

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Physiol., 25 January 2013
Sec. Membrane Physiology and Membrane Biophysics
Volume 4 - 2013 | https://doi.org/10.3389/fphys.2013.00002
This article is part of the Research TopicGasotransmitters: novel regulators of ion channels and transportersView all 10 articles
I remember as a neophyte academician in the early 80's the excitement and at times heated discussion generated by nitric oxide (NO). How could this toxic and relatively unheard-of gas possibly be a regulatory molecule? Even its discovery was the result of a laboratory mistake (Furchgott, 1995). As with any revolutionary idea, there were both zealous aficionados, naysayers and folks in between that “just wanted more proof.” As a result, it took nearly 10 years for this field to reach its log growth phase (Figure 1A); the fact that the number of yearly publications has now leveled off at approximately 7000 per year is probably more attributable to resource limitations than to enthusiasm. The physiological relevance of carbon monoxide (CO) was realized shortly after (Wu and Wang, 2005) but again there was a similar lag between discovery and acceptability as the scientific community tried to separate toxicology and pharmacology from physiology (Figure 1B).
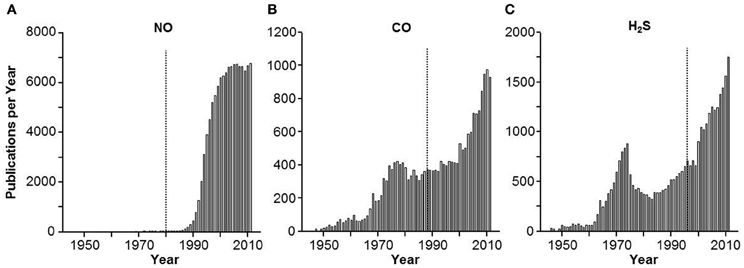
Figure 1. Frequency histogram of the number of yearly publications on (A) nitric oxide (NO), (B) carbon monoxide (CO) and (C) hydrogen sulfide H2S. Vertical lines indicate approximate year of the first description of a biological effect of the gas. Approximately 10 years passed before NO and CO caught on as biologically relevant signal molecules, acceptance of H2S was much quicker.
Hydrogen sulfide (H2S) was the third gas to be elevated to signaling status, i.e., a “gasotransmitter” (Wang, 2002). H2S has the same toxic pedigree as NO and CO (Guidotti, 2010) but it has not received the degree of initial skepticism that accompanied the latter two. As shown in Figure 1C, following the initial demonstrations of H2S signaling in the nervous and cardiovascular systems by Kimura's group (Abe and Kimura, 1996; Hosoki et al., 1997) the lag-time for H2S to catch on in the scientific community took about as long as the time for these two seminal papers to circulate. Within 3 years the log phase of H2S publications had begun, and it continues to this day.
It is not surprising that H2S was greeted with enthusiasm; gaseous signaling molecules were no longer novel, the implications of toxicity arising from excess signal were obvious, and a precedent for therapeutic potential (and profit) were firmly established. Add to this the fact that exogenous H2S appears to affect virtually every physiological system, it has potentially more biochemical diversity than the other gases, journals are eager for reviews (132 in 2011 and 93 as of the time of this writing in 2012), and away we go. The thought that the Emperor may not be wearing any clothes, and may even have a few warts, seems to be lost in the gala of the parade to the publisher and the patent office.
This is not to belittle the considerable potential of H2S research, there have been momentous gains, and undoubtedly more to come. Exogenous H2S affects virtually every organ system and physiological process to which it has been applied. Many of the effects of H2S are supported by compounds that are thought to inhibit or augment endogenous H2S production (See reviews; Caliendo et al., 2010; Kimura, 2010; Szabo, 2010; Olson, 2012b; Wang, 2012). The most notable among these are the cardiovascular system, where H2S mediates systemic vasodilation and pulmonary vasoconstriction, angiogenesis, oxygen sensing and is cardioprotective, the nervous system where H2S affects neuronal signaling, contributes to involved retinal function and may be involved in degenerative diseases, and the gastrointestinal (GI) tract where it contributes to signaling, insulin release, and metabolism. H2S appears to be anti-inflammatory in most tissues, although it may be inflammatory in some. Contrary to common thought, H2S itself does not readily react with reactive oxygen species (ROS) but it does stimulate glutathione production and augment reducing equivalents in the central nervous system (Kimura et al., 2010). Even novel signaling mechanisms whereby H2S interacts with the reduced (sulfhydration) or oxidized sulfur of cysteine to activate or inactivate proteins have been described (Mustafa et al., 2009; Tao et al., 2012). As well-illustrated in this issue, it is also becoming increasingly evident that the many of the biological effects of H2S are mediated directly through H2S interactions with ion channels (see also; Tang et al., 2010; Munaron et al., 2012; Peers et al., 2012).
