


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 06 August 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00311
This article is part of the Research TopicOxidant Stress in Atrial Fibrillation: Sources, impact and therapeutic significanceView all 8 articles
Atrial fibrillation (AF) is the most common arrhythmia that requires medical attention, and its incidence is increasing. Current ion channel blockade therapies and catheter ablation have significant limitations in treatment of AF, mainly because they do not address the underlying pathophysiology of the disease. Oxidative stress has been implicated as a major underlying pathology that promotes AF; however, conventional antioxidants have not shown impressive therapeutic effects. A more careful design of antioxidant therapies and better selection of patients likely are required to treat effectively AF with antioxidant agents. Current evidence suggest inhibition of prominent cardiac sources of reactive oxygen species (ROS) such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and targeting subcellular compartments with the highest levels of ROS may prove to be effective therapies for AF. Increased serum markers of oxidative stress may be an important guide in selecting the AF patients who will most likely respond to antioxidant therapy.
Atrial fibrillation (AF), which affects approximately 2.5 million individuals in the United States, is the most common arrhythmia that requires medical treatment. The incidence of AF is increasing with the increase in the elderly population. The incidence of AF increases with age from less than 0.5 per 1000 person-years in individuals younger than 50 years to approximately 10 per 1000 person-years in those older than 70 years (Krahn et al., 1995). Miyasaka and colleagues estimated that 15.9 million people in the United States will have AF by 2050 (Miyasaka et al., 2006). The most serious adverse effects of AF are increased risk of stroke and peripheral embolization caused by thrombus formation mainly in the left atrial appendage (LAA). The incidence of stroke after a diagnosis of AF is 1–5% annually, depending on the patient's age and the presence of other risk factors.
The pathophysiology of AF is not completely understood. Reentrant circuits and ectopic activities particularly around the muscular sleeves of the pulmonary veins have been identified as potential electrophysiological mechanisms of AF (Jais et al., 1997; Van Wagoner, 2007; Iwasaki et al., 2011); however, these arrhythmias are probably the final representations of the underlying pathophysiological events in AF. Targeting the focal activities and reentry circuits without addressing the underlying pathologies, for example as catheter ablation therapy does, is likely to have constrained success because of this. Current antiarrhythmic drugs that usually block one or a few ion channels in cardiomyocytes are not highly effective in the treatment of AF, and they have shown serious adverse effects (Gjesdal, 2009). A better understanding of the pathophysiologic events upstream to reentry, focal activity and ionic current abnormalities are required to identify effective therapeutic targets.
Excess reactive oxygen species (ROS) have been implicated in pathogenesis of AF by affecting ion channels and propagation of the action potential. Nevertheless, oxidative stress biology is complex, and general radical scavengers have not shown impressive therapeutic effects in clinical trials (Sesso et al., 2008; Van Wagoner, 2008). This review will consider this paradox in further detail.
Several limitations can be mentioned for the current available therapies for AF. The underlying reason for most of these shortcomings lies in the fact that the current therapies do not address the underlying pathophysiology of AF.
Current antiarrhythmic medications usually target one or a few ion channels and almost always block these channels. Table 1 summarizes the most common adverse effects of the antiarrhythmic drugs that are commonly used in the management of AF, and Table 2 provides a summary of most important clinical studies on proarrhythmic effects of antiarrhythmic drugs. Proarrhythmia is an adverse effect of all current antiarrhythmic agents, which suggests that proarrhythmia is an adverse effect of the current pharmacological approach rather than a side effect of a few of those drugs. The Cardiac Arrhythmia Suppression Trial (CAST) was a landmark clinical trial in which class IC antiarrhythmic agents were used to suppress premature ventricular contractions (PVCs) after myocardial infarction (MI) to reduce the risk of ventricular arrhythmias (CAST Investigators, 1989). The study rationale was that PVCs are associated with a higher rate of sudden arrhythmic death, and antiarrhythmic agents effectively suppress PVCs. Nevertheless, treatment with an antiarrhythmic drug in the CAST trial was associated with a higher cardiovascular mortality rate. The treatment strategy in CAST involved simply suppressing focal activity and blocking the sodium channel. Later, it was found that sodium channels are down-regulated in patients with heart failure (Santana et al., 2005), and therefore further blockade of those channels may promote arrhythmia.
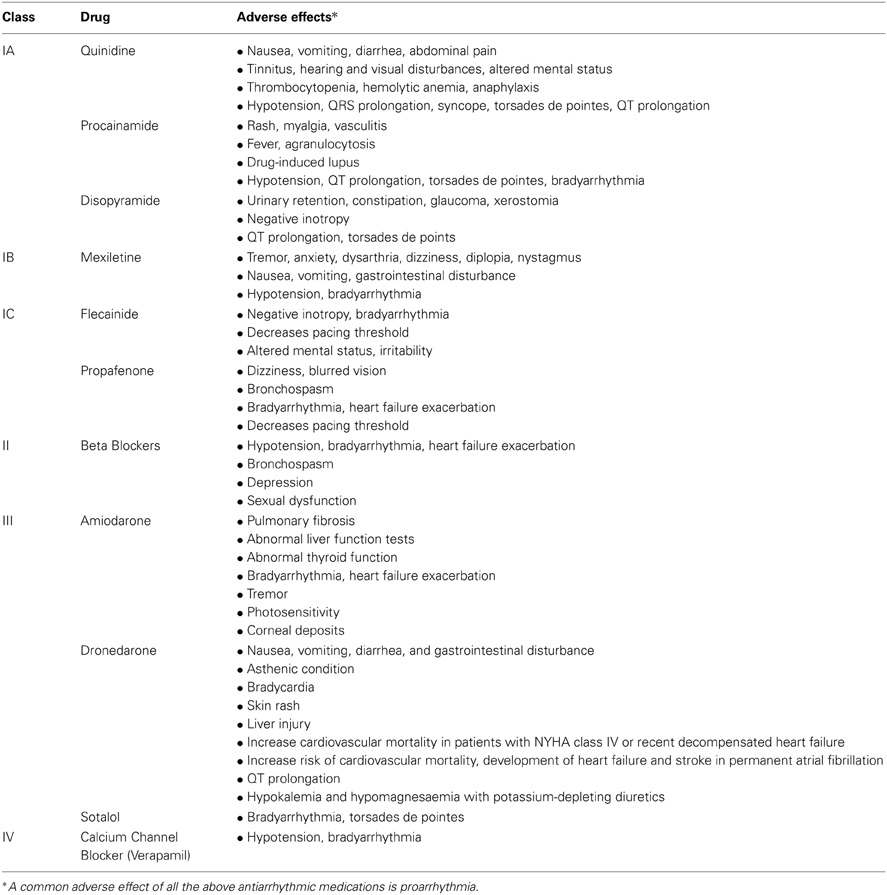
Table 1. The most common adverse effects of frequently used antiarrhythmic drugs in the management of atrial fibrillation.

Table 2. Clinical studies on the proarrhythmia of antiarrhythmic drugs.
Targeting ion channels as a therapeutic strategy carries the disadvantage of a narrow therapeutic index in which both low and high currents can cause arrhythmia. In addition, AF affects more than one ion current and blockade of one current may even potentiate the current imbalance toward arrhythmia.
Catheter ablation uses tissue destruction to block the propagation of the focal activity or to disrupt reentrant circuits. Catheter ablation has achieved considerable success in treating certain types of arrhythmia. Catheter ablation is an anatomically fixed treatment that may be the best choice for patients with an anatomically fixed substrate. For example, a bypass tract between the atria and the ventricles can be treated effectively with catheter ablation. Nevertheless, AF is often a complex arrhythmia with widespread and dynamic substrates. Thus, a line of ablation that cuts the current reentrant circuit may not be an effective treatment for future AF, since the substrate of arrhythmia may change location over time. The necessity to continue anticoagulation for prevention of stroke after catheter ablation of AF suggests the lack of complete suppression of AF by this treatment. In addition, the ablation-generated fibrotic scar tissue may provide an arrhythmogenic substrate and the procedure is associated with some immediate and long term complications (Maan et al., 2011).
The term ROS refers to a class of low molecular weight molecules that are partially reduced derivatives of molecular oxygen. ROS are wide range of molecules that include the superoxide radical anion (O2•−); hydrogen peroxide (H2O2); the hydroxyl radical (OH• + OH−); peroxynitrite (ONOO−), which is the product of the diffusion-controlled reaction between •NO and O2•−; and the derived radicals •NO2 and CO3•−. Low levels of ROS are necessary to mediate physiologic responses and to maintain homeostasis through the regulation of signal transduction events. Nevertheless, when cellular levels of ROS exceed the cell's ability to reduce excess free radicals, oxidative stress develops. The physiological concentration of ROS molecules may vary under different conditions and in different cellular compartments. In addition, methods of ROS measurement have certain limitations (Tarpey and Fridovich, 2001). It is generally thought that the intracellular concentration of superoxide rarely exceeds 1 nM (Brawn and Fridovich, 1980; Tarpey et al., 2004), and the normal physiological concentration of H2O2 is less than 15 μM (Tarpey and Fridovich, 2001; Kulagina and Michael, 2003; Liu et al., 2004; Tarpey et al., 2004). How much ROS increase in different pathological conditions may greatly vary; however, the known physiological range of ROS can provide a general guide to use relevant ROS concentrations and avoid extremely high levels of ROS in experimental studies.
Most ROS react with multiple biomolecules (proteins, deoxyribonucleic acid, ribonucleic acid, and lipids) and cause the loss of enzyme function, breaks in DNA strands, DNA mutations, lipid peroxidation, and cellular death. Protein cross-links, fragmentation, hydroxylation, nitration, halogenation, carboxylation, and reactive aldehyde formation are common outcomes of the interaction of proteins with various oxidants. Most of the ROS effects on proteins are irreversible and result in loss of function of those proteins, which eventually are degraded and removed by proteasomes (Levine, 2002). An aggressive ROS molecule such as hydroxyl radical can modify most amino acids (Halliwell et al., 1987); however, some amino acids such as cysteine, methionine, proline, arginine, tyrosine, and tryptophan have generally higher susceptibility to ROS modifications.
