
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol., 02 July 2012
Sec. Computational Physiology and Medicine
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00222
This article is part of the Research TopicSystems Biology Approaches to Understanding the Cause and Treatment of Heart, Lung, Blood, and Sleep DisordersView all 12 articles
 Thomas E. Dick1
Thomas E. Dick1 Yaroslav I. Molkov2
Yaroslav I. Molkov2 Gary Nieman3 Yee-Hsee Hsieh1 Frank J. Jacono1,4 John Doyle5,6
Gary Nieman3 Yee-Hsee Hsieh1 Frank J. Jacono1,4 John Doyle5,6 Jeremy D. Scheff7 Steve E. Calvano8
Jeremy D. Scheff7 Steve E. Calvano8 Ioannis P. Androulakis7 Gary An9
Ioannis P. Androulakis7 Gary An9 Yoram Vodovotz10,11*
Yoram Vodovotz10,11*Acute inflammation leads to organ failure by engaging catastrophic feedback loops in which stressed tissue evokes an inflammatory response and, in turn, inflammation damages tissue. Manifestations of this maladaptive inflammatory response include cardio-respiratory dysfunction that may be reflected in reduced heart rate and ventilatory pattern variabilities. We have developed signal-processing algorithms that quantify non-linear deterministic characteristics of variability in biologic signals. Now, coalescing under the aegis of the NIH Computational Biology Program and the Society for Complexity in Acute Illness, two research teams performed iterative experiments and computational modeling on inflammation and cardio-pulmonary dysfunction in sepsis as well as on neural control of respiration and ventilatory pattern variability. These teams, with additional collaborators, have recently formed a multi-institutional, interdisciplinary consortium, whose goal is to delineate the fundamental interrelationship between the inflammatory response and physiologic variability. Multi-scale mathematical modeling and complementary physiological experiments will provide insight into autonomic neural mechanisms that may modulate the inflammatory response to sepsis and simultaneously reduce heart rate and ventilatory pattern variabilities associated with sepsis. This approach integrates computational models of neural control of breathing and cardio-respiratory coupling with models that combine inflammation, cardiovascular function, and heart rate variability. The resulting integrated model will provide mechanistic explanations for the phenomena of respiratory sinus-arrhythmia and cardio-ventilatory coupling observed under normal conditions, and the loss of these properties during sepsis. This approach holds the potential of modeling cross-scale physiological interactions to improve both basic knowledge and clinical management of acute inflammatory diseases such as sepsis and trauma.
Sepsis is a significant public health concern, accounting for approximately 10% of total U.S. deaths annually (Angus et al., 2001; Martin et al., 2003; Vincent et al., 2006; Heron et al., 2009). For most infections, despite antibiotic treatments, death occurs primarily through the final common pathway of sepsis-induced multiple organ dysfunction syndrome (MODS).As the population ages, the preponderance of complex medical co-morbidities increases and the impact of sepsis is expected to increase (Anonymous, 1990; Angus et al., 2001; Martin et al., 2003; Weycker et al., 2003).
Despite a large body of scientific literature concerning individual mechanisms involved in sepsis – disordered endothelial activation (Aird, 2003; Ait-Oufella et al., 2010), organ dysfunction due to epithelial cell failure (Protti and Singer, 2006; Balestra et al., 2009), dysregulated inflammation and the associated complement, and coagulation networks (Rittirsch et al., 2008; Levi and van der Poll, 2010) – the primary challenge lies in integrating a large body of data into a cohesive whole that can guide novel therapies. Effectively modulating, or controlling, the inflammatory response in sepsis, without adverse effects, has proven daunting. Currently no drug is approved by the U.S. Food and Drug Administration (FDA) for MODS. A previously approved drug, recombinant human activated protein C, failed to offer a benefit over standard of care on a FDA-mandated repeat Phase III clinical trial and was removed from the market (Angus, 2011; Mitka, 2011).
We suggest that the rational development of new therapies for sepsis and other acute inflammatory diseases requires characterizing the functional architecture and control points of multi-compartment inflammation. Specifically, we suggest that MODS could be thought of as arising from containment failure (i.e., failure to contain pathogens as well as failure to contain inflammation appropriately) driven by multi-scale “tipping points” that drive a forward feedback loop of inflammation → tissue damage/dysfunction → inflammation (An et al., 2012). We use the term “tipping point” to refer to a functional threshold beyond which a particular compartment/subsystem/system undergoes a phase transition from one range of dynamic behaviors to another. Implicit in this concept is that the internal feedback control processes of a particular compartment/subsystem act to maintain its behavior within a physiological functional range in response to perturbation, but that eventually the persistence and magnitude of the perturbation can lead to control failure. This control feature leads to a shift in the behavioral domain of the subsystem that can in turn lead to disorders in associated compartments/subsystems. This conceptual architecture has the potential to allow: (1) identification of intra-compartment-specific metrics that can be used to determine how close that compartment is to its “tipping point;” (2) identification of inter-compartmental control structures and their associated metrics for efficacy of control; and (3) propose both intra- and inter-compartmental control points that can be targeted for therapeutic intervention.
Studies from our groups, which coalesced under the U.S. National Institute of Health Computational Biology Program1 and the Society for Complexity in Acute Illness2, have led us to hypothesize that these multi-scale inflammatory “tipping points,” subsequent containment failure, and forward feedback to further propagate inflammation are either centrally controlled by neural circuits, or that neural circuits are activated once inflammation is induced in the brain during sepsis. We hypothesize that the structural/functional (parenchymal) cells of a given organ, once stressed or injured in a process that accompanies the degradation of organ physiology, release damage-associated molecular patterns (DAMPs) that in turn induce resident inflammatory (non-parenchymal) cells to produce classical pro-inflammatory cytokines, which in turn cause further release of DAMPs. Thus, inflammation would be controlled locally until the threshold for local control is exceeded. Co-incident with the peripheral inflammation, the brain “maps” the peripheral inflammation through its own expression of inflammatory cytokines. Brain inflammation would trigger processes aimed at vagal control of inflammation, to maintain physiologic homeostasis (Piepoli et al., 1995; Annane et al., 1999; Korach et al., 2001).This is a key point: parenchymal cells have no way of “sensing” directly that the organ in which they are contained is functioning in- or out-side its optimal operating parameters; only the brain can “sense” this dysfunction. Moreover, parenchymal cells exhibit dysfunction locally through the release of DAMPs that are “sensed” by inflammatory cells. In this scheme, “tipping points” could occur in two distinct locations – a peripheral compartment in which the initial inflammation is manifest, and a “mirrored” representation of that compartment in the neural circuitry and tissue; both of these “tipping points” would affect – and be affected by – the physiologic function of a given organ. The implication of this hypothesis would be that centrally not only is there a loss of control but also dysfunctional control of other compartments, and that this dysfunction arises from the unique role of the brain as sensor and regulator of both organ function and inflammation.
