
 Jari M. Tuomi1†
Jari M. Tuomi1†

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 29 June 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00239
This article is part of the Research Topic Autonomic regulation of the heart - emerging regulators and arrhythmogenic mechanisms View all 8 articles
The heart receives sympathetic and parasympathetic efferent innervation as well as the ability to process information internally via an intrinsic cardiac autonomic nervous system (ICANS). For over a century, the role of the parasympathetics via vagal acetylcholine release was related to controlling primarily heart rate. Although in the late 1800s shown to play a role in atrial arrhythmia, the myocardium took precedence from the mid-1950s until in the last decade a resurgence of interest in the autonomics along with signaling cascades, regulators, and ion channels. Originally ignored as being benign and thus untreated, recent emphasis has focused on atrial arrhythmia as atrial fibrillation (AF) is the most common arrhythmia seen by the general practitioner. It is now recognized to have significant mortality and morbidity due to resultant stroke and heart failure. With the aging population, there will be an unprecedented increased burden on health care resources. Although it has been known for more than half a century that cholinergic stimulation can initiate AF, the classical concept focused on the M2 receptor and its signaling cascade including RGS4, as these had been shown to have predominant effects on nodal function (heart rate and conduction block) as well as contractility. However, recent evidence suggests that the M3 receptor may also playa role in initiation and perpetuation of AF and thus RGS2, a putative regulator of the M3 receptor, may be a target for therapeutic intervention. Mice lacking RGS2 (RGS2−/−), were found to have significantly altered electrophysiological atrial responses and were more susceptible to electrically induced AF. Vagally induced or programmed stimulation-induced AF could be blocked by the selective M3R antagonist, darifenacin. These results suggest a potential surgical target (ICANS) and pharmacological targets (M3R, RGS2) for the management of AF.
This “Special Topic” series explores how regulators of G protein signaling (RGS) proteins share common, yet distinct roles at multiple levels, controlling autonomic regulation of heart function, and what critical questions remain for translational researchers examining the role of these pathways in conditions such as atrial fibrillation (AF), where RGS proteins are currently being assessed as potential druggable targets. Although cholinergic AF has been investigated for decades, exact mechanisms are still controversial (Schotten et al., 2011). AF is highly complex involving multiple mechanisms for its initiation and sustaining the arrhythmia including genetic contributions, structural, ion handling, and autonomic alterations, the complexity of which results in beat-to-beat, second-to-second, and having age-dependent substrate changes. This paper provided some evidence on the role of RGS2 proteins in AF in the mouse. To put this into context, there is a brief introduction of the role of autonomics in AF. For more complete reviews, see a special review issue in Cardiovascular Research 2011 vol. 89 no. 4, and reviews by Dobrev and Nattel (2011), Schotten et al. (2011), Wakili et al. (2011).
The heart receives both sympathetic and parasympathetic efferent innervation as well as being able to process information via an intrinsic cardiac autonomic nervous system (ICANS). The ICANS may interact with the efferent nerve supply in a complex fashion to regulate cardiac function and it is thought to act as a “mini-brain on the heart.” The neurons of the ICANS are primarily located in five groups of atrial ganglionated plexi (GPs) which have been described by Arora et al. (2003) in the pig. These GPs may represent areas of local control of cardiac muscle (Cardinal et al., 2009). Although close proximity of the neurons to the muscle does not mean exclusive control of the adjacent area, it is an important consideration, and is analogous to the enteric nervous system, which serves as the “local brain” in the intestinal tract (Furness et al., 2004). Subpopulations of intrinsic cardiac neurons express multiple neurotransmitters (Hassall and Burnstock, 1987); however, in Guinea pig posterior ganglia, choline acetyltransferase immunostaining of all neurons indicates major parasympathetic cholinergic input to the myocardium (Mawe et al., 1996).
Atrial fibrillation, the most common clinical arrhythmia (Kannel et al., 1982; Benjamin et al., 1998), is characterized by having abnormal, irregularly irregular, rapid electrical activation. AF is a common health problem in the developed world (Savelieva and Camm, 2001). Its prevalence increases with age, from a prevalence of 0.02% of people of 18–39 years of age to 11.6% of people over 75 years of age (Nixon, 2011). As the population ages, AF will increase the burden on health care resources, both for its therapeutic management, and also due to its serious and costly complications: heart failure and stroke (Dries et al., 1998). Discussion of potential mechanisms for AF, as far back as 1870 (Efimov and Fedorov, 2005), has been separated into two basic themes; those stressing the primary role of the ICANS (referred to as neurogenic AF; Hoffa and Ludwig, 1850; Zipes et al., 1974; Coumel et al., 1978; Liu and Nattel, 1997; Chang et al., 2001; Nattel, 2004), or sometimes called “vagal AF” (Garrey, 1924) and those related exclusively to the role of the atrial muscle itself (referred to as myogenic AF). Both neurogenic (Hoffa and Ludwig, 1850; Zipes et al., 1974; Coumel et al., 1978; Liu and Nattel, 1997; Chang et al., 2001; Nattel, 2004) and myogenic (Vulpain, 1874; MacWilliam, 1887; Garrey, 1914; Moe and Abildskov, 1959; Moe, 1962; Moe et al., 1964; Allessie et al., 1985; Zipes, 1997; Jalife et al., 1998; Skanes et al., 1998; Mandapati et al., 2000) hypotheses have been proposed to account for arrhythmia mechanisms. The separation of AF mechanisms into strictly neurogenic and myogenic types is artificial as they certainly interact and share multiple mechanisms.
Since the studies of Moe and colleagues in the 1950s, AF was shown to be composed of multiple re-entrant wavelets (Nattel, 2002). However, optical mapping studies of AF in isolated sheep atria have found that AF may involve both a high frequency “mother rotor,” and fibrillatory conduction of multiple wavelets spawned from the deterministic driver (Kleber, 2000; Jalife, 2003). Remodeling of the atria may also promote the propagation of multiple wavelets as “AF begets AF,” indicating that atrial remodeling promotes sustained disorganized activity (Morillo et al., 1995; Shen et al., 2011). AF relies on the interaction of triggers, perpetuators, and a vulnerable substrate for initiation and maintenance of the arrhythmia (Allessie et al., 2001). Triggers combined with dynamic substrates of structural heterogeneities, reduced refractoriness, enhanced spatial dispersion of refractoriness and abnormal impulse conduction initiate and perpetuate the arrhythmia (Nattel et al., 2002). Focal activity originating from the pulmonary veins has been suggested to account for more than 90% of AF triggers (Haissaguerre et al., 2000).
Abnormal impulse initiation and conduction contribute to the mechanism of AT (Figure 1), but are also involved in the initiation and maintenance of AF. The rapid rates during AF (Nattel, 2002) promote remodeling of the atria that potentiates AF duration (AF begets AF). There are, however, a number of factors that cause remodeling and “prime” the initial state, promoting rapid triggers (e.g., pulmonary vein ectopy) and susceptibility to fibrillatory conduction necessary for AF. These “clinical risk factors” (Rosiak et al., 2010) for the onset and development of AF are listed in Figure 2, for more detail see Dobrev and Nattel (2011). Two factors not mentioned in the Rosiak article are mechanical stretch and hyperthyroidism (Klein and Ojamaa, 2001; Souza et al., 2012). Although usually associated with other contributing factors such as hypertension or valve disease, stretch and stretch-induced channel conduction can lead to electrical instability. Indeed, interactions between mechanical stretch and cholinergic stimulation has been shown (Yamazaki et al., 2009). Although in embryonic and not the adult atria, there is evidence of interaction between M3 receptors and TREK-2, a stretch activated K+ channel (Kang et al., 2006). There are also interactions between mechanical stretch and gap junctions (Wang et al., 2000). These interactions can be very complex. For instance, Cx40−/− mice had a lower pacing induced breakdown frequency of 1–1 right atrial impulse conduction than wild type mice but did not present with AF (Bagwe et al., 2005). This indicates that the factors that promote fibrillatory conduction may not necessarily contribute to the mechanism driving AF and may even oppose it. On the other hand, slowed conduction in patients with somatic connexin 40 (Gollob et al., 2006) or connexin 43 mutations (Thibodeau et al., 2010) or mice with mutant connexin 43 (Tuomi et al., 2011) were associated with enhanced arrhythmia susceptibility.
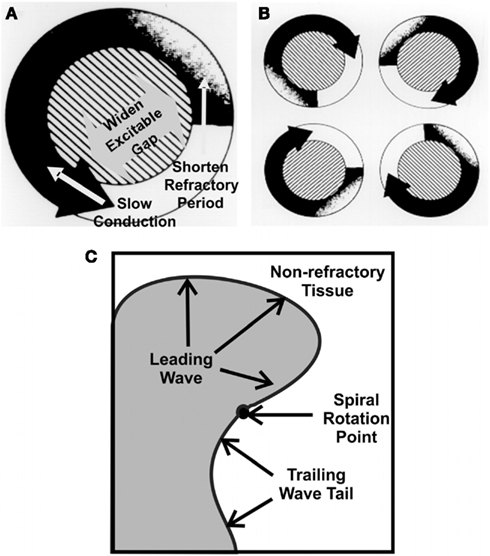
Figure 1. The leading circle, “wave length,” of re-entry in atrial fibrillation (AF). The size of a functional re-entry circuit [(A), right], depends on the wavelength (WL) which is a product of conduction velocity (CV) and refractory period (RP; WL = CV × RP). Short wavelengths allow multiple circuits [(B), left], favoring AF. The wave rotating around a fixed point, rotor [(C), lower; Jalife, 2003]. Factors such which slow conduction enhance the potential for multiple smaller waves. Enhanced cholinergics decrease conduction velocity and/or reduce effective refractoriness. Modified connexins also slow conduction. Either, or a combination of both could promote the initiation and sustaining of AF.
