

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 26 March 2013
Sec. Gastrointestinal Sciences
Volume 4 - 2013 | https://doi.org/10.3389/fphys.2013.00056
This article is part of the Research Topic Tumor-Stromal Interactions and Pancreatic Cancer View all 8 articles
Pancreatic cancer is a complex, aggressive, and heterogeneous malignancy driven by the multifaceted interactions within the tumor microenvironment. While it is known that the tumor microenvironment accommodates many cell types, each playing a key role in tumorigenesis, the major source of these stromal cells is not well-understood. This review examines the contribution of bone marrow-derived cells (BMDC) to pancreatic carcinogenesis, with respect to their role in constituting the tumor microenvironment. In particular, their role in supporting fibrosis, immunosuppression, and neovascularization will be discussed.
It is long known that adult stem cells have remarkable flexibility in their differentiation repertoires. This plasticity allows adult stem cells, particularly those of bone marrow origin, to engraft alternative non-haematopoietic locations and differentiate into cell types appropriate to their new niche. This is particularly evident when the recipient organ is damaged (Poulsom et al., 2002; Alison et al., 2006). Bone marrow derived cells (BMDC) can either engraft or fuse to adopt, or be reprogrammed, to the differentiated state of the particular epithelia (Jang et al., 2004) [reviewed in (Herzog et al., 2003)]. This suggests that the endogenous stem cell of an organ is not confined to each specific organ but may be a dynamic system involving circulating BMDC with stem cell niche environments regulating recruitment, proliferation, and differentiation (Alison et al., 2006; Diaz-Flores et al., 2006). This may have significant implications concerning the evolution of cancers in many solid organs, including the pancreas.
Much debate has loomed as to the questionable functional significance of BMDC that have differentiated into epithelial cells of solid organs (Krause et al., 2001; Wagers et al., 2002; Bruscia et al., 2006), while very few studies have investigated this phenomenon in the pancreas (Wang et al., 2006; Marrache et al., 2008). Most studies have focused on restoring endocrine function following islet cell injury (Choi et al., 2003; Hess et al., 2003; Ianus et al., 2003; Lechner et al., 2004; Mathews et al., 2004; Taneera et al., 2006; Hasegawa et al., 2007; Gao et al., 2008b), with few studies assessing the contribution of BMDC to the exocrine pancreas, in particular in pancreatic cancer (Pan et al., 2009; Scarlett et al., 2011). Wang et al. (2006) describe the contribution of BMDC to pancreatic duct formation in neonatal mice, Marrache et al. (2008), and Watanabe et al. (2009) demonstrate that BMDC contribute to the pancreatic stellate cell (PSC) population in chronic pancreatitis, while Pan et al. (2009) and Scarlett et al. (2011) identified a contribution of BMDC to the PSC population in rat and mouse models, respectively, of chemical carcinogenesis of the pancreas.
It is well-established that the pancreatic tumor microenvironment plays an integral role in driving pancreatic carcinogenesis and metastases, with the contribution of components of the pancreatic tumor microenvironment, such as PSC, myofibroblasts/fibroblasts, and inflammatory cells, reviewed elsewhere within this edition (Erkan et al., 2012; Evans and Costello, 2012). As such, this review will focus specifically on the contribution of BMDC in pancreatic carcinogenesis. In particular the significant role that BMDC has within the tumor microenvironment, and its implications for supporting pancreatic carcinogenesis, will be discussed. Table 1 and Figure 1 summarize the contribution of BMDC to the pancreatic tumor microenvironment.
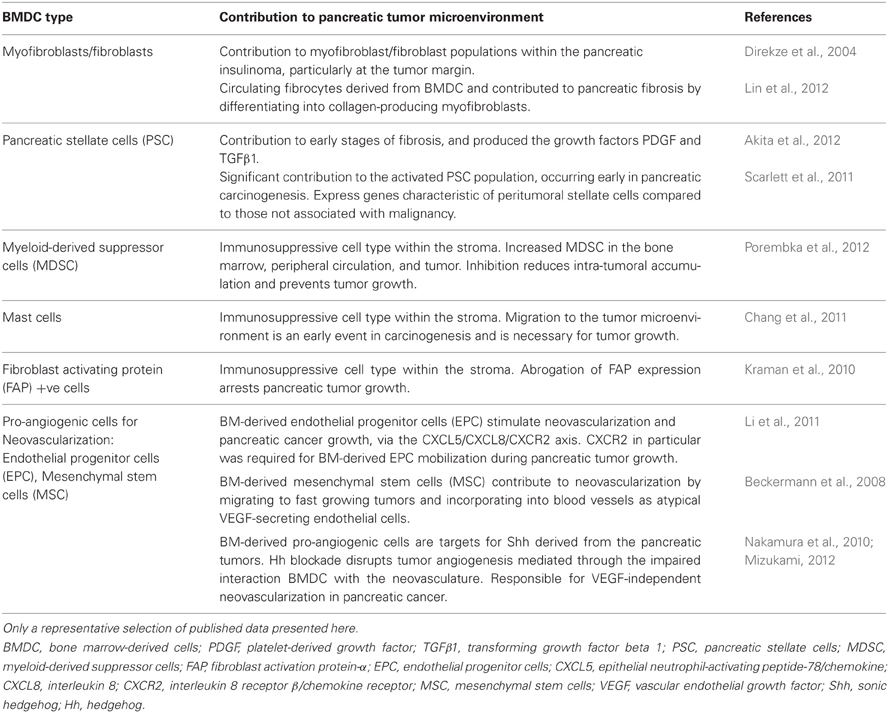
Table 1. Summary of bone marrow derived cell contribution to the pancreatic tumor microenvironment.
