 Sandy T. Liu1,2
Sandy T. Liu1,2 Hung Pham
Hung Pham Stephen J. Pandol
Stephen J. Pandol Andrzej Ptasznik
Andrzej Ptasznik- 1Department of Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, USA
- 2Department of Medicine, David Geffen School of Medicine, University of California-Los Angeles, CA, USA
- 3Department of Medicine, Veterans Affairs, Los Angeles, CA, USA
Although a causal link between chronic inflammation and cancer has been established, the exact molecular mechanism linking inflammation to cancer remains largely unknown. It was previously postulated that molecular switches responsible for cancer cell development, and for infiltration of inflammatory cells into cancer, were divided into a distinct set of intracellular proteins and signaling pathways. However, recent evidence suggests that both tumor cells and tumor-infiltrating immune cells utilize the same kinases, mostly that of Src family, to facilitate cancer development and progression. In the past few years several groups have found that Src activation both in cancer and inflammatory cells is mainly driven by pro-inflammatory cytokines within the tumor microenvironment. Here we evaluate the cross talks between Src kinase pathways in immune cells and cancer cells. We conclude that Src might serve as a critical mechanistic link between inflammation and cancer, mediating and propagating a cycle between immune and tissue cells that can ultimately lead to the development and progression of cancer.
Introduction
Inflammation is a vital defensive response that serve critical roles in a variety of physiological situations, and when dysregulated, can contribute to the pathogenesis of many diseases. Chronic inflammation is a well-documented risk for promoting cancer (Coussens and Werb, 2002; Balkwill et al., 2005; Mantovani et al., 2008), particularly in the pancreas and GI tract (Guerra et al., 2007; Terzić et al., 2010). Chronic pancreatitis is long-standing inflammation of the pancreas associated with an increased risk (~20-fold) for pancreatic cancer. This projects a serious clinical problem as pancreatic cancer is a highly lethal disease with the worst prognosis of all the major malignancies; for all stages combined, and a 5-year survival rate of 5% (Yadav et al., 2011). Similarly, uncontrolled inflammatory bowel disease poses a significant risk factor for colorectal cancer. When compared to the general population matched for age, sex, and years at risk, there is a 18-fold increase in Crohn's disease and a 19-fold increase in ulcerative colitis, (Bernstein et al., 2001; Eaden et al., 2001; Itzkowitz and Yio, 2004; Ullman and Itzkowitz, 2011). Interestingly, many environmental cancer risk factors, including alcohol overuse, smoking, chronic infections and obesity, can trigger some form of chronic inflammation, largely in the pancreas and colon (Trinchieri, 2012). These environmental risk factors seemingly facilitate the development and progression of cancer mostly through the induction of chronic persistent inflammation in these tissues.
Although many studies point to an association between inflammation and cancer, the mechanistic signaling basis of this linkage is not well understood. The importance of Src family kinases in colon and pancreatic cancer development is known for many years and is well established (Staley et al., 1997; Lutz et al., 1998; Aligayer et al., 2002). Recent evidence has shown that Src signaling network is also very important in movement and infiltration of immune cells into tumor (Balkwill, 2004; Kulbe et al., 2004). Several groups have found that Src activation in cancer and immune inflammatory cells are mediated by inflammatory cytokines within the tumor microenvironment. Given that Src is overactive in both tumor cells and in tumor-infiltrating immune cells, and is also involved in cytokine-mediated cross talk between cancer and inflammatory cells—Src may be a critical link between inflammation and cancer. We illustrate and expound on this concept using the model of chronic pancreatitis and pancreatic cancer.
Persistent Inflammation Increases Cancer Risk in Pancreas
Chronic pancreatitis highlights an important role for chronic inflammation in the development of cancer. Chronic pancreatitis is the most consistent risk factor for pancreatic cancer and alone increases the risk of developing pancreatic cancer by 10–20-fold (Dítě et al., 2012). Many of the environmental cancer risk factors can initially induce chronic inflammation that subsequently leads to pancreatic cancer. Recurrent pancreatic injury from alcohol abuse, smoking, high-fat diet, diabetes, and genetic predisposition, induces a pro-inflammatory environment consisting of various types of immune cells, cytokines, chemokines, and growth factors that, when dysregulated and persistent, can ultimately lead to the development and progression of cancer (Lowenfels et al., 2001; Shoelson et al., 2007; Pannala et al., 2009; Momi et al., 2012).
Alcohol abuse is a major cause of acute and chronic pancreatitis. The disease usually presents as an acute episode of pancreatitis and progress with additional exacerbations that can lead to chronic pancreatitis, characterized by a sequence of necrotic and fibrotic events. The initial tissue injures are associated with cytokine release during necro-inflammation and appears to include premature intracellular activation of digestive enzymes, leading to autodigestion. Alcohol metabolism causes release of endogenous hydrolases from pancreatic lysozymes, which are responsible for premature activation of trypsinogen leading to intrapancreatic autodigestion and inflammation (Talamini et al., 1999). Reactive oxygen species generated results in further pancreatic tissue injury, and further release of pro-inflammatory cytokines and chemokines (Shi et al., 2005).