We suggest that the use of data-driven and computational modeling, combined with experiments in relevant animals models of sepsis/MODS, is necessary to define and rationally modulate these “tipping points.” Time-domain signal-processing analysis has correlated alterations (primarily decreases) in physiologic variability with morbidity and mortality in critically ill patients (Pomeranz et al., 1985; Anonymous, 1996; Godin et al., 1996; Korach et al., 2001; Barnaby et al., 2002; Pontet et al., 2003; Kleiger et al., 2005; Chen and Kuo, 2007; Ahmad et al., 2009; Fairchild et al., 2009). To unify these diverse observations, we hypothesize that the progress of tissue-level failure toward MODS is accompanied by defined inflammatory networks in different organs, is controlled by inflammation “maps” in the brain, and manifests as decreased physiologic variability (Figure 1A).
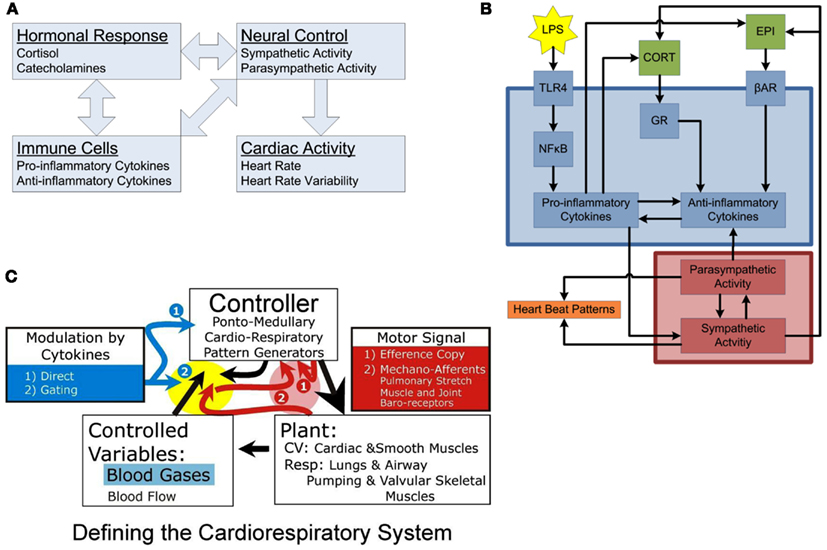
Figure 1. Conceptual models of organization. (A) Inter-compartmental communication. Immune cells provoke activation of hormonal responses and neural controls, systemically manifested in alterations in cardiac activity. The interconnected nature of the regulatory interactions among compartments leads to the emergence of complex systemic responses. (B) Intra-compartment dynamics. At the cellular level, LPS is recognized by TLR4, activating the NF-κB signaling, leading to release of pro-inflammatory cytokines which turn on the anti-inflammatory machinery eventually leading to release of hormones driving sympathetic/parasympathetic imbalance altering heart beat patterns leading to diminished heart rate variability. Each individual compartment is characterized by its own, embedded, feedback regulatory structures. (C) Basic compartments of a physiologic system: Plant, Controlled Variables, and Controller. The plant is the coupled cardio-respiratory system functioning as single physiologic system serving gas exchange. In this system, blood gases and flow (e.g., vascular resistance, heart rate) are the controlled variables in delivering oxygen to various tissues. The controller generates rhythmic respiratory sympathetic and parasympathetic activities. Pink highlight, these red arrows relate to variables in the red box (1) Efference copy and (2) Mechano-receptors [pulmonary stretch, muscle- and joint-, and baro-receptors] which provide input to controller regarding plant performance on a breath-by-breath or beat-by-beat basis for a given motor signal (large black arrow). Yellow highlight: these arrows relate to how the controlled variables and the mechano-receptor afferents are modulated by the controller and in the presence of cytokines. The yellow and pink highlight areas relate to the nTS and dl pons, respectively. In the dl pons, we hypothesize that mechano-receptor afferent project to the dl pons (via the nTS) and interact with an Efference Copy produced by the controller. Efference Copy is defined as a copy of the motor signal delivered to the plant. Differences between these dl pontine inputs (e.g., the magnitude and strength of the muscle contraction, the lung inflation, etc) are compared to the generated motor signal. Loss of variability in the activity pattern of the plant can result from a failure of the controller to adapt to disparities between sensory input and Efference Copy. In the nTS, cytokines are expressed during ALI and, we expect sepsis, and may affect how afferent input is relayed to the controller. We propose a gating mechanism; one in which afferent inputs are depolarized and neural transmission efficacy is diminished.
We have previously suggested a computational modeling approach to the dilemma of MODS that represents a conceptual departure from the current view of acute inflammation and MODS, and offers a new paradigm of MODS pathogenesis based on multifaceted, multi-compartment, and multi-scale inflammatory processes (An et al., 2012). This approach is based on the following premises:
• The recognition – suggested, driven, and linked by computational modeling – that the inflammatory response is both compartmentalized and tightly coupled with physiological processes.
• Given this architecture, that at a certain level of perturbation the reinforcing feedback loop of inflammation → damage/dysfunction → inflammation leads to failure of intra-compartment control. Such failure manifests as the crossing of compartment-specific “tipping points” that have “all-or-none” systemic and physiologic consequences (An et al., 2012).
• That the control structure for integrating inflammation and physiology is based on neural circuits, and thus disordered and failing control of effectively contained inflammation is manifest as altered physiological variability (Namas et al., 2012).
• Finally, that therapies should be targeted based on this structure and dynamic behavior.
Below, we discuss each of these points and suggest how they connect to drive a novel view of MODS.
Inflammation is a prototypical complex system, with multi-feedback interactions among its components, robustness to perturbation coupled with the potential for severe failure at key nodes, and system-level properties difficult to intuit from reductionist analysis (Csete and Doyle, 2002; Vodovotz and An, 2009). We assert that these properties have led to the ineffectiveness of mediator-targeting systemic therapies. As a result, the clinical approach to MODS is focused on providing supportive care of specific points of end-organ failure (e.g., fluids and drugs to alleviate hypotension, ventilation to support failing lungs, etc.). Unfortunately, these interventions often have unintended “ripple effects” that propagate the disordered systemic inflammatory response to the overall detriment of the patient (Ferguson et al., 2005; Sakr et al., 2005; Cotton et al., 2006). Therefore, a disconnect exists between the clinical needs of supporting physiology-level organ function and the need to control the dynamics of the generative cellular-molecular processes that eventually determine that function. This gulf between the cellular-molecular biology of inflammation and organ physiology seen in MODS is precisely the multi-scale barrier that must be traversed to provide a rational basis for potential therapies for MODS.