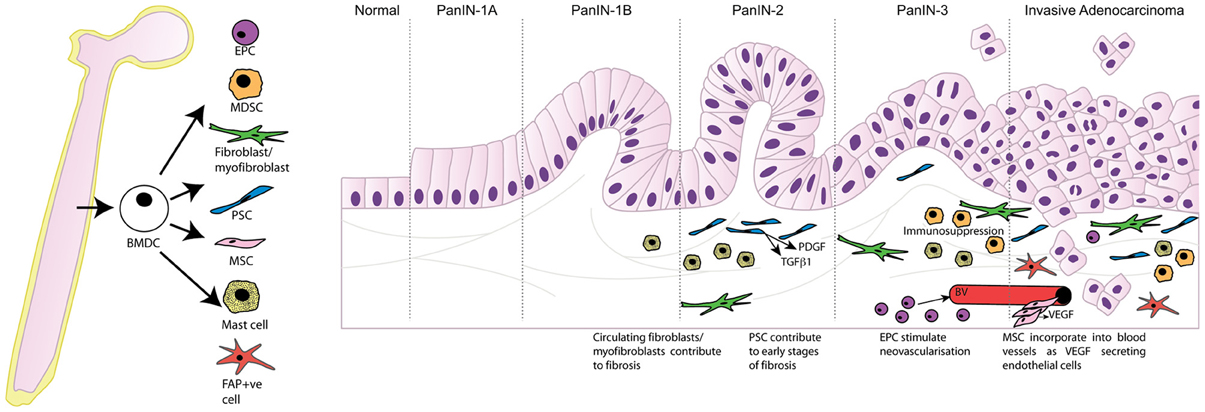
Figure 1. Bone marrow derived cells contribute to the tumor microenvironment following recruitment to the desmoplastic stroma and differentiation into multiple cell types promoting tumor progression. PanIN, pancreatic intraepithelial neoplasia; BMDC, bone marrow-derived cells; EPC, endothelial progenitor cells; MDSC, myeloid-derived suppressor cells; PSC, pancreatic stellate cells; MSC, mesenchymal stem cells; FAP, fibroblast activation protein-α; PDGF, platelet-derived growth factor; TGFβ1, transforming growth factor beta 1; VEGF, vascular endothelial growth factor; BV, blood vessel.
A novel paradigm of epithelial cancer development was postulated following the early investigations of Houghton et al. (2004), where in a model of Helicobacter felis induced gastric carcinogenesis, the development of metaplasia and dysplasia was linked to the engraftment and expansion of the BMDC population, eventually giving rise to gastric adenocarcinoma (Houghton et al., 2004). It was thus hypothesized that in response to tissue damage, BMDC could migrate to and lodge in the stem/progenitor cell niche, expand as a clonal unit and repopulate the epithelium, and that mutations in these engrafted BMDC then lead to carcinogenesis [reviewed in (Alison et al., 2006)].
Studies of the exocrine pancreas have failed to provide evidence for differentiation of BMDC into epithelial tumor cells directly. Pan et al. (2009) demonstrated in a rat model of carcinogenesis that BMDC could modulate pancreatic cancer growth via incorporation into the microenvironment, specifically the PSC within the stroma (Pan et al., 2009). These findings were strengthened in a more recent study by Scarlett et al. (2011), who also demonstrated a significant contribution of BMDC to the peritumoral stroma in a mouse model of pancreatic carcinogenesis (Scarlett et al., 2011). These data suggest that while BMDC do not contribute directly to pancreatic epithelial tumorigenesis, they play a significant and supportive role in promoting carcinogenesis via interactions within the tumor microenvironment. This will be discussed in more detail below.
