
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 19 August 2014
Sec. Mitochondrial Research
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00309
This article is part of the Research Topic Mitochondria: Hubs of cellular signaling, energetics and redox balance View all 18 articles
 Styliani Vakrou
Styliani Vakrou M. Roselle Abraham*
M. Roselle Abraham*Hypertrophic cardiomyopathy (HCM) has been recently recognized as the most common inherited cardiovascular disorder, affecting 1 in 500 adults worldwide. HCM is characterized by myocyte hypertrophy resulting in thickening of the ventricular wall, myocyte disarray, interstitial and/or replacement fibrosis, decreased ventricular cavity volume and diastolic dysfunction. HCM is also the most common cause of sudden death in the young. A large proportion of patients diagnosed with HCM have mutations in sarcomeric proteins. However, it is unclear how these mutations lead to the cardiac phenotype, which is variable even in patients carrying the same causal mutation. Abnormalities in calcium cycling, oxidative stress, mitochondrial dysfunction and energetic deficiency have been described constituting the basis of therapies in experimental models of HCM and HCM patients. This review focuses on evidence supporting the role of cellular metabolism and mitochondria in HCM.
Hypertrophic cardiomyopathy (HCM) was first recognized as a clinical entity, approximately 55 years ago (Brock, 1957; Teare, 1958; Cohen et al., 1964; Ross et al., 1966). It is the most common inherited cardiac disease with an estimated prevalence of 1: 500 in young individuals (Maron, 2002). Inheritance is autosomal dominant, with variable penetrance in 50–60% of patients; causal mutations have not been identified in 40–50% of HCM patients (Jarcho et al., 1989; Solomon et al., 1990; Marian and Roberts, 2001; Marian, 2002). Nine different chromosomal loci have been linked to HCM with the majority of genes encoding cardiac sarcomeric proteins (Jarcho et al., 1989; Geisterfer-Lowrance et al., 1990; Watkins et al., 1993; Thierfelder et al., 1994). The most common mutations occur in genes encoding for β-myosin heavy chain (35%), myosin binding protein C (20%), troponin T (5%) and α-tropomyosin (<3%), which have roles in cardiac excitation-contraction coupling (Maron and Maron, 2013).
Asymmetric ventricular hypertrophy and left ventricular outflow tract obstruction with normal or hyperdynamic systolic function are common morphologic manifestations of HCM (Maron et al., 2003). However, clinical phenotype is variable even among individuals carrying the same causal mutation due to effects of modifier genes, which are largely unknown (Seidman and Seidman, 2001). As a result degree and location (mid-ventricular, septal, apical and concentric) of hypertrophy and obstruction are variable in patients with HCM (Figure 1). Clinical presentation is also heterogeneous, spanning the spectrum from individuals who are largely asymptomatic, to patients with moderate to severe symptoms, ranging from angina, exercise intolerance to heart failure (requiring heart transplantation), atrial fibrillation and sudden cardiac death (Maron, 2002; Maron et al., 2002; Gersh et al., 2011). Irrespective of the causal mutation, pathologically, HCM is characterized by myocyte hypertrophy, myocyte disarray and fibrosis (Ho et al., 2010).
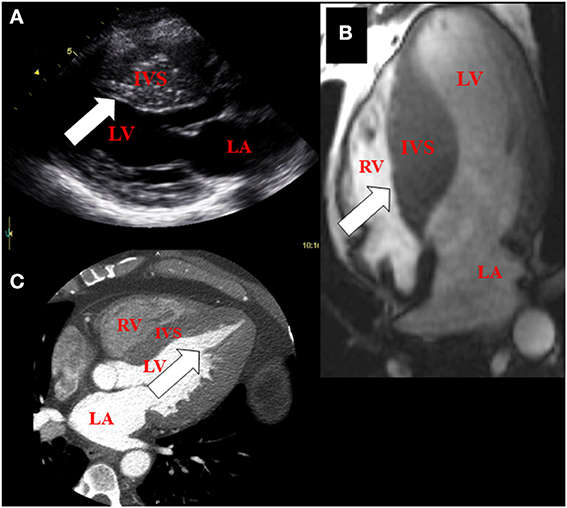
Figure 1. Imaging features of hypertrophic cardiomyopathy using multi-modality cardiac imaging in patients. (A) Basal hypertrophy of the inter-ventricular septum (arrow) using echocardiography (parasternal long axis view of the heart). (B) Mid-septal hypertrophy (arrow) using magnetic resonance imaging (4-chamber view of the heart). (C) Apical hypertrophy (arrow) using Computed tomography (4-chamber view of the heart). LA, left atrium; IVS, inter-ventricular septum; LV, left ventricle; RV, right ventricle.
Pioneering work by several groups has revealed the molecular genetics and biophysical mechanisms underlying HCM. A variety of functional defects, including altered Ca2+ sensitivity and/or affinity, myofibrillar ATPase activity, Ca2+ handling, cross-bridge dynamics, impaired energetics, oxidative stress and electrophysiologic abnormalities have been identified in experimental models (Straceski et al., 1994; Spindler et al., 1998; Blanchard et al., 1999; Gao et al., 1999; Georgakopoulos et al., 1999; Tardiff et al., 1999; Solaro et al., 2002; Javadpour et al., 2003; Adhikari et al., 2004; Szczesna-Cordary et al., 2004; Ertz-Berger et al., 2005; Hernandez et al., 2005; Robinson et al., 2007; Greenberg et al., 2009, 2010; Guinto et al., 2009; Mettikolla et al., 2011; Puglisi et al., 2014) and patients (Haq et al., 2001; Crilley et al., 2003; Nakamura et al., 2005; Dimitrow et al., 2009; Unno et al., 2009; Ho et al., 2010; Bravo et al., 2012; Coppini et al., 2013; Lin et al., 2013; Gruner et al., 2014). Since HCM-causing mutations increase the energetic cost of tension development, it has been hypothesized that excessive sarcomeric energy use leads to the HCM phenotype (Blair et al., 2001; Crilley et al., 2003; Abozguia et al., 2010). We (Abraham et al., 2013) and others (Jung et al., 1998, 2000; Crilley et al., 2003; Timmer et al., 2011) have shown reduced PCr/ATP ratios in HCM patients with both established left ventricular hypertrophy and in the pre-hypertrophic stage, which suggests that bioenergetic deficits may be a primary cause of myocardial remodeling.
31P NMR spectroscopy studies have demonstrated a reduction in ATP reserve in HCM mouse models following inotropic stimulation (Spindler et al., 1998; Javadpour et al., 2003). Evidence for energy deficit in HCM has also been obtained from patient studies revealing increased glucose uptake (Tadamura et al., 1996), reduction of PCr/ATP ratios in pre-hypertrophic patients (Crilley et al., 2003) and reduced coronary sinus pH despite non-limiting capillary oxygen pressures (possibly indicating up-regulation of glycolysis with lactate generation) (Tadamura et al., 1996; Jung et al., 1998; Ashrafian et al., 2003; Keren et al., 2008). However, it is not known whether the energy deficit paradigm can be generalized to all HCM patients, at all stages of the disease. Furthermore, the molecular basis of the energetic deficits in HCM and their attendant consequences has been understudied.
