 Stephanie E. Lindsey
Stephanie E. Lindsey Jonathan T. Butcher
Jonathan T. Butcher Huseyin C. Yalcin
Huseyin C. Yalcin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 21 August 2014
Sec. Biophysics
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00318
This article is part of the Research TopicMechanotransduction and Development of Cardiovascular Form and Function.View all 11 articles
Mechanical forces are essential contributors to and unavoidable components of cardiac formation, both inducing and orchestrating local and global molecular and cellular changes. Experimental animal studies have contributed substantially to understanding the mechanobiology of heart development. More recent integration of high-resolution imaging modalities with computational modeling has greatly improved our quantitative understanding of hemodynamic flow in heart development. Merging these latest experimental technologies with molecular and genetic signaling analysis will accelerate our understanding of the relationships integrating mechanical and biological signaling for proper cardiac formation. These advances will likely be essential for clinically translatable guidance for targeted interventions to rescue malforming hearts and/or reconfigure malformed circulations for optimal performance. This review summarizes our current understanding on the levels of mechanical signaling in the heart and their roles in orchestrating cardiac development.
The heart is the first functional organ to develop in the embryo, convecting nutrients to surrounding tissues to facilitate growth. As the embryo grows, the heart transforms from a linear valve-less tube to a multi-chambered structure complete with 4 fibrous valves (Srivastava and Olson, 2000; Bartman and Hove, 2005). Changes in pressure, strain and wall shear stress (WSS) accompany cardiac morphogenesis and orchestrate molecular and cellular responses that help coordinate downstream tissue changes. Congenital heart defects (CHDs) form when cardiac morphogenetic processes are disrupted. CHDs affect 1–2% of newborn children and are the leading cause of death in infants under 1 year of age. CHDs represent the single largest class of birth defects and account for approximately 25% of all human congenital abnormalities (Roger et al., 2011). Despite their prevalence, the etiology of many CHDs remains unknown. While clinical and experimental research has identified multiple genetic mutations that participate in the formation of CHDs, they fail to fully account for the disease phenotype. Extreme locus heterogeneity and lack of a distinct genotype–phenotype correlation have limited causative gene discovery (Yuan et al., 2013). Recent insights into the molecular mechanisms of heart development have shown that a given structural CHD is often linked to multiple loci. A variety of phenotypes is often observed in families with a specific gene mutation (Nemer, 2008). Family members with the same mutation may present with an atrial septal defect, tetralogy of fallot, or ventricular septal defect (Bruneau, 2008). Conversely, mutations in different genes may cause an identical malformation (Fahed et al., 2013). Zaidi and colleagues recently identified new point mutations in hundreds of genes that together may contribute to only approximately 10% of CHDs (Zaidi et al., 2013). Oyen et al. determined the risk of an infant presenting with a CHD in families with prior history of CHDs was only 2–4%, suggesting CHDs largely occur in families without history of disease (Oyen et al., 2009). In addition to genetic factors, environmental factors such as drug exposure and hemodynamic patterning contribute to the development of CHDs (Huang et al., 2010). Mechanical perturbation of blood flow can induce diseased phenotypes (Hogers et al., 1999; Sedmera et al., 1999, 2002; Miller et al., 2003; Reckova et al., 2003; Groenendijk et al., 2005; deAlmeida et al., 2007).
Blood flow exerts forces on surrounding tissue. These forces include, the force normal to the walls from blood pressure, the associated circumferential stress that occurs as the walls stretch in response to pressure and the frictional force exerted by flow along the walls, WSS (Gjorevski and Nelson, 2010; Samsa et al., 2013) (Figure 1). Local mechanical properties of cardiac tissue as well as its loading-induced residual stress govern structural deformation in response to mechanical stimuli. Mechanical signals induce gene expression and differentiation on a cellular level, translating molecular level events into tissue-level deformations that guide embryo development (Wang et al., 2009; Mammoto and Ingber, 2010; Yalcin et al., 2011; Bharadwaj et al., 2012; Wyczalkowski et al., 2012). This review summarizes cardiac morphogenesis and the role of mechanical signaling in its effectuation.
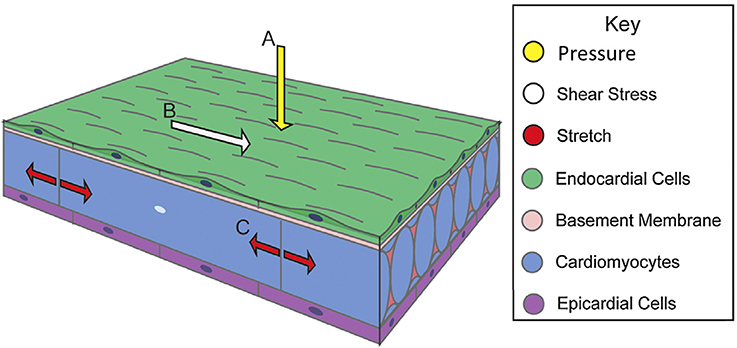
Figure 1. Biomechanical forces in cardiac wall maturation. Biomechanical forces are important for normal developmental patterning. Forces exerted on the wall from blood flow include (A) pressure, a force perpendicular to the vessel wall, (B) shear stress, the frictional force parallel to the vessel wall, and (C) circumferential stress that occurs as the walls stretch. Arrows indicate force vectors. Adapted from Samsa et al. (2013).
While cardiac morphogenesis varies across vertebrate species, major landmarks can be broken down into four basic steps, namely, heart tube formation, looping, trabeculation and valve formation/septation. Major developmental events are summarized below as well as in Table 1 and in Figure 2. Tables 2, 3 evaluate changes in heart rate and blood pressure in humans and in common, animal models of cardiovascular development.

Table 1. Comparative cardiovascular development across animal models.
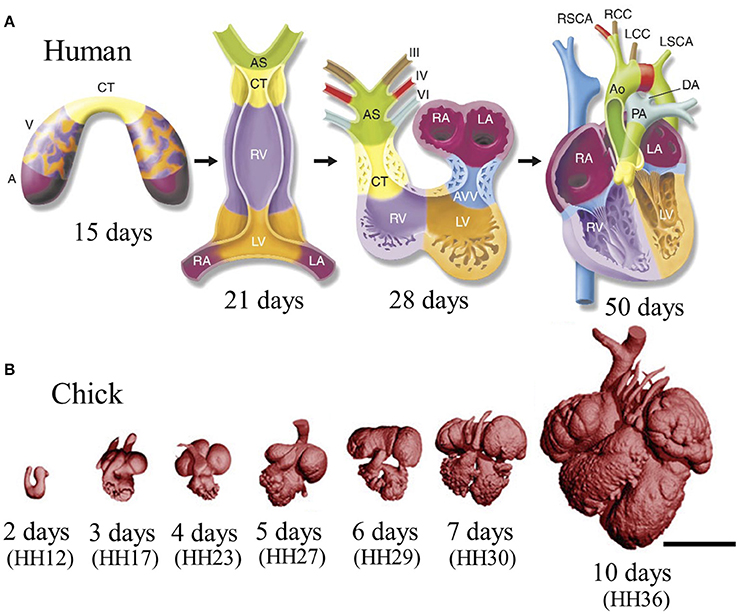
Figure 2. Stages of heart development. (A) Schematic of cardiac morphogenesis in human. Illustrations depict cardiac development with color coding of morphologically related regions, seen from a ventral view. Cardiogenic precursors form a crescent (far-left panel) that is specified to form specific segments of the linear heart tube, which is patterned along the anteroposterior axis to form the various regions and chambers of the looped and mature heart. Each cardiac chamber balloons out from the outer curvature of the looped heart tube in a segmental manner. Neural crest cells populate the bilaterally symmetric aortic arch arteries (III, IV, and VI) and aortic sac (AS) that together contribute to specific segments of the mature aortic arch. Mesenchymal cells form the cardiac valves from the contruncal (CT) and atrioventricular valve (AVV) segments. Corresponding days of human embryonic development are indicated. Abbreviations: A, atrium; V, ventricle; RV, right ventricle; LV, left ventricle; RA, right atrium; LA, left atrium; PA, pulmonary artery; Ao, aorta; DA, ductus arteriosus; RSCA, right subclavian artery; LSCA, left subclavian artery; RCC, right common carotid; LCC, left common carotid. Adapted from Srivastava and Olson (2000). (B) Scaled micro-CT images of chicken embryonic hearts at representative days. Corresponding Hamburger–Hamilton (HH) stages were shown in paranthesis. Images were generated by our group in our previous work. Scale bar is 1 mm.
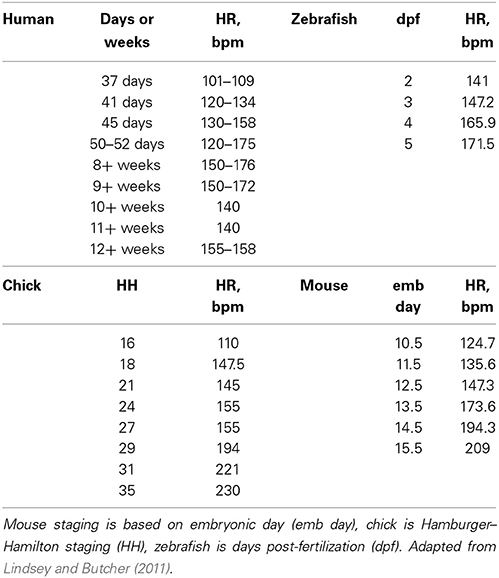
Table 2. Heart rate (HR) in beats per minute (bpm) across developmental animal models.
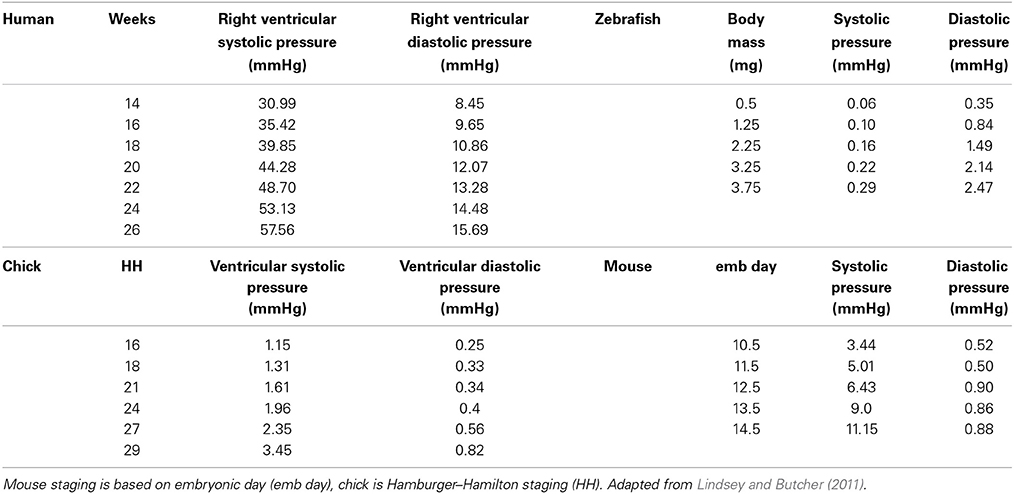
Table 3. Blood pressure across developmental animal models.
During early stages of cardiogenesis, two bilateral mesodermal heart forming fields, also known as the primary heart fields (PHF), fuse at the ventral midline, to form the tubular heart (Rana et al., 2013). At this stage, the cardiac wall is composed of an inner endocardial layer and outer myocardial sheath. Separating these two layers is a gelatinous acellular hyaline matrix called the cardiac jelly. The cells in this newly formed linear heart tube will primarily contribute to the developing atria, atrioventricular (AV) canal, and the left ventricle (Meilhac et al., 2004; Zaffran et al., 2004).
