxic amounts of H2O2 administered extracellularly have been shown to stimulate mitogen activated protein kinase (MAPK), that directly affects the inflammatory response, cell proliferation, differentiation, and survival in response to extracellular stimuli (Stevenson et al., 1994; Arbabi and Maier, 2002). Exogenous H2O2 has also been shown to activate JNK, (Finkel, 1998) p38, (Hensley et al., 2000) and NF-KB, a protein complex that induces expression of protective genes during inflammation and infection (Baeuerle and Henkel, 1994).
ROS participate in the host innate immune response. Immune cells undergo respiratory burst, producing high levels of free oxygen radicals during infection, a response that results toxic to pathogens (Spooner and Yilmaz, 2011). When a pathogen invades a cell, Leucine-Rich Repeat-containing family member receptor, NLRX1, moves to the mitochondria to stimulate the electron transport chain and initiate ROS production (Arnoult et al., 2009). Since ROS are produced by NADPH oxidases in mature phagocytic cells, many bacteria, like Francisellatularensis, interfere with NADPH oxidase assembly (Cirillo et al., 2009). Oxidative burst within macrophages has been shown to decrease urogenital infections associated with pathogen Chlamydia trachomatis (Boncompain et al., 2010).
In addition to fighting infections, ROS have also been implicated in cancer therapy since increased ROS levels may be lethal for tumor cells triggering apoptosis. Direct exposure of cancer cells to ROS generating agents like arsenic trioxide, or alternatively inhibiting antioxidant enzymes with2-methoxyestradiol, a SOD inhibitor, may trigger apoptosis in human leukemia (Zhou et al., 2003; Pelicano et al., 2004).
Overall, available evidence indicates that controlled levels of ROS represent effective signaling molecules, intervening in different cellular processes as diverse as communication, inflammation, immunity, and as therapeutic cancer agents. The role of RAS induced ROS in signaling pathways is discussed next.
EGF is required for Ang II effects on various tyrosine kinases, and its activation is initiated by NOX-induced ROS (Eguchi et al., 1998; Griendling and Ushio-Fukai, 2000; Ushio-Fukai et al., 2001). C-Src, a tyrosine kinase activated by Ang II, promotes multiple signaling events including various MAPKs. Ang II-induced activation of c-Src is redox-sensitive since its stimulation is inhibited by antioxidants (Abe et al., 1997; Ushio-Fukai et al., 2001). EGF receptor activation is needed for Ang II action on ERK1/2, a type of MAPK (Eguchi et al., 1996; Liao et al., 1997; Griendling and Ushio-Fukai, 2000). ERK1/2 has an important role in cell adhesion, cell cycle progression, migration, survival, differentiation, metabolism, and proliferation (Roskoski, 2012). Activation of ERK1/2 is said to be redox-sensitive or redox-insensitive in some studies (Sundaresan et al., 1995; Viedt et al., 2000).
MAPKs control various cellular activities like growth, apoptosis, and stress signals. Four main MAPKs are ERK1/2, c-Jun N terminal kinase (JNK), p38MAPKs, and big MAPK-1. Ang II can activate JNK, ERK1/2, and p38MAPK. Though there is an ambiguity as to whether ERK 1/2 is redox-sensitive or not, p38MAPK and JNK have been shown to be redox-sensitive when activated by Ang II. Ang II triggers ROS generation through NADPH oxidase activation, followed by JNK and p39MAPK stimulation (Griendling and Ushio-Fukai, 2000; Viedt et al., 2000). In VSMCs, Ang II activates Akt, a serine threonine kinase that plays a role in cell survival and protein synthesis. NADPH-derived ROS induces Akt which is associated with heat shock protein 27, also activated by H2O2 (Konishi et al., 1997; Coffer et al., 1998; Ushio-Fukai et al., 1999; Griendling and Ushio-Fukai, 2000).
NOS is another enzyme capable of producing O·−2 using tetrahydrobiopterin as a cofactor. Superoxide generated from NADPH oxidases can give rise to peroxynitrite (ONOO−) (Figure 1). ONOO− contributes to oxidation of tetrahydrobiopterin, leading to NOS uncoupling and more O·−2generation (Landmesser et al., 2003). A mitochondrial variant of NOS (mtNOS) can generate nitric oxide (NO·), a reactive nitrogen species (RNS). Mitochondrial respiration can be partially inhibited by NO·, through inactivation of cytochrome c oxidase from complex IV that can lead to increased ROS production and a vicious cycle of mitochondrial damage by ROS excess (Kang and Pervaiz, 2012). Excess NO· can react with O·−2 to form the highly reactive ONOO− which, like O·−2, is unable to permeate the mitochondrial membrane, can cause oxidative damage, nitration and nitrosation (Squadrito and Pryor, 1995; Kang and Pervaiz, 2012). MnSOD can be nitrated by ONOO−, decreasing its activity (Quijano et al., 2001). Ang II-treated endothelial cells released more H2O2, and exhibited mitochondrial loss of membrane potential, respiration impairment, and decreased GSH and NO· formation (Doughan et al., 2008; Daiber, 2010). The reduction of dihydrobiopterin to tetrahydrobiopterin is catalyzed by dihydrofolate kinase. Tetrahydrobiopterin is needed by eNOS for basal NO· production. Ang II can lead to eNOS uncoupling via H2O2 production in a NOX-dependent manner. This results in down-regulation of dihydrobiopterin kinase and diminished tetrahydrobiopterin cofactor and nNOS uncoupling. Dysfunctional eNOS results in decreased NO· bioavailability (Cai and Harrison, 2000; Sunggip et al., 2013). Ang II-induced O·−2 can react with endothelial NO· produced by eNOS, decreasing its concentration even more. Loss of NO· contributes to endothelial dysfunction initiating atherosclerosis (Nickenig and Harrison, 2002). In addition, expression of inflammatory molecules like MCP-1 and VCAM-1 appears to be indirectly regulated by Ang II, accelerating atherosclerosis (Nickenig and Harrison, 2002). Angiotensin II-induced phosphorylation of various kinases activates NOX and increases ROS generation.
XO is a major source of ROS. Xanthine oxidoreductase (XOR) degrades purines to generate uric acid. XOR is transcribed as xanthine dehydrogenase, which is converted to XO after oxidation of cysteine residues or proteolysis (Waud and Rajagopalan, 1976; Amaya et al., 1990; Kelley et al., 2010). XO generates both O·−2 and H2O2, the latter in greater amounts under low O2 and pH, e.g., inflammation, ischemia. Higher O·−2 levels can be detected under low xanthine concentrations, and high O2 tensions (Kelley et al., 2010). Landmesser et al. showed that Ang II administration elevates XO through ROS from NADPH oxidase. NADPH oxidase inhibition decreased XO and O·−2 levels suggesting that Ang II stimulation of NOX proteins is needed for XO stimulation. Additionally, XO inhibitors like oxypurinol and tungsten markedly reduced Ang II-induced endothelial O·−2, in agreement with the idea that Ang II stimulates XO. Furthermore, losartan (AT1R blocker) treatment decreased the endothelial levels of both XO and O·−2. Patients with coronary disease treated with AT1R blocker for 4 weeks displayed reduced endothelial XO activation compared to the placebo (Landmesser et al., 2007).
Other enzymes such as pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, glycerol-3-phosphate dehydrogenase, and from fatty acid β oxidation can also contribute to ROS generation (Kang and Pervaiz, 2012). Ang II can modulate the activity of these various enzymes. For instance, many studies have shown effects of Ang II on fatty acid oxidation and nonalcoholic fatty liver disease (Kurita et al., 2008; Toblli et al., 2008). Increased Ang II levels cause mitochondrial oxidative damage, which leads to impairment of beta oxidation causing hepatic steatosis. Treatment with AT1R blockers like valsartan produced substantial improvement of mitochondrial abnormalities (Monteiro et al., 2005). The mechanism by which fatty acid oxidation appears to be mediated by ROS generated from NADPH oxidase as well as decreased Cu, Zn SOD activity (Wei et al., 2009). Ang II has also been shown to promote pyruvate dehydrogenase complex acetylation, leading to decreased glucose oxidation (Mori et al., 2013). α-ketoglutarate dehydrogenase activity was shown to be increased by Ang II treatment. Increased cytoplasmic and mitochondrial free Ca+2 levels are associated with hepatic stimulation via Ang II. Since α-ketoglutarate dehydrogenase function is positively stimulated by Ca+2, its activity is indirectly influenced by Ang II (Exton, 1985; Williamson et al., 1985; Rashed et al., 1988).
Together, the evidence available indicates that RAS can induce ROS generation through direct—influencing various signaling pathways, as well as indirect—modulating activities of antioxidant enzymes, mechanisms.
Aging is associated with the accumulation of various deleterious changes in cells. According to the free radical and mitochondrial theory of aging, mitochondria initiate most of the deleterious changes in aging and govern life span (Harman, 1956, 2006; Cadenas and Davies, 2000; Cadenas, 2004). Three key mitochondrial functions that become dysregulated with aging are: (1) ROS production, (2) ATP synthesis, and (3) apoptosis (Conley et al., 2007). As proposed by the mitochondrial theory of aging, increased mitochondrial ROS generation precipitates mitochondrial DNA damage and mutations, which in turn leads to failed oxidative phosphorylation and diminished ADP/ATP reservoir (Harman, 1956, 2006), ultimately contributing to mitochondrial deterioration and activation of cell death pathways (Echtay et al., 2002; Dirks et al., 2006; Skulachev, 2006; Conley et al., 2007; Pandur et al., 2014). Moreover, mitochondrial morphodynamics through fusion and fission can also be altered with aging could potentially lead to their dysfunction (Bleazard et al., 1999; Chen et al., 2003; Yu et al., 2012). Chronically activated ROS has been implicated in mitochondrial energetic impairment along with the development and progression of a host of aging-related conditions including atherosclerosis, myocardial hypertrophy, vascular dysfunction, hypertension (Heymes et al., 1998; Wang et al., 2003; Kimura et al., 2005; Doughan et al., 2008; Fukai, 2009; Min et al., 2009; Widder et al., 2009), type 2 diabetes, frailty, heart failure, neurodegeneration (including Alzheimer's disease, Horan et al., 2012), and sarcopenia (Ballinger et al., 1994; Wallace, 2001, 2005, 2010, 2011; Loeb et al., 2005; Conley et al., 2007; Moore et al., 2010). Clinically, the onset of this mitochondrial failure is difficult to estimate, however, the accumulation of damaged mitochondria typically appears in humans by mid to late seventies and once established is thought to be irreversible (Aiken et al., 2002; Herbst et al., 2007). Recently, several studies have shed light on early mitochondrial changes in healthy subjects that predate the accumulation of damaged mitochondria by almost a decade. These studies have demonstrated that increased uncoupling leads to a reduction in mitochondrial efficiency in otherwise healthy people in their sixties (Greco et al., 2003; Hutter et al., 2004; Bua et al., 2006; Mogensen et al., 2006; Amara et al., 2007).