In the heart, ATP supply is tightly regulated to meet energetic demands of the myofilaments. The mechanisms by which cardiac energetics is finely tuned are still a matter of considerable debate, but there is emerging consensus on the importance of two regulators, Ca2+ and ADP (Cortassa et al., 2006; Saks et al., 2006; Balaban, 2009). During contraction, Ca2+-induced Ca2+ release from the sarcoplasmic reticulum floods the cytoplasm where it binds the thin filament regulatory protein Troponin C, thereby initiating contraction (Bers, 2002). Coordinate activation of ATP production arises because mitochondria, positioned close to the SR, take up Ca2+ via the mitochondrial calcium uniporter (MCU) (Maack and O'Rourke, 2007). Mitochondrial matrix calcium regulates 3 key enzymes in the tricarboxylic acid (TCA) cycle that harnesses the decarboxylation of acetyl-CoA to yield reduced nicotinamide adenine dinucleotide (NADH) which fuels the respiratory electron transport chain (ETC) and is converted to NADPH which plays a critical role in maintaining mitochondrial anti-oxidant capacity (McCormack and Denton, 1990; Hansford and Zorov, 1998; Liu et al., 2014); Mitochondrial Ca2+ can also directly stimulate respiratory complex activity, including the mitochondrial ATP synthase (F1F0 ATPase) (Territo et al., 2000). Thus, Ca2+ coordinately regulates ATP-consuming myofilaments and ATP-generating oxidative phosphorylation (Figure 2).
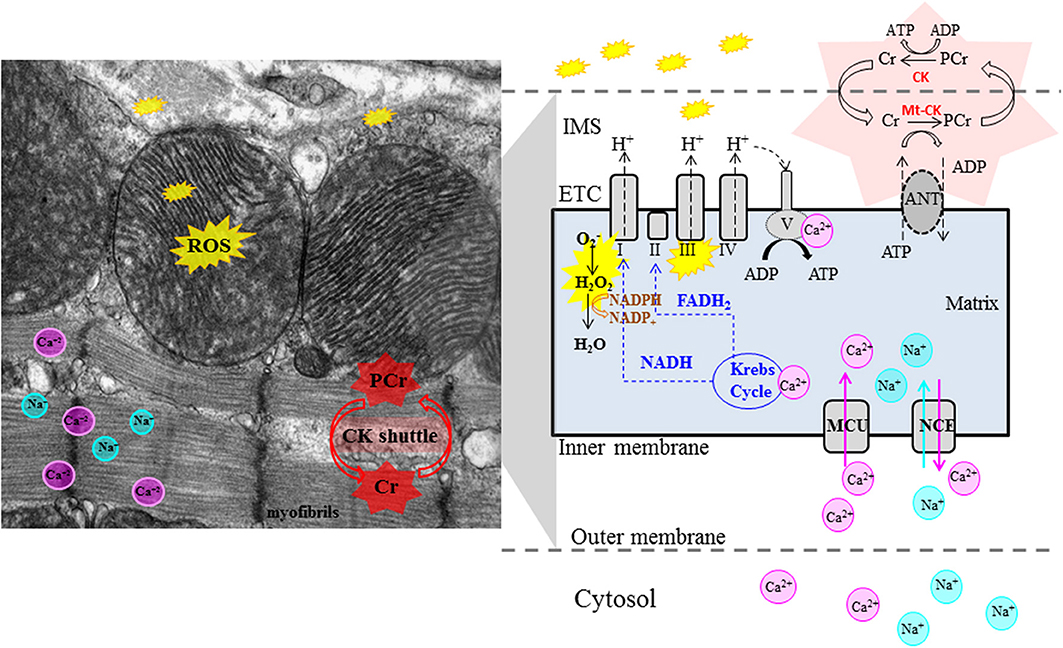
Figure 2. Role of Mitochondria in pathogenesis of cardiac phenotype in HCM. Left panel: electron microscopy image of mouse heart. Right panel: schematic illustrating mitochondrial physiology. The Krebs cycle generates reducing equivalents (NADH, FADH2) that drive proton pumping, establish the proton-motive force across the mitochondrial inner membrane and contribute to ROS scavenging. Mitochondrial ATP synthase (complex V) couples proton influx to ATP generation. Matrix concentrations of Ca2+ and Na+ play an important role in control of oxidative phosphorylation. Mitochondria are the main source of ATP generation and important source of ROS (from complexes I and III) in cardiac myocytes. Abnormalities in mitochondrial function, reduced CK flux, oxidative stress and impaired Ca2+ handling have been implicated in generation of the cardiac phenotype in HCM. Cr, creatine; PCr, creatine-phosphate, Mt-CK, mitochondrial creatine kinase; ANT, Adenine nucleotide translocator; ETC, electron transport chain; IMS, inter-membrane space; MCU, mitochondrial calcium uniporter; NCE, mitochondrial Na+−Ca2+ exchanger.
Ca2+ uptake by mitochondria is dependent on cytosolic Na+ levels, which has been demonstrated to be elevated in experimental models of heart failure (Liu and O'Rourke, 2008) and failing human hearts (Pieske and Houser, 2003). The O'Rourke group has demonstrated that elevated cytosolic Na+ increases the rate of the mitochondrial Na+−Ca2+ exchanger (mNCE), which promotes mitochondrial Ca2+ efflux and decreases the mitochondria's ability to accumulate Ca2+ during conditions of high demand (Maack et al., 2006; Liu and O'Rourke, 2013). Without Ca2+-induced Kreb's cycle stimulation, NADH and NADPH become more oxidized and are unable to recharge antioxidant systems, leading to ROS accumulation in the mitochondrial matrix and release into the cytosol (Kohlhaas et al., 2010; Gauthier et al., 2013; Liu and O'Rourke, 2013). Partial inhibition of mNCE by CGP-37157 attenuated adverse ventricular remodeling and was anti-arrhythmic in a guinea pig model of pressure overload (Liu et al., 2014). A recent study of Ranolazine, an inhibitor of late Na+ current, that is known to contribute to cytosolic Na+ overload revealed salutary effects on action potential duration and arrhythmias in cardiac myocytes of HCM patients who underwent myectomy (Coppini et al., 2013). Investigation of cytosolic Na+ levels (Gao et al., 2013) and mitochondrial Ca2+ handling is needed in order to assess whether altered mitochondrial Ca2+ dynamics contribute to energetic deficits and oxidative stress in HCM (Nakamura et al., 2005; Senthil et al., 2005; Marian et al., 2006; Dimitrow et al., 2009).
