



- 1Department of Paediatrics, Institute of Shaanxi Province Children's Cardiovascular Diseases, The Shaanxi Provincial People's Hospital of Xi'an Jiaotong University, Xi'an, PR of China
- 2Faculty of Medicine and Human Sciences, Institute of Cardiovascular Sciences, University of Manchester, Manchester, UK
- 3Physiological Laboratory, Faculty of Biology, University of Cambridge, Cambridge, UK
- 4Department of Biochemistry, University of Cambridge, Cambridge, UK
Ryanodine receptor type 2 (RyR2) mutations are implicated in catecholaminergic polymorphic ventricular tachycardia (CPVT) thought to result from altered myocyte Ca2+ homeostasis reflecting inappropriate “leakiness” of RyR2-Ca2+ release channels arising from increases in their basal activity, alterations in their phosphorylation, or defective interactions with other molecules or ions. The latter include calstabin, calsequestrin-2, Mg2+, and extraluminal or intraluminal Ca2+. Recent clinical studies additionally associate RyR2 abnormalities with atrial arrhythmias including atrial tachycardia (AT), fibrillation (AF), and standstill, and sinus node dysfunction (SND). Some RyR2 mutations associated with CPVT in mouse models also show such arrhythmias that similarly correlate with altered Ca2+ homeostasis. Some examples show evidence for increased Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of RyR2. A homozygotic RyR2-P2328S variant demonstrates potential arrhythmic substrate resulting from reduced conduction velocity (CV) in addition to delayed afterdepolarizations (DADs) and ectopic action potential (AP) firing. Finally, one model with an increased RyR2 activity in the sino-atrial node (SAN) shows decreased automaticity in the presence of Ca2+-dependent decreases in ICa, L and diastolic sarcoplasmic reticular (SR) Ca2+ depletion.
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is one of the most malignant cardiac channelopathies. It is characterized by episodic, life-threatening, arrhythmias provoked by stress and emotion in individuals with otherwise structurally normal hearts (Swan et al., 1999; Priori et al., 2001; Priori and Chen, 2011). Two genes have been implicated in CPVT. One, transmitted as an autosomal dominant trait, is caused by mutations in the ryanodine receptor type 2 (RyR2) gene (Laitinen et al., 2001; Priori et al., 2001). It accounts for 50–55% of the CPVT cases attributable to genetic abnormalities (Priori et al., 2002). The other, less common, recessive variant results from mutations in the cardiac-specific isoform of calsequestrin type 2 gene (CASQ2) (Lahat et al., 2001). There are currently more than 150 known pathological allelic variants involving RyR2 that are associated with CPVT (Priori and Chen, 2011). Ventricular arrhythmia has been attributed in such cases to altered myocyte Ca2+ homeostasis. This results in an inappropriate “leakiness” of RyR2-Ca2+ release channels owing to increases in their basal activity, altered phosphorylation status or defective interactions with other molecules or ions, including calstabin (FKBP12.6) (Lehnart et al., 2004b), CASQ2, or Mg2+, or abnormal activation by extra- or intraluminal Ca2+ (Wehrens et al., 2004). This in turn results in spontaneous Ca2+ waves of Ca2+-induced Ca2+ release (CICR) in turn producing the delayed afterdepolarisation (DADs) thought to be the major cause of RyR2-associated CPVT (George et al., 2007; Mohamed et al., 2007; Priori and Chen, 2011). Seven RyR2 mutations are additionally associated with atrial arrhythmic disorders that include atrial tachycardia (AT), fibrillation (AF), and standstill as well as sinus node dysfunction (SND) (Laitinen et al., 2001; Bhuiyan et al., 2007; Sumitomo et al., 2007; Beery et al., 2009; Marjamaa et al., 2009; Kazemian et al., 2011). RyR mutations can thus affect function in cardiac regions including sino-atrial node (SAN), atria, and atrioventricular (AV) node in addition to ventricular myocardium. This review surveys atrial arrhythmias and SNDs related to RyR2 mutations as revealed by investigations in genetically modified murine cardiac models.
Features of Ca2+ Homeostasis in Sino-Atrial and Atrial Cells have Implications for Rhythm Abnormalities
Atrial myocytes show differences from ventricular cells in their Ca2+ homeostasis in some important respects, with implications for the physiological consequences of alterations in their RyR2 function. Thus, atrial myocytes of small mammals lack extensive T-tubules (Mackenzie et al., 2001, 2004; Brette and Orchard, 2003). These are replaced by prominent, transversely-orientated, sarcoplasmic reticular (SR), Z-tubular, elements. Junctional RyR2–L-type Ca2+ channel (LTCC) clusters are confined to the cell peripheries in contrast to their dense distribution throughout the more extensive ventricular tubular system (Mackenzie et al., 2001). However, an abundant corbular SR contains non-junctional RyR2s (Jorgensen et al., 1993). Atrial activation is thus likely initiated by CICR following voltage-sensitive extracellular Ca2+ entry at the peripheral T-SR junctional elements. This initiates a centripetal propagation of this CICR process thereby increasing open probabilities in the corbular RyR2-Ca2+ release channels in the cell interior (Bootman et al., 2001; Mackenzie et al., 2001, 2004; Blatter et al., 2003; Sheehan and Blatter, 2003).