Expansion and elongation of the heart tube initiates the next stage of cardiac formation: looping. Elongation begins with the addition of cardiac progenitor cells to either end of the tubular heart. This population of highly proliferative mesodermal tissue is commonly called the second heart field (SHF) (Dyer and Kirby, 2009). At the arterial pole, these cells will contribute to the outflow tract (OFT), right ventricle, and the interventricular septum and at the venous pole, they contribute to the developing atria and the atrial septum (Verzi et al., 2005; Lockhart et al., 2011). Looping occurs in three phases: dextral looping or c-looping, formation of the primitive s-loop, and formation of the mature s-loop (Manner, 2004). Asynchronous contractions begin with the formation of the linear tubular heart, but become coordinated once looping starts to propel blood (Goenezen et al., 2012). At the C-loop stage, the once straight heart tube becomes a looped structure with a morphologically distinct primitive atrium, primitive ventricle, and primitive OFT. During S-looping, regional wall thickenings of cardiac jelly form endocardial cushions (ECs) in the AV canal and OFT regions. At this stage, ECs function as primitive valves by closing off the lumen during contraction, facilitating unidirectional pulsatile flow (Butcher et al., 2007).
Ventricular segment trabeculation is initiated toward the last stages of looping. During cardiac trabeculation, cardiac jelly is displaced from the ventricle's outer curvature and endocardial extensions grow toward the myocardial layer (Moorman and Christoffels, 2003). These extensions then form a network of luminal projections called trabeculae. Trabeculae consist of myocardial cells covered by an endocardial layer (Samsa et al., 2013); greatly increasing surface area, as well as myocardial mass, and wall stiffness (Yang et al., 1994). The increase in surface area boosts nutrition and oxygen uptake in the embryonic myocardium prior to coronary vascularization without increasing heart size. This enhancement leads to an increase in cardiac output as well (Liu et al., 2010). Trabecular compaction then follows, and contributes to the formation of the ventricular septum and to the thickening of the ventricular compact layer (Goenezen et al., 2012). As the compact layer grows in size and complexity, it replaces trabeculae as the major contractile force. Compact myocardium ultimately provides most of the myocardial mass and therefore contractile force in the mature heart (Wessels and Sedmera, 2003). During chamber growth, the ventricular conduction system differentiates as well. This differentiation is marked by an apparent reversal in the sequence of ventricular activation (Chuck et al., 1997). The immature base-to-apex pattern of epicardial activation switches to a mature apex-to-base pattern (Gourdie et al., 2003).
Septation of the atrium and the ventricle transforms the heart into a four-chambered organ. Septation occurs via trabecular compaction and the fusion of ECs (van den Berg and Moorman, 2009). ECs are protrusions inside the AV canal and OFT and work as primitive valves, performing a sphincter-like function until mature valves develop (Butcher et al., 2007). ECs consist of mesenchymal progenitor cells with overlying endocardial endothelial cells. The EC mesenchyme is derived from the AV canal and OFT endocardial endothelial cells after an endothelial-to-mesenchymal transition (Person et al., 2005), with minor and transient contributions from epicardial or neural crest derived cells. The AV ECs fuse and condense into mitral and tricuspid valve leaflets whereas the distal OFT ECs are remodeled into the semilunar valve leaflets (Combs and Yutzey, 2009).
The underlying mechanics of heart tube formation is not fully understood. Extracellular matrix (ECM) fiber interactions and cell adhesions are important early mechanical stimuli. Through fibronectin and mesodermal cell tracking, Zamir et al. showed that convective tissue motions contribute significantly to total cell displacement (Zamir et al., 2006). Cell autonomous displacements (i.e., cell movement relative to the ECM) decreased gradually after egression from the primitive streak, with caudual cells actively moving faster than cranial cells. The tracking of ECM tags relative to endocardial progenitors, however, revealed little active motility of endocardial cells relative to its surrounding ECM (Aleksandrova et al., 2012). In a study that investigated migration of the bilateral heart fields to the embryonic midline, Vamer et al. showed that endodermal shortening around the anterior intestinal portal is responsible for movement of cardiac mesoderm and endoderm toward the midline (Varner and Taber, 2012). Endodermal shortening is driven by cytoskeletal contraction, and can be arrested by blebbistatin, a myosin-II inhibitor. Shortening was shown to decrease both tissue stiffness and mechanical tension. These findings highlight the importance of large-scale tissue deformations in transporting cells to their appropriate positions during heart tube formation.
As a cell engages ECM and actively applies force on its surrounding matrix, it senses local elastic resistance of the ECM and nearby cells (Buxboim et al., 2010). ECM or tissue elasticity has an influential role in regulating cell functions such as migration and proliferation (Hadjipanayi et al., 2009; Winer et al., 2009). Cell migration and proliferation are modulated by cell-generated, actin-myosin forces that both depend on and influence matrix elasticity. Embryonic tissue elasticity may therefore be more strongly influenced by cellular cytoskeleton than in adult. Forces exerted on the cell by the ECM may affect the expression of matrix-sensitive genes (Buxboim et al., 2010). Cells can also respond to the stiffness of their surrounding matrix. For example, cardiomyocyte contraction and myocyte architecture development is optimized on substrate elasticity mimicking that of the developing myocardial environment (Engler et al., 2008; Majkut et al., 2013). This data suggests that, cell-ECM interactions are important to the placement of cardiac progenitor cells and their subsequent differentiation during cardiogenesis.
Forces initiating the furling of cells into the primitive heart tube are not well-understood. One hypothesis is that the cylindrical bending of a sheet of epithelial cells could result in the formation of a hollow tube (Taber, 1998). The key to this theoretical model is the asymmetric contraction of actin microfilaments surrounding the apices of the epithelial cells, causing the normally cylindrical cells to become more wedge shaped. This shape change eventually forces the apical surface of the sheet into the inner curvature of the developing tube. The ability of individual cells to resist apical–basal elongation during this contraction would result in a more substantial bending of the sheet (Bartman and Hove, 2005). Confocal laser scanning microscopic studies have revealed that the early cardiac epithelial cells do in fact have such a microfilament arrangement (Shiraishi et al., 1992), and pharmacological inhibition of the microfilament results in failed heart tube formation (Ettensohn, 1985).
Spontaneous action potentials can be detected in the newly formed chick heart tube at Hamburger–Hamilton (HH) stage 9 before myocyte contraction begins (Kamino et al., 1981). Starting around early HH10, the tubular heart beats irregularly and slowly without effective blood flow. By the end of stage HH10, a clear heart rhythm is present (Hogers et al., 1995). The frequency of this rhythm increases and blood flow can be detected as early as HH11 (Granados-Riveron and Brook, 2012). Effective blood flow starts during looping at HH12 and at which time a peristaltic wavelike contraction pattern is observed along the length of the heart tube, from the atrium to the OFT (Taber, 2006). By HH20, a vigorous circulation is established (Granados-Riveron and Brook, 2012).
One would think that the embryonic heart starts beating in order to pump blood for convective transport. However, chicken embryos do not show hemoglobin-mediated transport of oxygen until HH20 (Cirotto and Arangi, 1989). It can therefore be concluded that diffusion is a sufficient means of transport for oxygen, nutrients, metabolic wastes, and hormones in the early chick embryo (Burggren, 2004). Total elimination of cardiac ejection by complete ligation of the cardiac OFT in HH20–HH23 chick embryos has no significant effect on O2 consumption in the 4 h after surgical manipulation (Burggren et al., 2004). If it is not the nutrition transport, than what is the purpose of the initial heart beat?
Although the role of early cardiac contraction is unclear, recent evidence suggests that hemodynamics generated by contraction of cardiomyocytes acts to drive cardiogenesis on a molecular and cellular level (Granados-Riveron and Brook, 2012). Lucitti et al. showed that lower hematocrit results in lower shear stress and defective vessel remodeling in the embryonic mouse (Lucitti et al., 2007). These defects were subsequently rescued through the resotration of control viscosity. These results support that hemodynamic force is necessary and sufficient to induce vessel remodeling. To study the effects of hemodynamic forces on heart development, flow was occluded at either the cardiac inflow or OFT of zebrafish embryos [37 hours-post-fertilization (hpf), straight heart tube stage] in order to reduce shear stress in the AV canal (Hove et al., 2003). Inflow and outflow obstruction resulted in hearts with an abnormal third chamber, diminished looping and impaired valve formation at later stages. Hove et al. stipulate that the observed cardiac abnormalities were the result of altered intracardiac flow patterns that reduced local WSS acting on the endocardial cells. Bartman et al. investigated the respective roles of myocardial function (contraction) and blood flow induced shear stress in EC development for zebrafish embryos (Bartman et al., 2004). In their experiments, they varied the degree of myofibril inhibition using different concentrations of 2,3-butanedione monoxime and studied the resulting effects on blood flow. Myocardial function decreased in dose dependent manner with myofiber inhibition, and the percentage of embryos that formed endocardial rings at 48 hpf also decreased. Interestingly, at very high concentrations, blood flow was completely abolished and no shear stress existed though myocardial function persisted, with 58% of these embryos still forming an endocardial ring. These studies did not assess the potential for changes in secreted molecule profile from the myocardium, but the results support that both shear stress and myocardial function play a role in cardiogenesis (Mironov et al., 2005).
Manning and MacLachlan showed that isolated tubular hearts could bend but not rotate during the c-loop phase of cardiac development (Manning and McLachlan, 1990). Their results suggest the bending component of C-looping is intrinsic to the heart tube, while torsion is mainly driven by extrinsic forces (Wyczalkowski et al., 2012). Actin polymerization-driven myocardial cell shape changes have been found to contribute to the bending of the heart tube (Manasek et al., 1972; Latacha et al., 2005). The torsional component of C-looping is largely due to forces from its encapsulating membrane, the splanchnopleure (SPL) (Taber et al., 2010). When the SPL is removed from HH11 chick hearts, they no longer maintain their looped configuration (Voronov and Taber, 2002; Voronov et al., 2004). External forces may not be alone in guiding cardiac rotation. When the SPL is removed from embryonic chick hearts around the onset of looping (HH10), torsion is initially suppressed, but restored several hours later (Nerurkar et al., 2006). Delayed torsion was found to coincide with increased myocardial stiffness, suggesting that delayed torsion is caused by an abnormal cytoskeletal contraction. Overall, these results show that while intrinsic cytoskeletal forces contribute to the dextral bending component of C-looping, both intrinsic and extrinsic forces are required for the rotational component of C-looping.
In the tubular heart, cyclic generation of traveling mechanical waves sweep from the venous to arterial end generating a unidirectional blood flow. These traveling mechanical waves were traditionally considered myocardial peristaltic waves (Xavier-Neto et al., 2007) and tubular embryonic heart was accepted to work like a technical roller peristaltic pump (Manner et al., 2010). Experiments on zebrafish embryos offer an alternative theory (Forouhar et al., 2006): (I) While peak flow velocity generated by a roller peristaltic pump corresponds to the speed of a compression wave, peak ventricular inflow velocity of an embryonic zebrafish heart recorded exceeds the speed of a traveling contraction wave. (II) In the zebrafish embryo, the relationship between cardiac contraction frequency and the flow rate is non-linear and exceeds the maximum flow rate possible for a roller peristaltic pump. (III) Roller peristaltic pumps are marked by non-stationary sites of active compression that move in a uniform direction along the length of a flexible tube whereas the early embryonic zebrafish heart possesses a single stationary center of active myocardial contractions at its venous pole. Several other studies support these findings. The tubular embryonic heart does not function as a technical roller peristaltic pump (Hu and Clark, 1989; Butcher et al., 2007), but may be considered a valveless “Liebau pump” (also known as suction or impedance pump) (Manner et al., 2010). Valveless pumping can be achieved experimentally by periodically compressing the asymmetric site of a fluid filled tube made up of stiff and soft elastic sections (Liebau, 1955, 1956; Ottesen, 2003; Hickerson et al., 2005). Here, periodic compression at an asymmetric site in the soft elastic region leads to unidirectional flow. In order to be considered Liebau driven flow, the tube must have a flexible wall and a finite length with active compression stemming from a small asymmetric non-central section. Flow generated by Liebau pumps is typically pulsatile (Manner et al., 2010).