Conditions such as exercise that quickly elevate heart rate impose energetic demands that can quickly exceed Ca2+-regulated supply. In these cases, the by-product of myofilament ATPase activity, ADP, provides vital feedback stimulation of energy in two ways. Firstly the original work by Britton Chance and colleagues showed that the rate of ATP formation by Complex V is driven by the concentration of ADP. Myofilaments also possess a local ATP-buffering capacity maintained by cytosolic creatine kinase (CK). In high work conditions, ADP accumulation is sensed by CK, which catalyzes phosphoryl group transfer from phosphocreatine to regenerate ATP (Saks et al., 2006; Balaban, 2009) (Figure 2). Our studies in patients from a family carrying the R403Q mutation in myosin heavy chain (MHC) revealed a 43% reduction in forward CK flux at rest, indicating reduced metabolic reserve (Abraham et al., 2013). A recent study by Critoph et al. revealed reduced cardiac reserve secondary to blunted increase in cardiac output, in HCM patients undergoing exercise stress testing (Critoph et al., 2014). NMR studies in mice with R403Q-MHC (Spindler et al., 1998) and R92-TNT (Javadpour et al., 2003; He et al., 2007) mutations verified impaired myocardial energetics during inotropic stimulation. The decreased [PCr], increased [Pi], [ADP] and unchanged or decreased [ATP] can result in reduction in the calculated free energy release from ATP hydrolysis (lΔGl) (Spindler et al., 1998) which in turn can impair the function of cellular ATPases (e.g., myosin ATPase, Na+−K+-ATPase) and Ca2+ pumps like SERCA, leading to systolic and/or diastolic dysfunction, reduction or blunted increase in stroke volume, increased levels of cytosolic Na+, Ca2+ and arrhythmias under conditions of high work load, such as intense exercise (Unno et al., 2009; Ashrafian et al., 2011; Watkins et al., 2011).
The vital role of mitochondria as providers of energy for the high demands of cardiac contractility is well recognized, as is their contribution to necrotic and apoptotic cell death (Seddon et al., 2007). More, recently, the novel role of mitochondria as signaling organelles has emerged, primarily through their ability to produce reactive oxygen species (ROS) -including superoxide (O−2), hydrogen peroxide (H2O2) and hydroxyl radicals (.OH)- and reactive nitrogen species, including nitric oxide (NO) and peroxynitrite (ONOO−) (Balaban et al., 2005; Figueira et al., 2013). ROS are best known for the damage they cause by directly oxidizing proteins, lipids, and DNA, but recent evidence suggests that the controlled and carefully modulated release of ROS from the mitochondrial network can activate specific signaling pathways or mediate reversible post-translational modifications of target proteins with pronounced effects on function (Terentyev et al., 2008; Bayeva and Ardehali, 2010). Additionally, because mitochondria are major determinants of the redox potential of both the pyrimidine nucleotide (NADH, NADPH) and thiol (GSH, thioredoxin) pools, they are important regulators of myocyte function (Stanley et al., 2011; Kembro et al., 2013; Liu and O'Rourke, 2013; Liu et al., 2014). However, very little is known about myocyte and mitochondrial redox potential in HCM (Figure 2).
Impairment of mitochondrial function and morphological disorganization has been reported in mouse models (Tardiff et al., 1999; Lucas et al., 2003) and in HCM patients (Unno et al., 2009). However, a systematic study of mitochondrial function is lacking. It is also unclear whether mitochondrial abnormalities are a primary event or secondary event in HCM. Of note, patients with mutations in mitochondrial DNA (Obayashi et al., 1992; Rotig et al., 1997; Okajima et al., 1998; Elliott and McKenna, 2004) can have a similar cardiac phenotype as HCM patients with sarcomeric protein mutations, suggesting that energetic deficits can lead to the cardiac phenotype of HCM.
The normal heart relies primarily on fatty acid oxidation for ATP generation (Abozguia et al., 2006; Ingwall, 2009). Pathologic hypertrophy is known to be associated with a reduction in fatty acid oxidation and increased reliance on glucose for ATP generation (Abozguia et al., 2006; Coppini et al., 2013). Positron emission tomography (PET) using 11C-acetate and 18FDG have been employed to study glucose and fatty acid oxidation in HCM patients (Grover-McKay et al., 1989; Nienaber et al., 1993; Perrone-Filardi et al., 1993; Tadamura et al., 1996; Tuunanen et al., 2007): some studies have found decrease/no change or increase in glucose uptake and the same is true for fatty acid oxidation, in hypertrophied and non-hypertrophied walls, when compared to controls. This may be attributable to differences in clinical characteristics (stage of disease, degree of hypertrophy, presence of microvascular dysfunction) and mutation status of the small number of patients who were studied. Since enzymes for fatty acid oxidation are located in mitochondria, it is unclear whether abnormalities in fatty acid oxidation are secondary to mitochondrial dysfunction. Metabolomic studies (Mayr, 2008) are needed in mouse models and HCM patients to obtain insights into metabolic remodeling and its role in generation of the cardiac phenotype in HCM.
Sudden cardiac death is the most dreaded and tragic phenotype, as it is often the first manifestation of the disease and occurs in asymptomatic and apparently healthy young individuals (Maron and Maron, 2013). The enhanced ventricular arrhythmogenicity has been attributed to abnormal cardiomyocyte orientation and alignment (disarray), microvascular ischemia, and fibrosis (Coppini et al., 2013).
Sarcomeres are known to sequester Ca2+ (bound: free ratio is 100:1). It has been hypothesized that HCM mutations may increase “Ca2+ trapping,” and through altered on–off kinetics may lead to altered Ca2+ signaling and arrhythmogenesis (Semsarian et al., 2002; Ashrafian et al., 2011).
Another possibility that has not been explored is energetic deficits, because energy compromise would be most marked when the heart is subject to increased work load, as is the case during high intensity exercise. In fact, exercise-induced arrhythmias are common causes of sudden death and defibrillator discharges in HCM patients (Ommen and Gersh, 2009; Spirito et al., 2014). Since HCM is associated with high sarcomeric ATP consumption at rest, the ability of the heart to provide sufficient ATP for myosin ATPase, SERCA and membrane ATPases could be compromised during exercise, leading to cytosolic Na+ and Ca2+ overload and triggered activity leading to clinical arrhythmias (Watkins et al., 2011). Another possibility is the “metabolic sink hypothesis,” proposed by the O'Rourke group (Akar et al., 2005), wherein regional oxidative stress (ROS-induced ROS release) in mitochondria results in mitochondrial membrane depolarization, KATP−channel opening and reentrant arrhythmias (Zorov et al., 2000, 2006; Aon et al., 2003, 2006; O'Rourke et al., 2005; Zhou et al., 2009; Cortassa et al., 2014).