In addition, alcohol when combined with cigarette smoking exacerbates the chronic inflammatory process (Go et al., 2005; Maisonneuve et al., 2005; Wiśniewska et al., 2013). Cigarette smoking contributes to the development of chronic pancreatitis by inducing cytokine release and inflammation. Smoking is the major risk factor for the development of pancreatic cancer accounting for 20–30% of cases (Lowenfels et al., 2001). In experimental models, nicotine stimulated an acute inflammatory reaction in the pancreas, which progressed to chronic pancreatitis after repeated sessions of smoking-induced acute pancreatic inflammation. These nicotine-induced inflammatory events are clearly associated with the release of pro-inflammatory cytokines (Nordskog et al., 2003).
Both central and overall obesity are associated with increased risk for pancreatic cancer (Pannala et al., 2009). Although the exact mechanism of obesity to pancreatic cancer is unclear, the major issues revolve around chronic inflammation, glucose intolerance, hyperinsulinemia, insulin resistance, and oxidative stress. Inflammation, along with the immune system plays a vital role in the development of insulin resistance, diabetes, and ultimately pancreatic cancer. Adipose tissue is involved in the release of cytokines and chemokines including TNF-α, IL-6, MCP-1, CXCL12, CCL5, CCL20, that lead to the recruitment of pro-inflammatory cells into adipose tissue (Shoelson et al., 2007; Sell et al., 2012). Obese individuals also exhibit lower circulating levels of anti-inflammatory adipokines that sustains a low-grade systemic inflammation.
Hereditary pancreatitis is a rare autosomal dominant condition caused by gain-of-function mutations in the cationic trypsinogen gene (PRSS1) and is responsible for <1% of all forms of pancreatitis. Mutant PRSS1 gene causes premature activation or impairs the deactivation of trypsin leading to recurrent injury, cytokine release, and inflammation. The risk of developing pancreatic cancer is 53 times higher when compared to the risk in unaffected individuals. Of the patients who progress to chronic pancreatitis, the risk of developing pancreatic cancer by age 70 years is approximately 40%. Pancreatic inflammation also occurs at a much younger age in this group of patients. In addition, Lowenfels et al. reported a 2-fold increased risk of developing pancreatic cancer in smokers with hereditary pancreatitis as compared to non-smokers. Pancreatic cancer also developed 20 years earlier in smokers than in non-smokers (Howes et al., 2004; Rebours et al., 2009), suggesting that nicotine-induced release of cytokines and inflammation can rapidly accelerate the promotion and development of cancer in these patients.
Inflammatory Cells Infiltrate Tumor in Pancreas
Since the role of various immune cells (including lymphocytes, granulocytes, and macrophages) in pancreatic inflammation and cancer has been discussed elsewhere (Mantovani et al., 2008), this review will focus on studies of macrophages as Src kinase-dependent and cytokine-mediated linkage between inflammation and cancer seems most apparent in these cells. Tumor-associated macrophages are key players in pancreatic inflammation and cancer and an important source of cytokines (Feig et al., 2012; Liou et al., 2013). As described above, chronic pancreatitis is often initiated by environmental risk factors, leads to permanent damage of pancreas, and is a consistent risk factor for pancreatic cancer. Chronic pancreatitis is characterized by marked stroma formation with a high number of infiltrating macrophages and myofiroblastic-like stellate cells, which are believed to play a central role in initiating inflammation and disease progression (Erkan et al., 2012). In response to pancreatic injury (alcohol abuse, cigarette smoking, obesity, mutations in genetically predisposed persons, etc.), inflammatory signals and chemokines production are upregulated leading to infiltration of leukocytes and stellate cells to the damaged acinar cells. Inflammatory cells that are recruited in turn secrete several cytokines, including chemokines, interleukins, and interferons, that contribute to cancer growth, invasion, and metastasis (Figure 1, Table 1).
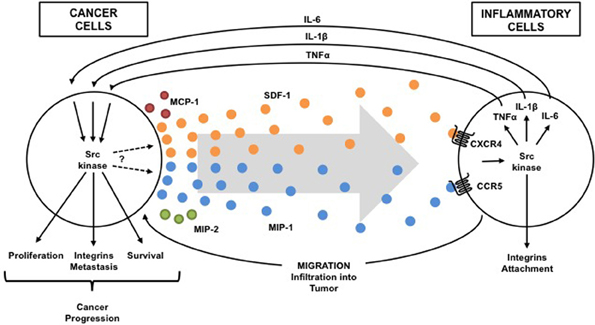
Figure 1. Src-dependent cross talk between inflammatory and cancer cells. Src activation is driven in both inflammatory cells and cancer cells by pro-inflammatory cytokines within the tumor microenvironment. Activation of Src kinases in immune cells by tumor-secreted cytokines (chemokines: SDF-1, MIP-1, MCP-1, MIP-2, etc.) induces production of cytokines (TNF-a, IL-1b, IL-6, etc.) that reciprocally activate Src in cancer cells to promote cancer progression and to induce more cytokine (chemokine) that attract more inflammatory cells into the tumor. Thus, a Src-dependent condition exists in which one problem causes another problem that makes the first problem worse (vicious cycle).