Forouhar et al. identified a single site of active myocardial contractions as well as a non-linear relationship between contraction frequency and flow rate in the embryonic zebrafish heart (Forouhar et al., 2006). They concluded that tubular embryonic hearts work as Liebau pumps rather than peristaltic pumps. Previous work by our group investigated pumping mechanisms at various stages of embryonic chick development (Butcher et al., 2007). At HH17 blood velocity exceeds that of tissue velocity, and wave propagation is initiated by a single myocardial source, supporting the Liebau pump hypothesis. At HH25 the embryo utilizes a piston-pumping mechanism. Piston pumping is defined as a volume change–driven propulsion of fluid, with the orifice working to throttle the outlet flow. HH21 embryos don't fall into one pumping category, but rather act as a transition stage between two pumping styles. This same transition can be seen in the pumping mechanism of zebrafish embryonic hearts at 36 hpf (Johnson et al., 2013).
Although the early embryonic heart does not function as a technical roller peristaltic pump, it still incorporates peristaltic mechanisms. Taber et al. showed that peristaltic heart tubes can generate pulsatile blood flow as a result of the presence of ECs at the inflow and outflow of the ventricular loop (Taber et al., 2007). Traveling contractile waves generate pressure and flow values an order of magnitude greater in tubes with valvular protrusions. In their peristalsis model with ECs, flow velocities exceeded the contractile wave speed by 75%. These results highlight the importance of ECs in pulsatile pump force generation.
Damon et al. measured a gradient of strain across the chamber wall, with strain greatest along inner layers and lowest in the compact myocardium (50% elongation vs. 20% elongation) (Figure 3A) (Damon et al., 2009). Cell proliferation was found to be greatest in the compact myocardium and lowest in the trabeculae (Sedmera and Thompson, 2011). A separate study using zebrafish embryos found that erbb2, a regulator of myocyte proliferation, contributes to ballooning and chamber growth during heart development (Liu et al., 2010). Together these results suggest strain magnitudes may alter proliferation rates, retarding proliferation with an increase in strain magnitude. Indeed WSS and strain play a major role in cardiogenesis. Many computational models modulate growth as a function of stresses and strains (Taber, 2009; Buskohl et al., 2012b; Rugonyi, 2013).
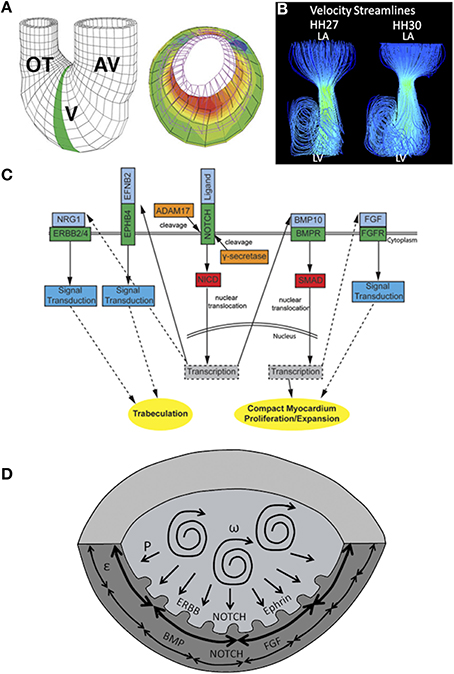
Figure 3. Mechanical forces driving chamber development. (A) A computational model showing that inner trabecular layers undergo greater strain that outer compact layer in ventricular chamber. AV, atrioventricular canal; V, ventricle; OT, outflow tract. Adapted from Damon et al. (2009). (B) Velocity streamlines in left AV canal for HH27 and for HH30 chicken embryos, generated with computational fluid dynamics. Strong vortices develop in the ventricular cavity, possibly necessary for the expansion of the chambers. Adapted from Yalcin et al. (2011). (C) Signaling pathways in ventricular chamber maturation. NOTCH, ERBB, and Ephrin play role in trabeculation whereas NOTCH, BMP, and FGF play role in compact myocardium development. Adapted from Samsa et al. (2013). (D) Proposed ventricular chamber development model. Circulatory flows cause expansion of the ventricles. Pressure induces a gradient in circumferential strain in the trabeculae and compact myocardium. This gradient cause a difference in myocyte proliferation as well as different gene expression profiles. P represents pressure, ω represents circulations, and ε represent circumferential strain.
Cell morphologies have also been shown to contribute to the ballooning process of heart chambers. Ballooning involves the expansion and bulging of the linear walls of the looped heart tube into bean-shaped chambers, with a convex outer curvature and a concave inner curvature (Gjorevski and Nelson, 2010). High-resolution imaging of the developing zebrafish hearts revealed that cells of the outer curvature flatten and elongate relative to those of the inner curvature, which maintain a cuboidal shape. The characteristic curves of cardiac chambers are due to these differences in cellular morphologies (Auman et al., 2007). Given that the inner curvature of the early heart is exposed to higher WSS than the outer curvature (Hierck et al., 2008), one may suppose shear stress levels affect cellular morphologies at these locations. Employing zebrafish mutants with functional deficiencies, Auman et al. showed that in addition to blood flow forces, contractility also independently regulate cell shape changes in the emerging ventricle (Auman et al., 2007). Blood flow, an external force, promotes ventricular cell enlargement and elongation whereas contractility, an internal force, restricts ventricular cell size and elongation. Thus, the acquisition of normal cardiomyocyte morphology and therefore chamber morphology, requires a balance between extrinsic and intrinsic physical forces. Recently, Chi et al. showed that, independent of contractile and hemodynamic forces, as an intrinsic force, electrical forces may also affect cardiomyocyte morphology and subsequent chamber morphology (Chi et al., 2010).
Finally, previous work by our group showed that, in addition to shear stress levels and profiles, blood flow patterns evolve within developing hearts. Specifically, flow remains laminar in the HH17 heart, and after septation at HH27, jet flow in the AV canal allows strong vortices to develop in ventricular cavities. The emergence of these vortices coincides with ventricular expansion, suggesting vortex formation may affect chamber maturation (Figure 3B) (Yalcin et al., 2011). A summary of gene expression pathways that play a role in chamber maturation is given in Figure 3C. Figure 3D proposes a model of chamber development based on the aforementioned studies.
In an effort to understand the contribution of hemodynamic forces on the development of CHDs, many animal models and experimental techniques have been developed. Two common models of flow manipulation in the chick embryo are conotruncal banding (CTB) and left atrial ligation (LAL). In CTB, a knot is tied around the conotruncus region of the OFT at HH18 in order to induce an outflow flow constriction (Clark et al., 1984). In LAL, the left atria is partially ligated at HH20–21, obstructing flow to the ventricle (Sedmera et al., 1999). CTB results in an immediate increase in pressure and is therefore considered an increased afterload model. LALs shift blood flow to the right side of the heart and are considered a model of increased right ventricular and decreased left ventricular preload (Sedmera et al., 1999).
Tobita et al. measured flow velocities in the AV canal and examined wall deformation patterns in the right and left ventricle (RV and LV, respectively) following LAL (Tobita and Keller, 2000). Decreases in max as well as average velocity were evident immediately following LAL. At HH31 (post-septation), average RV inflow velocity was found to be higher in LAL embryos, while maximum LV inflow velocity was lower. These results suggest flow is redirected to the right side of the heart after LAL. Increased right ventricular flow accelerated the onset of RV circumferential strain patterns, and decreased flow in LV abolished these patterns. Kowalski et al. investigated immediate alterations in blood flow patterns and WSS levels following LAL through the use of a computational fluid dynamics (CFD) model (Kowalski et al., 2014). Intracardiac flow patterns are immediately altered following LAL, resulting in a decrease in WSS levels at the left AV canal as well as at the left side of the common ventricle (Kowalski et al., 2014). WSS level decreased from 10 dynes/cm2 to 5 dynes/cm2 at these locations for LAL embryos. Using optical coherence tomography, Rugonyi et al. studied immediate changes in blood flow velocities and wall motions in the OFT region of HH18 chick embryos following CTB (Rugonyi et al., 2008). CTB resulted in an increase in the blood velocity in the banded part of the OFT due to flow constriction at that region. Strain patterns revealed that mechanically altered (banded) embryos exerted more energy to maintain growth.
Sedmera et al. investigated morphological abnormalities due to hemodynamic alterations resulting from LAL and CTB (Sedmera et al., 1999). Following CTB, a thickening and dilation of the compact myocardium and trabeculae in the LV was observed. LAL resulted in hypoplasia of the left heart structures, most likely due to decreased flow, with compensatory overdevelopment on the right side, most likely the result of increased flow. LAL was shown to decrease myocyte proliferation rates in LV (Sedmera et al., 2002). DeAlmeida et al. also showed that partial clipping of the right atrial appendage increases blood flow to LV leading to an increase in chamber volume and myocardial mass based on myocyte proliferation in both LAL and normal embryos (deAlmeida et al., 2007). Similarly, addition of fibroblast growth factor-2 (FGF2), a known stimulant of embryonic myocyte division was shown to increase cellular proliferation and abate the diseased phenotype (deAlmedia and Sedmera, 2009).
Effects of hemodynamic alterations from CTB and LAL on the developing conduction system were investigated by Reckova et al. (2003). CTB resulted in premature emergence of the mature apex-to-base activation sequence, whereas LAL resulted in delayed transition to a mature activation sequence. Biomechanics play a critical role in induction and patterning of the cardiac conduction system. Sedmera et al. studied the effects of hemodynamic alterations on the expression of endothelin converting enzyme (ECE) protein, which is involved in the inductive recruitment of Purkinje fibers in embryonic chick conduction systems (Sedmera et al., 2008). Following LAL, ECE expression was down-regulated in LVs with decreased preload and up-regulated in RVs with increased preload. Hearts cultured without hemodynamic loading showed decreased activation of the conduction pathway (Sankova et al., 2010). This primitive phenotype was rescued following artificial loading of the ventricles via a droplet of silicone oil. Together these findings suggest that loading is a necessary component of conduction system formation and maturation.
During cardiac looping, TBX3 (Moorman et al., 2004) and Notch1 (Luna-Zurita et al., 2010) act to localize myocardial-endocardial signals to the AV and OFT regions. In the first stage of this process, a subset of endocardial cells lining these two zones transform into a mesenchymal phenotype and invade the cardiac jelly, a process known as endocardial to mesenchymal transformation (EMT) (Runyan and Markwald, 1983; Eisenberg and Markwald, 1995; Camenisch et al., 2002; Tavares et al., 2006; Butcher and Markwald, 2007; Combs and Yutzey, 2009). The invasive, proliferating mesenchyme progressively remodels the hyaluronan matrix, replacing it with proteoglycans, matricellular proteins, and eventually structural proteins such as collagen I (Norris et al., 2004; Person et al., 2005). These amorphous, compliant, cellularized masses, now dubbed cushions, continue to grow and extend into the lumen space (Schroeder et al., 2003)
Two cushions (superior and inferior) form initially in the AV canal at HH16 (E9.5 in mouse), followed by the appearance of two mural/lateral cushions on the left and right side of the AV canal at HH19 (Snarr et al., 2008). The superior and inferior AV cushions fuse together by HH26 (E12 in mouse) forming a septation of the AV canal that joins with the ventricular septum and the protruding atrial cap. The lateral portions of this fused mass undergo continual remodeling to form valves, as do the left and right mural cushions. During the remodeling of primitive AV valves into fibrous leaflets, the AV myocardium forms a fold at its junction with the ventricular myocardium creating a substrate on which the AV cushions can extend. The cushions extend along their substrate through the expansion of a proliferation zone in the subepithelial portion of the AV cushions (Sugi et al., 2003). Fenestrations develop as a result of the elongating cushions and the ventricular tissue underneath the cushion tissue delaminates, resulting in primitive leaflets that are continuous with developing papillary muscles and simultaneous expansion of the ventricular OFT (Wenink and Gittenberger-de Groot, 1986). The myocardial tissue of the AV valves disappears and they condense into fibrous leaflets (de Lange et al., 2004). Thin strands of elongated muscle remain tethered to the valve tissue with thickened trabecular aspects on the ventricular myocardial wall. These structures become the tendinous chords and papillary muscles of the mature valve (Icardo and Colvee, 1995). Epicardially derived fibroblasts have also been shown to contribute to the mural AV leaflets in developing murine hearts (Wessels et al., 2012).