There is a need for therapies that prevent/reverse the cardiac phenotype in HCM (Force et al., 2010). Drugs such as beta-adrenergic antagonists (e.g., Metoprolol), L-type Ca2+ channel blockers (e.g., Diltiazem, Verapamil) (Semsarian et al., 2002; Elliott and McKenna, 2004; Spirito and Autore, 2006), angiotensin II receptor antagonists (e.g., Losartan), (Lim et al., 2001; Lombardi et al., 2009; Shimada et al., 2013) carnitine palmitoyltransferase-1/2 inhibitor (Perhexiline) (Abozguia et al., 2010), antiarrhythmics (e.g., Disopyramide, Amiodarone), surgical myectomy and alcohol septal ablation (Sorajja et al., 2012) have been used to treat symptomatic HCM (Gersh et al., 2011). Antioxidant therapy with L-NAC was shown to prevent hypertrophy and fibrosis in experimental models of HCM (Marian et al., 2006) and is now in clinical trials (HALT-HCM study). Recently, there has been interest in the use of Ranolazine, based on beneficial effects on action potential duration and arrhythmias, in cardiac myocytes derived from HCM patients undergoing myectomy that exhibited evidence of electrophysiologic remodeling (increased late Na+ and Ca2+ currents, reduced repolarizing K+ currents) (Coppini et al., 2013). Two studies are currently under way to test the efficacy of ranolazine on exercise tolerance and diastolic function in symptomatic HCM patients (RESTYLE-HCM, Germany, Menarini) and to treat chest pain or dyspnea in patients with HCM (RHYME, USA) (Spoladore et al., 2012). However, it is not known whether cytosolic Na+ is increased early in the course of the disease (pre-hypertrophic stage) and whether it leads to mitochondrial dysfunction in any/all HCM mutations, or whether high levels of cytosolic Na+ occur after the onset of myocyte hypertrophy and/or symptoms. Most importantly, none of the agents tested clinically have been demonstrated to change disease course in symptomatic patients (Nagueh et al., 2010). Possible reasons may be that the pathophysiology of myocyte hypertrophy is mutation-specific and the extent of hypertrophy (a common clinical endpoint) is only one determinant of prognosis. Another possibility is that all HCM is not created equal and consequently, individualized, mutation-specific therapies need to be developed.
Hypertrophy is a compensatory response to myocardial injury. While hypertension and HCM can both cause left ventricular hypertrophy which may be indistinguishable by clinical imaging, the molecular mechanisms underlying myocyte hypertrophy are probably different based on an early study of cyclosporine, which prevented left ventricular hypertrophy in the TAC (transverse aortic constriction) model (that simulates increased afterload caused by hypertension) (Sussman et al., 1998), but expedited hypertrophy in HCM mice with a mutation (R403Q) in the α-MHC gene (Teekakirikul et al., 2010). Hence there is need for further investigations to clarify the mechanisms underlying the cardiac phenotype in HCM in order to spur development of new therapeutic strategies and pre-clinical screening tests.
Identification of mutations has defined the genetic causes of HCM in 50–60% of HCM patients, but the molecular mechanisms underlying myocyte hypertrophy, fibrosis and ventricular arrhythmias have not been completely elucidated (Force et al., 2010). It is unclear to what extent genetic variants of HCM exhibit a common mechanism of pathogenesis and to what extent they differ. It is also unknown why certain sarcomeric mutations are well tolerated while others are particularly pernicious in patients, but not in animal models.
Based on positive results in animal models, clinical trials have investigated Ca2+ channel blockers and inhibitors of the renin-angiotensin-aldosterone system in the HCM population, with limited success -possible reasons include differences in disease pathophysiology between HCM-causing mutations and differences in physiology between mouse and human myocytes. Hence, studies in human myocytes are needed to confirm results obtained in mouse models and develop therapies that modify the clinical course of disease. Since human heart tissue can only be obtained by heart biopsy or during surgery, it has been difficult to conduct human studies of disease pathophysiology in large numbers of HCM patients at various stages of disease.
Advances in IPSC (induced pluripotent stem cell) technology permit derivation of human cardiac myocytes obtained by differentiation of human IPSCs derived from HCM patients (Matsa et al., 2014). A recent study by the Wu group at Stanford demonstrated that myocytes differentiated from IPSCs (IPSC-CMs) recapitulate the HCM disease phenotype and can serve as a platform to test therapies (Lan et al., 2013). Furthermore, mitochondrial dysfunction resulting from low levels of Frataxin was also reproduced in IPSC-CMs derived from patients with Friedrich's ataxia (Hick et al., 2013). Hence, IPSC-CMs derived from HCM patients could serve as human model systems of HCM to investigate mitochondrial function and molecular mechanisms underlying cardiac phenotype, develop individualized screening tests and drug therapies in HCM patients with known and unknown causal mutations.
HCM is caused by mutations in sarcomeric proteins in 50–60% of patients. These mutations have been shown to increase the energetic cost of tension development. However, it is unclear whether energetic deficits are involved in generation of the cardiac phenotype in all HCM patients and whether mitochondrial dysfunction precedes development of energetic deficits. Further investigation of mitochondrial function, metabolism and its relationship to cardiac function and electrophysiology in animal models of HCM and/or patient-derived myocytes is needed to clarify the molecular mechanisms underlying the cardiac phenotype in HCM and to design therapies that prevent, arrest and reverse the disease phenotype.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Styliani Vakrou was funded by a grant from the Hellenic Cardiology Society. M. Roselle Abraham was funded by the John Taylor Babbitt (JTB) Foundation.
Abozguia, K., Clarke, K., Lee, L., and Frenneaux, M. (2006). Modification of myocardial substrate use as a therapy for heart failure. Nat. Clin. Pract. Cardiovasc. Med. 3, 490–498. doi: 10.1038/ncpcardio0583
Abozguia, K., Elliott, P., McKenna, W., Phan, T. T., Nallur-Shivu, G., Ahmed, I., et al. (2010). Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 122, 1562–1569. doi: 10.1161/CIRCULATIONAHA.109.934059
Abraham, M. R., Bottomley, P. A., Dimaano, V. L., Pinheiro, A., Steinberg, A., Traill, T. A., et al. (2013). Creatine kinase adenosine triphosphate and phosphocreatine energy supply in a single kindred of patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 112, 861–866. doi: 10.1016/j.amjcard.2013.05.017
Adhikari, B. B., Regnier, M., Rivera, A. J., Kreutziger, K. L., and Martyn, D. A. (2004). Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys. J. 87, 1784–1794. doi: 10.1529/biophysj.103.039131
Akar, F. G., Aon, M. A., Tomaselli, G. F., and O'Rourke, B. (2005). The mitochondrial origin of postischemic arrhythmias. J. Clin. Invest. 115, 3527–3535. doi: 10.1172/JCI25371
Aon, M. A., Cortassa, S., Akar, F. G., and O'Rourke, B. (2006). Mitochondrial criticality: a new concept at the turning point of life or death. Biochim. Biophys. Acta 1762, 232–240. doi: 10.1016/j.bbadis.2005.06.008
Aon, M. A., Cortassa, S., Marban, E., and O'Rourke, B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744. doi: 10.1074/jbc.M302673200
Ashrafian, H., McKenna, W. J., and Watkins, H. (2011). Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ. Res. 109, 86–96. doi: 10.1161/CIRCRESAHA.111.242974
Ashrafian, H., Redwood, C., Blair, E., and Watkins, H. (2003). Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 19, 263–268. doi: 10.1016/S0168-9525(03)00081-7
Balaban, R. S. (2009). The role of Ca(2+) signaling in the coordination of mitochondrial atp production with cardiac work. Biochim. Biophys. Acta 1787, 1334–1341. doi: 10.1016/j.bbabio.2009.05.011
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Bayeva, M., and Ardehali, H. (2010). Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr. Hypertens. Rep. 12, 426–432. doi: 10.1007/s11906-010-0149-8
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Blair, E., Redwood, C., Ashrafian, H., Oliveira, M., Broxholme, J., Kerr, B., et al. (2001). Mutations in the gamma(2) subunit of amp-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 10, 1215–1220. doi: 10.1093/hmg/10.11.1215
Blanchard, E., Seidman, C., Seidman, J. G., Lewinter, M., and Maughan, D. (1999). Altered crossbridge kinetics in the alphamhc403/+ mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 84, 475–483. doi: 10.1161/01.RES.84.4.475
Bravo, P. E., Pinheiro, A., Higuchi, T., Rischpler, C., Merrill, J., Santaularia-Tomas, M., et al. (2012). Pet/Ct assessment of symptomatic individuals with obstructive and nonobstructive hypertrophic cardiomyopathy. J. Nucl. Med. 53, 407–414. doi: 10.2967/jnumed.111.096156
Brock, R. (1957). Functional obstruction of the left ventricle; acquired aortic subvalvar stenosis. Guys. Hosp. Rep. 106, 221–238.
