 Yedidya Saiman and Scott L. Friedman*
Yedidya Saiman and Scott L. Friedman*- Division of Liver Diseases, Department of Medicine, Mount Sinai School of Medicine, New York, NY, USA
Chemokines are small molecular weight proteins primarily known to drive migration of immune cell populations. In both acute and chronic liver injury, hepatic chemokine expression is induced resulting in inflammatory cell infiltration, angiogenesis, and cell activation and survival. During acute injury, massive parenchymal cell death due to apoptosis and/or necrosis leads to chemokine production by hepatocytes, cholangiocytes, Kupffer cells, hepatic stellate cells, and sinusoidal endothelial cells. The specific chemokine profile expressed during injury is dependent on both the type and course of injury. Hepatotoxicity by acetaminophen for example leads to cellular necrosis and activation of Toll-like receptors while the inciting insult in ischemia reperfusion injury produces reactive oxygen species and subsequent production of pro-inflammatory chemokines. Chemokine expression by these cells generates a chemoattractant gradient promoting infiltration by monocytes/macrophages, NK cells, NKT cells, neutrophils, B cells, and T cells whose activity are highly regulated by the specific chemokine profiles within the liver. Additionally, resident hepatic cells express chemokine receptors both in the normal and injured liver. While the role of these receptors in normal liver has not been well described, during injury, receptor up-regulation, and chemokine engagement leads to cellular survival, proliferation, apoptosis, fibrogenesis, and expression of additional chemokines and growth factors. Hepatic-derived chemokines can therefore function in both paracrine and autocrine fashions further expanding their role in liver disease. More recently it has been appreciated that chemokines can have diverging effects depending on their temporal expression pattern and the type of injury. A better understanding of chemokine/chemokine receptor axes will therefore pave the way for development of novel targeted therapies for the treatment of liver disease.
Introduction
Chemokines are small molecular weight proteins (8–13 kDa) initially identified by their ability to provide migratory cues to inflammatory cells (Wasmuth et al., 2010). Their expression is up-regulated in nearly all forms of injury in all tissues, leading to infiltration by immune cells. To date, more than 50 different chemokines and 20 chemokine receptors have been identified (Bromley et al., 2008). Most chemokine receptors engage more than one ligand leading to redundancy in chemokine signaling and divergent outcomes following signaling through a single receptor (Bromley et al., 2008).
Chemokines are subdivided into two broad categories based on their amino acid sequences. CC chemokines contain two adjacent N-terminus cysteine residues while the N-terminal cysteines in CXC chemokines are separated by a single amino acid. These cysteine residues form disulfide bonds with additional internal cysteines providing tertiary structure to the protein. Two additional categories, C- and CX3C-chemokines have either a single N-terminus cysteine or two cysteine residues separated by three amino acids, respectively. Additionally, CXC chemokines can be further subdivided based on specific sequence motifs (ELR positive/negative) that impart specific functional properties to the chemokine (Bajetto et al., 2002; Fernandez and Lolis, 2002; Oo et al., 2010; Wasmuth et al., 2010).
The field of chemokine biology has rapidly progressed and additional functions are recognized well beyond immune cell migration. Chemokine receptors have been identified on non-immune cells, and as a result, their roles have expanded to include organ homeostasis and non-inflammatory aspects of injury (Rossi et al., 1999; Shibuta et al., 2002; Zlotnik et al., 2011). The functions of chemokines have therefore expanded to include: cellular differentiation, survival, proliferation, and apoptosis in addition to the ability to modulate development, organ fibrogenesis, vascular angiogenesis, and tumor metastasis (Shibuta et al., 2002; Hong et al., 2009; Zlotnik et al., 2011; Mukaida and Baba, 2012).
For the purposes of this review we discuss chemokine responses in both immune cells and liver parenchymal cells. Although infiltrating immune cells secrete chemokines, the primary sources in the liver are hepatocytes, Kupffer cells, stellate cells, sinusoidal endothelial cells, and biliary epithelial cells. Together, these cells secrete an array of chemokines that drive immune cell infiltration, development of chronic inflammation, liver injury and regeneration, and progression and resolution of fibrosis (Karlmark et al., 2008; Oo et al., 2010; Wasmuth et al., 2010). The numerous and often disparate functions of chemokines in the liver reflect the divergent temporal expression of these molecules and their receptors by immune and resident hepatic cells.
The regulation of both chemokine and receptor expression is modulated by a range of stimuli including growth factors (Gerritsma et al., 1998), cytokines (Harvey et al., 2003), cellular stressors (bile acids, ROS, etc.; Friedman, 2008a,b; Steib et al., 2010), cellular activation by apoptotic bodies (Zernecke et al., 2009), and release of cellular debris from necrotic cells (Jaeschke et al., 2002). Chemokines function as paracrine signals and in autocrine loops, with both positive and negative feedback elements. The complexity of these networks is immense and therefore we highlight herein those aspects that are most instructive in clarifying chemokine biology in acute liver injury (ALI; Table 1).
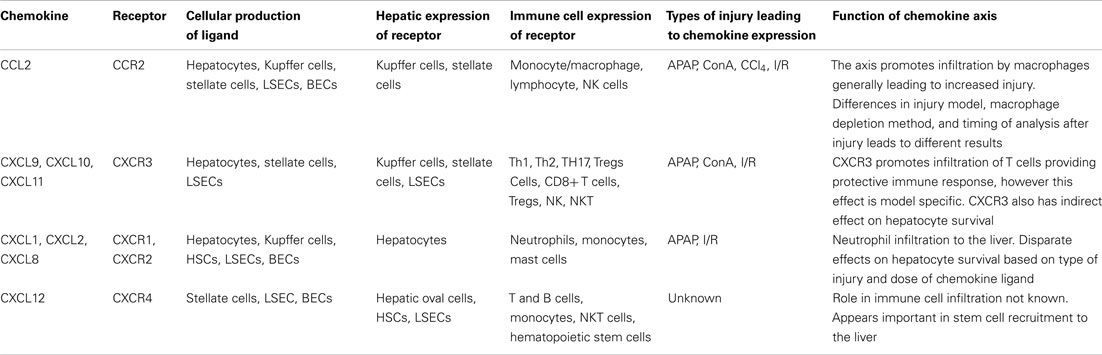
Table 1. Cellular expression and function of chemokines in acute liver injury.
CCL2 Promotes Aonocyte/Macrophage Recruitment and Cytokine Production during Acute Liver Injury
CCL2 (MCP-1) is among the most extensively studied chemokines in liver injury. Its primary role is the recruitment of monocytes and macrophages via its receptor, CCR2, but NK cells and lymphocytes also express CCR2 and migrate in response to CCL2 (Hokeness et al., 2005; Karlmark et al., 2008). Intrahepatic and serum levels of CCL2 are increased in patients with fulminant hepatic failure and also in murine models of acute liver failure (Possamai et al., 2010). Injured hepatocytes and activated Kupffer cells are thought to be the primary sources of hepatic CCL2, however, hepatic stellate cells and liver sinusoidal endothelial cells can also secrete this molecule (Leifeld et al., 2003; Friedman, 2008b; Kolios et al., 2008; Chen et al., 2010; Figure 1).
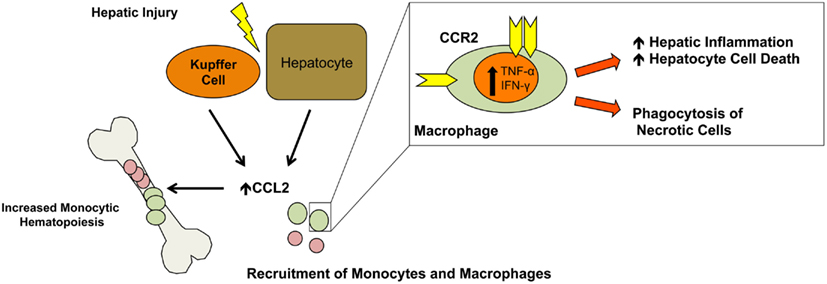
Figure 1. CCL2 monocyte/macrophage recruitment. In response to injury, hepatocytes and Kupffer cells secrete CCL2 leading to liver infiltration by monocytes and macrophages from the periphery. Additionally, CCL2 promotes monocytic hematopoiesis in the bone marrow increasing the pool of circulating monocytes/macrophages. In the liver, macrophages secrete TNF-α and IFN-γ promoting inflammation and hepatocyte cell death while also removing necrotic cells which is important for liver remodeling and return to normal function.
The role of CCL2 in liver injury is twofold. First, as CCL2 levels in the serum rise, it stimulates monocytic hematopoiesis and increased differentiation and production of the monocyte/macrophage lineages (Leifeld et al., 2003; Friedman, 2008b; Kolios et al., 2008; Chen et al., 2010). Furthermore, CCL2 levels in the liver are greater than in the blood, establishing a gradient driving macrophage egress from the bone marrow (Dambach et al., 2002; Tsou et al., 2007; Karlmark et al., 2009). Moreover, it has been shown that it predominantly recruits the inflammatory Gr1 high monocyte-derived macrophage subpopulation (Karlmark et al., 2009). Macrophages have distinctive roles in initiation, propagation, and resolution of ALI. Their involvement is critical during the initial inflammatory stages by phagocytosing necrotic cells, secreting cytokines and growth factors, and recruiting inflammatory cells (Duffield, 2003). Additionally, macrophages assist in the reparative phase leading to hepatic remodeling and the return to normal liver function (Duffield et al., 2005).