The OFT differs in that endocardial cells along the entire lumen undergo EMT. Paired bulges emanating in proximal (just outside the right ventricle) and distal zones (just after a “dogleg” bend in the OFT) become cushions around HH22/E10, while the rest of the cardiac jelly regresses. The proximal/conal cushions are alternatively referred to as the septal/sinistroventral and parietal/dorsodextral ridges (Ya et al., 1998; Qayyum et al., 2001). A third distal cushion ridge subsequently forms (HH25/E11). These growing cushions fuse at the midline, forming two tortuous lumens. Between HH26/E11.5 and HH30/E13, the distal dorsal cushion of the OFT aligns with the proximal left cushion along the inner heart curvature, continuous with the superior cushion of the AV canal. Simultaneously, a wishbone shaped ridge of mesenchyme invades the OFT in a spiraling pattern, separating it into left and right portions and dividing the outflow cushions into two groups of three. While the fused proximal cushions myocardialize and form the muscular infundibulum, which separates the right and left ventricular outlets, the distal cushions become the rudiments of the pulmonary and aortic outlet valves (Qayyum et al., 2001). For the remodeling of primitive outflow valves into fibrous leaflets, unlike the AV valves, which formed through delamination from the muscular walls, valves of the OFT form through a process of excavation or hollowing of the cushion's aortic side. Cushion excavation begins at HH29/ED13 with a small depression in the arterial face of the cusps. The endothelium lining the aortic surface of the valves becomes thickened with rounded cells that flake and undergo apoptosis, while the ventricular epithelium remains flat and elongated (Garcia-Martinez et al., 1991). The deepening furrow condenses the fibrous matrix around it, creating thin cusps of tissue that are attached in an arc pattern called the commissures (Butcher and Markwald, 2007).
Blood flow guides cardiac morphogenesis, sculpting tissue by promoting growth in response to increased demands. Less than 48 h after incubation, the presence of two blood streams is apparent in the embryonic chicken heart. A spiraling complex is created, as the force of the larger stream pulls the smaller stream around it, changing the mechanical environment of the developing heart (Jaffee, 1965). The rapid growth of the endothelial tube inside the early heart is a result of an increase in blood pressure (Chang, 1932). In this way blood flow, shear stress and stretching forces are thought to influence the duration of vessel growth and their morphological characteristics (Taber and Eggers, 1996). During this time of heart formation, shear stress is greatest in the inner curvature and sites of lumen constrictions, corresponding to the AV canal and OFT where the ECs form and develop into functioning valves (Groenendijk et al., 2004). The velocity profile of flow through the primitive valves begins with a Poiseuille parabola (HH17), before resembling plug flow; cushions arise shortly after peak inflow velocity and extend perpendicularly to the direction of flow (Figures 4A,B, left) (Yalcin et al., 2011; Bharadwaj et al., 2012). As the heart continues to grow, the energy extended in pulsatile flow increases from one-third to two-thirds of total energy between HH18 and 29 (Clark et al., 1986). A jet profile characterizes the AV region of HH27 chick embryos (Figure 4A, right). Figure 4A, right, highlights circulating eddies, which form beneath the cushion surface. WSS levels increase from 80 dynes/cm2 at HH23 to 250 dynes/cm2 at HH27 (Figure 4B). As the embryo's pressure, cardiac output and WSS increase, the cushions elongate to form thin fibrous leaflets with increased ECM proteins and greater mechanical stiffness (Butcher et al., 2007; Biechler et al., 2010; Buskohl et al., 2012a). Strain energy density increases linearly with AV valve leaflet length (Buskohl et al., 2012a).
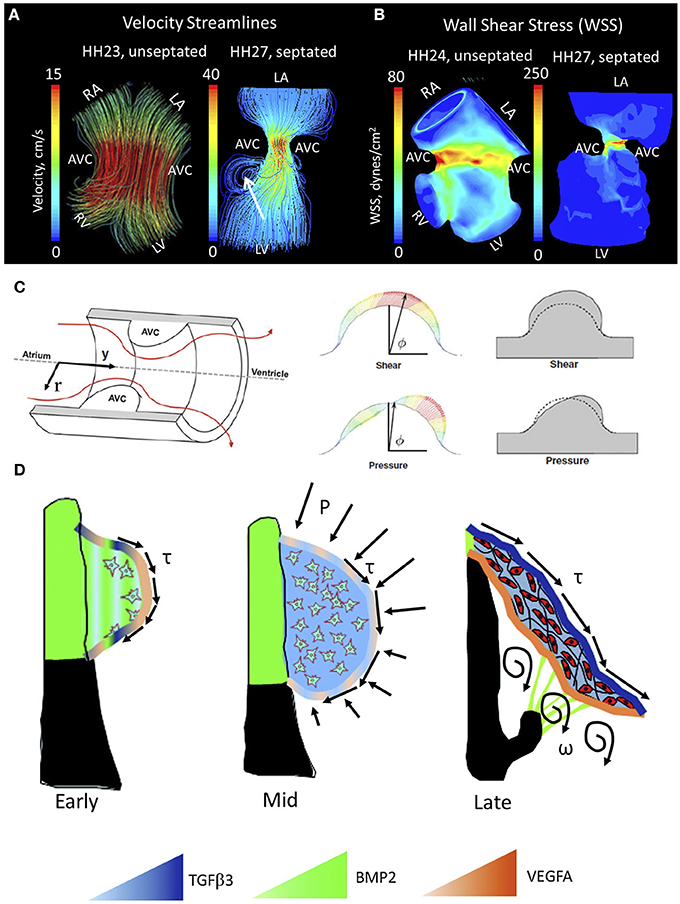
Figure 4. Mechanical forces driving AV valve morphogenesis. (A) Velocity streamlines in AV canal for HH23 (unseptated) and for HH27 (septated) chicken embryos, generated with computational fluid dynamics. In HH23, flow profile is laminar parallel streamlines, whereas in HH27 it is jet flow with circulating eddies beneath AV cushions (shown with arrrow). LA, left atria; RA, right atria; LV, left ventricle; RV, right ventricle; AVC, atrioventricular cushion (B) WSS levels for HH23 and HH27 embryos. High stress levels are localized to the mid cushion region. Adapted form Yalcin et al. (2011). (C) Hemodynamic-driven AV cushion growth and remodeling computational model. Based on this model, the pressure distribution on the AV cushion cause leaflet-like elongation in the direction of flow, whereas shear tractions regulated the remodeling of tissue near the cushion surface suggesting WSS is important in mechanotransduction. Adapted from Buskohl et al. (2012b). (D) Proposed AV valve morphogenesis model. WSS on cushion surface regulate gene expression for valve development. Pressure on the cushion surface cause leaflet-like elongation and circulatory flows beneath the cushion helps detachment of the leaflet from the myocardial wall. Growth factor expression levels were indicated via color gradation as shown with colored triangles. Adapted from Chiu et al. (2010). τ represents shear stress, P represents pressure, and ω represents circulations.
While the importance of biomechanics in the formation of the valve leaflets has been acknowledged for some time, only recently have individual roles for pressure and WSS been proposed. Buskohl et al. created a computational model in which a finite element model was coupled to a fluid dynamics model, highlighting interaction between the two (Figure 4C) (Buskohl et al., 2012b). Buskhol's iterative computational approach involved determining pressure and velocity profiles, transferring flow-induced pressure and shear tractions on the AV cushion surface, and updating the fluid simulations after inelastic deformation had taken place. Fluid shear tractions were found not to significantly alter cushion volume, but rather functioned as the driver for cushion surface remodeling. While pressure was found to be negligible in terms of tissue deformation, shear traction forces from the top-center cushion region greatly contributed to tissue elongation. Pressure was responsible for tissue resorption on the inflow side of AV cushions and expansion on the outflow side (Buskohl et al., 2012b). Regulation of the cushion surface, particularly the leading edge of valve leaflets, may be heavily influenced by mechanotransduction.
Altered hemodynamic flow patterns during critical periods of development have been shown to lead to a variety of cardiac abnormalities, many of which influence valve formation (Hogers et al., 1997; Sedmera et al., 1999; Hove et al., 2003). Maintenance of circulatory energy efficiency and pressure are critical for development (Lucitti et al., 2005). Sedmera et al. found that alterations in ventricular load influenced AV valve morphogenesis as well as trabeculation patterns (Sedmera et al., 1999). In embryos with increased ventricular afterload (CTB), right AV valve morphology no longer took the form of a muscular flap but rather resembled a bicuspid structure. Previous work by our group showed that photoablation of the superior AV cushions of HH24 chick embryos immediately altered AV canal hemodynamics (i.e., increased regurgitation), resulting in stunted ventricular and valvular growth in 48 h (Yalcin et al., 2010). Shear stress and shear stress-induced or repressed gene expression are important factors in remodeling of the cardiac cushions (Hove et al., 2003; Groenendijk et al., 2005; Vermot et al., 2009). Hove et al. attributed abnormal third chamber development in mechanically perturbed zebrafish embryos to a reduction in shear stress on ECs (Hove et al., 2003). Bartman et al. argue observed phenotypes were not the result of a decrease in shear alone. Their 2004 study found that 58% of zebrafish embryos treated with a myofibril inhibitor (2,3-butanedione monoxime) were still able to form an endocardial ring, affirming that blood flow is not required for the initial steps of cushion formation (Bartman et al., 2004).
While hemodynamics undoubtedly plays a large role in valvulogenesis, the role of hemodynamic signaling remains a point of contention. Though changes in shear stress have been found to presage the development of cardiac malformations (Hogers et al., 1997, 1999; Hove et al., 2003; Yashiro et al., 2007), other studies claim myocardial function surpasses shear stress as a major epigenetic factor (Bartman et al., 2004). Endocardial cells lining the luminal cushion surface may promote valvular morphogenesis by coupling mechanical stimuli and molecular signaling pathways (Butcher et al., 2007).