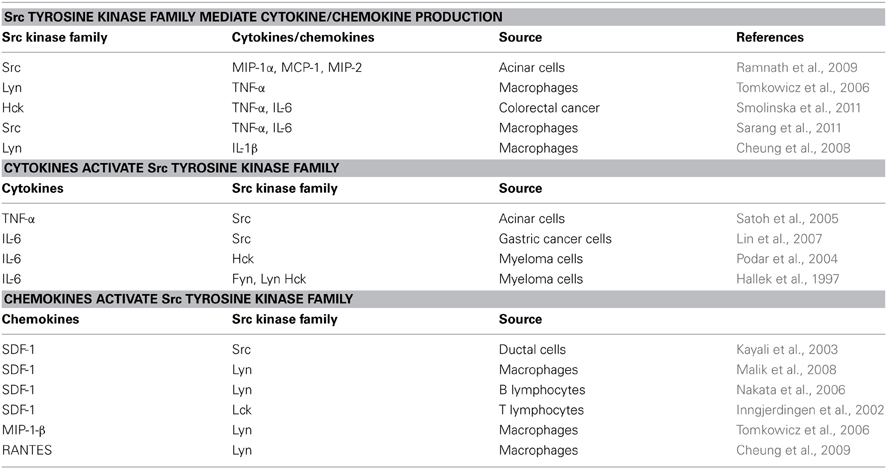
Table 1. Src tyrosine kinase family and cytokine/chemokine interaction in immune and cancer cells.
Numerous experimental studies have suggested an important role of macrophages in generating the microenvironment for both chronic pancreatitis and tumor cells, thus highlighting a similarity between stroma composition in chronic pancreatitis and pancreatic cancer. Macrophages are derived from circulating peripheral monocytes mostly in response to chemokine monocyte chemoattractant protein 1 (MCP-1). Several other chemokines, including MIP-1, MIP-2, and SDF-1, are also increased at the site of inflammation attracting leukocytes and tissue precursors to the injured pancreas (Spaeth et al., 2008). In turn, macrophages, other leukocytes, and stellate cells, which all infiltrate the tumor, release cytokines, including IL-1, IL-6 and TNF that directly effect cancer cell proliferation, and movement/attachment. This process promotes cancer development and progression (Figure 1, Table 1). It is also possible that the cytokine-mediated persistent activation of certain key intracellular signaling pathways, which occurs during chronic inflammation, might inhibit apoptosis and prevent the elimination of genetically altered, precancerous and cancerous cells.
Src Activation Contributes to Both Inflammation and Cancer in Pancreas
Src was the first transforming protein discovered and isolated (Rous, 1911; Stehelin et al., 1976; Brugge and Erikson, 1997) and was also the first gene product with protein tyrosine kinase activity (Hunter and Sefton, 1980). The Src family kinases comprise of nine non-receptor protein tyrosine kinases that share similar structure and function. Src family kinases have a critical role in cell adhesion, proliferation, survival, and invasion, including cell movement, and activation of cytokine receptors. Numerous groups have found that hyper-activation and/or overexpression of Src family kinases are critical to various types of cancers.
Expression of several members of the Src-family tyrosine kinases, including Src, Fyn, Yes, Fgr and Lyn has been demonstrated in pancreatic cancer cell lines and primary cells. The expression of Lyn kinase is the most abundant in these cells (Fu et al., 2006). Numerous studies have shown that elevated Src-family kinase activity in human pancreatic carcinomas (when compared to normal pancreatic cells) not only contributes to pancreatic cancer growth, but also to invasion and metastasis (Lutz et al., 1998; Trevino et al., 2006; Yokoi et al., 2011). Src kinases and oncogenic Ras, PI3K, p38MAPK and Dynamin-2 have been shown to co-operatively stimulate the growth, metastatic migration and invasion of pancreatic carcinoma (Summy et al., 2005; Shields et al., 2011).
Src activation has been observed in circulating blood monocytes and tissue macrophages in chronic pancreatitis, as well as in tumor-associated macrophages and acinar cells in pancreatic cancer (Yokoi et al., 2011). Elevated level of activity of Src in inflammatory monocytes/macrophages was proposed as a biomarker for pancreatic cancer (Coppola, 2000; Yokoi et al., 2011). However, no oncogenic mutations responsible for Src activation in inflammatory and cancer cells in the pancreas have yet been identified. Thus, Src activation is likely a result of underlying inflammation and the consequence of a cytokine-mediated inflammatory microenvironment during malignant transformation and progression. It seems that the signal activating Src kinases is within the inflammatory microenvironment without the necessity of the Src mutation. Consequently, several groups have found that Src activation is driven by pro-inflammatory cytokines, and inversely, the cytokine production is driven by Src kinases, in various types of cancer and inflammatory cells, as summarized in Table 1.