We view the inflammatory response as compartmentalized both structurally and across multiple scales of organization (Boujoukos et al., 1993; Schein et al., 1996; Molina et al., 2001; Cavaillon and Annane, 2006). Why focus on compartments, when inflammation in sepsis manifests systemically? Despite the attractiveness of purely systemic therapy, the ineffectiveness of such interventions has been demonstrated in multiple studies in animal models of sepsis in which suppressing systemic inflammation resulted in immunosuppression and elevated morbidity and mortality due to overwhelming infection (Remick et al., 1995; Natanson, 1997). Similarly, an insufficient systemic inflammatory response is associated with elevated morbidity and mortality even in nominally sterile trauma/hemorrhage in both humans and swine (Namas et al., 2009a). Thus, systemic-level interventions may lead to inappropriate damping of a necessary inflammatory response in those tissues and compartments that have not approached their “tipping point.” We have begun to refine the dynamic relationships between inflammatory compartments to develop a roadmap toward the translational engineering of therapeutic interventions. We employ progressive use of traditional in vivo experiments and platforms, augmented by computational models to explore, explain, and bridge the fundamental aspects of multi-compartment inflammation.
A central aspect of our interdisciplinary approach to deciphering the inflammatory response involves augmenting laboratory studies with computational models that can integrate, suggest, explain, and potentially predict biological knowledge and data. These computational models include both traditional mathematical models based on ordinary differential equations, as well as agent-based and rules-based models (Vodovotz et al., 2004, 2008, 2009; An et al., 2008, 2009; Foteinou et al., 2009b; Vodovotz and An, 2009; Mi et al., 2010; Namas et al., 2012). We initially discerned inflammatory “tipping points” using a multi-scale, multi-tissue, and multi-organ agent-based model (ABM) of the gut-lung axis of systemic inflammation (An, 2008). In this ABM, both organs are represented by spatially distinct, aggregated populations of epithelial and endothelial cells that interact with circulating inflammatory cells and mediators. Simulations of gut ischemia demonstrated a clear gut ischemia threshold, or “tipping point,” beyond which MODS could be discerned: first ARDS, then systemic hypoxia, and ultimately in silico “death” (An, 2008). Simulation of ventilatory support allowed the system to tolerate more severe gut ischemia, but the “tipping point” persisted. Despite the abstraction of this ABM, it did provide early evidence of the role of compartmental inflammation on the generation of inflammatory “tipping points” and subsequent MODS, and suggested that interventions for sepsis might need to be targeted at the compartment level rather than systemically, or as an adjunct to systemic therapy.
In a similar vein, we created a two-compartment mathematical model of porcine endotoxemia (Nieman et al., 2012), based on an existing mathematical model of mouse endotoxemia (Chow et al., 2005; Lagoa et al., 2006; Prince et al., 2006; Torres et al., 2009). This previous single-compartment mathematical model of inflammation was capable of making qualitative and quantitative predictions with regard to endotoxin-induced inflammation and blood pressure in genetically identical mice (Chow et al., 2005; Lagoa et al., 2006; Prince et al., 2006; Torres et al., 2009). Without compartmentalization, we realized that key meta-behaviors of inflammation were absent, and thus multi-compartment models would be necessary to address the role of inflammatory “tipping.” As in the gut/lung ABM described above, this equation-based model was extended to support clinical interventions such as a fluid resuscitation and mechanical ventilation (Nieman et al., 2012). Importantly, this model was capable of addressing individual variations in the porcine inflammatory and pathophysiologic response to endotoxin, including correlation with clinically useful indices such as the Oxygen Index (Nieman et al., 2012).
To define inflammatory networks that drive compartment-specific “tipping points,” we have applied Dynamic Network Analysis (DyNA) algorithm (Mi et al., 2011), with a more recently developed Dynamic Bayesian Network (DyBN) algorithm (adapted from; Grzegorczyk and Husmeier, 2011). We utilized the DyBN method to examine the connectivity of inflammation in multiple organs in endotoxemic mice. These studies suggested a high degree of connectivity as well as feed-forward behavior for multiple chemokines, as well as the presence of indirect positive feedback loops, all of which appeared to induce the cytokine IL-6 (a biomarker of dysregulated inflammation in multiple contexts; Abraham and Singer, 2007; Namas et al., 2009b).Based on these studies, we suggest that the compartment-specific response to inflammatory stimuli initially remains within a given compartment, helping to coordinate responses appropriate to a given stimulus. However, when the magnitude or duration of an inflammatory stimulus exceeds certain (likely genetically encoded) thresholds, the response spills over into other compartments. This process could occur via both the systemic and lymphatic circulation, leading to progressive organ dysfunction. We hypothesize that this dysfunction, in turn, further aggravates inflammation.
As noted above, one of our goals is to create multi-scale, multi-compartment computational models of inflammation, and (patho) physiology applicable across species, to transcend the barrier between pre-clinical and clinical studies (An et al., 2010). Accordingly, to facilitate comparison to computational modeling studies calibrated against data in mice (Chow et al., 2005), rats (Daun et al., 2008), and swine (Nieman et al., 2012), we have studied human systemic inflammation in the setting of endotoxemia.Even though species-specific thresholds of responsiveness to microbial product differ, each of the animal models share a common mammalian inflammatory response (Parker and Watkins, 2001). Endotoxemia triggers Toll-like receptor (TLR)4 signaling, and subsequent events propagate through a network of intimately connected and interacting compartments with an end result of either resolution of the inflammatory response, or a self-sustaining imbalance that, we hypothesize, drives inflammatory “tipping points” that manifest in organ dysfunction (Figure 1B).
In addition we performed data-driven analyses of high-dimensional leukocyte microarray data taken from human endotoxemia experiments (Calvano et al., 2005), identifying critical transcriptional responses to endotoxemia through a novel clustering approach (Yang et al., 2009). These responses were quantitatively linked through physicochemical modeling, producing an initial computational model of the transcriptional response to human endotoxemia (Foteinou et al., 2009a).
To account for hormonal modulation of the inflammatory response (Figure 1B), pharmacokinetic/pharmacodynamic models linking the dynamics of epinephrine, cortisol, and endotoxemia were proposed (Foteinou et al., 2009a). This approach provided fundamental information, so we could explore the impact of hormonal rhythmicity on host fitness, specifically Circadian rhythms (Coogan and Wyse, 2008). We developed a model to assess the impacts of circadian rhythmicity, as imposed by the suprachiasmatic nucleus (SCN) through circadian hormones, on the response to endotoxemia (Scheff et al., 2010). This model predicted a differential response to identical inflammatory inputs depending on the circadian phase, thus illustrating the importance in circadian rhythmicity in determining the response to endotoxemia.