The detailed mechanisms leading to AF are poorly understood. Nevertheless, increasing evidence implicates alterations in intracellular Ca2+ signaling in its associated focal firing, substrate evolution and remodeling processes (Hove-Madsen et al., 2004; Vest et al., 2005; Dobrev and Nattel, 2008; Liang et al., 2008; Qi et al., 2008; Chelu et al., 2009; Dobrev, 2010). Acute atrial arrhythmogenesis in intact hearts in turn is related to diastolic Ca2+ events in atrial myocytes. These depend upon finite SR Ca2+ stores and diastolic CICR processes both of which ultimately depend upon extracellular Ca2+ entry. Thus, ectopic activity results from afterdepolarisation and triggered activity, whether arising from increased action potential duration (APD) resulting in early afterdepolarisation (EAD) or SR Ca2+ release resulting in DAD phenomena, and may trigger re-entrant activity or drive atrial rhythm at rapid rate resulting in fibrillatory conduction. However, reentry requires a vulnerable substrate, likely determined by the balance of conduction velocity (CV) and refractory period (Comtois et al., 2005; Dobrev and Nattel, 2008; Nattel et al., 2008).
Atrial myocytes from chronic AF patients similarly show increased frequencies of pro-arrhythmic spontaneous Ca2+ release events (Hove-Madsen et al., 2004; Vest et al., 2005). These took place despite reduced L-type voltage-dependent Ca2+ currents (Bosch et al., 1999; Van Wagoner et al., 1999; Workman et al., 2001; Christ et al., 2004) and have accordingly been attributed to increased RyR2-mediated Ca2+ release activity (De Bakker et al., 2002; Vest et al., 2005). Thus, RyR2-single-channel open probabilities were increased in canine hearts with persistent AF. AF is also associated with increased protein kinase A (PKA)-mediated RyR2 phosphorylation at S2808 and decreased calstabin binding to RyR2 (Vest et al., 2005). Such changes would be expected to cause a failure of RyR2 channel closure resulting in a potentially arrhythmogenic Ca2+ leak from the SR (Wehrens et al., 2004; Balasubramaniam et al., 2005; Chelu and Wehrens, 2007). However, the extent to which such a mechanism might be directly applicable to AF remains under discussion (Venetucci et al., 2007; Eckstein et al., 2008).
These general principles are likely to be applicable to atria in murine hearts, which have previously also provided useful models for studies of ventricular arrhythmic phenomena. Introduction of caffeine transiently induced diastolic Ca2+ waves in regularly stimulated atrial myocytes. This could result either from RyR2 sensitization to cytosolic Ca2+ or increased intracellular cAMP levels resulting from phosphodiesterase inhibition (Daly, 2007). However, this effect was inhibited by prior inhibition of either Ca2+-ATPase activity by cyclopiazonic acid or of extracellular Ca2+ entry by nifedipine. These latter findings in turn directly correlated with the induction of atrial arrhythmic tendency or its inhibition by the same agents in intact hearts (Zhang et al., 2009, 2011).
Possible roles for RyR2-Ca2+ release channels in normal SAN pacemaker activity remain under discussion (Mangoni and Nargeot, 2008; Lakatta and Difrancesco, 2009). The SAN contains both RyR2 and RyR3 as well as SR Ca2+ stores (Masumiya et al., 2003). Release of intracellularly stored Ca2+ tends to activate depolarizing inward Na+/Ca2+ exchange current (Rubenstein and Lipsius, 1989; Ju and Allen, 1998; Terrar and Rigg, 2000; Bogdanov et al., 2001; Lakatta et al., 2010). Should this occur at the end of diastolic depolarization it could potentially facilitate action potential (AP) firing. Such findings have given rise to suggestions for a “Ca2+ clock” (Bogdanov et al., 2001; Vinogradova et al., 2004). Certainly, SAN cells show high basal levels of cAMP that could increase PKA-dependent RyR2 phosphorylation modulating its release of Ca2+ and thereby potentially influencing SAN pacemaker function (Vinogradova et al., 2006). Ryanodine-mediated RyR2 block indeed reduces SAN beating frequency (Rubenstein and Lipsius, 1989; Ju and Allen, 1998; Bogdanov et al., 2001). However, others have suggested that such SR Ca2+ release is not a predominant factor in normal SAN pacemaker activity (Honjo et al., 2003; Mangoni and Nargeot, 2008; Lakatta and Difrancesco, 2009; Himeno et al., 2011).