The major mechanism by which dysfunction occurs is via mtDNA mutations. Unlike nuclear genome, mitochondrial genome is circular, and is not condensed around histones or packed tightly. This makes it less protected and more easily damageable than nuclear DNA (nDNA) (Croteau et al., 1999). mtDNA damage is mostly attributed to ROS. AngII can stimulate mitochondrial ROS production via activation of cytoplasmic NOX-derived O·−2and through direct effects on mitochondria as well. Administration of antioxidants inhibited Ang II effects on AP-1 signaling pathway (Puri et al., 1995; Xia et al., 1998; de Cavanagh et al., 2007). Further evidence in support of Ang II action on mitochondria is given by research showing AT2R co-localization with this organelle likely in inner membrane, in various tissues. Ang II and AT2R are likely generally present in the inner membrane of mitochondria (Inagami, 2011). Ang II stimulates production of NO· via activation of calcium/calmodulin-dependent eNOS, including mRNA and protein expression levels (Brown, 1999; Yan et al., 2003). mtDNA codes for critical proteins participating in oxidative phosphorylation (Croteau et al., 1999). In addition, mtDNA has less repair mechanisms compared to nDNA; for example, mitochondria lack nucleotide excision repair mechanisms (Larsen et al., 2005). mtDNA mutation rate is shown to increase with age, affecting liver, skeletal muscles, and cardiac muscles (Katayama et al., 1991; Corral-Debrinski et al., 1992; Marin-Garcia et al., 2002; Druzhyna et al., 2008). Excess ROS generation may overwhelm antioxidant enzymes, thereby preventing the mitochondria from protecting themselves (Golden and Melov, 2001). With chronic exposure to elevated ROS and decline in repair mechanisms, mtDNA mutations accumulate during aging (Lin and Beal, 2006). Under RAS over-activation- as seen in diabetes, hypertension and aging- Ang II induced ROS plays a significant role in tissue damage. Ang II induced mtROS generation has been implicated in atherosclerotic lesions and impairment of cardiac respiration and TCA cycle function leading to disease (Pueyo et al., 2000; de Cavanagh et al., 2007).
In cardiovascular tissue, mitochondrial ROS contribute to senescence of endothelial cells and chronic low-grade vascular inflammation (Ungvari et al., 2007). These endothelial cells can contribute to atherosclerosis by suppressing regeneration and angiogenesis of endothelium in the vascular wall leading to cardiovascular aging (Dai et al., 2012) and the development of hypertension (Heymes et al., 2003; Shiomi et al., 2004; Wang et al., 2010; Sugamura and Keaney, 2011; Dikalov and Ungvari, 2013).
Development and progression of several neurodegenerative disorders has also been linked to mitochondrial dysfunction (Beal, 2007). A significant decrease in mitochondrial coupling efficiency in primary hippocampal neurons, reduced steady state basal respiration, and decreased ATP turnover were noted in several neurodegenerative disorders (Horan et al., 2012). In Alzheimer's disease, several studies have demonstrated that oxidative damage appears to increase development of intracellular Aβ plaques. ROS activation of c-Jun N terminal kinase and p38 mitogen activated protein kinase lead to increased activity of β secretase, causing increased Aβ levels (Nishida et al., 2006; Beal, 2007). Parkinson's disease is caused by a recessive mutation of DJ1 that leads to hypersensitivity to MPTP and oxidative stress since DJ1 protects against oxidative stress-induced cell death. Studies show that treatment with SOD1 and vitamin E decrease degeneration of dopaminergic neurons (Bonifati et al., 2003; Kim et al., 2005; Wang et al., 2006a). Huntington's disease (HD) is also associated with oxidative stress since reduced activity of Complex II and III is seen in basal ganglia and cortex. In addition, HD patients show decreased PGC1α, which suppresses ROS by activating ROS scavenging enzymes (St-Pierre et al., 2006; Weydt et al., 2006; Beal, 2007). ROS are implicated not only in neurodegenerative disorders but also in various other diseases like age-related musculoskeletal disorders. In sarcopenia, a disease characterized by reduced skeletal muscle mass with aging, electron transport system abnormalities were seen in muscle fibers with “ragged red” phenotype and these changes were associated with loss of muscle mass (Bua et al., 2002). Patients with Type 2 diabetes show decreased antioxidant capacity and possibly an increase in ROS generation by leukocytes (Mohanty et al., 2000). Studies show increased expression of ROS markers in pancreatic islet cells under diabetic conditions. Furthermore, β cells are much more sensitive to ROS due to decreased antioxidant capacity. Chronic hyperglycemia increases ROS levels while decreasing binding of the transcription factors PDX-1 and Maf-A from pancreas that exacerbates suppression of insulin synthesis and release. In addition, treatment with antioxidants such as N-acetyl-L-cysteine and taurine, diminished insulin resistance due to hyperglycemia (Kaneto et al., 2010).
The overlap between age related changes in RAS and mitochondria and the implications of this overlap on age-related diseases are quite complex. Studies shows that Ang II contributes to plaque rupture by initiation of VSMC apoptosis, which could be prevented by AT1R blockers (Lemay et al., 2000). Ang II induces metalloproteinase activity, which is involved in collagen breakdown and matrix degradation, indirectly through ROS (Shah and Galis, 2001; Nickenig and Harrison, 2002). Ang II can induce interleukin-6 (IL6), leukemia inhibitory factor, and cardiotrophin-1 in cardiac fibroblasts (Sano et al., 2001). IL-6 is an inflammatory marker and a high level of which is associated with mortality due to increased progression of cardiovascular disease (Volpato et al., 2001). Leukemia inhibitory factor is a key player in cardiac hypertrophy (Kodama et al., 1997). Cardiotrophin-1 plays a role in heart failure since it can induce ventricular remodeling by activating cardiomyocyte hypertrophy and collagen synthesis (Calabro et al., 2009).
Many studies highlighted the role that RAS plays in hypertension and cardiovascular disease (Marchesi et al., 2008). By activating local mediators, like vascular endothelial growth factor (VEGF) and prostaglandins, such as leukotriene C4, PGE2, and PGI2, Ang II plays a critical role in regulating vascular permeability during hypertension (Harris et al., 2004). Ang II-stimulated release of these local factors in VSMCs leads to angiogenesis, vascular permeability, and inflammation. In particular, studies have shown that AT1R activation causes VEGF secretion (Suzuki et al., 2003). Hypertension is a condition that results from ROS of vascular origin produced by elevated levels of Ang II. Chronic administration of Ang II in mice, triggered elevated O·−2 from mitochondria as compared to controls (Widder et al., 2009). O·−2 and H2O2 produced due to elevated Ang II can carry out various actions in VSMCs—phosphorylation of MAP kinases, induction of proto-oncogenes, and activation of AP-1; H2O2 can induce PDGF stimulation of STATS. All these signaling events contribute to vascular wall remodeling and thickening seen in hypertension (Touyz, 2000). Ang II via AT1R also affects brain tissue by increasing neuronal firing rate and activity. In neurons, NADPH oxidase-derived ROS produced due to Ang II signaling increase intracellular Ca+2 concentration (Wang et al., 2006b), which in turn can stimulate mitochondrial O·−2 stimulates mitochondrial superoxide generation (Hongpaisan et al., 2004). In rostral ventrolateral medulla, these effects may influence blood pressure, causing baroreflex abnormalities in chronic hypertension (Nozoe et al., 2008).
RAS has been implicated in many pathological conditions. Ang II can precipitate mild to severe mitochondrial dysfunction in addition to ROS generation. Amelioration of age-related renal mitochondrial dysfunction under hypertensive conditions, and ischemic injury, has been described in patients treated with angiotensin receptor blockers (Doughan et al., 2008). Losartan treatment prevents mitochondrial dysfunction and structural changes in the kidney while up regulating antioxidant enzymes, maintaining GSH and MnSOD levels, and attenuating uncoupling proteins. These effects were not apparent when patients were treated with a Ca2+ channel blocker allowing to conclude that Ang II must play a role in mitochondrial dysfunction (de Cavanagh et al., 2006).
Since Ang II is linked to NOX, Wosniak et al. (2009) examined the effects of mild mitochondrial uncoupling and Ang II stimulation of NOX isoform in VSMCs. Ethidium bromide was used to induce mild mitochondrial stress, which the investigators described as “neither rapidly lethal nor promoting profound redox derangements” (Wosniak et al., 2009). Results showed that Ang II-induced NOX activation can be completely eliminated with mild mitochondrial uncoupling. Ang II up regulates NOX 1 expression and down regulates NOX4 expression and with mild mitochondrial dysfunction there was a decrease in NOX1 and increase in NOX4. Therefore, these authors concluded that functional mitochondria are required for Ang II-induced NOX activation. Wosniak et al. also noted that mitochondrial function influences activation of growth factor receptors involved in NOX signaling (Wosniak et al., 2009). In addition, when Ang II was administered, increased mtDNA damage was only observed in those cells that had existing dysfunctional mitochondria. Cells with functional mitochondria did not show any markers of mtDNA damage. The researchers speculated that mitochondria might be acting as a switch between normal to pathological effects of Ang II (Wosniak et al., 2009). The relationship between angiotensin-related mitochondrial ROS and NADPH oxidase is still a novel area of research that can potentially provide insight into pathophysiology of many diseases and may pave the way for new therapeutic approaches.
