 Patrick F. Connolly
Patrick F. Connolly Richard Jäger2
Richard Jäger2 Howard O. Fearnhead
Howard O. Fearnhead
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 16 April 2014
Sec. Striated Muscle Physiology
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00149
This article is part of the Research Topic Frontiers in Skeletal Muscle Wasting, Regeneration and Stem Cells View all 24 articles
It has become increasingly clear that caspases, far from being merely cell death effectors, have a much wider range of functions within the cell. These functions are as diverse as signal transduction and cytoskeletal remodeling, and caspases are now known to have an essential role in cell proliferation, migration, and differentiation. There is also evidence that apoptotic cells themselves can direct the behavior of nearby cells through the caspase-dependent secretion of paracrine signaling factors. In some processes, including the differentiation of skeletal muscle myoblasts, both caspase activation in differentiating cells as well as signaling from apoptotic cells has been reported. Here, we review the non-apoptotic outcomes of caspase activity in a range of different model systems and attempt to integrate this knowledge.
Caspases are intracellular cysteine proteases that cut after specific aspartic acid residues. In mammals, 18 caspases have been characterized (Table 1), the majority of them playing roles in mediating apoptotic cell-demolition (Cohen, 1997). During apoptosis, these “killer caspases” cleave numerous cellular proteins, and they are the primary effectors responsible for taking apart the cell during apoptosis, playing a major proteolytic role in the disassembly of the nucleus and the cytoskeletal structure (Lüthi and Martin, 2007).
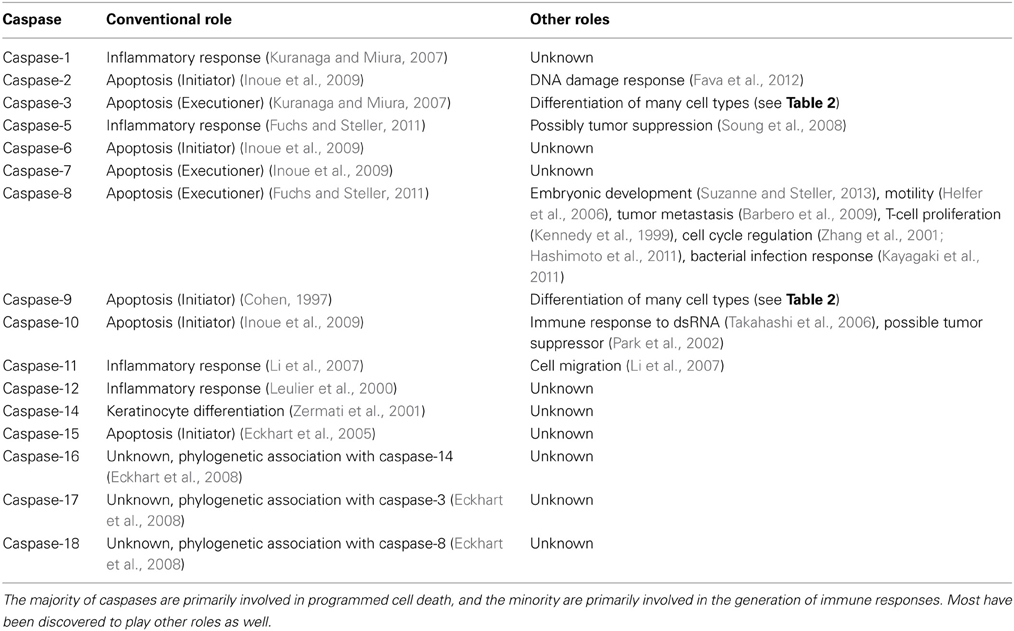
Table 1. Roles of caspases.
Caspases are present in all cells as inactive zymogens, called procaspases. They are activated through cleavage to generate the subunits that form an active caspase (Pop and Salvesen, 2009). At the apex of the activation cascade are the so-called initiator caspases (Table 1). Upon exposure to an apoptotic stimulus they become recruited to specific adaptor proteins which then assemble into activation platforms, which are large multimeric protein complexes mediating activation of the initiator caspases (reviewed in Mace and Riedl, 2010). Initiators activate downstream effectors which rapidly disassemble the cell. The ability of these caspases to kill cells is controlled, in part, by the inhibitor of apoptosis proteins (IAPs) which bind to active caspases and either inhibit proteolytic activity or induce ubiquitin-mediated caspase degradation (Mace et al., 2010).
Different activation platforms classify the main apoptotic pathways. The extrinsic pathway is initiated at the cell membrane by ligands of receptors of the tumor necrosis factor (TNF) receptor family. Ligand-binding leads to assembly of the death-inducing signaling complex (DISC) containing these receptors. Caspase-8 and -10 are recruited to the DISC and then activated, in a process that requires the adaptor protein FADD. The intrinsic pathway is initiated by mitochondria, whose outer membranes become permeable to cytochrome c upon certain cellular stresses. Released cytochrome c then binds to the adaptor protein APAF-1 which subsequently assembles into a large heptameric protein complex, the so-called apoptosome, which is the activation platform for caspase-9. The release of cytochrome c is controlled by proteins of the Bcl-2 family (Tait and Green, 2010). Concomitant with release of cytochrome c, other small proteins may be released, some of which block IAPs allowing for unrestrained caspase activation.
The majority of studies of caspases have focused on their roles as cell killers. There were some notable exceptions describing caspase-dependent cellular differentiation processes that involve denucleation or other degenerative events (Fernando and Megeney, 2007), but these appear to represent a limited or frustrated apoptosis, rather than a fundamentally different process. More recently, the study of apoptotic caspases has broadened to include caspase-dependent paracrine signaling from apoptotic cells to explain how apoptotic cells alter the behavior of surrounding cells (Li et al., 2010a).
However, it has also emerged that apoptosis-associated caspases play non-apoptotic roles and that they are not simply destructive. Examples of these are cell differentiation (Fernando and Kelly, 2002), embryonic development (Miura, 2012; Suzanne and Steller, 2013), motility (Barbero et al., 2009), and compensatory proliferation (Fan and Bergmann, 2008). Within these processes, the “killer” caspases clearly do not cause cell demolition. This gives caspases an entirely new role in determining the fate or behavior of cells. Thus, caspases drive in a far wider range of cellular behaviors than previously known.
There are several theories to explain how apoptotic caspases can lead to non-apoptotic outcomes (Figure 1). In one mechanism, the “cell-autonomous” or “direct” model, caspase activity leads to altered cell behavior through the modulation of regulatory networks, such as through cleavage of cell cycle repressors to alter cell proliferation (Schwerk and Schulze-Osthoff, 2003; Woo et al., 2003), activation of gene transcription to induce skeletal muscle differentiation (Larsen and Megeney, 2010) or cleavage of cytoskeletal proteins to influence cell motility (Helfer et al., 2006). In these cases the caspase activity is “autonomous” in that the entire process of caspase activation, cleavage of substrates, and downstream effects on cell behavior all occur within the same cell.
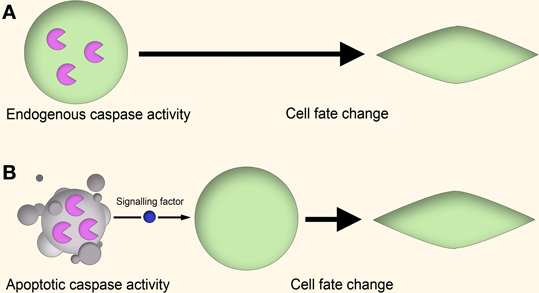
Figure 1. Cell-autonomous vs. non-cell-autonomous models of caspase signaling. (A) Cell-autonomous model. In the proliferating cell, a non-apoptotic caspase signaling pathway leads to a change in cell behavior through modulation of gene expression, cytoskeletal restructuring, or other means. This signaling is endogenous to the proliferating cell itself, and no apoptotic cell signaling is involved. (B) Non-cell-autonomous model. In this model, caspase activity within apoptotic cells lead to the generation of signaling factors which stimulate the cell behavior change of nearby cells in a paracrine fashion.