Sulfide salts, such as NaHS and Na2S, have been historically used to rapidly generate H2S but this occurs at an uncontrolled rate and their purity may be problematic. These salts are gradually being replaced by the next generation of compounds that slowly release H2S. The clinical applicability these true H2S “releasing” drugs is now becoming evident and they offer additional advantages in that they can be combined with other, unrelated drugs and provide a multi-focal therapeutic approach (Caliendo et al., 2010; Olson, 2011; Kashfi and Olson, 2012). Already, H2S-releasing drugs have been combined with a variety of compounds including virtually all non-steroidal anti-inflammatory drugs (NSAIDs), sildenafil, levodopa, the anti-glaucoma drug lantanoprost, and the angiotensin-1 receptor antagonist losartan to name a few. The ability of some of these H2S-releasing drugs to uniquely counter the adverse effects of aspirin and other NSAIDs on the GI tract or to treat other inflammatory process such as inflammatory bowel disease (IBD), irritable bowel syndrome (IBS) and acute and chronic joint pain are nearing or already in initial clinical trials. A third generation of compounds that can be combined with drugs such as NSAIDs and release both H2S and NO, the so-called “NOSH” compounds may further increase potency and versatility; preliminary studies suggest they may be especially effective in the treatment of cancer (Kashfi and Olson, 2012).
A better understanding of H2S biochemistry and metabolism has also lead to promising therapy (Viscomi et al., 2010; Drousiotou et al., 2011). Ethylmalonic encephalopathy (EE) is an autosomal recessive deletion in the ETHE1 gene that encodes a mitochondrial sulfur dioxygenase. EE is typically fatal in the first decade of life. Patients with EE present with greatly elevated urinary and tissue thiosulfate concentrations and increased tissue H2S, presumably due to the inability of cells to efficiently metabolize H2S. This condition has recently been treated by oral administration of the bactericide metronidazole, to inhibit H2S production by colonic bacteria, and N-acetylcysteine. The latter acts as a membrane-permeable precursor of reduced glutathione (GSH) and appears to combine with H2S to form a non-toxic persulfide, GSSH. Targeting another mitochondrial enzyme, sulfur quinone oxidoreductase (SQR), which catalyzes the first step in H2S oxidation, may be a future therapeutic option (Linden et al., 2010).
The intent of this opinion is not to be a nayser, but just to request more proof that while we are going full-speed ahead we are also going in the right direction and not out of control. Clearly, we don't need proof that exogenous H2S activates/inhibits numerous biological systems, or that these systems can be modulated by fiddling with H2S metabolism, or that H2S has specific molecular targets, there are at least 225 reviews to substantiate these. However, we do need to critically examine a number of practical and technical issues. These include the promiscuity of many “selective” inhibitors of H2S biosynthesis, separating physiological from pharmacological effects of H2S, dosing issues, and measuring H2S in tissues and blood. Obviously, all are related; I will focus on the latter two.
H2S is a gas and we need some justification for ignoring its ephemeral nature; the half-life of H2S in open containers is a few scant minutes, even less when aerated (DeLeon et al., 2011). When we expose cells and tissues to H2S for hours and days in tissue culture or organ baths what are we looking at? Is this why we need to expose tissues to excessive H2S concentrations that can't possibly exist naturally? And where do we separate physiology from toxicology?