As previously discussed, in response to pancreatic injury, chemokine production is upregulated leading to infiltration of leukocytes and stellate cells to the injured acinar cells. Rather limited information is available on the exact role of Src kinases in chemokine production in pancreatic inflammation and cancer (note the question mark in the Figure 1). However, Src kinases involvement in the secretion of several chemokines was demonstrated in pancreatic acinar cells (Ramnath et al., 2009) and ductal cells (Ungefroren et al., 2011). The pretreatment of pancreatic acini with Src kinase inhibitors markedly decreased MCP-1, MIP-1, and MIP-2 production after stimulation with the substance-P (Ramnath et al., 2009). Substance-P is known to play a role in pathogenesis of cerulein-induced pancreatitis and pancreatic cancer invasion (Ramnath and Bhatia, 2006; Ito et al., 2007).
Accordingly, it also has been shown that the expression of CCR5 receptor for MIP-1, MCP-2 and RANTES, is upregulated in chronic pancreatitis in human tissue, as compared with the healthy pancreas, and the majority of CCR5-positive cells were infiltrating macrophages (Goecke et al., 2000). Similarly, the expression of the CCR5 chemokine receptor and its ligands (MIP-1, MCP-2, RANTES) was significantly increased in the mouse pancreas during cerulein-induced pancreatitis (Goecke et al., 2000; Duell et al., 2006). On the other hand, the SDF-1 chemokine signaling in pancreas and in the other tissues is also dependent on Src family kinases (Takatomo et al., 2000; Nakata et al., 2006; Malik et al., 2008). Src family kinases are downstream intracellular targets of CXCR4 receptor, and are required for the SDF-1—mediated cell movement and attachment (Nakata et al., 2006; Malik et al., 2008). The SDF-1-CXCR4 ligand receptor axis induces pancreatic cancer cell invasion, and the Src-mediated SDF-1 signaling is also an obligatory component of pancreatic regeneration (Takatomo et al., 2000; Kayali et al., 2003; Gao et al., 2010).
In addition, we have previously shown the SDF-1-CXCR4-Src signaling axis is crucial for the movement and invasiveness of inflammatory leukocytes, in a variety of pathological contexts ranging from inflammation to cancer (Nakata et al., 2006; Chen et al., 2008; Malik et al., 2008). Several studies have shown that in human primary leukocytes, Src family members, particularly Lyn and Lck, are required for CXCR4-dependent cell movement and infiltration into various inflamed tissues (Inngjerdingen et al., 2002; Malik et al., 2008). The SDF-1-mediated activation of Lyn kinase in monocytes, modifies integrin activity through inside-out signaling, and transiently destabilizes monocyte/endothelial cell interactions, facilitating full monocyte detachment from endothelium and penetration into inflamed tissue (Nakata et al., 2006; Chen et al., 2008; Malik et al., 2008). Importantly, Lyn is also required for TNF-α and IL-1β production in inflammatory macrophages during stimulation with the CCR5 receptor ligands (Tomkowicz et al., 2006; Cheung et al., 2008, 2009). The other Src family members, Src and Hck, have been shown to play a critical role in IL-6 production in osteoblasts and inflammatory macrophages, respectively (Smolinska et al., 2011; Peruzzi et al., 2012). IL-6 is required for the maintenance and progression of pancreatic cancer precursor lesions, and thus is required for pancreatic cancer growth (Zhang et al., 2013).
In summary, Src family kinases have been demonstrated to be important in the activation of macrophage, dendritic cells, neutrophils and natural killer cells in normal tissues (Ptasznik et al., 1995, 1996; Abram and Lowell, 2008; Malik et al., 2008). It has also been shown to control production of cytokine TNF-alpha stimulated by LPS in normal cells (Orlicek et al., 1999; Sarang et al., 2011; Okenwa et al., 2013). Thus, Src affects both innate and adaptive immune responses in normal cells. Consequently, the elevated and dysregulated Src activity may play a key role in initiation of the invasive cell phenotype both in infiltrating immune cells and precancerous cells. However, its most robust effects are from the production of cytokines and alterations of cell movement/attachment. In fact, the Src family kinase signaling network is the go between that relay crucial cytokine signals from inflammatory cells to cancer cells, and conversely, within the tumor microenvironment (Figure 1). The Src-mediated stimulatory effects on malignant cell proliferation and inhibitory effect on cell death, leads to the accumulation of malignant cells and thus increases the total mass of the tumor. Consequently, this elevates the production of pro-inflammatory cytokines, including chemokines, by the tumor which further leads to the recruitment and activation of additional leukocytes that results in a cycle (as depicted in the Figure 1) leading to cancer development and progression.