In this cell autonomous model, it is as yet unclear how apoptotic caspase activity is harnessed for non-apoptotic purposes without killing the cell, although work in Drosophila melanogaster has begun to unravel this problem. For example, a recent report of caspase activation in Drosophila proposes a model in which both the magnitude and rate of caspase activation is controlled, which can give rise to high (apoptotic) levels of caspase activity as well as low (non-apoptotic) levels of activity (Florentin and Arama, 2012). It is also possible that, unlike the traditional model where executioner caspases are only activated upon receipt of a cell stress signal, there is a constant basal level of activated caspases within the cell, but these are normally held in check by inhibitory mechanisms. Such basal levels of caspase activity have been found in the context of cell behavior changes in glioblastoma cells, where low levels of constitutively-active caspase-8 and -3 are found to be necessary for cell migration and invasion (Gdynia et al., 2007). Along with this, relatively high levels of caspases activity may be tolerated if they can be sequestered within their target organelle or sub-cellular region, as is observed in the dendritic pruning of neurons (Williams et al., 2006), in spermatid individualization in Drosophila (Arama et al., 2007; Kaplan et al., 2010), and in the nuclear degradation of keratinocytes (Weil et al., 1999).
In the “non-autonomous” or “indirect” model to explain the role of caspases in non-apoptotic processes, the caspase activity is localized within apoptotic cells, catalyzing the generation of secretory paracrine signaling factors or enabling cell surface-mediated signaling (Hochreiter-Hufford et al., 2013). This model is indirect in that the caspase activity is associated with one cell, while the downstream effect is induced in another cell by an inter-cellular signaling event. In this model the caspase-mediated non-apoptotic effects do not necessarily require the survival of the “caspase-active” cell, as apoptotic cells are still quite capable of signaling to their environment (Jäger and Fearnhead, 2012).
Here, we review the major non-apoptotic roles of caspases discovered to date, and discuss these findings in light of the direct and indirect theories of caspase signaling, with a particular focus on skeletal muscle. This is a rapidly advancing field of study, and a summation of the current state of the field is necessary.
Caspases are key players in the homeostatic balance between apoptosis and regeneration used to maintain tissue structure and function. In response to injury, dead cells engage in a signaling behavior which drives the proliferation of cells at the periphery of the site of injury until damaged portion of tissue is replaced with a new section of the same size and shape (Figure 2) (Bergmann and Steller, 2010). The role of caspases in repair and regeneration has been demonstrated in several different experimental models.
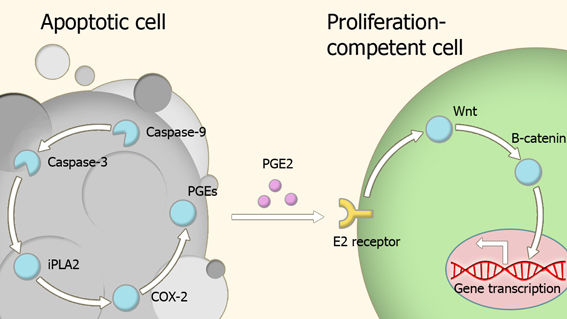
Figure 2. Model of compensatory proliferation. Caspase activity within apoptotic cells leads to the activation of the prostaglandin E2-synthesis pathway. Secreted prostaglandin E2 binds to E2 receptors on proliferation-competent cells, leading to changes in gene expression which trigger proliferation. Abbreviations: iPLA2, Phospholipase A2; COX-2, Cyclooxygenase-2; PGEs, Prostaglandin E synthase; PGE2, Prostaglandin E2.
In the simple metazoan Hydra, surgical-induced injury produces an apoptotic response which stimulates a compensatory proliferative mechanism in surrounding progenitor cells. Treatment with pan-caspase inhibitors abolishes this regenerative response (Cikala et al., 1999; Chera et al., 2009). Regeneration in the amphibian Xenopus requires caspase-mediated events (Tseng et al., 2007), as does tissue regeneration in planaria (Fuchs and Steller, 2011), and the regeneration of newt forelimbs (Vlaskalin et al., 2004).
Regeneration of mammalian tissue is never so dramatic but some tissues, like the liver, can undergo remarkable regeneration after injury (Taub, 2004). Liver regeneration and the healing of skin wounds is impaired in caspase-3 and -7 deficient mice, showing that the role of caspases in regenerative processes is conserved in mammals (Li et al., 2010a).
Paracrine molecules secreted by the apoptotic cells appear to be important in caspase-dependent regeneration. Prostaglandin E2 is one such a molecule, as its production pathway is directly controlled by caspase-3 (Boland et al., 2013), and has a wide number of roles in regeneration and proliferation (Castellone et al., 2005; Goessling et al., 2009; Morata et al., 2011; Beaulieu et al., 2012; Boland et al., 2013). This effect is exerted through transient activation of the Wnt-β-catenin pathway via binding to members of the EP receptor family (Goessling et al., 2009).
Lysophosphatidylcholine (LPC) is another molecule which mediates caspase-activity-induced regenerative responses. It is produced by apoptotic cells, and its presence in the interstitial medium acts as an attraction signal to phagocytes (Lauber et al., 2003). Moreover, LPC induces the differentiation of keratinocytes, which is a necessary step in the wound healing response in skin (Ryborg et al., 2004).
Sphingosine-1-phosphate is another molecule that is secreted by apoptotic cells and is a chemoattractant signal for immune cells (Brecht et al., 2011). It is produced by the enzyme ceramidase, and is a signal that drives growth arrest and differentiation, as well as cell migration and adhesion (Mao and Obeid, 2008), all of which are involved in wound-healing responses.
Fractalkine (CX3CL1) is a large peptide that engages in pro-survival functions in many cell types (White and Greaves, 2012) and is released from apoptotic cells in a caspase-dependant process (Truman et al., 2008). Fractalkine is normally associated with immune cell chemotaxis (Chazaud et al., 2003), but it is also known that soluble fractalkine promotes both migration of endothelial cells and differentiation of osteoblasts (Koizumi et al., 2009). This makes it another potential paracrine signaling factor released by apoptotic cells to modulate tissue regeneration.
From the examples described, it is seen that caspase activity in a dying cell can indirectly induce compensatory proliferation of neighboring cells as part of the regenerative response to injury. Thus, regenerative processes conform to the indirect, apoptotic cell-driven model of caspase function in non-apoptotic processes. However, caspases can also regulate cell proliferation in a cell-autonomous manner through the cleavage of cell cycle regulators.
There are hundreds of confirmed caspase substrates (Lüthi and Martin, 2007; Johnson and Kornbluth, 2008), although the functional significance of cleavage is often uncertain. Among this large group there are several key cell cycle regulators (Hashimoto et al., 2011) and a series of studies have implicated both initiator and effector caspases in the control of the cell cycle of lymphocytes. Because of this, abnormal caspase activity can lead to either depressed or hyperactive cell proliferation.
Through the DISC adaptor protein FADD, caspases-8 and -10 play a role in cell proliferation (Imtiyaz et al., 2009). Peripheral T-cells from FADD deficient mice show a profound impairment of proliferation once they are activated by mitogens or antigens, leading to a reduced number of mature T-cells (Zhang et al., 1998). This inhibition of proliferation results from a failure to enter the cell cycle at the beginning of S-phase due to abnormal expression of cyclin-dependent kinases (Zhang et al., 2001). Pharmacological inhibition of caspases prevents T-cell proliferation in vitro supporting the idea that caspase activity is required for proliferation (Kennedy et al., 1999). The cell cycle role for caspases may not be limited to T-cells as impaired T-cell, B-cell, and NK-cell proliferation is seen in immune-deficient humans with caspase-8 defects (Chun et al., 2002).