Macrophage derived chemokines, cytokines, and growth factors can influence hepatocyte function and recruitment of additional immune cell populations. TNF-α which leads to hepatocyte cell death, is increased early in acetaminophen (APAP) injury and is secreted predominantly by macrophages but also hepatocytes and other immune cells (Hassan et al., 2007). Additionally, IFN-γ, a pro-inflammatory cytokine released by macrophages as well as CCR2-dependent T cells and NKT cells, may promote injury by stimulating hepatic inflammation and amplifying liver damage (Hogaboam et al., 2000; Dambach et al., 2002). Loss of IFN-γ signaling in both KO mice or mice treated with IFN-γ neutralizing antibodies demonstrates a protective effect in APAP or ConA induced ALI (Jaruga et al., 2004; Dong et al., 2005). Interestingly, IFN-γ is important in regulation of the CXCL9–11/CXCR3 chemokine axis, which is reviewed below.
Given the dependence of macrophage recruitment on the CCL2/CCR2 axis, many groups have utilized monocyte/macrophage depletion studies to establish the relevance of the CCL2/CCR2 axis. While the overall role of CCL2 recruited macrophages in ALI appears to be protective, conflicting reports have made it difficult to clearly define the role of CCL2 during injury (Laskin et al., 1995; Michael et al., 1999; Hogaboam et al., 2000; Dambach et al., 2002; Ju et al., 2002; Holt et al., 2008; Karlmark et al., 2009). The method of macrophage depletion (clodronate, gadolinium chloride, CD11b DTR), the type of injury (APAP, CCl4, Ischemia reperfusion), and the extent of injury all influence the interpretation of how macrophages affect liver function.
One technical concern is that many macrophage depletion methods affect other cell lineages including dendritic cells and bone marrow hematopoietic cells. Additionally, as a result of strain differences and the dependence of bone marrow stem cells egress on CCL2/CCR2, global knockouts of this axis have failed to delineate the specific role of hepatic-derived CCL2 during injury. Overall, it appears that loss of CCL2 function perturbs monocyte/macrophage infiltration into the injured liver with a concomitant dampening of pro-inflammatory cytokine production. Therefore, future studies will need to utilize conditional CCL2 or CCR2 knockouts from specific hepatic and immune cell populations in order to determine the cellular populations involved in ALI.
Macrophages recruited by CCL2 are involved in both the initial stages of injury during ALI and the reparative stages. However, little data exists on the role of macrophages during liver regeneration or during restoration after acute injury. In murine models of chronic liver injury and fibrosis, there exist two waves of macrophage infiltration responsible for both disease progression and regression (although this is not CCL2-dependent; Duffield et al., 2005). Similar studies have not been undertaken in ALI models, which might shed additional light on the role of CCL2 and infiltrating macrophages in ALI.
The concept of multiple waves of macrophage has also been shown in patients with ALI. After APAP-induced acute liver failure there is an increase in hepatic CCL2 which directly correlated with clinical outcomes. Furthermore, there is a specific and marked reduction in circulating monocytes which negatively correlated with the hepatic CCL2 levels indicating that their recruitment into the inflamed liver is via CCR2. In areas of liver necrosis there was a predominance proliferating resident Kupffer cells. This study indicates that the appearance of macrophages early after injury are derived from circulating monocytes and resident proliferating Kupffer cells represent a second wave implicated in organ resolution (Antoniades et al., 2012). While not definitive, these results impressively mimic rodent models of macrophage recruitment in rodent models.
While the majority of studies of CCL2 have focused on its effect on monocytes and macrophages, it can also recruit/activate T cell and NKT cell populations, albeit to a lesser extent. Neutralization of the CCL2/CCR2 axis in the T cell-mediated ConA injury model leads to an increase in hepatic injury, indicating that, in this model, CCL2 may be anti-inflammatory (Ajuebor et al., 2003). Mice treated with CCR2 neutralizing serum showed a decrease in TNF-α and IFN-γ, but worse hepatitis and an unexpected and significant increase in IL-4, representing a shift from a Th1 to a Th2 response. ConA treatment increased CCR2 expression on resident hepatic NKT cells and receptor engagement dampened their secretion of IL-4. Therefore, blocking this axis actually increased the expression of IL-4, indicating that CCL2 may also function in an anti-inflammatory manner.
The only resident liver cells to express CCR2 are Kupffer cells and hepatic stellate cells (Friedman, 2008b; Krohn et al., 2009). As noted previously, Kupffer cells are an important source of CCL2 whose production is stimulated by ROS. The specific role of resident hepatic cell CCR2 in ALI has not been demonstrated, but in models of chronic liver injury, the stellate cell’s response to CCL2 is important in the production of ROS, cellular chemotaxis, and the acquisition of a pro-fibrogenic phenotype. Given the limited data regarding the role of hepatic stellate cells during ALI, additional work is required to determine the role of CCL2 on resident hepatic cell function during acute injury.
CXCL9–11 Recruits T Cell Populations Involved in Immune Modulation
The CXCL9–11/CXCR3 axis is unique based on the alternative regulation of its three ligands, which are all induced by IFN-γ. CXCR3 is primarily expressed on CD4+ Th1 helper cells, CD8+ cytotoxic lymphocytes, and innate lymphocytes including NK cells, NKT cells, and dendritic cell subsets. During activation by professional antigen presenting cells, CXCR3 is rapidly up-regulated on leukocyte populations (Groom and Luster, 2011; Figure 2).
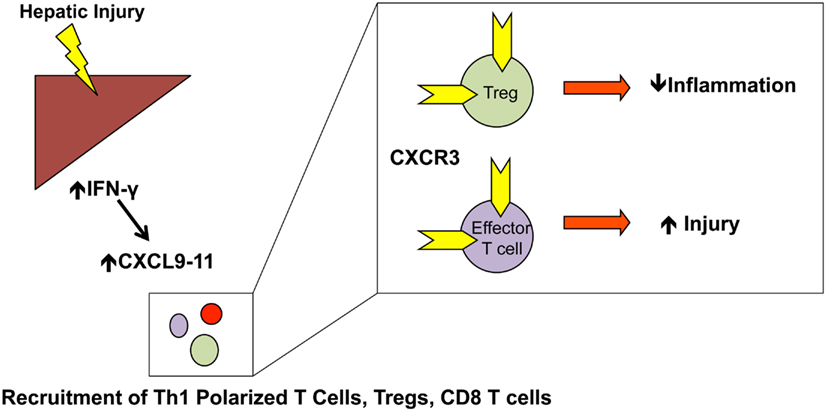
Figure 2. CXCL9–11 T cell recruitment: CXCL9–11 expression is increased in an IFN-γ dependent manner. Numerous T cell populations, including Th1-polarized T cells, Tregs, and effector T cells are recruited to the liver in a CXCR3-dependent manner. The specific type of injury will determine the relative recruitment of Tregs vs. effector T cells and the protective/injurious role of the CXCL9–11/CXCR3 axis.
Similar to other chemokine axes, CXCL9–11 signaling is characterized by significant redundancy, however each of the chemokines has a distinctive role. Expression of all three ligands is mediated by IFN-γ, but the identification of unique promoter elements for each ligand leads to a distinct spatial and temporal expression pattern. Briefly, CXCL9 is dependent solely on IFN-γ whereas CXCL11 can also be induced by IFN-β and CXCL10 by both IFN-α/IFN-β and NF-κB activation. Hepatocytes, stellate cells, sinusoidal endothelial cells, and activated infiltrating lymphocytes all secrete CXCR3 ligands in response to these stimuli. Differences in the sensitivity of each cell type to IFN-γ signaling along with the differences in the ligand promoters leads to the differential expression patterns seen in liver disease (Luster et al., 1985; Farber, 1990;Ohmori and Hamilton, 1993, 1997;Ohmori et al., 1993, 1997; Cole et al., 1998; Majumder et al., 1998; Tensen et al., 1999; Medoff et al., 2006; Yang et al., 2007).
In patients with chronic liver disease, hepatic levels of both CXCR3 and its ligands are increased, and CXCR3 ligand levels correlate with the severity of disease (Apolinario et al., 2002). Additionally, in mouse models of chronic disease, CXCR3 is protective through several mechanisms. While no human data exists on the role of CXCR3 in ALI, CXCR3-deficient mice point to its being hepatoprotective (Hokeness et al., 2007; Zaldivar et al., 2012). Th1-polarized T cells, which provide a protective immune response, are rapidly recruited to the liver after injury in a CXCR3-dependent manner. Additionally, a large subset of Foxp3+/CD25+ regulatory T cells (Tregs) found in the liver during injury are CXCR3+, and this population is diminished in CXCR3-deficient mice. Loss of CXCR3 therefore affects recruitment of both effector T cell populations and T regulatory cells (Erhardt et al., 2011). The balance between these populations influences the outcome following blockade of this axis and may explain the contrasting results seen in different experimental models.
Ischemia reperfusion (I/R) injury is a major concern following liver transplantation and is an important contributor to liver rejection in this setting. After I/R, levels of CXCL9–11 are increased early after injury leading to infiltration of CXCR3+ T cells. Blocking this interaction in rats decreases hepatocellular damage and increases survival. Similarly, studies of cardiac and islet cell allografts indicate that blocking CXCL10 and CXCL11 prolongs survival and prevents acute allograft rejection by inhibiting the recruitment of effector T cells (Horiguchi et al., 2002).
Tregs have important functions in controlling immune cell activation within the liver. Patients with autoimmune hepatitis, as well as autoimmune liver disease, have decreased numbers of Tregs. In the T cell-mediated ConA injury model, CXCL9–11 expression is increased and mice deficient for CXCR3 are significantly more susceptible to ConA injury. Similar to other models, the total number of CXCR3+ T cells was decreased as were the number of hepatic Tregs. However, in this model, the decrease in infiltrating CXCR3+ effector T cells does not compensate for the loss of hepatic Tregs, and nonetheless leads to increased injury and lethality. Consistent with this finding, the number of Tregs in the spleen and lymph nodes were increased, indicating that they failed to recruit to the liver in the absence of CXCR3 (Erhardt et al., 2011). Similarly, loss of IFN-γ (and subsequently CXCL9–11 up-regulation) or neutralization of CXCL10 leads to decreased hepatic Tregs and enhanced hepatic inflammation. This effect is partially mediated by resident liver NKT cells, which express high levels of IFN-γ in response to injury. Mice deficient in NKT cells therefore show a marked decrease in Treg recruitment, as do mice deficient for IFN-γ.