Concluding Remarks
The Src kinases-dependent signaling that link immune system with normal tissue plays a vital role in regulating and coordinating immune defense responses. The cross talk between Src kinase pathways in immune cells and Src kinase-mediated pathways in target tissue cells is mediated via cytokine signals elicited by these cells. These Src-dependent signaling pathways, when hyper-activated and dysregulated, can lead to the development of chronic inflammation that predispose to cancer. Src activation both in infiltrating immune cells and cancer precursor lesions is driven by pro-inflammatory cytokines within tumor-promoting microenvironment. This leads to a vicious cycle in which Src activation increases cytokine production that again induces Src activation, leading to invasive inflammatory cell and cancer cell phenotypes. Thus, elucidating the Src-dependent cross talk signaling mechanisms that link inflammatory cells with cancer cells, may facilitate the design of new pharmacological agents for the concurrent treatment of tumor-promoting inflammation and cancer. Pancreatic cancer, because of its robust cytokine mediated interactions between the tumor cells and tumor microenvironment, can be used in designing new agents for the inhibition of the linkage between inflammation and cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
LPS, lipopolysaccharides; TNF-α, tumor necrosis factor; IL-1, interleukine 1; IL-6, interleukine 6; MCP-1, monocyte chemoattractant protein-1; MIP-1, macrophage inflammatory protein 1; MIP-2, macrophage inflammatory protein 2; SDF-1, stromal cell-derived factor 1; PI3K, phosphatidylinositol-3 kinase.
References
Abram, C. L., and Lowell, C. A. (2008). The diverse functions of Src family kinases in macrophages. Front. Biosci. 13, 4426–4450. doi: 10.2741/3015
Aligayer, H., Boyd, D. D., Heiss, M. M., Abdalla, E. K., Curley, S. A., and Gallick, G. E. (2002). Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer 94, 344–351. doi: 10.1002/cncr.10221
Balkwill, F. (2004). The significance of cancer cell expression of the chemokine receptor CXCR4. Semin. Cancer Biol. 14, 171–179. doi: 10.1016/j.semcancer.2003.10.003
Balkwill, F., Charles, K. A., and Mantovani, A. (2005). Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7, 211–217. doi: 10.1016/j.ccr.2005.02.013
Bernstein, C. N., Blanchard, J. F., Kliewer, E., and Wajda, A. (2001). Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 91, 854–862. doi: 10.1002/1097-0142(20010215)91:4<854::AID-CNCR1073>3.0.CO;2-Z
Brugge, J. S., and Erikson, R. L. (1997). Identification of a transformation-specific antigen induced by an avian sarcome virus. Nature 269, 346–348. doi: 10.1038/269346a0
Chen, Y. Y., Malik, M., Tomkowicz, B. E., Collman, R. G., and Ptasznik, A. (2008). BCR-ABL1 alters SDF-1alpha-mediated adhesive responses through the beta2 integrin LFA-1 in leukemia cells. Blood 111, 5182–5186. doi: 10.1182/blood-2007-10-117705
Cheung, R., Malik, M., Ravyn, V., Tomkowicz, B., Ptasznik, A., and Collman, R. G. (2009). An arrestin-dependent multi-kinase signaling complex mediates MIP-1beta/CCL4 signaling and chemotaxis of primary human macrophages. J. Leukoc. Biol. 86, 833–845. doi: 10.1189/jlb.0908551
Cheung, R., Ravyn, V., Wang, L., Ptasznik, A., and Collman, R. G. (2008). Signaling mechanism of HIV-1 gp120 and virion-induced IL-1beta release in primary human macrophages. J. Immunol. 180, 6675–6684.
Coussens, L. M., and Werb, Z. (2002). Inflammation and cancer. Nature 420, 860–867. doi: 10.1038/nature01322
Dítě, P., Hermanová, M., Trna, J., Novotný, I., Růžièka, M., Liberda, M., et al. (2012). The role of chronic inflammation: chronic pancreatitis as a risk factor of pancreatic cancer. Dig. Dis. 30, 277–283. doi: 10.1159/000336991
Duell, E. J., Casella, D. P., Burk, R. D., Kelsey, K. T., and Holly, E. A. (2006). Inflammation, genetic polymorphisms in proinflammatory genes TNF-A, RANTES, and CCR5, and risk of pancreatic adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 15, 726–731. doi: 10.1158/1055-9965.EPI-05-0797
Eaden, J. A., Abrams, K. R., and Mayberry, J. F. (2001). The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48, 526–535. doi: 10.1136/gut.48.4.526
Erkan, M., Hausmann, S., Michalski, C. W., Fingerle, A. A., Dobritz, M., Kleeff, J., et al. (2012). The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 9, 454–467. doi: 10.1038/nrgastro.2012.115
Feig, C., Gopinathan, A., Neesse, A., Chan, D. S., Cook, N., and Tuveson, D. A. (2012). The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276. doi: 10.1158/1078-0432.CCR-11-3114
Fu, Y., Zagozdzon, R., Avraham, R., and Avraham, H. K. (2006). CHK negatively regulates Lyn kinase and suppresses pancreatic cancer invasion. Int. J. Oncol. 29, 1453–1458.