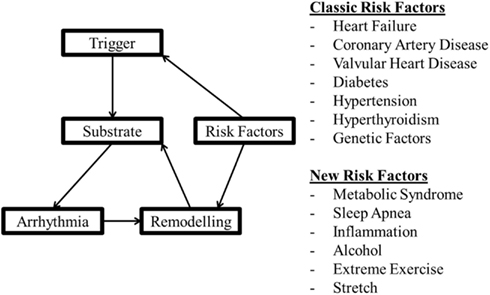
Figure 2. A model of factors involved in arrhythmogenesis and the effect of remodeling with the classical and more recent factors thought to be involved in arrhythmia risk.
Atrial fibrillation causes remodeling of the atria that promotes long lasting or even permanent AF (Thijssen et al., 2000). AF can be classified based on its duration and therapeutic history (Nixon, 2011; Yan and Kowey, 2011). Patterns of AF following the ACC/AHA/ESC guidelines include: (1) Paroxysmal (self-terminating), episodes that generally last less than or equal to 7 days (most less than 24 h); (2) Persistent (not self-terminating), usually more than 7 days, and (3) Permanent, cardioversion failed or not attempted. Either paroxysmal or persistent AF may be recurrent (Fuster et al., 2001).
Persistent AF is associated with diffuse macroscopic and histological atrial myocardial changes, i.e., remodeling. Atrial arrhythmia can also cause ventricular remodeling and, if protracted, cardiac failure. This in turn, can feed back on the atria and produce irreversible interstitial atrial fibrosis (Kalifa et al., 2003).
Lone or paroxysmal AF has been shown to be primarily initiated from the pulmonary veins and posterior left atrial regions (Morillo et al., 1995; Haissaguerre et al., 1998), where most of the GP are in contact with the left atrium. Both alterations in vagal responses and a significant increase in AF control (Pappone et al., 2000, 2004; Nademanee et al., 2004; Platt et al., 2004; Nakagawa et al., 2005a,b) were found in patients when the GPs were targeted (Sakamoto et al., 2010) or inadvertently modified during catheter ablation of AF sources in the pulmonary veins. Also, selective ablation of the GPs in pigs abolished the ability to acutely induce AF (Jones et al., 2008b) and reduced the acute inducibility in dogs (Nishida et al., 2011), which persisted in some animals up to 4 weeks following ablation in dogs (Nishida et al., 2011). These observations demonstrate the importance of the ICANS in AF, i.e., neurogenic AF (Hoffa and Ludwig, 1850; Zipes et al., 1974; Coumel et al., 1978; Liu and Nattel, 1997; Chang et al., 2001; Nattel, 2004). Neurogenic or sometimes called “vagal AF” (Garrey, 1924) relies extensively on the action of acetylcholine. The roles of acetylcholine in setting the stage for rotor development and stabilization of the leading sources of AF have been reviewed (Sarmast et al., 2003; Efimov and Fedorov, 2005; Dobrev and Nattel, 2011; Schotten et al., 2011; Wakili et al., 2011). Acetylcholine promotes left to right frequency gradients and enhances the dominant frequency of the AF driver (Mansour et al., 2001). These responses were also dependent on acetylcholine concentration, with higher concentrations resulting in faster dominant frequencies. Previous studies on AF mechanisms commonly focus on dependence on acetylcholine concentration and the expression profiles of receptors and downstream effectors, while ignoring the regulators of the G protein signaling cascades.
Five different subtypes of muscarinic acetylcholine receptors have been cloned. M1, M3, and M5 receptors are Gαq coupled receptors, while M2 and M4 receptors are Gαi/o coupled. The classical notion is that the cardiac muscarinic acetylcholine receptors are exclusively of the M2 subtype as M2 receptors are the most plentiful. However, there is increasing evidence supporting an important functional role of M3 receptors (Shi et al., 2004; Wang et al., 2004). Of interest, even in the recent review (Schotten et al., 2011), differential effects of cholinergic stimulation, despite its presence in the heart, overlook M3 and are solely related to M2-cholinergic innervation using archival information based on vagal stimulation and atropine blockade “… heterogeneity in the distribution of… M2-cholinoceptors (4, 343).” Reference 4 is Alessi et al. (1958). Reference 343 is Liu and Nattel (1997). Both of these references are prior to the description of M3 receptors in the heart. More recently however, novel M2 blockade with NTC-801 was found to block experimental AF, although its effects on M3 receptors were not assessed (Machida et al., 2011). M3 muscarinic receptors were first identified in human atria in Hellgren et al. (2000). Early quantification of M1–M5 muscarinic subtype mRNAs in rat atria utilizing competitive RT-PCR indicated that M2 mRNA represented more than 90% of the total muscarinic mRNA in the atria, while M3 was less than 3% (Krejci and Tucek, 2002). More recently, quantitative PCR showed low level expression of M1, M4, and M5 receptors in the atria, but this may have been due to low level DNA contamination as RNA in most studies was not treated with DNAse (Kitazawa et al., 2009). The functional consequence of co-expression of multiple muscarinic receptor types has been the focus of many studies discussed below, the most compelling of which use M2 and M3 receptor knockout mice.
After 1 week of rapid pacing, Yeh et al. (2007) found approximately a 50% decrease in levels of mRNA encoding M2, M3, and M4 receptor subtypes in both the left atrial appendix and left pulmonary vein region. In contrast, Voigt et al. (2010) found there were differential alterations in samples from atrial appendices of patients with chronic or paroxysmal AF. In paroxysmal AF, basal current was ~2-fold larger in the left versus right atrial appendix, indicating a left-to-right atrial gradient. In both atria, Kir2.1 (IK1) expression was ~2-fold greater in chronic AF but comparable in paroxysmal AF versus patients in sinus rhythm. Kir2.3 levels (IK, ACh) were unchanged in chronic AF and the right atrial appendix in paroxysmal AF but were 51% lower in the left atrial appendix in paroxysmal AF. In sinus rhythm carbachol-activated IK, ACh was 70% greater in RA versus LA. This right-to-left atrial gradient was decreased in paroxysmal and chronic AF caused by a selective reduction in IK, ACh in right atrium. Similarly, in sinus rhythm, Kir3.1 and Kir3.4 proteins were greater in right versus left atrium and decreased in the right atrium of paroxysmal and chronic AF. Kir3.1 and Kir3.4 expression was unchanged in the left atrium of paroxysmal and chronic AF (Voigt et al., 2010).
In the heart, M2 receptors inhibit adenylyl cyclase (AC) activity via Gαi and induce activation of the G protein coupled inward rectifier K+ channel (GIRK3.1/3.4 or IK, ACh; Kovoor et al., 2001) by Gβγ released from Gαi. Inhibition of adenylyl cyclase production of cAMP reduces the activity of If (a cAMP dependent current) and the L-type Ca2+ current (ICa, L) through Gαo. ICa, L is also regulated by Gαi and nitric oxide (Burger et al., 2009a,b). Adenylyl cyclase 5 deficient mice have reduced ICa, L activity, while Gαi2 knockout mice lack muscarinic regulation of ICa, L (Chen et al., 2001). M2 receptor inhibition of Gαi decreases cAMP production, which reduces protein kinase A (PKA) activation (Kovoor et al., 2001). PKA, through phosphorylation, increases the activity of ICa, L and the rapid delayed rectifier potassium channel (IKr), which promotes repolarization from the plateau phase of cardiac action potential (Tamargo et al., 2004). The inhibition of IKr prolongs AP duration (Clark et al., 2004).
In the atria, activation of IK, ACh increases membrane K+ permeability, facilitating an outward current that hyperpolarizes the cell. There is a gradient of IK, ACh current in the mouse atria that, when combined with the heterogeneous distribution of parasympathetic ganglia, may augment the dispersion of atrial refractoriness (Lomax et al., 2003). In mice (Nygren et al., 2004), optically mapped action potential duration was shorter in the left than the right atrium (Nygren et al., 2004) and the cholinergic agonist, carbachol, reduced durations in all areas except the left atrial appendage. GIRK4 knock out (GIRK4−/−) mice which lack functional IK, ACh channels have abnormal heart rate regulation and are resistant to carbachol-induced AF (Kovoor et al., 2001). In the SA node, IK, ACh hyperpolarizes the membrane potential, and decreases the pacemaker pre-potential slope, which contributes ~50% to the in vivo bradycardic response. In summary, the functions of the M2 receptor include activation of IK, ACh (hyperpolarization), inhibition of IKr (prolonging repolarization), and inhibition of If (negative chronotropy; Hashimoto et al., 2006).
On the other hand, M3 muscarinic receptors couple to Gαq to activate PLC, PKC, and IK, M3 (Shi et al., 2004). To date, IK, M3 is the only identified K+ channel activated by Gαq. IK, M3 current is highly selective for K+ and has delayed rectifier properties with a relatively slow activation time constant (~150 ms at +50 mV), which is followed by a partial and slower decay (Wang et al., 2004). The tail current is characterized by an initial rapid rising phase followed by a slow decay with a mean time constant of ~170 ms (Shi et al., 2003). The waveform is similar to that of IKr; however, it is insensitive to the IKr blockers dofetilide and E-4031, and to the slow delayed rectifier K+ current (IKs) inhibitor, chromanol 293B. IK, M3 is blocked by the non-selective muscarinic antagonist, atropine (Wang et al., 2004), and the M3 selective antagonist, darifenacin (Wang et al., 2004, 2007a). M3 content is also altered in patients with chronic AF associated with mitral stenosis (Zhao et al., 2008) and in experimental chronic tachycardia in dogs (Yeh et al., 2007).