There is increasing evidence that the tumor microenvironment influences tumor proliferation and survival, metastasis, resistance to therapy and escape from immune control [reviewed in (Feig et al., 2012)]. The desmoplastic stroma associated with pancreatic cancer is made up of a heterogeneous population of cells including immune cells, stellate cells, blood vessels, extracellular matrix (ECM), fibroblasts, and myofibroblasts. However, the major source of pancreatic stromal cells is not well-understood. While conversion of resident cells within normal tissue stroma are obvious candidates, increasing evidence is emerging that BMDCs are a source of non-resident stromal cells that contribute significantly to the stroma, and thus aid in the progression and invasion of pancreatic malignancy (Luo et al., 2012).
The role of myofibroblasts in fibrosis is well-documented in numerous solid tumors (Direkze et al., 2004), and along with fibroblasts and ECM proteins they produce, are key components of the desmoplastic response to tumors (De Wever and Mareel, 2003). Early studies by Direkze et al. (2004) demonstrated in a mouse model of pancreatic insulinoma that BMDC contribute to both myofibroblast and fibroblast populations within the tumors, particularly at the tumor margin. This study raised many questions, and suggested that the development of the pancreatic tumor stroma may be a less localized phenomenon than first thought (Direkze et al., 2004).
More recently, Lin et al. (2012) demonstrated in a mouse model of caerulein-induced pancreatitis that circulating fibrocytes were indeed derived from BMDC. These fibrocytes could engraft to the pancreas from peripheral circulation, and contributed to pancreatic fibrosis in part by differentiating into collagen-producing myofibroblasts. Further, when genetically modifying the BMDC, the severity of the fibrosis within the pancreas could be altered, suggesting that BMDC can function as fibrogenic cells (Lin et al., 2012).
PSC are resident myofibroblast-like cells existing in the periacinar space of the exocrine pancreas. In a healthy pancreas, PSC (quiescent) comprise approximately 4% of pancreatic cells, and exist in a periacinar dissemination. There is increasing evidence to suggest that PSC are key participants in the pathogenesis of pancreatic exocrine diseases, particularly in the production of the abundant fibrous stroma, which is a feature of pancreatic cancer (Apte et al., 1998, 2004; Bachem et al., 1998; Hwang et al., 2008; Vonlaufen et al., 2008; Phillips, 2012).
Akita et al. (2012) demonstrated in a rat model of pancreatic fibrosis that bone marrow derived activated PSC contribute to the early stages of fibrosis, and produced the growth factors PDGF and TGFβ1, key growth factors involved in the cross-talk between pancreatic tumor cells and PSC that contribute to tumor invasion and metastasis (Vonlaufen et al., 2008; Akita et al., 2012).
In a recent study investigating the potential role for BMDC in pancreatic injury, regeneration and carcinogenesis, Scarlett et al. (2011) observed that there was significant BMDC recruitment to the inflammatory infiltrate at the time of pancreatic injury (caerulein-induced chronic pancreatitis) consistent with previous reports (Minami et al., 2005; Sparmann et al., 2010; Scarlett et al., 2011), which was transient as cell numbers diminished over time to low levels when exocrine regeneration was complete. Interestingly, PSC remained amongst the residual population of BMDC, suggesting that BMDC play a role in supporting the regenerative process, but do not transform to contribute to the regenerative epithelium itself. As observed for chronic pancreatitis, increased recruitment of BMDC to the pancreas occurred following DMBA-induced carcinogenesis, which predominantly included inflammatory infiltrate, while activated PSC were also present (Scarlett et al., 2011). In an earlier study, Erkan et al. (2010) used transcript profiling to identify markers to differentiate PSC associated with chronic pancreatitis against those of pancreatic cancer, with the ultimate aim of subtyping PSC into either inflammation- or tumor-associated-PSC. Erkan et al. identified Pre-B-cell leukemia transcription factor 1 (PBX1) to be upregulated in inflammation-associated PSC compared to tumor-associated PSC, while cadherin EGF LAG seven-pass G-type receptor 3 (CELSR3) expression was upregulated in tumor-associated PSC compared to that of inflammation-associated PSC (Erkan et al., 2010). In the study of Scarlett et al. (2011), expression of CELSR3 in tumor-associated BMDC suggests that there was modification of these PSC by the tumor microenvironment, and that tumor associated BMDC PSC may be retained in the peri-tumoral stroma, whilst those associated with pancreatitis are not. This is supported by studies where bone marrow-derived mesenchymal stem cells (MSC) preferentially localize to regions of pancreatic tumor growth (Kallifatidis et al., 2008) and have been shown to transform into tumor-associated myofibroblasts in insulinomas (Direkze et al., 2004).