As well as caspases-8 and -10, caspase-3 has also been found to play a role in regulating cell proliferation. Proliferative brain cells were found to contain active caspase-3, localized in the nucleus (Oomman et al., 2005). In lymphoid cells, caspase-3 supports the proliferation through cleavage of the CDK inhibitor p27 (Frost et al., 2001). In these examples, caspase-mediated stimulation of proliferation appears to be a cell-autonomous event. On the other hand, proliferating cells utilize caspases in a non-apoptotic capacity to downregulate cell cycle inhibitors which normally keep the cell in a quiescent state (Zhang et al., 2001; Woo et al., 2003; Lamkanfi et al., 2007). For example, caspase-3 can exert a strongly anti-proliferative effect in B-cells through cleavage of p21 and caspase-3 knockout mice show a hyperproliferative phenotype in their B-cells (Woo et al., 2003). Thus, killer caspases seem to be able to exert both positive and negative regulation of cell proliferation through selective cleavage of cell cycle regulators without necessarily inducing apoptosis.
Caspase-1, which is involved in toxin-sensing (Li et al., 1995; Franchi et al., 2009), and caspase-11, involved in the production of inflammatory factors (Kayagaki et al., 2011) have long been associated with inflammation and immunity and are not central to cell death processes. However, it has more recently been discovered that apoptotic caspases also have roles in immunity. This can be through their role in the differentiation programs of immune cells, such as with caspase-8 paralog Dredd (Leulier et al., 2000), or through the modulation of the immune response itself, such as with caspase-12 (Saleh et al., 2004) and caspase-3 paralog ced-3 (Aballay and Ausubel, 2001). Such modulation may occur through the generation of inflammatory and anti-inflammatory factors (Kuranaga and Miura, 2007) or through their role in the apoptosis of infected cells.
Caspases engage in irreversible signal transduction (Kuranaga, 2012). Such irreversible signaling mechanisms are suitable for guiding cell fate choices, such as differentiation. Indeed, such caspase signaling has been found to play important roles in the terminal differentiation programs of several cell types, both in early development, and in tissue regeneration.
The first cell types in which caspases were found to have a direct role in differentiation had one feature in common: their differentiation programs bore a strong resemblance to apoptosis. For example, during terminal differentiation of the lens fiber cells degenerative processes including organelle degradation, chromatin condensation, and DNA fragmentation all occur, and are mediated by the activity of caspases (Ishizaki et al., 1998). The time required for this apoptosis-like process is much longer than that required for caspase-driven cell death, suggesting a more controlled and meticulous version of the same general procedure. Soon after this, erythrocytes and keratinocytes were also found to utilize caspase activity in their terminal differentiation programs (Eckhart et al., 2000; Zermati et al., 2001).
Subsequently, it was found that caspases also play roles in differentiation programs that bore no major similarity to apoptosis. An example of this is the differentiation of peripheral blood monocytes into macrophages, which requires the activation of the caspases-3, -8, and -9 for differentiation (Sordet et al., 2002). Deletion of caspase-3 limits the cytokine-induced differentiation of hematopoietic stem cells (Janzen et al., 2008) and the differentiation ability of iPSCs is enhanced by transient induction of caspase activity (Li et al., 2010b). Several other caspase-dependent cell differentiation programs have been discovered, including those of skeletal myoblasts, osteoblasts, spermatids, placental trophoblast, and embryonic and neural stem cells (Table 2).
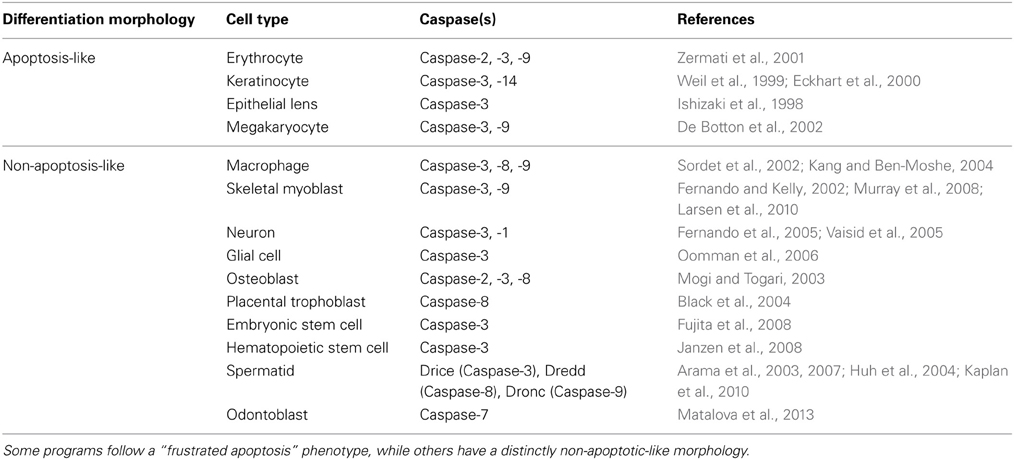
Table 2. Caspase involvement in the differentiation programs of several cell types.
A well-studied example of caspase-stimulated in vitro differentiation is that of mouse muscle myoblasts into multinucleated myotubes (Fernando and Kelly, 2002; Murray et al., 2008; Larsen et al., 2010). In this model capase-3-mediates activation in differentiating mouse myoblasts of a specific DNase called CAD (Larsen et al., 2010). Normally CAD (DFF40 in humans) is bound to a chaperone called ICAD (DFF45) that inhibits the nuclease activity of CAD. Caspase-3 mediated cleavage of ICAD releases CAD, allowing it to cleave DNA. This is a key step in the generation of oligonucleosomal DNA fragments seen in apoptosis. Perhaps surprisingly, activation of CAD occurs in differentiating myoblasts and RNAi directed against CAD causes profound inhibition of myoblast differentiation. Larsen et al. propose that CAD-dependent activation of p21 expression is the key event explaining this defect as p21 expression is an early and necessary event in myoblast differentiation (Larsen et al., 2010). In other words, it is proposed that caspase-3 drives non-apoptotic outcomes by inducing expression of specific genes. In addition, Fernando et al. showed that microinjection of active caspase-3 induced expression of muscle specific genes (Fernando and Kelly, 2002). These two reports support the idea that caspase activity is present in the differentiating myoblast (the direct/autonomous model). Caspase-3 activation during differentiation requires caspase-9 and is blocked by overexpression of Bcl-XL (Murray et al., 2008), which implicates the intrinsic or mitochondrial apoptotic pathway, although the role of cytochrome c release or Apaf-1 in this differentiation has not been conclusively demonstrated.
Although this seems like strong evidence for cell-autonomous model of caspase action, there are also data to support non-autonoumous roles for caspase activity in muscle differentiation. It has been found that myoblast fusion is driven by apoptotic cells through a phosphatidylserine-mediated activation of the BAI1 receptor. During apoptosis, caspase-dependent presentation of phosphatidylserine (PS) on the surface of dying cells is an important “eat me” signal for phagocytes and so plays a central role in the clearance of apoptotic bodies. Adding apoptotic cells to cultures where caspase activity has been abolished with pharmacological inhibitors restores myoblast fusion, and adding annexin V, a PS-binding protein, blocks myoblast fusion (Hochreiter-Hufford et al., 2013). This finding thus supports the non-cell-autonomous model, in that fusion is driven by caspase-mediated presentation of cell-surface signaling factors on apoptotic cells. It may be that both cell autonomous and non-autonomous roles for caspases are important in myoblast differentiation.