Of note, M3 receptor distribution in cardiac tissue was found to be mostly confined to the intercalated disk region (Wang et al., 2004), and thus was also suggested to regulate ventricular gap junction-mediated cell-to-cell conduction (Wang et al., 2007a). Physical and functional interactions between M3 and connexin 43 (Cx43) have been found in rat ventricular myocytes, which were impaired during myocardial ischemia (Yue et al., 2006). Gap junction remodeling is an essential component promoting atrial as well as lethal ventricular tachyarrhythmias (Dupont et al., 2001a,b; Gutstein et al., 2001; Danik et al., 2004; Shiroshita-Takeshita et al., 2007). The functions of the cardiac M3 receptors suggested so far include: (1) depressed inotropy; (2) protection against myocardial ischemia; (3) regulation of cell-to-cell communication, and (4) participation in the generation and maintenance of AF (Wang et al., 2004). Using the M3 receptor antagonist 4-DAMP Wang et al. (2004), concluded that the M3 receptor also mediates, in part, bradycardia elicited by vagal stimulation. However, the pKb of 4-DAMP for M3 is 8.9–9.3, only one order of magnitude greater the pKb for M2 receptors (7.8–8.4). Perhaps the weak inhibition of vagal bradycardia with high dose 4-DAMP used in Wang’s study was due to blockade of M2 rather than M3 receptors. On the other hand, vagally induced bradycardia was completely abolished in M2 muscarinic receptor (M2−/−) deficient mice but was not affected in M3 muscarinic receptor (M3−/−) deficient mice (Fisher et al., 2004), which does not support a role for M3 in bradycardia in the mouse. However, this does not exclude a role for M3-mediated modulation of atrial or AVN function, as analysis of other atrial or AVN functions was not done.
The functional consequence of co-expression of both M2 and M3 receptors in the atria was studied in M2−/−, M3−/−, and M2/M3 double knockout (M2−/−/M3−/−) mice (Kitazawa et al., 2009). In wild type mouse atria, carbachol-induced a negative inotropic response followed by a sustained positive inotropic response. M3−/− mice were devoid of the positive inotropic phase while M2−/− mice had only a positive inotropic response. M2−/−/M3−/− double knockout mice were devoid of any inotropic response (Kitazawa et al., 2009). The susceptibility to arrhythmia was not studied in these mice. However, these results strongly indicate the functional nature of M3 muscarinic receptors present in the atria.
G protein coupled receptors (GPCRs) are seven transmembrane domain cell surface receptors that bind extracellular ligands (hormones, neurotransmitters, and drugs) and transduce signals into the intracellular environment. Most sympathetic and parasympathetic receptors are members of the GPCR superfamily which couple to various downstream signaling cascades including those controlling adenylyl cyclase, Ca2+ homeostasis, and ion channels (Wieland et al., 2007). In the heart, GPCRs control the chronotropic (rate of contraction), inotropic (strength of contraction), lusitropic (rate of relaxation), and dromotropic (velocity of conduction) responses to acetylcholine and adrenaline (Wieland et al., 2007). Ligand binding stabilizes or induces a conformational change in its GPCR that activates a coupled intracellular heterotrimeric G protein (Gαβγ). There are four families of mammalian heterotrimeric G proteins including Gsα, Gi/oα, Gqα, and G12/13α with various subtypes, effectors, and expression profiles (Milligan and Kostenis, 2006).
Regulators of G Protein Signaling proteins were first identified as GTPase activating proteins (GAPs), accelerating the intrinsic GTPase activity of Gα subunits, as the duration of G protein activation is primarily controlled by GTP hydrolysis. Also, RGS proteins can interfere with G protein binding to effector proteins, separate from their regulation of hydrolysis (Abramow-Newerly et al., 2006). Thus RGS proteins are important limiters of GPCR signaling. Currently, 20 different genes encoding RGS proteins have been identified and divided into four subfamilies, based on their sequence and structural similarities: RZ/A (RGS17, 19, and 20); R4/B (RGS1–5, 8, 13, 16, and 21); R7/C (RGS6, 7, 9, and 11), and R12/D (RGS10, 12, and 14; Bansal et al., 2007). The majority of RGS proteins are GAPs for Gαi/o and many also act on Gαq/1189; however, none are GAPs for Gαs but some RGS-like proteins such as p115RhoGEF do have GAP activity on the Gα11/12 subfamily. RGS2, 3, 4, 6, 10, RGSZ2, and GAIP are all found in atrial cardiomyocytes (Doupnik et al., 2001).
The ongoing control of cardiac function by GPCRs is itself modulated by RGS proteins. RGS-insensitive GαoG184S homozygous knock-in (Gαi2GS/GS) embryonic stem cell-derived cardiocytes had enhanced responses to M2 muscarinic but not A1 adenosine receptor stimulation, while analogous GαoGS/GS cardiocytes had enhanced A1 adenosine and M2 receptor mediated responses (Fu et al., 2006). Gαs stimulation of the intrinsic beating rate was almost completely abolished in Gαi2GS/GS cells, likely due to a failure of AC activation resulting from an increase in countervailing inhibitory activity. Gαi2GS/GS insensitive mutant mice also had pronounced (five-fold greater) muscarinic mediated bradycardia and third degree AV nodal block (Fu et al., 2007). These mutant mouse data indicate that in the SA and AV node, M2 receptors primarily couple through Gαi2 to inhibit cAMP production and activation of PKA (Fu et al., 2006, 2007). Adenosine 1 and 3 receptor overexpressing mice have also have prolonged AVN conduction (Kirchhof et al., 2003; Fabritz et al., 2004), which may involve receptor mediated inhibition of adenylyl cyclase 5/6 (AC5/6), primarily coupling through Gαo (Fu et al., 2006, 2007). Susceptibility to atrial arrhythmia has not been examined in these mice. It is currently believed that specific RGS proteins may couple to specific signaling pathways within the cell.
RGS2, a member of the R4/B subfamily, the second most highly expressed RGS protein in the heart (Doupnik et al., 2001), is a selective GAP for Gαq (Zou et al., 2006). This selectivity does not reflect any exceptional affinity for the latter but rather a relatively weak RGS2 affinity for Gαi/o (Ingi et al., 1998; Cladman and Chidiac, 2002) which is due to the substitution of three conserved amino acid residues in its RGS domain that decrease binding affinity to Gαi/o but not Gαq (Heximer et al., 1999). RGS2 also produces a poorly understood inhibitory effect on Gs-activated AC signaling (Sinnarajah et al., 2001; Roy et al., 2006b). The mechanism by which such inhibition occurs does not appear to involve a GAP effect, as RGS2 fails to alter the GTPase activity of either free (Ingi et al., 1998) or receptor-activated Gs (Roy et al., 2003). Some evidence suggests that the inhibitory effect may involve a physical interaction of the N-terminus of RGS2 with the C1 catalytic loop of AC5 (Salim et al., 2003; Beazely and Watts, 2006); however other studies suggest that RGS2 may bind directly to Gαs (Tseng and Zhang, 1998; Ko et al., 2001). In HEK293 cells, GFP-RGS2 tended to localize to the nucleus but was recruited to the plasma membrane by the co-expression of either Gαs or most adenylyl cyclase isoforms, and BRET (bioluminescence resonance energy transfer) interactions between RGS2 and Gαs are not inhibited by adenylyl cyclase (Roy et al., 2006a). RGS2 appears to be important in the desensitization of Gq- and Gs-mediated GPCR receptor signals as it is upregulated by both and moreover it can mediate cross-desensitization between Gαq and Gαs (Sinnarajah et al., 2001; Roy et al., 2006b).
Removal of RGS2’s inhibitory effect on Gαq signaling increases Gαq-dependent signaling activity in cardiomyocytes (Zhang et al., 2006). Interestingly, RGS2 has also been shown to be an important target in cardiovascular disease (cardiac hypertrophy and hypertension; Tsang et al., 2010). RGS2−/− mice have a mild to moderate hypertensive phenotype (Heximer et al., 2003; Gross et al., 2005; Tank et al., 2007) that is suggested to be mediated by vascular changes, possibly through angiotensin II, endothelin and/or α-adrenergic receptors, rather than being due to cardiac alterations, although cardiac “contractility,” measured by left ventricular dP/dt, was not determined (Oliveira-dos-Santos et al., 2000; Heximer et al., 2003). Altered autonomic function and effects mediated via the kidneys may also contribute to elevated blood pressure in these animals (Gu et al., 2009). An increase in mean arterial pressure of ~10 mm Hg was found with Telemetry, while heart rate was unchanged, indicating a resetting of the baroreceptor reflex (Gross et al., 2005). This was confirmed by directly measuring baroreflex sensitivity calculated by cross-spectral analysis of heart rate variability. However, heart rate variability was not different in RGS2−/− compared to RGS2+/+ mice (Gross et al., 2005), indicating that the cardiac phenotype was not explained by a baroreflex mediated enhancement of vagal nerve activity. Since RGS2 is a selective regulator of Gαq signaling that can inhibit signaling via M3 receptors in other cell types (Tovey and Willars, 2004; Karakoula et al., 2008), it follows that RGS2 may be an important regulator of M3 muscarinic signaling in the atria. Notably, the scaffolding protein spinophilin, which is present in cardiomyocytes (Bers, 2004), binds to both RGS2 and M3 muscarinic receptors, and thus it may facilitate the inhibition of M3 muscarinic receptor-activated Gq signals, as has been observed with analogous α1-adrenergic signals (Wang et al., 2007b).
RGS4 regulates SA nodal function (Cifelli et al., 2008) via inhibition of parasympathetic signaling and IK, ACh activity. In mice expressing LacZ under the control of the RGS4 promoter, high expression of LacZ-RGS4 was found in the SAN will little expression was observed in the surrounding right atrium. RGS4-null (RGS4−/− mice had lower baseline heart rates and greater increases in heart rate following atropine blockade. High expression of RGS4 (demonstrated by LacZ staining), was also seen in the AV node and RGS4−/− mice were highly susceptible to carbachol-induced AV nodal conduction block when treated with isoproterenol. However, there is limited electrophysiological data describing the location of conduction block, and results were obtained principally in Langendorff hearts (removed from in vivo autonomic influences).
As RGS4 was found only in the SA and AV nodal region, the RGS protein regulating parasympathetic signaling and IK, ACh activity in the atrium remained, until recently, unknown. RGS6-deficient (RGS6−/−) atrial myocytes exhibited a significant reduction in the time course of IK, ACh activation and deactivation. RGS6−/− mice also displayed bradycardia and AV nodal conduction block indicating that there is at least partial redundancy in RGS protein function in vivo.