While most tumor associated activated PSC are thought to arise from endogenous quiescent PSC, evidence now exists that a proportion of these are bone marrow derived, and display different phenotypes depending on whether they are recruited to an inflammatory or a carcinogenic pancreas. In pancreatic carcinogenesis, and in chronic pancreatitis, BMDC contribute significantly to the activated PSC population, with evidence suggesting that this process occurs as an early event in pancreatic carcinogenesis (Scarlett et al., 2011). Those associated with pancreatic cancer express genes characteristic of peritumoral stellate cells as compared to those not associated with malignancy, providing further evidence that BMDC play an important role in supporting pancreatic carcinogenesis, the mechanisms of which remain to be elucidated.
Of interest are the mechanisms by which BMDC are recruited to the stroma. Are they recruited by resident PSC themselves? Or do they migrate to regions of fibrosis via inflammatory signaling? Or both? Within the early stages of pancreatic injury, resident quiescent PSC become activated and promote fibrosis via the expression of ECM proteins, cytokines and growth factors such as PDGF and TGFβ1. These growth factors are known activators of PSC and in an autocrine manner leads to persistent PSC activation, promoting further fibrosis. A consequence of fibrosis is the increased recruitment of BMDC to the stroma, including BM derived PSC (Akita et al., 2012). This suggests a role for tissue repair, however, depending on the signals that the migrating BMDC receive (such as those observed in a tumor-associated microenvironment), this may lead to an environment supporting tumor progression as opposed to tissue repair, as described above (Erkan et al., 2010) and [reviewed in (Evans and Costello, 2012)]. Clearly there is more complexity involved than just an inflammation, PSC activation, BMDC recruitment and differentiation loop occurring in the fibrotic microenvironment. Delineating the exact mechanisms by which this occurs will lead to novel therapeutic strategies targeting the fibrotic stroma.
It is long known that inflammation is crucially linked to pancreatic cancer development, as evidenced by chronic pancreatitis being a major risk factor (Lowenfels et al., 1993). The mechanisms by which inflammation exacerbates an individual's risk of developing pancreatic cancer are yet to be fully understood. It is becoming increasingly evident that immune cells make up approximately 50% of the tumor mass (tumor and stroma) (Clark et al., 2007; Scarlett et al., 2011), with immunosuppressive cells, such as Myeloid-derived suppressor cells (MDSC) and mast cells, predominant.
MDSC are a heterogeneous population of undifferentiated and immature myeloid immunosuppressive cells. Tumor-induced alterations in bone-marrow myelopoiesis are driven by growth factors and cytokines secreted by the tumor, which leads to expansion and mobilization of MDSC. Once mobilized to the stroma, MDSC promotes tumor growth, invasion, and tumor-induced immunosuppression and host immune evasion by inhibiting lymphocyte activation and antigen recognition (Serafini et al., 2006; Marigo et al., 2008; Gabrilovich and Nagaraj, 2009). Of importance, Porembka et al. (2012) recently demonstrated that patients with pancreatic adenocarcinoma exhibited increased MDSC in the bone marrow and peripheral circulation, as well as within the tumor itself, while inhibition of MDSC with the aminobisphosphonate zoledronic acid, reduces intra-tumoral accumulation of MDSC preventing tumor growth and increasing T-cell recruitment (Porembka et al., 2012).
Mast cells regulate adaptive immune responses via the release of cytokines and other immunomodulatory factors, which promote immune suppression and potentially tumor progression (Galli et al., 2005; Kalesnikoff and Galli, 2008). Chang et al. (2011) demonstrated that migration of bone-marrow derived mast cells to the tumor microenvironment is an early event in carcinogenesis and is necessary for tumor growth, the mechanisms of which remain to be elucidated (Chang et al., 2011).
The importance of an additional immunosuppressive cell type within the stroma was further defined through a recent study by Kraman et al. (2010), who demonstrated that a sub-population of stromal cells that express fibroblast activation protein (FAP) suppress the immune response and that abrogation of FAP expression arrests the growth of pancreatic tumors, potentially by removing their inhibitory effect on the host's immune response. (Kraman et al., 2010; Schreiber and Rowley, 2010). Thus, novel immunostimulatory therapeutic strategies targeting these immunosuppressive mechanisms may prove effective against pancreatic adenocarcinomas.