Elevated Ang II produces increased levels of ROS (by activating NADPH oxidase and various other enzymes) that contribute to various pathological conditions. AT1R blockade decreases RAS-mediated activation of NADPH oxidase and oxidative stress, leading to reduced left ventricular fibrosis and mitochondrial remodeling (Whaley-Connell et al., 2008). Losartan treatment also reverses left ventricular hypertrophy, reduces fibrosis, ultimately causing an overall improvement of cardiac function (Khaper and Singal, 2001). Various studies have shown a lower rate of mortality from cardiovascular disease in patients treated with angiotensin receptor antagonists. Beneficial effects of angiotensin receptor blockers include lowered cerebral lesion incidence, reduced cardiac hypertrophy, and reduced glomerulosclerosis. Eprosartan was shown to be effective in preventing cardiac remodeling, renal failure, and decreasing mortality (Takemori et al., 2005). Interestingly, insulin treatment induces AT1R overexpression, and in diabetics, ARBs have been shown to effectively preserve renal function and reduce cardiovascular endpoints (Nickenig et al., 1998; Nickenig, 2004). ARBs reduces Ang-II induced lipolysis and adipocyte dysfunction (Takemori et al., 2013). Contrary to AT1R, the effects of AT2R are considered protective. ARBs may increase activation of AT2 receptor augmenting end-organ protection (Carey et al., 2000; Unger, 2002). More research on ARBs and activation of AT2 receptor might yield promising results in treating various pathological conditions.
AngII receptor blockers have several features in common: high affinity for AT1R and almost no affinity for AT2 receptors; high protein binding capacity behaving as competitive inhibitors with slow dissociation (Burnier, 2001). There are six common types of ARBs used currently as treatment for hypertension—losartan, valsartan, candesartan cilexetil, irbesartan, eprosartan, and telmisartan. These ARBs have been shown to protect against various disease states. For example, in rats with myocardial infarction, losartan improves function of several antioxidant enzymes, significantly reducing oxidative stress. In particular, losartan increases the activity of GSH peroxidase (Khaper and Singal, 2001) while decreasing levels of vascular O·−2 due to its antagonist activity on AT1 receptors (Kurz et al., 1999). It also restores NO· synthesis and availability playing its role as an antihypertensive drug protecting endothelial cells (Qadri et al., 2001). Valsartan is effective in improving heart mitochondrial function under acute ischemia (Monteiro et al., 2005). Long-term treatment of rabbit heart with valsartan, after infarction, showed reduced lipid peroxide levels; an improvement in postinfarct ventricular remodeling and coronary endothelial dysfunction was also seen (Kuno et al., 2002). In diabetic patients, candesartan cilexetil has been shown to improve pancreatic β cell function (Qadri et al., 2001) and irbesartan was effective against postprandial hyperglycemia and hypertriglyceridemia on endothelial function (Ceriello et al., 2005). Telmisartan can inhibit apoptosis, oxidative stress, and neuro-inflammation in addition to its antihypertensive functions (Beckman et al., 1990; Butler et al., 2003; Monteiro et al., 2005; Shao et al., 2006). In hypertensive patients, ARBs were shown to improve normal retinal perfusion and endothelium-dependent vasodilation in coronary and renal circulation (Delles et al., 2004). In addition, ARBs have been associated with a reduction in inflammatory markers. This is of particular importance since low grade chronic inflammation is associated with many neurodegenerative diseases like Alzheimer's and Parkinson's diseases, amyotrophic lateral sclerosis, multiple sclerosis, Huntington's disease and frailty (Tracy, 2003; Gao and Hong, 2008; Holmes et al., 2009). ARBs are also able to reverse early myocardial impairment (Cadeddu et al., 2010), improve insulin sensitivity, decrease incidence of type 2 diabetes (Saitoh et al., 2009) and prevent renal fibrosis (Shao et al., 2006).
Mitochondrial dysfunction and oxidative stress underlie many pathologies and constitute primary theories of aging. Understanding how age-related mitochondrial dysfunction might be mitigated or exacerbated is critical to advancing this research field. The RAS is currently regarded as a physiological system of vital importance because of its links to both mitochondrial function/dysfunction and a host of age-related diseases. Although many prior studies have advanced our understanding in each of these aging-relevant biological systems, the progress in delineating the molecular mechanisms involved has been rather slow. Given the availability of selective, and relatively safe blockers of RAS, studies focusing on the interface between mitochondria, RAS, and aging may prove to be very important in clinical translation of research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the Johns Hopkins Older Americans Independence Center National Institute on Aging Grant P30 AG021334, National Institute on Aging Grants 1R01AG046441 and K23 AG035005, and Nathan Shock in Aging Scholarship Award (to Peter Abadir) and Medical Student in Aging Research (MSTAR) (to Ramya Vajapey)
Abadir, P. M. (2011). The frail renin-angiotensin system. Clin. Geriatr. Med. 27, 53–65. doi: 10.1016/j.cger.2010.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abadir, P. M., Foster, D. B., Crow, M., Cooke, C. A., Rucker, J. J., Jain, A., et al. (2011). Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. U.S.A. 108, 14849–14854. doi: 10.1073/pnas.1101507108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abadir, P. M., Walston, J. D., and Carey, R. M. (2012). Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides 38, 437–445. doi: 10.1016/j.peptides.2012.09.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Abe, J., Takahashi, M., Ishida, M., Lee, J. D., and Berk, B. C. (1997). c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J. Biol. Chem. 272, 20389–20394. doi: 10.1074/jbc.272.33.20389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aiken, J., Bua, E., Cao, Z., Lopez, M., Wanagat, J., McKenzie, D., et al. (2002). Mitochondrial DNA deletion mutations and sarcopenia. Ann. N.Y. Acad. Sci. 959, 412–423. doi: 10.1111/j.1749-6632.2002.tb02111.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A. M., and Cook, J. L. (1999). Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078. doi: 10.1074/jbc.274.37.26071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amara, C. E., Shankland, E. G., Jubrias, S. A., Marcinek, D. J., Kushmerick, M. J., and Conley, K. E. (2007). Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 1057–1062. doi: 10.1073/pnas.0610131104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Amaya, Y., Yamazaki, K., Sato, M., Noda, K., Nishino, T., and Nishino, T. (1990). Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J. Biol. Chem. 265, 14170–14175.
Andrukhiv, A., Costa, A. D., West, I. C., and Garlid, K. D. (2006). Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am. J. Physiol. Heart Circ. Physiol. 291, H2067–H2074. doi: 10.1152/ajpheart.00272.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aon, M. A., Cortassa, S., Maack, C., and O'Rourke, B. (2007). Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 282, 21889–21900. doi: 10.1074/jbc.M702841200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aon, M. A., Cortassa, S., Marban, E., and O'Rourke, B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744. doi: 10.1074/jbc.M302673200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aon, M. A., Cortassa, S., and O'Rourke, B. (2010). Redox-optimized ROS balance: a unifying hypothesis. Biochim. Biophys. Acta 1797, 865–877. doi: 10.1016/j.bbabio.2010.02.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aon, M. A., Stanley, B. A., Sivakumaran, V., Kembro, J. M., O'Rourke, B., Paolocci, N., et al. (2012). Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J. Gen. Physiol. 139, 479–491. doi: 10.1085/jgp.201210772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Apel, K., and Hirt, H. (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399. doi: 10.1146/annurev.arplant.55.031903.141701
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arbabi, S., and Maier, R. V. (2002). Mitogen-activated protein kinases. Crit. Care Med. 30, S74–S79. doi: 10.1097/00003246-200201001-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arnoult, D., Soares, F., Tattoli, I., Castanier, C., Philpott, D. J., and Girardin, S. E. (2009). An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 122(Pt 17), 3161–3168. doi: 10.1242/jcs.051193
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baeuerle, P. A., and Henkel, T. (1994). Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 12, 141–179. doi: 10.1146/annurev.iy.12.040194.001041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baker, K. M., Chernin, M. I., Schreiber, T., Sanghi, S., Haiderzaidi, S., Booz, G. W., et al. (2004). Evidence of a novel intracrine mechanism in angiotensin II-induced cardiac hypertrophy. Regul. Pept. 120, 5–13. doi: 10.1016/j.regpep.2004.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ballinger, S. W., Shoffner, J. M., Gebhart, S., Koontz, D. A., and Wallace, D. C. (1994). Mitochondrial diabetes revisited. Nat. Genet. 7, 458–459. doi: 10.1038/ng0894-458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barja, G., and Herrero, A. (1998). Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J. Bioenerg. Biomembr. 30, 235–243. doi: 10.1023/A:1020592719405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bayraktutan, U., Draper, N., Lang, D., and Shah, A. M. (1998). Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc. Res. 38, 256–262. doi: 10.1016/S0008-6363(98)00003-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beal, M. F. (2007). Mitochondria and neurodegeneration. Novartis Found. Symp. 287, 183–192. doi: 10.1002/9780470725207.ch13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bechara, R. I., Pelaez, A., Palacio, A., Joshi, P. C., Hart, C. M., Brown, L. A., et al. (2005). Angiotensin II mediates glutathione depletion, transforming growth factor-beta1 expression, and epithelial barrier dysfunction in the alcoholic rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L363–L370. doi: 10.1152/ajplung.00141.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624. doi: 10.1073/pnas.87.4.1620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benigni, A., Corna, D., Zoja, C., Sonzogni, A., Latini, R., Salio, M., et al. (2009). Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 119, 524–530. doi: 10.1172/JCI36703
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bergelson, S., Pinkus, R., and Daniel, V. (1994). Induction of AP-1 (Fos/Jun) by chemical agents mediates activation of glutathione S-transferase and quinone reductase gene expression. Oncogene 9, 565–571.
Bleazard, W., McCaffery, J. M., King, E. J., Bale, S., Mozdy, A., Tieu, Q., et al. (1999). The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1, 298–304. doi: 10.1038/13014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boncompain, G., Schneider, B., Delevoye, C., Kellermann, O., Dautry-Varsat, A., and Subtil, A. (2010). Production of reactive oxygen species is turned on and rapidly shut down in epithelial cells infected with Chlamydia trachomatis. Infect. Immun. 78, 80–87. doi: 10.1128/IAI.00725-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brand, M. D., Affourtit, C., Esteves, T. C., Green, K., Lambert, A. J., Miwa, S., et al. (2004). Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 37, 755–767. doi: 10.1016/j.freeradbiomed.2004.05.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brewer, A. C., Murray, T. V., Arno, M., Zhang, M., Anilkumar, N. P., Mann, G. E., et al. (2011). Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 51, 205–215. doi: 10.1016/j.freeradbiomed.2011.04.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Briones, A. M., Tabet, F., Callera, G. E., Montezano, A. C., Yogi, A., He, Y., et al. (2011). Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J. Am. Soc. Hypertens. 5, 137–153. doi: 10.1016/j.jash.2011.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, G. C. (1999). Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta 1411, 351–369. doi: 10.1016/S0005-2728(99)00025-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bua, E., Johnson, J., Herbst, A., Delong, B., McKenzie, D., Salamat, S., et al. (2006). Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480. doi: 10.1086/507132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bua, E. A., McKiernan, S. H., Wanagat, J., McKenzie, D., and Aiken, J. M. (2002). Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J. Appl. Physiol. (1985) 92, 2617–2624.