Gao, Z., Wang, X., Wu, K., Zhao, Y., and Hu, G. (2010). Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology 10, 186–193. doi: 10.1159/000236012
Go, V. L., Gukovskaya, A., and Pandol, S. J. (2005). Alcohol and pancreatic cancer. Alcohol 35, 205–211. doi: 10.1016/j.alcohol.2005.03.010
Goecke, H., Forssmann, U., Uguccioni, M., Friess, H., Conejo-Garcia, J. R., Zimmermann, A., et al. (2000). Macrophages infiltrating the tissue in chronic pancreatitis express the chemokine receptor CCR5. Surgery 128, 806–814. doi: 10.1067/msy.2000.108613
Guerra, C., Schuhmacher, A. J., Cañamero, M., Grippo, P. J., Verdaguer, L., Pérez-Gallego, L., et al. (2007). Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302. doi: 10.1016/j.ccr.2007.01.012
Hallek, M., Neumann, C., Schäffer, M., Danhauser-Riedl, S., von Bubnoff, N., de Vos, G., et al. (1997). Signal transduction of interleukin-6 involves tyrosine phosphorylation of multiple cytosolic proteins and activation of Src-family kinases Fyn, Hck, and Lyn in multiplemyeloma cell lines. Exp. Hematol. 25, 1367–1377.
Howes, N., Lerch, M. M., Greenhalf, W., Stocken, D. D., Ellis, I., Simon, P., et al. (2004). Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2, 252–261. doi: 10.1016/S1542-3565(04)00013-8
Hunter, T., and Sefton, B. M. (1980). Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc. Natl. Acad. Sci. U.S.A. 77, 1311–1315. doi: 10.1073/pnas.77.3.1311
Inngjerdingen, M., Torgersen, K. M., and Maghazachi, A. A. (2002). Lck is required for stromal cell-derived factor 1 alpha (CXCL12)-induced lymphoid cell chemotaxis. Blood. 15, 99</page>, <page>4318–4325. doi: 10.1182/blood.V99.12.4318
Ito, Y., Lugea, A., Pandol, S. J., and McCuskey, R. S. (2007). Substance P mediates cerulein-induced pancreatic microcirculatory dysfunction in mice. Pancreas 34, 138–143. doi: 10.1097/01.mpa.0000246663.30751.24
Itzkowitz, S. H., and Yio, X. (2004). Inflammation and cancer IV. colorectal cancer in inflammatory bowel disease: the role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G7–G17. doi: 10.1152/ajpgi.00079.2004
Kayali, A. G., Van Gunst, K., Campbell, I. L., Stotland, A., Kritzik, M., Liu, G., et al. (2003). The stromal cell-derived factor-1alpha/CXCR4 ligand-receptor axis is critical for progenitor survival and migration in the pancreas. J. Cell Biol. 163, 859–869. doi: 10.1083/jcb.200304153
Kulbe, H., Levinson, N. R., Balkwill, F., and Wilson, J. L. (2004). The chemokine network in cancer–much more than directing cell movement. Int. J. Dev. Biol. 48, 489–496. doi: 10.1387/ijdb.041814hk
Lin, M. T., Lin, B. R., Chang, C. C., Chu, C. Y., Su, H. J., Chen, S. T., et al. (2007). IL-6 induces AGS gastric cancer cell invasion via activation of the c-Src/RhoA/ROCK signaling pathway. Int. J. Cancer. 120, 2600–2608. doi: 10.1002/ijc.22599
Liou, G. Y., Döppler, H., Necela, B., Krishna, M., Crawford, H. C., Raimondo, M., et al. (2013). Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κ B and MMPs. J. Cell Biol. 202, 563–577. doi: 10.1083/jcb.201301001
Lowenfels, A. B., Maisonneuve, P., Whitcomb, D. C., Lerch, M. M., and DiMagno, E. P. (2001). Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. JAMA 286, 169–170. doi: 10.1001/jama.286.2.169
Lutz, M. P., Esser, I. B., Flossmann-Kast, B. B., Vogelmann, R., Lührs, H., Friess, H., et al. (1998). Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem. Biophys. Res. Commun. 243, 503–508. doi: 10.1006/bbrc.1997.8043
Maisonneuve, P., Lowenfels, A. B., Müllhaupt, B., Cavallini, G., Lankisch, P. G., Andersen, J. R., et al. (2005). Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 54, 510–514. doi: 10.1136/gut.2004.039263
Malik, M., Chen, Y. Y., Kienzle, M. F., Tomkowicz, B. E., Collman, R. G., and Ptasznik, A. (2008). Monocyte migration and LFA-1-mediated attachment to brain microvascular endothelia is regulated by SDF-1 alpha through Lyn kinase. J. Immunol. 181, 4632–4637.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008). Cancer-related inflammation. Nature 454, 436–444. doi: 10.1038/nature07205
Momi, N., Kaur, S., Ponnusamy, M. P., Kumar, S., Wittel, U. A., and Batra, S. K. (2012). Interplay between smoking-induced genotoxicity and altered signaling in pancreatic carcinogenesis. Carcinogenesis 33, 1617–1628. doi: 10.1093/carcin/bgs186
Nakata, Y., Tomkowicz, B., Gewirtz, A. M., and Ptasznik, A. (2006). Integrin inhibition through Lyn-dependent cross talk from CXCR4 chemokine receptors in normal human CD34+ marrow cells. Blood 107, 4234–4239. doi: 10.1182/blood-2005-08-3343
Nordskog, B. K., Blixt, A. D., Morgan, W. T., Fields, W. R., and Hellmann, G. M. (2003). Matrix-degrading and pro-inflammatory changes in human vascular endothelial cells exposed to cigarette smoke condensate. Cardiovasc. Toxicol. 3, 101–117. doi: 10.1385/CT:3:2:101