Cohen, J., Effat, H., Goodwin, J. F., Oakley, C. M., and Steiner, R. E. (1964). Hypertrophic obstructive cardiomyopathy. Br. Heart J. 26, 16–32. doi: 10.1136/hrt.26.1.16
Coppini, R., Ferrantini, C., Yao, L., Fan, P., Del Lungo, M., Stillitano, F., et al. (2013). Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127, 575–584. doi: 10.1161/CIRCULATIONAHA.112.134932
Cortassa, S., Aon, M. A., O'Rourke, B., Jacques, R., Tseng, H. J., Marban, E., et al. (2006). A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys. J. 91, 1564–1589. doi: 10.1529/biophysj.105.076174
Cortassa, S., O'Rourke, B., and Aon, M. A. (2014). Redox-optimized ros balance and the relationship between mitochondrial respiration and ros. Biochim. Biophys. Acta 1837, 287–295. doi: 10.1016/j.bbabio.2013.11.007
Crilley, J. G., Boehm, E. A., Blair, E., Rajagopalan, B., Blamire, A. M., Styles, P., et al. (2003). Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 41, 1776–1782. doi: 10.1016/S0735-1097(02)03009-7
Critoph, C. H., Patel, V., Mist, B., and Elliott, P. M. (2014). Cardiac output response and peripheral oxygen extraction during exercise among symptomatic hypertrophic cardiomyopathy patients with and without left ventricular outflow tract obstruction. Heart 100, 639–646. doi: 10.1136/heartjnl-2013-304914
Dimitrow, P. P., Undas, A., Wolkow, P., Tracz, W., and Dubiel, J. S. (2009). Enhanced oxidative stress in hypertrophic cardiomyopathy. Pharmacol. Rep. 61, 491–495. doi: 10.1016/S1734-1140(09)70091-X
Elliott, P., and McKenna, W. J. (2004). Hypertrophic cardiomyopathy. Lancet 363, 1881–1891. doi: 10.1016/S0140-6736(04)16358-7
Ertz-Berger, B. R., He, H., Dowell, C., Factor, S. M., Haim, T. E., Nunez, S., et al. (2005). Changes in the chemical and dynamic properties of cardiac troponin t cause discrete cardiomyopathies in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 102, 18219–18224. doi: 10.1073/pnas.0509181102
Figueira, T. R., Barros, M. H., Camargo, A. A., Castilho, R. F., Ferreira, J. C., Kowaltowski, A. J., et al. (2013). Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid. Redox Signal. 18, 2029–2074. doi: 10.1089/ars.2012.4729
Force, T., Bonow, R. O., Houser, S. R., Solaro, R. J., Hershberger, R. E., Adhikari, B., et al. (2010). Research priorities in hypertrophic cardiomyopathy: report of a working group of the national heart, lung, and blood institute. Circulation 122, 1130–1133. doi: 10.1161/CIRCULATIONAHA.110.950089
Gao, W. D., Perez, N. G., Seidman, C. E., Seidman, J. G., and Marban, E. (1999). Altered cardiac excitation-contraction coupling in mutant mice with familial hypertrophic cardiomyopathy. J. Clin. Invest. 103, 661–666. doi: 10.1172/JCI5220
Gao, Z., Rasmussen, T. P., Li, Y., Kutschke, W., Koval, O. M., Wu, Y., et al. (2013). Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ. Res. 112, 309–317. doi: 10.1161/CIRCRESAHA.111.300193
Gauthier, L. D., Greenstein, J. L., O'Rourke, B., and Winslow, R. L. (2013). An integrated mitochondrial ros production and scavenging model: implications for heart failure. Biophys. J. 105, 2832–2842. doi: 10.1016/j.bpj.2013.11.007
Geisterfer-Lowrance, A. A., Kass, S., Tanigawa, G., Vosberg, H. P., McKenna, W., Seidman, C. E., et al. (1990). A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 62, 999–1006. doi: 10.1016/0092-8674(90)90274-I
Georgakopoulos, D., Christe, M. E., Giewat, M., Seidman, C. M., Seidman, J. G., and Kass, D. A. (1999). The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation. Nat. Med. 5, 327–330. doi: 10.1038/6549
Gersh, B. J., Maron, B. J., Bonow, R. O., Dearani, J. A., Fifer, M. A., Link, M. S., et al. (2011). 2011 Accf/Aha guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the american college of cardiology foundation/american heart association task force on practice guidelines. developed in collaboration with the american association for thoracic surgery, american society of echocardiography, American society of nuclear cardiology, heart failure society of america, heart rhythm society, society for cardiovascular angiography and interventions, and society of thoracic surgeons. J. Am. Coll. Cardiol. 58, E212–E260. doi: 10.1016/j.jacc.2011.06.011
Greenberg, M. J., Kazmierczak, K., Szczesna-Cordary, D., and Moore, J. R. (2010). Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc. Natl. Acad. Sci. U.S.A. 107, 17403–17408. doi: 10.1073/pnas.1009619107
Greenberg, M. J., Watt, J. D., Jones, M., Kazmierczak, K., Szczesna-Cordary, D., and Moore, J. R. (2009). Regulatory light chain mutations associated with cardiomyopathy affect myosin mechanics and kinetics. J. Mol. Cell. Cardiol. 46, 108–115. doi: 10.1016/j.yjmcc.2008.09.126
Grover-McKay, M., Schwaiger, M., Krivokapich, J., Perloff, J. K., Phelps, M. E., and Schelbert, H. R. (1989). Regional myocardial blood flow and metabolism at rest in mildly symptomatic patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 13, 317–324. doi: 10.1016/0735-1097(89)90505-6
Gruner, C., Chan, R. H., Crean, A., Rakowski, H., Rowin, E. J., Care, M., et al. (2014). Significance of left ventricular apical-basal muscle bundle identified by cardiovascular magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Eur. Heart J. doi: 10.1093/eurheartj/ehu154. [Epub ahead of print].