Burnier, M. (2001). Angiotensin II type 1 receptor blockers. Circulation 103, 904–912. doi: 10.1161/01.CIR.103.6.904
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Butler, A. E., Janson, J., Bonner-Weir, S., Ritzel, R., Rizza, R. A., and Butler, P. C. (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110. doi: 10.2337/diabetes.52.1.102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Byrne, J. A., Grieve, D. J., Bendall, J. K., Li, J. M., Gove, C., Lambeth, J. D., et al. (2003). Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 93, 802–805. doi: 10.1161/01.RES.0000099504.30207.F5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cadeddu, C., Piras, A., Mantovani, G., Deidda, M., Dessi, M., Madeddu, C., et al. (2010). Protective effects of the angiotensin II receptor blocker telmisartan on epirubicin-induced inflammation, oxidative stress, and early ventricular impairment. Am. Heart J. 160, 487. doi: 10.1016/j.ahj.2010.05.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cadenas, E. (2004). Mitochondrial free radical production and cell signaling. Mol. Aspects. Med. 25, 17–26. doi: 10.1016/j.mam.2004.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cai, H., Griendling, K. K., and Harrison, D. G. (2003). The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 24, 471–478. doi: 10.1016/S0165-6147(03)00233-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cai, H., and Harrison, D. G. (2000). Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 87, 840–844. doi: 10.1161/01.RES.87.10.840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Calabro, P., Limongelli, G., Riegler, L., Maddaloni, V., Palmieri, R., Golia, E., et al. (2009). Novel insights into the role of cardiotrophin-1 in cardiovascular diseases. J. Mol. Cell. Cardiol. 46, 142–148. doi: 10.1016/j.yjmcc.2008.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carey, R. M. (2012). Functional intracellular renin-angiotensin systems: potential for pathophysiology of disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R479–R481. doi: 10.1152/ajpregu.00656.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carey, R. M., Wang, Z. Q., and Siragy, H. M. (2000). Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 35(1 Pt 2), 155–163. doi: 10.1161/01.HYP.35.1.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carlstrom, M., Lai, E. Y., Ma, Z., Steege, A., Patzak, A., Eriksson, U. J., et al. (2010). Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension 56, 907–913. doi: 10.1161/HYPERTENSIONAHA.110.159301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ceriello, A., Assaloni, R., Da, R. R., Maier, A., Piconi, L., Quagliaro, L., et al. (2005). Effect of atorvastatin and irbesartan, alone and in combination, on postprandial endothelial dysfunction, oxidative stress, and inflammation in type 2 diabetic patients. Circulation 111, 2518–2524. doi: 10.1161/01.CIR.0000165070.46111.9F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandel, N. S. (2014). Mitochondria as signaling organelles. BMC Biol. 12, 34–12. doi: 10.1186/1741-7007-12-34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandel, N. S., Maltepe, E., Goldwasser, E., Mathieu, C. E., Simon, M. C., and Schumacker, P. T. (1998). Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 95, 11715–11720. doi: 10.1073/pnas.95.20.11715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandel, N. S., McClintock, D. S., Feliciano, C. E., Wood, T. M., Melendez, J. A., Rodriguez, A. M., et al. (2000a). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138. doi: 10.1074/jbc.M001914200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandel, N. S., Schumacker, P. T., and Arch, R. H. (2001). Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J. Biol. Chem. 276, 42728–42736. doi: 10.1074/jbc.M103074200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandel, N. S., Trzyna, W. C., McClintock, D. S., and Schumacker, P. T. (2000b). Role of oxidants in NF-kappa B activation and TNF-alpha gene transcription induced by hypoxia and endotoxin. J. Immunol. 165, 1013–1021. doi: 10.4049/jimmunol.165.2.1013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chose, O., Sansilvestri-Morel, P., Badier-Commander, C., Bernhardt, F., Fabiani, J. N., Rupin, A., et al. (2008). Distinct role of nox1, nox2, and p47phox in unstimulated versus angiotensin II-induced NADPH oxidase activity in human venous smooth muscle cells. J. Cardiovasc. Pharmacol. 51, 131–139. doi: 10.1097/FJC.0b013e31815d781d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cirillo, S. L., Subbian, S., Chen, B., Weisbrod, T. R., Jacobs, W. R. Jr., and Cirillo, J. D. (2009). Protection of Mycobacterium tuberculosis from reactive oxygen species conferred by the mel2 locus impacts persistence and dissemination. Infect. Immun. 77, 2557–2567. doi: 10.1128/IAI.01481-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clempus, R. E., Sorescu, D., Dikalova, A. E., Pounkova, L., Jo, P., Sorescu, G. P., et al. (2007). Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 27, 42–48. doi: 10.1161/01.ATV.0000251500.94478.18
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coffer, P. J., Jin, J., and Woodgett, J. R. (1998). Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 335 (Pt 1), 1–13.
Combs, A. P. (2010). Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem. 53, 2333–2344. doi: 10.1021/jm901090b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conley, K. E., Marcinek, D. J., and Villarin, J. (2007). Mitochondrial dysfunction and age. Curr. Opin. Clin. Nutr. Metab. Care 10, 688–692. doi: 10.1097/MCO.0b013e3282f0dbfb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conti, S., Cassis, P., and Benigni, A. (2012). Aging and the renin-angiotensin system. Hypertension 60, 878–883. doi: 10.1161/HYPERTENSIONAHA.110.155895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cook, J. L., and Re, R. N. (2012). Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R482–R493. doi: 10.1152/ajpregu.00493.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cook, J. L., Zhang, Z., and Re, R. N. (2001). In vitro evidence for an intracellular site of angiotensin action. Circ. Res. 89, 1138–1146. doi: 10.1161/hh2401.101270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., Beal, M. F., and Wallace, D. C. (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329. doi: 10.1038/ng1292-324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortassa, S., Aon, M. A., O'Rourke, B., Jacques, R., Tseng, H. J., Marban, E., et al. (2006). A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys. J. 91, 1564–1589. doi: 10.1529/biophysj.105.076174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortassa, S., O'Rourke, B., and Aon, M. A. (2014). Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim. Biophys. Acta 1837, 287–295. doi: 10.1016/j.bbabio.2013.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cristovam, P. C., Arnoni, C. P., de Andrade, M. C., Casarini, D. E., Pereira, L. G., Schor, N., et al. (2008). ACE-dependent and chymase-dependent angiotensin II generation in normal and glucose-stimulated human mesangial cells. Exp. Biol. Med. (Maywood) 233, 1035–1043. doi: 10.3181/0708-RM-229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Croteau, D. L., Stierum, R. H., and Bohr, V. A. (1999). Mitochondrial DNA repair pathways. Mutat. Res. 434, 137–148. doi: 10.1016/S0921-8777(99)00025-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dai, D. F., Rabinovitch, P. S., and Ungvari, Z. (2012). Mitochondria and cardiovascular aging. Circ. Res. 110, 1109–1124. doi: 10.1161/CIRCRESAHA.111.246140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dai, D. F., Santana, L. F., Vermulst, M., Tomazela, D. M., Emond, M. J., MacCoss, M. J., et al. (2009). Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119, 2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Daiber, A. (2010). Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797, 897–906. doi: 10.1016/j.bbabio.2010.01.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Cavanagh, E. M., Inserra, F., Ferder, M., and Ferder, L. (2007). From mitochondria to disease: role of the renin-angiotensin system. Am. J. Nephrol. 27, 545–553. doi: 10.1159/000107757
de Cavanagh, E. M., Toblli, J. E., Ferder, L., Piotrkowski, B., Stella, I., and Inserra, F. (2006). Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R1616–R1625. doi: 10.1152/ajpregu.00615.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delles, C., Michelson, G., Harazny, J., Oehmer, S., Hilgers, K. F., and Schmieder, R. E. (2004). Impaired endothelial function of the retinal vasculature in hypertensive patients. Stroke 35, 1289–1293. doi: 10.1161/01.STR.0000126597.11534.3b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Mello, W. C., and Frohlich, E. D. (2011). On the local cardiac renin angiotensin system. Basic and clinical implications. Peptides 32, 1774–1779. doi: 10.1016/j.peptides.2011.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Mello, W. C., and Gerena, Y. (2008). Eplerenone inhibits the intracrine and extracellular actions of angiotensin II on the inward calcium current in the failing heart. On the presence of an intracrine renin angiotensin aldosterone system. Regul. Pept. 151, 54–60. doi: 10.1016/j.regpep.2008.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di, L. F., Canton, M., Menabo, R., Kaludercic, N., and Bernardi, P. (2007). Mitochondria and cardioprotection. Heart Fail. Rev. 12, 249–260. doi: 10.1007/s10741-007-9028-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dikalov, S. I., Dikalova, A. E., Bikineyeva, A. T., Schmidt, H. H., Harrison, D. G., and Griendling, K. K. (2008). Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 45, 1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dikalov, S. I., and Nazarewicz, R. R. (2013). Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 19, 1085–1094. doi: 10.1089/ars.2012.4604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dikalov, S. I., and Ungvari, Z. (2013). Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 305, H1417–H1427. doi: 10.1152/ajpheart.00089.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dikalova, A., Clempus, R., Lassegue, B., Cheng, G., McCoy, J., Dikalov, S., et al. (2005). Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112, 2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dirks, A. J., Hofer, T., Marzetti, E., Pahor, M., and Leeuwenburgh, C. (2006). Mitochondrial DNA mutations, energy metabolism and apoptosis in aging muscle. Ageing Res. Rev. 5, 179–195. doi: 10.1016/j.arr.2006.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496. doi: 10.1161/CIRCRESAHA.107.162800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drose, S. (2013). Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim. Biophys. Acta 1827, 578–587. doi: 10.1016/j.bbabio.2013.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Druzhyna, N. M., Wilson, G. L., and LeDoux, S. P. (2008). Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 129, 383–390. doi: 10.1016/j.mad.2008.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Echtay, K. S., Roussel, D., St-Pierre, J., Jekabsons, M. B., Cadenas, S., Stuart, J. A., et al. (2002). Superoxide activates mitochondrial uncoupling proteins. Nature 415, 96–99. doi: 10.1038/415096a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eguchi, S., Matsumoto, T., Motley, E. D., Utsunomiya, H., and Inagami, T. (1996). Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells. Possible requirement of Gq-mediated p21ras activation coupled to a Ca2+/calmodulin-sensitive tyrosine kinase. J. Biol. Chem. 271, 14169–14175. doi: 10.1074/jbc.271.24.14169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eguchi, S., Numaguchi, K., Iwasaki, H., Matsumoto, T., Yamakawa, T., Utsunomiya, H., et al. (1998). Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J. Biol. Chem. 273, 8890–8896. doi: 10.1074/jbc.273.15.8890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ellis, B., Li, X. C., Miguel-Qin, E., Gu, V., and Zhuo, J. L. (2012). Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R494–R509. doi: 10.1152/ajpregu.00487.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eto, K., Ohya, Y., Nakamura, Y., Abe, I., and Iida, M. (2002). Intracellular angiotensin II stimulates voltage-operated Ca(2+) channels in arterial myocytes. Hypertension 39(2 Pt 2), 474–478. doi: 10.1161/hy02t2.102961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Exton, J. H. (1985). Mechanisms involved in alpha-adrenergic phenomena. Am. J. Physiol. 248(6 Pt 1), E633–E647.