Hormonal rhythmicity at time scales faster than circadian, called ultradian rhythms, are also increasingly being seen as partaking in important physiological functions (Desvergne and Heligon, 2009; Lightman and Conway-Campbell, 2010). Prominent among the implications of pulsatile secretion of cortisol is the potential role of these ultradian rhythms in regulating the appropriate homeostatic expression of glucocorticoid-responsive genes, including components of the peripheral circadian clock and inflammatory mediators. To investigate the impacts of ultradian rhythms further, we developed a model linking the ultradian production of cortisol in the hypothalamic-pituitary-adrenal (HPA) axis with its downstream effects, which showed the importance of rhythmic hormone patterns in signal transduction (Scheff et al., 2011b). This work was extended to consider the same system in the stress response. One of our key findings was that the homeostatic amplitude of ultradian rhythms was predictive of the responsiveness of the HPA axis to stress (Scheff et al., 2012). This is an example of how characteristics embedded in physiologic rhythms can contain information about the state of a biological system. However, analysis at the level of heart rate variability (HRV) allows for much more significant translational applications through the potential to incorporate hormonal rhythms into single clinically accessible metric.
A central hypothesis underlying our work involves the cross-regulation of inflammation and physiology via neural control mechanisms. The basic elements of a physiologic system defined in engineering terms are the plant, controller, and sensor (Figure 1C). Generally, the plant is the end organ whose function is regulated to maintain controlled variables; the controller, a neural network modulating the plant; and the sensor, specialized cells monitoring levels of the controlled variables. Here (Figure 1C), the plant is the cardio-pulmonary system; the controller, the coupled cardio-respiratory central pattern generator (CPG; for details see; Baekey et al., 2010; Molkov et al., 2010, 2011); and the controlled variables, blood gases, and flow for gas exchange.
Fundamental questions, which we are beginning to address, are: (1) How does cardio-respiratory control system “sense” peripheral dysfunction? (2) How does central expression of cytokines affect the control of a peripheral compartment and neural function itself? and (3) How are compartments organized in the brainstem? To sense dysfunction, we propose an interaction between efference copy and sensory input [Figure 1C (1 and 2, in red highlight)]. Efference copy is the expected effect, specifically, a “copy” of the motor output produced by the controller. This is compared to the sensory input, which is the effective output. Central expression of cytokines could modulate the controller within this functional loop (Figure 1C2, in yellow highlight) and by directly affecting the function of neurons in the CPG (Figure 1C1, no highlight). Further, the time-dependent properties of the expression of cytokines in this reduced control circuit are unknown; for instance, do cytokines become expressed sequentially or co-incidentally in different control nuclei? Further, cytokines definitely elicit “sickness” behavior, which will affect the output of automatic nuclei. Finally, cytokines evoke various second-messenger systems from glia and neurons and many of these second-messengers, including nitric oxide, act extracellularly. Control of other compartments may be elicited through spread of cytokines themselves, or of their second-order messengers.
As a dynamic controller, the respiratory CPG is capable of producing a wide repertoire of “output patterns” and a particular output pattern depends on peripheral and central inputs but also on prior experience and local internal milieu (Rybak et al., 2008). Without peripheral or central inputs, the respiratory CPG provides an output that is rhythmic and regular, i.e., characterized by low deterministic variability (Dhingra et al., 2011). Under normal conditions the intact cardio-respiratory system exhibits chaotic dynamics with deterministic variability (Sammon and Bruce, 1991). We theorize that alterations in the non-linear properties of cardio-respiratory variability associated with disease depend, at least partially, on cytokine-modulation of sensory feedback, and ponto-sensory interactions. In the presence of elevated levels of cytokines, the efficacy of sensory input is reduced, minimizing the non-linear deterministic properties of pattern variability (Figure 1C; Dhingra et al., 2011). Early models of breathing pattern during heart failure have demonstrated that increased gain and delayed feedback lead to sensor over-response and results in a waxing and waning pattern (Cherniack et al., 1966; Cherniack and Longobardo, 2006). In addition, pressure support ventilation (which effectively facilitates a gain of lung stretch receptors feedback) helps to maintain normal breathing pattern variability, which reduces when a patient is separated from ventilation (Wysocki et al., 2006).
We speculate that changes in deterministic variability in the cardio-respiratory patterns associated with systemic inflammation (Figure 2B3) are related to expression of cytokines in the nucleus tractus solitarius (nTS); which result from vagal input and which “maps” peripheral inflammation. We have preliminary evidence that acute lung injury results in specific expression of IL-1β in the commissural subnucleus in the nTS (Jacono et al., 2011; Figure 2A, serology in lower panel and Figure 2C, red fluorescent staining). This expression occurred in the absence of significant increases IL-1β, TNFα, and IL-6 in the plasma (Jacono et al., 2011). We have focused on the vagal afferent limb in controlling biologic pattern variability but this neural feedback lies in the context of its own control loop. The respiratory CPG, a major determinant of vagal efferent activity, modulates variability but may also regulate the expression of cytokines. Vagal efferent activity decreases peripheral levels of cytokines, acting as a negative feedback modulating the magnitude of the inflammatory response (Andersson and Tracey, 2012). However, the brainstem connectivity between the CPG network and the immunologic loop is unknown. In particular, it is unknown if the network that mediates biologic variability also modulates the magnitude of the inflammatory response. In defining this network, we speculate that this loop is affected by central expression of cytokines and, thus, has a role in determining the “tipping point” and systemic dysfunction.
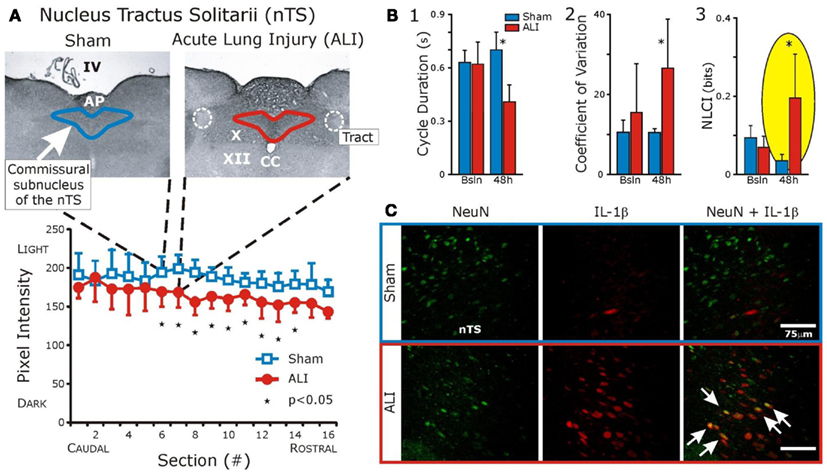
Figure 2. Interleukin-1β expression is increased in the commissural subnucleus of the nTS in the setting of acute lung injury (ALI) and altered ventilatory pattern. Bleomycin (three units) was instilled intratracheally causing ALI. Ventilatory pattern was measured and tissues were obtained 48 h later. (A) Histologic examination identified a significant increase in IL-1β in the commissural subnucleus of the nucleus Tractus Solitarius (nTS) in the dorsomedial medulla (shown). Abbreviations: AP, area postrema; CC, central canal; IV, fourth ventricle; X, dorsal motor nucleus of the vagus; XII, hypoglossal motor nucleus; white dashed line, solitary tract. (B) Compares ventilatory patterns of sham (blue) and ALI (red) rats at baseline and 48 h. After ALI: (1) Increased respiratory rate (significant decrease in cycle duration, TTOT), (2) Increased coefficient of variation (CV) of respiratory cycle length (CV of TTOT), and (3) Increased deterministic non-linear variability of the ventilatory pattern, as measured by a non-linear complexity index (NLCI, yellow highlight) computed using surrogate data analysis. (C) Fluorescent staining: IL-1β co-localized with nTS neurons (white arrows) identified using antibodies against the neuronal specific nuclear protein NeuN. (Adapted from; Wysocki et al., 2006).