These results point to the difficulty of establishing whether a single chemokine is protective or injurious. Undoubtedly, the specific disease process and chemokine/cytokine signature will determine the relative contribution of each cell type, and whether inhibition of the axis will be protective or injurious, further highlighting the multidimensional approach required to understanding the function of any chemokine axis (Ajuebor et al., 2007).
In models of ALI, CXLC10, and CXCR3 are up-regulated upon APAP administration, whereas recombinant CXCL10 is hepatoprotective. The effect of CXCL10 is indirect by increasing hepatocyte expression of CXCR2, which is protective in certain injury models, and blocking CXCR2 abrogates the effect of exogenously administered CXCL10. These results are particularly interesting, as hepatocytes do not express CXCR3, and therefore the up-regulation of CXCR2 on hepatocytes must be regulated by an independent yet unidentified signaling axis (Bone-Larson et al., 2001).
Within the liver CXCR3 is expressed by hepatic stellate cells and liver sinusoidal endothelial cells, and is anti-fibrotic in chronic injury (Shields et al., 1999; Crosby et al., 2009; Wasmuth et al., 2009). CXCR3 ligands have seemingly opposing effects on hepatic stellate cells. CXCL9 decreases collagen mRNA expression leading to a less activated phenotype, while CXCL10 promotes hepatic stellate cell migration (Wasmuth et al., 2009). Such opposing effects may reflect different roles stellate cells play during liver injury or could categorize subpopulations of these cells with divergent functions. Furthermore, CXCL9–11 ligands are angiostatic, and administration of CXCL9 in vivo inhibits neoangiogenisis and prevents development of fibrosis (Sahin et al., 2012).
As a final note, it is important to recognize that there are mouse strain differences in CXCL9–11 expression. C57Bl/6 mice do not express CXCL11 due to a point mutation leading to an early stop codon. Bl/6 mice are still responsive to exogenous CXCL11, but because CXCL9 and CXLC10 knockout mice were backcrossed onto a Bl/6 background it is difficult to distinguish between the function of each ligand (Sahin et al., 2012).
CXCR1/CXCR2 Ligands Promote Neutrophil Migration to the Liver and Regulate Hepatocyte Survival and Proliferation
The CXCL1, CXCL2, and CXCL8 chemokine axis is especially important in acute injury given the dependence of neutrophil and macrophage chemotaxis on CXCR1/CXCR2 ligands (Chen et al., 2006; Ishida et al., 2006; Kobayashi, 2008). Several ligands signal through CXCR1/CXCR2, including CXCL8 (IL-8), found only in humans, and CXCL1 and CXCL2 (KC and MIP-2) present in both humans and mice. In patients, CXCL8 levels are increased in alcoholic hepatitis, as well as in APAP overdose, which are predictive of hepatocellular damage (James et al., 2001; Zimmermann et al., 2011). Additionally, post-liver transplantation, patients with elevated serum CXCL8 have higher serum transaminases (Ilmakunnas et al., 2010). In view of the direct correlation between CXCL8 levels and hepatic function after acute injury, much research has focused on the role of CXCR1/CXCR2 in I/R and APAP injury models. Furthermore, this axis is of great interest due to the expression of CXCR1/CXCR2 by hepatocytes, enabling these chemokines to directly affect both acute inflammation and hepatocyte survival/function (Figure 3).
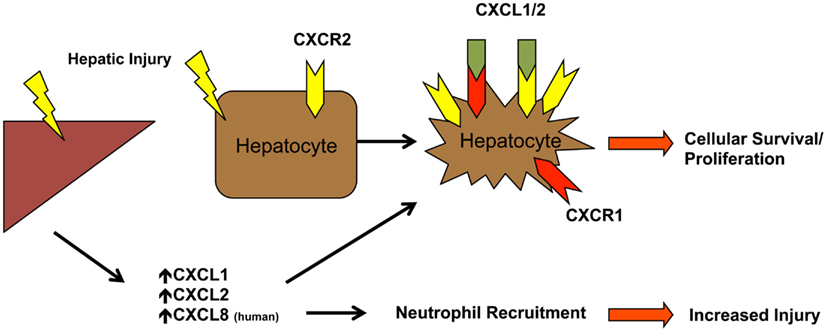
Figure 3. CXCL1/CXCL2/CXCL8 neutrophil recruitment and hepatocyte proliferation. Neutrophils are recruited from the periphery via CXCR2 in response to increased hepatic levels of CXCL1, CXCL2, and CXCL8 (in humans). Hepatic injury also promotes expression of CXCR1 and increased expression of CXCR2 on hepatocytes. Receptor engagement leads to changes in hepatocyte survival and proliferation in a dose dependent mechanism.
During injury most hepatic cells express CXCR1/CXCR2 ligands including hepatocytes, Kupffer cells, stellate cells, endothelial cells, and biliary epithelial cells (Kuboki et al., 2008). After I/R injury, Kupffer cell activation leads to a release of ROS and subsequent activation of hepatocyte-derived chemokines CXCL1 and CXCL2 (Jaeschke et al., 1991; Jaeschke and Farhood, 2002; Kuboki et al., 2008). Hepatocyte injury due to exposure to ethanol, IL-1, and TNF-α has also been shown to result in CXCL8 expression by human hepatocytes (Stefanovic and Stefanovic, 2006).
Neutrophils express CXCR1 and CXCR2, and increased ligand expression after injury is associated with neutrophil infiltration (Kobayashi, 2008). CXCL8 binds with high affinity to CXCR1 and with lower affinity to CXCR2, which binds an additional six chemokines. Both receptors drive neutrophil chemotaxis, but a neutrophil respiratory burst occurs only through CXCR1, indicating distinct roles for each receptor. A feature of all chemokines, which has been extensively studied in CXCL8, is that they can exist as a monomer, dimer, or a mixture of the two under physiological conditions. These two forms of CXCL8 differentially regulate CXCR1 phosphorylation, desensitization, and receptor internalization, but not that of CXCR2. In general, monomeric CXCL8 shows increased activity via CXCR1, and that in models of lung injury the ability of CXCL8 to reversibly exist as both a monomer and dimer regulates neutrophil chemotaxis and function (Nasser et al., 2009).
The development of effective chemokine gradients is dependent on the interactions between tissue-expressed glycosaminoglycans (GAGs) and chemokines. Monomeric and dimeric chemokine forms have different binding affinities for GAGs, and changes in the monomer/dimer equilibrium will affect the chemokine gradients established within an organ. Chemokine dimerization therefore adds an additional level of regulation to chemokine axes (Gangavarapu et al., 2012).
In models of I/R, receptor inhibition by neutralizing antibodies leads to decreased neutrophil infiltration and less damage overall. Similarly, CXCR2 deficient mice exhibit decreased neutrophil accumulation at 24 h, but not at 96 h, indicating that CXCR2 might be important specifically in early neutrophil infiltration. Knockout mice also exhibit less injury (ALT/AST) and hepatic necrosis (Kuboki et al., 2008). While there is a decrease in neutrophil infiltration, expression levels of CXCR2 chemokines are increased, indicating a potential negative feedback loop through CXCR2. Similarly, in models of APAP-induced injury, loss of CXCR2 leads to decreased neutrophil and macrophage infiltration and less injury (Hogaboam et al., 1999a; Hu and Colletti, 2010). However, when only neutrophils are depleted using a neutrophil specific depleting antibody, the protective effect is even greater. The authors contend that production of iNOS by neutrophils leads to increased injury, but that macrophage derived heme-oxygenase (HO)-1 is protective. CXCR2 KO mice exhibit increased injury compared with the neutrophil depleted mice due to the additional loss of macrophage derived HO-1 (Ishida et al., 2006).
Among the most interesting aspects of this axis is CXCR1/CXCR2 expression by hepatocytes and their role in survival and proliferation. While CXCR2 is expressed under normal physiological conditions and is up-regulated with injury, hepatocyte expression of CXCR1 is detected only after injury. After I/R injury, signaling through CXCR2 is detrimental to hepatocyte proliferation and regeneration. Loss of CXCR2 function leads to an increase in STAT3 and NK-κB signaling which regulates liver regeneration. Furthermore, CXCR1 expression is increased during I/R injury, and while inhibition of CXCR1 does not mitigate early liver injury and neutrophil infiltration, it appears to be involved in the reparative and regenerative phase after I/R injury (Kuboki et al., 2008; Clarke et al., 2011).
Alternatively, in models of APAP injury and partial hepatectomy, CXCR2 activation by recombinant CXCL2 is mitogenic and promotes liver regeneration, while loss of CXCR2 leads to marked liver necrosis and hemorrhaging (Sakai et al., 2011). Currently, the only available treatment for APAP toxicity includes administration of N-acetylcysteine, however, the window of efficacy is only within 8 h after APAP ingestion. Conversely, administration of CXCL2 is only effective if administered 10 h after the initial injury, indicating that its role is to support hepatocyte survival and proliferation after the initial injury (Hu and Colletti, 2010). In another model, adenoviral administration of CXCL2 inhibits neutrophil influx while promoting hepatocyte proliferation after APAP, while CXCL2 inhibition increased liver injury and overall mortality (Hogaboam et al., 1999b).