More importantly, however, we really need proof that what we are measuring in tissues and blood is actually H2S. This has profound implications in perhaps the most exciting avenue of H2S research, its therapeutic potential. My biggest concern is that if we do not do this right it can have disastrous consequences resulting in missed diagnoses and inappropriate therapies. I will give three examples, one where a simple calculation can be used to tell us if tissue H2S values are realistic, a second that will (hopefully) red-flag plasma and blood H2S measurements and a third that serves as an excellent example of how critical thinking can put the brake on a potentially dangerous clinical application.
First, lets look at tissue H2S concentrations. Most studies report tissue H2S concentrations anywhere from 1 nmol/mg protein to up to values well in excess of 10 μmol/mg protein. As I have previously calculated (Olson, 2012a) if one assumes a cell is 15% protein and 70% water then 1 μmol H2S/mg protein would result in a cytosolic concentration of 214 mmol/l! Even 1 nmol/mg protein in a cell is still be more than five times the toxic level. How can these values be so high? There are at least two reasons. First, most methods require incubating the tissue for 30 min or more under anoxic conditions. These conditions prevent normal H2S oxidation while H2S production continues unabated. Second, the most commonly used methods, methylene blue, monobromobimane and ion-selective electrodes (ISE) measure more than just H2S; and it's generally not clear what many of these other molecules are (Olson, 2012a). The alkaline antioxident buffer used with the ISE generates H2S from cysteine in serum albumin (Whitfield et al., 2008).
Even more troubling are H2S measurements in plasma and blood. H2S is commonly measured in plasma using the above methods or in headspace gas (typically after equilibration for 30 min or more under anoxic conditions) and concentrations from 1–30 μmol/l are common, although some go as high as 300 μmol/l. These are unrealistic because of the reasons described above, and as so elegantly shown by Furne et al. (2008), the human nose can detect 1 μmol/l; blood obviously doesn't smell like rotten eggs. While these erroneous values are a detraction in laboratory experiments, they are potentially dangerous when used diagnostically. And the latter is becoming more common. I offer a simple challenge to any laboratory that is interested in, or currently measuring H2S in blood; in addition to making your standard curve in buffer, make another standard curve in whole blood and compare the two. I guarantee the values obtained in the plasma will not correlate with buffer values and, in fact, there will be no measurable H2S in the former because red blood cells avidly remove H2S (Whitfield et al., 2008).
There is considerable, and alarming interest in using blood H2S measurements for clinical diagnosis and a variety of pathological conditions have been correlated with altered plasma H2S. Here I will give a single example. Goslar et al. (2011) used the standard methylene blue method to measure total plasma sulfide in patients admitted to the intensive care unit (ICU) with various forms of shock. Presuming plasma pH is 7.4 this would be ~20% H2S gas; in acidosis the percent H2S gas will increase. Goslar et al. (2011) reported that plasma sulfide was lower in survivors than non-survivors (13 vs. 32 μ M) and they concluded that plasma sulfide could be used as a predictor of mortality in the ICU. This concept was critically tested in a rat model of hemorrhagic shock by Van de Louw and Haouzi (2012) who found no evidence for such a correlation and, in fact, the methylene blue method measured plasma turbidity, not sulfide.
This Opinion is not meant to discourage research in this exciting field but to encourage critical thinking about how we are generating information and interpreting it. Clearly, these are exciting times. However, it may be necessary now and then to apply the brakes on exuberance to keep things from getting out of control. These points are further detailed in several recent reviews (Olson, 2009, 2011, 2012a; Linden et al., 2010).
The author wishes to thank the numerous students and colleagues for their effort and critical thought. The author also thanks the National Science Foundation for past and current (IOS 1051627) support on the biology of H2S.
Abe, K., and Kimura, H. (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071.
Caliendo, G., Cirino, G., Santagada, V., and Wallace, J. L. (2010). Synthesis and biological effects of hydrogen sulfide (H2S): development of H2S-releasing drugs as pharmaceuticals. J. Med. Chem. 9, 6275–6286.
DeLeon, E. R., Stoy, G. F., and Olson, K. R. (2011). Passive loss of hydrogen sulfide in biological experiments. Anal. Biochem. 421, 203–207.