A number of studies have demonstrated the shear sensitivity of valvular morphogens in vivo. Endothelin-1 (ET-1) and endothelial nitric oxide synthase (NOS-3) are shear stress responsive genes linked to cardiovascular development by knockout mice that display a wide range of cardiovascular defects (Yanagisawa et al., 1998). Critical periods of cardiovascular remodeling (HH20–HH30) were marked by ET-1 and KLF2/NOS-3 restriction to narrow sites (Groenendijk et al., 2004). ET-1 was downregulated by shear stress, while KLF2 and NOS-3 were upregulated by shear. Observed patterns changed with vitelline vein obstruction and cardiovascular malformations ensued (Groenendijk et al., 2005). In addition to the aforementioned genes, the expression patterns of potent valvular morphogens, such as transforming growth factor β (TGFβ), bone morphogenetic protein (BMP), and vascular endothelial growth factor (VEGF) are spatially and temporally restricted in a manner that suggests hemodynamic regulation (Figure 4D) (Butcher and Nerem, 2007; Chiu et al., 2010). TGFβ appears in the endocardium of the valve forming regions around HH20, when flow transitions from its laminar poiseuille flow to more plug-like flow and rapidly increases in velocity (Yalcin et al., 2011). In vitro analyses suggest that TGFβ3 induces cell migration, invasion, and matrix condensation; BMP2 induces invasion; VEGFA inhibits invasion but increases migration (Tan et al., 2011). TGFβ3 was found to induce myofibroblastic differentiation in a dose-dependent manner, whereas VEGFA and BMP2 did not, suggesting that hemodynamic forces work through these transcription factors in the remodeling of the AV valves. Using an in vitro system, Tan et al. were able to show the flow-regulated development of the fibrous AV valves is dependent on rhoA expression (Tan et al., 2013). This body of work is depicted in Figure 4D.
The great vessels are derived from the pharyngeal arch arteries. A total of six arch pairs sequentially emerge, regress, or remodel throughout OFT development. While arch arteries I and II ultimately regress, the third arch pair forms the mature brachiocephalic arteries. The mature aortic arch is derived from the right fourth arch in chicken and the left fourth arch in mammalian embryos (Wang et al., 2009). The sixth arch pair forms pulmonary artery and ductus arteriosus. The formation of the truncus arteriosus separates the pulmonary artery and right ventricular OFT from the aorta and left ventricular OFT posteriorly (Qayyum et al., 2001; Hu et al., 2009).
Hemodynamic forces initiate extensive remodeling of the symmetric aortic arch system in a highly asymmetric fashion (Hu et al., 2009). Yashiro et al. investigated the mechanisms behind regression of the right VI aortic arch and persistence of left VI arch in mice (Yashiro et al., 2007). Using a variety of experimental and genetic mutant models, they found that, the genetic programme, including the expression of Pitx2, induces a dynamic morphological change in the OFT, which in turn generates a differential distribution of blood flow. Mice lacking the asymmetric enhancer PITX2 failed to adopt the spiral structure of the OFT and in these embryos OFT remained linear. Changes in OFT geometry led to differential flow distribution, successive signaling abnormalities and further changes in geometry. In normal embryos, increased blood flow in the left VI arch sufficiently induces PDGFR and VEGFR2 signaling and subsequently maintains arterial structure. Decreased flow in the right VI arch artery leads to vessel regression. Wang et al. discovered that increases in WSS corresponded with increases in arch artery diameters in day 3 (HH18) and day 4 (HH24) chick embryos (Wang et al., 2009). CFD analysis revealed that HH21 embryos had elevated, rather than intermediate WSS compared to both HH18 and HH24 embryos (Kowalski et al., 2013) suggesting periods of vascular remodeling may be preceded by acute increases in WSS. Hu et al. showed that altered hemodynamic flow in the aortic arch arteries due to LAL was associated with a range of cardiac defects (Hu et al., 2009). Overall, these studies suggest hemodynamic forces play a significant role in asymmetric remodeling of aortic arch network. Characterization of aberrant hemodynamic flow may be a resource in understanding OFT and arch network abnormalities, which account for 50% of infants with a CHD (Roger et al., 2011).
The embryonic heart adapts ventricular geometry and function to optimize mechanical efficiency (Lin and Taber, 1995). Distinctions between gene and hemodynamic-related abnormalities are not well defined. Though chromosomes linked to specific defects have been identified, they rarely provide a full picture, with only 10–15 percent of ventricular outflow malformations loosely associated to a chromosomal abnormality (McBride et al., 2009). While genetic mutations are associated with CHDs, genes cannot be manipulated to produce CHDs in the same way as flow can be perturbed to produce disease phenotypes. The use of computational modeling in the study of abnormal development has led to a more thorough analysis of disease etiologies and their underlying mechanisms. In this section, the pathology of two major CHDs associated with abnormal hemodynamic pattering and valve formation are explored: bicuspid aortic valve (BAV) and hypoplastic left heart syndrome (HLHS).
BAV is the most common congenital anomaly of the heart, marked by two aortic valve leaflets rather than three. This two cusped configuration constrains the patient, as the free edges of the bicuspid valve are more straight than round and offer limited mobility. The leaflets are usually of unequal size with a raphe, or seam-like union, apparent in the larger leaflet (Yener et al., 2002). BAV is frequently associated with aortic valve stenosis, regurgitation and endocarditis, though these symptoms develop well after valve formation. Excessive length of one or both cusps results in abnormal contact which in turn leads to fibrous thickening that will later become diffuse and calcified. Stenosis usually develops in bicuspid valves containing no redundant cusp tissue, while valve incompetence is associated with redundancy and endocarditis. The large calcific deposits associated with BAV are unusual before the age of 30 and very prevalent thereafter (Roberts, 1970). In a 2003 study of 44 bicuspid aortic valves, BAV patients without significant stenosis or regurgitation were found to have a larger aortic annulus, aortic sinus and proximal ascending aorta when compared to normal tricuspid valves. The peak aortic velocity (Nkomo et al., 2003) and peak systolic wall velocity in the anterolateral region of the ascending aorta (Bauer et al., 2006) were also found to be higher in BAV patients than controls. This flow has been classified as helical, or flow composed of a forward component along the long axis of the aorta and a rotational component, moving circumferentially along that same axis (Bissell et al., 2013; Lorenz et al., 2014). A significant increase in absolute peak helicity is present during systole of BAV patients, with a substantially greater distribution of mean helicity in the aorta (Lorenz et al., 2014).
Recent advances in cardiac imaging, have allowed scientists to map these regions and collect 3D spatial visualizations of flow patterns over time (Bissell et al., 2013; Lorenz et al., 2014). In this way, temporal evolution of complex flow patterns over time can be studied and linked to aortic function. Bissell et al. found that patients with BAV presented with predominantly abnormal right-handed helical flow in the ascending aorta, larger ascending aortas, higher helical flow, elevated systolic angle and elevated systolic WSS. In their study of 69 BAV patients, left-handed helical flow, normal flow, and complex flow occurred in 4, 11, and 13%, respectively, whereas right-handed helical flow occurred in 72% of patients. Although flow patterns differed, distensibility, aortic strain, and pulse wave velocity of the aorta were similar across all groups. Flow abnormalities initiate aortopathy as a means of maintaining optimal WSS values (Bissell et al., 2013). Through the use of fluid structure interaction models, (Chandra et al., 2012) degree of leaflet calcification is linked to orifice area, oscillatory shear index and temporal shear magnitude. While the regular tricuspid and non-coronary BAV leaflets shared similar shear stress characteristics, the base of fused BAV leaflet fibrosa differed greatly. The temporal shear index of fused leaflets was heavily modulated by degree of calcification, with 6- to 16-fold increases seen over BAVs ranging from normal to severely calcified. Results support a mechano-sensored model of calcified aortic valve disease in the BAV patients (Chandra et al., 2012).
HLHS is characterized by acute underdevelopment of the left ventricle. No strong genetic correlation exists. In a study of 83 HLHS patients, nine had underlying chromosomal abnormalities, four had single gene defects, 10 had one or more extracardiac anomaly and two were patients of insulin-dependent mothers (Natowicz et al., 1988). Disease formation is thought to result from diminished flow to the left ventricle and aortic OFT. Retrograde aortic flow may play a role in impaired development of the aortic root and ascending aorta (Simpson and Sharland, 1997). Cardiac defects associated with HLHS include mitral valve hypoplasia or mitral stenosis coincident with left heart obstruction, hypoplastic left ventricle, aortic atresia, hypoplastic aorta, and coarctation of the aorta. Out of 96 HLHS patients, 12.5 percent exhibited dysplastic aortic valvular stenosis, 37.5 percent were found to have malfunctioning aortic and mitral valves, 50 percent presented with abnormal AV valves (Ilbawi et al., 2007). To date, the LAL remains the only experimental model to fully recapulate these disease phenotypes (Sedmera et al., 1999, 2002; Tobita et al., 2002). Changes in WSS as shown by Kowalski et al. may be responsible for underdevelopment of left heart structures (Kowalski et al., 2013). Variations in heart rate and AV inflow velocity were acute to non-existent in these models (Tobita et al., 2002). Mechanical manipulation of ventricular filling dates back to Harh et al. (1973). Harh et al. inserted a nylon device into the left AV canal, thereby reducing ejection volume from the ventricle to ascending aorta (Harh et al., 1973). Failure of the cushions to differentiate into fibrous leaflets led to hypoplasia. Narrowing resulted in a reversed atrial shunt. Though Harh et al. observed disease phenotypes, they were unable to fully characterize mechanical changes. Combining experimental results and computational models presents a promising way of understand the mechanisms through which changes may arise.
Mechanical forces are essential drivers of cardiac morphogenesis, transforming the linear heart tube into a multichambered unidirectional machine capable of adapting to environmental demands. CHDs arise when the heart is prevented from following the normal pathways of development; understanding the intricate mechanisms involved in heart development is necessary for the advancement of clinical solutions. The majority of CHDs result from improper positioning of the cardiac OFT, impaired remodeling of the cushions into valve leaflets, or abnormal remodeling of the arches into great vessels. Quantitative imaging modalities provide a gateway into elucidating CHD formation. Integrating these imaging modalities with CFD and growth modeling will greatly strengthen our inference of causative mechanical forces of CHDs, such as BAV and HLHS. Targeted surgical techniques, including laser ablation, can help scientists distinguish between the effects of flow and tissue deformation alone. In vitro 3D culture and bioreactor studies work to further our understanding of how specific mechanical forces influence multi-scale biological responses. As our insight into CHD etiology improves, so will our ability to effectively and targetedly restore proper remodeling of cardiac tissues.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by funding from the American Heart Association, the National Science Foundation, the LeDucq Foundation, and The Hartwell Foundation as well as by a Marie Curie International Reintegration Grant within the 7th European Community Framework Programme, (IRG276987 to Huseyin C. Yalcin), and by The Scientific and Technological Research Council of Turkey, TUBITAK (112M148 to Huseyin C. Yalcin and 112M895 to Huseyin C. Yalcin).