In both RGS2−/− and C57Bl\6 wild type (WT, control) anesthetized mice (Tuomi et al., 2010) with body temperature maintained within the normal physiological range (36.5–38°C; Connolly and Lynch, 1981) and His bundle recording (Figure 3) used to validate correct positioning of the electrode (Figure 4), atrial effective refractory periods (AERPs) were heterogeneous, being longer in the high right compared to the mid right atrium. In addition, AERPs were significantly lower in RGS2−/− mice compared to WT mice. M2 receptor and M3 receptor mRNA was not different in RGS2−/− compared to WT mice. Carbachol reduced AERPs in both strains but strain differences persisted. In contrast, the AERPs were increased to similar levels in both strains with atropine. The selective M3 blocker, darifenacin, increased the AERP for the mid atrium of RGS2−/− and WT mice, and eliminated the strain-related differences (Tuomi et al., 2010), supporting a role of RGS2 in atrial function. Of interest, in paroxysmal AF there is a frequency gradient but in chronic AF, the frequency gradient in the left atrium has been shown to be absent (Lazar et al., 2006).

Figure 3. A photograph of an octapolar electrode catheter (CIB’ER Mouse®, NuMED, Inc., Hopkinton, NY, USA) used for recording from the atrium, His bundle region and ventricle of the anesthetized mouse. The small marks on the ruler are 1 mm spacing.
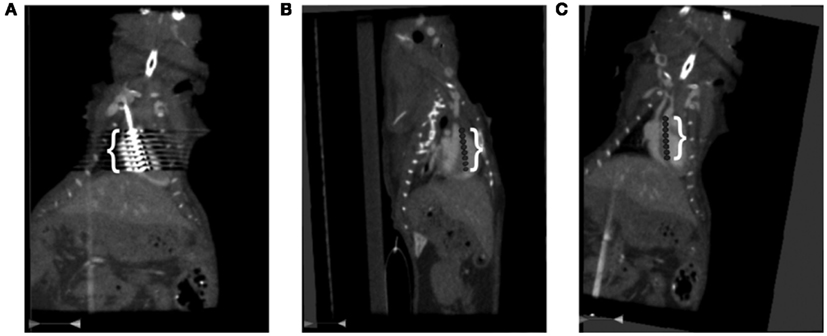
Figure 4. Computed Tomography (CT) of the catheter position in the right heart of the anesthetized mouse. The mouse’s head is at the top and tail, off the bottom of each image. (A) The white bracket delineates the region of interference from catheter’s eight electrodes shown as very bright echoes extending laterally from the catheter. (B) A sagittal view of the digitally inserted catheter. The white bracket delineates the position of the catheter’s eight electrodes. The bright dots to the left of the torso are from the ribs and those on the left are from the spinal column. (C) A coronal view of catheter placement with electrodes three and four across the tricuspid valve. The white bracket delineates the catheter’s eight electrodes. The bright dots to the left and right of the torso are from the ribs.
Since vagally induced bradycardia is mediated via M2 but not M3 receptors (Fisher et al., 2004), we compared the effects of cholinergic blockade using darifenacin and atropine on heart rate changes following vagus nerve stimulation. Stimulating the vagus decreased heart rate by 38 ± 6.8% in WT animals. This decrease was abolished by atropine but was unaltered in the presence of darifenacin (Tuomi et al., 2010). This provided evidence that this dose of darifenacin did not affect M2 signaling. Of note, this dose of darifenacin also terminated electrically induced fibrillation in connexin 43 mutant mice (Tuomi et al., 2011).
Both programmed electrical stimulation (Jones et al., 2001) and burst pacing (Jones et al., 2008a) were used to determine susceptibility to atrial arrhythmia induction. A single atrial stimulus induced AF (Figure 5). Overall, RGS2−/− mice were more susceptible to electrically induced arrhythmia (Table 1; Tuomi et al., 2010).
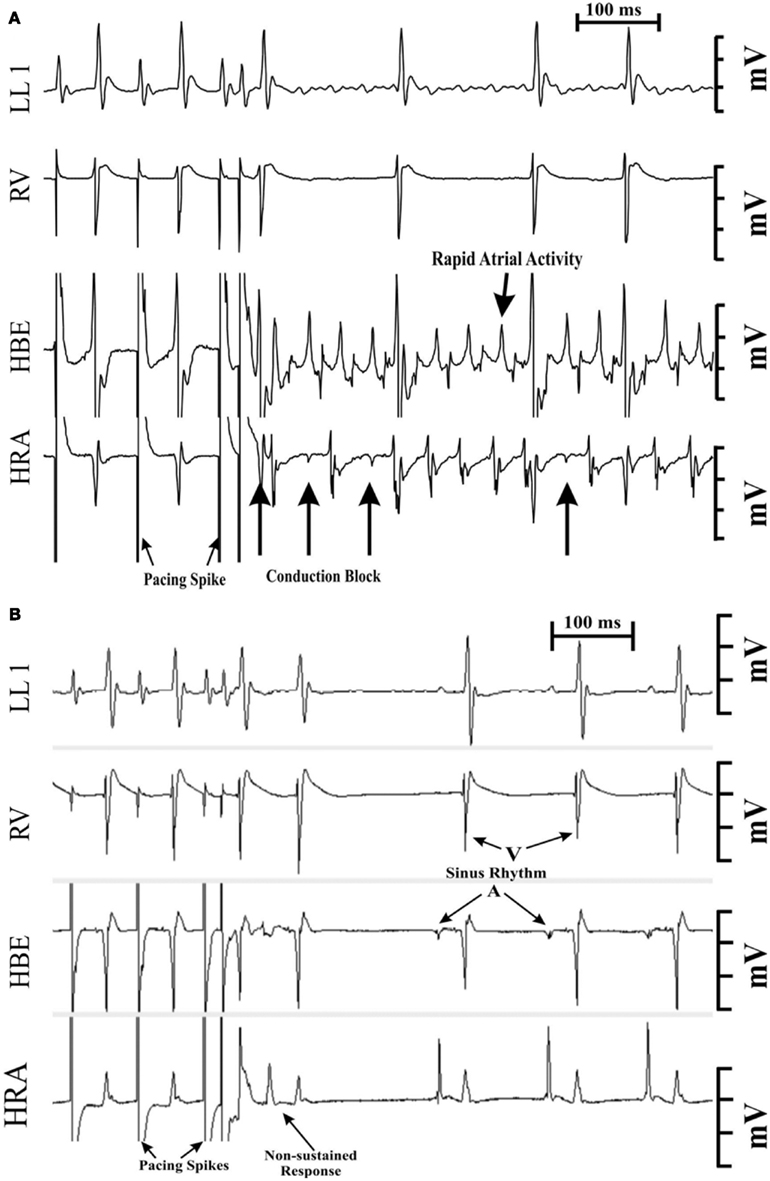
Figure 5. Programmed electrical stimulation-induced atrial fibrillation (AF) in a 1-month-old RGS2−/−mouse (A). Note the conduction block into the high right atrial (HRA) region denoted by the far-field (low amplitude) potential recordings during block. The duration of the arrhythmia was >200 s with an average atrial rate of ~1600 bpm. A similar stimulation protocol failed to induce atrial fibrillation in wild type mice (B). LL1, limb lead 1.RV, right ventricular electrogram. HBE, electrogram from the His bundle region. HRA, recording from the high right atrium.
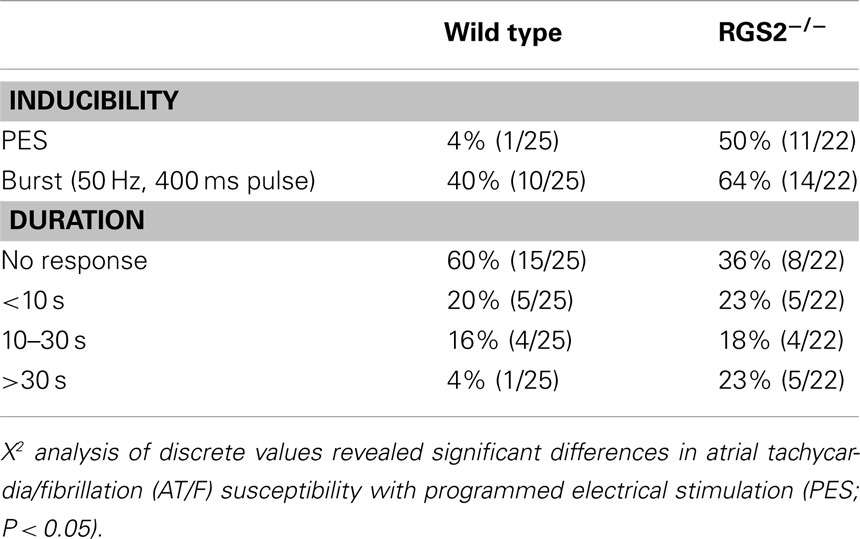
Table 1. Incidence of AT/F induced by PES or burst pacing and maximum duration of induced AT/F with either PES or burst pacing induction.
Atrial fibrillation is very rarely induced by single extrastimuli in large animal and human studies and was thus anticipated to be even less provocable in mice, due to their small size. Thus sensitivity to single extrastimuli in the mouse implies a highly vulnerable myocardial substrate. The duration of induced arrhythmia is also important with “sustained” AF being defined in large animal and human electrophysiological studies as lasting >30 s. Scaling of electrophysiological variables has been related to the BM0.25 (Noujaim et al., 2004). Using this relationship, the duration equivalent is in the second range. However as there is no gold standard: indeed not even a convention, we concluded it best to use ranges. Hence, the data were analyzed based on susceptibility to pacing modality and grouped by the duration of the induced arrhythmia, lasting: <10 s; between 10 and 30 s, and >30 s. As expected, WT mice were virtually non-inducible with a single stimulus (4%, 1/25), compared to burst pacing (40%, 10/25). Carbachol increased the susceptibility to both single extrastimuli (33%, 4/12) and burst pacing (58%, 7/12). Carbachol also prolonged the fibrillation duration with only 2 of 12 untreated mice being sustained, while 5 of the 12 were sustained after carbachol. The atrial stimulation site also affected arrhythmia susceptibility. Burst pacing more readily induced arrhythmia from the mid right atrium of WT mice (40%, 10/25) compared to those from the high right atrial region (8%, 2/25, P < 0.05; Tuomi et al., 2010). This likely reflects the regional AERPs being significantly shorter in the mid-compared to the high right atrium. Electrical heterogeneity may be an important factor in providing a permissive substrate for wave break and re-entry initiation in the atrium.