Drousiotou, A., DiMeo, I., Mineri, R., Georgiou, T., Stylianidou, G., and Tiranti, V. (2011). Ethylmalonic encephalopathy: application of improved biochemical and molecular diagnostic approaches. Clin. Genet. 79, 385–390.
Furchgott, R. F. (1995). A research trail over half a century. Annu. Rev. Pharmacol. Toxicol. 35, 1–27.
Furne, J., Saeed, A., and Levitt, M. D. (2008). Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1479–R1485.
Goslar, T., Mars, T., and Podbregar, M. (2011). Total plasma sulfide as a marker of shock severity in nonsurgical adult patients. Shock 36, 350–355.
Guidotti, T. L. (2010). Hydrogen sulfide: advances in understanding human toxicity. Int. J. Toxicol. 29, 569–581.
Hosoki, R., Matsuki, N., and Kimura, H. (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237, 527–531.
Kashfi, K., and Olson, K. R. (2012). Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. doi: 10.1016/j.bcp.2012.10.019. [Epub ahead of print].
Kimura, H. (2010). Hydrogen sulfide: its production, release and functions. Amino. Acids 41, 113–121.
Kimura, Y., Goto, Y., and Kimura, H. (2010). Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 12, 1–13.
Linden, D. R., Levitt, M. D., Farrugia, G., and Szurszewski, J. H. (2010). Endogenous production of H2S in the gastrointestinal tract: still in search of a physiologic function. Antioxid. Redox Signal. 12, 1135–1146.
Munaron, L., Avanzato, D., Moccia, F., and Mancardi, D. (2012). Hydrogen sulfide as a regulator of calcium channels. Cell Calcium doi: 10.1016/j.ceca.2012.07.001. [Epub ahead of print].
Mustafa, A. K., Gadalla, M. M., and Snyder, S. H. (2009). Signaling by gasotransmitters. Sci. Signal. 2, 1–8.
Olson, K. R. (2009). Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim. Biophys. Acta 1787, 856–863.
Olson, K. R. (2011). The therapeutic potential of hydrogen sulfide: separating hype from hope. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R297–R312.
Olson, K. R. (2012a). A practical look at the chemistry and biology of hydrogen sulfide. Antioxid. Redox Signal. 17, 32–44.
Olson, K. R. (2012b). Hydrogen sulfide as an oxygen sensor. Clin. Chem. Lab. Med. doi: 10.1515/cclm-2012-0551. [Epub ahead of print].
Peers, C., Bauer, C. C., Boyle, J. P., Scragg, J. L., and Dallas, M. L. (2012). Modulation of ion channels by hydrogen sulfide. Antioxid. Redox Signal. 17, 95–105.
Szabo, C. (2010). Gaseotransmitters: new frontiers for translational science. Sci. Transl. Med. 2, 1–7.
Tang, G., Wu, L., and Wang, R. (2010). Interaction of hydrogen sulfide with ion channels. Clin. Exp. Pharmacol. Physiol. 37, 753–763.
Tao, B. B., Liu, S. Y., Zhang, C. C., Fu, W., Cai, W. J., Wang, Y., et al. (2012). VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid. Redox Signal. doi: 10.1089/ars.2012.4565. [Epub ahead of print].
Van de Louw, A., and Haouzi, P. (2012). Oxygen deficit and H2S in hemorrhagic shock in rats. Crit. Care 16, R178.
Viscomi, C., Burlina, A. B., Dweikat, I., Savoiardo, M., Lamperti, C., Hildebrandt, T., et al. (2010). Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 16, 869–871.
Wang, R. (2002). Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 16, 1792–1798.
Wang, R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896.
Whitfield, N. L., Kreimier, E. L., Verdial, F. C., Skovgaard, N., and Olson, K. R. (2008). Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1930–R1937.
Citation: Olson KR (2013) Hydrogen sulfide: both feet on the gas and none on the brake? Front. Physio. 4:2. doi: 10.3389/fphys.2013.00002
Received: 28 December 2012; Accepted: 06 January 2013;
Published online: 25 January 2013.
Edited by:
Mike Althaus, Justus-Liebig University of Giessen, GermanyReviewed by:
Mike Althaus, Justus-Liebig University of Giessen, GermanyCopyright © 2013 Olson. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence:a29sc29uQG5kLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.