Aleksandrova, A., Czirok, A., Szabo, A., Filla, M. B., Hossain, M. J., Whelan, P. F., et al. (2012). Convective tissue movements play a major role in avian endocardial morphogenesis. Dev. Biol. 363, 348–361. doi: 10.1016/j.ydbio.2011.12.036
Auman, H. J., Coleman, H., Riley, H. E., Olale, F., Tsai, H. J., and Yelon, D. (2007). Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 5:e53. doi: 10.1371/journal.pbio.0050053
Bartman, T., and Hove, J. (2005). Mechanics and function in heart morphogenesis. Dev. Dyn. 233, 373–381. doi: 10.1002/dvdy.20367
Bartman, T., Walsh, E. C., Wen, K. K., McKane, M., Ren, J., Alexander, J., et al. (2004). Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol. 2:E129. doi: 10.1371/journal.pbio.0020129
Bauer, M., Siniawski, H., Pasic, M., Schaumann, B., and Hetzer, R. (2006). Different hemodynamic stress of the ascending aorta wall in patients with bicuspid and tricuspid aortic valve. J. Card. Surg. 21, 218–220. doi: 10.1111/j.1540-8191.2006.00219.x
Bharadwaj, K. N., Spitz, C., Shekhar, A., Yalcin, H. C., and Butcher, J. T. (2012). Computational fluid dynamics of developing avian outflow tract heart valves. Ann. Biomed. Eng. 40, 2212–2227. doi: 10.1007/s10439-012-0574-8
Biechler, S. V., Potts, J. D., Yost, M. J., Junor, L., Goodwin, R. L., and Weidner, J. W. (2010). Mathematical modeling of flow-generated forces in an in vitro system of cardiac valve development. Ann. Biomed. Eng. 38, 109–117. doi: 10.1007/s10439-009-9824-9
Bissell, M. M., Hess, A. T., Biasiolli, L., Glaze, S. J., Loudon, M., Pitcher, A., et al. (2013). Aortic dilation in bicuspid aortic valve disease: flow pattern is a major contributor and differs with valve fusion type. Circ. Cardiovasc. Imaging 6, 499–507. doi: 10.1161/CIRCIMAGING.113.000528
Bruneau, B. G. (2008). The developmental genetics of congenital heart disease. Nature 451, 943–948. doi: 10.1038/nature06801
Burggren, W., Khorrami, S., Pinder, A., and Sun, T. (2004). Body, eye, and chorioallantoic vessel growth are not dependent on cardiac output level in day 3-4 chicken embryos. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1399–R1406. doi: 10.1152/ajpregu.00086.2004
Burggren, W. W. (2004). What is the purpose of the embryonic heart beat? or how facts can ultimately prevail over physiological dogma. Physiol. Biochem. Zool. 77, 333–345. doi: 10.1086/422230
Buskohl, P. R., Gould, R. A., and Butcher, J. T. (2012a). Quantification of embryonic atrioventricular valve biomechanics during morphogenesis. J. Biomech. 45, 895–902. doi: 10.1016/j.jbiomech.2011.11.032
Buskohl, P. R., Jenkins, J. T., and Butcher, J. T. (2012b). Computational simulation of hemodynamic-driven growth and remodeling of embryonic atrioventricular valves. Biomech. Model. Mechanobiol. 11, 1205–1217. doi: 10.1007/s10237-012-0424-5
Butcher, J. T., and Markwald, R. R. (2007). Valvulogenesis: the moving target. Philos. Trans. R. Soc. Lond. B Biol. Sci. 362, 1489–1503. doi: 10.1098/rstb.2007.2130
Butcher, J. T., McQuinn, T. C., Sedmera, D., Turner, D., and Markwald, R. R. (2007). Transitions in early embryonic atrioventricular valvular function correspond with changes in cushion biomechanics that are predictable by tissue composition. Circ. Res. 100, 1503–1511. doi: 10.1161/CIRCRESAHA.107.148684
Butcher, J. T., and Nerem, R. M. (2007). Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 362, 1445–1457. doi: 10.1098/rstb.2007.2127
Buxboim, A., Ivanovska, I. L., and Discher, D. E. (2010). Matrix elasticity, cytoskeletal forces and physics of the nucleus: how deeply do cells “feel” outside and in? J. Cell Sci. 123, 297–308. doi: 10.1242/jcs.041186
Camenisch, T. D., Molin, D. G. M., Person, A., Runyan, R. B., Gittenberger-de Groot, A. C., McDonald, J. A., et al. (2002). Temporal and distinct TGFβ ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 248, 170–181. doi: 10.1006/dbio.2002.0731
Chandra, S., Rajamannan, N. M., and Sucosky, P. (2012). Computational assessment of bicuspid aortic valve wall-shear stress: implications for calcific aortic valve disease. Biomech. Model. Mechanobiol. 11, 1085–1096. doi: 10.1007/s10237-012-0375-x
Chang, C. (1932). On the reaction of the endocardium to the blood stream in the embryonic heart, with special reference to the endocardial thickenings in the atrioventricular canal and the bulbus cordis. Anat. Rec. 51, 253–265. doi: 10.1002/ar.1090510305
Chi, N. C., Bussen, M., Brand-Arzamendi, K., Ding, C., Olgin, J. E., Shaw, R. M., et al. (2010). Cardiac conduction is required to preserve cardiac chamber morphology. Proc. Natl. Acad. Sci. U.S.A. 107, 14662–14667. doi: 10.1073/pnas.0909432107
Chiu, Y. N., Norris, R. A., Mahler, G., Recknagel, A., and Butcher, J. T. (2010). Transforming growth factor beta, bone morphogenetic protein, and vascular endothelial growth factor mediate phenotype maturation and tissue remodeling by embryonic valve progenitor cells: relevance for heart valve tissue engineering. Tissue Eng. Part A 16, 3375–3383. doi: 10.1089/ten.tea.2010.0027
Chuck, E. T., Freeman, D. M., Watanabe, M., and Rosenbaum, D. S. (1997). Changing activation sequence in the embryonic chick heart. implications for the development of the his-purkinje system. Circ. Res. 81, 470–476. doi: 10.1161/01.RES.81.4.470
Cirotto, C., and Arangi, I. (1989). How do avian embryos breathe? oxygen transport in the blood of early chick embryos. Comp. Biochem. Physiol. A Comp. Physiol. 94, 607–613.
Clark, E. B., Hu, N., Dummett, J. L., Vandekieft, G. K., Olson, C., and Tomanek, R. (1986). Ventricular function and morphology in chick embryo from stages 18 to 29. Am. J. Physiol. 250, H407–H413.
Clark, E. B., Hu, N., and Rosenquist, G. C. (1984). Effect of conotruncal constriction on aortic-mitral valve continuity in the stage 18, 21 and 24 chick embryo. Am. J. Cardiol. 53, 324–327. doi: 10.1016/0002-9149(84)90447-8
Combs, M. D., and Yutzey, K. E. (2009). Heart valve development: regulatory networks in development and disease. Circ. Res. 105, 408–421. doi: 10.1161/CIRCRESAHA.109.201566
Damon, B. J., Remond, M. C., Bigelow, M. R., Trusk, T. C., Xie, W., Perucchio, R., et al. (2009). Patterns of muscular strain in the embryonic heart wall. Dev. Dyn. 238, 1535–1546. doi: 10.1002/dvdy.21958
deAlmeida, A., McQuinn, T., and Sedmera, D. (2007). Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ. Res. 100, 1363–1370. doi: 10.1161/01.RES.0000266606.88463.cb
deAlmedia, A., and Sedmera, D. (2009). Fibroblast growth factor-2 regulates proliferation of cardiac myocytes in normal and hypoplastic left ventricles in the developing chick. Cardiol. Young 19, 159–169. doi: 10.1017/S1047951109003552
de Lange, F. J., Moorman, A. F., Anderson, R. H., Manner, J., Soufan, A. T., de Gier-de Vries, C., et al. (2004). Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 95, 645–654. doi: 10.1161/01.RES.0000141429.13560.cb
Dyer, L. A., and Kirby, M. L. (2009). The role of secondary heart field in cardiac development. Dev. Biol. 336, 137–144. doi: 10.1016/j.ydbio.2009.10.009
Eisenberg, L. M., and Markwald, R. R. (1995). Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 77, 1–6. doi: 10.1161/01.RES.77.1.1
Engler, A. J., Carag-Krieger, C., Johnson, C. P., Raab, M., Tang, H. Y., Speicher, D. W., et al. (2008). Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J. Cell Sci. 121, 3794–3802. doi: 10.1242/jcs.029678
Ettensohn, C. A. (1985). Mechanisms of epithelial invagination. Q. Rev. Biol. 60, 289–307. doi: 10.1086/414426
Fahed, A. C., Gelb, B. D., Seidman, J. G., and Seidman, C. E. (2013). Genetics of congenital heart disease: the glass half empty. Circ. Res. 112, 707–720. doi: 10.1161/CIRCRESAHA.112.300853
Forouhar, A. S., Liebling, M., Hickerson, A., Nasiraei-Moghaddam, A., Tsai, H. J., Hove, J. R., et al. (2006). The embryonic vertebrate heart tube is a dynamic suction pump. Science 312, 751–753. doi: 10.1126/science.1123775
Garcia-Martinez, V., Sanchez-Quintana, D., and Hurle, J. M. (1991). Histochemical and ultrastructural changes in the extracellular matrix of the developing chick semilunar heart valves. Acta Anat. 142, 87–96. doi: 10.1159/000147166
Gjorevski, N., and Nelson, C. M. (2010). The mechanics of development: models and methods for tissue morphogenesis. Birth Defects Res. C Embryo Today 90, 193–202. doi: 10.1002/bdrc.20185
Goenezen, S., Rennie, M. Y., and Rugonyi, S. (2012). Biomechanics of early cardiac development. Biomech. Model. Mechanobiol. 11, 1187–1204. doi: 10.1007/s10237-012-0414-7
Gourdie, R. G., Harris, B. S., Bond, J., Justus, C., Hewett, K. W., O'Brien, T. X., et al. (2003). Development of the cardiac pacemaking and conduction system. Birth Defects Res. C Embryo Today 69, 46–57. doi: 10.1002/bdrc.10008
Granados-Riveron, J. T., and Brook, J. D. (2012). The impact of mechanical forces in heart morphogenesis. Circ. Cardiovasc. Genet. 5, 132–142. doi: 10.1161/CIRCGENETICS.111.961086
Groenendijk, B. C., Hierck, B. P., Gittenberger-De Groot, A. C., and Poelmann, R. E. (2004). Development-related changes in the expression of shear stress responsive genes KLF-2, ET-1, and NOS-3 in the developing cardiovascular system of chicken embryos. Dev. Dyn. 230, 57–68. doi: 10.1002/dvdy.20029
Groenendijk, B. C., Hierck, B. P., Vrolijk, J., Baiker, M., Pourquie, M. J., Gittenberger-de Groot, A. C., et al. (2005). Changes in shear stress-related gene expression after experimentally altered venous return in the chicken embryo. Circ. Res. 96, 1291–1298. doi: 10.1161/01.RES.0000171901.40952.0d
Hadjipanayi, E., Mudera, V., and Brown, R. A. (2009). Close dependence of fibroblast proliferation on collagen scaffold matrix stiffness. J. Tissue Eng. Regen. Med. 3, 77–84. doi: 10.1002/term.136
Harh, J. Y., Paul, M. H., Gallen, W. J., Friedberg, D. Z., and Kaplan, S. (1973). Experimental production of hypoplastic left heart syndrome in the chick embryo. Am. J. Cardiol. 31, 51–56. doi: 10.1016/0002-9149(73)90810-2
Hickerson, A., Rinderknecht, D., and Gharib, M. (2005). Experimental behavior of a valveless impedance pump. Exp. Fluids 38, 534–540. doi: 10.1007/s00348-005-0946-z
Hierck, B. P., Van der Heiden, K., Poelma, C., Westerweel, J., and Poelmann, R. E. (2008). Fluid shear stress and inner curvature remodeling of the embryonic heart. choosing the right lane!. ScientificWorldJournal 8, 212–222. doi: 10.1100/tsw.2008.42
Hogers, B., DeRuiter, M. C., Baasten, A. M., Gittenberger-de Groot, A. C., and Poelmann, R. E. (1995). Intracardiac blood flow patterns related to the yolk sac circulation of the chick embryo. Circ. Res. 76, 871–877. doi: 10.1161/01.RES.76.5.871
Hogers, B., DeRuiter, M. C., Gittenberger-de Groot, A. C., and Poelmann, R. E. (1997). Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ. Res. 80, 473–481. doi: 10.1161/01.RES.80.4.473
Hogers, B., DeRuiter, M. C., Gittenberger-de Groot, A. C., and Poelmann, R. E. (1999). Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc. Res. 41, 87–99. doi: 10.1016/S0008-6363(98)00218-1
Hove, J. R., Koster, R. W., Forouhar, A. S., Acevedo-Bolton, G., Fraser, S. E., and Gharib, M. (2003). Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421, 172–177. doi: 10.1038/nature01282
Hu, N., Christensen, D. A., Agrawal, A. K., Beaumont, C., Clark, E. B., and Hawkins, J. A. (2009). Dependence of aortic arch morphogenesis on intracardiac blood flow in the left atrial ligated chick embryo. Anat. Rec. 292, 652–660. doi: 10.1002/ar.20885
Hu, N., and Clark, E. B. (1989). Hemodynamics of the stage 12 to stage 29 chick embryo. Circ. Res. 65, 1665–1670. doi: 10.1161/01.RES.65.6.1665
Huang, J. B., Liu, Y. L., Sun, P. W., Lv, X. D., Du, M., and Fan, X. M. (2010). Molecular mechanisms of congenital heart disease. Cardiovasc. Pathol. 19, e183–e193. doi: 10.1016/j.carpath.2009.06.008
Icardo, J. M., and Colvee, E. (1995). Atrioventricular valves of the mouse: III. collagenous skeleton and myotendinous junction. Anat. Rec. 243, 367–375. doi: 10.1002/ar.1092430311
Ilbawi, A. M., Spicer, D. E., Bharati, S., Cook, A., and Anderson, R. H. (2007). Morphologic study of the ascending aorta and aortic arch in hypoplastic left hearts: surgical implications. J. Thorac. Cardiovasc. Surg. 134, 99–105. doi: 10.1016/j.jtcvs.2007.01.070
Jaffee, O. C. (1965). Hemodynamic factors in the development of the chick embryo heart. Anat. Rec. 151, 69–75. doi: 10.1002/ar.1091510107
Johnson, B. M., Garrity, D. M., and Dasi, L. P. (2013). The transitional cardiac pumping mechanics in the embryonic heart. Cardiovasc. Eng. Technol. 4, 246–255. doi: 10.1007/s13239-013-0120-3
Kamino, K., Hirota, A., and Fujii, S. (1981). Localization of pacemaking activity in early embryonic heart monitored using voltage-sensitive dye. Nature 290, 595–597. doi: 10.1038/290595a0
Kowalski, W. J., Dur, O., Wang, Y., Patrick, M. J., Tinney, J. P., Keller, B. B., et al. (2013). Critical transitions in early embryonic aortic arch patterning and hemodynamics. PLoS ONE 8:e60271. doi: 10.1371/journal.pone.0060271
Kowalski, W. J., Teslovich, N. C., Menon, P. G., Tinney, J. P., Keller, B. B., and Pekkan, K. (2014). Left atrial ligation alters intracardiac flow patterns and the biomechanical landscape in the chick embryo. Dev. Dyn. 243, 652–662. doi: 10.1002/dvdy.24107
Latacha, K. S., Remond, M. C., Ramasubramanian, A., Chen, A. Y., Elson, E. L., and Taber, L. A. (2005). Role of actin polymerization in bending of the early heart tube. Dev. Dyn. 233, 1272–1286. doi: 10.1002/dvdy.20488
Liebau, G. (1956). Possibility of movement of blood in cardiovascular system without cardiac and venous valve function. Verh. Dtsch. Ges. Kreislaufforsch. 22, 354–359.