Ferder, M., Inserra, F., Manucha, W., and Ferder, L. (2013). The world pandemic of vitamin D deficiency could possibly be explained by cellular inflammatory response activity induced by the renin-angiotensin system. Am. J. Physiol. Cell Physiol. 304, C1027–C1039. doi: 10.1152/ajpcell.00403.2011
Finkel, T. (1998). Oxygen radicals and signaling. Curr. Opin. Cell Biol. 10, 248–253. doi: 10.1016/S0955-0674(98)80147-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fukai, T. (2009). Mitochondrial Thioredoxin: novel regulator for NADPH oxidase and angiotensin II-induced hypertension. Hypertension 54, 224–225. doi: 10.1161/HYPERTENSIONAHA.109.134403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fulton, D. J. (2009). Nox5 and the regulation of cellular function. Antioxid. Redox Signal. 11, 2443–2452. doi: 10.1089/ars.2009.2587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, H. M., and Hong, J. S. (2008). Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365. doi: 10.1016/j.it.2008.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, J., Chao, J., Parbhu, K. J., Yu, L., Xiao, L., Gao, F., et al. (2012). Ontogeny of angiotensin type 2 and type 1 receptor expression in mice. J. Renin Angiotensin Aldosterone Syst. 13, 341–352. doi: 10.1177/1470320312443720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garcia, I. M., Altamirano, L., Mazzei, L., Fornes, M., Molina, M. N., Ferder, L., et al. (2012). Role of mitochondria in paricalcitol-mediated cytoprotection during obstructive nephropathy. Am. J. Physiol. Renal Physiol. 302, F1595–F1605. doi: 10.1152/ajprenal.00617.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geiszt, M., Kopp, J. B., Varnai, P., and Leto, T. L. (2000). Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. U.S.A. 97, 8010–8014. doi: 10.1073/pnas.130135897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Golden, T. R., and Melov, S. (2001). Mitochondrial DNA mutations, oxidative stress, and aging. Mech. Ageing Dev. 122, 1577–1589. doi: 10.1016/S0047-6374(01)00288-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greco, M., Villani, G., Mazzucchelli, F., Bresolin, N., Papa, S., and Attardi, G. (2003). Marked aging-related decline in efficiency of oxidative phosphorylation in human skin fibroblasts. FASEB J. 17, 1706–1708.
Griendling, K. K., Murphy, T. J., and Alexander, R. W. (1993). Molecular biology of the renin-angiotensin system. Circulation 87, 1816–1828. doi: 10.1161/01.CIR.87.6.1816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Griendling, K. K., and Ushio-Fukai, M. (2000). Reactive oxygen species as mediators of angiotensin II signaling. Regul. Pept. 91, 21–27. doi: 10.1016/S0167-0115(00)00136-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gwathmey, T. M., Alzayadneh, E. M., Pendergrass, K. D., and Chappell, M. C. (2012). Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R518–R530. doi: 10.1152/ajpregu.00525.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. doi: 10.1093/geronj/11.3.298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harman, D. (2006). Free radical theory of aging: an update: increasing the functional life span. Ann. N.Y. Acad. Sci. 1067, 10–21. doi: 10.1196/annals.1354.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Harris, R. C., Zhang, M. Z., and Cheng, H. F. (2004). Cyclooxygenase-2 and the renal renin-angiotensin system. Acta Physiol. Scand. 181, 543–547. doi: 10.1111/j.1365-201X.2004.01329.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heinzel, F. R., Luo, Y., Li, X., Boengler, K., Buechert, A., Garcia-Dorado, D., et al. (2005). Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ. Res. 97, 583–586. doi: 10.1161/01.RES.0000181171.65293.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hensley, K., Robinson, K. A., Gabbita, S. P., Salsman, S., and Floyd, R. A. (2000). Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 28, 1456–1462. doi: 10.1016/S0891-5849(00)00252-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herbst, A., Pak, J. W., McKenzie, D., Bua, E., Bassiouni, M., and Aiken, J. M. (2007). Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J. Gerontol. A Biol. Sci. Med. Sci. 62, 235–245. doi: 10.1093/gerona/62.3.235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heymes, C., Bendall, J. K., Ratajczak, P., Cave, A. C., Samuel, J. L., Hasenfuss, G., et al. (2003). Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 41, 2164–2171. doi: 10.1016/S0735-1097(03)00471-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heymes, C., Silvestre, J. S., Llorens-Cortes, C., Chevalier, B., Marotte, F., Levy, B. I., et al. (1998). Cardiac senescence is associated with enhanced expression of angiotensin II receptor subtypes. Endocrinology 139, 2579–2587.
Hoek, J. B., and Rydstrom, J. (1988). Physiological roles of nicotinamide nucleotide transhydrogenase. Biochem. J. 254, 1–10.
Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., et al. (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. doi: 10.1212/WNL.0b013e3181b6bb95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hongpaisan, J., Winters, C. A., and Andrews, S. B. (2004). Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J. Neurosci. 24, 10878–10887. doi: 10.1523/JNEUROSCI.3278-04.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Horan, M. P., Pichaud, N., and Ballard, J. W. (2012). Review: quantifying mitochondrial dysfunction in complex diseases of aging. J. Gerontol. A Biol. Sci. Med. Sci. 67, 1022–1035. doi: 10.1093/gerona/glr263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hornig, B., Landmesser, U., Kohler, C., Ahlersmann, D., Spiekermann, S., Christoph, A., et al. (2001). Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: role of superoxide dismutase. Circulation 103, 799–805. doi: 10.1161/01.CIR.103.6.799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hunt, M. K., Ramos, S. P., Geary, K. M., Norling, L. L., Peach, M. J., Gomez, R. A., et al. (1992). Colocalization and release of angiotensin and renin in renal cortical cells. Am. J. Physiol. 263, F363–F373.
Hutter, E., Renner, K., Pfister, G., Stockl, P., Jansen-Durr, P., and Gnaiger, E. (2004). Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem. J. 380, 919–928. doi: 10.1042/BJ20040095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inagami, T. (2011). Mitochondrial angiotensin receptors and aging. Circ. Res. 109, 1323–1324. doi: 10.1161/RES.0b013e31823f05e0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inagami, T., Mizuno, K., Nakamaru, M., Pandey, K. N., Naruse, M., Naruse, K., et al. (1988). The renin-angiotensin system: an overview of its intracellular function. Cardiovasc. Drugs Ther. 2, 453–458. doi: 10.1007/BF00051182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inagami, T., Mizuno, K., Naruse, K., Okamura, T., and Kawamura, M. (1990). Intracellular formation and release of angiotensins from juxtaglomerular cells. Kidney Int. Suppl. 30, S33–S37.
Inagami, T., Nakamaru, M., Pandey, K. N., Naruse, M., Naruse, K., Misono, K., et al. (1986). Intracellular action of renin, angiotensin production and release. J. Hypertens. Suppl. 4, S11–S16.
Jackson, J. B. (2003). Proton translocation by transhydrogenase. FEBS Lett. 545, 18–24. doi: 10.1016/S0014-5793(03)00388-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, S. N., Yang, W. K., Kim, J., Kim, H. S., Kim, E. J., Yun, H., et al. (2008). Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 29, 713–721. doi: 10.1093/carcin/bgn032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaneto, H., Katakami, N., Matsuhisa, M., and Matsuoka, T. A. (2010). Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010:453892. doi: 10.1155/2010/453892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, J., and Pervaiz, S. (2012). Mitochondria: redox metabolism and dysfunction. Biochem. Res. Int. 2012:896751. doi: 10.1155/2012/896751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katayama, M., Tanaka, M., Yamamoto, H., Ohbayashi, T., Nimura, Y., and Ozawa, T. (1991). Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem. Int. 25, 47–56.
Kelley, E. E., Khoo, N. K., Hundley, N. J., Malik, U. Z., Freeman, B. A., and Tarpey, M. M. (2010). Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 48, 493–498. doi: 10.1016/j.freeradbiomed.2009.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kembro, J. M., Cortassa, S., and Aon, M. A. (2014). “Mitochondrial reactive oxygen species (ROS) and arrhythmias,” in Systems Biology of Free Radicals and Anti Oxidants, ed I. Laher (Berlin; Heidelberg: Springer-Verlag), 1047–1076.