One explanation for the discrepancies in assessing the effects of this axis on hepatocyte proliferation is that high doses of CXCR2 ligands are hepatotoxic while low doses are hepatoprotective. In vitro studies indicate that hepatocyte treatment with equal concentration of CXCR2 ligands, CXCL1 and CXCL2, may act synergistically and adjusting the relative concentration of each chemokine alters the overall effect (Kuboki et al., 2008). As numerous chemokines signal through CXCR2, differences in injury models may induce unique expression levels of CXCR2 binding chemokines leading to divergent outcomes.
CXCL12 Promotes Hematopoietic Stem Cell Recruitment to the Liver and Neutrophil Egress from the Bone Marrow
CXCL12 (SDF-1α), which binds to the CXCR4 receptor, regulates organ homeostasis and several pathological responses (Nagasawa et al., 1996). CXCL12 is crucial in early embryogenesis, hematopoiesis, and angiogenesis, as well as maintenance of the bone marrow stem cell niche. A unique feature of CXCL12 is its high expression levels even in normal tissues that is further up-regulated with injury. In the uninjured liver, biliary epithelial cells constitutively express CXCL12 and in patients with chronic injury and fibrosis, CXCL12 levels increase in parallel with the extent of fibrotic injury (Wald et al., 2004). In distinction to many other chemokines in the liver, CXCL12 is not expressed by hepatocytes and Kupffer cells, but is localized to biliary epithelial cells, cells of the ductular reaction, hepatic stellate cells, and sinusoidal endothelial cells (Sawitza et al., 2009). Dissecting the specific role of the CXCL12/CXCR4 axis has proven difficult given the lack of a suitable animal model. As mentioned, CXCL12 is crucial for embryogenesis and mice deficient in either CXCL12 or its receptor CXCR4 are not viable due to a lack of cardiac development and a failure of hematopoiesis (Nagasawa et al., 1996; Figure 4).
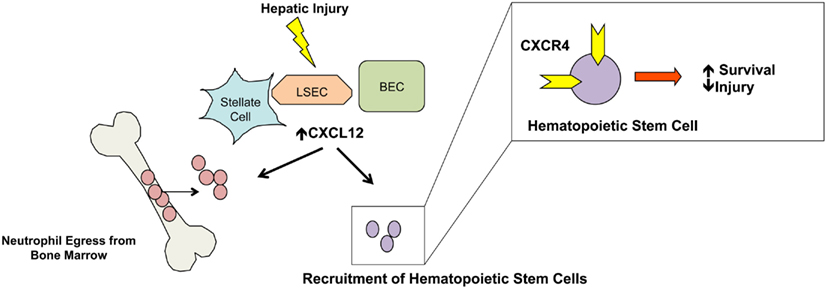
Figure 4. CXCL12 stem cell mobilization and neutrophil egress. Increased expression of CXCL12 by stellate cells, endothelial cells, and biliary epithelial cells promotes migration of bone marrow stem cells to the liver. Additionally, neutrophil egress from the bone marrow is regulated by CXCL12 and increased levels of hepatic and serum levels promotes neutrophil egress. While not depicted, nearly 50% of liver infiltrating cells are CXCR4 positive and localized around CXCL12-rich periportal regions.
Most inflammatory cells, including neutrophils, monocytes, and B and T lymphocytes express CXCR4 and locally produced CXCL12 recruits immune cells to the injured site and promotes angiogenesis (Liekens et al., 2010). Neutrophil regulation and egress from the bone marrow is predominantly CXCR4-dependent and in chronic liver injury over 50% of liver infiltrating cells are CXCR4 positive. Neutrophils contain large cytoplasmic stores of CXCR4, which can be rapidly expressed on the cell surface in response to cytokines, growth factors, or injurious stimuli (Link, 2005; Christopher and Link, 2007; Ramaiah and Jaeschke, 2007). During injury, bone marrow G-CSF levels increase leading to changes in the bone marrow CXCL12/CXCR4 axis and subsequent release of neutrophils (Lei et al., 2010). No study has directly examined the effect of CXCR4-dependent neutrophil migration to the liver during acute injury, but based on studies of CXCR2-dependent neutrophil migration, the inhibition of CXCR4 driven neutrophil migration would be protective (Cao et al., 2000). Alternatively, inhibition of this axis alone may lead to an unfavorable outcome due to increased neutrophil egress from the bone marrow that migrate to the liver.
CXCL12 also plays a role in lymphocyte adhesion, migration, and extravasation into the liver parenchyma. Treatment of lymphocytes with CXCL12 increases ICAM-1 dependent tethering and can enhance tethering and firm adhesion required for transendothelial migration, suggesting a role for endothelial cell-derived CXCL12 (Campbell et al., 1998; Peled et al., 1999; Goddard et al., 2001). Additionally, liver sinusoidal endothelial cells can present abluminal CXCL12 on their luminal cell surface via transcytosis, which leads to T cell adhesion and migration across endothelial cell membranes (Schrage et al., 2008).
The role of bone marrow derived cells in ALI has been of great interest, with considerable progress made. It is unclear if bone marrow cells can directly replenish hepatocytes, however, injection of autologous bone marrow cells, or increased mobilization of bone marrow cells, are protective after ALI (Li et al., 2010; Possamai et al., 2010; Shi et al., 2010; Inagaki and Higashiyama, 2012; Takami et al., 2012; Zhao et al., 2012). In both animal models and clinical studies, delivery of bone marrow cells or agents that increase circulating bone marrow cells are advantageous. Whole bone marrow and different subpopulations of cells including CD34+, CD133+, mononuclear cells, mesenchymal stem cells, and endothelial stem cells are all reportedly beneficial. The specific bone marrow-derived cell type that is responsible for protection is controversial, but the CXCL12/CXCR4 axis is implicated in many of these models (Shafritz et al., 2006; Khurana and Mukhopadhyay, 2007; Oertel and Shafritz, 2008;Jin et al., 2009, 2010; Karlmark et al., 2009; Baldo et al., 2010).
Myeloid cells alone are sufficient to generate hepatocytes, but this only occurs in models of severe hepatic disease. In more mild models including chronic CCl4 and bile duct ligation, these cells do not directly contribute to hepatocytes, but may be beneficial by secreting chemokines, cytokines, and growth factors thereby establishing a specific reparative niche (Kisseleva et al., 2010).
Rats administered a sublethal dose of acute CCl4 along with G-CSF and AMD3100, have increased survival and accumulation of CD34+ cells around CXCL12-rich periportal areas (Mark et al., 2010). G-CSF releases bone marrow progenitor cells by decreasing expression of CXCL12 by bone marrow endothelium, and by activating osteoclasts, inducing their expression of proteolytic activity which cleaves cell surface CXCR4, further releasing hematopoietic stem cells into the periphery (Damon, 2009). Furthermore, in NOD/SCID mice injected with human CD34+ stem cells, liver engraftment is dependent on the CXCL12/CXCR4 axis. Neutralization of CXCR4 with a specific antibody inhibits engraftment, while hepatic administration of recombinant CXCL12 leads to increased engraftment. The location of the CD34+ cells around the CXCL12-rich periportal areas further demonstrates their dependence on CXCL12. Finally, induction of either acute or chronic liver injury with CCl4 increases CXCR4 expression on human CD34+ cells and a greater degree of hepatic engraftment (Kollet et al., 2003).
CXCL12 can also bind the CXCR7 chemokine receptor. CXCR7, which also binds CXCL11, is classically thought to be a scavenging receptor capable of binding ligand, but unable to generate downstream signals, effectively sequestering the chemokine and preventing it from signaling through CXCR4 (Maksym et al., 2009; Luker et al., 2012; Sartina et al., 2012). Other receptors including DARC and D6 have similar scavenging properties, and their role in liver disease is apparent from mouse models deficient in the D6 receptor. D6 knockout mice exhibit prolonged liver damage after CCl4 injury associated with increased hepatic levels of chemokines CCL2, CCL3, and CCL5 (Berres et al., 2009). Increased inflammatory chemokines resulted in greater inflammation highlighting the role of chemokine sequestration in the control of inflammation.
As discussed above, inflammatory cell recruitment by CXCR4 and other chemokine receptors is largely regulated by chemokine ligand expression. However, chemokine receptor expression on infiltrating cells is also tightly controlled and finely tunable. Cell surface expression of CXCR4 in particular is highly regulated by factors including TGF-β, TNF-α, bacterial glycoproteins, and hypoxia.
Receptor expression can be controlled through transcriptional regulation and receptor internalization. TNF-α, IFN-γ, and LPS specifically reduces cell surface CXCR4 expression on neutrophils, but not lymphocytes, via receptor internalization in a time and dose dependent manner (Nagase et al., 2002; Kim et al., 2007). Paradoxically, however, CXCR4 mRNA levels are actually increased after stimulation, indicating that the change in cell surface CXCR4 expression is due to receptor internalization (Kim et al., 2007).
TGF-β is implicated in all forms of liver injury and has many functions in controlling immune cell differentiation and chemokine receptor expression. In both neutrophils and macrophages, TGF-β1 increases mRNA levels and cell surface expression of CXCR4 and enhances the effect of CXCL12 stimulation (Nagase et al., 2002; Chen et al., 2005). Furthermore, treatment of both immature and mature dendritic cells with TGF-β decreases the expression of CCR7 and increases expression of CCR1, CCR3, CCR5, and CXCR4 leading to a chemokine receptor profile that preferentially migrates toward sites of inflammation (Sato et al., 2000).
The liver with its dual arterial and venous blood supply has a low oxygen tension, which is worsened during injury, leading to a hypoxic environment. Monocytes and macrophages increase mRNA and cell surface expression of CXCR4 in response to hypoxic conditions in a HIF-1α-dependent mechanism, whereas transcript levels of CCR5 are unaffected (Schioppa et al., 2003). CXCL12 production by endothelial cells is induced by hypoxia, further increasing the effect of CXCL12/CXCR4 axis during hypoxia (Hitchon et al., 2002; Santiago et al., 2011).