One important way in which ROS exert their effect is by modifying the thiol group of proteins (cysteine amino acid contains thiol groups; Chen et al., 2003) often times interfering with signal transduction cascades what leads to low pKa thiol phosphatase inhibition with consequent augmentation of kinase activity (Sommer et al., 2002; Connor et al., 2005). In addition to direct oxidation of thiol groups of proteins, ROS can oxidize low molecular weight biomolecules such as glutathione generating secondary oxidative products that then may react with protein thiols (Eaton, 2006). Methionine residues can be oxidized to methionine-S-sulfoxides and methionine-R-sulfoxides, which may be a reversible process; however, further oxidation of methionine residues to methionine-S-sulfone seems to be irreversible (Weissbach et al., 2002). An additional effect of ROS on amino acids is reactive aldehyde formation. It has been shown that ROS (and particularly HOCl) oxidize almost all amino acids commonly found in the plasma to a corresponding family of aldehydes in high yield (Hazen et al., 1998a,b). The reactive aldehydes have been shown to mediate the effect of ROS in cardiovascular disorders such as atherosclerosis and in diabetes (Uchida, 2000). Carbonylation of proline, lysine, threonine, and arginine is another important protein modification by ROS (Levine, 2002). In failing explanted human hearts, an increase in the carbonylation of actin and tropomyosin, and an increase in the dimerization and nitrosation of tropomyosin have been reported as evidence of the oxidative modifications of important cardiac proteins (Canton et al., 2011). Table 3 summarizes some of the important modifications of amino acids by oxidative stress.

Table 3. Some important amino acid modifications by reactive oxygen/nitrogen species.
ROS reactions can also lead to the formation of lipid hydroperoxides, which are oxygenated products of the primordial lipid radical. Lipid peroxidation is self-perpetuating and thus amplifies several-fold the initial damage of ROS-induced oxidation. The accumulation of reactive lipid peroxides and lipid-derived aldehydes also contributes to oxidant-mediated signaling and cell damage. DNA is a frequent target of ROS. The most common ROS-induced modifications to DNA include single-strand breaks. Double-strand breaks are potentially hazardous to cells and are repaired via two main pathways: homologous recombination and nonhomologous end-joining. In addition to strand breakage, hydroxylation, adduct formation, and the nitration of bases can damage the DNA. Repair mechanisms exist that are largely dependent on base excision, replacement, and relegation (Riis and Poulsen, 2005).
Of the numerous cellular sources of ROS generation, mitochondria, the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and uncoupled NOS are considered the major ROS production systems in the human heart. Those sources of cardiac ROS are interrelated and often activation of one results in activation of the others (Doughan et al., 2008; Zinkevich and Gutterman, 2011; Figure 1). For example, mitochondrial function differentially modulates NADPH oxidase expression and activity (Wosniak et al., 2009; Kuroda et al., 2010).
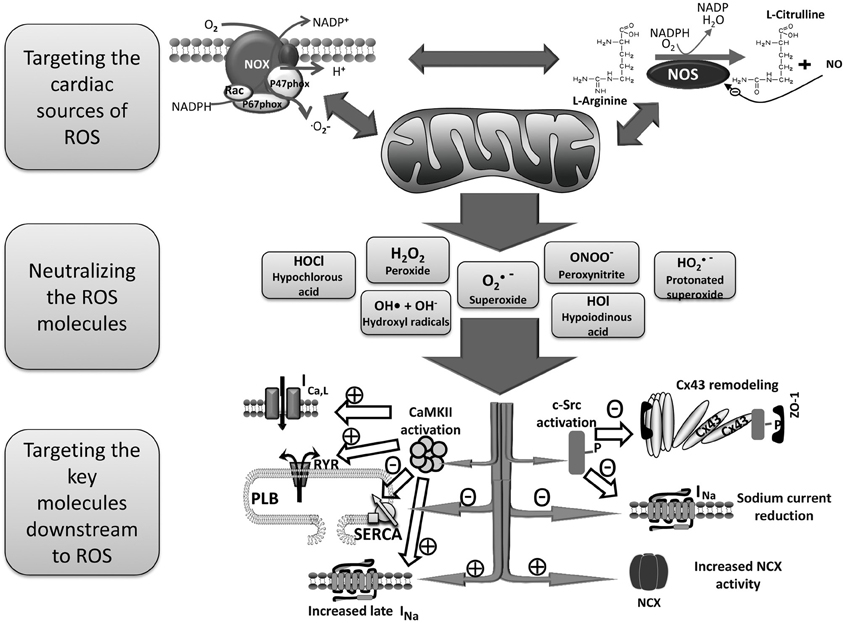
Figure 1. Schema for the treatment of AF by reducing ROS. Mitochondria, NADPH oxidase and uncoupled NOS are probably the most important cardiac sources of excess ROS. There are positive feedbacks among these sources in a way that activation of one results in increased activity and ROS production of the others. A variety of ROS molecules are produced as a result of activation of those sources of cardiac ROS which then oxidize proteins and lipids resulting in arrhythmia in several ways. Three main therapeutic strategies to prevent ROS induced arrhythmia are targeting the main cardiac sources of ROS, neutralizing ROS molecules, and searching for the key molecules that mediate the arrhythmogenic effects of ROS. AF, atrial fibrillation; CaMKII, Ca2+/calmodulin-dependent protein kinases II; CX43, connexin43; NADPH, nicotinamide adenine dinucleotide phosphate; NCX, Na+/Ca2+ exchanger; NOS, nitric oxide synthase; PLB, phospholamban; ROS, reactive oxygen species; RYR, ryanodine receptor, SERCA, sarco/endoplasmic reticulum Ca2+-ATPase.
In mitochondria, when electrons flow from nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide to molecular oxygen via complex shuttle mechanisms, partially reduced semiquinone intermediates are produced and can react directly with O2 to produce O2•−. The coenzyme Q semiquinone is believed to be the major point of electron leakage in mitochondria.
The NAD(P)H oxidase is an enzyme that uses NAD(P)H to reduce molecular oxygen and produces large amounts of superoxide radicals. ROS are generated as a primary product of the NAD(P)H oxidase system, rather than a byproduct as they are in the mitochondrial system. Although NAD(P)H oxidase activity was first described in macrophages, a number of NAD(P)H oxidase isoforms (the Nox family) have been discovered in variety of nonphagocytic cells and Nox1, Nox2, and Nox4 have been detected in the heart. Nox activity is regulated in part by angiotensin II (Sorescu et al., 2002).
Uncoupled or dysfunctional nitric oxide synthases (NOSs) are other important sources of ROS. There are three major isoforms of NOS enzymes: inducible NOS (iNOS), endothelial NOS (eNOS), and neuronal NOS (nNOS). Their basic function is oxidizing the terminal guanidine nitrogen atom of L-arginine by using electrons from NADPH to produce NO., as shown below:
NO• increases the cGMP level. It reversibly binds to and inhibits complex IV in mitochondria (Brown and Cooper, 1994), and it may nitrosate some thiols (Stamler and Hausladen, 1998). The oxidation of NO• leads to the formation of •NO2, which is also involved in redox signaling and direct tissue damage. NO• signaling is diverse. One important interaction is the reaction of NO• with superoxide, the product of which is peroxynitrite (ONOO−/ONOOH). The formation of ONOO−/ONOOH can directly or via the depletion of tetrahydrobiopterin (BH4) lead to NOS uncoupling, which further reduces NO• production. Increased superoxide production has been shown to reduce NO• signaling with detrimental effects on endothelial cell homeostasis (Freeman et al., 1995).
In addition to mitochondria, NAD(P)H oxidase, and NOS systems, there are other sources of ROS (e.g., peroxidases, xanthine oxidase, and aldehyde oxidase), the roles of which in cardiac oxidative stress are less well-documented than these three sources.
AF is associated with several clinical risk factors including some cardiac and respiratory disorders, aging, and surgeries. Table 4 shows the main clinical risk factors of AF. An elevated level of ROS has been linked with increasing age, heart failure, diabetes mellitus, coronary artery disease, obesity, and alcohol intoxication, all conditions associated with AF (Sovari and Dudley, 2010). In addition, multiple studies have shown an increase in markers of oxidative stress in humans with AF. The activity of myofibrillar creatine kinase (MM-CK), which is redox sensitive, can be used as an indirect marker of oxidative stress (Mihm et al., 2001). MM-CK activity is decreased, and immunodetectable 3-nitrotyrosine, a marker for the presence of peroxynitrite, is increased in right atrial appendage (RAA) of patients with AF compared to those in normal sinus rhythm (Mihm et al., 2001). Moreover, coronary artery bypass surgery, a procedure that is associated with AF in a considerable percentage of patients, is linked with an increase in oxidized glutathione and lipid peroxidation (De Vecchi et al., 1998). Derivatives of reactive oxidative metabolites (DROMs) and ratios of oxidized to reduced glutathione [E(h) GSH] and cysteine [E(h) CySH] that quantify oxidative stress in patients are increased in patients with AF (Neuman et al., 2007). The increase in the odds ratios for AF for an elevated E(h) GSH, E(h) CySH, or DROMs are 6.1 (95% CI, 1.3–28.3; P = 0.02), 13.6 (95% CI, 2.5–74.1; P = 0.01), and 15.9 (95% CI, 1.7–153.9; P = 0.02), respectively (Neuman et al., 2007).