Khaper, N., and Singal, P. K. (2001). Modulation of oxidative stress by a selective inhibition of angiotensin II type 1 receptors in MI rats. J. Am. Coll. Cardiol. 37, 1461–1466. doi: 10.1016/S0735-1097(01)01126-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, R. H., Smith, P. D., Aleyasin, H., Hayley, S., Mount, M. P., Pownall, S., et al. (2005). Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220. doi: 10.1073/pnas.0501282102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kimura, S., Zhang, G. X., Nishiyama, A., Shokoji, T., Yao, L., Fan, Y. Y., et al. (2005). Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 45, 438–444. doi: 10.1161/01.HYP.0000157169.27818.ae
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodama, H., Fukuda, K., Pan, J., Makino, S., Baba, A., Hori, S., et al. (1997). Leukemia inhibitory factor, a potent cardiac hypertrophic cytokine, activates the JAK/STAT pathway in rat cardiomyocytes. Circ. Res. 81, 656–663. doi: 10.1161/01.RES.81.5.656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Konishi, H., Matsuzaki, H., Tanaka, M., Takemura, Y., Kuroda, S., Ono, Y., et al. (1997). Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 410, 493–498. doi: 10.1016/S0014-5793(97)00541-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kregel, K. C., and Zhang, H. J. (2007). An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R18–R36. doi: 10.1152/ajpregu.00327.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, R., Singh, V. P., and Baker, K. M. (2007). The intracellular renin-angiotensin system: a new paradigm. Trends Endocrinol. Metab. 18, 208–214. doi: 10.1016/j.tem.2007.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, R., Singh, V. P., and Baker, K. M. (2008). The intracellular renin-angiotensin system: implications in cardiovascular remodeling. Curr. Opin. Nephrol. Hypertens. 17, 168–173. doi: 10.1097/MNH.0b013e3282f521a8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kumar, R., Thomas, C. M., Yong, Q. C., Chen, W., and Baker, K. M. (2012). The intracrine renin-angiotensin system. Clin. Sci. (Lond.) 123, 273–284. doi: 10.1042/CS20120089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuno, A., Miura, T., Tsuchida, A., Hasegawa, T., Miki, T., Nishino, Y., et al. (2002). Blockade of angiotensin II type 1 receptors suppressed free radical production and preserved coronary endothelial function in the rabbit heart after myocardial infarction. J. Cardiovasc. Pharmacol. 39, 49–57. doi: 10.1097/00005344-200201000-00006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurita, S., Takamura, T., Ota, T., Matsuzawa-Nagata, N., Kita, Y., Uno, M., et al. (2008). Olmesartan ameliorates a dietary rat model of non-alcoholic steatohepatitis through its pleiotropic effects. Eur. J. Pharmacol. 588, 316–324. doi: 10.1016/j.ejphar.2008.04.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurz, S., Hink, U., Nickenig, G., Borthayre, A. B., Harrison, D. G., and Munzel, T. (1999). Evidence for a causal role of the renin-angiotensin system in nitrate tolerance. Circulation 99, 3181–3187. doi: 10.1161/01.CIR.99.24.3181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landmesser, U., Dikalov, S., Price, S. R., McCann, L., Fukai, T., Holland, S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209. doi: 10.1172/JCI200314172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Landmesser, U., Spiekermann, S., Preuss, C., Sorrentino, S., Fischer, D., Manes, C., et al. (2007). Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 27, 943–948. doi: 10.1161/01.ATV.0000258415.32883.bf
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larkin, J. E., Frank, B. C., Gaspard, R. M., Duka, I., Gavras, H., and Quackenbush, J. (2004). Cardiac transcriptional response to acute and chronic angiotensin II treatments. Physiol. Genomics 18, 152–166. doi: 10.1152/physiolgenomics.00057.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Larsen, N. B., Rasmussen, M., and Rasmussen, L. J. (2005). Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion 5, 89–108. doi: 10.1016/j.mito.2005.02.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lassegue, B., Sorescu, D., Szocs, K., Yin, Q., Akers, M., Zhang, Y., et al. (2001). Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 88, 888–894. doi: 10.1161/hh0901.090299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemay, J., Hamet, P., and deBlois, D. (2000). Losartan-induced apoptosis as a novel mechanism for the prevention of vascular lesion formation after injury. J. Renin Angiotensin Aldosterone Syst. 1, 46–50. doi: 10.3317/jraas.2000.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, X. C., Hopfer, U., and Zhuo, J. L. (2012). Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am. J. Physiol. Renal Physiol. 303, F1617–F1628. doi: 10.1152/ajprenal.00219.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liao, D. F., Monia, B., Dean, N., and Berk, B. C. (1997). Protein kinase C-zeta mediates angiotensin II activation of ERK1/2 in vascular smooth muscle cells. J. Biol. Chem. 272, 6146–6150. doi: 10.1074/jbc.272.10.6146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y., Fiskum, G., and Schubert, D. (2002). Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80, 780–787. doi: 10.1046/j.0022-3042.2002.00744.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Loeb, L. A., Wallace, D. C., and Martin, G. M. (2005). The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 18769–18770. doi: 10.1073/pnas.0509776102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ma, J., Nishimura, H., Fogo, A., Kon, V., Inagami, T., and Ichikawa, I. (1998). Accelerated fibrosis and collagen deposition develop in the renal interstitium of angiotensin type 2 receptor null mutant mice during ureteral obstruction. Kidney Int. 53, 937–944. doi: 10.1111/j.1523-1755.1998.00893.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malhotra, R., Sadoshima, J., Brosius, F. C. 3rd., and Izumo, S. (1999). Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes In vitro. Circ. Res. 85, 137–146. doi: 10.1161/01.RES.85.2.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchesi, C., Paradis, P., and Schiffrin, E. L. (2008). Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 29, 367–374. doi: 10.1016/j.tips.2008.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marin-Garcia, J., Zoubenko, O., and Goldenthal, M. J. (2002). Mutations in the cardiac mitochondrial DNA control region associated with cardiomyopathy and aging. J. Card. Fail. 8, 93–100. doi: 10.1054/jcaf.2002.32501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mercure, C., Ramla, D., Garcia, R., Thibault, G., Deschepper, C. F., and Reudelhuber, T. L. (1998). Evidence for intracellular generation of angiotensin II in rat juxtaglomerular cells. FEBS Lett. 422, 395–399. doi: 10.1016/S0014-5793(98)00052-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Min, L. J., Mogi, M., Iwai, M., and Horiuchi, M. (2009). Signaling mechanisms of angiotensin II in regulating vascular senescence. Ageing Res. Rev. 8, 113–121. doi: 10.1016/j.arr.2008.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mitchell, P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. doi: 10.1038/191144a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mogensen, M., Bagger, M., Pedersen, P. K., Fernstrom, M., and Sahlin, K. (2006). Cycling efficiency in humans is related to low UCP3 content and to type I fibres but not to mitochondrial efficiency. J. Physiol. 571, 669–681. doi: 10.1113/jphysiol.2005.101691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mohanty, P., Hamouda, W., Garg, R., Aljada, A., Ghanim, H., and Dandona, P. (2000). Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J. Clin. Endocrinol. Metab. 85, 2970–2973. doi: 10.1210/jcem.85.8.6854
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monteiro, P., Duarte, A. I., Goncalves, L. M., and Providencia, L. A. (2005). Valsartan improves mitochondrial function in hearts submitted to acute ischemia. Eur. J. Pharmacol. 518, 158–164. doi: 10.1016/j.ejphar.2005.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moore, A. Z., Biggs, M. L., Matteini, A., O'Connor, A., McGuire, S., Beamer, B. A., et al. (2010). Polymorphisms in the mitochondrial DNA control region and frailty in older adults. PLoS ONE 5:e11069. doi: 10.1371/journal.pone.0011069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mori, J., Alrob, O. A., Wagg, C. S., Harris, R. A., Lopaschuk, G. D., and Oudit, G. Y. (2013). ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: a critical role of PDK4. Am. J. Physiol. Heart Circ. Physiol. 304, H1103–H1113. doi: 10.1152/ajpheart.00636.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murtaza, I., Wang, H. X., Feng, X., Alenina, N., Bader, M., Prabhakar, B. S., et al. (2008). Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 283, 5996–6004. doi: 10.1074/jbc.M706466200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nemoto, S., Takeda, K., Yu, Z. X., Ferrans, V. J., and Finkel, T. (2000). Role for mitochondrial oxidants as regulators of cellular metabolism. Mol. Cell. Biol. 20, 7311–7318. doi: 10.1128/MCB.20.19.7311-7318.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen Dinh, C. A., Montezano, A. C., Burger, D., and Touyz, R. M. (2013). Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 19, 1110–1120. doi: 10.1089/ars.2012.4641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nguyen, T., Nioi, P., and Pickett, C. B. (2009). The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291–13295. doi: 10.1074/jbc.R900010200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nickel, A., Kohlhaas, M., and Maack, C. (2014). Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 73C, 26–33. doi: 10.1016/j.yjmcc.2014.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nickenig, G. (2004). Should angiotensin II receptor blockers and statins be combined? Circulation 110, 1013–1020. doi: 10.1161/01.CIR.0000139857.85424.45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nickenig, G., and Harrison, D. G. (2002). The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part II: AT(1) receptor regulation. Circulation 105, 530–536. doi: 10.1161/hc0402.102619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nickenig, G., Roling, J., Strehlow, K., Schnabel, P., and Bohm, M. (1998). Insulin induces upregulation of vascular AT1 receptor gene expression by posttranscriptional mechanisms. Circulation 98, 2453–2460. doi: 10.1161/01.CIR.98.22.2453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishida, Y., Yokota, T., Takahashi, T., Uchihara, T., Jishage, K., and Mizusawa, H. (2006). Deletion of vitamin E enhances phenotype of Alzheimer disease model mouse. Biochem. Biophys. Res. Commun. 350, 530–536. doi: 10.1016/j.bbrc.2006.09.083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nozoe, M., Hirooka, Y., Koga, Y., Araki, S., Konno, S., Kishi, T., et al. (2008). Mitochondria-derived reactive oxygen species mediate sympathoexcitation induced by angiotensin II in the rostral ventrolateral medulla. J. Hypertens. 26, 2176–2184. doi: 10.1097/HJH.0b013e32830dd5d3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pain, T., Yang, X. M., Critz, S. D., Yue, Y., Nakano, A., Liu, G. S., et al. (2000). Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ. Res. 87, 460–466. doi: 10.1161/01.RES.87.6.460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Panahpour, H., and Dehghani, G. A. (2012). Attenuation of focal cerebral ischemic injury following post-ischemic inhibition of angiotensin converting enzyme (ACE) activity in normotensive rat. Iran. Biomed. J. 16, 202–208.
Pandur, S., Ravuri, C., Moens, U., and Huseby, N. E. (2014). Combined incubation of colon carcinoma cells with phorbol ester and mitochondrial uncoupling agents results in synergic elevated reactive oxygen species levels and increased gamma-glutamyltransferase expression. Mol. Cell. Biochem. 388, 149–156. doi: 10.1007/s11010-013-1906-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pelicano, H., Carney, D., and Huang, P. (2004). ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 7, 97–110. doi: 10.1016/j.drup.2004.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Persson, P., Palm, F., and Friederich-Persson, M. (in press). The effects of angiotensin ii on mitochondrial respiration: a role of normoglycemia versus hyperglycemia. Mitochondr. Physiol. Netw. 8–18.