Further, improved understanding of the feedback loops involved in the neural control of inflammation will serve to identify novel targets for modulation of the immune response. For example, imposing rhythmic vagal afferent activity by mechanical ventilation may impact the neural and peripheral immune responses. Thus, one potential benefit of biologically variable ventilation is that it imposes a sensory input related to lung inflation that artificially replicates a variable “normal” signal. This may predispose the CPG to produce “normal” pattern that, perhaps, reduces inflammation. If biologic variability of rhythmic patterns decreases the cytokine concentration as compared to the “autonomous regime” then cardio-respiratory coupling and the variability in the bursting pattern of its activity, may prevent MODS and the inflammatory response from reaching its “tipping point.”
Variability is a property of the biological systems we have studied, that in and of itself is neither good nor bad, but may reflect a “state” of the system that is adaptable and responsive (within boundaries). HRV is diminished in various inflammatory conditions, including experimental human endotoxemia. Recent studies have investigated HRV as a predictive metric in trauma patients (Morris et al., 2007; Cancio et al., 2008; Riordan et al., 2009; Batchinsky et al., 2010) and sepsis (Ahmad et al., 2009; Moorman et al., 2011). Although correlations of HRV with disease state have produced many important insights, the physiological processes linking inflammation with systemic changes are not well defined, motivating the development of mathematical models linking inflammation, and HRV (Buchman, 2009; Foteinou et al., 2010; Scheff et al., 2011a; Namas et al., 2012). We linked multilevel dynamics of the molecular and cellular patterns occurring in human endotoxemia to cardiac function through a model that outputs discrete heart beats as modulated by physiological rhythms, which can then be post-processed to assess HR and HRV (Scheff et al., 2011a). Action potentials at the sinoatrial (SA) node of the heart initiate the contraction of cardiac tissue, producing rhythmic heart beats. Sympathetic and parasympathetic nerves converging at the SA node result in fluctuating levels of autonomic neurotransmitters which modulate the firing pattern of SA node cells. Variability in the firing of the SA node, and thus variability in the beating of the heart, is regulated by autonomic output. Thus, we proposed a model of autonomic activity at the SA node, influenced by the inflammatory mediators in our model of human endotoxemia, to modulate the HRV (Scheff et al., 2011a). Our discrete modeling of heart beats allowed the calculation of HRV metrics used in the clinical setting. The diversity of data supporting this modeling work at transcriptional, hormonal, and systemic levels was matched by appropriate computational modeling techniques to allow for the assessment of autonomic dysfunction, manifested in altered HR rhythmicity, in human endotoxemia. We have demonstrated that stress induces alterations in the homeostatic dynamics of the feedback structures such that some level of perturbation permanently disrupts these regulatory structures; i.e., “tipping points.” Progressive crossing of these “tipping points” would lead to cascading systems failure and the clinical syndrome of MODS/sepsis.
We suggest that our evolving mathematical models will help identify and predict potential bioactive interventions, and validate those predictions in further experiments in rodents, swine, and, ultimately, in clinical trials based on our in silico studies. The multi-compartment/multi-scale computational models could be used to test interventions targeted at effector organs, such as ventilation strategies aimed at manipulating the lung. Importantly, despite their limitations of abstraction of relevant biological interactions, mechanistic mathematical models have the potential to allow us to bridge the gap from rodent studies to large-animal studies and eventually to clinical studies (Vodovotz et al., 2006; An et al., 2010). Trans-species and trans-compartment mechanistic and data-driven modeling will also allow us to integrate those inflammatory networks that help drive “tipping point” behavior.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge the support of NIH/NHLBI Exploratory Program in Systems Biology. Many of the researchers who are co-authors on this review chapter were supported by this program and specific awards include R33HL087377 (Thomas E. Dick, Yaroslav I. Molkov, Yee-Hsee Hsieh, Frank J. Jacono.) and R33-HL-089082 (Gary Nieman, Yoram Vodovotz). In addition, the authors would like to acknowledge additional grant support from NIH (R01GM67240, P50GM53789, R01HL080926, R01HL76157, UO1DK072146, RO1GM082974, RO1GM34695); National Science Foundation grant 0830-370-V601; Award No. I01BX000873 from the Biomedical Laboratory Research and Development Service of the VA Office of Research and Development; a Shared University Research Award from IBM, Inc.; and grants from the Commonwealth of Pennsylvania, the Pittsburgh Lifesciences Greenhouse, and the Pittsburgh Tissue Engineering Initiative/Department of Defense. The authors would like to acknowledge Dr. IlyaRybak (Drexel University, Department of Neurobiology and Anatomy) for helpful discussion and seminar contributions to the modeling of neural control of breathing patterns.
Abraham, E., and Singer, M. (2007). Mechanisms of sepsis-induced organ dysfunction. Crit. Care Med. 35, 2408.
Ahmad, S., Ramsay, T., Huebsch, L., Flanagan, S., McDiarmid, S., Batkin, I., McIntyre, L., Sundaresan, S. R., Maziak, D. E., Shamji, F. M., Hebert, P., Fergusson, D., Tinmouth, A., and Seely, A. J. (2009). Continuous multi-parameter heart rate variability analysis heralds onset of sepsis in adults. PLoS ONE 4, e6642. doi:10.1371/journal.pone.0006642
Aird, W. C. (2003). The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101, 3765.
Ait-Oufella, H., Maury, E., Lehoux, S., Guidet, B., and Offenstadt, G. (2010). The endothelium: physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 36, 1286.
An, G. (2008). Introduction of a agent based multi-scale modular architecture for dynamic knowledge representation of acute inflammation. Theor. Biol. Med. Model. 5. doi:10.1186/1742-4682-5-11
An, G., Bartels, J., and Vodovotz, Y. (2010). In silico augmentation of the drug development pipeline: examples from the study of acute inflammation. Drug Dev. Res. 72, 1.
An, G., Faeder, J., and Vodovotz, Y. (2008). Translational systems biology: introduction of an engineering approach to the pathophysiology of the burn patient. J. Burn Care Res. 29, 277.
An, G., Mi, Q., Dutta-Moscato, J., Solovyev, A., and Vodovotz, Y. (2009). Agent-based models in translational systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 159.
An, G., Nieman, G., and Vodovotz, Y. (2012). Computational and systems biology in trauma and sepsis: current state and future perspectives. Int. J. Burns Trauma 2, 1.