RyR2 Mutations are Associated with Clinical Atrial Arrhythmic and SND Phenotypes
A number of RyR2 mutations associated with CPVT are also associated with atrial arrhythmias and SND (Table 1). However, there have been no reported cases of lone AF or SAN dysfunction in the absence of CPVT. Large deletions encompassing exon 3 in RyR2 NH2-terminal regions are relatively frequent. This may reflecting their containing Alu repeats predisposing to deletions resulting from large Alu repeat–mediated genomic rearrangements resulting from polymerase slippage (Gu et al., 2008). Alu sequences occur in intron 2, 190 bp upstream from exon 3 and also 536 bp downstream in intron 3. They are related to the 1.1 kb deletion containing part of intron 2, exon 3, and part of intron 3, with a Alu-Alu recombination in which exon 2 is recombined with exon 4 and exon 3 is completely deleted (Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros-Domingo et al., 2009). However, crystal structure studies revealed that exon 3 deletion causes a structural rescue whereby a flexible loop inserts itself into the β trefoil domain. This increases the thermal stability of the RyR2 channel. As a result the mutation is neither lethal nor confers loss of function (Lobo et al., 2011). In the HEK293 expression system, it results in a marked reduction in the luminal Ca2+ threshold at which Ca2+ release terminates thereby increasing fractional Ca2+ release (Tang et al., 2012). The RyR2 NH2-terminal region may thus be an important determinant for termination of Ca2+ release, abnormalities in which are common in RyR2-associated cardiomyopathies (Gu et al., 2008; Tang et al., 2012).
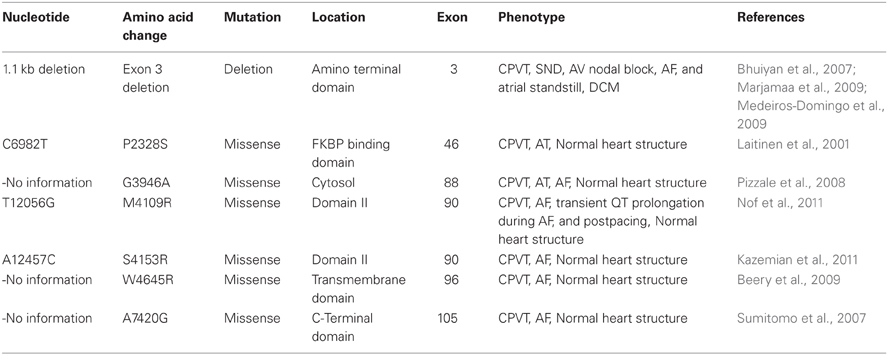
Table 1. Summary of RyR2 mutations associated with atrial and/or sinoatrial node dysfunction.
A large exon 3 deletion has been reported in a number of unrelated families. It has been associated with a broad range of atrial, ventricular and SND phenotypes. Thus, several unrelated families showed a large deletion encompassing part of intron 2, exon 3, and part of intron 3, expected to result in an in-frame deletion of 35 amino acids (p.Asn57-Gly91) (NM-001035) (Bhuiyan et al., 2007; Marjamaa et al., 2009; Medeiros-Domingo et al., 2009). This deletion was associated with a spectrum of phenotypes including SND, AF, atrioventricular block (AVB), decreased left ventricular function, increased trabeculation, CPVT, and dilated cardiomyopathy (DCM). Two further families showing CPVT with large deletions in the region did not show the association with DCM (Marjamaa et al., 2009). Mutation carriers in one of these families showed CPVT, AF, sinus bradycardia, and AV conduction abnormalities without structural cardiovascular abnormalities. Patients from the other family showed increased left ventricular trabeculation suggestive of non-compaction cardiomyopathy (Marjamaa et al., 2009). An additional report of a large large 3.6 Kb exon 3 deletion was not accompanied by a description of the corresponding patient phenotype (Medeiros-Domingo et al., 2009).
Some single CPVT-associated RyR2 mutations are also associated with atrial arrhythmias. Two such families, including one with RyR-P2328S, contained 14 CPVT patients (Swan et al., 1999; Laitinen et al., 2001). All carriers showed polymorphic ventricular tachycardia (PVT) and/or ventricular fibrillation (VF) of which three patients also showed non-sustained AT. Three further case reports described a further three RyR2 mutations all of which showed AT and/or AF in addition to CPVT without abnormalities in cardiac structure (Sumitomo et al., 2007; Pizzale et al., 2008; Kazemian et al., 2011). Thus, a patient with the RyR2-G3946A variant who had an implantable cardioverter defibrillator (ICD) for CPVT showed AT/AF both during exercise and on ambulatory electrocardiographic (ECG) recording (Pizzale et al., 2008). The RyR2-S4153R variant was identified in a women presenting with a cardiac arrest due to VF; she showed AF on 12-lead ECG immediately following emergency defibrillation. Over a 12 month follow-up period, in addition to experiencing three ICD shocks related to appropriately detected PVTs, she showed several exercise-induced self-terminating episodes of AF with rapid ventricular responses (Kazemian et al., 2011). Finally, a RyR2-A7420G variant was identified in a patient with both clinical and induced AF as well as frequent episodes of paroxysmal AF during walking or with mental stress despite normal sinus node recovery times (SNRT) and cardiac structure (Sumitomo et al., 2007).
In a further clinical situation, a preceding rapid AF could initiate CPVT, after a marked transient QT prolongation following a long RR interval that in turn followed a sequence of short R-R beats. This was associated with two heterozygous nucleotide substitutions, RyR2-M4109R and RyR2-I406T. All the RyR2-M4109R but none of the RyR2-I606T carriers showed prominent postpacing QT prolongation during electrophysiological study, although both showed normal QT intervals during regular rhythm (Nof et al., 2011). Finally, VT and AT could be induced by isoproterenol infusion, an effect blocked by propranolol administration, in a mother and son both having a RyR2-W4645R mutation and clinical diagnoses of PVT and AT in an absence of detectable significant underlying cardiac disease (Beery et al., 2009).