Peters, J. (2008). Secretory and cytosolic (pro)renin in kidney, heart, and adrenal gland. J. Mol. Med. 86, 711–714. doi: 10.1007/s00109-008-0328-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peters, J. (2012). Local renin-angiotensin systems in the adrenal gland. Peptides 34, 427–432. doi: 10.1016/j.peptides.2012.01.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prathapan, A., Vineetha, V. P., and Raghu, K. G. (2014). Protective effect of Boerhaavia diffusa L. against mitochondrial dysfunction in angiotensin II induced hypertrophy in H9c2 cardiomyoblast cells. PLoS ONE 9:e96220. doi: 10.1371/journal.pone.0096220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pueyo, M. E., Gonzalez, W., Nicoletti, A., Savoie, F., Arnal, J. F., and Michel, J. B. (2000). Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol. 20, 645–651. doi: 10.1161/01.ATV.20.3.645
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Puri, P. L., Avantaggiati, M. L., Burgio, V. L., Chirillo, P., Collepardo, D., Natoli, G., et al. (1995). Reactive oxygen intermediates mediate angiotensin II-induced c-Jun.c-Fos heterodimer DNA binding activity and proliferative hypertrophic responses in myogenic cells. J. Biol. Chem. 270, 22129–22134. doi: 10.1074/jbc.270.38.22129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qadri, F., Arens, T., Schwartz, E. C., Hauser, W., and Dominiak, P. (2001). Angiotensin-converting enzyme inhibitors and AT1-receptor antagonist restore nitric oxide synthase (NOS) activity and neuronal NOS expression in the adrenal glands of spontaneously hypertensive rats. Jpn. J. Pharmacol. 85, 365–369. doi: 10.1254/jjp.85.365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quijano, C., Hernandez-Saavedra, D., Castro, L., McCord, J. M., Freeman, B. A., and Radi, R. (2001). Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem. 276, 11631–11638. doi: 10.1074/jbc.M009429200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quinlan, C. L., Gerencser, A. A., Treberg, J. R., and Brand, M. D. (2011). The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 286, 31361–31372. doi: 10.1074/jbc.M111.267898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rashed, H. M., Waller, F. M., and Patel, T. B. (1988). Hormonal regulation of the alpha-ketoglutarate dehydrogenase complex in the isolated perfused rat liver. J. Biol. Chem. 263, 5700–5706.
Re, R. N., Vizard, D. L., Brown, J., and Bryan, S. E. (1984). Angiotensin II receptors in chromatin fragments generated by micrococcal nuclease. Biochem. Biophys. Res. Commun. 119, 220–227. doi: 10.1016/0006-291X(84)91641-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Redmond, E. M., and Cahill, P. A. (2012). The NOX-ROS connection: targeting Nox1 control of N-cadherin shedding in vascular smooth muscle cells. Cardiovasc. Res. 93, 386–387. doi: 10.1093/cvr/cvs020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ricci, C., Pastukh, V., and Schaffer, S. W. (2005). Involvement of the mitochondrial permeability transition pore in angiotensin II-mediated apoptosis. Exp. Clin. Cardiol. 10, 160–164.
Rich, P. R., and Marechal, A. (2010). The mitochondrial respiratory chain. Essays Biochem. 47, 1–23. doi: 10.1042/bse0470001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robertson, A. L. Jr., and Khairallah, P. A. (1971). Angiotensin II: rapid localization in nuclei of smooth and cardiac muscle. Science 172, 1138–1139. doi: 10.1126/science.172.3988.1138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez-Iturbe, B., Sepassi, L., Quiroz, Y., Ni, Z., Wallace, D. C., and Vaziri, N. D. (2007). Association of mitochondrial SOD deficiency with salt-sensitive hypertension and accelerated renal senescence. J. Appl. Physiol. (1985) 102, 255–260. doi: 10.1152/japplphysiol.00513.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roskoski, R. Jr. (2012). ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol. Res. 66, 105–143. doi: 10.1016/j.phrs.2012.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sadoshima, J., Xu, Y., Slayter, H. S., and Izumo, S. (1993). Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 75, 977–984. doi: 10.1016/0092-8674(93)90541-W
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saitoh, Y., Hongwei, W., Ueno, H., Mizuta, M., and Nakazato, M. (2009). Telmisartan attenuates fatty-acid-induced oxidative stress and NAD(P)H oxidase activity in pancreatic beta-cells. Diabetes Metab. 35, 392–397. doi: 10.1016/j.diabet.2009.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sano, M., Fukuda, K., Sato, T., Kawaguchi, H., Suematsu, M., Matsuda, S., et al. (2001). ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 89, 661–669. doi: 10.1161/hh2001.098873
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schroder, K., Zhang, M., Benkhoff, S., Mieth, A., Pliquett, R., Kosowski, J., et al. (2012). Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 110, 1217–1225. doi: 10.1161/CIRCRESAHA.112.267054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shah, P. K., and Galis, Z. S. (2001). Matrix metalloproteinase hypothesis of plaque rupture: players keep piling up but questions remain. Circulation 104, 1878–1880.
Shao, J., Iwashita, N., Ikeda, F., Ogihara, T., Uchida, T., Shimizu, T., et al. (2006). Beneficial effects of candesartan, an angiotensin II type 1 receptor blocker, on beta-cell function and morphology in db/db mice. Biochem. Biophys. Res. Commun. 344, 1224–1233. doi: 10.1016/j.bbrc.2006.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sherrod, M., Liu, X., Zhang, X., and Sigmund, C. D. (2005). Nuclear localization of angiotensinogen in astrocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R539–R546. doi: 10.1152/ajpregu.00594.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shigenaga, M. K., Hagen, T. M., and Ames, B. N. (1994). Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U.S.A. 91, 10771–10778. doi: 10.1073/pnas.91.23.10771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shiomi, T., Tsutsui, H., Matsusaka, H., Murakami, K., Hayashidani, S., Ikeuchi, M., et al. (2004). Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 109, 544–549. doi: 10.1161/01.CIR.0000109701.77059.E9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, H., Stefani, E., and Toro, L. (2012). Intracellular BK(Ca) (iBK(Ca)) channels. J. Physiol. (Lond). 590, 5937–5947. doi: 10.1113/jphysiol.2011.215533
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Skulachev, V. P. (2006). Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 11, 473–485. doi: 10.1007/s10495-006-5881-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sovari, A. A., Rutledge, C. A., Jeong, E. M., Dolmatova, E., Arasu, D., Liu, H., et al. (2013). Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ. Arrhythm. Electrophysiol. 6, 623–631. doi: 10.1161/CIRCEP.112.976787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spooner, R., and Yilmaz, O. (2011). The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 12, 334–352. doi: 10.3390/ijms12010334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Squadrito, G. L., and Pryor, W. A. (1995). The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem. Biol. Interact 96, 203–206. doi: 10.1016/0009-2797(94)03591-U
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevenson, M. A., Pollock, S. S., Coleman, C. N., and Calderwood, S. K. (1994). X-irradiation, phorbol esters, and H2O2 stimulate mitogen-activated protein kinase activity in NIH-3T3 cells through the formation of reactive oxygen intermediates. Cancer Res. 54, 12–15.
St-Pierre, J., Drori, S., Uldry, M., Silvaggi, J. M., Rhee, J., Jager, S., et al. (2006). Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408. doi: 10.1016/j.cell.2006.09.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugamura, K., and Keaney, J. F. Jr. (2011). Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 51, 978–992. doi: 10.1016/j.freeradbiomed.2011.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., and Finkel, T. (1995). Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 270, 296–299. doi: 10.1126/science.270.5234.296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sunggip, C., Kitajima, N., and Nishida, M. (2013). Redox control of cardiovascular homeostasis by angiotensin II. Curr. Pharm. Des. 19, 3022–3032. doi: 10.2174/1381612811319170008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, Y., Ruiz-Ortega, M., Lorenzo, O., Ruperez, M., Esteban, V., and Egido, J. (2003). Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 35, 881–900. doi: 10.1016/S1357-2725(02)00271-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Szanto, I., Rubbia-Brandt, L., Kiss, P., Steger, K., Banfi, B., Kovari, E., et al. (2005). Expression of NOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol. 207, 164–176. doi: 10.1002/path.1824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takac, I., Schroder, K., and Brandes, R. P. (2012). The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr. Hypertens. Rep. 14, 70–78. doi: 10.1007/s11906-011-0238-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takemori, K., Inoue, T., and Ito, H. (2013). Effects of angiotensin II type 1 receptor blocker and adiponectin on adipocyte dysfunction in stroke-prone spontaneously hypertensive rats. Lipids Health Dis. 12, 108–112. doi: 10.1186/1476-511X-12-108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takemori, K., Ishida, H., and Ito, H. (2005). Continuous inhibition of the renin-angiotensin system and protection from hypertensive end-organ damage by brief treatment with angiotensin II type 1 receptor blocker in stroke-prone spontaneously hypertensive rats. Life Sci. 77, 2233–2245. doi: 10.1016/j.lfs.2004.12.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tan, W. Q., Wang, K., Lv, D. Y., and Li, P. F. (2008). Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J. Biol. Chem. 283, 29730–29739. doi: 10.1074/jbc.M805514200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanito, M., Nakamura, H., Kwon, Y. W., Teratani, A., Masutani, H., Shioji, K., et al. (2004). Enhanced oxidative stress and impaired thioredoxin expression in spontaneously hypertensive rats. Antioxid. Redox Signal. 6, 89–97. doi: 10.1089/152308604771978381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thareja, S., Aggarwal, S., Bhardwaj, T. R., and Kumar, M. (2012). Protein tyrosine phosphatase 1B inhibitors: a molecular level legitimate approach for the management of diabetes mellitus. Med. Res. Rev. 32, 459–517. doi: 10.1002/med.20219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thrasher, A. J., Keep, N. H., Wientjes, F., and Segal, A. W. (1994). Chronic granulomatous disease. Biochim. Biophys. Acta 1227, 1–24. doi: 10.1016/0925-4439(94)90100-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toblli, J. E., Munoz, M. C., Cao, G., Mella, J., Pereyra, L., and Mastai, R. (2008). ACE inhibition and AT1 receptor blockade prevent fatty liver and fibrosis in obese Zucker rats. Obesity (Silver. Spring). 16, 770–776. doi: 10.1038/oby.2007.114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Touyz, R. M. (2000). Oxidative stress and vascular damage in hypertension. Curr. Hypertens. Rep. 2, 98–105. doi: 10.1007/s11906-000-0066-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Touyz, R. M. (2004). Reactive oxygen species and angiotensin II signaling in vascular cells—implications in cardiovascular disease. Braz. J. Med. Biol. Res. 37, 1263–1273. doi: 10.1590/S0100-879X2004000800018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Touyz, R. M., and Berry, C. (2002). Recent advances in angiotensin II signaling. Braz. J. Med. Biol. Res. 35, 1001–1015. doi: 10.1590/S0100-879X2002000900001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tracy, R. P. (2003). Emerging relationships of inflammation, cardiovascular disease and chronic diseases of aging. Int. J. Obes. Relat. Metab. Disord. 27(Suppl. 3), S29–S34. doi: 10.1038/sj.ijo.0802497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turrens, J. (2003). F. Mitochondrial formation of reactive oxygen species. J. Physiol. 552(Pt 2), 335–344. doi: 10.1113/jphysiol.2003.049478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Turrens, J. F., and Boveris, A. (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 191, 421–427.