Andersson, U., and Tracey, K. J. (2012). Reflex principles of immunological homeostasis. Annu. Rev. Immunol. 30, 313–335.
Angus, D. C. (2011). The search for effective therapy for sepsis: back to the drawing board? JAMA 306, 2614.
Angus, D. C., Linde-Zwirble, W. T., Lidicker, J., Clermont, G., Carcillo, J., and Pinsky, M. R. (2001). Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303.
Annane, D., Trabold, F., Sharshar, T., Jarrin, I., Blanc, A. S., Raphael, J. C., and Gajdos, P. (1999). Inappropriate sympathetic activation at onset of septic shock: a spectral analysis approach. Am. J. Respir. Crit Care Med. 160, 458.
Anonymous. (1990). Increase in National Hospital Discharge Survey rates for septicemia – United States, 1979–1987. MMWR Morb. Mortal. Wkly. Rep. 39, 31.
Anonymous. (1996). Heart rate variability. Standards of measurement, physiological interpretation, and clinical use. Task Force of the European Society of Cardiology, and the North American Society of Pacing, and Electrophysiology. Eur. Heart J. 17, 354.
Baekey, D. M., Molkov, Y. I., Paton, J. F., Rybak, I. A., and Dick, T. E. (2010). Effect of baroreceptor stimulation on the respiratory pattern: insights into respiratory-sympathetic interactions. Respir. Physiol. Neurobiol. 174, 135.
Balestra, G. M., Legrand, M., and Ince, C. (2009). Microcirculation and mitochondria in sepsis: getting out of breath. Curr. Opin. Anaesthesiol. 22, 184.
Barnaby, D., Ferrick, K., Kaplan, D. T., Shah, S., Bijur, P., and Gallagher, E. J. (2002). Heart rate variability in emergency department patients with sepsis. Acad. Emerg. Med. 9, 661.
Batchinsky, A. I., Skinner, J. E., Necsoiu, C., Jordan, B. S., Weiss, D., and Cancio, L. C. (2010). New measures of heart-rate complexity: effect of chest trauma and hemorrhage. J. Trauma 68, 1178.
Boujoukos, A. J., Martich, G. D., Supinski, E., and Suffredini, A. F. (1993). Compartmentalization of the acute cytokine response in humans after intravenous endotoxin administration. J. Appl. Physiol. 74, 3027.
Buchman, T. G. (2009). The digital patient: predicting physiologic dynamics with mathematical models. Crit. Care Med. 37, 1167.
Calvano, S. E., Xiao, W., Richards, D. R., Felciano, R. M., Baker, H. V., Cho, R. J., Chen, R. O., Brownstein, B. H., Cobb, J. P., Tschoeke, S. K., Miller-Graziano, C., Moldawer, L. L., Mindrinos, M. N., Davis, R. W., Tompkins, R. G., Lowry, S. F., Large Scale Collab. Res. Program. (2005). A network-based analysis of systemic inflammation in humans. Nature 437, 1032.
Cancio, L. C., Batchinsky, A. I., Salinas, J., Kuusela, T., Convertino, V. A., Wade, C. E., and Holcomb, J. B. (2008). Heart-rate complexity for prediction of prehospital lifesaving interventions in trauma patients. J. Trauma 65, 813.
Cavaillon, J. M., and Annane, D. (2006). Compartmentalization of the inflammatory response in sepsis and SIRS. J. Endotoxin Res. 12, 151.
Chen, W. L., and Kuo, C. D. (2007). Characteristics of heart rate variability can predict impending septic shock in emergency department patients with sepsis. Acad. Emerg. Med. 14, 392.
Cherniack, N. S., and Longobardo, G. S. (2006). Mathematical models of periodic breathing and their usefulness in understanding cardiovascular and respiratory disorders. Exp. Physiol. 91, 295.
Cherniack, N. S., Longobardo, G. S., Levine, O. R., Mellins, R., and Fishman, A. P. (1966). Periodic breathing in dogs. J. Appl. Physiol. 21, 1847.
Chow, C. C., Clermont, G., Kumar, R., Lagoa, C., Tawadrous, Z., Gallo, D., Betten, B., Bartels, J., Constantine, G., Fink, M. P., Billiar, T. R., and Vodovotz, Y. (2005). The acute inflammatory response in diverse shock states. Shock 24, 74.
Coogan, A. N., and Wyse, C. A. (2008). Neuroimmunology of the circadian clock. Brain Res. 1232, 104.
Cotton, B. A., Guy, J. S., Morris, J. A. Jr., and Abumrad, N. N. (2006). The cellular, metabolic, and systemic consequences of aggressive fluid resuscitation strategies. Shock 26, 115.
Csete, M. E., and Doyle, J. C. (2002). Reverse engineering of biological complexity. Science 295, 1664.
Daun, S., Rubin, J., Vodovotz, Y., Roy, A., Parker, R., and Clermont, G. (2008). An ensemble of models of the acute inflammatory response to bacterial lipopolysaccharide in rats: results from parameter space reduction. J. Theor. Biol. 253, 843.
Desvergne, B., and Heligon, C. (2009). Steroid hormone pulsing drives cyclic gene expression. Nat. Cell Biol. 11, 1051.
Dhingra, R. R., Jacono, F. J., Fishman, M., Loparo, K. A., Rybak, I. A., and Dick, T. E. (2011). Vagal-dependent nonlinear variability in the respiratory pattern of anesthetized, spontaneously breathing rats. J. Appl. Physiol. 111, 272.
Fairchild, K. D., Saucerman, J. J., Raynor, L. L., Sivak, J. A., Xiao, Y., Lake, D. E., and Moorman, J. R. (2009). Endotoxin depresses heart rate variability in mice: cytokine and steroid effects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1019.
Ferguson, N. D., Frutos-Vivar, F., Esteban, A., Anzueto, A., Alia, I., Brower, R. G., Stewart, T. E., Apezteguia, C., Gonzalez, M., Soto, L., Abroug, F., and Brochard, L. (2005). Airway pressures, tidal volumes, and mortality in patients with acute respiratory distress syndrome. Crit. Care Med. 33, 21.
Foteinou, P. T., Calvano, S. E., Lowry, S. F., and Androulakis, I. P. (2009a). Modeling endotoxin-induced systemic inflammation using an indirect response approach. Math. Biosci. 217, 27.
Foteinou, P., Calvano, S., Lowry, S., and Androulakis, I. P. (2009b). Translational potential of systems-based models of inflammation. Clinical and Translational Science 2, 85–89.
Foteinou, P. T., Calvano, S. E., Lowry, S. F., and Androulakis, I. P. (2010). Multiscale model for the assessment of autonomic dysfunction in human endotoxemia. Physiol. Genomics 42, 5.
Godin, P. J., Fleisher, L. A., Eidsath, A., Vandivier, R. W., Preas, H. L., Banks, S. M., Buchman, T. G., and Suffredini, A. F. (1996). Experimental human endotoxemia increases cardiac regularity: results from a prospective, randomized, crossover trial. Crit. Care Med. 24, 1117.