Additional Chemokines in Acute Liver Injury
Two additional chemokines whose expression is induced during ALI are CCL5 (RANTES) and CX3CL1 (Fractalkine). CCL5 which binds to both CCR1 and CCR5 is of particular interest given the availability of maraviroc, a CCR5 small molecule inhibitor (Proudfoot et al., 2010). NK and NKT cells, CD4+ T cells, macrophages, and hepatic stellate cells all express CCR5 and in models of ConA induced hepatitis CCR5 deficiency promotes liver failure by preventing NKT cell activation-induced apoptosis (Ajuebor et al., 2006). Mice deficient for CCR5 show an unexpected increase in CCL5 ligand, perhaps due to loss of a negative feedback through CCR5, and an increase in CCR1-dependent NK cell recruitment and worse injury (Ajuebor et al., 2007). Within the liver CCR5 is expressed by stellate cells and is important in promoting their migration during chronic injury (Schwabe et al., 2003; Seki et al., 2009). The role of CCR5 on stellate cells has not been elucidated in models of acute injury.
CX3CL1, the only member of the CX3C family, is unique in that it is synthesized as a transmembrane protein and can be released by metalloproteinease cleavage. CX3CR1 is increased in patients with either chronic or acute liver disease specifically in areas of inflammation and in regenerating bile duct epithelia (Efsen et al., 2002). Similar to CCR5, loss of CX3CR1 leads to greater injury and delayed recovery after acute and chronic CCl4. Loss of CX3CR1 on macrophages promotes increased apoptosis and a more pro-inflammatory phenotype (Aoyama et al., 2010). As different waves of macrophages are important in injury and resolution of ALI, similar to CCL2, CX3CL1 may be important in the different stages of macrophage recruitment to the liver.
Therapeutic Potential of Chemokine Receptor Inhibition
The development of small molecule chemokine inhibitors and neutralizing antibodies provides promise for modulating immune cell infiltration during inflammatory diseases. A number of trials have examined the use of small molecule inhibitors in autoimmune and infectious diseases (Proudfoot et al., 2010); however, to date no clinical trial has examined the use of small molecule inhibitors directly in liver disease. Despite the large amount of data generated from knockout mice, inhibition of these axes in human disease states has not recapitulated pre-clinical data. Blocking CCR2 or inhibiting CCL2 in patients with rheumatoid arthritis (RA) did not achieve clinical endpoints (Haringman et al., 2006; Vergunst et al., 2008). Similarly, trials targeting CCR1 for RA, psoriasis, or multiple sclerosis have failed despite strong pre-clinical data (Trebst et al., 2001; Proudfoot et al., 2010).
There are numerous hurdles in the development of such inhibitors. Chemokine redundancy, with most receptors binding numerous ligands and receptor heterodimerization ensures that a single molecule will not achieve full inhibition of a given ligand. Additionally, the timing of chemokine inhibition during the disease process may be crucial and the window of effective inhibition narrow. For instance, the CXCR1/CXCR2 inhibitor, reparixin, has no effect on hepatic I/R injury when administered immediately after the injury, yet when administered 24 h after injury led to a worse outcome (Clarke et al., 2011). Similarly, in murine models of cardiac ischemia, AMD3100 can either increase or decrease the extent of injury based on the dosing protocol (Dai et al., 2010; Jujo et al., 2010). Finally, the choice of disease may determine if an inhibitor is successful. Mozobil (AMD3100), a CXCR4 inhibitor, is currently used for hematopoietic stem cell mobilization, yet failed as an HIV-entry inhibitor. Despite these difficulties successful chemokine inhibitors have been developed including maraviroc (CCR5 HIV-entry inhibitor) which may also show beneficial effects in patients with liver disease (Macias et al., 2012).
Conclusion
Chemokines are a large family of molecules whose function has expanded to now include immune cell infiltration, cellular survival and proliferation, vascular angiogenesis, organ fibrogenesis, and tumor metastasis. The interactions between chemokines, cytokines, and growth factors add additional levels of complexity to the networks and allow for spatial and temporal regulation. While most studies have focused on their adverse role in acute liver disease, a growing number of studies have revealed the beneficial role that they may play during injury as well. The availability of small molecule inhibitors for many chemokine axes makes them promising targets for therapeutics. However, given the many roles of chemokines during disease, a greater understanding of the underlying mechanisms is mandated.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the author’s laboratory is supported by RO1-DK56621, NIDDK F30-DK090986 to Yedidya Saiman and MSTP Training Grant T32-GM007280.
References
Ajuebor, M. N., Carey, J. A., and Swain, M. G. (2006). CCR5 in T cell-mediated liver diseases: what’s going on? J. Immunol. 177, 2039–2045.
Ajuebor, M. N., Hogaboam, C. M., Le, T., and Swain, M. G. (2003). C-C chemokine ligand 2/monocyte chemoattractant protein-1 directly inhibits NKT cell IL-4 production and is hepatoprotective in T cell-mediated hepatitis in the mouse. J. Immunol. 170, 5252–5259.
Ajuebor, M. N., Wondimu, Z., Hogaboam, C. M., Le, T., Proudfoot, A. E., and Swain, M. G. (2007). CCR5 deficiency drives enhanced natural killer cell trafficking to and activation within the liver in murine T cell-mediated hepatitis. Am. J. Pathol. 170, 1975–1988.
Antoniades, C. G., Quaglia, A., Taams, L. S., Mitry, R. R., Hussain, M., Abeles, R., Possamai, L. A., Bruce, M., Mcphail, M., Starling, C., Wagner, B., Barnardo, A., Pomplun, S., Auzinger, G., Bernal, W., Heaton, N., Vergani, D., Thursz, M. R., and Wendon, J. (2012). Source and characterisation of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology. [Epub ahead of print]. doi: 10.1002/hep.25657.
Aoyama, T., Inokuchi, S., Brenner, D. A., and Seki, E. (2010). CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 52, 1390–1400.
Apolinario, A., Majano, P. L., Alvarez-Perez, E., Saez, A., Lozano, C., Vargas, J., and Garcia-Monzon, C. (2002). Increased expression of T cell chemokines and their receptors in chronic hepatitis C: relationship with the histological activity of liver disease. Am. J. Gastroenterol. 97, 2861–2870.
Bajetto, A., Bonavia, R., Barbero, S., and Schettini, G. (2002). Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J. Neurochem. 82, 1311–1329.
Baldo, G., Giugliani, R., Uribe, C., Belardinelli, M. C., Duarte, M. E., Meurer, L., Da Silveira, T. R., and Matte, U. (2010). Bone marrow mononuclear cell transplantation improves survival and induces hepatocyte proliferation in rats after CCl(4) acute liver damage. Dig. Dis. Sci. 55, 3384–3392.
Berres, M. L., Trautwein, C., Zaldivar, M. M., Schmitz, P., Pauels, K., Lira, S. A., Tacke, F., and Wasmuth, H. E. (2009). The chemokine scavenging receptor D6 limits acute toxic liver injury in vivo. Biol. Chem. 390, 1039–1045.
Bone-Larson, C. L., Hogaboam, C. M., Evanhoff, H., Strieter, R. M., and Kunkel, S. L. (2001). IFN-gamma-inducible protein-10 (CXCL10) is hepatoprotective during acute liver injury through the induction of CXCR2 on hepatocytes. J. Immunol. 167, 7077–7083.
Bromley, S. K., Mempel, T. R., and Luster, A. D. (2008). Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat. Immunol. 9, 970–980.
Campbell, J. J., Hedrick, J., Zlotnik, A., Siani, M. A., Thompson, D. A., and Butcher, E. C. (1998). Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 279, 381–384.
Cao, X., Zhang, W., Wan, T., He, L., Chen, T., Yuan, Z., Ma, S., Yu, Y., and Chen, G. (2000). Molecular cloning and characterization of a novel CXC chemokine macrophage inflammatory protein-2 gamma chemoattractant for human neutrophils and dendritic cells. J. Immunol. 165, 2588–2595.
Chen, S., Tuttle, D. L., Oshier, J. T., Knot, H. J., Streit, W. J., Goodenow, M. M., and Harrison, J. K. (2005). Transforming growth factor-beta1 increases CXCR4 expression, stromal-derived factor-1alpha-stimulated signalling and human immunodeficiency virus-1 entry in human monocyte-derived macrophages. Immunology 114, 565–574.
Chen, X., Xu, C., Zhang, F., and Ma, J. (2010). Comparative analysis of expression profiles of chemokines, chemokine receptors, and components of signaling pathways mediated by chemokines in eight cell types during rat liver regeneration. Genome 53, 608–618.
Chen, Y. X., Sato, M., Kawachi, K., and Abe, Y. (2006). Neutrophil-mediated liver injury during hepatic ischemia-reperfusion in rats. HBPD INT 5, 436–442.
Christopher, M. J., and Link, D. C. (2007). Regulation of neutrophil homeostasis. Curr. Opin. Hematol. 14, 3–8.
Clarke, C., Kuboki, S., Sakai, N., Kasten, K. R., Tevar, A. D., Schuster, R., Blanchard, J., Caldwell, C. C., Edwards, M. J., and Lentsch, A. B. (2011). CXC chemokine receptor-1 is expressed by hepatocytes and regulates liver recovery after hepatic ischemia/reperfusion injury. Hepatology 53, 261–271.
Cole, K. E., Strick, C. A., Paradis, T. J., Ogborne, K. T., Loetscher, M., Gladue, R. P., Lin, W., Boyd, J. G., Moser, B., Wood, D. E., Sahagan, B. G., and Neote, K. (1998). Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 187, 2009–2021.