A question that arises with the cell-autonomous model of caspase activity; how is this activity prevented from progressing to apoptosis? The convention is that activation of apoptotic caspases is an irreversible threshold event, leading to a runaway process of proteolytic cleavage, culminating in apoptotic cell death. If caspases truly are activated within differentiating cells themselves, there must be some mechanism for restraining, sequestering, or otherwise preventing this activity from killing the cell. Members of the IAP family are important caspase regulators (Mace et al., 2010), but there is so far little evidence that any of these proteins regulates caspase activity during muscle differentiation. Kaplan et al. showed that in spermatids there is a gradient of the giant IAP protein, dBRUCE, that establishes a gradient of caspase activity during the process of spermatid individualization in Drosophila (Kaplan et al., 2010). There is also other evidence for caspase localization being important during differentiation (Table 3). Interestingly, myogenin expressing satellite cells from young donors display active caspase-3 only at the nucleus, whereas myogenin expressing satellite cells from aged donors contain active caspase-3 both at the nucleus and at the cytoplasm. The satellite cells from aged donors also show a higher level of apoptotic cell death and together these data suggest a model in which the failure to properly localize active caspase-3 leads to satellite cell death and impaired muscle regeneration as we age (Fulle et al., 2013).
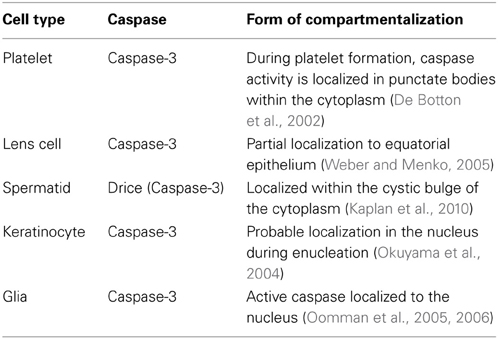
Table 3. Compartmentalization of caspases has been found to be used in the differentiation programs of several cell types.
Moving to in vivo models of muscle differentiation, the role of caspases in regeneration becomes less clear. In caspase-9 (Hakem et al., 1998; Kuida et al., 1998) and caspase-3 (Kuida et al., 1996) knock-out mice embryonic myogenesis appears normal so the role of caspase-9 and caspase-3 in muscle differentiation in vivo at first appears unlikely. However, besides prenatal myogenesis, there is a distinct postnatal muscle development process as well as repair and regeneration processes in adult muscle that have not been evaluated in the caspase knock-out mice. Defects in these processes underlie a range of muscular dystrophies and age-related sarcopenia. In some instances, defects that have profound effects on muscle regeneration do not affect embryonic muscle development. For example, mice lacking caveolin-3 or expressing a Pro104Leu mutation in caveolin-3 (a model for human Limb Girdle Muscular Dystrophy 1C) show normal muscle development but muscle degeneration after 8 weeks of age (Hagiwara et al., 2000; Sunada et al., 2001). It is therefore possible that caspase-driven processes are important primarily in regeneration of adult muscle rather than muscle development but defects have not been observed in caspase deficient mice because of the perinatal lethality associated with these knock outs.
Activation of caspase-8 by TNF induces apoptosis and blocks muscle regeneration in in vivo models of cachexia (Moresi et al., 2008, 2009), data that also appears inconsistent with a model in which caspase activity is required for differentiation. The conflicting reports of the role of caspases in muscle differentiation may be reconciled by a model in which caspase-8 induces high levels of effector caspase activity that kill cells while differentiation is associated with lower effector caspase activity. Just such a switch between death and differentiation has been reported in Drosophila models (Florentin and Arama, 2012). Alternatively, TNF-dependent caspase activation may result in a different localization of active caspases compared to caspase activation associated with differentiation as discussed above for young and aged satellite cells (Fulle et al., 2013).
It is also possible that a particular cellular differentiation process involves more than one caspase-driven step. Muscle differentiation may represent an example of this, with caspase signaling from apoptotic cells as well as caspase activity in the differentiating cells. Differentiating myoblasts may even rely on their caspase activity to drive more than one process during differentiation. Larsen et al. present compelling evidence that caspase-3 mediated DNA damage drives changes in gene expression that are required for myoblast differentiation (Larsen et al., 2010). Others have argued that primary consequence of preventing caspase activation in differentiation is a failure of myoblast fusion (Murray et al., 2008). It is possible that caspases contribute to myoblast fusion by influencing cell motility, as this is required for both muscle development (Brand-Saberi et al., 1996; Molkentin and Olson, 1996) and regeneration (Seale and Rudnicki, 2000).
Cellular locomotion essentially involves the continuous deformation and manipulation of the cytoskeleton to achieve movement. Caspases are the major manipulators of cytoskeletal structure during apoptosis, so it is conceivable that caspases could also have a role in enabling cell motility. In support of this model, in vitro studies have shown that caspase-8-knockout mouse embryonic fibroblasts are both motility-defective, and unable to form proper lamellipodia (Helfer et al., 2006). It is thought that caspase-8 engages in a multiprotein complex with calpain to cleave focal adhesion substrates (Helfer et al., 2006).
Additionally, the embryonic lethality of caspase-8 homozygous knockout mice has been attributed to the failure to develop a functional circulatory system through a defect in endothelial cell migration (Kang and Ben-Moshe, 2004). It is as yet unknown how caspase-8 mediates migration. It could be mediated through activation of downstream effector caspases like caspase-3, leading to modification of the cytoskeleton, or it could act through a separate pathway that does not involve executioner caspases. There is even evidence suggesting that the catalytic activity of caspase-8 is not required for its effects on cell motility (Senft et al., 2007). In addition to caspase-8, caspase-3 has also been implicated in cell motility. Pharmacological inhibition of caspase-3 activity reduces cancer cell motility and invasiveness (Gdynia et al., 2007).
Metastasis of tumor cells involves cellular migration and invasion of tissues, and the subsequent growth of secondary tumors at distant sites. Normally, cells are unable to escape into systemic circulation, as detachment from their basement matrix induces cell death through anoikis or amorphosis (Mehlen and Puisieux, 2006). However, when apoptosis is compromised through silencing of the downstream effector caspase-3, caspase-8 can act to promote metastasis. In this state, caspase-8 enters into a complex with FAK and CPN2, engaging a signaling pathway which induces cell migration (Barbero et al., 2009).
In a Drosophila model of tumor invasion, a non-apoptotic effector caspase pathway is utilized to activate the key invasion protein Mmp1 via JNK signaling (Rudrapatna et al., 2013) (Figure 3). It has been proposed that this cell invasion is achieved through co-opting functions of apoptotic caspase such as cytoskeletal modification. This could be extrapolated as a general feature of non-apoptotic caspase activities in different processes. Together, this suggests a new way of thinking about caspases. Perhaps it is more constructive to think of caspases as cell-structure modifying enzymes rather than as just cell death effectors. This idea is consistent with emerging data showing the role of caspases in neuronal plasticity.
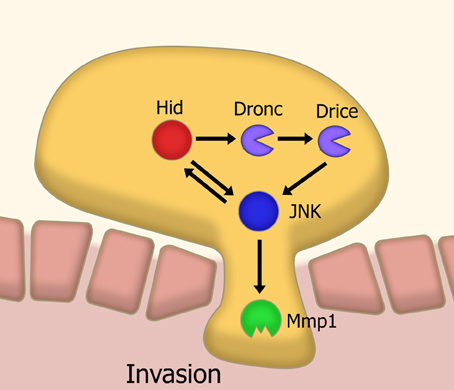
Figure 3. Model of caspase-mediated tissue invasion based on Drosophila studies. A sub-apoptotic level of caspase activation leads to the activation, via JNK signaling, of matrix metalloproteases. This metalloprotease activity is a necessary step in the invasion of tissues. Abbreviations: JNK, Jun kinase; Mmp1, Matrix metalloprotease 1; Hid, head involution defective; Dronc, Drosophila Nedd2-like caspase; Drice, Drosophila ICE.