A consequence of the excessive desmoplastic stroma is a profound alteration of the tumor vasculature, with the subsequent vascular dysfunction presenting a significant barrier to the delivery of chemotherapeutic agents to the tumor cells. As such, strategies targeting elements within the stroma have become more extensively studied (Olive et al., 2009; Provenzano et al., 2012). The mysteries surrounding hypovascularity in pancreatic cancer have been reviewed elsewhere (Feig et al., 2012), with many questions remaining unanswered at present. This section will focus solely on the contribution of BMDC to tumor neovascularization, and the implications therein.
Most solid tumors require a vascular supply to provide oxygen and nutrients to enhance tumor progression and invasion. The tumor can either exploit existing vessels, or recruit and mobilize BMDC to induce neovascularization. BMDC are increasingly valued to be important contributors to the expansion of the tumor vasculature, however, the mechanisms of BMDC recruitment and mobilization by the tumors are yet to be fully understood (Lyden et al., 2001; Shojaei et al., 2007; Chan et al., 2009).
Endothelial progenitor cells (EPCs) originate from BMDC and possess the capacity to differentiate into mature endothelial cells, contributing to the complex process of tumor neovascularization. There is evidence that tumor angiogenesis can be stimulated by tumor cell secreted CXC chemokine ligands CXCL5 and CXCL8, via their common receptor CXCR2 (Wente et al., 2006; Raman et al., 2007). Li et al. (2011) recently demonstrated a role for BM-derived EPCs in stimulating neovascularization and pancreatic cancer growth, with EPCs mobilized by the pancreatic cancer cells, and that targeting the CXCL5/CXCL8/CXCR2 axis impaired EPC mobilization, proliferation, differentiation and neovascularization. CXCR2 in particular was required for EPC mobilization during pancreatic tumor growth, indicating a critical role for CXCR2 in the regulation of BM-derived progenitor cells with respect to tumor angiogenesis (Li et al., 2011).
BM-derived MSC have also been shown to migrate toward tumor hypoxia-induced secretion of growth factors (VEGF, PDGF, EGF) in pancreatic tumors, and contribute to neovascularization by homing to fast growing tumors and incorporating into blood vessels as atypical VEGF-secreting endothelial cells (Beckermann et al., 2008). For pancreatic cancer in particular, anti-vascular therapies have failed to show a survival benefit, most likely due to the heterogeneous tumor microenvironment that creates a hypovascular milieu. It is clear that the tumor microenvironment plays a critical role in pancreatic tumor development and progression and that circulating BMDC have an inherent capacity to differentiate into endothelial cells to form new vasculature (Gao et al., 2008a). Recently, Nakamura et al. (2010) identified that BM-derived pro-angiogenic cells are potential targets for Sonic Hedgehog (Shh), derived from the pancreatic tumors, and that Hh blockade can disrupt tumor angiogenesis in vivo, which is mediated through the impaired interaction BMDC with the neovasculature in pancreatic cancer (Nakamura et al., 2010). As such, it was speculated that induction of angiogenesis via Shh may mediate VEGF-independent neovascularization in pancreatic cancer, thereby serving as a potential mechanism for resistance to anti-VEGF therapy (Mizukami, 2012).
There is mounting evidence that BMDC contribute to the neovascularization in pancreatic cancer, however, pancreatic cancer remains a hypovascular tumor with significant perfusion impairment. With a distinct lack of efficacy of anti-angiogenic therapies for pancreatic cancer [reviewed in (Feig et al., 2012)], alternative approaches targeting the pancreatic cancer vasculature are required.
In conclusion, pancreatic carcinogenesis is an extremely aggressive, and complex malignancy with very few effective therapeutic modalities. Pancreatic cancer is characterized by an extensive desmoplastic stroma that interacts with the tumor cells, where together they drive tumor progression, invasion and metastasis. In addition, the stroma provides a physical barrier, denying the tumor cells access to chemotherapeutic agents. Future research priorities should be focused on further understanding the complex tumor-stroma relationships, which is key for facilitating the development of potential novel therapeutic strategies. Such strategies may include reducing tumor neovascularization by targeting the ablation of BM derived EPC (Li et al., 2011); or genetic manipulation of (1) BMDC to block Hh signaling and regress the tumor vasculature (Nakamura et al., 2010), or (2) BM derived PSC by using the hGFAP promotor to develop targeted gene/drug therapy (Ding et al., 2009); or by unmasking the host's immune response by targeting BMD stromal fibroblasts that contribute to immunosuppression and tumor progression (Kraman et al., 2010). This review discussed the role of BMDC in pancreatic carcinogenesis, in particular their contribution to the tumor microenvironment, where they play a significant role in fibrosis, immunosuppression and neovascularization, and may provide opportunities for novel therapeutic strategies for pancreatic cancer.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Christopher J. Scarlett acknowledges the following funding support: Ramaciotti Foundation (ES2012/0104); Cancer Australia and Cure Cancer Australia Foundation (1033781); Cancer Institute NSW (09/CDF/2-40).