Rapid focal activity may initiate rotors due to the interaction of a high frequency propagating wave fronts with the refractory tail of the previous wave (Jalife and Pandit, 2005; Vaquero et al., 2008). Immediately after arrhythmia induction, local cycle lengths were often identical in the high right and His bundle regions. However, sometimes at onset, there was regional conduction block into the high right atrium (Figure 5), which could be accompanied by heterogeneity in local cycle lengths in RGS2−/− mice, with His bundle electrograms being shorter than those of the high right atrium. Over time, these intervals converged (Tuomi et al., 2010). This pattern is consistent with the concept of a drifting rotor that rapidly became anchored, although it may also indicate a tachycardia with a rapid rate at onset that slowed down as the driving mechanism stabilized. However, rapid onset may combine with the regional refractory heterogeneity to produce functional conduction block/slowing that eventually recovers at slower rates. Of note, the cycle length during arrhythmia was faster than the measured intrinsic AERP in the high right atrial region, suggesting the possibility of electronic interactions from a rotor core causing reduced refractoriness (Vaquero et al., 2008).
In summary, in addition to the previous focus on the role of M2 receptors in atrial arrhythmia, there is evidence that arrhythmia inducibility may be due to parasympathetic M3 receptors and their regulation by RGS2 knockout. RGS2 accelerates the intrinsic GTPase activity of Gαq, thus limiting the lifetime of the activated state, thereby regulating the Gαq coupled M3 receptor and its associated current, IK, M3. These data are the first we know that demonstrate a role for RGS proteins in atrial arrhythmia. The RGS2−/− mice were more susceptible to electrically induced AF, and there was a greater percentage of RGS2−/− mice with sustained AF. This strain dependent difference was maintained in the presence of carbachol, while atropine abolished the strain dependent differences. These findings suggest an alteration in muscarinic receptor-gated K+ flux evidently due to an increase in M3 muscarinic receptor response per se.
With the aging of the population, there is an increasing drive to identify novel treatments for AF. This will require increased understanding of the signaling pathways and molecular regulators involved in arrhythmia induction, perpetuation, and atrial remodeling. Current medical management of cardiac arrhythmias depends primarily on ion channel blockade, most of which (~70%) are controlled by GPCRs; however, these agents may be proarrhythmic (The Cardiac Arrhythmia Suppression Trial (CAST) Investigators, 1989) and reduce the quality of life of the patients. Thus developing selective agents for cholinergic signaling may provide an effective treatment for AF initiation and perpetuation. Mutations in, or altered expression/function of a variety of RGS proteins could be involved in AF mechanisms in patients, and indeed, changes in RGS2 have been identified in several human cardiovascular phenotypes. Thus, targeting RGS proteins may be important for drug development (Roman et al., 2009). Selective M3R blockade, alone, or in combination with other anti-arrhythmic agents, may be useful for patients with AF. However, in mice, oral darifenacin exerted only transient binding to cardiac muscarinic receptors (Yamada et al., 2006), thus its use may be limited.
While these results indicate a role for RGS2 and the M3 muscarinic receptor in promoting AF in the mouse, additional studies will need to directly determine the role of IK, M3 activity. Also, it would be beneficial to examine the effects of novel IK, ACh antagonists NTC-801 (Machida et al., 2011) and IKur antagonist, acacetin (Li et al., 2008). Atrial vulnerability may also be due to a balance of autonomics (Scherlag et al., 2006; Jones et al., 2007). RGS2−/− mice have also been shown to have reduced renal sympathetic nerve activity compared to WT mice (Tank et al., 2007). Although, the lack of a heart rate difference between RGS2−/− and WT mice found in their earlier study (Gross et al., 2005) does not assist in establishing the role of the sympathetic nervous system in atrial susceptibility, the role of the balance between sympathetics and M3 responses remains to be determined. It is also recognized that it may not be possible to directly extrapolate from mouse experiments to the human.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by the Heart and Stroke Foundation of Ontario (NA6403). Jari Micheal Tuomi was the recipient of an Ontario Graduate Scholarship in Science and Technology and the Canadian Institutes for Health Research CGS Doctoral Research Award. Peter Chidiac holds a Career Investigator Award from the Heart and Stroke Foundation of Ontario. The authors thank Maria Drangova for assistance with the coordinating experiments involving CT Scans and Sarah Detombe for performing the scans.
Abramow-Newerly, M., Roy, A. A., Nunn, C., and Chidiac, P. (2006). RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell. Signal. 18, 579–591.
Alessi, R., Nusynowitz, M., Abildskov, J. A., and Moe, G. K. (1958). Nonuniform distribution of vagal effects on the atrial refractory period. Am. J. Physiol. 194, 406–410.
Allessie, M. A., Boyden, P. A., Camm, A. J., Kleber, A. G., Lab, M. J., Legato, M. J., Rosen, M. R., Schwartz, P. J., Spooner, P. M., Van Wagoner, D. R., and Waldo, A. L. (2001). Pathophysiology and prevention of atrial fibrillation. Circulation 103, 769–777.
Allessie, M. A., Lammers, W., and Bonke, F. (1985). “Experimental evaluation of Moe’s multiple wavelet hypothesis of atrial fibrillation,” in Cardiac Electrophysiology and Arrhythmias, eds D. P. Zipes and J. Jarife (New York, NY: Grune & Stratton Inc.), 265–275.
Arora, R. C., Waldmann, M., Hopkins, D. A., and Armour, J. A. (2003). Porcine intrinsic cardiac ganglia. Anat. Rec. 271A, 249–258.
Bagwe, S., Berenfeld, O., Vaidya, D., Morley, G. E., and Jalife, J. (2005). Altered right atrial excitation and propagation in connexin 40 knockout mice. Circulation 112, 2245–2253.
Bansal, G., Druey, K. M., and Xie, Z. (2007). R4 RGS proteins: regulation of G-protein signaling and beyond. Pharmacol. Ther. 116, 473–495.
Beazely, M. A., and Watts, V. J. (2006). Regulatory properties of adenylate cyclases type 5 and 6: a progress report. Eur. J. Pharmacol. 535, 1–12.
Benjamin, E. J., Wolf, P. A., D’Agostino, R. B., Silbershatz, H., Kannel, W. B., and Levy, D. (1998). Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 98, 946–952.
Bers, D. M. (2004). Macromolecular complexes regulating cardiac ryanodine receptor function. J. Mol. Cell. Cardiol. 37, 417–429.
Burger, D. E., Lu, X., Lei, M., Xiang, F. L., Hammoud, L., Jiang, M., Wang, H., Jones, D. L., Sims, S. M., and Feng, Q. (2009a). Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation 120, 1345–1354.
Burger, D. E., Xiang, F. L., Hammoud, L., Jones, D. L., and Feng, Q. (2009b). Erythropoietin protects the heart from ventricular arrhythmia during ischemia and reperfusion via neuronal nitric-oxide synthase. J. Pharmacol. Exp. Ther. 329, 900–907.
Cardinal, R., Page, P., Vermeulen, M., Ardell, J. L., and Armour, J. A. (2009). Spatially divergent cardiac responses to nicotinic stimulation of ganglionated plexus neurons in the canine heart. Auton. Neurosci. 145, 55–62.
Chang, C. M., Wu, T. J., Zhou, S., Doshi, R. N., Lee, M. H., Ohara, T., Fishbein, M. C., Karagueuzian, H. S., Chen, P. S., and Chen, L. S. (2001). Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation 103, 22–25.
Chen, F., Spicher, K., Jiang, M., Birnbaumer, L., and Wetzel, G. T. (2001). Lack of muscarinic regulation of Ca(2+) channels in G(i2)alpha gene knockout mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 280, H1989–H1995.
Cifelli, C., Rose, R. A., Zhang, H., Voigtlaender-Bolz, J., Bolz, S. S., Backx, P. H., and Heximer, S. P. (2008). RGS4 Regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535.
Cladman, W., and Chidiac, P. (2002). Characterization and comparison of RGS2 and RGS4 as GTPase-activating proteins for m2 muscarinic receptor-stimulated G(i). Mol. Pharmacol. 62, 654–659.
Clark, R. B., Mangoni, M. E., Lueger, A., Couette, B., Nargeot, J., and Giles, W. R. (2004). A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 286, H1757–H1766.
Connolly, M. S., and Lynch, C. B. (1981). Circadian variation of strain differences in body temperature and activity in mice. Physiol. Behav. 27, 1045–1049.
Coumel, P., Attuel, P., Lavallee, J., Flammang, D., Leclercq, J. F., and Slama, R. (1978). The atrial arrhythmia syndrome of vagal origin. Arch. Mal. Coeur Vaiss. 71, 645–656.
Danik, S. B., Liu, F., Zhang, J., Suk, H. J., Morley, G. E., Fishman, G. I., and Gutstein, D. E. (2004). Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 95, 1035–1041.
Dobrev, D., and Nattel, S. (2011). New insights into the molecular basis of atrial fibrillation: mechanistic and therapeutic implications. Cardiovasc. Res. 89, 689–691.
Doupnik, C. A., Xu, T., and Shinaman, J. M. (2001). Profile of RGS expression in single rat atrial myocytes. Biochim. Biophys. Acta 1522, 97–107.
Dries, D. L., Exner, D. V., Gersh, B. J., Domanski, M. J., Waclawiw, M. A., and Stevenson, L. W. (1998). Atrial fibrillation is associated with an increased risk for mortality and heart failure progression in patients with asymptomatic and symptomatic left ventricular systolic dysfunction: a retrospective analysis of the SOLVD trials. Studies of left ventricular dysfunction. J. Am. Coll. Cardiol. 32, 695–703.