During early development, live imaging of caspase activity in the brain shows a complex pattern of expression and subcellular localization, occurring in discrete waves (Oomman et al., 2006). These waves of activity correspond to specific periods of brain maturation. Here, we look at the roles of this non-apoptotic caspase activity in neuronal network pruning, synaptic plasticity, signal modulation, and axonal guidance (Hyman and Yuan, 2012).
Pruning of axons and dendrites are the mechanisms through which undesired neural connections are removed. Neural network pruning during larval development in Drosophila is carried out through severing the connection between the outgrowth and the cell body, by means of localized executioner caspase activity, which is mediated by the spatially-restricted degradation of IAP proteins through caspase-3-like activity. An essential step in the process is degradation of DIAP1, a key inhibitor of caspase activity. Inhibition of the caspase-3-ortholog Dronc prevents this pruning process (Kuo et al., 2006) (Figure 4).
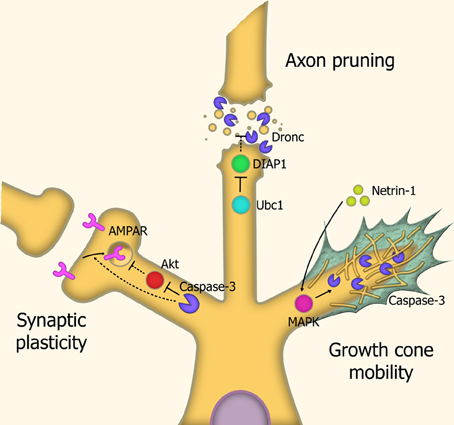
Figure 4. Caspases in neuronal restructuring and signaling. Synaptic plasticity: Through cleavage of Akt, caspase-3 is involved in the endocytosis of AMPA receptors, leading to Long-Term Depression (LTD) of synaptic signaling. Axonal pruning: Localized proteosomal degradation of Inhibitor of Apoptosis proteins (IAPs) within axons lead to a localized caspase activity which shears the axon from the cell body. This process does not kill the parent neuron. Growth cone mobility: Netrin-1 acts as a chemoattractant, signaling MAP Kinase-mediated activation of caspase-3, which remodels the cytoskeletal structure within the growth cone, allowing axonal migration down the chemotrophic gradient. Abbreviations: AMPAR, AMPA receptor; Ubc1, Ubiquitin-conjugating enzyme E2 1; DIAP1, Drosophila Inhibitor of Apoptosis 1; MAPK, Mitogen-activated protein kinase.
In mammals, caspases were found to modulate synaptic plasticity through localized activation within synaptic terminals and neurites in response to stressors. Caspase-3 activity leads to dephosphorylation and internalization of AMPA-type receptors. The loss of these receptors causes degradation of the local dendritic spine. This leads to overall modulation of glutamate signaling. Consequentially, caspases have a role in long-term depression (LTD) of neurons, and overexpression of the anti-apoptotic proteins XIAP or Bcl-xL prevent this LTD (Li et al., 2010c).
Axonal guidance is carried out through the diffusion of molecular signals by the target site, generating a chemotrophic gradient for the axon. Caspases also contribute to this chemotrophic migration by regulating the growth of neurites, through localized proteolytic activity within growth cone structures. Caspase-3 activation is required for this response, as LPA-induced growth cone collapse and netrin-1-induced growth cone attraction are both blocked by caspase-3 inhibitors (Campbell and Holt, 2003). This caspase-3-mediated effect does not require caspase-9 activation, suggesting a distinct, non-canonical activation pathway. It has been speculated that caspase-3 mediated modulation of growth cones is carried out through degradation of cytoskeletal structural elements such as actin and gelsolin (Campbell and Holt, 2003) and rock-1 (Riento and Ridley, 2003).
Such caspase-mediated modulation of synaptic plasticity, axon pruning, and growth cone mobility appear to be cell-autonomous events, in that all utilize localized caspase activity within the target cell, likely through spatially-restricted degradation of inhibitors of caspase activity.
Here, we reviewed the major non-apoptotic roles of caspases discovered to date. We discussed such roles in terms of different cell behaviors such as differentiation, migration, and cell signaling, and presented evidence for the cell autonomous and non-cell-autonomous models of caspase signaling.
In some systems, it seems rather clear that a cell autonomous event is occurring. This is the case in, for example, axonal pruning, where a defined cell autonomous pathway of caspase activation has been elucidated. Other systems appear to be examples of the non-cell-autonomous model. An example of this is the process of compensatory proliferation, whereby caspase-generated signals from apoptotic cells stimulate the proliferation of nearby cells in an intercellular, receptor mediated fashion. Finally, there are systems where the evidence is conflicting. This includes the process of myoblast differentiation, in which there appears to be an essential role for both cell membrane contact with apoptotic, PS exposing cells, and for the cell autonomous caspase activation of nucleases to enable the transcription of myogenic genes. Some further approaches that may prove fruitful for this field include the live imaging of caspase activity in individual cells undergoing differentiation, the identification of soluble mitogenic signaling factors from apoptotic cells, and investigation of the interplay between caspase signaling pathways and other signaling pathways.