Guinto, P. J., Haim, T. E., Dowell-Martino, C. C., Sibinga, N., and Tardiff, J. C. (2009). Temporal and mutation-specific alterations in ca2+ homeostasis differentially determine the progression of Ctnt-related cardiomyopathies in murine models. Am. J. Physiol. Heart Circ. Physiol. 297, H614–H626. doi: 10.1152/ajpheart.01143.2008
Hansford, R. G., and Zorov, D. (1998). Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell. Biochem. 184, 359–369. doi: 10.1023/A:1006893903113
Haq, S., Choukroun, G., Lim, H., Tymitz, K. M., Del Monte, F., Gwathmey, J., et al. (2001). Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation 103, 670–677. doi: 10.1161/01.CIR.103.5.670
He, H., Javadpour, M. M., Latif, F., Tardiff, J. C., and Ingwall, J. S. (2007). R-92l And R-92w mutations in cardiac troponin t lead to distinct energetic phenotypes in intact mouse hearts. Biophys. J. 93, 1834–1844. doi: 10.1529/biophysj.107.107557
Hernandez, O. M., Szczesna-Cordary, D., Knollmann, B. C., Miller, T., Bell, M., Zhao, J., et al. (2005). F110i And R278c troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J. Biol. Chem. 280, 37183–37194. doi: 10.1074/jbc.M508114200
Hick, A., Wattenhofer-Donze, M., Chintawar, S., Tropel, P., Simard, J. P., Vaucamps, N., et al. (2013). Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich's Ataxia. Dis. Model. Mech. 6, 608–621. doi: 10.1242/dmm.010900
Ho, C. Y., Lopez, B., Coelho-Filho, O. R., Lakdawala, N. K., Cirino, A. L., Jarolim, P., et al. (2010). Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N. Engl. J. Med. 363, 552–563. doi: 10.1056/NEJMoa1002659
Ingwall, J. S. (2009). Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 81, 412–419. doi: 10.1093/cvr/cvn301
Jarcho, J. A., McKenna, W., Pare, J. A., Solomon, S. D., Holcombe, R. F., Dickie, S., et al. (1989). Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 321, 1372–1378. doi: 10.1056/NEJM198911163212005
Javadpour, M. M., Tardiff, J. C., Pinz, I., and Ingwall, J. S. (2003). Decreased energetics in murine hearts bearing the R92q mutation in cardiac troponin T. J. Clin. Invest. 112, 768–775. doi: 10.1172/JCI15967
Jung, W. I., Hoess, T., Bunse, M., Widmaier, S., Sieverding, L., Breuer, J., et al. (2000). Differences in cardiac energetics between patients with familial and nonfamilial hypertrophic cardiomyopathy. Circulation 101, E121. doi: 10.1161/01.CIR.101.12.e121
Jung, W. I., Sieverding, L., Breuer, J., Hoess, T., Widmaier, S., Schmidt, O., et al. (1998). 31p Nmr spectroscopy detects metabolic abnormalities in asymptomatic patients with hypertrophic cardiomyopathy. Circulation 97, 2536–2542. doi: 10.1161/01.CIR.97.25.2536
Kembro, J. M., Aon, M. A., Winslow, R. L., O'Rourke, B., and Cortassa, S. (2013). Integrating mitochondrial energetics, redox and ros metabolic networks: a two-compartment model. Biophys. J. 104, 332–343. doi: 10.1016/j.bpj.2012.11.3808
Keren, A., Syrris, P., and McKenna, W. J. (2008). Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat. Clin. Pract. Cardiovasc. Med. 5, 158–168. doi: 10.1038/ncpcardio1110
Kohlhaas, M., Liu, T., Knopp, A., Zeller, T., Ong, M. F., Bohm, M., et al. (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911
Lan, F., Lee, A. S., Liang, P., Sanchez-Freire, V., Nguyen, P. K., Wang, L., et al. (2013). Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113. doi: 10.1016/j.stem.2012.10.010
Lim, D.-S., Lutucuta, S., Bachireddy, P., Youker, K., Evans, A., Entman, M., et al. (2001). Angiotensin Ii blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation 103, 789–791. doi: 10.1161/01.CIR.103.6.789
Lin, X., Liang, H. Y., Pinheiro, A., Dimaano, V., Sorensen, L., Aon, M., et al. (2013). Electromechanical relationship in hypertrophic cardiomyopathy. J. Cardiovasc. Transl. Res. 6, 604–615. doi: 10.1007/s12265-013-9481-0
Liu, T., and O'Rourke, B. (2008). Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 103, 279–288. doi: 10.1161/CIRCRESAHA.108.175919
Liu, T., and O'Rourke, B. (2013). Regulation of the Na+/Ca2+ exchanger by pyridine nucleotide redox potential in ventricular myocytes. J. Biol. Chem. 288, 31984–31992. doi: 10.1074/jbc.M113.496588
Liu, T., Takimoto, E., Dimaano, V. L., Demazumder, D., Kettlewell, S., Smith, G. L., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ. Res. 115, 44–54. doi: 10.1161/CIRCRESAHA.115.303062
Lombardi, R., Rodriguez, G., Chen, S. N., Ripplinger, C. M., Li, W., Chen, J., et al. (2009). Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol-sensitive mechanisms. Circulation 119, 1398–1407. doi: 10.1161/CIRCULATIONAHA.108.790501
Lucas, D. T., Aryal, P., Szweda, L. I., Koch, W. J., and Leinwand, L. A. (2003). Alterations in mitochondrial function in a mouse model of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 284, H575–H583. doi: 10.1152/ajpheart.00619.2002
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O'Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182. doi: 10.1161/01.RES.0000232546.92777.05
Maack, C., and O'Rourke, B. (2007). Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 102, 369–392. doi: 10.1007/s00395-007-0666-z
Marian, A. J. (2002). Modifier genes for hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 17, 242–252. doi: 10.1097/00001573-200205000-00006
Marian, A. J., and Roberts, R. (2001). The molecular genetic basis for hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 33, 655–670. doi: 10.1006/jmcc.2001.1340
Marian, A. J., Senthil, V., Chen, S. N., and Lombardi, R. (2006). Antifibrotic effects of antioxidant N-acetylcysteine in a mouse model of human hypertrophic cardiomyopathy mutation. J. Am. Coll. Cardiol. 47, 827–834. doi: 10.1016/j.jacc.2005.10.041
Maron, B. J. (2002). Hypertrophic cardiomyopathy: a systematic review. JAMA 287, 1308–1320. doi: 10.1001/jama.287.10.1308
Maron, B. J., and Maron, M. S. (2013). Hypertrophic cardiomyopathy. Lancet 381, 242–255. doi: 10.1016/S0140-6736(12)60397-3