Turrens, J. F., Freeman, B. A., Levitt, J. G., and Crapo, J. D. (1982). The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch. Biochem. Biophys. 217, 401–410. doi: 10.1016/0003-9861(82)90518-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Unger, T. (2002). The role of the renin-angiotensin system in the development of cardiovascular disease. Am. J. Cardiol. 89, 3A–9A. doi: 10.1016/S0002-9149(01)02321-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ungvari, Z., Orosz, Z., Labinskyy, N., Rivera, A., Xiangmin, Z., Smith, K., et al. (2007). Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 293, H37–H47. doi: 10.1152/ajpheart.01346.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ushio-Fukai, M., Alexander, R. W., Akers, M., Yin, Q., Fujio, Y., Walsh, K., et al. (1999). Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 274, 22699–22704. doi: 10.1074/jbc.274.32.22699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ushio-Fukai, M., Griendling, K. K., Becker, P. L., Hilenski, L., Halleran, S., and Alexander, R. W. (2001). Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 21, 489–495. doi: 10.1161/01.ATV.21.4.489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valente, A. J., Yoshida, T., Murthy, S. N., Sakamuri, S. S., Katsuyama, M., Clark, R. A., et al. (2012). Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 303, H282–H296. doi: 10.1152/ajpheart.00231.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Venkatesan, B., Mahimainathan, L., Das, F., Ghosh-Choudhury, N., and Ghosh, C. G. (2007). Downregulation of catalase by reactive oxygen species via PI 3 kinase/Akt signaling in mesangial cells. J. Cell. Physiol. 211, 457–467. doi: 10.1002/jcp.20953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vidotti, D. B., Casarini, D. E., Cristovam, P. C., Leite, C. A., Schor, N., and Boim, M. A. (2004). High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. Am. J. Physiol. Renal Physiol. 286, F1039–F1045. doi: 10.1152/ajprenal.00371.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Viedt, C., Soto, U., Krieger-Brauer, H. I., Fei, J., Elsing, C., Kubler, W., et al. (2000). Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler. Thromb. Vasc. Biol. 20, 940–948. doi: 10.1161/01.ATV.20.4.940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vila-Porcile, E., and Corvol, P. (1998). Angiotensinogen, prorenin, and renin are Co-localized in the secretory granules of all glandular cells of the rat anterior pituitary: an immunoultrastructural study. J. Histochem. Cytochem. 46, 301–311. doi: 10.1177/002215549804600303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Volpato, S., Guralnik, J. M., Ferrucci, L., Balfour, J., Chaves, P., Fried, L. P., et al. (2001). Cardiovascular disease, interleukin-6, and risk of mortality in older women: the women's health and aging study. Circulation 103, 947–953. doi: 10.1161/01.CIR.103.7.947
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wallace, D. C. (2001). A mitochondrial paradigm for degenerative diseases and ageing. Novartis Found. Symp. 235, 247–263. doi: 10.1002/0470868694.ch20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wallace, D. C. (2010). Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 51, 440–450.
Wallace, D. C. (2011). Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 1–16. doi: 10.1101/sqb.2011.76.010462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, D., Qian, L., Xiong, H., Liu, J., Neckameyer, W. S., Oldham, S., et al. (2006a). Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 103, 13520–13525. doi: 10.1073/pnas.0604661103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, G., Anrather, J., Glass, M. J., Tarsitano, M. J., Zhou, P., Frys, K. A., et al. (2006b). Nox2, Ca2+, and protein kinase C play a role in angiotensin II-induced free radical production in nucleus tractus solitarius. Hypertension 48, 482–489. doi: 10.1161/01.HYP.0000236647.55200.07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, G., Hamid, T., Keith, R. J., Zhou, G., Partridge, C. R., Xiang, X., et al. (2010). Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 121, 1912–1925. doi: 10.1161/CIRCULATIONAHA.109.905471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, M., Takagi, G., Asai, K., Resuello, R. G., Natividad, F. F., Vatner, D. E., et al. (2003). Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 41, 1308–1316. doi: 10.1161/01.HYP.0000073843.56046.45
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wangler, N. J., Santos, K. L., Schadock, I., Hagen, F. K., Escher, E., Bader, M., et al. (2012). Identification of membrane-bound variant of metalloendopeptidase neurolysin (EC 3.4.24.16) as the non-angiotensin type 1 (non-AT1), non-AT2 angiotensin binding site. J. Biol. Chem. 287, 114–122. doi: 10.1074/jbc.M111.273052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warnholtz, A., Nickenig, G., Schulz, E., Macharzina, R., Brasen, J. H., Skatchkov, M., et al. (1999). Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation 99, 2027–2033. doi: 10.1161/01.CIR.99.15.2027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waud, W. R., and Rajagopalan, K. V. (1976). The mechanism of conversion of rat liver xanthine dehydrogenase from an NAD+-dependent form (type D) to an O2-dependent form (type O). Arch. Biochem. Biophys. 172, 365–379. doi: 10.1016/0003-9861(76)90088-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, Y., Clark, S. E., Thyfault, J. P., Uptergrove, G. M., Li, W., Whaley-Connell, A. T., et al. (2009). Oxidative stress-mediated mitochondrial dysfunction contributes to angiotensin II-induced nonalcoholic fatty liver disease in transgenic Ren2 rats. Am. J. Pathol. 174, 1329–1337. doi: 10.2353/ajpath.2009.080697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weydt, P., Pineda, V. V., Torrence, A. E., Libby, R. T., Satterfield, T. F., Lazarowski, E. R., et al. (2006). Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 4, 349–362. doi: 10.1016/j.cmet.2006.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Whaley-Connell, A., Habibi, J., Cooper, S. A., Demarco, V. G., Hayden, M. R., Stump, C. S., et al. (2008). Effect of renin inhibition and AT1R blockade on myocardial remodeling in the transgenic Ren2 rat. Am. J. Physiol. Endocrinol. Metab. 295, E103–E109. doi: 10.1152/ajpendo.00752.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Widder, J. D., Fraccarollo, D., Galuppo, P., Hansen, J. M., Jones, D. P., Ertl, G., et al. (2009). Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 54, 338–344. doi: 10.1161/HYPERTENSIONAHA.108.127928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wilkinson-Berka, J. L., Deliyanti, D., Rana, I., Miller, A. G., Agrotis, A., Armani, R., et al. (2014). NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxid. Redox Signal. 20, 2726–2740. doi: 10.1089/ars.2013.5357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Williamson, J. R., Cooper, R. H., Joseph, S. K., and Thomas, A. P. (1985). Inositol trisphosphate and diacylglycerol as intracellular second messengers in liver. Am. J. Physiol. 248(3 Pt 1), C203–C216.
Wingler, K., Wunsch, S., Kreutz, R., Rothermund, L., Paul, M., and Schmidt, H. H. (2001). Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic. Biol. Med. 31, 1456–1464. doi: 10.1016/S0891-5849(01)00727-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wood, E. J. (2006). Marks' basic medical biochemistry: a clinical approach (second edition). Biochem. Mol. Biol. Educ. 34, 395. doi: 10.1002/bmb.2006.494034052660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wosniak, J. Jr., Santos, C. X., Kowaltowski, A. J., and Laurindo, F. R. (2009). Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 11, 1265–1278. doi: 10.1089/ars.2009.2392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xia, Y., Buja, L. M., and McMillin, J. B. (1998). Activation of the cytochrome c gene by electrical stimulation in neonatal rat cardiac myocytes. Role of NRF-1 and c-Jun. J. Biol. Chem. 273, 12593–12598. doi: 10.1074/jbc.273.20.12593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiong, S., Salazar, G., San, M. A., Ahmad, M., Patrushev, N., Hilenski, L., et al. (2010). PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 285, 2474–2487. doi: 10.1074/jbc.M109.065235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, C., Kim, D., Aizawa, T., and Berk, B. C. (2003). Functional interplay between angiotensin II and nitric oxide: cyclic GMP as a key mediator. Arterioscler. Thromb. Vasc. Biol. 23, 26–36. doi: 10.1161/01.ATV.0000046231.17365.9D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, E., Mercer, J., and Bennett, M. (2012). Mitochondria in vascular disease. Cardiovasc. Res. 95, 173–182. doi: 10.1093/cvr/cvs111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zaobornyj, T., and Ghafourifar, P. (2012). Strategic localization of heart mitochondrial NOS: a review of the evidence. Am. J. Physiol. Heart Circ. Physiol. 303, H1283–H1293. doi: 10.1152/ajpheart.00674.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, G. X., Lu, X. M., Kimura, S., and Nishiyama, A. (2007). Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc. Res. 76, 204–212. doi: 10.1016/j.cardiores.2007.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, M., Brewer, A. C., Schroder, K., Santos, C. X., Grieve, D. J., Wang, M., et al. (2010). NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 18121–18126. doi: 10.1073/pnas.1009700107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, Y., Hileman, E. O., Plunkett, W., Keating, M. J., and Huang, P. (2003). Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 101, 4098–4104. doi: 10.1182/blood-2002-08-2512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier, J. L., and Sollott, S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014. doi: 10.1084/jem.192.7.1001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mitochondria, angiotensin II, angiotensin II type 1 receptor blockers, mitochondrial angiotensin system, redox regulation, aging, frailty
Citation: Vajapey R, Rini D, Walston J and Abadir P (2014) The impact of age-related dysregulation of the angiotensin system on mitochondrial redox balance. Front. Physiol. 5:439. doi: 10.3389/fphys.2014.00439
Received: 25 August 2014; Accepted: 27 October 2014;
Published online: 24 November 2014.
Edited by:
Miguel A. Aon, Johns Hopkins University School of Medicine, USAReviewed by:
Roy Sutliff, Emory University, USACopyright © 2014 Vajapey, Rini, Walston and Abadir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Abadir, Johns Hopkins University, 5501 Hopkins Bayview Circle, Rm. 1A62, Baltimore, MD 21224, USA e-mail:cGFiYWRpcjFAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.