Crosby, H. A., Lalor, P. F., Ross, E., Newsome, P. N., and Adams, D. H. (2009). Adhesion of human haematopoietic (CD34+) stem cells to human liver compartments is integrin and CD44 dependent and modulated by CXCR3 and CXCR4. J. Hepatol. 51, 734–749.
Dai, S., Yuan, F., Mu, J., Li, C., Chen, N., Guo, S., Kingery, J., Prabhu, S. D., Bolli, R., and Rokosh, G. (2010). Chronic AMD3100 antagonism of SDF-1alpha-CXCR4 exacerbates cardiac dysfunction and remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 49, 587–597.
Dambach, D. M., Watson, L. M., Gray, K. R., Durham, S. K., and Laskin, D. L. (2002). Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology 35, 1093–1103.
Damon, L. E. (2009). Mobilization of hematopoietic stem cells into the peripheral blood. Expert Rev. Hematol. 2, 717–733.
Dong, Z., Zhang, C., Wei, H., Sun, R., and Tian, Z. (2005). Impaired NK cell cytotoxicity by high level of interferon-gamma in concanavalin A-induced hepatitis. Can. J. Physiol. Pharmacol. 83, 1045–1053.
Duffield, J. S. (2003). The inflammatory macrophage: a story of Jekyll and Hyde. Clin. Sci. 104, 27–38.
Duffield, J. S., Forbes, S. J., Constandinou, C. M., Clay, S., Partolina, M., Vuthoori, S., Wu, S., Lang, R., and Iredale, J. P. (2005). Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 115, 56–65.
Efsen, E., Grappone, C., Defranco, R. M., Milani, S., Romanelli, R. G., Bonacchi, A., Caligiuri, A., Failli, P., Annunziato, F., Pagliai, G., Pinzani, M., Laffi, G., Gentilini, P., and Marra, F. (2002). Up-regulated expression of fractalkine and its receptor CX3CR1 during liver injury in humans. J. Hepatol. 37, 39–47.
Erhardt, A., Wegscheid, C., Claass, B., Carambia, A., Herkel, J., Mittrucker, H. W., Panzer, U., and Tiegs, G. (2011). CXCR3 deficiency exacerbates liver disease and abrogates tolerance in a mouse model of immune-mediated hepatitis. J. Immunol. 186, 5284–5293.
Farber, J. M. (1990). A macrophage mRNA selectively induced by gamma-interferon encodes a member of the platelet factor 4 family of cytokines. Proc. Natl. Acad. Sci. U.S.A. 87, 5238–5242.
Fernandez, E. J., and Lolis, E. (2002). Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42, 469–499.
Friedman, S. L. (2008b). Hepatic stellate cells – protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 88, 125–172.
Gangavarapu, P., Rajagopalan, L., Kolli, D., Guerrero-Plata, A., Garofalo, R. P., and Rajarathnam, K. (2012). The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 91, 259–265.
Gerritsma, J. S., Van Kooten, C., Gerritsen, A. F., Van Es, L. A., and Daha, M. R. (1998). Transforming growth factor-beta 1 regulates chemokine and complement production by human proximal tubular epithelial cells. Kidney Int. 53, 609–616.
Goddard, S., Williams, A., Morland, C., Qin, S., Gladue, R., Hubscher, S. G., and Adams, D. H. (2001). Differential expression of chemokines and chemokine receptors shapes the inflammatory response in rejecting human liver transplants. Transplantation 72, 1957–1967.
Groom, J. R., and Luster, A. D. (2011). CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215.
Haringman, J. J., Gerlag, D. M., Smeets, T. J., Baeten, D., Van Den Bosch, F., Bresnihan, B., Breedveld, F. C., Dinant, H. J., Legay, F., Gram, H., Loetscher, P., Schmouder, R., Woodworth, T., and Tak, P. P. (2006). A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 54, 2387–2392.
Harvey, C. E., Post, J. J., Palladinetti, P., Freeman, A. J., Ffrench, R. A., Kumar, R. K., Marinos, G., and Lloyd, A. R. (2003). Expression of the chemokine IP-10 (CXCL10) by hepatocytes in chronic hepatitis C virus infection correlates with histological severity and lobular inflammation. J. Leukoc. Biol. 74, 360–369.
Hassan, M., Selimovic, D., Ghozlan, H., and Abdel-Kader, O. (2007). Induction of high-molecular-weight (HMW) tumor necrosis factor(TNF) alpha by hepatitis C virus (HCV) non-structural protein 3 (NS3) in liver cells is AP-1 and NF-kappaB-dependent activation. Cell. Signal. 19, 301–311.
Hitchon, C., Wong, K., Ma, G., Reed, J., Lyttle, D., and El-Gabalawy, H. (2002). Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 46, 2587–2597.
Hogaboam, C. M., Bone-Larson, C. L., Steinhauser, M. L., Lukacs, N. W., Colletti, L. M., Simpson, K. J., Strieter, R. M., and Kunkel, S. L. (1999a). Novel CXCR2-dependent liver regenerative qualities of ELR-containing CXC chemokines. FASEB J. 13, 1565–1574.
Hogaboam, C. M., Simpson, K. J., Chensue, S. W., Steinhauser, M. L., Lukacs, N. W., Gauldie, J., Strieter, R. M., and Kunkel, S. L. (1999b). Macrophage inflammatory protein-2 gene therapy attenuates adenovirus- and acetaminophen-mediated hepatic injury. Gene Ther. 6, 573–584.
Hogaboam, C. M., Bone-Larson, C. L., Steinhauser, M. L., Matsukawa, A., Gosling, J., Boring, L., Charo, I. F., Simpson, K. J., Lukacs, N. W., and Kunkel, S. L. (2000). Exaggerated hepatic injury due to acetaminophen challenge in mice lacking C-C chemokine receptor 2. Am. J. Pathol. 156, 1245–1252.
Hokeness, K. L., Deweerd, E. S., Munks, M. W., Lewis, C. A., Gladue, R. P., and Salazar-Mather, T. P. (2007). CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J. Virol. 81, 1241–1250.
Hokeness, K. L., Kuziel, W. A., Biron, C. A., and Salazar-Mather, T. P. (2005). Monocyte chemoattractant protein-1 and CCR2 interactions are required for IFN-alpha/beta-induced inflammatory responses and antiviral defense in liver. J. Immunol. 174, 1549–1556.
Holt, M. P., Cheng, L., and Ju, C. (2008). Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J. Leukoc. Biol. 84, 1410–1421.
Hong, F., Tuyama, A., Lee, T. F., Loke, J., Agarwal, R., Cheng, X., Garg, A., Fiel, M. I., Schwartz, M., Walewski, J., Branch, A., Schecter, A. D., and Bansal, M. B. (2009). Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1alpha-mediated stellate cell activation. Hepatology 49, 2055–2067.
Horiguchi, K., Kitagawa-Sakakida, S., Sawa, Y., Li, Z. Z., Fukushima, N., Shirakura, R., and Matsuda, H. (2002). Selective chemokine and receptor gene expressions in allografts that develop transplant vasculopathy. J. Heart Lung Transplant. 21, 1090–1100.
Hu, B., and Colletti, L. M. (2010). CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. Hepatology 52, 691–702.
Ilmakunnas, M., Hockerstedt, K., Makisalo, H., Siitonen, S., Repo, H., and Pesonen, E. J. (2010). Hepatic IL-8 release during graft procurement is associated with impaired graft function after human liver transplantation. Clin. Transplant. 24, 29–35.
Inagaki, Y., Nemoto, T., Kushida, M., Sheng, Y., Higashi, K., Ikeda, K., Kawada, N., Shirasaki, F., Takehara, K., Sugiyama, K., Fujii, M., Yamauchi, H., Nakao, A., De Crombrugghe, B., Watanabe, T., and Okazaki, I. (2003). Interferon alfa down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis in mice. Hepatology 38, 890–899.
Ishida, Y., Kondo, T., Kimura, A., Tsuneyama, K., Takayasu, T., and Mukaida, N. (2006). Opposite roles of neutrophils and macrophages in the pathogenesis of acetaminophen-induced acute liver injury. Eur. J. Immunol. 36, 1028–1038.
Jaeschke, H., Bautista, A. P., Spolarics, Z., and Spitzer, J. J. (1991). Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic. Res. Commun. 15, 277–284.
Jaeschke, H., and Farhood, A. (2002). Kupffer cell activation after no-flow ischemia versus hemorrhagic shock. Free Radic. Biol. Med. 33, 210–219.
Jaeschke, H., Gores, G. J., Cederbaum, A. I., Hinson, J. A., Pessayre, D., and Lemasters, J. J. (2002). Mechanisms of hepatotoxicity. Toxicol. Sci. 65, 166–176.
James, L. P., Farrar, H. C., Darville, T. L., Sullivan, J. E., Givens, T. G., Kearns, G. L., Wasserman, G. S., Simpson, P. M., and Hinson, J. A. (2001). Elevation of serum interleukin 8 levels in acetaminophen overdose in children and adolescents. Clin. Pharmacol. Ther. 70, 280–286.
Jaruga, B., Hong, F., Kim, W. H., and Gao, B. (2004). IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G1044–G1052.
Jin, S. Z., Meng, X. W., Han, M. Z., Sun, X., Sun, L. Y., and Liu, B. R. (2009). Stromal cell derived factor-1 enhances bone marrow mononuclear cell migration in mice with acute liver failure. World J. Gastroenterol. 15, 2657–2664.
Jin, S. Z., Meng, X. W., Sun, X., Han, M. Z., Liu, B. R., Wang, X. H., Sun, L. Y., Huang, Q., Zhao, R. B., Ban, X., Yu, H. Y., and Yu, H. W. (2010). Granulocyte colony-stimulating factor enhances bone marrow mononuclear cell homing to the liver in a mouse model of acute hepatic injury. Dig. Dis. Sci. 55, 2805–2813.