Dupont, E., Ko, Y., Rothery, S., Coppen, S. R., Baghai, M., Haw, M., and Severs, N. J. (2001a). The gap-junctional protein connexin 40 is elevated in patients susceptible to postoperative atrial fibrillation. Circulation 103, 842–849.
Dupont, E., Matsushita, T., Kaba, R. A., Vozzi, C., Coppen, S. R., Khan, N., Kaprielian, R., Yacoub, M. H., and Severs, N. J. (2001b). Altered connexin expression in human congestive heart failure. J. Mol. Cell. Cardiol. 33, 359–371.
Efimov, I. R., and Fedorov, V. V. (2005). Chessboard of atrial fibrillation: reentry or focus? Single or multiple source(s)? Neurogenic or myogenic? Am. J. Physiol. Heart Circ. Physiol. 289, H977–H979.
Fabritz, L., Kirchhof, P., Fortmuller, L., Auchampach, J. A., Baba, H. A., Breithardt, G., Neumann, J., Boknik, P., and Schmitz, W. (2004). Gene dose-dependent atrial arrhythmias, heart block, and brady-cardiomyopathy in mice overexpressing A(3) adenosine receptors. Cardiovasc. Res. 62, 500–508.
Fisher, J. T., Vincent, S. G., Gomeza, J., Yamada, M., and Wess, J. (2004). Loss of vagally mediated bradycardia and bronchoconstriction in mice lacking M2 or M3 muscarinic acetylcholine receptors. FASEB J. 18, 711–713.
Fu, Y., Huang, X., Piao, L., Lopatin, A. N., and Neubig, R. R. (2007). Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am. J. Physiol. Heart Circ. Physiol. 292, H2532–H2539.
Fu, Y., Huang, X., Zhong, H., Mortensen, R. M., D’Alecy, L. G., and Neubig, R. R. (2006). Endogenous RGS proteins and Galpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ. Res. 98, 659–666.
Furness, J. B., Jones, C., Nurgali, K., and Clerc, N. (2004). Intrinsic primary afferent neurons and nerve circuits within the intestine. Prog. Neurobiol. 72, 143–164.
Fuster, V., Ryden, L. E., Asinger, R. W., Cannom, D. S., Crijns, H. J., Frye, R. L., Halperin, J. L., Kay, G. N., Klein, W. W., Levy, S., McNamara, R. L., Prystowsky, E. N., Wann, L. S., Wyse, D. G., Gibbons, R. J., Antman, E. M., Alpert, J. S., Faxon, D. P., Fuster, V., Gregoratos, G., Hiratzka, L. F., Jacobs, A. K., Russell, R. O., Smith, S. C. Jr., Klein, W. W., Alonso-Garcia, A., Blomstrom-Lundqvist, C., De, B. G., Flather, M., Hradec, J., Oto, A., Parkhomenko, A., Silber, S., and Torbicki, A. (2001). ACC/AHA/ESC Guidelines for the Management of Patients With Atrial Fibrillation: Executive Summary A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines and Policy Conferences (Committee to Develop Guidelines for the Management of Patients With Atrial Fibrillation) Developed in Collaboration With the North American Society of Pacing and Electrophysiology. Circulation 104, 2118–2150.
Garrey, W. E. (1914). The nature of fibrillary contraction of the heart. Its relations to tissue mass and form. Am. J. Physiol. 33, 397–414.
Gollob, M. H., Jones, D. L., Krahn, A. D., Danis, L., Gong, X. Q., Shao, Q., Liu, X., Veinot, J. P., Tang, A. S., Stewart, A. F., Tesson, F., Klein, G. J., Yee, R., Skanes, A. C., Guiraudon, G. M., Ebihara, L., and Bai, D. (2006). Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677–2688.
Gross, V., Tank, J., Obst, M., Plehm, R., Blumer, K. J., Diedrich, A., Jordan, J., and Luft, F. C. (2005). Autonomic nervous system and blood pressure regulation in RGS2-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1134–R1142.
Gu, S., Cifelli, C., Wang, S., and Heximer, S. P. (2009). RGS proteins: identifying new GAPs in the understanding of blood pressure regulation and cardiovascular function. Clin. Sci. (Lond.) 116, 391–399.
Gutstein, D. E., Morley, G. E., Tamaddon, H., Vaidya, D., Schneider, M. D., Chen, J., Chien, K. R., Stuhlmann, H., and Fishman, G. I. (2001). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88, 333–339.
Haissaguerre, M., Jais, P., Shah, D. C., Takahashi, A., Hocini, M., Quiniou, G., Garrigue, S., Le Mouroux, A., Le Metayer, P., and Clementy, J. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666.
Haissaguerre, M., Shah, D. C., Jais, P., Hocini, M., Yamane, T., Deisenhofer, I., Garrigue, S., and Clementy, J. (2000). Mapping-guided ablation of pulmonary veins to cure atrial fibrillation. Am. J. Cardiol. 86, 9K–19K.
Hashimoto, N., Yamashita, T., and Tsuruzoe, N. (2006). Tertiapin, a selective IKACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol. Res. 54, 136–141.
Hassall, C. J., and Burnstock, G. (1987). Immunocytochemical localisation of neuropeptide Y and 5-hydroxytryptamine in a subpopulation of amine-handling intracardiac neurones that do not contain dopamine beta-hydroxylase in tissue culture. Brain Res. 422, 74–82.
Hellgren, I., Mustafa, A., Riazi, M., Suliman, I., Sylven, C., and Adem, A. (2000). Muscarinic M3 receptor subtype gene expression in the human heart. Cell. Mol. Life Sci. 57, 175–180.
Heximer, S. P., Knutsen, R. H., Sun, X., Kaltenbronn, K. M., Rhee, M. H., Peng, N., Oliveira-dos-Santos, A., Penninger, J. M., Muslin, A. J., Steinberg, T. H., Wyss, J. M., Mecham, R. P., and Blumer, K. J. (2003). Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Invest. 111, 445–452.
Heximer, S. P., Srinivasa, S. P., Bernstein, L. S., Bernard, J. L., Linder, M. E., Hepler, J. R., and Blumer, K. J. (1999). G protein selectivity is a determinant of RGS2 function. J. Biol. Chem. 274, 34253–34259.
Hoffa, M., and Ludwig, C. (1850). Einge neueversuche uber herzbewegung. Zeitschrift Rationellen Medizin 9, 107–144.
Ingi, T., Krumins, A. M., Chidiac, P., Brothers, G. M., Chung, S., Snow, B. E., Barnes, C. A., Lanahan, A. A., Siderovski, D. P., Ross, E. M., Gilman, A. G., and Worley, P. F. (1998). Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J. Neurosci. 18, 7178–7188.
Jalife, J. (2003). Rotors and spiral waves in atrial fibrillation. J. Cardiovasc. Electrophysiol. 14, 776–780.
Jalife, J., Berenfeld, O., Skanes, A., and Mandapati, R. (1998). Mechanisms of atrial fibrillation: mother rotors or multiple daughter wavelets, or both? J. Cardiovasc. Electrophysiol. 9(Suppl. 8), S2–S12.
Jalife, J., and Pandit, S. V. (2005). Ionic mechanisms of wavebreak in fibrillation. Heart Rhythm 2, 660–663.
Jones, D. L., Guiraudon, G. M., Skanes, A. C., and Guiraudon, C. M. (2008a). Anatomical pitfalls during encircling cryoablation of the left atrium for atrial fibrillation therapy in the pig. J. Interv. Card. Electrophysiol. 21, 187–193.
Jones, D. L., Tuomi, J., Ramsay, D., Guiraudon, C. M., Cardinal, R., Page, P., and Guiraudon, G. M. (2008b). Left atrial neuroablation for atrial fibrillation therapy: a feasibility study. J. Interv. Card. Electro Physiol. 21, 171–172.
Jones, D. L., Petrie, J. P., and Li, H. G. (2001). Spontaneous, electrically, and cesium chloride induced arrhythmia and afterdepolarizations in the rapidly paced dog heart. Pacing Clin. Electrophysiol. 24(Pt 1), 474–485.
Jones, D. L., Tuomi, J., Ramsay, D., Guiraudon, C. M., Armour, J. A., Cardinal, R., Page, P., and Guiraudon, G. M. (2007). Left atrial neuroablation for atrial fibrillation: a feasibility study. Can. J. Cardiol. 23, 225C.
Kalifa, J., Jalife, J., Zaitsev, A. V., Bagwe, S., Warren, M., Moreno, J., Berenfeld, O., and Nattel, S. (2003). Intra-atrial pressure increases rate and organization of waves emanating from the superior pulmonary veins during atrial fibrillation. Circulation 108, 668–671.
Kang, D., Han, J., and Kim, D. (2006). Mechanism of inhibition of TREK-2 (K2P10.1) by the Gq-coupled M3 muscarinic receptor. Am. J. Physiol. Cell Physiol. 291, C649–C656.
Kannel, W. B., Abbott, R. D., Savage, D. D., and McNamara, P. M. (1982). Epidemiologic features of chronic atrial fibrillation: the Framingham study. N. Engl. J. Med. 306, 1018–1022.
Karakoula, A., Tovey, S. C., Brighton, P. J., and Willars, G. B. (2008). Lack of receptor-selective effects of either RGS2, RGS3 or RGS4 on muscarinic M3- and gonadotropin-releasing hormone receptor-mediated signalling through G alpha q/11. Eur. J. Pharmacol. 587, 16–24.
Kirchhof, P., Fabritz, L., Fortmuller, L., Matherne, G. P., Lankford, A., Baba, H. A., Schmitz, W., Breithardt, G., Neumann, J., and Boknik, P. (2003). Altered sinus nodal and atrioventricular nodal function in freely moving mice overexpressing the A1 adenosine receptor. Am. J. Physiol. Heart Circ. Physiol. 285, H145–H153.
Kitazawa, T., Asakawa, K., Nakamura, T., Teraoka, H., Unno, T., Komori, S., Yamada, M., and Wess, J. (2009). M3 muscarinic receptors mediate positive inotropic responses in mouse atria: a study with muscarinic receptor knockout mice. J. Pharmacol. Exp. Ther. 330, 487–493.