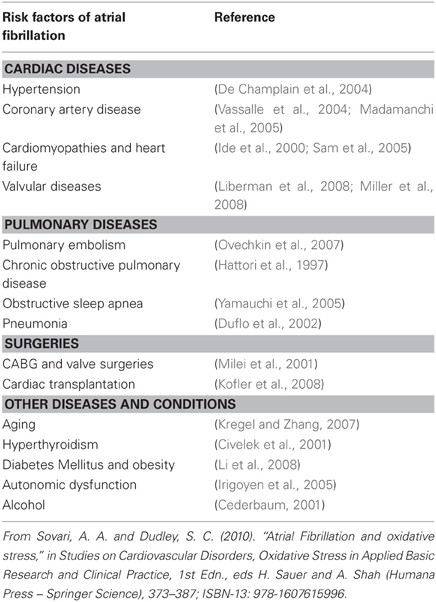
Table 4. Main clinical risk factors of atrial fibrillation and their association with oxidative stress.
Kim and colleagues measured NAD(P)H-stimulated superoxide production in RAA, and plasma markers of lipid and protein oxidation (thiorbabituric acid-reactive substances, 8-isoprostane, and protein carbonyls) in 170 patients undergoing coronary artery bypass surgery. They found that NAD(P)H oxidase activity was the strongest independent predictor of postoperative AF (odds ratio 2.41; 95% confidence interval 1.71–3.40, p < 0.0001; Kim et al., 2008).
In addition, genetic studies have shown that the gene expression pattern of atrial tissue in patients with AF is associated with oxidative stress. AF in human has been shown to be associated with a significant reduction in the gene expression of antioxidant genes as well as a significant increase in the gene expression of five genes related to ROS, supporting a clear shift toward pro-oxidation state in AF (Kim et al., 2003). The gene expression of glutathione peroxidase-1 and heme oxygenase-2 are decreased while the gene expression of flavin containing monooxygenase-1, monoamine oxidase-B, uniquin specific protease-8, tyrosine-related protein-1, and tyrosine 3-monooxygenase are increased in atrial tissue of AF patients (Kim et al., 2003). For a more complete list of gene expression and protein level changes of pro-oxidant and antioxidant enzymes in AF patients please see Table 5.
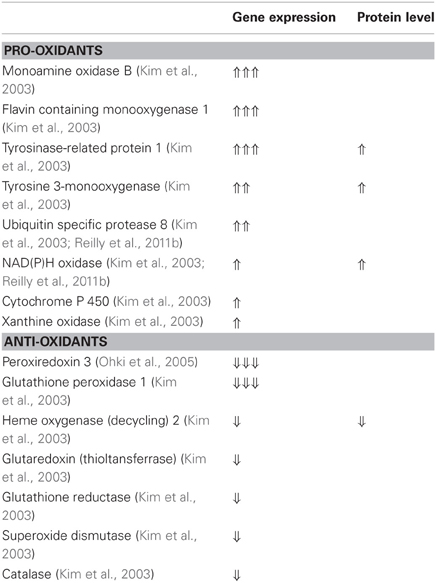
Table 5. Gene expression and protein level of pro-oxidants and antioxidants in the right atrial appendage of AF patients compared to patients in sinus rhythm.
Recent studies on the effect of omega-3 fatty acids on ROS production have produced conflicting results (Kowey et al., 2010; Liu et al., 2011), and it was shown that some forms of polyunsaturated fatty acids may even increase oxidative stress (Kimura et al., 2012), which may provide an explanation for some of the associated proarrhythmic effects that have been seen with omega-3 fatty acids (Billman et al., 2011).
The data supporting the association of excess ROS with human AF is in the form of increased oxidized glutathione, oxidized cysteine, DROMs, superoxide, peroxynitrite, and NAD(P)H oxidase. Therefore, these studies have not established conclusively cause and effect nor have they provided mechanistic insight (Carnes et al., 2007; Neuman et al., 2007; Antoniades et al., 2012). Therefore, most of our knowledge about the possible mechanisms by which excess ROS can induce arrhythmia is from experimental and isolated cellular studies, which may not be applicable in clinical AF.
H2O2 prolongs the action potential duration (APD) and induces triggered activity (TA) via early afterdepolarization (EAD) and delayed afterdepolarization (DAD) mechanisms in myocytes (Beresewicz and Horackova, 1991). Perfusion of H2O2 (0.1–1 mM) into fibrotic rat and rabbit hearts in the Langendorff setting induces EADs, TAs, and subsequent arrhythmia (Morita et al., 2009). One of the mechanisms of H2O2-induced APD prolongation and EAD formation is by the development of an enhanced late sodium (Na+) current (Song et al., 2006). Treatment with H2O2 and angiotensin II enhance the late Na+ current but decreases the overall Na+ current in isolated myocytes through the down-regulation of SCN5A transcription (Shang et al., 2008). The antiarrhythmic effect of the late Na+ current blocker, ranolazine supports a role for the late Na+ current in mediating the genesis of EADs by oxidative stress (Morita et al., 2011). Nevertheless, one should consider that ranolazine has multiple other effects including its metabolic effects and blockade of Ikr current and that the role of late Na+ current in AF is not universally accepted (Schotten et al., 2010). While increase in late Na+ current may result in arrhythmia via an EAD mechanism, the reduction in total Na+ current caused by ROS may cause a reduction in CV and provide a substrate for reentry. We have shown that ROS can downregulate cardiac Na+ channels, and mitochondrial antioxidants can reverse this effect (Liu et al., 2010). ROS also can directly stimulate the L-type Ca2+ current, which results in abnormal intracellular calcium cycling in myocytes and facilitates EADs (Thomas et al., 1998). The studies on the net effect of ROS on L-type Ca2+ current have shown conflicting results, however. For example, L-type Ca2+ was found to be decreased in isolated atrial myocytes of patients with AF, probably via S-nitrosation of the calcium channels, and the current was restored to normal level by using N-acetylcysteine (Carnes et al., 2007). Hydroxyl radicals increase the open probability of cardiac ryanodine receptors, which control the Ca2+ release from the sarcoplasmic reticulum (SR) to the cytoplasm (Anzai et al., 1998). Excess ROS also increase Ito current probably via an increase in expression of the regulatory β-subunit KChlP2 (Sridhar et al., 2009).
A key factor in arrhythmogenesis is reduction of the repolarization reserve. This refers to the balance of inward depolarizing currents such as Na+ and Ca2+ and to the outward repolarizing currents such as potassium (K+) during the second and third phases of the cardiac action potential. Decreasing the repolarization reserve occurs when the balance is shifted away from repolarizing currents and results in a prolonged action potential and increased the likelihood of EADs and TA. The repolarization reserve and the cytoplasmic Ca2+ level of cardiac myocytes are affected by the rate of Ca2+ uptake by the SR. Exposure to OH− significantly decreases SR Ca2+ uptake, which leads to an increased Ca2+ level in myocytes during diastole (Morris and Sulakhe, 1997). This short-term effect on Ca2+ transport is likely because of the OH−-mediated peroxidation of lipid membranes and protein sulfhydryl formation, which leads to an indirect effect on the SR Ca2+ transporter (Morris and Sulakhe, 1997).
ROS also affect gap junctions. Gap junctions form connections between cells through aggregation of connexin (Cx) proteins into hemichannels that meet to form conductive channels at cardiomyocyte interactions. Ventricular gap junctions are formed primarily from Cx43, however, a significant portion of gap junctions in the atria are formed by Cx40. c-Src is known to be activated by ROS. In an animal model of MI, the up-regulation of c-Src tyrosine kinase and an increase in the level of phosphorylated Tyr 416 c-Src (the active form of c-Src) resulted in the down-regulation of connexin43 (Cx43) via competition between phosphorylated c-Src and Cx43 for a binding site at zonula occludens-1, an intercalated disk scaffolding protein (Kieken et al., 2009). Other mechanisms for the reduction of Cx43 activity via the up-regulation of c-Src have also been suggested. They include tyrosine phosphorylation of Cx43 by c-Src, which also impairs gap junction function (Toyofuku et al., 1999). Inhibition of c-Src prevents Cx43 remodeling and ventricular arrhythmia caused by of angiotensin II activation and oxidative stress (Iravanian et al., 2011; Sovari et al., 2011b).
AF is multifactorial. Inflammation, myocardial fibrosis, and oxidative stress are important pathologic events that promote AF (Negi et al., 2010; Sovari and Dudley, 2010). Activated inflammatory cells such as monocytes, neutrophils, eosinophils, and macrophages produce ROS and lysosomal hydrolytic enzymes at sites of inflammation (Morel et al., 1991). Also, ROS enhance the inflammatory response partially via the activation of signaling events that mediate the expression of inflammatory genes (Suzuki et al., 1997) in part through activation of nuclear factor-kappa B (NF-κB; Kabe et al., 2005). NF-κB, a family of related transcription factors that act as principal regulators of inflammation, are activated by various stimuli (such as ROS) after MI, in ischemic states, and during reperfusion (Lu et al., 2004; Seddon et al., 2007). We recently showed that the elevation of ROS levels by angiotensin II can activate NF-κB; this in turn transcriptionally downregulates Na+ currents (Shang et al., 2008). Nevertheless, whether the cardiac Na+ current is actually decreased in AF is controversial (Bosch et al., 1999; Sossalla et al., 2010; Schotten et al., 2011).
ROS enhance fibroblast proliferation and type I collagen gene expression (Murrell et al., 1990). Antioxidant therapy reduces fibrosis by decreasing the level of transforming growth factor-β, which is the major cytokine that promotes cardiac fibrosis (Koli et al., 2008; Cu et al., 2009). In addition, Ca2+/calmodulin-dependent protein kinases II (CaMKII) has been recently identified as one of the mediators of fibroblast proliferation in response to angiotensin II (Bellocci et al., 2007). Because CaMKII activity increases in the presence of oxidative stress, CaMKII activation may be a pathway by which oxidative stress stimulates fibroblast proliferation within the myocardium.