Atrial Arrhythmic and Ca2+ Homeostatic Abnormalities in Mouse Models Harboring RyR2 Mutations
Increased diastolic Ca2+ release has been implicated in generation of ventricular arrhythmias in genetically modified mouse models harboring CPVT-related RyR2 mutations (Cerrone et al., 2005; Kannankeril et al., 2006; Goddard et al., 2008; Lehnart et al., 2008; Suetomi et al., 2011). Recently, a few mouse models have similarly shown atrial arrhythmias and SND and similarly demonstrated evidence for diastolic SR Ca2+ release through “leaky” RyR2s (Chelu et al., 2009; Suetomi et al., 2011; Zhang et al., 2011; Neco et al., 2012; Shan et al., 2012).
RyR2-R176Q/+ mice (Kannankeril et al., 2006) did not show spontaneous AF but nevertheless showed increased vulnerabilities to AF during rapid atrial pacing when compared to wild-type (WT). This was associated with increased Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of RyR2. Conversely, pharmacological inhibition of CaMKII prevented this AF inducibility. So did genetic inhibition of CaMKII-mediated RyR2 phosphorylation in RyR2-S2814A mice studied in a vagotonic AF model. Atrial biopsies from mice with atrial enlargement and spontaneous AF, goats with lone AF, and patients with chronic AF similarly showed increased CaMKII phosphorylation of RyR2. These findings implicate a RyR2-dependent Ca2+ release resulting from increased CaMKII activity in susceptibility to AF (Chelu et al., 2009).
The RyR2-P2328S mutation has been clinically related to atrial in addition to ventricular arrhythmic phenotypes (Swan et al., 1999; Laitinen et al., 2001). Heterozygotic and homozygotic RyR2-P2328S mouse atria both showed acute arrhythmogenic properties in the absence of structural abnormalities with a severity related to gene dose, consistent with previous results in expression systems (Lehnart et al., 2004a). Intact anaesthetized hetero- or homozygotic RyR2-P2328S showed normal electrocardiographic parameters both before and after isoproterenol addition apart from increased heart rates. Nevertheless, bipolar electrogram and monophasic AP recordings from either regular or programmed stimulation demonstrated higher atrial arrhythmogenic incidences in isolated perfused homozygotic but not heterozygotic RyR2-P2328S relative to WT. Isoproterenol increased these incidences in all three groups despite unchanged AP durations at 90% recovery (APD90) and atrial effective refractory periods (AERP) (Figure 1A). These findings correlated with occurrences of diastolic Ca2+ release phenomena in regularly stimulated, isolated, fluo-3-loaded RyR2S/S, but not RyR2+/S or WT, atrial myocytes, whose frequency was increased by isoproterenol (Figure 1B) (Zhang et al., 2011). Finally, microelectrode recordings in isolated perfused homozygotic RyR2-P2328S atria showed DADs and ectopic APs not found in WT that could potentially result from such Ca2+ release phenomena (King et al., 2013). These also further implicated atrial arrhythmic substrate resulting from reduced CV in agreement with similar results bearing on ventricular conduction (Zhang et al., 2013). This was reflected in their increased interatrial conduction delays, reduced epicardial CVs and reduced maximum rates of AP depolarization [(dV/dt)max], despite similar effective refractory periods, AP durations and AP amplitudes.
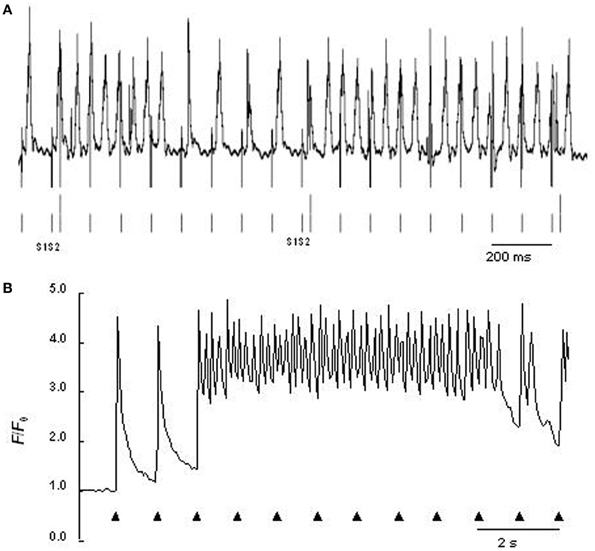
Figure 1. (A) Atrial monophasic action potential recording of arrhythmic events provoked by programmed electrical stimulation involving extrasystolic S2 stimulation (long vertical bars beneath trace) following series of pacing S1 stimuli (short vertical bars beneath trace) from isolated perfused homozygotic RyR2-P2328S heart. (B) Ca2+ transients from regularly stimulated fluo-3-loaded RyR2-P2328S atrial myocytes under confocal microscopy, both following introduction of isoproterenol [From Figures 5D, 6F of Zhang et al. (2011)].