Kleber, A. G. (2000). The fibrillating atrial myocardium. What can the detection of wave breaks tell us? Cardiovasc. Res. 48, 181–184.
Klein, I., and Ojamaa, K. (2001). Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 344, 501–509.
Ko, J. K., Choi, K. H., Kim, I. S., Jung, E. K., and Park, D. H. (2001). Inducible RGS2 is a cross-talk regulator for parathyroid hormone signaling in rat osteoblast-like UMR106 cells. Biochem. Biophys. Res. Commun. 287, 1025–1033.
Kovoor, P., Wickman, K., Maguire, C. T., Pu, W., Gehrmann, J., Berul, C. I., and Clapham, D. E. (2001). Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143.
Krejci, A., and Tucek, S. (2002). Quantitation of mRNAs for M(1) to M(5) subtypes of muscarinic receptors in rat heart and brain cortex. Mol. Pharmacol. 61, 1267–1272.
Lazar, S., Dixit, S., Callans, D. J., Lin, D., Marchlinski, F. E., and Gerstenfeld, E. P. (2006). Effect of pulmonary vein isolation on the left-to-right atrial dominant frequency gradient in human atrial fibrillation. Heart Rhythm 3, 889–895.
Li, G. R., Wang, H. B., Qin, G. W., Jin, M. W., Tang, Q., Sun, H. Y., Du, X. L., Deng, X. L., Zhang, X. H., Chen, J. B., Chen, L., Xu, X. H., Cheng, L. C., Chiu, S. W., Tse, H. F., Vanhoutte, P. M., and Lau, C. P. (2008). Acacetin, a natural flavone, selectively inhibits human atrial repolarization potassium currents and prevents atrial fibrillation in dogs. Circulation 117, 2449–2457.
Liu, L., and Nattel, S. (1997). Differing sympathetic and vagal effects on atrial fibrillation in dogs: role of refractoriness heterogeneity. Am. J. Physiol. 273(Pt 2), H805–H816.
Lomax, A. E., Rose, R. A., and Giles, W. R. (2003). Electrophysiological evidence for a gradient of G protein-gated K+ current in adult mouse atria. Br. J. Pharmacol. 140, 576–584.
Machida, T., Hashimoto, N., Kuwahara, I., Ogino, Y., Matsuura, J., Yamamoto, W., Itano, Y., Zamma, A., Matsumoto, R., Kamon, J., Kobayashi, T., Ishiwata, N., Yamashita, T., Ogura, T., and Nakaya, H. (2011). Effects of a highly selective acetylcholine-activated K+ channel blocker on experimental atrial fibrillation. Circ. Arrhythm. Electrophysiol. 4, 94–102.
Mandapati, R., Skanes, A., Chen, J., Berenfeld, O., and Jalife, J. (2000). Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation 101, 194–199.
Mansour, M., Mandapati, R., Berenfeld, O., Chen, J., Samie, F. H., and Jalife, J. (2001). Left-to-right gradient of atrial frequencies during acute atrial fibrillation in the isolated sheep heart. Circulation 103, 2631–2636.
Mawe, G. M., Talmage, E. K., Lee, K. P., and Parsons, R. L. (1996). Expression of choline acetyltransferase immunoreactivity in guinea pig cardiac ganglia. Cell Tissue Res. 285, 281–286.
Milligan, G., and Kostenis, E. (2006). Heterotrimeric G-proteins: a short history. Br. J. Pharmacol. 147(Suppl. 1), S46–S55.
Moe, G. K. (1962). On the multiple wavelet hypothesis of atrial fibrillation. Arch. Int. Pharmacodyn. Ther. 40, 183–189.
Moe, G. K., and Abildskov, J. A. (1959). Atrial fibrillation as a self-sustaining arrhythmia independent of focal discharge. Am. Heart J. 58, 59–70.
Moe, G. K., Rheinboldt, W. C., and Abildskov, J. A. (1964). A computer model of atrial fibrillation. Am. Heart J. 67, 200–220.
Morillo, C. A., Klein, G. J., Jones, D. L., and Guiraudon, C. M. (1995). Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 91, 1588–1595.
Nademanee, K., McKenzie, J., Kosar, E., Schwab, M., Sunsaneewitayakul, B., Vasavakul, T., Khunnawat, C., and Ngarmukos, T. (2004). A new approach for catheter ablation of atrial fibrillation: mapping of the electrophysiologic substrate. J. Am. Coll. Cardiol. 43, 2044–2053.
Nakagawa, H., Sherlag, B. J., Lockwood, D., Wolf, R. K., Peyton, M., Wu, R., Yokoyama, K., Po, S. S., Herring, L., Lazzara, R., Jackmann, W. K., and Armour, J. A. (2005a). Localization of left atrial gamglionated plexuses using endocardial and epicardial high frequency stimulation in patients with atrial fibrillation. Heart Rhythm 2, S10–S11.
Nakagawa, H., Sherlag, B. J., Wu, A. W., Lockwood, D., Yokoyama, K., Herring, L., Lazzara, R., and Jackman, W. M. (2005b). Addition of selective ablation of autonomic ganglia to pulmonary vein antrum ablation for treatment of paroxysmal and persistent atrial fibrillation. Circulation 110, III-543.
Nattel, S. (2004). Age, gender, and supraventricular arrhythmias: roles of ion channels, connexins, and tissue architecture? Heart Rhythm 1, 397–398.
Nattel, S., Allessie, M., and Haissaguerre, M. (2002). Spotlight on atrial fibrillation-the “complete arrhythmia.” Cardiovasc. Res. 54, 197–203.
Nishida, K., Maguy, A., Sakabe, M., Comtois, P., Inoue, H., and Nattel, S. (2011). The role of pulmonary veins vs. autonomic ganglia in different experimental substrates of canine atrial fibrillation. Cardiovasc. Res. 89, 825–833.
Noujaim, S. F., Lucca, E., Munoz, V., Persaud, D., Berenfeld, O., Meijler, F. L., and Jalife, J. (2004). From mouse to whale: a universal scaling relation for the PR interval of the electrocardiogram of mammals. Circulation 110, 2802–2808.
Nygren, A., Lomax, A. E., and Giles, W. R. (2004). Heterogeneity of action potential durations in isolated mouse left and right atria recorded using voltage-sensitive dye mapping. Am. J. Physiol. Heart Circ. Physiol. 287, H2634–H2643.
Oliveira-dos-Santos, A. J., Matsumoto, G., Snow, B. E., Bai, D., Houston, F. P., Whishaw, I. Q., Mariathasan, S., Sasaki, T., Wakeham, A., Ohashi, P. S., Roder, J. C., Barnes, C. A., Siderovski, D. P., and Penninger, J. M. (2000). Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc. Natl. Acad. Sci. U.S.A. 97, 12272–12277.
Pappone, C., Rosanio, S., Oreto, G., Tocchi, M., Gugliotta, F., Vicedomini, G., Salvati, A., Dicandia, C., Mazzone, P., Santinelli, V., Gulletta, S., and Chierchia, S. (2000). Circumferential radiofrequency ablation of pulmonary vein ostia: a new anatomic approach for curing atrial fibrillation. Circulation 102, 2619–2628.
Pappone, C., Santinelli, V., Manguso, F., Vicedomini, G., Gugliotta, F., Augello, G., Mazzone, P., Tortoriello, V., Landoni, G., Zangrillo, A., Lang, C., Tomita, T., Mesas, C., Mastella, E., and Alfieri, O. (2004). Pulmonary vein denervation enhances long-term benefit after circumferential ablation for paroxysmal atrial fibrillation. Circulation 109, 327–334.
Platt, M., Mandapati, R., Sherlag, B. J., Yamanashi, W. S., Nagakawa, H., Lazzara, R., and Jackman, W. M. (2004). Limiting the number and extend of radiofrequency applications to terminate atrial fibrillation and subsequently prevent its inducibility. Heart Rhythm. 1, S11.
Roman, D. L., Ota, S., and Neubig, R. R. (2009). Polyplexed flow cytometry protein interaction assay: a novel high-throughput screening paradigm for RGS protein inhibitors. J. Biomol. Screen. 14, 610–619.
Rosiak, M., Dziuba, M., Chudzik, M., Cygankiewicz, I., Bartczak, K., Drozdz, J., and Wranicz, J. K. (2010). Risk factors for atrial fibrillation: not always severe heart disease, not always so “lonely.” Cardiol. J. 17, 437–442.
Roy, A. A., Baragli, A., Bernstein, L. S., Hepler, J. R., Hebert, T. E., and Chidiac, P. (2006a). RGS2 interacts with Gs and adenylyl cyclase in living cells. Cell. Signal. 18, 336–348.
Roy, A. A., Nunn, C., Ming, H., Zou, M. X., Penninger, J., Kirshenbaum, L. A., Dixon, S. J., and Chidiac, P. (2006b). Up-regulation of endogenous RGS2 mediates cross-desensitization between Gs and Gq signaling in osteoblasts. J. Biol. Chem. 281, 32684–32693.
Roy, A. A., Lemberg, K. E., and Chidiac, P. (2003). Recruitment of RGS2 and RGS4 to the plasma membrane by G proteins and receptors reflects functional interactions. Mol. Pharmacol. 64, 587–593.
Sakamoto, S., Schuessler, R. B., Lee, A. M., Aziz, A., Lall, S. C., and Damiano, R. J. Jr. (2010). Vagal denervation and reinnervation after ablation of ganglionated plexi. J. Thorac. Cardiovasc. Surg. 139, 444–452.
Salim, S., Sinnarajah, S., Kehrl, J. H., and Dessauer, C. W. (2003). Identification of RGS2 and type V adenylyl cyclase interaction sites. J. Biol. Chem. 278, 15842–15849.
Sarmast, F., Kolli, A., Zaitsev, A., Parisian, K., Dhamoon, A. S., Guha, P. K., Warren, M., Anumonwo, J. M., Taffet, S. M., Berenfeld, O., and Jalife, J. (2003). Cholinergic atrial fibrillation: I(K,ACh) gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc. Res. 59, 863–873.