Therefore, excess amount of ROS can promote AF by effects on ion channels and by enhancing myocardial fibrosis. The level of excess ROS may vary in different conditions and may be a factor to determine the specific downstream effects of ROS; a matter that requires more studies.
Virchow's triad of blood stasis, endothelial dysfunction, and a hypercoagulable state are the main etiologic factors determining thrombus formation. The presence of left atrial spontaneous echo contrast and chamber enlargement, both of which are evidence of blood flow stasis in patients with AF, are strongly associated with an increased risk for cerebral ischemic events in this condition (Celermajer et al., 1994; Jones et al., 1996). The proclivity for thrombus formation in the LAA, emphasizes the central role of stasis in thromboembolism associated with AF. Oxidative stress can potentially contribute to the risk of thrombus formation in AF by causing endocardial dysfunction, and endocardial dysfunction has been demonstrated in AF. AF is associated with the downregulation of NO• and the upregulation of superoxide production in the left atrium (Radomski et al., 1987), which shifts the endocardial balance toward thrombogenicity with overexpression of the prothombotic protein, plasminogen activator inhibitor-1, and increased expression of adhesion molecules on the endothelial surface (Bouchie et al., 1998; Carnes et al., 2001; Cai et al., 2002).
Antioxidant therapeutic agents for management of AF can be designed against at least three categories of targets: (1) ROS molecules, (2) the sources of ROS production, and (3) the key signaling molecules that mediate the arrhythmogenic effect of ROS (Sovari et al., 2011a; Figure 1).
One therapeutic approach for suppression of ROS is by using oxygen-radical scavengers such as vitamin E, vitamin C, N-acetyl-cysteine, ebselen, and tempol, which neutralize ROS molecules. Administration of vitamin C may reduce the incidence of postoperative AF (Carnes et al., 2001). Nevertheless, oxygen-radical scavengers have failed to show an impressive therapeutic effect for cardiovascular disorders in most clinical trials (Sesso et al., 2008; Song et al., 2009). ROS usually are highly reactive molecules, and conventional antioxidants may not be able to neutralize the ROS molecules before they exert their effect on proteins and lipids. In addition, ROS include a wide range of molecules, which may be generated from other ROS in reactions that are catalyzed by a wide range of enzymes. Table 5 reviews some of the known changes in important pro-oxidant or antioxidant enzymes in AF. Table 6 provides a review of some of the known downstream arrhythmogenic effects of only a few of the most important ROS molecules. A ROS scavenger may not effectively neutralize all ROS molecules to prevent the arrhythmogenic downstream effects. This may explain the lack of therapeutic effects of conventional antioxidants.

Table 6. A review of some of the known arrhythmogenic molecular targets of ROS molecules.
A more effective therapeutic approach may involve inhibition of the sources of excess cardiac ROS. NAD(P)H oxidase activity has been shown to increase in AF (Dudley et al., 2005). The NAD(P)H oxidase is upregulated in early stages of AF but not in chronic AF (Reilly et al., 2011a). Therefore, inhibition of the NAD(P)H oxidase is most likely to be an effective treatment for primary prevention of AF and for postoperative AF (Sovari et al., 2008; Sovari, 2011). The association of NAD(P)H oxidase activity with development of postoperative AF supports the idea of using NAD(P)H oxidase inhibitors for prevention of AF following surgeries (Kim et al., 2008).
The effectiveness of NOS inhibitors such as NG-Nitro-L-arginine methyl ester (L-NAME) in treating arrhythmias has been tested in various experiments. In a model of occlusion-reperfusion arrhythmia in cats, it was shown that repeated injections of L-NAME decreased the incidence of occlusion arrhythmias by 40%, eliminated reperfusion-induced ventricular arrhythmias, and reduced the latency of occlusion arrhythmias (Kukushkina et al., 1999). More studies are required to evaluate the effect of NOS inhibitors on AF. One potential problem with NOS inhibition is the simultaneous reduction of NO with inhibition of ROS produced by uncoupled NOS. A more effective way to prevent ROS production by uncoupled NOS may be prevention of uncoupling by providing the required coenzyme, tetrahydrobiopterin. In a canine model of nonischemic heart failure with increased propensity to AF, treatment with tetrahydrobioptein and L-arginine prevented oxidative stress in the atrial tissue (Nishijima et al., 2011).
Mitochondrial ROS seems like an attractive therapeutic target. A large portion of cardiomyocytes are occupied by mitochondria, and they are major sources of cardiac ROS (O'Rourke et al., 2005). We tested the effect of seven different antioxidant therapies on prevention of ventricular arrhythmia in an angiotensin II activation mouse model with increased levels of ROS (Sovari et al., 2011c). ROS were highly compartmentalized in mitochondria and a mitochondria-targeted antioxidant prevented spontaneous and pacing induced ventricular arrhythmia. Whether this result can be applied to AF remains to be tested.
When an antioxidant agent that inhibits sources of ROS is designed, several important factors must be considered. Various sources of ROS can be activated under different pathologic conditions; therefore, an antioxidant against a specific source of ROS may be effective in the prevention of arrhythmia only under certain conditions. In addition, because of positive feedback loops among the sources of cardiac ROS, the simultaneous targeting of several important sources of ROS may prove to be an effective therapy.
A third therapeutic strategy may target signals that are downstream from ROS. For example, CaMKII inhibition may prevent many of the ROS-mediated effects on Ca2+ and Na+ channels or on the promotion of the fibrosis that causes arrhythmia. Ranolazine, a late Na+ current blocker, may inhibit some of the arrhythmogenic effects of ROS. We showed that the inhibition of c-Src tyrosine kinase prevents the effects of angiotensin II and ROS on Cx43 remodeling (Sovari et al., 2010, 2011b), and c-Src is activated by ROS (Sovari et al., 2011d). Thus c-Src may be another example of antiarrhythmic therapeutic targets that are probably downstream from oxidative stress.
In addition to the aforementioned three categories of antioxidant therapy, it is also possible to target the upstream pathologies that result in activation of sources of ROS. For example, angiotensin II is known to increase ROS production by activation of NAD(P)H oxidase and probably by increasing ROS production in mitochondria (Iravanian et al., 2008; Dikalov, 2011; Sovari et al., 2011c; Jeong et al., 2012). Angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) exert antioxidant and antiarrhythmic effect (Kober et al., 1995; Schramm et al., 2012). Nevertheless, there are numerous pathological insults that may activate sources of ROS such as ischemia-reperfusion, inflammation, aging, diabetes, tachycardia, mechanical stretch and sheer stress, and there are probably more unknown causes of oxidative stress (Molyneux et al., 2002; Boldt et al., 2003; De Champlain et al., 2004; Issac et al., 2007; Kregel and Zhang, 2007; Li et al., 2008; Van Wagoner, 2008; Ahmed et al., 2010; Morita et al., 2011; Sovari et al., 2012). Many of those pathological processes are complex, and no effective therapy is available for them. Moreover, some of the available therapies for these upstream targets are not completely effective. For example, ACE inhibitors and ARBs do not completely suppress the elevated levels of angiotensin II (Jorde et al., 2000; van de Wal et al., 2006).
An important consideration in the treatment of AF is early intervention. In later stages of chronic AF, the disease may be associated with multiple pathological processes, and some of the remodeling may not be reversible. For example, ACE inhibitors and ARBs are effective in primary prevention of AF; however, they may not be effective in the management of chronic AF (Disertori et al., 2009; Goette et al., 2012; Khatib et al., 2012). Similarly, antioxidant therapy may be much more effective if it is applied in early stages of AF and in primary prevention. Postoperative AF is an example for opportunities to test the efficacy of an early intervention. Statin drugs that exert some antioxidant activity by Rac1-mediated suppression of NAD(P)H oxidase have been shown to be effective in prevention of postoperative AF (Reilly et al., 2011b; Antoniades et al., 2012).
While chronic AF with significant structural and electrical remodeling of the heart is almost certainly a complex and multifactorial disease, the AF may be multifactorial from the onset. For example, the cause of a new onset AF in an elderly patient with diabetes and cardiomyopathy may be because of a combination of increased myocardial fibrosis, abnormal intracellular Ca2+ handling, autonomic dysfunction, and multiple abnormalities in ionic currents. Oxidative stress may be only one of the underlying pathologies. Therefore, the best antioxidant therapy may not completely prevent AF in all patients because, in some AF patients, other arrhythmogenic processes independent from oxidative stress exist.
Important guides in selecting those AF patients who will most likely benefit from antioxidant therapy are markers of oxidative stress. There are numerous serum markers of oxidative stress available such as thiobarbituric acid-reacting substances, superoxide dismutase and glutathione peroxidase activities (Hartnett et al., 2000), thioredoxin, ischemia-modified albumin (Lambrinoudaki et al., 2009), carotenoids, oxidized low density lipoproteins, oxidized low density lipoprotein antibodies (Suzuki et al., 2003), oxidized to reduced glutathione and cysteine, and DROMs (Neuman et al., 2007; Shimano et al., 2009). Some of these serum markers of oxidative stress have been shown to be elevated in AF patients; however, however, more studies are required to identify the best serum markers for patients at risk of AF and for those AF patients who possibly will respond to antioxidant treatment.
Treatments that do not address the underlying pathophysiology of AF are likely to have limits to their efficacy. There is considerable evidence that oxidative stress has an important role in genesis of AF. The pathological link remains to be proven, however, and designing an effective antioxidant therapy requires a better understanding of the complex biology of oxidative stress.
Current evidence suggests that NAD(P)H oxidase inhibitors may be effective in primary prevention of AF and in postoperative AF. Mitochondria-targeted antioxidants may prove to be most effective antioxidant therapeutic intervention in persistent AF. Targeting important molecules downstream from ROS such as c-Src and CaMKII may provide additional antiarrhythmic effect. Effective antioxidant therapy may also reduce the risk of thromboembolism in patients with AF by improving endocardial dysfunction.