Finally, AF could be induced by an intra-esophageal burst pacing protocol in mice harboring the RyR2-R2474S+/−, RyR2-N2386I+/−, and RyR2-L433P+/− mutations known to be associated with human CPVT. Their isolated atrial myocytes showed significant diastolic SR Ca2+ leaks. Atrial RyR2s from RyR2-R2474S+/− mice were oxidized and their RyR2 complexes depleted of the calstabin2 unit known to stabilize the RyR2 closed state. The Rycal agent S107, known to stabilize RyR2-calstabin2 interactions and inhibit the oxidation/phosphorylation-induced dissociation of calstabin2 from the channel, rescued the resulting increase in Ca2+ release, and prevented burst pacing–induced AF in vivo. It did not do so in calstabin2-deficient mice, implicating calstabin2 in these arrhythmic properties (Shan et al., 2012).
Paradoxical Sinus Node Disorder in Mouse Models Harboring RyR2 Mutations
In contrast to experimental evidence implicating a Ca2+ clock involving RyR2 in pacemaker activity (see section on Features of Ca2+ homeostasis in sino-atrial and atrial cells have implications for rhythm abnormalities), CPVT is associated with high incidences of SAN dysrhythmia in the form of reduced SAN automaticity resulting in basal bradycardia, sinus pauses, and impaired chronotropic responses to β-adrenergic stimulation (Leenhardt et al., 1995; Sumitomo et al., 2003; Postma et al., 2005; Katz et al., 2009). Although they do not show a basal bradycardia, RyR2-R4496C mice studied using in vivo telemetric recordings similarly showed sinus pauses overcome by atrial and junctional escapes triggered by catecholamines, and impaired SAN automaticity following isoproterenol injection. These features paralleled post-exercise findings in CPVT patients (Neco et al., 2012). Measurements of spontaneous [Ca2+]i transients reflecting pacemaker activity in SAN cells by confocal microscopy correspondingly demonstrated slower pacemaker activity and impaired chronotropic responses to β-adrenergic stimulation, and sinus pauses in 75% of the cases. These changes were associated with 50% reductions in L-type Ca2+ current (ICa, L) density in whole-cell patch-clamped RyR2-R4496C SAN cells. Isoproterenol dramatically increased the frequency of Ca2+ sparks and Ca2+ waves by ~5 and ~10-fold (Neco et al., 2012).
Conclusion
Enhanced RyR2 activity associated with RyR2 mutations can be associated with atrial arrhythmic tendency and SND. This is potentially attributable to increased RyR2-mediated release of intracellularly stored, SR, Ca2+ release and altered interactions with CaMKII and calstabin2. Increased RyR2 activity in the SAN leads to an unanticipated decrease in its automaticity accompanied by a Ca2+-dependent decrease of ICa, L and diastolic SR Ca2+ depletion.
Funding Source
This work was supported by the Medical Research Council, Wellcome Trust, British Heart Foundation, the Biotechnology and Biological Sciences Research Council, and Natural Science Foundation of China (30371571, 30830051, and 30672209).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Balasubramaniam, R., Chawla, S., Grace, A. A., and Huang, C. L. (2005). Caffeine-induced arrhythmias in murine hearts parallel changes in cellular Ca2+ homeostasis. Am. J. Physiol. Heart Circ. Physiol. 289, H1584–H1593. doi: 10.1152/ajpheart.01250.2004
Beery, T. A., Shah, M. J., and Benson, D. W. (2009). Genetic characterization of familial CPVT after 30 years. Biol. Res. Nurs. 11, 66–72. doi: 10.1177/1099800409333369
Bhuiyan, Z. A., Van Den Berg, M. P., Van Tintelen, J. P., Bink-Boelkens, M. T., Wiesfeld, A. C., Alders, M., et al. (2007). Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation 116, 1569–1576. doi: 10.1161/CIRCULATIONAHA.107.711606
Blatter, L. A., Kockskamper, J., Sheehan, K. A., Zima, A. V., Huser, J., and Lipsius, S. L. (2003). Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J. Physiol. 546, 19–31. doi: 10.1113/jphysiol.2002.025239
Bogdanov, K. Y., Vinogradova, T. M., and Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ. Res. 88, 1254–1258. doi: 10.1161/hh1201.092095
Bootman, M. D., Collins, T. J., Peppiatt, C. M., Prothero, L. S., Mackenzie, L., De Smet, P., et al. (2001). Calcium signalling–an overview. Semin. Cell Dev. Biol. 12, 3–10. doi: 10.1006/scdb.2000.0211
Bosch, R. F., Zeng, X., Grammer, J. B., Popovic, K., Mewis, C., and Kuhlkamp, V. (1999). Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 44, 121–131. doi: 10.1016/S0008-6363(99)00178-9
Brette, F., and Orchard, C. (2003). T-tubule function in mammalian cardiac myocytes. Circ. Res. 92, 1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD
Cerrone, M., Colombi, B., Santoro, M., Di Barletta, M. R., Scelsi, M., Villani, L., et al. (2005). Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ. Res. 96, e77–e82. doi: 10.1161/01.RES.0000169067.51055.72
Chelu, M. G., Sarma, S., Sood, S., Wang, S., Van Oort, R. J., Skapura, D. G., et al. (2009). Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951.