Regardless of how caspases are regulated in these models, it seems clear that caspases do indeed have roles other than as effectors of cell death. This new understanding suggests unexpected complications in situations where caspase-dependent cell death is considered desitable, such as in response to cancer chemotherapy. The newfound alternative roles of caspases present the possibility that chemotherapy drugs may induce a wide range of cell behaviors such as increased migration and compensatory proliferation of cancer cells (Jäger and Zwacka, 2010) that are both unexpected and unwanted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aballay, A., and Ausubel, F. M. (2001). Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc. Natl. Acad. Sci. U.S.A. 98, 2735–2739. doi: 10.1073/pnas.041613098
Arama, E., Agapite, J., and Steller, H. (2003). Caspase activity and a specific cytochrome C are required for sperm differentiation in Drosophila. Dev. Cell 4, 687–697. doi: 10.1016/S1534-5807(03)00120-5
Arama, E., Bader, M., Rieckhof, G. E., and Steller, H. (2007). A ubiquitin ligase complex regulates caspase activation during sperm differentiation in Drosophila. PLoS Biol. 5:e251. doi: 10.1371/journal.pbio.0050251
Barbero, S., Mielgo, A., Torres, V., and Teitz, T. (2009). Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 69, 3755–3763. doi: 10.1158/0008-5472.CAN-08-3937
Beaulieu, D., Thebault, P., Pelletier, R., Chapdelaine, P., Tarnopolsky, M., Furling, D., et al. (2012). Abnormal prostaglandin E2 production blocks myogenic differentiation in myotonic dystrophy. Neurobiol. Dis. 45, 122–129. doi: 10.1016/j.nbd.2011.06.014
Bergmann, A., and Steller, H. (2010). Apoptosis, stem cells, and tissue regeneration. Sci. Signal. 3, 1–16. doi: 10.1126/scisignal.3145re8.Apoptosis
Black, S., Kadyrov, M., Kaufmann, P., Ugele, B., Emans, N., and Huppertz, B. (2004). Syncytial fusion of human trophoblast depends on caspase 8. Cell Death Differ. 11, 90–98. doi: 10.1038/sj.cdd.4401307
Boland, K., Flanagan, L., and Prehn, J. (2013). Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. 4, e725. doi: 10.1038/cddis.2013.250
Brand-Saberi, B., Müller, T. S., Wilting, J., Christ, B., and Birchmeier, C. (1996). Scatter factor/hepatocyte growth factor (SF/HGF) induces emigration of myogenic cells at interlimb level in vivo. Dev. Biol. 179, 303–308. doi: 10.1006/dbio.1996.0260
Brecht, K., Weigert, A., Hu, J., Popp, R., Fisslthaler, B., Korff, T., et al. (2011). Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 25, 2408–2417. doi: 10.1096/fj.10-179473
Campbell, D. S., and Holt, C. E. (2003). Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37, 939–952. doi: 10.1016/S0896-6273(03)00158-2
Castellone, M. D., Teramoto, H., Williams, B. O., Druey, K. M., and Gutkind, J. S. (2005). Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310, 1504–1510. doi: 10.1126/science.1116221
Chazaud, B., Sonnet, C., Lafuste, P., Bassez, G., Rimaniol, A. C., Poron, F., et al. (2003). Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 163, 1133–1143. doi: 10.1083/jcb.200212046
Chera, S., Ghila, L., Dobretz, K., Wenger, Y., Bauer, C., Buzgariu, W., et al. (2009). Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 17, 279–289. doi: 10.1016/j.devcel.2009.07.014
Chun, H. J., Zheng, L., Ahmad, M., Wang, J., Speirs, C. K., Siegel, R. M., et al. (2002). Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419, 395–399. doi: 10.1038/nature01063
Cikala, M., Wilm, B., Hobmayer, E., Böttger, A., and David, C. N. (1999). Identification of caspases and apoptosis in the simple metazoan Hydra. Curr. Biol. 9, 959–962. doi: 10.1016/S0960-9822(99)80423-0
De Botton, S., Sabri, S., Daugas, E., Zermati, Y., Guidotti, J. E., Hermine, O., et al. (2002). Platelet formation is the consequence of caspase activation within megakaryocytes. Blood 100, 1310–1317. doi: 10.1182/blood-2002-03-0686
Eckhart, L., Ballaun, C., Hermann, M., Vandeberg, J. L., Sipos, W., Uthman, A., et al. (2008). Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human caspase repertoire. Mol. Biol. Evol. 25, 831–841. doi: 10.1093/molbev/msn012
Eckhart, L., Ballaun, C., Uthman, A., Kittel, C., Stichenwirth, M., Buchberger, M., et al. (2005). Identification and characterization of a novel mammalian caspase with proapoptotic activity. J. Biol. Chem. 280, 35077–35080. doi: 10.1074/jbc.C500282200
Eckhart, L., Declercq, W., Ban, J., Rendl, M., Lengauer, B., Mayer, C., et al. (2000). Terminal differentiation of human keratinocytes and stratum corneum formation is associated with caspase-14 activation. J. Invest. Dermatol. 115, 1148–1151. doi: 10.1046/j.1523-1747.2000.00205.x
Fan, Y., and Bergmann, A. (2008). Apoptosis-induced compensatory proliferation. The Cell is dead. Long live the Cell! Trends Cell Biol. 18, 467–473. doi: 10.1016/j.tcb.2008.08.001
Fava, L. L., Bock, F. J., Geley, S., and Villunger, A. (2012). Caspase-2 at a glance. J. Cell Sci. 125, 5911–5915. doi: 10.1242/jcs.115105.95
Fernando, P., Brunette, S., and Megeney, L. A. (2005). Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J. 19, 1671–1673. doi: 10.1096/fj.04-2981fje
Fernando, P., and Kelly, J. (2002). Caspase 3 activity is required for skeletal muscle differentiation. Proc. Natl. Acad. Sci. U.S.A. 99, 11025–11030. doi: 10.1073/pnas.162172899
Fernando, P., and Megeney, L. A. (2007). Is caspase-dependent apoptosis only cell differentiation taken to the extreme? FASEB J. 21, 8–17. doi: 10.1096/fj.06-5912hyp
Florentin, A., and Arama, E. (2012). Caspase levels and execution efficiencies determine the apoptotic potential of the cell. J. Cell Biol. 196, 513–527. doi: 10.1083/jcb.201107133
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R., and Nuñez, G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247. doi: 10.1038/ni.1703
Frost, V., Al-Mehairi, S., and Sinclair, A. J. (2001). Exploitation of a non-apoptotic caspase to regulate the abundance of the cdkI p27(KIP1) in transformed lymphoid cells. Oncogene 20, 2737–2748. doi: 10.1038/sj.onc.1204367
Fuchs, Y., and Steller, H. (2011). Programmed cell death in animal development and disease. Cell 147, 742–758. doi: 10.1016/j.cell.2011.10.033
Fujita, J., Crane, A. M., Souza, M. K., Dejosez, M., Kyba, M., Flavell, R. A., et al. (2008). Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2, 595–601. doi: 10.1016/j.stem.2008.04.001
Fulle, S., Sancilio, S., Mancinelli, R., Gatta, V., and Di Pietro, R. (2013). Dual role of the caspase enzymes in satellite cells from aged and young subjects. Cell Death Dis. 4, e955. doi: 10.1038/cddis.2013.472
Gdynia, G., Grund, K., Eckert, A., Böck, B. C., Funke, B., Macher-Goeppinger, S., et al. (2007). Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol. Cancer Res. 5, 1232–1240. doi: 10.1158/1541-7786.MCR-07-0343
Goessling, W., North, T. E., Loewer, S., Lord, A. M., Lee, S., Stoick-Cooper, C. L., et al. (2009). Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration Cell 136, 1136–1147. doi: 10.1016/j.cell.2009.01.015
Hagiwara, Y., Sasaoka, T., Araishi, K., Imamura, M., Yorifuji, H., Nonaka, I., et al. (2000). Caveolin-3 deficiency causes muscle degeneration in mice. Hum. Mol. Genet. 9, 3047–3054. doi: 10.1093/hmg/9.20.3047
Hakem, R., Hakem, A., Duncan, G. S., Henderson, J. T., Woo, M., Soengas, M. S., et al. (1998). Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94, 339–352. doi: 10.1016/S0092-8674(00)81477-4
Hashimoto, T., Kikkawa, U., and Kamada, S. (2011). Contribution of caspase(s) to the cell cycle regulation at mitotic phase. PLoS ONE 6:e18449. doi: 10.1371/journal.pone.0018449
Helfer, B., Boswell, B. C., Finlay, D., Cipres, A., Vuori, K., Bong Kang, T., et al. (2006). Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 66, 4273–4278. doi: 10.1158/0008-5472.CAN-05-4183
Hochreiter-Hufford, A. E., Lee, C. S., Kinchen, J. M., Sokolowski, J. D., Arandjelovic, S., Call, J. A., et al. (2013). Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497, 263–267. doi: 10.1038/nature12135
Huh, J. R., Vernooy, S. Y., Yu, H., Yan, N., Shi, Y., Guo, M., et al. (2004). Multiple apoptotic caspase cascades are required in nonapoptotic roles for Drosophila spermatid individualization. PLoS Biol. 2:E15. doi: 10.1371/journal.pbio.0020015
Hyman, B. T., and Yuan, J. (2012). Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 13, 395–406. doi: 10.1038/nrn3228
Imtiyaz, H. Z., Zhou, X., Zhang, H., Chen, D., Hu, T., and Zhang, J. (2009). The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation. J. Biol. Chem. 284, 9917–9926. doi: 10.1074/jbc.M900249200
Inoue, S., Browne, G., Melino, G., and Cohen, G. M. (2009). Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ. 16, 1053–1061. doi: 10.1038/cdd.2009.29
Ishizaki, Y., Jacobson, M. D., and Raff, M. C. (1998). A role for caspases in lens fiber differentiation. J. Cell Biol. 140, 153–158.