Grzegorczyk, M., and Husmeier, D. (2011). Improvements in the reconstruction of time-varying gene regulatory networks: dynamic programming and regularization by information sharing among genes. Bioinformatics 27, 693–699.
Heron, M., Hoyert, D. L., Murphy, S. L., Xu, J., Kochanek, K. D., and Tejada-Vera, B. (2009). Deaths: final data for 2006. Natl. Vital Stat. Rep. 57, 1.
Jacono, F. J., Mayer, C. A., Hsieh, Y. H., Wilson, C. G., and Dick, T. E. (2011). Lung and brainstem cytokine levels are associated with breathing pattern changes in a rodent model of acute lung injury. Respir. Physiol. Neurobiol. 178, 429.
Kleiger, R. E., Stein, P. K., and Bigger, J. T. Jr. (2005). Heart rate variability: measurement and clinical utility. Ann. Noninvasive Electrocardiol. 10, 88.
Korach, M., Sharshar, T., Jarrin, I., Fouillot, J. P., Raphael, J. C., Gajdos, P., and Annane, D. (2001). Cardiac variability in critically ill adults: influence of sepsis. Crit. Care Med. 29, 1380.
Lagoa, C. E., Bartels, J., Baratt, A., Tseng, G., Clermont, G., Fink, M. P., Billiar, T. R., and Vodovotz, Y. (2006). The role of initial trauma in the host’s response to injury and hemorrhage: insights from a comparison of mathematical simulations and hepatic transcriptomic analysis. Shock 26, 592.
Lightman, S. L., and Conway-Campbell, B. L. (2010). The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat. Rev. Neurosci. 11, 710.
Martin, G. S., Mannino, D. M., Eaton, S., and Moss, M. (2003). The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546.
Mi, Q., Constantine, G., Ziraldo, C., Solovyev, A., Torres, A., Namas, R., Bentley, T., Billiar, T. R., Zamora, R., Puyana, J. C., and Vodovotz, Y. (2011). A dynamic view of trauma/hemorrhage-induced inflammation in mice: principal drivers and networks. PLoS ONE 6, e19424. doi:10.1371/journal.pone.0019424
Mi, Q., Li, N. Y. K., Ziraldo, C., Ghuma, A., Mikheev, M., Squires, R., Okonkwo, D. O., Verdolini Abbott, K., Constantine, G., An, G., and Vodovotz, Y. (2010). Translational systems biology of inflammation: potential applications to personalized medicine. Per. Med. 7, 549.
Mitka, M. (2011). Drug for severe sepsis is withdrawn from market, fails to reduce mortality. JAMA 306, 2439.
Molina, P. E., Bagby, G. J., and Stahls, P. (2001). Hemorrhage alters neuroendocrine, hemodynamic, and compartment-specific TNF responses to LPS. Shock 16, 459.
Molkov, Y. I., Abdala, A. P., Bacak, B. J., Smith, J. C., Paton, J. F., and Rybak, I. A. (2010). Late-expiratory activity: emergence and interactions with the respiratory CpG. J. Neurophysiol. 104, 2713.
Molkov, Y. I., Zoccal, D. B., Moraes, D. J., Paton, J. F., MacHado, B. H., and Rybak, I. A. (2011). Intermittent hypoxia-induced sensitization of central chemoreceptors contributes to sympathetic nerve activity during late expiration in rats. J. Neurophysiol. 105, 3080.
Moorman, J. R., Carlo, W. A., Kattwinkel, J., Schelonka, R. L., Porcelli, P. J., Navarrete, C. T., Bancalari, E., Aschner, J. L., Whit, W. M., Perez, J. A., Palmer, C., Stukenborg, G. J., Lake, D. E., and Michael, O. T. (2011). Mortality reduction by heart rate characteristic monitoring in very low birth weight neonates: a randomized trial. J. Pediatr. 159, 900.
Morris, J. A. Jr., Norris, P. R., Waitman, L. R., Ozdas, A., Guillamondegui, O. D., and Jenkins, J. M. (2007). Adrenal insufficiency, heart rate variability, and complex biologic systems: a study of 1,871 critically ill trauma patients. J. Am. Coll. Surg. 204, 885.
Namas, R., Ghuma, A., Torres, A., Polanco, P., Gomez, H., Barclay, D., Gordon, L., Zenker, S., Kim, H. K., Hermus, L., Zamora, R., Rosengart, M. R., Clermont, G., Peitzman, A., Billiar, T. R., Ochoa, J., Pinsky, M. R., Puyana, J. C., and Vodovotz, Y. (2009a). An adequately robust early TNF-α response is a hallmark of survival following trauma/hemorrhage. PLoS ONE 4, e8406. doi:10.1371/journal.pone.0008406
Namas, R., Ghuma, A., Hermus, L., Zamora, R., Okonkwo, D. O., Billiar, T. R., and Vodovotz, Y. (2009b). The acute inflammatory response in trauma/hemorrhage and traumatic brain injury: current state and emerging prospects. Libyan J. Med. 4, 136.
Namas, R., Zamora, R., Namas, R., An, G., Doyle, J., Dick, T. E., Jacono, F. J., Androulakis, I. P., Chang, S., Billiar, T. R., Kellum, J. A., Angus, D. C., and Vodovotz, Y. (2012). Sepsis: something old, something new, and a systems view. J. Crit. Care 27, 314e1–314e11.
Natanson, C. (1997). “Re-evaluation of anti-inflammatory trials in sepsis: a meta-analysis,” in Cytokines and Pulmonary Infection. Part II: The Role of Cytokines in Systemic and Pulmonary Medicine, ed. M. R. Pratter (Chicago: American Thoracic Society), 7–18.
Nieman, G., Brown, D., Sarkar, J., Kubiak, B., Ziraldo, C., Vieau, C., Barclay, D., Gatto, L., Maier, K., Zamora, R., Mi, Q., Chang, S., and Vodovotz, Y. (2012). A two-compartment mathematical model of endotoxin-induced inflammatory and physiologic alterations in swine. Crit. Care Med. 40, 1052–1063.
Parker, S. J., and Watkins, P. E. (2001). Experimental models of gram-negative sepsis. Br. J. Surg. 88, 22.
Piepoli, M., Garrard, C. S., Kontoyannis, D. A., and Bernardi, L. (1995). Autonomic control of the heart and peripheral vessels in human septic shock. Intensive Care Med. 21, 112.
Pomeranz, B., MacAulay, R. J., Caudill, M. A., Kutz, I., Adam, D., Gordon, D., Kilborn, K. M., Barger, A. C., Shannon, D. C., and Cohen, R. J. (1985). Assessment of autonomic function in humans by heart rate spectral analysis. Am. J. Physiol. 248, H151.