Dr. Dudley has applied for patents: (1) Method for Predicting Onset/Risk of Atrial Fibrillation, (2) Prevention of sudden death by modulation of Src family, (3) Modulating mitochondrial reactive oxygen species to increase cardiac sodium channel current and mitigate sudden death, (4) Biomarkers for Prediction of Stroke Risk in Atrial Fibrillation, and (5) Mitochondrial anti-oxidants for prevention of sudden death by raising connexin43 levels.
RO1 HL1024025, T32 HL072742, P01 HL058000, R01 HL106592, a VA MERIT grant, and an American Heart Association Midwest Affiliate Postdoctoral Fellowship # AHA10POST4450037.
Adachi, T., Weisbrod, R. M., Pimentel, D. R., Ying, J., Sharov, V. S., Schoneich, C., and Cohen, R. A. (2004). S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207.
Ahmed, M. I., Gladden, J. D., Litovsky, S. H., Lloyd, S. G., Gupta, H., Inusah, S., Denney, T. Jr., Powell, P., McGiffin, D. C., and Dell'italia, L. J. (2010). Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitral regurgitation and ejection fraction >60%. J. Am. Coll. Cardiol. 55, 671–679.
Akhtar, M., Gilbert, C. J., and Shenasa, M. (1981). Effect of lidocaine on atrioventricular response via the accessory pathway in patients with Wolff-Parkinson-White syndrome. Circulation 63, 435–441.
Alvarez, B., and Radi, R. (2003). Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25, 295–311.
Antoniades, C., Demosthenous, M., Reilly, S., Margaritis, M., Zhang, M. H., Antonopoulos, A., Marinou, K., Nahar, K., Jayaram, R., Tousoulis, D., Bakogiannis, C., Sayeed, R., Triantafyllou, C., Koumallos, N., Psarros, C., Miliou, A., Stefanadis, C., Channon, K. M., and Casadei, B. (2012). Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. J. Am. Coll. Cardiol. 59, 60–70.
Anzai, K., Ogawa, K., Kuniyasu, A., Ozawa, T., Yamamoto, H., and Nakayama, H. (1998). Effects of hydroxyl radical and sulfhydryl reagents on the open probability of the purified cardiac ryanodine receptor channel incorporated into planar lipid bilayers. Biochem. Biophys. Res. Commun. 249, 938–942.
Barford, D. (2004). The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 14, 679–686.
Bellocci, F., Biasucci, L. M., Gensini, G. F., Padeletti, L., Raviele, A., Santini, M., Giubilato, G., Landolina, M., Biondi-Zoccai, G., Raciti, G., Sassara, M., Castro, A., Kheir, A., and Crea, F. (2007). Prognostic role of post-infarction C-reactive protein in patients undergoing implantation of cardioverter-defibrillators: design of the C-reactive protein Assessment after Myocardial Infarction to GUide Implantation of DEfibrillator (CAMI GUIDE) study. J. Cardiovasc. Med. (Hagerstown) 8, 293–299.
Beresewicz, A., and Horackova, M. (1991). Alterations in electrical and contractile behavior of isolated cardiomyocytes by hydrogen peroxide: possible ionic mechanisms. J. Mol. Cell Cardiol. 23, 899–918.
Billman, G. E., Harris, W. S., Carnes, C. A., Adamson, P. B., Vanoli, E., and Schwartz, P. J. (2011). Dietary omega-3 fatty acids and susceptibility to ventricular fibrillation: lack of protection and a proarrhythmic effect. Circ. Arrhythm. Electrophysiol. 300, H2288–H2299.
Blaustein, M. P., and Lederer, W. J. (1999). Sodium/calcium exchange: its physiological implications. Physiol. Rev. 79, 763–854.
Boldt, A., Wetzel, U., Weigl, J., Garbade, J., Lauschke, J., Hindricks, G., Kottkamp, H., Gummert, J. F., and Dhein, S. (2003). Expression of angiotensin II receptors in human left and right atrial tissue in atrial fibrillation with and without underlying mitral valve disease. J. Am. Coll. Cardiol. 42, 1785–1792.
Bosch, R. F., Zeng, X., Grammer, J. B., Popovic, K., Mewis, C., and Kuhlkamp, V. (1999). Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 44, 121–131.
Bouchie, J. L., Hansen, H., and Feener, E. P. (1998). Natriuretic factors and nitric oxide suppress plasminogen activator inhibitor-1 expression in vascular smooth muscle cells. Role of cGMP in the regulation of the plasminogen system. Arterioscler. Thromb. Vasc. Biol. 18, 1771–1779.
Brawn, K., and Fridovich, I. (1980). Superoxide radical and superoxide dismutases: threat and defense. Acta Physiol. Scand. Suppl. 492, 9–18.
Brown, G. C., and Cooper, C. E. (1994). Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 356, 295–298.
Brumell, J. H., Burkhardt, A. L., Bolen, J. B., and Grinstein, S. (1996). Endogenous reactive oxygen intermediates activate tyrosine kinases in human neutrophils. J. Biol. Chem. 271, 1455–1461.
Cai, H., Li, Z., Goette, A., Mera, F., Honeycutt, C., Feterik, K., Wilcox, J. N., Dudley, S. C. Jr., Harrison, D. G., and Langberg, J. J. (2002). Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation 106, 2854–2858.
Cairns, J. A., Connolly, S. J., Roberts, R., and Gent, M. (1997). Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT. Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Lancet 349, 675–682.
Canton, M., Menazza, S., Sheeran, F. L., Polverino de Laureto, P., Di Lisa, F., and Pepe, S. (2011). Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 57, 300–309.
Carnes, C. A., Chung, M. K., Nakayama, T., Nakayama, H., Baliga, R. S., Piao, S., Kanderian, A., Pavia, S., Hamlin, R. L., McCarthy, P. M., Bauer, J. A., and Van Wagoner, D. R. (2001). Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 89, E32–E38.
Carnes, C. A., Janssen, P. M., Ruehr, M. L., Nakayama, H., Nakayama, T., Haase, H., Bauer, J. A., Chung, M. K., Fearon, I. M., Gillinov, A. M., Hamlin, R. L., and Van Wagoner, D. R. (2007). Atrial glutathione content, calcium current, and contractility. J. Biol. Chem. 282, 28063–28073.
CAST Investigators. (1989). Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N. Engl. J. Med. 321, 406–412.
Cederbaum, A. I. (2001). Introduction-serial review: alcohol, oxidative stress and cell injury. Free Radic. Biol. Med. 31, 1524–1526.
Celermajer, D. S., Sorensen, K. E., Spiegelhalter, D. J., Georgakopoulos, D., Robinson, J., and Deanfield, J. E. (1994). Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J. Am. Coll. Cardiol. 24, 471–476.
Chen, K., Thomas, S. R., and Keaney, J. F. Jr. (2003). Beyond LDL oxidation: ROS in vascular signal transduction. Free Radic. Biol. Med. 35, 117–132.
Chesnais, J. M., Fischmeister, R., and Mery, P. F. (1999). Peroxynitrite is a positive inotropic agent in atrial and ventricular fibres of the frog heart. J. Physiol. 521 (Pt 2), 375–388.
Civelek, S., Seymen, O., Seven, A., Yigit, G., Hatemi, H., and Burcak, G. (2001). Oxidative stress in heart tissue of hyperthyroid and iron supplemented rats. J. Toxicol. Environ. Health A 64, 499–506.
Connor, K. M., Subbaram, S., Regan, K. J., Nelson, K. K., Mazurkiewicz, J. E., Bartholomew, P. J., Aplin, A. E., Tai, Y. T., Aguirre-Ghiso, J., Flores, S. C., and Melendez, J. A. (2005). Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280, 16916–16924.
Coplen, S. E., Antman, E. M., Berlin, J. A., Hewitt, P., and Chalmers, T. C. (1990). Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion. A meta-analysis of randomized control trials. Circulation 82, 1106–1116.
Cu, A., Ye, Q., Sarria, R., Nakamura, S., Guzman, J., and Costabel, U. (2009). N-acetylcysteine inhibits TNF-α, sTNFR, and TGF-β 1 release by alveolar macrophages in idiopathic pulmonary fibrosis in vitro. Sarcoidosis Vasc. Diffuse Lung Dis. 26, 147–154.
De Champlain, J., Wu, R., Girouard, H., Karas, M., EL Midaoui, A., Laplante, M. A., and Wu, L. (2004). Oxidative stress in hypertension. Clin. Exp. Hypertens. 26, 593–601.
De Vecchi, E., Pala, M. G., Di Credico, G., Agape, V., Paolini, G., Bonini, P. A., Grossi, A., and Paroni, R. (1998). Relation between left ventricular function and oxidative stress in patients undergoing bypass surgery. Heart 79, 242–247.
Di Wang, H., Hope, S., Du, Y., Quinn, M. T., Cayatte, A., Pagano, P. J., and Cohen, R. A. (1999). Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension 33, 1225–1232.
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301.
Disertori, M., Latini, R., Barlera, S., Franzosi, M. G., Staszewsky, L., Maggioni, A. P., Lucci, D., Di, P. G., and Tognoni, G. (2009). Valsartan for prevention of recurrent atrial fibrillation. N. Engl. J. Med. 360, 1606–1617.
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496.
Dremina, E. S., Sharov, V. S., Davies, M. J., and Schoneich, C. (2007). Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem. Res. Toxicol. 20, 1462–1469.
Dudley, S. C. Jr., Hoch, N. E., McCann, L. A., Honeycutt, C., Diamandopoulos, L., Fukai, T., Harrison, D. G., Dikalov, S. I., and Langberg, J. (2005). Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112, 1266–1273.
Duflo, F., Debon, R., Goudable, J., Chassard, D., and Allaouchiche, B. (2002). Alveolar and serum oxidative stress in ventilator-associated pneumonia. Br. J. Anaesth. 89, 231–236.