Akita, S., Kubota, K., Kobayashi, A., Misawa, R., Shimizu, A., Nakata, T., et al. (2012). Role of bone marrow cells in the development of pancreatic fibrosis in a rat model of pancreatitis induced by a choline-deficient/ethionine-supplemented diet. Biochem. Biophys. Res. Commun. 420, 743–749.
Alison, M. R., Lovell, M. J., Direkze, N. C., Wright, N. A., and Poulsom, R. (2006). Stem cell plasticity and tumour formation. Eur. J. Cancer 42, 1247–1256.
Apte, M. V., Haber, P. S., Applegate, T. L., Norton, I. D., McCaughan, G. W., Korsten, M. A., et al. (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43, 128–133.
Apte, M. V., Park, S., Phillips, P. A., Santucci, N., Goldstein, D., Kumar, R. K., et al. (2004). Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 29, 179–187.
Bachem, M. G., Schneider, E., Gross, H., Weidenbach, H., Schmid, R. M., Menke, A., et al. (1998). Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115, 421–432.
Beckermann, B. M., Kallifatidis, G., Groth, A., Frommhold, D., Apel, A., Mattern, J., et al. (2008). VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br. J. Cancer 99, 622–631.
Bruscia, E. M., Ziegler, E. C., Price, J. E., Weiner, S., Egan, M. E., and Krause, D. S. (2006). Engraftment of donor-derived epithelial cells in multiple organs following bone marrow transplantation into newborn mice. Stem Cells 24, 2299–2308.
Chan, D. A., Kawahara, T. L., Sutphin, P. D., Chang, H. Y., Chi, J. T., and Giaccia, A. J. (2009). Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 15, 527–538.
Chang, D. Z., Ma, Y., Ji, B., Wang, H., Deng, D., Liu, Y., et al. (2011). Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 17, 7015–7023.
Choi, J. B., Uchino, H., Azuma, K., Iwashita, N., Tanaka, Y., Mochizuki, H., et al. (2003). Little evidence of transdifferentiation of bone marrow-derived cells into pancreatic beta cells. Diabetologia 46, 1366–1374.
Clark, C. E., Hingorani, S. R., Mick, R., Combs, C., Tuveson, D. A., and Vonderheide, R. H. (2007). Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 67, 9518–9527.
De Wever, O., and Mareel, M. (2003). Role of tissue stroma in cancer cell invasion. J. Pathol. 200, 429–447.
Diaz-Flores, L. Jr., Madrid, J. F., Gutierrez, R., Varela, H., Valladares, F., Alvarez-Arguelles, H., et al. (2006). Adult stem and transit-amplifying cell location. Histol. Histopathol. 21, 995–1027.
Ding, Z., Maubach, G., Masamune, A., and Zhuo, L. (2009). Glial fibrillary acidic protein promoter targets pancreatic stellate cells. Dig. Liver Dis. 41, 229–236.
Direkze, N. C., Hodivala-Dilke, K., Jeffery, R., Hunt, T., Poulsom, R., Oukrif, D., et al. (2004). Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 64, 8492–8495.
Erkan, M., Hausmann, S., Michalski, C. W., Schlitter, A. M., Fingerle, A. A., Dobritz, M., et al. (2012). How fibrosis influences imaging and surgical decisions in pancreatic cancer. Front. Physiol. 3:389. doi: 10.3389/fphys.2012.00389
Erkan, M., Weis, N., Pan, Z., Schwager, C., Samkharadze, T., Jiang, X., et al. (2010). Organ-, inflammation- and cancer specific transcriptional fingerprints of pancreatic and hepatic stellate cells. Mol. Cancer 9, 88.
Evans, A., and Costello, E. (2012). The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 3:270. doi: 10.3389/fphys.2012.00270
Feig, C., Gopinathan, A., Neesse, A., Chan, D. S., Cook, N., and Tuveson, D. A. (2012). The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276.
Gabrilovich, D. I., and Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174.
Galli, S. J., Nakae, S., and Tsai, M. (2005). Mast cells in the development of adaptive immune responses. Nat. Immunol. 6, 135–142.
Gao, D., Nolan, D. J., Mellick, A. S., Bambino, K., McDonnell, K., and Mittal, V. (2008a). Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319, 195–198.