Maron, B. J., Olivotto, I., Bellone, P., Conte, M. R., Cecchi, F., Flygenring, B. P., et al. (2002). Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 39, 301–307. doi: 10.1016/S0735-1097(01)01727-2
Maron, M. S., Olivotto, I., Betocchi, S., Casey, S. A., Lesser, J. R., Losi, M. A., et al. (2003). Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N. Engl. J. Med. 348, 295–303. doi: 10.1056/NEJMoa021332
Matsa, E., Burridge, P. W., and Wu, J. C. (2014). Human stem cells for modeling heart disease and for drug discovery. Sci. Transl. Med. 6, 239ps6. doi: 10.1126/scitranslmed.3008921
Mayr, M. (2008). Metabolomics: ready for the prime time? Circ. Cardiovasc. Genet. 1, 58–65. doi: 10.1161/CIRCGENETICS.108.808329
McCormack, J. G., and Denton, R. M. (1990). The role of mitochondrial Ca2+ transport and matrix Ca2+ in signal transduction in mammalian tissues. Biochim. Biophys. Acta 1018, 287–291. doi: 10.1016/0005-2728(90)90269-A
Mettikolla, P., Calander, N., Luchowski, R., Gryczynski, I., Gryczynski, Z., Zhao, J., et al. (2011). Cross-bridge kinetics in myofibrils containing familial hypertrophic cardiomyopathy R58q mutation in the regulatory light chain of myosin. J. Theor. Biol. 284, 71–81. doi: 10.1016/j.jtbi.2011.06.014
Nagueh, S. F., Lombardi, R., Tan, Y., Wang, J., Willerson, J. T., and Marian, A. J. (2010). Atorvastatin and cardiac hypertrophy and function in hypertrophic cardiomyopathy: a pilot study. Eur. J. Clin. Invest. 40, 976–983. doi: 10.1111/j.1365-2362.2010.02349.x
Nakamura, K., Kusano, K. F., Matsubara, H., Nakamura, Y., Miura, A., Nishii, N., et al. (2005). Relationship between oxidative stress and systolic dysfunction in patients with hypertrophic cardiomyopathy. J. Card. Fail. 11, 117–123. doi: 10.1016/j.cardfail.2004.05.005
Nienaber, C. A., Gambhir, S. S., Mody, F. V., Ratib, O., Huang, S. C., Phelps, M. E., et al. (1993). Regional myocardial blood flow and glucose utilization in symptomatic patients with hypertrophic cardiomyopathy. Circulation 87, 1580–1590. doi: 10.1161/01.CIR.87.5.1580
Obayashi, T., Hattori, K., Sugiyama, S., Tanaka, M., Tanaka, T., Itoyama, S., et al. (1992). Point mutations in mitochondrial dna in patients with hypertrophic cardiomyopathy. Am. Heart J. 124, 1263–1269. doi: 10.1016/0002-8703(92)90410-W
Okajima, Y., Tanabe, Y., Takayanagi, M., and Aotsuka, H. (1998). A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (melas). Heart 80, 292–295.
Ommen, S. R., and Gersh, B. J. (2009). Sudden cardiac death risk in hypertrophic cardiomyopathy. Eur. Heart J. 30, 2558–2559. doi: 10.1093/eurheartj/ehp307
O'Rourke, B., Cortassa, S., and Aon, M. A. (2005). Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda) 20, 303–315. doi: 10.1152/physiol.00020.2005
Perrone-Filardi, P., Bacharach, S. L., Dilsizian, V., Panza, J. A., Maurea, S., and Bonow, R. O. (1993). Regional systolic function, myocardial blood flow and glucose uptake at rest in hypertrophic cardiomyopathy. Am. J. Cardiol. 72, 199–204. doi: 10.1016/0002-9149(93)90160-E
Pieske, B., and Houser, S. R. (2003). [Na+]I Handling in the failing human heart. Cardiovasc. Res. 57, 874–886. doi: 10.1016/S0008-6363(02)00841-6
Puglisi, J. L., Goldspink, P. H., Gomes, A. V., Utter, M. S., Bers, D. M., and Solaro, R. J. (2014). Influence of a constitutive increase in myofilament Ca(2+)-sensitivity on Ca(2+)-fluxes and contraction of mouse heart ventricular myocytes. Arch. Biochem. Biophys. 552–553, 50–59. doi: 10.1016/j.abb.2014.01.019
Robinson, P., Griffiths, P. J., Watkins, H., and Redwood, C. S. (2007). Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273. doi: 10.1161/CIRCRESAHA.107.156380
Ross, J. Jr. Braunwald, E., Gault, J. H., Mason, D. T., and Morrow, A. G. (1966). The mechanism of the intraventricular pressure gradient in idiopathic hypertrophic subaortic stenosis. Circulation 34, 558–578. doi: 10.1161/01.CIR.34.4.558
Rotig, A., De Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., et al. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in friedreich ataxia. Nat. Genet. 17, 215–217. doi: 10.1038/ng1097-215
Saks, V., Dzeja, P., Schlattner, U., Vendelin, M., Terzic, A., and Wallimann, T. (2006). Cardiac system bioenergetics: metabolic basis of the frank-starling law. J. Physiol. 571, 253–273. doi: 10.1113/jphysiol.2005.101444
Seddon, M., Looi, Y. H., and Shah, A. M. (2007). Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93, 903–907. doi: 10.1136/hrt.2005.068270
Seidman, J. G., and Seidman, C. (2001). The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104, 557–567. doi: 10.1016/S0092-8674(01)00242-2
Semsarian, C., Ahmad, I., Giewat, M., Georgakopoulos, D., Schmitt, J. P., McConnell, B. K., et al. (2002). The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J. Clin. Invest. 109, 1013–1020. doi: 10.1172/JCI200214677
Senthil, V., Chen, S. N., Tsybouleva, N., Halder, T., Nagueh, S. F., Willerson, J. T., et al. (2005). Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ. Res. 97, 285–292. doi: 10.1161/01.RES.0000177090.07296.ac
Shimada, Y. J., Passeri, J. J., Baggish, A. L., O'Callaghan, C., Lowry, P. A., Yannekis, G., et al. (2013). Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 1, 480–487. doi: 10.1016/j.jchf.2013.09.001
Solaro, R. J., Varghese, J., Marian, A. J., and Chandra, M. (2002). Molecular mechanisms of cardiac myofilament activation: modulation by ph and a troponin t mutant R92q. Basic Res. Cardiol. 97 (Suppl. 1), I102–I110. doi: 10.1007/s003950200038
Solomon, S. D., Jarcho, J. A., McKenna, W., Geisterfer-Lowrance, A., Germain, R., Salerni, R., et al. (1990). Familial hypertrophic cardiomyopathy is a genetically heterogeneous disease. J. Clin. Invest. 86, 993–999. doi: 10.1172/JCI114802
Sorajja, P., Ommen, S. R., Holmes, D. R. Jr. Dearani, J. A., Rihal, C. S., Gersh, B. J., et al. (2012). Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 126, 2374–2380. doi: 10.1161/CIRCULATIONAHA.111.076257