Eaton, P. (2006). Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic. Biol. Med. 40, 1889–1899.
Erickson, J. R., He, B. J., Grumbach, I. M., and Anderson, M. E. (2011). CaMKII in the cardiovascular system: sensing redox states. Physiol. Rev. 91, 889–915.
Fauconnier, J., Thireau, J., Reiken, S., Cassan, C., Richard, S., Matecki, S., Marks, A. R., and Lacampagne, A. (2010). Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 107, 1559–1564.
Freeman, B. A., White, C. R., Gutierrez, H., Paler-Martinez, A., Tarpey, M. M., and Rubbo, H. (1995). Oxygen radical-nitric oxide reactions in vascular diseases. Adv. Pharmacol. 34, 45–69.
Gautier, M., Zhang, H., and Fearon, I. M. (2008). Peroxynitrite formation mediates LPC-induced augmentation of cardiac late sodium currents. J. Mol. Cell Cardiol. 44, 241–251.
Gjesdal, K. (2009). Non-investigational antiarrhythmic drugs: long-term use and limitations. Expert Opin. Drug Saf. 8, 345–355.
Goette, A., Schon, N., Kirchhof, P., Breithardt, G., Fetsch, T., Hausler, K. G., Klein, H. U., Steinbeck, G., Wegscheider, K., and Meinertz, T. (2012). Angiotensin II-antagonist in paroxysmal atrial fibrillation (ANTIPAF) trial. Circ. Arrhythm. Electrophysiol. 5, 43–51.
Halliwell, B., Gutteridge, J. M., and Aruoma, O. I. (1987). The deoxyribose method: a simple “test-tube” assay for determination of rate constants for reactions of hydroxyl radicals. Anal. Biochem. 165, 215–219.
Hartnett, M. E., Stratton, R. D., Browne, R. W., Rosner, B. A., Lanham, R. J., and armstrong, D. (2000). Serum markers of oxidative stress and severity of diabetic retinopathy. Diabetes Care 23, 234–240.
Hattori, Y., Atsushi, S., Hiroaki, F., and Toyama, J. (1997). Effects of cilazapril on ventricular arrhythmia in patients with congestive heart failure. Clin. Ther. 19, 481–486.
Hazen, S. L., d'Avignon, A., Anderson, M. M., Hsu, F. F., and Heinecke, J. W. (1998a). Human neutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system to oxidize α-amino acids to a family of reactive aldehydes. Mechanistic studies identifying labile intermediates along the reaction pathway. J. Biol. Chem. 273, 4997–5005.
Hazen, S. L., Hsu, F. F., d'Avignon, A., and Heinecke, J. W. (1998b). Human neutrophils employ myeloperoxidase to convert α-amino acids to a battery of reactive aldehydes: a pathway for aldehyde generation at sites of inflammation. Biochemistry 37, 6864–6873.
Ide, T., Tsutsui, H., Kinugawa, S., Suematsu, N., Hayashidani, S., Ichikawa, K., Utsumi, H., Machida, Y., Egashira, K., and Takeshita, A. (2000). Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ. Res. 86, 152–157.
Iravanian, S., and Dudley, S. C. Jr. (2008). The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm. 5, S12–S17.
Iravanian, S., Sovari, A. A., Lardin, H. A., Liu, H., Xiao, H. D., Dolmatova, E., Jiao, Z., Harris, B. S., Witham, E. A., Gourdie, R. G., Duffy, H. S., Bernstein, K. E., and Dudley, S. C. Jr. (2011). Inhibition of renin-angiotensin system (RAS) reduces ventricular tachycardia risk by altering connexin43. J. Mol. Med. (Berl.) 89, 677–687.
Irigoyen, M. C., Paulini, J., Flores, L. J., Flues, K., Bertagnolli, M., Moreira, E. D., Consolim-Colombo, F., Bello-Klein, A., and De, A. K. (2005). Exercise training improves baroreflex sensitivity associated with oxidative stress reduction in ovariectomized rats. Hypertension 46, 998–1003.
Issac, T. T., Dokainish, H., and Lakkis, N. M. (2007). Role of inflammation in initiation and perpetuation of atrial fibrillation: a systematic review of the published data. J. Am. Coll. Cardiol. 50, 2021–2028.
Iwasaki, Y. K., Nishida, K., Kato, T., and Nattel, S. (2011). Atrial fibrillation pathophysiology: implications for management. Circulation 124, 2264–2274.
Jais, P., Haissaguerre, M., Shah, D. C., Chouairi, S., Gencel, L., Hocini, M., and Clementy, J. (1997). A focal source of atrial fibrillation treated by discrete radiofrequency ablation. Circulation 95, 572–576.
Jeong, E. M., Liu, M., Sturdy, M., Gao, G., Varghese, S. T., Sovari, A. A., and Dudley, S. C. (2012). Metabolic stress, reactive oxygen species, and arrhythmia. J. Mol. Cell Cardiol. 52, 454–463.
Jones, E. F., Calafiore, P., McNeil, J. J., Tonkin, A. M., and Donnan, G. A. (1996). Atrial fibrillation with left atrial spontaneous contrast detected by transesophageal echocardiography is a potent risk factor for stroke. Am. J. Cardiol. 78, 425–429.
Jorde, U. P., Ennezat, P. V., Lisker, J., Suryadevara, V., Infeld, J., Cukon, S., Hammer, A., Sonnenblick, E. H., and Le Jemtel, T. H. (2000). Maximally recommended doses of angiotensin-converting enzyme (ACE) inhibitors do not completely prevent ACE-mediated formation of angiotensin II in chronic heart failure. Circulation 101, 844–846.
Julian, D. G., Camm, A. J., Frangin, G., Janse, M. J., Munoz, A., Schwartz, P. J., and Simon, P. (1997). Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. European Myocardial Infarct Amiodarone Trial Investigators. Lancet 349, 667–674.
Kabe, Y., Ando, K., Hirao, S., Yoshida, M., and Handa, H. (2005). Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal. 7, 395–403.
Kawakami, M., and Okabe, E. (1998). Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol. Pharmacol. 53, 497–503.
Khatib, R., Joseph, P., Briel, M., Yusuf, S., and Healey, J. (2012). Blockade of the renin-angiotensin-aldosterone system (RAAS) for primary prevention of non-valvular atrial fibrillation: a systematic review and meta analysis of randomized controlled trials. Int. J. Cardiol. doi: 10.1016/j.ijcard.2012.02.009. [Epub ahead of print].
Kieken, F., Mutsaers, N., dolmatova, E., Virgil, K., Wit, A. L., Kellezi, A., Hirst-Jensen, B. J., Duffy, H. S., and Sorgen, P. L. (2009). Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ. Res. 104, 1103–1112.
Kim, Y. M., Kattach, H., Ratnatunga, C., Pillai, R., Channon, K. M., and Casadei, B. (2008). Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 51, 68–74.
Kim, Y. H., Lim, D. S., Lee, J. H., Shim, W. J., Ro, Y. M., Park, G. H., Becker, K. G., Cho-Chung, Y. S., and Kim, M. K. (2003). Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp. Mol. Med. 35, 336–349.
Kimura, Y., Sato, M., Kurotani, K., Nanri, A., Kawai, K., Kasai, H., Imaizumi, K., and Mizoue, T. (2012). PUFAs in serum cholesterol ester and oxidative DNA damage in Japanese men and women. Am. J. Clin. Nutr. 95, 1209–1214.
Kober, L., Torp-Pedersen, C., Carlsen, J. E., Bagger, H., Eliasen, P., Lyngborg, K., Videbaek, J., Cole, D. S., Auclert, L., and Pauly, N. C. (1995). A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. N. Engl. J. Med. 333, 1670–1676.
Kofler, S., Petrakopoulou, P., Grimm, C., Kaczmarek, I., Meiser, B. M., and Weis, M. (2008). Graft-infiltrating dendritic cells and coronary endothelial dysfunction after human heart transplantation. J. Heart Lung Transplant. 27, 387–393.
Koli, K., Myllarniemi, M., Keski-Oja, J., and Kinnula, V. L. (2008). Transforming growth factor-β activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid. Redox Signal. 10, 333–342.
Kowey, P. R., Reiffel, J. A., Ellenbogen, K. A., Naccarelli, G. V., and Pratt, C. M. (2010). Efficacy and safety of prescription omega-3 fatty acids for the prevention of recurrent symptomatic atrial fibrillation: a randomized controlled trial. JAMA 304, 2363–2372.
Krahn, A. D., Manfreda, J., Tate, R. B., Mathewson, F. A., and Cuddy, T. E. (1995). The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba Follow-Up Study. Am. J. Med. 98, 476–484.
Kregel, K. C., and Zhang, H. J. (2007). An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R18–R36.
Kukushkina, O. I., Fedotkina, L. K., Balashov, V. P., Balykova, L. A., and Sosunov, A. A. (1999). Effect of NO-synthetase inhibitor L-NAME on occlusive and reperfusion arrhythmias in cats. Biull. Eksp. Biol. Med. 127, 509–511.
Kulagina, N. V., and Michael, A. C. (2003). Monitoring hydrogen peroxide in the extracellular space of the brain with amperometric microsensors. Anal. Chem. 75, 4875–4881.
Kuroda, J., Ago, T., Matsushima, S., Zhai, P., Schneider, M. D., and Sadoshima, J. (2010). NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 107, 15565–15570.
Lambrinoudaki, I. V., Augoulea, A., Christodoulakos, G. E., Economou, E. V., Kaparos, G., Kontoravdis, A., Papadias, C., and Creatsas, G. (2009). Measurable serum markers of oxidative stress response in women with endometriosis. Fertil. Steril. 91, 46–50.
Levine, R. L. (2002). Carbonyl modified proteins in cellular regulation, aging, and disease. Free Radic. Biol. Med. 32, 790–796.