Gao, X., Song, L., Shen, K., Wang, H., Niu, W., and Qin, X. (2008b). Transplantation of bone marrow derived cells promotes pancreatic islet repair in diabetic mice. Biochem. Biophys. Res. Commun. 371, 132–137.
Hasegawa, Y., Ogihara, T., Yamada, T., Ishigaki, Y., Imai, J., Uno, K., et al. (2007). Bone marrow (BM) transplantation promotes beta-cell regeneration after acute injury through BM cell mobilization. Endocrinology 148, 2006–2015.
Herzog, E. L., Chai, L., and Krause, D. S. (2003). Plasticity of marrow-derived stem cells. Blood 102, 3483–3493.
Hess, D., Li, L., Martin, M., Sakano, S., Hill, D., Strutt, B., et al. (2003). Bone marrow-derived stem cells initiate pancreatic regeneration. Nat. Biotechnol. 21, 763–770.
Houghton, J., Stoicov, C., Nomura, S., Rogers, A. B., Carlson, J., Li, H., et al. (2004). Gastric cancer originating from bone marrow-derived cells. Science 306, 1568–1571.
Hwang, R. F., Moore, T., Arumugam, T., Ramachandran, V., Amos, K. D., Rivera, A., et al. (2008). Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926.
Ianus, A., Holz, G. G., Theise, N. D., and Hussain, M. A. (2003). In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J. Clin. Invest. 111, 843–850.
Jang, Y. Y., Collector, M. I., Baylin, S. B., Diehl, A. M., and Sharkis, S. J. (2004). Hematopoietic stem cells convert into liver cells within days without fusion. Nat. Cell Biol. 6, 532–539.
Kalesnikoff, J., and Galli, S. J. (2008). New developments in mast cell biology. Nat. Immunol. 9, 1215–1223.
Kallifatidis, G., Beckermann, B. M., Groth, A., Schubert, M., Apel, A., Khamidjanov, A., et al. (2008). Improved lentiviral transduction of human mesenchymal stem cells for therapeutic intervention in pancreatic cancer. Cancer Gene Ther. 15, 231–240.
Kraman, M., Bambrough, P. J., Arnold, J. N., Roberts, E. W., Magiera, L., Jones, J. O., et al. (2010). Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330, 827–830.
Krause, D. S., Theise, N. D., Collector, M. I., Henegariu, O., Hwang, S., Gardner, R., et al. (2001). Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105, 369–377.
Lechner, A., Yang, Y. G., Blacken, R. A., Wang, L., Nolan, A. L., and Habener, J. F. (2004). No evidence for significant transdifferentiation of bone marrow into pancreatic beta-cells in vivo. Diabetes 53, 616–623.
Li, A., Cheng, X. J., Moro, A., Singh, R. K., Hines, O. J., and Eibl, G. (2011). CXCR2-dependent endothelial progenitor cell mobilization in pancreatic cancer growth. Transl. Oncol. 4, 20–28.
Lin, W. R., Inatomi, O., Lee, C. Y., Kallis, Y. N., Otto, W. R., Jeffery, R., et al. (2012). Bone marrow-derived cells contribute to cerulein-induced pancreatic fibrosis in the mouse. Int. J. Exp. Pathol. 93, 130–138.
Lowenfels, A. B., Maisonneuve, P., Cavallini, G., Ammann, R. W., Lankisch, P. G., Andersen, J. R., et al. (1993). Pancreatitis and the risk of pancreatic cancer. N. Engl. J. Med. 328, 1433–1437.
Luo, G., Long, J., Zhang, B., Liu, C., Xu, J., Ni, Q., et al. (2012). Stroma and pancreatic ductal adenocarcinoma: an interaction loop. Biochim. Biophys. Acta 1826, 170–178.
Lyden, D., Hattori, K., Dias, S., Costa, C., Blaikie, P., Butros, L., et al. (2001). Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7, 1194–1201.
Marigo, I., Dolcetti, L., Serafini, P., Zanovello, P., and Bronte, V. (2008). Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 222, 162–179.
Marrache, F., Pendyala, S., Bhagat, G., Betz, K. S., Song, Z., and Wang, T. C. (2008). Role of bone marrow-derived cells in experimental chronic pancreatitis. Gut 57, 1113–1120.
Mathews, V., Hanson, P. T., Ford, E., Fujita, J., Polonsky, K. S., and Graubert, T. A. (2004). Recruitment of bone marrow-derived endothelial cells to sites of pancreatic beta-cell injury. Diabetes 53, 91–98.
Minami, E., Laflamme, M. A., Saffitz, J. E., and Murry, C. E. (2005). Extracardiac progenitor cells repopulate most major cell types in the transplanted human heart. Circulation 112, 2951–2958.