Chelu, M. G., and Wehrens, X. H. (2007). Sarcoplasmic reticulum calcium leak and cardiac arrhythmias. Biochem. Soc. Trans. 35, 952–956. doi: 10.1042/BST0350952
Christ, T., Boknik, P., Wohrl, S., Wettwer, E., Graf, E. M., Bosch, R. F., et al. (2004). L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110, 2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A
Comtois, P., Kneller, J., and Nattel, S. (2005). Of circles and spirals: bridging the gap between the leading circle and spiral wave concepts of cardiac reentry. Europace 7(Suppl. 2), 10–20. doi: 10.1016/j.eupc.2005.05.011
Daly, J. W. (2007). Caffeine analogs: biomedical impact. Cell. Mol. Life Sci. 64, 2153–2169. doi: 10.1007/s00018-007-7051-9
De Bakker, J. M., Ho, S. Y., and Hocini, M. (2002). Basic and clinical electrophysiology of pulmonary vein ectopy. Cardiovasc. Res. 54, 287–294. doi: 10.1016/S0008-6363(01)00532-6
Dobrev, D. (2010). Atrial Ca2+ signaling in atrial fibrillation as an antiarrhythmic drug target. Naunyn Schmiedebergs Arch. Pharmacol. 381, 195–206. doi: 10.1007/s00210-009-0457-1
Dobrev, D., and Nattel, S. (2008). Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J. Cardiovasc. Pharmacol. 52, 293–299. doi: 10.1097/FJC.0b013e318171924d
Eckstein, J., Verheule, S., De Groot, N. M., Allessie, M., and Schotten, U. (2008). Mechanisms of perpetuation of atrial fibrillation in chronically dilated atria. Prog. Biophys. Mol. Biol. 97, 435–451. doi: 10.1016/j.pbiomolbio.2008.02.019
George, C. H., Jundi, H., Thomas, N. L., Fry, D. L., and Lai, F. A. (2007). Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J. Mol. Cell. Cardiol. 42, 34–50. doi: 10.1016/j.yjmcc.2006.08.115
Goddard, C. A., Ghais, N. S., Zhang, Y., Williams, A. J., Colledge, W. H., Grace, A. A., et al. (2008). Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol. (Oxf.) 194, 123–140. doi: 10.1111/j.1748-1716.2008.01865.x
Gu, W., Zhang, F., and Lupski, J. R. (2008). Mechanisms for human genomic rearrangements. Pathogenetics 1, 4. doi: 10.1186/1755-8417-1-4
Himeno, Y., Toyoda, F., Satoh, H., Amano, A., Cha, C. Y., Matsuura, H., et al. (2011). Minor contribution of cytosolic Ca2+ transients to the pacemaker rhythm in guinea pig sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 300, H251–H261. doi: 10.1152/ajpheart.00764.2010
Honjo, H., Inada, S., Lancaster, M. K., Yamamoto, M., Niwa, R., Jones, S. A., et al. (2003). Sarcoplasmic reticulum Ca2+ release is not a dominating factor in sinoatrial node pacemaker activity. Circ. Res. 92, e41–e44. doi: 10.1161/01.RES.0000055904.21974.BE
Hove-Madsen, L., Llach, A., Bayes-Genis, A., Roura, S., Rodriguez Font, E., Aris, A., et al. (2004). Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 110, 1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87
Jorgensen, A. O., Shen, A. C., Arnold, W., McPherson, P. S., and Campbell, K. P. (1993). The Ca2+-release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J. Cell. Biol. 120, 969–980. doi: 10.1083/jcb.120.4.969
Ju, Y. K., and Allen, D. G. (1998). Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. J. Physiol. 508, 153–166. doi: 10.1111/j.1469-7793.1998.153br.x
Kannankeril, P. J., Mitchell, B. M., Goonasekera, S. A., Chelu, M. G., Zhang, W., Sood, S., et al. (2006). Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 103, 12179–12184. doi: 10.1073/pnas.0600268103
Katz, G., Arad, M., and Eldar, M. (2009). Catecholaminergic polymorphic ventricular tachycardia from bedside to bench and beyond. Curr. Probl. Cardiol. 34, 9–43. doi: 10.1016/j.cpcardiol.2008.09.002
Kazemian, P., Gollob, M. H., Pantano, A., and Oudit, G. Y. (2011). A novel mutation in the RYR2 gene leading to catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation: dose-dependent arrhythmia-event suppression by beta-blocker therapy. Can. J. Cardiol. 27, 870.e7–870.e10. doi: 10.1016/j.cjca.2011.02.003
King, J. H., Zhang, Y., Lei, M., Grace, A. A., Huang, C. L., and Fraser, J. A. (2013). Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol. (Oxf.). 207, 308–323. doi: 10.1111/apha.12006
Lahat, H., Pras, E., Olender, T., Avidan, N., Ben-Asher, E., Man, O., et al. (2001). A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 69, 1378–1384. doi: 10.1086/324565
Laitinen, P. J., Brown, K. M., Piippo, K., Swan, H., Devaney, J. M., Brahmbhatt, B., et al. (2001). Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490. doi: 10.1161/01.CIR.103.4.485
Lakatta, E. G., and Difrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both. J. Mol. Cell. Cardiol. 47, 157–170. doi: 10.1016/j.yjmcc.2009.03.022