Lin, I. E., and Taber, L. A. (1995). A model for stress-induced growth in the developing heart. J. Biomech. Eng. 117, 343–349. doi: 10.1115/1.2794190
Lindsey, S. E., and Butcher, J. T. (2011). The cycle of form and function in cardiac valvulogenesis. Aswan Heart Cent. Sci. Pract Ser. 2, 10. doi: 10.5339/ahcsps.2011.10
Liu, J., Bressan, M., Hassel, D., Huisken, J., Staudt, D., Kikuchi, K., et al. (2010). A dual role for ErbB2 signaling in cardiac trabeculation. Development 137, 3867–3875. doi: 10.1242/dev.053736
Lockhart, M., Wirrig, E., Phelps, A., and Wessels, A. (2011). Extracellular matrix and heart development. Birth Defects Res. Part A Clin. Mol. Teratol. 91, 535–550. doi: 10.1002/bdra.20810
Lorenz, R., Bock, J., Barker, A. J., von Knobelsdorff-Brenkenhoff, F., Wallis, W., Korvink, J. G., et al. (2014). 4D flow magnetic resonance imaging in bicuspid aortic valve disease demonstrates altered distribution of aortic blood flow helicity. Magn. Reson. Med. 71, 1542–1553. doi: 10.1002/mrm.24802
Lucitti, J. L., Jones, E. A., Huang, C., Chen, J., Fraser, S. E., and Dickinson, M. E. (2007). Vascular remodeling of the mouse yolk sac requires hemodynamic force. Development 134, 3317–3326. doi: 10.1242/dev.02883
Lucitti, J. L., Tobita, K., and Keller, B. B. (2005). Arterial hemodynamics and mechanical properties after circulatory intervention in the chick embryo. J. Exp. Biol. 208, 1877–1885. doi: 10.1242/jeb.01574
Luna-Zurita, L., Prados, B., Grego-Bessa, J., Luxan, G., del Monte, G., Benguria, A., et al. (2010). Integration of a notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J. Clin. Invest. 120, 3493–3507. doi: 10.1172/JCI42666
Majkut, S., Idema, T., Swift, J., Krieger, C., Liu, A., and Discher, D. E. (2013). Heart-specific stiffening in early embryos parallels matrix and myosin expression to optimize beating. Curr. Biol. 23, 2434–2439. doi: 10.1016/j.cub.2013.10.057
Mammoto, T., and Ingber, D. E. (2010). Mechanical control of tissue and organ development. Development 137, 1407–1420. doi: 10.1242/dev.024166
Manasek, F. J., Burnside, M. B., and Waterman, R. E. (1972). Myocardial cell shape changes as a mechanism of embryonic heart looping. Dev. Biol. 29, 349–371. doi: 10.1016/0012-1606(72)90077-2
Manner, J. (2004). On rotation, torsion, lateralization, and handedness of the embryonic heart loop: new insights from a simulation model for the heart loop of chick embryos. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 278, 481–492. doi: 10.1002/ar.a.20036
Manner, J., Wessel, A., and Yelbuz, T. M. (2010). How does the tubular embryonic heart work? looking for the physical mechanism generating unidirectional blood flow in the valveless embryonic heart tube. Dev. Dyn. 239, 1035–1046. doi: 10.1002/dvdy.22265
Manning, A., and McLachlan, J. C. (1990). Looping of chick embryo hearts in vitro. J. Anat. 168, 257–263.
McBride, K. L., Zender, G. A., Fitzgerald-Butt, S. M., Koehler, D., Menesses-Diaz, A., Fernbach, S., et al. (2009). Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur. J. Hum. Genet. 17, 811–819. doi: 10.1038/ejhg.2008.255
Meilhac, S. M., Esner, M., Kelly, R. G., Nicolas, J. F., and Buckingham, M. E. (2004). The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev. Cell 6, 685–698. doi: 10.1016/S1534-5807(04)00133-9
Miller, C. E., Wong, C. L., and Sedmera, D. (2003). Pressure overload alters stress-strain properties of the developing chick heart. Am. J. Physiol. Heart Circ. Physiol. 285, H1849–H1856. doi: 10.1152/ajpheart.00384.2002
Mironov, V., Visconti, R. P., and Markwald, R. R. (2005). On the role of shear stress in cardiogenesis. Endothelium 12, 259–261. doi: 10.1080/10623320500476708
Moorman, A. F., and Christoffels, V. M. (2003). Cardiac chamber formation: development, genes, and evolution. Physiol. Rev. 83, 1223–1267. doi: 10.1152/physrev.00006.2003
Moorman, A. F. M., Soufan, A. T., Hagoort, J., De Boer, P. A. J., and Christoffels, V. M. (2004). Development of the building plan of the heart. Ann. N.Y. Acad. Sci. 1015, 171–181. doi: 10.1196/annals.1302.014
Natowicz, M., Chatten, J., Clancy, R., Conard, K., Glauser, T., Huff, D., et al. (1988). Genetic disorders and major extracardiac anomalies associated with the hypoplastic left heart syndrome. Pediatrics 82, 698–706.
Nemer, M. (2008). Genetic insights into normal and abnormal heart development. Cardiovasc. Pathol. 17, 48–54. doi: 10.1016/j.carpath.2007.06.005
Nerurkar, N. L., Ramasubramanian, A., and Taber, L. A. (2006). Morphogenetic adaptation of the looping embryonic heart to altered mechanical loads. Dev. Dyn. 235, 1822–1829. doi: 10.1002/dvdy.20813
Nkomo, V. T., Enriquez-Sarano, M., Ammash, N. M., Melton, L. J. 3rd., Bailey, K. R., Desjardins, V., et al. (2003). Bicuspid aortic valve associated with aortic dilatation: a community-based study. Arterioscler. Thromb. Vasc. Biol. 23, 351–356. doi: 10.1161/01.ATV.0000055441.28842.0A
Norris, R. A., Kern, C. B., Wessels, A., Moralez, E. I., Markwald, R. R., and Mjaatvedt, C. H. (2004). Identification and detection of the periostin gene in cardiac development. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 281A, 1227–1233. doi: 10.1002/ar.a.20135
Ottesen, J. T. (2003). Valveless pumping in a fluid-filled closed elastic tube-system:one-dimensional theory with experimental validation. J. Math. Biol. 46, 309–332. doi: 10.1007/s00285-002-0179-1
Oyen, N., Poulsen, G., Boyd, H. A., Wohlfahrt, J., Jensen, P. K., and Melbye, M. (2009). Recurrence of congenital heart defects in families. Circulation 120, 295–301. doi: 10.1161/CIRCULATIONAHA.109.857987
Person, A. D., Klewer, S. E., and Runyan, R. B. (2005). Cell biology of cardiac cushion development. Int. Rev. Cytol. 243, 287–335. doi: 10.1016/S0074-7696(05)43005-3
Qayyum, S. R., Webb, S., Anderson, R. H., Verbeek, F. J., Brown, N. A., and Richardson, M. K. (2001). Septation and valvar formation in the outflow tract of the embryonic chick heart. Anat. Rec. 264, 273–283. doi: 10.1002/ar.1162
Rana, M. S., Christoffels, V. M., and Moorman, A. F. (2013). A molecular and genetic outline of cardiac morphogenesis. Acta Physiol. 207, 588–615. doi: 10.1111/apha.12061
Reckova, M., Rosengarten, C., deAlmeida, A., Stanley, C. P., Wessels, A., Gourdie, R. G., et al. (2003). Hemodynamics is a key epigenetic factor in development of the cardiac conduction system. Circ. Res. 93, 77–85. doi: 10.1161/01.RES.0000079488.91342.B7
Roberts, W. C. (1970). The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am. J. Cardiol. 26, 72–83. doi: 10.1016/0002-9149(70)90761-7
Roger, V. L., Go, A. S., Lloyd-Jones, D. M., Adams, R. J., Berry, J. D., Brown, T. M., et al. (2011). Heart disease and stroke statistics–2011 update: a report from the american heart association. Circulation 123, e18–e209. doi: 10.1161/CIR.0b013e3182009701
Rugonyi, S. (2013). Strain-induced tissue growth laws: applications to embryonic cardiovascular development. J. Appl. Mech. Eng. S11:001. doi: 10.4172/2168-9873.S11-001
Rugonyi, S., Shaut, C., Liu, A., Thornburg, K., and Wang, R. K. (2008). Changes in wall motion and blood flow in the outflow tract of chick embryonic hearts observed with optical coherence tomography after outflow tract banding and vitelline-vein ligation. Phys. Med. Biol. 53, 5077–5091. doi: 10.1088/0031-9155/53/18/015
Runyan, R. B., and Markwald, R. R. (1983). Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev. Biol. 95, 108–114. doi: 10.1016/0012-1606(83)90010-6
Samsa, L. A., Yang, B., and Liu, J. (2013). Embryonic cardiac chamber maturation: trabeculation, conduction, and cardiomyocyte proliferation. Am. J. Med. Genet. C Semin. Med. Genet. 163C, 157–168. doi: 10.1002/ajmg.c.31366
Sankova, B., Machalek, J., and Sedmera, D. (2010). Effects of mechanical loading on early conduction system differentiation in the chick. Am. J. Physiol. Heart Circ. Physiol. 298, H1571–H1576. doi: 10.1152/ajpheart.00721.2009
Schroeder, J. A., Jackson, L. F., Lee, D. C., and Camenisch, T. D. (2003). Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J. Mol. Med. 81, 392–403. doi: 10.1007/s00109-003-0456-5
Sedmera, D., Harris, B. S., Grant, E., Zhang, N., Jourdan, J., Kurkova, D., et al. (2008). Cardiac expression patterns of endothelin-converting enzyme (ECE): implications for conduction system development. Dev. Dyn. 237, 1746–1753. doi: 10.1002/dvdy.21572
Sedmera, D., Hu, N., Weiss, K. M., Keller, B. B., Denslow, S., and Thompson, R. P. (2002). Cellular changes in experimental left heart hypoplasia. Anat. Rec. 267, 137–145. doi: 10.1002/ar.10098
Sedmera, D., Pexieder, T., Rychterova, V., Hu, N., and Clark, E. B. (1999). Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 254, 238–252.