Spindler, M., Saupe, K. W., Christe, M. E., Sweeney, H. L., Seidman, C. E., Seidman, J. G., et al. (1998). Diastolic dysfunction and altered energetics in the Alphamhc403/+ mouse model of familial hypertrophic cardiomyopathy. J. Clin. Invest. 101, 1775–1783. doi: 10.1172/JCI1940
Spirito, P., and Autore, C. (2006). Management of hypertrophic cardiomyopathy. BMJ 332, 1251–1255. doi: 10.1136/bmj.332.7552.1251
Spirito, P., Autore, C., Formisano, F., Assenza, G. E., Biagini, E., Haas, T. S., et al. (2014). Risk of sudden death and outcome in patients with hypertrophic cardiomyopathy with benign presentation and without risk factors. Am. J. Cardiol. 113, 1550–1555. doi: 10.1016/j.amjcard.2014.01.435
Spoladore, R., Maron, M. S., D'Amato, R., Camici, P. G., and Olivotto, I. (2012). Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur. Heart J. 33, 1724–1733. doi: 10.1093/eurheartj/ehs150
Stanley, B. A., Sivakumaran, V., Shi, S., McDonald, I., Lloyd, D., Watson, W. H., et al. (2011). Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J. Biol. Chem. 286, 33669–33677. doi: 10.1074/jbc.M111.284612
Straceski, A. J., Geisterfer-Lowrance, A., Seidman, C. E., Seidman, J. G., and Leinwand, L. A. (1994). Functional analysis of myosin missense mutations in familial hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 91, 589–593. doi: 10.1073/pnas.91.2.589
Sussman, M. A., Lim, H. W., Gude, N., Taigen, T., Olson, E. N., Robbins, J., et al. (1998). Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science 281, 1690–1693. doi: 10.1126/science.281.5383.1690
Szczesna-Cordary, D., Guzman, G., Ng, S. S., and Zhao, J. (2004). Familial hypertrophic cardiomyopathy-linked alterations In Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J. Biol. Chem. 279, 3535–3542. doi: 10.1074/jbc.M307092200
Tadamura, E., Tamaki, N., Matsumori, A., Magata, Y., Yonekura, Y., Nohara, R., et al. (1996). Myocardial metabolic changes in hypertrophic cardiomyopathy. J. Nucl. Med. 37, 572–577.
Tardiff, J. C., Hewett, T. E., Palmer, B. M., Olsson, C., Factor, S. M., Moore, R. L., et al. (1999). Cardiac troponin t mutations result in allele-specific phenotypes in a mouse model for hypertrophic cardiomyopathy. J. Clin. Invest. 104, 469–481. doi: 10.1172/JCI6067
Teare, D. (1958). Asymmetrical hypertrophy of the heart in young adults. Br. Heart J. 20, 1–8. doi: 10.1136/hrt.20.1.1
Teekakirikul, P., Eminaga, S., Toka, O., Alcalai, R., Wang, L., Wakimoto, H., et al. (2010). Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-Beta. J. Clin. Invest. 120, 3520–3529. doi: 10.1172/JCI42028
Terentyev, D., Gyorke, I., Belevych, A. E., Terentyeva, R., Sridhar, A., Nishijima, Y., et al. (2008). Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 103, 1466–1472. doi: 10.1161/CIRCRESAHA.108.184457
Territo, P. R., Mootha, V. K., French, S. A., and Balaban, R. S. (2000). Ca(2+) Activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-Atpase. Am. J. Physiol. Cell Physiol. 278, C423–C435.
Thierfelder, L., Watkins, H., Macrae, C., Lamas, R., McKenna, W., Vosberg, H. P., et al. (1994). Alpha-tropomyosin and cardiac troponin t mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77, 701–712. doi: 10.1016/0092-8674(94)90054-X
Timmer, S. A., Germans, T., Brouwer, W. P., Lubberink, M., Van Der Velden, J., Wilde, A. A., et al. (2011). Carriers of the hypertrophic cardiomyopathy Mybpc3 mutation are characterized by reduced myocardial efficiency in the absence of hypertrophy and microvascular dysfunction. Eur. J. Heart Fail. 13, 1283–1289. doi: 10.1093/eurjhf/hfr135
Tuunanen, H., Kuusisto, J., Toikka, J., Jaaskelainen, P., Marjamaki, P., Peuhkurinen, K., et al. (2007). Myocardial perfusion, oxidative metabolism, and free fatty acid uptake in patients with hypertrophic cardiomyopathy attributable to the asp175asn mutation in the alpha-tropomyosin gene: a positron emission tomography study. J. Nucl. Cardiol. 14, 354–365. doi: 10.1016/j.nuclcard.2006.12.329
Unno, K., Isobe, S., Izawa, H., Cheng, X. W., Kobayashi, M., Hirashiki, A., et al. (2009). Relation of functional and morphological changes in mitochondria to myocardial contractile and relaxation reserves in asymptomatic to mildly symptomatic patients with hypertrophic cardiomyopathy. Eur. Heart J. 30, 1853–1862. doi: 10.1093/eurheartj/ehp184
Watkins, H., Ashrafian, H., and Redwood, C. (2011). Inherited cardiomyopathies. N. Engl. J. Med. 364, 1643–1656. doi: 10.1056/NEJMra0902923
Watkins, H., Macrae, C., Thierfelder, L., Chou, Y. H., Frenneaux, M., McKenna, W., et al. (1993). A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat. Genet. 3, 333–337. doi: 10.1038/ng0493-333
Zhou, L., Cortassa, S., Wei, A. C., Aon, M. A., Winslow, R. L., and O'Rourke, B. (2009). Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea Pig cardiomyocytes. Biophys. J. 97, 1843–1852. doi: 10.1016/j.bpj.2009.07.029
Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier, J. L., and Sollott, S. J. (2000). Reactive oxygen species (ros)-induced ros release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014. doi: 10.1084/jem.192.7.1001
Keywords: hypertrophic cardiomyopathy, mitochondria, calcium handling, bioenergetic deficit, induced pluripotent stem cells (iPSCs)
Citation: Vakrou S and Abraham MR (2014) Hypertrophic cardiomyopathy: a heart in need of an energy bar? Front. Physiol. 5:309. doi: 10.3389/fphys.2014.00309
Received: 11 June 2014; Accepted: 30 July 2014;
Published online: 19 August 2014.
Edited by:
Miguel A. Aon, Johns Hopkins University School of Medicine, USAReviewed by:
David F. Stowe, Medical College of Wisconsin, USACopyright © 2014 Vakrou and Abraham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Roselle Abraham, Division of Cardiology, School of Medicine, Johns Hopkins University, 720 Rutland Ave., Ross 871, Baltimore, 21205 MD, USA e-mail:bWFicmFoYTNAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.