Jäger, R., and Fearnhead, H. O. (2012). “Dead Cells Talking”: the silent form of cell death is not so quiet. Biochem. Res. Int. 2012:453838. doi: 10.1155/2012/453838
Jäger, R., and Zwacka, R. M. (2010). The enigmatic roles of caspases in tumor development. Cancers (Basel) 2, 1952–1979. doi: 10.3390/cancers2041952
Janzen, V., Fleming, H. E., Riedt, T., Karlsson, G., Riese, M. J., Lo Celso, C., et al. (2008). Hematopoietic stem cell responsiveness to exogenous signals is limited by caspase-3. Cell Stem Cell 2, 584–594. doi: 10.1016/j.stem.2008.03.012
Johnson, C. E., and Kornbluth, S. (2008). Caspase cleavage is not for everyone. Cell 134, 720–721. doi: 10.1016/j.cell.2008.08.019
Kang, T., and Ben-Moshe, T. (2004). Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 173, 2976–2984.
Kaplan, Y., Gibbs-Bar, L., Kalifa, Y., Feinstein-Rotkopf, Y., and Arama, E. (2010). Gradients of a ubiquitin E3 ligase inhibitor and a caspase inhibitor determine differentiation or death in spermatids. Dev. Cell 19, 160–173. doi: 10.1016/j.devcel.2010.06.009
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kennedy, N. J., Kataoka, T., Tschopp, J., and Budd, R. C. (1999). Caspase activation is required for T cell proliferation. J. Exp. Med. 190, 1891–1896. doi: 10.1084/jem.190.12.1891
Koizumi, K., Saitoh, Y., Minami, T., Takeno, N., Tsuneyama, K., Miyahara, T., et al. (2009). Role of CX3CL1/fractalkine in osteoclast differentiation and bone resorption. J. Immunol. 183, 7825–7831. doi: 10.4049/jimmunol.0803627
Kuida, K., Haydar, T. F., Kuan, C. Y., Gu, Y., Taya, C., Karasuyama, H., et al. (1998). Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94, 325–337. doi: 10.1016/S0092-8674(00)81476-2
Kuida, K., Zheng, T. S., Na, S., Kuan, C., Yang, D., Karasuyama, H., et al. (1996). Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384, 368–372.
Kuo, C. T., Zhu, S., Younger, S., Jan, L. Y., and Jan, Y. N. (2006). Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 51, 283–290. doi: 10.1016/j.neuron.2006.07.014
Kuranaga, E. (2012). Beyond apoptosis: caspase regulatory mechanisms and functions in vivo. Genes Cells 17, 83–97. doi: 10.1111/j.1365-2443.2011.01579.x
Kuranaga, E., and Miura, M. (2007). Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol. 17, 135–144. doi: 10.1016/j.tcb.2007.01.001
Lamkanfi, M., Festjens, N., Declercq, W., Vanden-Berghe, T., and Vandenabeele, P. (2007). Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 14, 44–55. doi: 10.1038/sj.cdd.4402047
Larsen, B. D., and Megeney, L. A. (2010). Parole terms for a killer: directing caspase3/CAD induced DNA strand breaks to coordinate changes in gene expression. Cell Cycle 9, 2940–2945. doi: 10.4161/cc.9.15.12335
Larsen, B. D., Rampalli, S., Burns, L. E., Brunette, S., Dilworth, F. J., and Megeney, L. A. (2010). Caspase 3/caspase-activated DNase promotes cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci. U.S.A. 107, 4230–4235. doi: 10.1073/pnas.0913089107
Lauber, K., Bohn, E., Kröber, S., and Xiao, Y. (2003). Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730. doi: 10.1016/S0092-8674(03)00422-7
Leulier, F., Rodriguez, A., Khush, R. S., Abrams, J. M., and Lemaitre, B. (2000). The Drosophila caspase Dredd is required to resist gram-negative bacterial infection. EMBO Rep. 1, 353–358. doi: 10.1093/embo-reports/kvd073
Li, F., He, Z., Shen, J., Huang, Q., Li, W., Liu, X., et al. (2010b). Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 7, 508–520. doi: 10.1016/j.stem.2010.09.003
Li, F., Huang, Q., Chen, J., Peng, Y., Roop, D. R., and Bedford, J. S. (2010a). Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, 13. doi: 10.1126/scisignal.2000634
Li, J., Brieher, W. M., Scimone, M. L., Kang, S. J., Zhu, H., Yin, H., et al. (2007). Caspase-11 regulates cell migration by promoting Aip1-Cofilin-mediated actin depolymerization. Nat. Cell Biol. 9, 276–286. doi: 10.1038/ncb1541
Li, P., Allen, H., Banerjee, S., Frankli, S., Herzog, L., Johnston, C., et al. (1995). Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80, 401–411.
Li, Z., Jo, J., Jia, J. M., Lo, S. C., Whitcomb, D. J., Jiao, S., et al. (2010c). Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871. doi: 10.1016/j.cell.2010.03.053
Lüthi, A. U., and Martin, S. J. (2007). The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 14, 641–650. doi: 10.1038/sj.cdd.4402103
Mace, P. D., and Riedl, S. J. (2010). Molecular cell death platforms and assemblies. Curr. Opin. Cell Biol. 22, 828–836. doi: 10.1016/j.ceb.2010.08.004
Mace, P. D., Shirley, S., and Day, C. L. (2010). Assembling the building blocks: structure and function of inhibitor of apoptosis proteins. Cell Death Differ. 17, 46–53. doi: 10.1038/cdd.2009.45
Mao, C., and Obeid, L. M. (2008). Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 1781, 424–434. doi: 10.1016/j.bbalip.2008.06.002
Matalova, E., Lesot, H., Svandova, E., Vanden Berghe, T., Sharpe, P. T., Healy, C., et al. (2013). Caspase-7 participates in differentiation of cells forming dental hard tissues. Dev. Growth Differ. 55, 615–621. doi: 10.1111/dgd.12066
Mehlen, P., and Puisieux, A. (2006). Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458. doi: 10.1038/nrc1886
Miura, M. (2012). Apoptotic and nonapoptotic caspase functions in animal development. Cold Spring Harb. Perspect. Biol. 4, 1–16. doi: 10.1101/cshperspect.a008664
Mogi, M., and Togari, A. (2003). Activation of caspases is required for osteoblastic differentiation. J. Biol. Chem. 278, 47477–47482. doi: 10.1074/jbc.M307055200
Molkentin, J. D., and Olson, E. N. (1996). Defining the regulatory networks for muscle development. Curr. Opin. Genet. Dev. 6, 445–453. doi: 10.1016/S0959-437X(96)80066-9
Morata, G., Shlevkov, E., and Pérez-Garijo, A. (2011). Mitogenic signaling from apoptotic cells in Drosophila. Dev. Growth Differ. 53, 168–176. doi: 10.1111/j.1440-169X.2010.01225.x
Moresi, V., Garcia-Alvarez, G., Pristerà, A., Rizzuto, E., Albertini, M. C., Rocchi, M., et al. (2009). Modulation of caspase activity regulates skeletal muscle regeneration and function in response to vasopressin and tumor necrosis factor. PLoS ONE 4:e5570. doi: 10.1371/journal.pone.0005570
Moresi, V., Pristerà, A., Scicchitano, B. M., Molinaro, M., Teodori, L., Sassoon, D., et al. (2008). Tumor necrosis factor-alpha inhibition of skeletal muscle regeneration is mediated by a caspase-dependent stem cell response. Stem Cells 26, 997–1008. doi: 10.1634/stemcells.2007-0493