Sedmera, D., and Thompson, R. P. (2011). Myocyte proliferation in the developing heart. Dev. Dyn. 240, 1322–1334. doi: 10.1002/dvdy.22650
Shiraishi, I., Takamatsu, T., Minamikawa, T., and Fujita, S. (1992). 3-D observation of actin filaments during cardiac myofibrinogenesis in chick embryo using a confocal laser scanning microscope. Anat. Embryol. 185, 401–408. doi: 10.1007/BF00188551
Simpson, J. M., and Sharland, G. K. (1997). Natural history and outcome of aortic stenosis diagnosed prenatally. Heart 77, 205–210. doi: 10.1136/hrt.77.3.205
Snarr, B. S., Kern, C. B., and Wessels, A. (2008). Origin and fate of cardiac mesenchyme. Dev. Dyn. 237, 2804–2819. doi: 10.1002/dvdy.21725
Srivastava, D., and Olson, E. N. (2000). A genetic blueprint for cardiac development. Nature 407, 221–226. doi: 10.1038/35025190
Sugi, Y., Ito, N., Szebenyi, G., Myers, K., Fallon, J. F., Mikawa, T., et al. (2003). Fibroblast growth factor (FGF)-4 can induce proliferation of cardiac cushion mesenchymal cells during early valve leaflet formation. Dev. Biol. 258, 252–263. doi: 10.1016/S0012-1606(03)00099-X
Taber, L. A. (1998). Mechanical aspects of cardiac development. Prog. Biophys. Mol. Biol. 69, 237–255. doi: 10.1016/S0079-6107(98)00010-8
Taber, L. A. (2006). Biophysical mechanisms of cardiac looping. Int. J. Dev. Biol. 50, 323–332. doi: 10.1387/ijdb.052045lt
Taber, L. A. (2009). Towards a unified theory for morphomechanics. Philos. Trans. A Math. Phys. Eng. Sci. 367, 3555–3583. doi: 10.1098/rsta.2009.0100
Taber, L. A., and Eggers, D. W. (1996). Theoretical study of stress-modulated growth in the aorta. J. Theor. Biol. 180, 343–357. doi: 10.1006/jtbi.1996.0107
Taber, L. A., Voronov, D. A., and Ramasubramanian, A. (2010). The role of mechanical forces in the torsional component of cardiac looping. Ann. N.Y. Acad. Sci. 1188, 103–110. doi: 10.1111/j.1749-6632.2009.05089.x
Taber, L. A., Zhang, J., and Perucchio, R. (2007). Computational model for the transition from peristaltic to pulsatile flow in the embryonic heart tube. J. Biomech. Eng. 129, 441–449. doi: 10.1115/1.2721076
Tan, H., Biechler, S., Junor, L., Yost, M. J., Dean, D., Li, J., et al. (2013). Fluid flow forces and rhoA regulate fibrous development of the atrioventricular valves. Dev. Biol. 374, 345–356. doi: 10.1016/j.ydbio.2012.11.023
Tan, H., Junor, L., Price, R. L., Norris, R. A., Potts, J. D., and Goodwin, R. L. (2011). Expression and deposition of fibrous extracellular matrix proteins in cardiac valves during chick development. Microsc. Microanal. 17, 91–100. doi: 10.1017/S1431927610094365
Tavares, A. L. P., Mercado-Pimentel, M. E., Runyan, R. B., and Kitten, G. T. (2006). TGF?-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev. Dyn. 235, 1589–1598. doi: 10.1002/dvdy.20771
Tobita, K., and Keller, B. B. (2000). Right and left ventricular wall deformation patterns in normal and left heart hypoplasia chick embryos. Am. J. Physiol. Heart Circ. Physiol. 279, H959–H969.
Tobita, K., Schroder, E. A., Tinney, J. P., Garrison, J. B., and Keller, B. B. (2002). Regional passive ventricular stress-strain relations during development of altered loads in chick embryo. Am. J. Physiol. Heart Circ. Physiol. 282, H2386–H2396. doi: 10.1152/ajpheart.00879.2001
van den Berg, G., and Moorman, A. F. (2009). Concepts of cardiac development in retrospect. Pediatr. Cardiol. 30, 580–587. doi: 10.1007/s00246-008-9369-y
Varner, V. D., and Taber, L. A. (2012). Not just inductive: a crucial mechanical role for the endoderm during heart tube assembly. Development 139, 1680–1690. doi: 10.1242/dev.073486
Vermot, J., Forouhar, A. S., Liebling, M., Wu, D., Plummer, D., Gharib, M., et al. (2009). Reversing blood flows act through klf2a to ensure normal valvulogenesis in the developing heart. PLoS Biol. 7:e1000246. doi: 10.1371/journal.pbio.1000246
Verzi, M. P., McCulley, D. J., De Val, S., Dodou, E., and Black, B. L. (2005). The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 287, 134–145. doi: 10.1016/j.ydbio.2005.08.041
Voronov, D. A., Alford, P. W., Xu, G., and Taber, L. A. (2004). The role of mechanical forces in dextral rotation during cardiac looping in the chick embryo. Dev. Biol. 272, 339–350. doi: 10.1016/j.ydbio.2004.04.033
Voronov, D. A., and Taber, L. A. (2002). Cardiac looping in experimental conditions: effects of extraembryonic forces. Dev. Dyn. 224, 413–421. doi: 10.1002/dvdy.10121
Wang, Y., Dur, O., Patrick, M. J., Tinney, J. P., Tobita, K., Keller, B. B., et al. (2009). Aortic arch morphogenesis and flow modeling in the chick embryo. Ann. Biomed. Eng. 37, 1069–1081. doi: 10.1007/s10439-009-9682-5
Wenink, A. C., and Gittenberger-de Groot, A. C. (1986). Embryology of the mitral valve. Int. J. Cardiol. 11, 75–84. doi: 10.1016/0167-5273(86)90201-9
Wessels, A., and Sedmera, D. (2003). Developmental anatomy of the heart: a tale of mice and man. Physiol. Genomics 15, 165–176. doi: 10.1152/physiolgenomics.00033.2003
Wessels, A., van den Hoff, M. J. B., Adamo, R. F., Phelps, A. L., Lockhart, M. M., Sauls, K., et al. (2012). Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 366, 111–124. doi: 10.1016/j.ydbio.2012.04.020
Winer, J. P., Janmey, P. A., McCormick, M. E., and Funaki, M. (2009). Bone marrow-derived human mesenchymal stem cells become quiescent on soft substrates but remain responsive to chemical or mechanical stimuli. Tissue Eng. Part A 15, 147–154. doi: 10.1089/ten.tea.2007.0388
Wyczalkowski, M. A., Chen, Z., Filas, B. A., Varner, V. D., and Taber, L. A. (2012). Computational models for mechanics of morphogenesis. Birth Defects Res. C Embryo Today 96, 132–152. doi: 10.1002/bdrc.21013
Xavier-Neto, J., Castro, R. A., Sampaio, A. C., Azambuja, A. P., Castillo, H. A., Cravo, R. M., et al. (2007). Parallel avenues in the evolution of hearts and pumping organs. Cell. Mol. Life Sci. 64, 719–734. doi: 10.1007/s00018-007-6524-1
Ya, J., van den Hoff, M. J., de Boer, P. A., Tesink-Taekema, S., Franco, D., Moorman, A. F., et al. (1998). Normal development of the outflow tract in the rat. Circ. Res. 82, 464–472. doi: 10.1161/01.RES.82.4.464
Yalcin, H. C., Shekhar, A., McQuinn, T. C., and Butcher, J. T. (2011). Hemodynamic patterning of the avian atrioventricular valve. Dev. Dyn. 240, 23–35. doi: 10.1002/dvdy.22512
Yalcin, H. C., Shekhar, A., Nishimura, N., Rane, A. A., Schaffer, C. B., and Butcher, J. T. (2010). Two-photon microscopy-guided femtosecond-laser photoablation of avian cardiogenesis: noninvasive creation of localized heart defects. Am. J. Physiol. Heart Circ. Physiol. 299, H1728–H1735. doi: 10.1152/ajpheart.00495.2010
Yanagisawa, H., Yanagisawa, M., Kapur, R. P., Richardson, J. A., Williams, S. C., Clouthier, D. E., et al. (1998). Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development 125, 825–836.
Yang, M., Taber, L. A., and Clark, E. B. (1994). A nonliner poroelastic model for the trabecular embryonic heart. J. Biomech. Eng. 116, 213–223. doi: 10.1115/1.2895722
Yashiro, K., Shiratori, H., and Hamada, H. (2007). Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 450, 285–288. doi: 10.1038/nature06254
Yener, N., Oktar, G. L., Erer, D., Yardimci, M. M., and Yener, A. (2002). Bicuspid aortic valve. Ann. Thorac. Cardiovasc. Surg. 8, 264–267.
Yuan, S., Zaidi, S., and Brueckner, M. (2013). Congenital heart disease: emerging themes linking genetics and development. Curr. Opin. Genet. Dev. 23, 352–359. doi: 10.1016/j.gde.2013.05.004
Zaffran, S., Kelly, R. G., Meilhac, S. M., Buckingham, M. E., and Brown, N. A. (2004). Right ventricular myocardium derives from the anterior heart field. Circ. Res. 95, 261–268. doi: 10.1161/01.RES.0000136815.73623.BE
Zaidi, S., Choi, M., Wakimoto, H., Ma, L., Jiang, J., Overton, J. D., et al. (2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223. doi: 10.1038/nature12141
Keywords: hemodynamics, congenital heart defects, heart development, mechanotransduction, shear stress
Citation: Lindsey SE, Butcher JT and Yalcin HC (2014) Mechanical regulation of cardiac development. Front. Physiol. 5:318. doi: 10.3389/fphys.2014.00318
Received: 15 May 2014; Accepted: 03 August 2014;
Published online: 21 August 2014.
Edited by:
Michiko Watanabe, Case Western Reserve University School of Medicine, USAReviewed by:
Indika Rajapakse, University of Michigan, USACopyright © 2014 Lindsey, Butcher and Yalcin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huseyin C. Yalcin, Department of Mechanical Engineering, Dogus University, G Blok 508B, Acibadem Zeamet Sokak, Kadikoy 34722, Istanbul, Turkey e-mail:eWFsY2luY2FnYXRheUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.