Lakatta, E. G., Maltsev, V. A., and Vinogradova, T. M. (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ. Res. 106, 659–673. doi: 10.1161/CIRCRESAHA.109.206078
Leenhardt, A., Lucet, V., Denjoy, I., Grau, F., Ngoc, D. D., and Coumel, P. (1995). Catecholaminergic polymorphic ventricular tachycardia in children. a 7-year follow-up of 21 patients. Circulation 91, 1512–1519. doi: 10.1161/01.CIR.91.5.1512
Lehnart, S. E., Mongillo, M., Bellinger, A., Lindegger, N., Chen, B. X., Hsueh, W., et al. (2008). Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Invest. 118, 2230–2245. doi: 10.1172/JCI35346
Lehnart, S. E., Wehrens, X. H., Laitinen, P. J., Reiken, S. R., Deng, S. X., Cheng, Z., et al. (2004a). Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 109, 3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC
Lehnart, S. E., Wehrens, X. H., and Marks, A. R. (2004b). Calstabin deficiency, ryanodine receptors, and sudden cardiac death. Biochem. Biophys. Res. Commun. 322, 1267–1279. doi: 10.1016/j.bbrc.2004.08.032
Liang, X., Xie, H., Zhu, P. H., Hu, J., Zhao, Q., Wang, C. S., et al. (2008). Ryanodine receptor-mediated Ca2+ events in atrial myocytes of patients with atrial fibrillation. Cardiology 111, 102–110. doi: 10.1159/000119697
Lobo, P. A., Kimlicka, L., Tung, C. C., and Van Petegem, F. (2011). The deletion of exon 3 in the cardiac ryanodine receptor is rescued by beta strand switching. Structure 19, 790–798. doi: 10.1016/j.str.2011.03.016
Mackenzie, L., Bootman, M. D., Berridge, M. J., and Lipp, P. (2001). Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J. Physiol. 530, 417–429. doi: 10.1111/j.1469-7793.2001.0417k.x
Mackenzie, L., Roderick, H. L., Berridge, M. J., Conway, S. J., and Bootman, M. D. (2004). The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J. Cell Sci. 117, 6327–6337. doi: 10.1242/jcs.01559
Mangoni, M. E., and Nargeot, J. (2008). Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982. doi: 10.1152/physrev.00018.2007
Marjamaa, A., Laitinen-Forsblom, P., Lahtinen, A. M., Viitasalo, M., Toivonen, L., Kontula, K., et al. (2009). Search for cardiac calcium cycling gene mutations in familial ventricular arrhythmias resembling catecholaminergic polymorphic ventricular tachycardia. BMC Med. Genet. 10:12. doi: 10.1186/1471-2350-10-12
Masumiya, H., Yamamoto, H., Hemberger, M., Tanaka, H., Shigenobu, K., Chen, S. R., et al. (2003). The mouse sino-atrial node expresses both the type 2 and type 3 Ca2+ release channels/ryanodine receptors. FEBS Lett. 553, 141–144. doi: 10.1016/S0014-5793(03)00999-2
Medeiros-Domingo, A., Bhuiyan, Z. A., Tester, D. J., Hofman, N., Bikker, H., Van Tintelen, J. P., et al. (2009). The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol. 54, 2065–2074. doi: 10.1016/j.jacc.2009.08.022
Mohamed, U., Napolitano, C., and Priori, S. G. (2007). Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 18, 791–797. doi: 10.1111/j.1540-8167.2007.00766.x
Nattel, S., Burstein, B., and Dobrev, D. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73. doi: 10.1161/CIRCEP.107.754564
Neco, P., Torrente, A. G., Mesirca, P., Zorio, E., Liu, N., Priori, S. G., et al. (2012). Paradoxical effect of increased diastolic Ca2+ release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 126, 392–401. doi: 10.1161/CIRCULATIONAHA.111.075382
Nof, E., Belhassen, B., Arad, M., Bhuiyan, Z. A., Antzelevitch, C., Rosso, R., et al. (2011). Postpacing abnormal repolarization in catecholaminergic polymorphic ventricular tachycardia associated with a mutation in the cardiac ryanodine receptor gene. Heart Rhythm 8, 1546–1552. doi: 10.1016/j.hrthm.2011.05.016
Pizzale, S., Gollob, M. H., Gow, R., and Birnie, D. H. (2008). Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J. Cardiovasc. Electrophysiol. 19, 1319–1321. doi: 10.1111/j.1540-8167.2008.01211.x
Postma, A. V., Denjoy, I., Kamblock, J., Alders, M., Lupoglazoff, J. M., Vaksmann, G., et al. (2005). Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 42, 863–870. doi: 10.1136/jmg.2004.028993
Priori, S. G., and Chen, S. R. (2011). Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 108, 871–883. doi: 10.1161/CIRCRESAHA.110.226845
Priori, S. G., Napolitano, C., Memmi, M., Colombi, B., Drago, F., Gasparini, M., et al. (2002). Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106, 69–74. doi: 10.1161/01.CIR.0000020013.73106.D8
Priori, S. G., Napolitano, C., Tiso, N., Memmi, M., Vignati, G., Bloise, R., et al. (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200. doi: 10.1161/01.CIR.103.2.196
Qi, X. Y., Yeh, Y. H., Xiao, L., Burstein, B., Maguy, A., Chartier, D., et al. (2008). Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ. Res. 103, 845–854. doi: 10.1161/CIRCRESAHA.108.175463