Mizukami, Y. (2012). Bone marrow-derived proangiogenic cells in pancreatic cancer. J. Gastroenterol. Hepatol. 27(Suppl. 2), 23–26.
Nakamura, K., Sasajima, J., Mizukami, Y., Sugiyama, Y., Yamazaki, M., Fujii, R., et al. (2010). Hedgehog promotes neovascularization in pancreatic cancers by regulating Ang-1 and IGF-1 expression in bone-marrow derived pro-angiogenic cells. PLoS ONE 5:e8824. doi: 10.1371/journal.pone.0008824
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., McIntyre, D., Honess, D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461.
Pan, J. J., Oh, S. H., Lee, W. C., and Petersen, B. E. (2009). Bone marrow-derived progenitor cells could modulate pancreatic cancer tumorigenesis via peritumoral microenvironment in a rat model. Oncol. Res. 17, 339–345.
Phillips, P. (2012). “Pancreatic stellate cells and fibrosis,” in Pancreatic Cancer and Tumor Microenvironment, eds P. J. Grippo and H. G. Munshi (Trivandrum: Transworld Research Network), 29–53.
Porembka, M. R., Mitchem, J. B., Belt, B. A., Hsieh, C. S., Lee, H. M., Herndon, J., et al. (2012). Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol. Immunother. 61, 1373–1385.
Poulsom, R., Alison, M. R., Forbes, S. J., and Wright, N. A. (2002). Adult stem cell plasticity. J. Pathol. 197, 441–456.
Provenzano, P. P., Cuevas, C., Chang, A. E., Goel, V. K., Von Hoff, D. D., and Hingorani, S. R. (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429.
Raman, D., Baugher, P. J., Thu, Y. M., and Richmond, A. (2007). Role of chemokines in tumor growth. Cancer Lett. 256, 137–165.
Scarlett, C. J., Colvin, E. K., Pinese, M., Chang, D. K., Morey, A. L., Musgrove, E. A., et al. (2011). Recruitment and activation of pancreatic stellate cells from the bone marrow in pancreatic cancer: a model of tumor-host interaction. PLoS ONE 6:e26088. doi: 10.1371/journal.pone.0026088
Serafini, P., Borrello, I., and Bronte, V. (2006). Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 16, 53–65.
Shojaei, F., Wu, X., Zhong, C., Yu, L., Liang, X. H., Yao, J., et al. (2007). Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450, 825–831.
Sparmann, G., Kruse, M. L., Hofmeister-Mielke, N., Koczan, D., Jaster, R., Liebe, S., et al. (2010). Bone marrow-derived pancreatic stellate cells in rats. Cell Res. 20, 288–298.
Taneera, J., Rosengren, A., Renstrom, E., Nygren, J. M., Serup, P., Rorsman, P., et al. (2006). Failure of transplanted bone marrow cells to adopt a pancreatic beta-cell fate. Diabetes 55, 290–296.
Vonlaufen, A., Joshi, S., Qu, C., Phillips, P. A., Xu, Z., Parker, N. R., et al. (2008). Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093.
Wagers, A. J., Sherwood, R. I., Christensen, J. L., and Weissman, I. L. (2002). Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 297, 2256–2259.
Wang, X., Ge, S., Gonzalez, I., McNamara, G., Rountree, C. B., Xi, K. K., et al. (2006). Formation of pancreatic duct epithelium from bone marrow during neonatal development. Stem Cells 24, 307–314.
Watanabe, T., Masamune, A., Kikuta, K., Hirota, M., Kume, K., Satoh, K., et al. (2009). Bone marrow contributes to the population of pancreatic stellate cells in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G1138–G1146.
Keywords: bone marrow derived stem cells, pancreatic cancer, stroma, pancreatic stellate cells, fibrosis, immunosuppression, neovascularization
Citation: Scarlett CJ (2013) Contribution of bone marrow derived cells to the pancreatic tumor microenvironment. Front. Physiol. 4:56. doi: 10.3389/fphys.2013.00056
Received: 21 January 2013; Paper pending published: 05 February 2013;
Accepted: 08 March 2013; Published online: 26 March 2013.
Edited by:
Phoebe Phillips, University of New South Wales, AustraliaReviewed by:
Phoebe Phillips, University of New South Wales, AustraliaCopyright © 2013 Scarlett. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Christopher J. Scarlett, Food Bioactives and Pancreatic Cancer Biology Group, School of Environmental and Life Sciences, University of Newcastle, Brush Road, Ourimbah, NSW 2258, Australia. e-mail:Yy5zY2FybGV0dEBuZXdjYXN0bGUuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.