Pontet, J., Contreras, P., Curbelo, A., Medina, J., Noveri, S., Bentancourt, S., and Migliaro, E. R. (2003). Heart rate variability as early marker of multiple organ dysfunction syndrome in septic patients. J. Crit. Care 18, 156.
Prince, J. M., Levy, R. M., Bartels, J., Baratt, A., Kane, J. M. III, Lagoa, C., Rubin, J., Day, J., Wei, J., Fink, M. P., Goyert, S. M., Clermont, G., Billiar, T. R., and Vodovotz, Y. (2006). In silico and in vivo approach to elucidate the inflammatory complexity of CD14-deficient mice. Mol. Med. 12, 88.
Protti, A., and Singer, M. (2006). Bench-to-bedside review: potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit. Care 10, 228.
Remick, D., Manohar, P., Bolgos, G., Rodriguez, J., Moldawer, L., and Wollenberg, G. (1995). Blockade of tumor necrosis factor reduces lipopolysaccharide lethality, but not the lethality of cecal ligation and puncture. Shock 4, 89.
Riordan, W. P. Jr., Norris, P. R., Jenkins, J. M., and Morris, J. A. Jr. (2009). Early loss of heart rate complexity predicts mortality regardless of mechanism, anatomic location, or severity of injury in 2178 trauma patients. J. Surg. Res. 156, 283.
Rittirsch, D., Flierl, M. A., and Ward, P. A. (2008). Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8, 776.
Rybak, I. A., O’Connor, R., Ross, A., Shevtsova, N. A., Nuding, S. C., Segers, L. S., Shannon, R., Dick, T. E., Dunin-Barkowski, W. L., Orem, J. M., Solomon, I. C., Morris, K. F., and Lindsey, B. G. (2008). Reconfiguration of the pontomedullary respiratory network: a computational modeling study with coordinated in vivo experiments. J. Neurophysiol. 100, 1770.
Sakr, Y., Vincent, J. L., Reinhart, K., Groeneveld, J., Michalopoulos, A., Sprung, C. L., Artigas, A., and Ranieri, V. M. (2005). High tidal volume and positive fluid balance are associated with worse outcome in acute lung injury. Chest 128, 3098.
Sammon, M. P., and Bruce, E. N. (1991). Vagal afferent activity increases dynamical dimension of respiration in rats. J. Appl. Physiol. 70, 1748.
Scheff, J. D., Calvano, S. E., Lowry, S. F., and Androulakis, I. P. (2010). Modeling the influence of circadian rhythms on the acute inflammatory response. J. Theor. Biol. 264, 1068.
Scheff, J. D., Calvano, S. E., Lowry, S. F., and Androulakis, I. P. (2012). Transcriptional implications of ultradian glucocorticoid secretion in homeostasis and in the acute stress response. Physiol. Genomics 44, 121.
Scheff, J. D., Mavroudis, P. D., Calvano, S. E., Lowry, S. F., and Androulakis, I. P. (2011a). Modeling autonomic regulation of cardiac function and heart rate variability in human endotoxemia. Physiol. Genomics 43, 951.
Scheff, J. D., Kosmides, A. K., Calvano, S. E., Lowry, S. F., and Androulakis, I. P. (2011b). Pulsatile glucocorticoid secretion: origins and downstream effects. IEEE Trans. Biomed. Eng. 58, 3504.
Schein, M., Wittmann, D. H., Holzheimer, R., and Condon, R. E. (1996). Hypothesis: compartmentalization of cytokines in intraabdominal infection [review]. Surgery 119, 694.
Torres, A., Bentley, T., Bartels, J., Sarkar, J., Barclay, D., Namas, R., Constantine, G., Zamora, R., Puyana, J. C., and Vodovotz, Y. (2009). Mathematical modeling of post-hemorrhage inflammation in mice: studies using a novel, computer-controlled, closed-loop hemorrhage apparatus. Shock 32, 172.
Vincent, J. L., Sakr, Y., Sprung, C. L., Ranieri, V. M., Reinhart, K., Gerlach, H., Moreno, R., Carlet, J., Le Gall, J. R., and Payen, D. (2006). Sepsis in European intensive care units: results of the SOAP study. Crit. Care Med. 34, 344.
Vodovotz, Y., and An, G. (2009). “Systems biology and inflammation,” in Systems Biology in Drug Discovery and Development: Methods and Protocols, ed. Q. Yan (Totowa, NJ: Springer Science and Business Media), 181–201.
Vodovotz, Y., Chow, C. C., Bartels, J., Lagoa, C., Prince, J., Levy, R., Kumar, R., Day, J., Rubin, J., Constantine, G., Billiar, T. R., Fink, M. P., and Clermont, G. (2006). In silico models of acute inflammation in animals. Shock 26, 235.
Vodovotz, Y., Clermont, G., Chow, C., and An, G. (2004). Mathematical models of the acute inflammatory response. Curr.Opin.Crit. Care 10, 383.
Vodovotz, Y., Constantine, G., Rubin, J., Csete, M., Voit, E. O., and An, G. (2009). Mechanistic simulations of inflammation: current state and future prospects. Math. Biosci. 217, 1.
Vodovotz, Y., Csete, M., Bartels, J., Chang, S., and An, G. (2008). Translational systems biology of inflammation. PLoS. Comput. Biol. 4, e1000014. doi:10.1371/journal.pcbi.10000141
Weycker, D., Akhras, K. S., Edelsberg, J., Angus, D. C., and Oster, G. (2003). Long-term mortality and medical care charges in patients with severe sepsis. Crit. Care Med. 31, 2316.
Wysocki, M., Cracco, C., Teixeira, A., Mercat, A., Diehl, J. L., Lefort, Y., Derenne, J. P., and Similowski, T. (2006). Reduced breathing variability as a predictor of unsuccessful patient separation from mechanical ventilation. Crit. Care Med. 34, 2076.
Keywords: mathematical model, inflammation, physiologic variability, heart rate variability, neural control
Citation: Dick TE, Molkov YI, Nieman G, Hsieh Y-H, Jacono FJ, Doyle J, Scheff JD, Calvano SE, Androulakis IP, An G and Vodovotz Y (2012) Linking inflammation, cardiorespiratory variability, and neural control in acute inflammation via computational modeling. Front. Physio. 3:222. doi: 10.3389/fphys.2012.00222
Received: 29 February 2012; Accepted: 03 June 2012;
Published online: 02 July 2012.
Edited by:
Raimond L. Winslow, Johns Hopkins University, USAReviewed by:
Zhihui Wang, School of Medicine, University of New Mexico, USACopyright: © 2012 Dick, Molkov, Nieman, Hsieh, Jacono, Doyle, Scheff, Calvano, Androulakis, An and Vodovotz. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Yoram Vodovotz, Department of Surgery, University of Pittsburgh, W944 Starzl Biomedical Sciences Tower, 200 Lothrop Street, Pittsburgh, PA 15213, USA. e-mail:dm9kb3ZvdHp5QHVwbWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.