Rubenstein, D. S., and Lipsius, S. L. (1989). Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ. Res. 64, 648–657. doi: 10.1161/01.RES.64.4.648
Shan, J., Xie, W., Betzenhauser, M., Reiken, S., Chen, B. X., Wronska, A., et al. (2012). Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 111, 708–717. doi: 10.1161/CIRCRESAHA.112.273342
Sheehan, K. A., and Blatter, L. A. (2003). Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation-contraction coupling in cat atrial myocytes. J. Physiol. 546, 119–135. doi: 10.1113/jphysiol.2002.026963
Suetomi, T., Yano, M., Uchinoumi, H., Fukuda, M., Hino, A., Ono, M., et al. (2011). Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca2+release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation 124, 682–694. doi: 10.1161/CIRCULATIONAHA.111.023259
Sumitomo, N., Harada, K., Nagashima, M., Yasuda, T., Nakamura, Y., Aragaki, Y., et al. (2003). Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 89, 66–70. doi: 10.1136/heart.89.1.66
Sumitomo, N., Sakurada, H., Taniguchi, K., Matsumura, M., Abe, O., Miyashita, M., et al. (2007). Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ. J. 71, 1606–1609. doi: 10.1253/circj.71.1606
Swan, H., Piippo, K., Viitasalo, M., Heikkila, P., Paavonen, T., Kainulainen, K., et al. (1999). Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J. Am. Coll. Cardiol. 34, 2035–2042. doi: 10.1016/S0735-1097(99)00461-1
Tang, Y., Tian, X., Wang, R., Fill, M., and Chen, S. R. (2012). Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ. Res. 110, 968–977. doi: 10.1161/CIRCRESAHA.111.256560
Terrar, D., and Rigg, L. (2000). What determines the initiation of the heartbeat. J. Physiol. 524, 316. doi: 10.1111/j.1469-7793.2000.00316.x
Van Wagoner, D. R., Pond, A. L., Lamorgese, M., Rossie, S. S., McCarthy, P. M., and Nerbonne, J. M. (1999). Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 85, 428–436. doi: 10.1161/01.RES.85.5.428
Venetucci, L. A., Trafford, A. W., and Eisner, D. A. (2007). Increasing ryanodine receptor open probability alone does not produce arrhythmogenic calcium waves: threshold sarcoplasmic reticulum calcium content is required. Circ. Res. 100, 105–111. doi: 10.1161/01.RES.0000252828.17939.00
Vest, J. A., Wehrens, X. H., Reiken, S. R., Lehnart, S. E., Dobrev, D., Chandra, P., et al. (2005). Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 111, 2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C
Vinogradova, T. M., Lyashkov, A. E., Zhu, W., Ruknudin, A. M., Sirenko, S., Yang, D., et al. (2006). High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 98, 505–514. doi: 10.1161/01.RES.0000204575.94040.d1
Vinogradova, T. M., Zhou, Y. Y., Maltsev, V., Lyashkov, A., Stern, M., and Lakatta, E. G. (2004). Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 94, 802–809. doi: 10.1161/01.RES.0000122045.55331.0F
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., Deng, S. X., Vest, J. A., Cervantes, D., et al. (2004). Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304, 292–296. doi: 10.1126/science.1094301
Workman, A. J., Kane, K. A., and Rankin, A. C. (2001). The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc. Res. 52, 226–235. doi: 10.1016/S0008-6363(01)00380-7
Zhang, Y., Fraser, J. A., Jeevaratnam, K., Hao, X., Hothi, S. S., Grace, A. A., et al. (2011). Acute atrial arrhythmogenicity and altered Ca2+ homeostasis in murine RyR2-P2328S hearts. Cardiovasc. Res. 89, 794–804. doi: 10.1093/cvr/cvq229
Zhang, Y., Schwiening, C., Killeen, M. J., Ma, A., Lei, M., Grace, A. A., et al. (2009). Pharmacological changes in cellular Ca2+ homeostasis parallel initiation of atrial arrhythmogenesis in murine Langendorff-perfused hearts. Clin. Exp. Pharmacol. Physiol. 36, 969–980. doi: 10.1111/j.1440-1681.2009.05170.x
Keywords: RyR2, mutation, sinus node dysfunction, atrial arrhythmias, mouse models
Citation: Zhang Y, Matthews GDK, Lei M and Huang CL-H (2013) Abnormal Ca2+ homeostasis, atrial arrhythmogenesis, and sinus node dysfunction in murine hearts modeling RyR2 modification. Front. Physiol. 4:150. doi: 10.3389/fphys.2013.00150
Received: 21 April 2013; Accepted: 05 June 2013;
Published online: 25 June 2013.
Edited by:
Ian N. Sabir, King's College London, UKReviewed by:
Henggui Zhang, The University of Manchester, UKSøren P. Olesen, University of Copenhagen, Denmark
Copyright © 2013 Zhang, Matthews, Lei and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Yanmin Zhang, Institute of Cardiovascular Sciences, University of Manchester, CTF Building, 46 Grafton Street, Manchester M13 9NT, UK e-mail:eWFubWluLnpoYW5nQG1hbmNoZXN0ZXIuYWMudWs=
†Joint senior authors for this paper.