Savelieva, I., and Camm, A. J. (2001). Clinical trends in atrial fibrillation at the turn of the millenium. J. Intern. Med. 250, 369–372.
Scherlag, B. J., Patterson, E., and Po, S. S. (2006). The neural basis of atrial fibrillation. J. Electrocardiol. 39(Suppl. 4), S180–S183.
Schotten, U., Verheule, S., Kirchhof, P., and Goette, A. (2011). Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol. Rev. 91, 265–325.
Shen, M. J., Choi, E. K., Tan, A. Y., Han, S., Shinohara, T., Maruyama, M., Chen, L. S., Shen, C., Hwang, C., Lin, S. F., and Chen, P. S. (2011). Patterns of baseline autonomic nerve activity and the development of pacing-induced sustained atrial fibrillation. Heart Rhythm 8, 583–589.
Shi, H., Wang, H., Yang, B., Xu, D., and Wang, Z. (2004). The M3 receptor-mediated K(+) current (IKM3), a G(q) protein-coupled K(+) channel. J. Biol. Chem. 279, 21774–21778.
Shi, H., Yang, B., Xu, D., Wang, H., and Wang, Z. (2003). Electrophysiological characterization of cardiac muscarinic acetylcholine receptors: different subtypes mediate different potassium currents. Cell. Physiol. Biochem. 13, 59–74.
Shiroshita-Takeshita, A., Sakabe, M., Haugan, K., Hennan, J. K., and Nattel, S. (2007). Model-dependent effects of the gap junction conduction-enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation 115, 310–318.
Sinnarajah, S., Dessauer, C. W., Srikumar, D., Chen, J., Yuen, J., Yilma, S., Dennis, J. C., Morrison, E. E., Vodyanoy, V., and Kehrl, J. H. (2001). RGS2 regulates signal transduction in olfactory neurons by attenuating activation of adenylyl cyclase III. Nature 409, 1051–1055.
Skanes, A. C., Mandapati, R., Berenfeld, O., Davidenko, J. M., and Jalife, J. (1998). Spatiotemporal periodicity during atrial fibrillation in the isolated sheep heart. Circulation 98, 1236–1248.
Souza, M. V., Duarte, M. M., Coeli, C. M., and Vaisman, M. (2012). Atrial fibrillation and hyperthyroidism: relation between transoesophageal markers of a thrombogenic milieu and clinical risk factors for thromboembolism. Clin. Endocrinol. (Oxf.) 76, 448–453.
Tamargo, J., Caballero, R., Gomez, R., Valenzuela, C., and Delpon, E. (2004). Pharmacology of cardiac potassium channels. Cardiovasc. Res. 62, 9–33.
Tank, J., Obst, M., Diedrich, A., Brychta, R. J., Blumer, K. J., Heusser, K., Jordan, J., Luft, F. C., and Gross, V. (2007). Sympathetic nerve traffic and circulating norepinephrine levels in RGS2-deficient mice. Auton. Neurosci. 136, 52–57.
The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. (1989). Preliminary report: effect of encainide, and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N. Engl. J Med. 321, 406–412.
Thibodeau, I. L., Xu, J., Li, Q., Liu, G., Lam, K., Veinot, J. P., Birnie, D. H., Jones, D. L., Krahn, A. D., Lemery, R., Nicholson, B. J., and Gollob, M. H. (2010). Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation 122, 236–244.
Thijssen, V. L., Ausma, J., Liu, G. S., Allessie, M. A., van Eys, G. J., and Borgers, M. (2000). Structural changes of atrial myocardium during chronic atrial fibrillation. Cardiovasc. Pathol. 9, 17–28.
Tovey, S. C., and Willars, G. B. (2004). Single-cell imaging of intracellular Ca2+ and phospholipase C activity reveals that RGS 2, 3, and 4 differentially regulate signaling via the Galphaq/11-linked muscarinic M3 receptor. Mol. Pharmacol. 66, 1453–1464.
Tsang, S., Woo, A. Y., Zhu, W., and Xiao, R. P. (2010). Deregulation of RGS2 in cardiovascular diseases. Front. Biosci. (Schol. Ed.) 2, 547–557.
Tseng, C. C., and Zhang, X. Y. (1998). Role of regulator of G protein signaling in desensitization of the glucose-dependent insulinotropic peptide receptor. Endocrinology 139, 4470–4475.
Tuomi, J. M., Chidiac, P., and Jones, D. L. (2010). Evidence for enhanced M3 muscarinic receptor function and sensitivity to atrial arrhythmia in the RGS2-deficient mouse. Am. J. Physiol. Heart Circ. Physiol. 298, H554–H561.
Tuomi, J. M., Tyml, K., and Jones, D. L. (2011). Atrial tachycardia/fibrillation in the connexin 43 G60S mutant (Oculodentodigital dysplasia) mouse. Am. J. Physiol. Heart Circ. Physiol. 300, H1402–H1411.
Vaquero, M., Calvo, D., and Jalife, J. (2008). Cardiac fibrillation: from ion channels to rotors in the human heart. Heart Rhythm 5, 872–879.
Voigt, N., Trausch, A., Knaut, M., Matschke, K., Varro, A., Van Wagoner, D. R., Nattel, S., Ravens, U., and Dobrev, D. (2010). Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ. Arrhythm. Electrophysiol. 3, 472–480.
Vulpain, A. (1874). Note sur les effets de la faradisation directe des ventriculaes du coeur le chien. Arch. Physiol. i, :975.
Wakili, R., Voigt, N., Kaab, S., Dobrev, D., and Nattel, S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin. Invest. 121, 2955–2968.
Wang, H., Lu, Y., and Wang, Z. (2007a). Function of cardiac M3 receptors. Auton. Autacoid. Pharmacol. 27, 1–11.
Wang, X., Zeng, W., Kim, M. S., Allen, P. B., Greengard, P., and Muallem, S. (2007b). Spinophilin/neurabin reciprocally regulate signaling intensity by G protein-coupled receptors. EMBO J. 26, 2768–2776.
Wang, T. L., Tseng, Y. Z., and Chang, H. (2000). Regulation of connexin 43 gene expression by cyclical mechanical stretch in neonatal rat cardiomyocytes. Biochem. Biophys. Res. Commun. 267, 551–557.
Wang, Z., Shi, H., and Wang, H. (2004). Functional M3 muscarinic acetylcholine receptors in mammalian hearts. Br. J. Pharmacol. 142, 395–408.
Wieland, T., Lutz, S., and Chidiac, P. (2007). Regulators of G protein signalling: a spotlight on emerging functions in the cardiovascular system. Curr. Opin. Pharmacol. 7, 201–207.
Yamada, S., Maruyama, S., Takagi, Y., Uchida, S., and Oki, T. (2006). In vivo demonstration of M3 muscarinic receptor subtype selectivity of darifenacin in mice. Life Sci. 80, 127–132.
Yamazaki, M., Vaquero, L. M., Hou, L., Campbell, K., Zlochiver, S., Klos, M., Mironov, S., Berenfeld, O., Honjo, H., Kodama, I., Jalife, J., and Kalifa, J. (2009). Mechanisms of stretch-induced atrial fibrillation in the presence and the absence of adrenocholinergic stimulation: interplay between rotors and focal discharges. Heart Rhythm 6, 1009–1017.
Yeh, Y. H., Qi, X., Shiroshita-Takeshita, A., Liu, J., Maguy, A., Chartier, D., Hebert, T., Wang, Z., and Nattel, S. (2007). Atrial tachycardia induces remodelling of muscarinic receptors and their coupled potassium currents in canine left atrial and pulmonary vein cardiomyocytes. Br. J. Pharmacol. 152, 1021–1032.
Yue, P., Zhang, Y., Du, Z., Xiao, J., Pan, Z., Wang, N., Yu, H., Ma, W., Qin, H., Wang, W. H., Lin, D. H., and Yang, B. (2006). Ischemia impairs the association between connexin 43 and M3 subtype of acetylcholine muscarinic receptor (M3-mAChR) in ventricular myocytes. Cell. Physiol. Biochem. 17, 129–136.
Zhang, W., Anger, T., Su, J., Hao, J., Xu, X., ZHU, M., Gach, A., Cui, L., Liao, R., and Mende, U. (2006). Selective loss of fine tuning of Gq/11 signaling by RGS2 protein exacerbates cardiomyocyte hypertrophy. J. Biol. Chem. 281, 5811–5820.
Zhao, Q., Huang, C., Jiang, H., Okello, E., Tang, Y. H., Wang, Z., and Wu, Z. (2008). M2 and M3-muscarinic acetylcholine receptors remodelling in patients with a dilated atrium. Acta Cardiol 63, 166–170.
Zipes, D. P. (1997). The seventh annual Gordon K. Moe Lecture. Atrial fibrillation: from cell t bedside. J. Cardiovasc. Electrophysiol. 8, 927–938.
Zipes, D. P., Mihalick, M. J., and Robbins, G. T. (1974). Effects of selective vagal and stellate ganglion stimulation of atrial refractoriness. Cardiovasc. Res. 8, 647–655.
Keywords: RGS proteins, autonomic nervous system, cholinergic, arrhythmia, atrial fibrillation, intrinsic cardiac autonomic nervous system, heart, M3 muscarinic receptor
Citation: Jones DL, Tuomi JM and Chidiac P (2012) Role of cholinergic innervation and RGS2 in atrial arrhythmia. Front. Physio. 3:239. doi: 10.3389/fphys.2012.00239
Received: 27 December 2011; Accepted: 12 June 2012;
Published online: 29 June 2012.
Edited by:
Craig Doupnik, University of South Florida College of Medicine, USAReviewed by:
Dobromir Dobrev, University of Dresden, GermanyCopyright: © 2012 Jones, Tuomi and Chidiac. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Douglas L. Jones, Department of Physiology and Pharmacology, University of Western Ontario, London, ON, Canada N6A 5C1. e-mail:ZG91Zy5qb25lc0BzY2h1bGljaC51d28uY2E=
†Present address: Jari M. Tuomi, Northern Ontario Medical School, Sudbury, ON, Canada.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.