Okenwa, C., Kumar, A., Rego, D., Konarski, Y., Nilchi, L., Wright, K., et al. (2013). SHP-1-Pyk2-Src protein complex and p38 MAPK pathways independently regulate IL-10 production in lipopolysaccharide-stimulated macrophages. J. Immunol. 191, 2589–2603. doi: 10.4049/jimmunol.1300466
Orlicek, S. L., Hanke, J. H., and English, B. K. (1999). The src family-selective tyrosine kinase inhibitor PP1 blocks LPS and IFN-gamma-mediated TNF and iNOS production in murine macrophages. Shock 12, 350–354. doi: 10.1097/00024382-199911000-00004
Pannala, R., Basu, A., Petersen, G. M., and Chari, S. T. (2009). New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 10, 88–95. doi: 10.1016/S1470-2045(08)70337-1
Peruzzi, B., Cappariello, A., Del Fattore, A., Rucci, N., De Benedetti, F., and Teti, A. (2012). c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat. Commun. 3, 630. doi: 10.1038/ncomms1651
Podar, K., Mostoslavsky, G., Sattler, M., Tai, Y. T., Hayashi, T., Catley, L. P., et al. (2004). Critical role for hematopoietic cell kinase (Hck)-mediated phosphorylation of Gab1 and Gab2 docking proteins in interleukin 6-induced proliferation and survival of multiple myeloma cells. J. Biol. Chem. 279, 21658–21665. doi: 10.1074/jbc.M305783200
Ptasznik, A., Prossnitz, E. R., Yoshikawa, D., Smrcka, A., Traynor-Kaplan, A. E., and Bokoch, G. M. (1996). A tyrosine kinase signaling pathway accounts for the majority of phosphatidylinositol 3,4,5-trisphosphate formation in chemoattractant-stimulated human neutrophils. J. Biol. Chem. 271, 25204–25207. doi: 10.1074/jbc.271.41.25204
Ptasznik, A., Traynor-Kaplan, A., and Bokoch, G. M. (1995). G protein-coupled chemoattractant receptors regulate Lyn tyrosine kinase.Shc adapter protein signaling complexes. J. Biol. Chem. 270, 19969–19973. doi: 10.1074/jbc.270.34.19969
Ramnath, R. D., and Bhatia, M. (2006). Substance P treatment stimulates chemokine synthesis in pancreatic acinar cells via the activation of NF-kappaB. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G1113–G1119. doi: 10.1152/ajpgi.00177.2006
Ramnath, R. D., Sun, J., and Bhatia, M. (2009). Involvement of SRC family kinases in substance P-induced chemokine production in mouse pancreatic acinar cells and its significance in acute pancreatitis. J. Pharmacol. Exp. Ther. 329, 418–428. doi: 10.1124/jpet.108.148684
Rebours, V., Boutron-Ruault, M. C., Schnee, M., Férec, C., Le Maréchal, C., Hentic, O., et al. (2009). The natural history of hereditary pancreatitis: a national series. Gut 58, 97–103. doi: 10.1136/gut.2008.149179
Rous, P. (1911). A sarcoma of the fowl transmissible by an aget seperable from the tumor cells. J Exp. Med. 13, 397–411. doi: 10.1084/jem.13.4.397
Sarang, Z., Koroskenyi, K., Pallai, A., Duro, E., Melino, G., Griffin, M., et al. (2011). Translutaminase 2 null macrophages respond to lipopolysaccharide stimulation by elevated proinflammatory ctyokine production due to an enhanced ανβ3 integrin-induced Src tyrosine kinase sinaling. Immunol. Lett. 138, 71–78. doi: 10.1016/j.imlet.2011.03.004
Satoh, A., Gukovskaya, A. S., Edderkaoui, M., Daghighian, M. S., Reeve, J. R. Jr, Shimosegawa, T., et al. (2005). Tumor necrosis factor-alpha mediates pancreatitis responses in acinar cells via protein kinase C and proline-rich tyrosine kinase 2. Gastroenterology 129, 639–651. doi: 10.1016/j.gastro.2005.05.005
Sell, H., Habich, C., and Eckel, J. (2012). Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 8, 709–716. doi: 10.1038/nrendo.2012.114
Shi, C., Andersson, R., Zhao, X., and Wang, X. (2005). Potential role of reactive oxygen species in pancreatitis-associated multiple organ dysfunction. Pancreatology 5, 492–500. doi: 10.1159/000087063
Shields, D. J., Murphy, E. A., Desgrosellier, J. S., Mielgo, A., Lau, S. K., Barnes, L. A., et al. (2011). Oncogenic Ras/Src cooperativity in pancreatic neoplasia. Oncogene 30, 2123–2134. doi: 10.1038/onc.2010.589
Shoelson, S. E., Herrero, L., and Naaz, A. (2007). Obesity, inflammation, and insulin resistance. Gastroenterology 132, 2169–2180. doi: 10.1053/j.gastro.2007.03.059
Smolinska, M. J., Page, T. H., Urbaniak, A. M., Mutch, B. E., and Horwood, N. J. (2011). Hck tyrosine kinase regulates TLR4-induced TNF and IL-6 production via AP-1. J. Immunol. 187, 6043–6051. doi: 10.4049/jimmunol.1100967
Spaeth, E., Klopp, A., Dembinski, J., Andreeff, M., and Marini, F. (2008). Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 15, 730–738. doi: 10.1038/gt.2008.39
Staley, C. A., Parikh, N. U., and Gallick, G. E. (1997). Decreased tumorigenicity of a human colon adenocarcinoma cell line by an antisense expression vector specific for c-Src. Cell Growth Differ. 8, 269–274.