Li, N., Frigerio, F., and Maechler, P. (2008). The sensitivity of pancreatic β-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem. Soc. Trans. 36, 930–934.
Liberman, M., Bassi, E., Martinatti, M. K., Lario, F. C., Wosniak, J. Jr., Pomerantzeff, P. M., and Laurindo, F. R. (2008). Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler. Thromb. Vasc. Biol. 28, 463–470.
Liu, M., Liu, H., and Dudley, S. C. Jr. (2010). Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 107, 967–974.
Liu, R. M., Liu, Y., Forman, H. J., Olman, M., and Tarpey, M. M. (2004). Glutathione regulates transforming growth factor-β-stimulated collagen production in fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 286, L121–L128.
Liu, T., Korantzopoulos, P., Shehata, M., Li, G., Wang, X., and Kaul, S. (2011). Prevention of atrial fibrillation with omega-3 fatty acids: a meta-analysis of randomised clinical trials. Heart 97, 1034–1040.
Lu, L., Chen, S. S., Zhang, J. Q., Ramires, F. J., and Sun, Y. (2004). Activation of nuclear factor-kappaB and its proinflammatory mediator cascade in the infarcted rat heart. Biochem. Biophys. Res. Commun. 321, 879–885.
Ma, J. H., Luo, A. T., and Zhang, P. H. (2005). Effect of hydrogen peroxide on persistent sodium current in guinea pig ventricular myocytes. Acta Pharmacol. Sin. 26, 828–834.
Maan, A., Shaikh, A. Y., Mansour, M., Ruskin, J. N., and Heist, E. K. (2011). Complications from catheter ablation of atrial fibrillation: a systematic review. Crit. Path. Cardiol. 10, 76–83.
Madamanchi, N. R., Vendrov, A., and Runge, M. S. (2005). Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 29–38.
Mallet, R. T. (2005). Hypoxic modulation of cardiac L-type Ca2+ current: interaction of reactive oxygen species and β-adrenergic signaling. Cardiovasc. Res. 67, 578–580.
Mihm, M. J., Yu, F., Carnes, C. A., Reiser, P. J., McCarthy, P. M., Van Wagoner, D. R., and Bauer, J. A. (2001). Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104, 174–180.
Milei, J., Ferreira, R., Grana, D. R., and Boveris, A. (2001). Oxidative stress and mitochondrial damage in coronary artery bypass graft surgery: effects of antioxidant treatments. Compr. Ther. 27, 108–116.
Miller, J. D., Chu, Y., Brooks, R. M., Richenbacher, W. E., Pena-Silva, R., and Heistad, D. D. (2008). Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J. Am. Coll. Cardiol. 52, 843–850.
Miyasaka, Y., Barnes, M. E., Gersh, B. J., Cha, S. S., Bailey, K. R., Abhayaratna, W. P., Seward, J. B., and Tsang, T. S. (2006). Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 114, 119–125.
Molyneux, C. A., Glyn, M. C., and Ward, B. J. (2002). Oxidative stress and cardiac microvascular structure in ischemia and reperfusion: the protective effect of antioxidant vitamins. Microvasc. Res. 64, 265–277.
Morel, F., Doussiere, J., and Vignais, P. V. (1991). The superoxide-generating oxidase of phagocytic cells. Physiological, molecular and pathological aspects. Eur. J. Biochem. 201, 523–546.
Morganroth, J., and Goin, J. E. (1991). Quinidine-related mortality in the short-to-medium-term treatment of ventricular arrhythmias. A meta-analysis. Circulation 84, 1977–1983.
Morita, N., Lee, J. H., Xie, Y., Sovari, A., Qu, Z., Weiss, J. N., and Karagueuzian, H. S. (2011). Suppression of re-entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J. Am. Coll. Cardiol. 57, 366–375.
Morita, N., Sovari, A. A., Xie, Y., Fishbein, M. C., Mandel, W. J., Garfinkel, A., Lin, S. F., Chen, P. S., Xie, L. H., Chen, F., Qu, Z., Weiss, J. N., and Karagueuzian, H. S. (2009). Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 297, H1594–H1605.
Morris, T. E., and Sulakhe, P. V. (1997). Sarcoplasmic reticulum Ca2+-pump dysfunction in rat cardiomyocytes briefly exposed to hydroxyl radicals. Free Radic. Biol. Med. 22, 37–47.
Murrell, G. A., Francis, M. J., and Bromley, L. (1990). Modulation of fibroblast proliferation by oxygen free radicals. Biochem. J. 265, 659–665.
Negi, S., Sovari, A. A., and Dudley, S. C. Jr. (2010). Atrial fibrillation: the emerging role of inflammation and oxidative stress. Cardiovasc. Hematol. Disord. Drug Targets. 10, 262–268.
Neuman, R. B., Bloom, H. L., Shukrullah, I., Darrow, L. A., Kleinbaum, D., Jones, D. P., and Dudley, S. C. Jr. (2007). Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 53, 1652–1657.
Nishijima, Y., Sridhar, A., Bonilla, I., Velayutham, M., Khan, M., Terentyeva, R., Li, C., Kuppusamy, P., Elton, T. S., Terentyev, D., Gyorke, S., Zweier, J. L., Cardounel, A. J., and Carnes, C. A. (2011). Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc. Res. 91, 71–79.
O'Rourke, B., Cortassa, S., and Aon, M. A. (2005). Mitochondrial ion channels: gatekeepers of life and death. Physiology (Bethesda.) 20, 303–315.
Ohki, R., Yamamoto, K., Ueno, S., Mano, H., Misawa, Y., Fuse, K., Ikeda, U., and Shimada, K. (2005). Gene expression profiling of human atrial myocardium with atrial fibrillation by DNA microarray analysis. Int. J. Cardiol. 102, 233–238.
Ovechkin, A. V., Lominadze, D., Sedoris, K. C., Robinson, T. W., Tyagi, S. C., and Roberts, A. M. (2007). Lung ischemia-reperfusion injury: implications of oxidative stress and platelet-arteriolar wall interactions. Arch. Physiol. Biochem. 113, 1–12.
Pu, M., Akhand, A. A., Kato, M., Hamaguchi, M., Koike, T., Iwata, H., Sabe, H., Suzuki, H., and Nakashima, I. (1996). Evidence of a novel redox-linked activation mechanism for the Src kinase which is independent of tyrosine 527-mediated regulation. Oncogene 13, 2615–2622.
Radomski, M. W., Palmer, R. M., and Moncada, S. (1987). Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br. J. Pharmacol. 92, 181–187.
Reilly, S. N., Jayaram, R., Nahar, K., Antoniades, C., Verheule, S., Channon, K. M., Alp, N. J., Schotten, U., and Casadei, B. (2011a). Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation 124, 1107–1117.
Reilly, S. N., Jayaram, R., Nahar, K., Antoniades, C., Verheule, S., Channon, K. M., Alp, N. J., Schotten, U., and Casadei, B. (2011b). Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation 124, 1107–1117.
Riis, B., and Poulsen, H. E. (2005). “DNA repair: mechanisms and measurments,” in Oxidant and Antioxidants Defense Systems, ed T. Grune (Berlin: Springer), 152–169.
Sam, F., Kerstetter, D. L., Pimental, D. R., Mulukutla, S., Tabaee, A., Bristow, M. R., Colucci, W. S., and Sawyer, D. B. (2005). Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card Fail. 11, 473–480.
Santana, L. F., Nunez-Duran, H., Dilly, K. W., and Lederer, W. J. (2005). Sodium current and arrhythmogenesis in heart failure. Heart Fail. Clin. 1, 193–205.
Schotten, U., Verheule, S., Kerfant, B. G., and Greiser, M. (2010). Enhanced late Na+ currents in atrial fibrillation new drug target or just an epiphenomenon? J. Am. Coll. Cardiol. 55, 2343–2345.
Schotten, U., Verheule, S., Kirchhof, P., and Goette, A. (2011). Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol. Rev. 91, 265–325.
Schramm, A., Matusik, P., Osmenda, G., and Guzik, T. J. (2012). Targeting NADPH oxidases in vascular pharmacology. Vascul. Pharmacol. 56, 216–231.
Seddon, M., Looi, Y. H., and Shah, A. M. (2007). Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93, 903–907.
Sesso, H. D., Buring, J. E., Christen, W. G., Kurth, T., Belanger, C., MacFadyen, J., Bubes, V., Manson, J. E., Glynn, R. J., and Gaziano, J. M. (2008). Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians' Health Study II randomized controlled trial. JAMA 300, 2123–2133.
Shan, J., Kushnir, A., Betzenhauser, M. J., Reiken, S., Li, J., Lehnart, S. E., Lindegger, N., Mongillo, M., Mohler, P. J., and Marks, A. R. (2010). Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Invest. 120, 4388–4398.
Shang, L. L., Sanyal, S., Pfahnl, A. E., Jiao, Z., Allen, J., Liu, H., and Dudley, S. C. Jr. (2008). NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am. J. Physiol. Cell Physiol. 294, C372–C379.
Shimano, M., Shibata, R., Inden, Y., Yoshida, N., Uchikawa, T., Tsuji, Y., and Murohara, T. (2009). Reactive oxidative metabolites are associated with atrial conduction disturbance in patients with atrial fibrillation. Heart Rhythm 6, 935–940.
Shirotani, K., Katsura, M., Higo, A., Takesue, M., Mohri, Y., Shuto, K., Tarumi, C., and Ohkuma, S. (2001). Suppression of Ca2+ influx through L-type voltage-dependent calcium channels by hydroxyl radical in mouse cerebral cortical neurons. Brain Res. Mol. Brain Res. 92, 12–18.
Siebels, J., Cappato, R., Ruppel, R., Schneider, M. A., and Kuck, K. H. (1993). Preliminary results of the Cardiac Arrest Study Hamburg (CASH). CASH Investig. Am. J. Cardiol. 72, 109F–113F.