Ju, C., Reilly, T. P., Bourdi, M., Radonovich, M. F., Brady, J. N., George, J. W., and Pohl, L. R. (2002). Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem. Res. Toxicol. 15, 1504–1513.
Jujo, K., Hamada, H., Iwakura, A., Thorne, T., Sekiguchi, H., Clarke, T., Ito, A., Misener, S., Tanaka, T., Klyachko, E., Kobayashi, K., Tongers, J., Roncalli, J., Tsurumi, Y., Hagiwara, N., and Losordo, D. W. (2010). CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc. Natl. Acad. Sci. U.S.A. 107, 11008–11013.
Karlmark, K. R., Wasmuth, H. E., Trautwein, C., and Tacke, F. (2008). Chemokine-directed immune cell infiltration in acute and chronic liver disease. Expert Rev. Gastroenterol. Hepatol. 2, 233–242.
Karlmark, K. R., Weiskirchen, R., Zimmermann, H. W., Gassler, N., Ginhoux, F., Weber, C., Merad, M., Luedde, T., Trautwein, C., and Tacke, F. (2009). Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 50, 261–274.
Khurana, S., and Mukhopadhyay, A. (2007). Characterization of the potential subpopulation of bone marrow cells involved in the repair of injured liver tissue. Stem Cells 25, 1439–1447.
Kim, H. K., Kim, J. E., Chung, J., Han, K. S., and Cho, H. I. (2007). Surface expression of neutrophil CXCR4 is down-modulated by bacterial endotoxin. Int. J. Hematol. 85, 390–396.
Kisseleva, T., Gigante, E., and Brenner, D. A. (2010). Recent advances in liver stem cell therapy. Curr. Opin. Gastroenterol. 26, 395–402.
Kolios, G., Valatas, V., Manousou, P., Xidakis, C., Notas, G., and Kouroumalis, E. (2008). Nitric oxide and MCP-1 regulation in LPS activated rat Kupffer cells. Mol. Cell. Biochem. 319, 91–98.
Kollet, O., Shivtiel, S., Chen, Y. Q., Suriawinata, J., Thung, S. N., Dabeva, M. D., Kahn, J., Spiegel, A., Dar, A., Samira, S., Goichberg, P., Kalinkovich, A., Arenzana-Seisdedos, F., Nagler, A., Hardan, I., Revel, M., Shafritz, D. A., and Lapidot, T. (2003). HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J. Clin. Invest. 112, 160–169.
Krohn, N., Kapoor, S., Enami, Y., Follenzi, A., Bandi, S., Joseph, B., and Gupta, S. (2009). Hepatocyte transplantation-induced liver inflammation is driven by cytokines-chemokines associated with neutrophils and Kupffer cells. Gastroenterology 136, 1806–1817.
Kuboki, S., Shin, T., Huber, N., Eismann, T., Galloway, E., Schuster, R., Blanchard, J., Edwards, M. J., and Lentsch, A. B. (2008). Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 48, 1213–1223.
Laskin, D. L., Gardner, C. R., Price, V. F., and Jollow, D. J. (1995). Modulation of macrophage functioning abrogates the acute hepatotoxicity of acetaminophen. Hepatology 21, 1045–1050.
Lei, Y., Liu, Z., Han, Q., Kang, W., Zhang, L., and Lou, S. (2010). G-CSF enhanced SDF-1 gradient between bone marrow and liver associated with mobilization of peripheral blood CD34+ cells in rats with acute liver failure. Dig. Dis. Sci. 55, 285–291.
Leifeld, L., Dumoulin, F. L., Purr, I., Janberg, K., Trautwein, C., Wolff, M., Manns, M. P., Sauerbruch, T., and Spengler, U. (2003). Early up-regulation of chemokine expression in fulminant hepatic failure. J. Pathol. 199, 335–344.
Li, N., Zhang, L., Li, H., and Fang, B. (2010). Human CD34+ cells mobilized by granulocyte colony-stimulating factor ameliorate radiation-induced liver damage in mice. Stem Cell Res. Ther. 1, 22.
Liekens, S., Schols, D., and Hatse, S. (2010). CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 16, 3903–3920.
Link, D. C. (2005). Neutrophil homeostasis: a new role for stromal cell-derived factor-1. Immunol. Res. 32, 169–178.
Luker, K. E., Lewin, S. A., Mihalko, L. A., Schmidt, B. T., Winkler, J. S., Coggins, N. L., Thomas, D. G., and Luker, G. D. (2012). Scavenging of CXCL12 by CXCR7 promotes tumor growth and metastasis of CXCR4-positive breast cancer cells. Oncogene. [Epub ahead of print]. doi: 10.1038/onc.2011.633.
Luster, A. D., Unkeless, J. C., and Ravetch, J. V. (1985). Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature 315, 672–676.
Macias, J., Viloria, M. M., Rivero, A., De Los Santos, I., Marquez, M., Portilla, J., Di Lello, F., Camacho, A., Sanz-Sanz, J., Ojeda, G., Mata, R., Gomez-Mateos, J., and Pineda, J. A. (2012). Lack of short-term increase in serum mediators of fibrogenesis and in non-invasive markers of liver fibrosis in HIV/hepatitis C virus-coinfected patients starting maraviroc-based antiretroviral therapy. Eur. J. Clin. Microbiol. Infect. Dis. [Epub ahead of print]. doi:10.1007/s10096-012-1546-5.
Majumder, S., Zhou, L. Z., Chaturvedi, P., Babcock, G., Aras, S., and Ransohoff, R. M. (1998). p48/STAT-1alpha-containing complexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with TNF-alpha. J. Immunol. 161, 4736–4744.
Maksym, R. B., Tarnowski, M., Grymula, K., Tarnowska, J., Wysoczynski, M., Liu, R., Czerny, B., Ratajczak, J., Kucia, M., and Ratajczak, M. Z. (2009). The role of stromal-derived factor-1–CXCR7 axis in development and cancer. Eur. J. Pharmacol. 625, 31–40.
Mark, A. L., Sun, Z., Warren, D. S., Lonze, B. E., Knabel, M. K., Melville Williams, G. M., Locke, J. E., Montgomery, R. A., and Cameron, A. M. (2010). Stem cell mobilization is life saving in an animal model of acute liver failure. Ann. Surg. 252, 591–596.
Medoff, B. D., Wain, J. C., Seung, E., Jackobek, R., Means, T. K., Ginns, L. C., Farber, J. M., and Luster, A. D. (2006). CXCR3 and its ligands in a murine model of obliterative bronchiolitis: regulation and function. J. Immunol. 176, 7087–7095.
Michael, S. L., Pumford, N. R., Mayeux, P. R., Niesman, M. R., and Hinson, J. A. (1999). Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology 30, 186–195.
Mukaida, N., and Baba, T. (2012). Chemokines in tumor development and progression. Exp. Cell Res. 318, 95–102.
Nagasawa, T., Hirota, S., Tachibana, K., Takakura, N., Nishikawa, S., Kitamura, Y., Yoshida, N., Kikutani, H., and Kishimoto, T. (1996). Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382, 635–638.
Nagase, H., Miyamasu, M., Yamaguchi, M., Imanishi, M., Tsuno, N. H., Matsushima, K., Yamamoto, K., Morita, Y., and Hirai, K. (2002). Cytokine-mediated regulation of CXCR4 expression in human neutrophils. J. Leukoc. Biol. 71, 711–717.
Nasser, M. W., Raghuwanshi, S. K., Grant, D. J., Jala, V. R., Rajarathnam, K., and Richardson, R. M. (2009). Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 183, 3425–3432.
Oertel, M., and Shafritz, D. A. (2008). Stem cells, cell transplantation and liver repopulation. Biochim. Biophys. Acta 1782, 61–74.
Ohmori, Y., and Hamilton, T. A. (1993). Cooperative interaction between interferon (IFN) stimulus response element and kappa B sequence motifs controls IFN gamma- and lipopolysaccharide-stimulated transcription from the murine IP-10 promoter. J. Biol. Chem. 268, 6677–6688.
Ohmori, Y., and Hamilton, T. A. (1997). IL-4-induced STAT6 suppresses IFN-gamma-stimulated STAT1-dependent transcription in mouse macrophages. J. Immunol. 159, 5474–5482.
Ohmori, Y., Schreiber, R. D., and Hamilton, T. A. (1997). Synergy between interferon-gamma and tumor necrosis factor-alpha in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor kappaB. J. Biol. Chem. 272, 14899–14907.
Ohmori, Y., Wyner, L., Narumi, S., Armstrong, D., Stoler, M., and Hamilton, T. A. (1993). Tumor necrosis factor-alpha induces cell type and tissue-specific expression of chemoattractant cytokines in vivo. Am. J. Pathol. 142, 861–870.
Oo, Y. H., Shetty, S., and Adams, D. H. (2010). The role of chemokines in the recruitment of lymphocytes to the liver. Dig. Dis. 28, 31–44.
Peled, A., Grabovsky, V., Habler, L., Sandbank, J., Arenzana-Seisdedos, F., Petit, I., Ben-Hur, H., Lapidot, T., and Alon, R. (1999). The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J. Clin. Invest. 104, 1199–1211.
Possamai, L. A., Antoniades, C. G., Anstee, Q. M., Quaglia, A., Vergani, D., Thursz, M., and Wendon, J. (2010). Role of monocytes and macrophages in experimental and human acute liver failure. World J. Gastroenterol. 16, 1811–1819.
Proudfoot, A. E., Power, C. A., and Schwarz, M. K. (2010). Anti-chemokine small molecule drugs: a promising future? Expert Opin. Investig. Drugs 19, 345–355.