Stehelin, D., Varmus, H. E., Bishop, J. M., and Vogt, P. K. (1976). DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 260, 170–173. doi: 10.1038/260170a0
Summy, J. M., Trevino, J. G., Baker, C. H., and Gallick, G. E. (2005). c-Src regulates constitutive and EGF-mediated VEGF expression in pancreatic tumor cells through activation of phosphatidyl inositol-3 kinase and p38 MAPK. Pancreas 31, 263–274. doi: 10.1097/01.mpa.0000178280.50534.0c
Takatomo, K., Ryo, H., Yoshiharu, M., Jun, I., Shoichiro, T., Sanae, N., et al. (2000). Expression of stromal cell-derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: a possible role for tumor progression. Clin. Cancer Res. 6, 3530–3535.
Talamini, G., Bassi, C., Falconi, M., Sartori, N., Salvia, R., Rigo, L., et al. (1999). Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig. Dis. Sci. 44, 1303–1311. doi: 10.1023/A:1026670911955
Terzić, J., Grivennikov, S., Karin, E., and Karin, M. (2010). Inflammation and colon cancer. Gastroenterology 138, 2101–2114. doi: 10.1053/j.gastro.2010.01.058
Tomkowicz, B., Lee, C., Ravyn, V., Cheung, R., Ptasznik, A., and Collman, R. G. (2006). The Src kinase Lyn is required for CCR5 signaling in response to MIP-1beta and R5 HIV-1 gp120 in human macrophages. Blood 108, 1145–1150. doi: 10.1182/blood-2005-12-012815
Trevino, J. G., Summy, J. M., Lesslie, D. P., Parikh, N. U., Hong, D. S., Lee, F. Y., et al. (2006). Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. Am. J. Pathol. 168, 962–972. doi: 10.2353/ajpath.2006.050570
Trinchieri, G. (2012). Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 30, 677–706. doi: 10.1146/annurev-immunol-020711-075008
Ullman, T. A., and Itzkowitz, S. H. (2011). Intestinal inflammation and cancer. Gastroenterology 140, 1807–1816. doi: 10.1053/j.gastro.2011.01.057
Ungefroren, H., Sebens, S., Groth, S., Gieseler, F., and Fändrich, F. (2011). Differential roles of Src in transforming growth factor-ß regulation of growth arrest, epithelial-to-mesenchymal transition and cell migration in pancreatic ductal adenocarcinoma cells. Int. J. Oncol. 38, 797–805. doi: 10.3892/ijo.2011.897
Wiśniewska, E., Dylik, A., Kulza, M., Florek, E., Piekoszewski, W., Señczuk-Przybylowska, M., et al. (2013). Exposure to ethanol and tobacco smoke in relation to level of PCNA antigen expression in pancreatic and hepatic rat cells. Pharmacol. Rep. 65, 914–926.
Yadav, D., Timmons, L., Benson, J. T., Dierkhising, R. A., and Chari, S. T. (2011). Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am. J. Gastroenterol. 106, 2192–2199. doi: 10.1038/ajg.2011.328
Yokoi, K., Hawke, D., Oborn, C. J., Jang, J. Y., Nishioka, Y., Fan, D., et al. (2011). Identification and validation of SRC and phospho-SRC family proteins in circulating mononuclear cells as novel biomarkers for pancreatic cancer. Transl. Oncol. 4, 83–91.
Keywords: inflammation, cancer, Src, cytokines, chronic pancreatitis, pancreatic cancer
Citation: Liu ST, Pham H, Pandol SJ and Ptasznik A (2014) Src as the link between inflammation and cancer. Front. Physiol. 4:416. doi: 10.3389/fphys.2013.00416
Received: 14 December 2013; Paper pending published: 30 December 2013;
Accepted: 31 December 2013; Published online: 16 January 2014.
Edited by:
Mouad Edderkaoui, University of California Los Angeles, USAReviewed by:
Kennichi Satoh, Miyagi Cancer Research Institute, JapanMasaki Ohmuraya, Kumamoto University, Japan
Copyright © 2014 Liu, Pham, Pandol and Ptasznik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrzej Ptasznik, Department of Medicine, Cedars-Sinai Medical Center, Davis Bldg., Office #3099-A, 8700 West Beverly Blvd, Los Angeles, CA 90048-1804, USA e-mail:YW5kcnplai5wdGFzem5pa0Bjc2hzLm9yZw==