Murray, T. V., McMahon, J. M., Howley, B. H., Stanley, A., Ritter, T., Mohr, A., et al. (2008). A non-apoptotic role for caspase-9 in muscle differentiation. J. Cell Sci. 121, 3786–3793. doi: 10.1242/jcs.024547
Okuyama, R., Nguyen, B. C., Talora, C., Ogawa, E., Tommasi di Vignano, A., Lioumi, M., et al. (2004). High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev. Cell 6, 551–562. doi: 10.1016/S1534-5807(04)00098-X
Oomman, S., Strahlendorf, H., Dertien, J., and Strahlendorf, J. (2006). Bergmann glia utilize active caspase-3 for differentiation. Brain Res. 1078, 19–34. doi: 10.1016/j.brainres.2006.01.041
Oomman, S., Strahlendorf, H., Finckbone, V., and Strahlendorf, J. (2005). Non-lethal active caspase-3 expression in Bergmann glia of postnatal rat cerebellum. Brain Res. Dev. Brain Res. 160, 130–145. doi: 10.1016/j.devbrainres.2005.07.010
Park, W. S., Lee, J. H., Shin, M. S., Park, J. Y., Kim, H. S., Lee, J. H., et al. (2002). Inactivating mutations of the caspase-10 gene in gastric cancer. Oncogene 21, 2919–2925. doi: 10.1038/sj.onc.1205394
Pop, C., and Salvesen, G. S. (2009). Human caspases: activation, specificity, and regulation. J. Biol. Chem. 284, 21777–21781. doi: 10.1074/jbc.R800084200
Riento, K., and Ridley, A. J. (2003). Rocks: multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 4, 446–456. doi: 10.1038/nrm1128
Rudrapatna, V. A., Bangi, E., and Cagan, R. L. (2013). Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep. 14, 172–177. doi: 10.1038/embor.2012.217
Ryborg, A. K., Johansen, C., Iversen, L., and Kragballe, K. (2004). Lysophosphatidylcholine induces keratinocyte differentiation and upregulation of AP-1- and NF-kappaB DNA-binding activity. Acta Derm. Venereol. 84, 433–438. doi: 10.1080/00015550410016930
Saleh, M., Vaillancourt, J. P., Graham, R. K., and Huyck, M. (2004). Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429, 75–79. doi: 10.1038/nature02502.1
Schwerk, C., and Schulze-Osthoff, K. (2003). Non-apoptotic functions of caspases in cellular proliferation and differentiation. Biochem. Pharmacol. 66, 1453–1458. doi: 10.1016/S0006-2952(03)00497-0
Seale, P., and Rudnicki, M. A. (2000). A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 218, 115–124. doi: 10.1006/dbio.1999.9565
Senft, J., Helfer, B., and Frisch, S. M. (2007). Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 67, 11505–11509. doi: 10.1158/0008-5472.CAN-07-5755
Sordet, O., Rébé, C., Plenchette, S., Zermati, Y., Hermine, O., Vainchenker, W., et al. (2002). Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 100, 4446–4453. doi: 10.1182/blood-2002-06-1778
Soung, Y. H., Jeong, E. G., Ahn, C. H., Kim, S. S., Song, S. Y., Yoo, N. J., et al. (2008). Mutational analysis of caspase 1, 4, and 5 genes in common human cancers. Hum. Pathol. 39, 895–900. doi: 10.1016/j.humpath.2007.10.015
Sunada, Y., Ohi, H., Hase, A., Ohi, H., Hosono, T., Arata, S., et al. (2001). Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity. Hum. Mol. Genet. 10, 173–178. doi: 10.1093/hmg/10.3.173
Suzanne, M., and Steller, H. (2013). Shaping organisms with apoptosis. Cell Death Differ. 20, 669–675. doi: 10.1038/cdd.2013.11
Tait, S. W. G., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Takahashi, K., Kawai, T., Kumar, H., Sato, S., Yonehara, S., and Akira, S. J. (2006). Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J. Immunol. 176, 4520–4524.
Taub, R. (2004). Liver regeneration: from myth to mechanism. Nat. Rev. Mol. Cell Biol. 5, 836–847. doi: 10.1038/nrm1489
Truman, L. A., Ford, C. A., Pasikowska, M., Pound, J. D., Wilkinson, S. J., Dumitriu, I. E., et al. (2008). CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036. doi: 10.1182/blood-2008-06-162404
Tseng, A. S., Adams, D. S., Qiu, D., Koustubhan, P., and Levin, M. (2007). Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev. Biol. 301, 62–69. doi: 10.1016/j.ydbio.2006.10.048
Vaisid, T., Kosower, N. S., and Barnoy, S. (2005). Caspase-1 activity is required for neuronal differentiation of PC12 cells: cross-talk between the caspase and calpain systems. Biochim. Biophys. Acta 1743, 223–230. doi: 10.1016/j.bbamcr.2005.01.001
Vlaskalin, T., Wong, C. J., and Tsilfidis, C. (2004). Growth and apoptosis during larval forelimb development and adult forelimb regeneration in the newt (Notophthalmus viridescens). Dev. Genes Evol. 214, 423–431. doi: 10.1007/s00427-004-0417-1
Weber, G. F., and Menko, A. S. (2005). The canonical intrinsic mitochondrial death pathway has a non-apoptotic role in signaling lens cell differentiation. J. Biol. Chem. 280, 22135–22145. doi: 10.1074/jbc.M414270200
Weil, M., Raff, M. C., and Braga, V. M. (1999). Caspase activation in the terminal differentiation of human epidermal keratinocytes. Curr. Biol. 9, 361–364. doi: 10.1016/S0960-9822(99)80162-6
White, G. E., and Greaves, D. R. (2012). Fractalkine: a survivor's guide: chemokines as antiapoptotic mediators. Arterioscler. Thromb. Vasc. Biol. 32, 589–594. doi: 10.1161/ATVBAHA.111.237412
Williams, D. W., Kondo, S., Krzyzanowska, A., Hiromi, Y., and Truman, J. W. (2006). Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236. doi: 10.1038/nn1774
Woo, M., Hakem, R., Furlonger, C., Hakem, A., Duncan, G. S., Sasaki, T., et al. (2003). Caspase-3 regulates cell cycle in B cells: a consequence of substrate specificity. Nat. Immunol. 4, 1016–1022. doi: 10.1038/ni976
Zermati, Y., Garrido, C., Amsellem, S., Fishelson, S., Bouscary, D., Valensi, F., et al. (2001). Caspase activation is required for terminal erythroid differentiation. J. Exp. Med. 193, 247–254. doi: 10.1084/jem.193.2.247
Zhang, J., Cado, D., Chen, A., Kabra, N., and Winoto, A. (1998). Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392, 296–300. doi: 10.1038/32681
Keywords: caspase, apoptosis, myogenesis, proliferation, differentiation, non-apoptotic roles
Citation: Connolly PF, Jäger R and Fearnhead HO (2014) New roles for old enzymes: killer caspases as the engine of cell behavior changes. Front. Physiol. 5:149. doi: 10.3389/fphys.2014.00149
Received: 17 December 2013; Accepted: 28 March 2014;
Published online: 16 April 2014.
Edited by:
Carlos Hermano J. Pinheiro, University of São Paulo, BrazilReviewed by:
Sergio Adamo, Sapienza University of Rome, ItalyCopyright © 2014 Connolly, Jäger and Fearnhead. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Howard O. Fearnhead, National University of Ireland Galway, Biological Sciences Building, University Road, Galway, Ireland e-mail:aG93YXJkLmZlYXJuaGVhZEBudWlnYWx3YXkuaWU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.