Ramaiah, S. K., and Jaeschke, H. (2007). Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol. Pathol. 35, 757–766.
Rossi, D. L., Hurst, S. D., Xu, Y., Wang, W., Menon, S., Coffman, R. L., and Zlotnik, A. (1999). Lungkine, a novel CXC chemokine, specifically expressed by lung bronchoepithelial cells. J. Immunol. 162, 5490–5497.
Sahin, H., Borkham-Kamphorst, E., Kuppe, C., Zaldivar, M. M., Grouls, C., Al-Samman, M., Nellen, A., Schmitz, P., Heinrichs, D., Berres, M. L., Doleschel, D., Scholten, D., Weiskirchen, R., Moeller, M. J., Kiessling, F., Trautwein, C., and Wasmuth, H. E. (2012). The chemokine Cxcl9 attenuates liver fibrosis-associated angiogenesis in mice. Hepatology. [Epub ahead of print]. doi: 10.1002/hep.25545.
Sakai, N., Kuboki, S., Van Sweringen, H. L., Tevar, A. D., Schuster, R., Blanchard, J., Edwards, M. J., and Lentsch, A. B. (2011). CXCR1 deficiency does not alter liver regeneration after partial hepatectomy in mice. Transplant. Proc. 43, 1967–1970.
Santiago, B., Calonge, E., Del Rey, M. J., Gutierrez-Canas, I., Izquierdo, E., Usategui, A., Galindo, M., Alcami, J., and Pablos, J. L. (2011). CXCL12 gene expression is upregulated by hypoxia and growth arrest but not by inflammatory cytokines in rheumatoid synovial fibroblasts. Cytokine 53, 184–190.
Sartina, E., Suguihara, C., Ramchandran, S., Nwajei, P., Rodriguez, M., Torres, E., Hehre, D., Devia, C., Walters, M. J., Penfold, M. E., and Young, K. C. (2012). Antagonism of CXCR7 attenuates chronic hypoxia-induced pulmonary hypertension. Pediatr. Res. 71, 682–688.
Sato, K., Kawasaki, H., Nagayama, H., Enomoto, M., Morimoto, C., Tadokoro, K., Juji, T., and Takahashi, T. A. (2000). TGF-beta 1 reciprocally controls chemotaxis of human peripheral blood monocyte-derived dendritic cells via chemokine receptors. J. Immunol. 164, 2285–2295.
Sawitza, I., Kordes, C., Reister, S., and Haussinger, D. (2009). The niche of stellate cells within rat liver. Hepatology 50, 1617–1624.
Schioppa, T., Uranchimeg, B., Saccani, A., Biswas, S. K., Doni, A., Rapisarda, A., Bernasconi, S., Saccani, S., Nebuloni, M., Vago, L., Mantovani, A., Melillo, G., and Sica, A. (2003). Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 198, 1391–1402.
Schrage, A., Wechsung, K., Neumann, K., Schumann, M., Schulzke, J. D., Engelhardt, B., Zeitz, M., Hamann, A., and Klugewitz, K. (2008). Enhanced T cell transmigration across the murine liver sinusoidal endothelium is mediated by transcytosis and surface presentation of chemokines. Hepatology 48, 1262–1272.
Schwabe, R. F., Bataller, R., and Brenner, D. A. (2003). Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G949–G958.
Seki, E., De Minicis, S., Gwak, G. Y., Kluwe, J., Inokuchi, S., Bursill, C. A., Llovet, J. M., Brenner, D. A., and Schwabe, R. F. (2009). CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 119, 1858–1870.
Shafritz, D. A., Oertel, M., Menthena, A., Nierhoff, D., and Dabeva, M. D. (2006). Liver stem cells and prospects for liver reconstitution by transplanted cells. Hepatology 43, S89–S98.
Shi, X. L., Gu, J. Y., Han, B., Xu, H. Y., Fang, L., and Ding, Y. T. (2010). Magnetically labeled mesenchymal stem cells after autologous transplantation into acutely injured liver. World J. Gastroenterol. 16, 3674–3679.
Shibuta, K., Mori, M., Shimoda, K., Inoue, H., Mitra, P., and Barnard, G. F. (2002). Regional expression of CXCL12/CXCR4 in liver and hepatocellular carcinoma and cell-cycle variation during in vitro differentiation. Jpn. J. Cancer Res. 93, 789–797.
Shields, P. L., Morland, C. M., Salmon, M., Qin, S., Hubscher, S. G., and Adams, D. H. (1999). Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J. Immunol. 163, 6236–6243.
Stefanovic, L., and Stefanovic, B. (2006). Mechanism of direct hepatotoxic effect of KC chemokine: sequential activation of gene expression and progression from inflammation to necrosis. J. Interferon Cytokine Res. 26, 760–770.
Steib, C. J., Bilzer, M., Hartl, J. M., Beitinger, F., Gulberg, V., Goke, B., and Gerbes, A. L. (2010). Kupffer cell activation by hydrogen peroxide: a new mechanism of portal pressure increase. Shock 33, 412–418.
Takami, T., Terai, S., and Sakaida, I. (2012). Stem cell therapy in chronic liver disease. Curr. Opin. Gastroenterol. 28, 203–208.
Tensen, C. P., Flier, J., Rampersad, S. S., Sampat-Sardjoepersad, S., Scheper, R. J., Boorsma, D. M., and Willemze, R. (1999). Genomic organization, sequence and transcriptional regulation of the human CXCL 11(1) gene. Biochim. Biophys. Acta 1446, 167–172.
Trebst, C., Sorensen, T. L., Kivisakk, P., Cathcart, M. K., Hesselgesser, J., Horuk, R., Sellebjerg, F., Lassmann, H., and Ransohoff, R. M. (2001). CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am. J. Pathol. 159, 1701–1710.
Tsou, C. L., Peters, W., Si, Y., Slaymaker, S., Aslanian, A. M., Weisberg, S. P., Mack, M., and Charo, I. F. (2007). Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 117, 902–909.
Vergunst, C. E., Gerlag, D. M., Lopatinskaya, L., Klareskog, L., Smith, M. D., Van Den Bosch, F., Dinant, H. J., Lee, Y., Wyant, T., Jacobson, E. W., Baeten, D., and Tak, P. P. (2008). Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 58, 1931–1939.
Wald, O., Pappo, O., Safadi, R., Dagan-Berger, M., Beider, K., Wald, H., Franitza, S., Weiss, I., Avniel, S., Boaz, P., Hanna, J., Zamir, G., Eid, A., Mandelboim, O., Spengler, U., Galun, E., and Peled, A. (2004). Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis B virus. Eur. J. Immunol. 34, 1164–1174.
Wasmuth, H. E., Lammert, F., Zaldivar, M. M., Weiskirchen, R., Hellerbrand, C., Scholten, D., Berres, M. L., Zimmermann, H., Streetz, K. L., Tacke, F., Hillebrandt, S., Schmitz, P., Keppeler, H., Berg, T., Dahl, E., Gassler, N., Friedman, S. L., and Trautwein, C. (2009). Antifibrotic effects of CXCL9 and its receptor CXCR3 in livers of mice and humans. Gastroenterology 137, 309-319; 319.e301–319.e303.
Wasmuth, H. E., Tacke, F., and Trautwein, C. (2010). Chemokines in liver inflammation and fibrosis. Semin. Liver Dis. 30, 215–225.
Yang, C. H., Wei, L., Pfeffer, S. R., Du, Z., Murti, A., Valentine, W. J., Zheng, Y., and Pfeffer, L. M. (2007). Identification of CXCL11 as a STAT3-dependent gene induced by IFN. J. Immunol. 178, 986–992.
Zaldivar, M. M., Berres, M. L., Sahin, H., Nellen, A., Heinrichs, D., Schmitz, P., Gassler, N., Streetz, K. L., Trautwein, C., and Wasmuth, H. E. (2012). The chemokine receptor CXCR3 limits injury after acute toxic liver damage. Lab. Invest. 92, 724–734.
Zernecke, A., Bidzhekov, K., Noels, H., Shagdarsuren, E., Gan, L., Denecke, B., Hristov, M., Koppel, T., Jahantigh, M. N., Lutgens, E., Wang, S., Olson, E. N., Schober, A., and Weber, C. (2009). Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2, ra81.
Zhao, W., Li, J. J., Cao, D. Y., Li, X., Zhang, L. Y., He, Y., Yue, S. Q., Wang, D. S., and Dou, K. F. (2012). Intravenous injection of mesenchymal stem cells is effective in treating liver fibrosis. World J. Gastroenterol. 18, 1048–1058.
Zimmermann, H. W., Seidler, S., Gassler, N., Nattermann, J., Luedde, T., Trautwein, C., and Tacke, F. (2011). Interleukin-8 is activated in patients with chronic liver diseases and associated with hepatic macrophage accumulation in human liver fibrosis. PLoS ONE 6, e21381. doi:10.1371/journal.pone.0021381
Keywords: chemokines, acute liver injury
Citation: Saiman Y and Friedman SL (2012) The role of chemokines in acute liver injury. Front. Physio. 3:213. doi: 10.3389/fphys.2012.00213
Received: 26 March 2012; Paper pending published: 09 April 2012;
Accepted: 30 May 2012; Published online: 20 June 2012.
Edited by:
Ali Canbay, University Hospital Essen, GermanyReviewed by:
Frank Tacke, University Hospital Aachen, GermanyLeo A. Van Grunsven, Vrije Universiteit Brussel, Belgium
Copyright: © 2012 Saiman and Friedman. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Scott L. Friedman, Division of Liver Diseases, Mount Sinai School of Medicine, Box 1123, 1425 Madison Avenue, Room 11-70C, New York, NY 10029-6574, USA. e-mail: scott.friedman@mssm.edu