Singh, S. N., Fletcher, R. D., Fisher, S. G., Singh, B. N., Lewis, H. D., Deedwania, P. C., Massie, B. M., Colling, C., and Lazzeri, D. (1995). Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N. Engl. J. Med. 333, 77–82.
Soliman, D., Hamming, K. S., Matemisz, L. C., and Light, P. E. (2009). Reactive oxygen species directly modify sodium-calcium exchanger activity in a splice variant-dependent manner. J. Mol. Cell Cardiol. 47, 595–602.
Sommer, D., Coleman, S., Swanson, S. A., and Stemmer, P. M. (2002). Differential susceptibilities of serine/threonine phosphatases to oxidative and nitrosative stress. Arch. Biochem. Biophys. 404, 271–278.
Song, Y., Cook, N. R., Albert, C. M., Van, D. M., and Manson, J. E. (2009). Effects of vitamins C and E and β-carotene on the risk of type 2 diabetes in women at high risk of cardiovascular disease: a randomized controlled trial. Am. J. Clin. Nutr. 90, 429–437.
Song, Y., Shryock, J. C., Wagner, S., Maier, L. S., and Belardinelli, L. (2006). Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 318, 214–222.
Sorescu, D., Weiss, D., Lassegue, B., Clempus, R. E., Szocs, K., Sorescu, G. P., Valppu, L., Quinn, M. T., Lambeth, J. D., Vega, J. D., Taylor, W. R., and Griendling, K. K. (2002). Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 105, 1429–1435.
Sossalla, S., Kallmeyer, B., Wagner, S., Mazur, M., Maurer, U., Toischer, K., Schmitto, J. D., Seipelt, R., Schondube, F. A., Hasenfuss, G., Belardinelli, L., and Maier, L. S. (2010). Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 55, 2330–2342.
Sovari, A. A. (2011). Antioxidant therapy for atrial fibrillation: what is the next step? Cardiol. Res. Pract. 2011, 429537.
Sovari, A. A., and Dudley, S. C. (2010). “Atrial fibrillation and oxidative stress,” in Studies in Cardiovascular Disorders, eds H. Sauer, M. S. Ajay, and F. R. Laurindo (New York, NY: Humana Press), 373–389.
Sovari, A. A., Bonini, M. G., and Dudley, S. C. (2011a). Effective antioxidant therapy for the management of arrhythmia. Expert. Rev. Cardiovasc. Ther. 9, 797–800.
Sovari, A. A., Iravanian, S., Dolmatova, E., Jiao, Z., Liu, H., Zandieh, S., kumar, V., Wang, K., Bernstein, K. E., Bonini, M. G., Duffy, H. S., and Dudley, S. C. (2011b). Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J. Am. Coll. Cardiol. 58, 2332–2339.
Sovari, A. A., Iravanian, S., Mitchell, D., Jeong, E. M., Gu, L., Bonini, M. G., and Dudley, S. C. (2011c). Mitochondria-targeted antioxidant, mito-TEMPO, prevents angiotensin II mediated connexin 43 remodeling and sudden cardiac death. J. Investig. Med. 59, 692–730.
Sovari, A. A., Jeong, E. M., Dolmatova, E., Tan, A. Y., Zandieh, S., Liu, M., Gu, L., and Arasu, D. (2011d). c-Src tyrosine kinase mediates the effect of mitochondrial oxidative stress on gap junctional remodeling and ventricular tachycardia. Circulation 124, A14385.
Sovari, A. A., Jiao, Z., Dolmatova, E., kumar, V., Wang, K., Bonini, M. G., Duffy, H., and Dudley, S. C. (2010). c-Src tyrosine kinase is a potential therapeutic target to prevent angiotensin-II mediated connexin43 remodeling and ventricular arrhythmia. Circulation 122, A17524.
Sovari, A. A., Morita, N., and Karagueuzian, H. S. (2008). Apocynin: a potent NADPH oxidase inhibitor for the management of atrial fibrillation. Redox. Rep. 13, 242–245.
Sovari, A. A., Shroff, A., and Kocheril, A. G. (2012). Postinfarct cardiac remodeling and the substrate for sudden cardiac death: role of oxidative stress and myocardial fibrosis. Expert. Rev. Cardiovasc. Ther. 10, 267–270.
Sridhar, A., Nishijima, Y., Terentyev, D., Khan, M., Terentyeva, R., Hamlin, R. L., Nakayama, T., Gyorke, S., Cardounel, A. J., and Carnes, C. A. (2009). Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc. Res. 84, 227–236.
Stadtman, E. R., Moskovitz, J., and Levine, R. L. (2003). Oxidation of methionine residues of proteins: biological consequences. Antioxid. Redox Signal. 5, 577–582.
Stamler, J. S., and Hausladen, A. (1998). Oxidative modifications in nitrosative stress. Nat. Struct. Biol. 5, 247–249.
Suzuki, K., Ito, Y., Ochiai, J., Kusuhara, Y., Hashimoto, S., Tokudome, S., Kojima, M., Wakai, K., Toyoshima, H., Tamakoshi, K., Watanabe, Y., Hayakawa, N., Maruta, M., Watanabe, M., Kato, K., Ohta, Y., and Tamakoshi, A. (2003). Relationship between obesity and serum markers of oxidative stress and inflammation in Japanese. Asian Pac. J. Cancer Prev. 4, 259–266.
Suzuki, Y. J., Forman, H. J., and Sevanian, A. (1997). Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 22, 269–285.
Tarpey, M. M., and Fridovich, I. (2001). Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circ. Res. 89, 224–236.
Tarpey, M. M., Wink, D. A., and Grisham, M. B. (2004). Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R431–R444.
Thomas, G. P., Sims, S. M., Cook, M. A., and Karmazyn, M. (1998). Hydrogen peroxide-induced stimulation of L-type calcium current in guinea pig ventricular myocytes and its inhibition by adenosine A1 receptor activation. J. Pharmacol. Exp. Ther. 286, 1208–1214.
Tong, X., Evangelista, A., and Cohen, R. A. (2010). Targeting the redox regulation of SERCA in vascular physiology and disease. Curr. Opin. Pharmacol. 10, 133–138.
Toyofuku, T., Yabuki, M., Otsu, K., Kuzuya, T., Tada, M., and Hori, M. (1999). Functional role of c-Src in gap junctions of the cardiomyopathic heart. Circ. Res. 85, 672–681.
Tu, H., Liu, J., Zhu, Z., Zhang, L., Pipinos, I. I., and Li, Y. L. (2012). Mitochondria-derived superoxide and voltage-gated sodium channels in baroreceptor neurons from chronic heart-failure rats. J. Neurophysiol. 107, 591–602.
Uchida, K. (2000). Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 28, 1685–1696.
van de Wal, R. M., Plokker, H. W., Lok, D. J., Boomsma, F., van der Horst, F. A., van Veldhuisen, D. J., van Gilst, W. H., and Voors, A. A. (2006). Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int. J. Cardiol. 106, 367–372.
Van Wagoner, D. R. (2007). Recent insights into the pathophysiology of atrial fibrillation. Semin. Thorac. Cardiovasc. Surg. 19, 9–15.
Van Wagoner, D. R. (2008). Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J. Cardiovasc. Pharmacol. 52, 306–313.
Vassalle, C., Petrozzi, L., Botto, N., Andreassi, M. G., and Zucchelli, G. C. (2004). Oxidative stress and its association with coronary artery disease and different atherogenic risk factors. J. Intern. Med. 256, 308–315.
Waldo, A. L., Camm, A. J., de Ruyter, H., Friedman, P. L., MacNeil, D. J., Pauls, J. F., Pitt, B., Pratt, C. M., Schwartz, P. J., and Veltri, E. P. (1996). Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet 348, 7–12.
Weissbach, H., Etienne, F., Hoshi, T., Heinemann, S. H., Lowther, W. T., Matthews, B., St, J. G., Nathan, C., and Brot, N. (2002). Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch. Biochem. Biophys. 397, 172–178.
Wong, C. M., Marcocci, L., Liu, L., and Suzuki, Y. J. (2010). Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 12, 393–404.
Wosniak, J. Jr., Santos, C. X., Kowaltowski, A. J., and Laurindo, F. R. (2009). Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox Signal. 11, 1265–1278.
Yamauchi, M., Nakano, H., Maekawa, J., Okamoto, Y., Ohnishi, Y., Suzuki, T., and Kimura, H. (2005). Oxidative stress in obstructive sleep apnea. Chest 127, 1674–1679.
Yoshizumi, M., Abe, J., Haendeler, J., Huang, Q., and Berk, B. C. (2000). Src and Cas mediate JNK activation but not ERK1/2 and p38 kinases by reactive oxygen species. J. Biol. Chem. 275, 11706–11712.
Ziegelstein, R. C., Zweier, J. L., Mellits, E. D., Younes, A., Lakatta, E. G., Stern, M. D., and Silverman, H. S. (1992). Dimethylthiourea, an oxygen radical scavenger, protects isolated cardiac myocytes from hypoxic injury by inhibition of Na+-Ca2+ exchange and not by its antioxidant effects. Circ. Res. 70, 804–811.
Keywords: antioxidants, atrial fibrillation, mitochondria, NADPH oxidase, nitric oxide synthase, therapy
Citation: Sovari AA and Dudley SC Jr (2012) Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physio. 3:311. doi: 10.3389/fphys.2012.00311
Received: 16 February 2012; Accepted: 15 July 2012;
Published online: 06 August 2012.
Edited by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USAReviewed by:
Sandeep Pandit, University of Michigan, Ann Arbor, USACopyright © 2012 Sovari and Dudley. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Samuel C. Dudley Jr., Section of Cardiology, University of Illinois at Chicago, 840 S. Wood Street, MC 715, Chicago, IL 60612, USA. e-mail:c2NkdWRsZXlAdWljLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.