 Veronica L. Massey
Veronica L. Massey Gavin E. Arteel*
Gavin E. Arteel*- Department of Pharmacology and Toxicology, University of Louisville Alcohol Research Center, University of Louisville Health Sciences Center, Louisville, KY, USA
Alcohol consumption is customary in most cultures and alcohol abuse is common worldwide. For example, more than 50% of Americans consume alcohol, with an estimated 23.1% of Americans participating in heavy and/or binge drinking at least once a month. A safe and effective therapy for alcoholic liver disease (ALD) in humans is still elusive, despite significant advances in our understanding of how the disease is initiated and progresses. It is now clear that acute alcohol binges not only can be acutely toxic to the liver, but also can contribute to the chronicity of ALD. Potential mechanisms by which acute alcohol causes damage include steatosis, dysregulated immunity and inflammation, and altered gut permeability. Recent interest in modeling acute alcohol exposure has yielded new insights into potential mechanisms of acute injury, which also may well be relevant for chronic ALD. Recent work by this group on the role of PAI-1 and fibrin metabolism in mediating acute alcohol-induced liver damage serve as an example of possible new targets that may be useful for alcohol abuse, be it acute or chronic.
Introduction
Alcohol consumption is customary in most cultures and alcohol abuse is common worldwide. For example, more than 50% of Americans consume alcohol, with an estimated 23.1% of Americans participating in heavy and/or binge drinking at least once a month (Substance Abuse and Mental Health Services Administration, 2011). The detrimental effects of drinking alcohol, particularly heavy and/or chronic consumption, are well established. The most commonly recognized symptoms of alcohol consumption are associated with chronic alcoholism, and it is a causal/risk factor in over 60 major types of diseases. These and other effects of alcohol consumption have made alcohol the third leading risk factor globally for disease and disability (World Health Organization, 2011). The cost of medical consequences of alcohol abuse have been projected to be more than 26 billion dollars in the US in 1998, making it a significant financial burden for the health care system (U. S. Department of Health and Human Services, 2000). These estimates take into account only direct medical expenses; it is estimated that the cost of lost productivity due to alcohol-related illness was ∼90 billion dollars in the US in 1998. Thus, the medical consequences of alcohol consumption have significant societal consequences in addition to the effects on the individual.
The liver is the main site of alcohol metabolism and a major target organ of alcohol-induced injury. The susceptibility of the liver to alcohol-induced toxicity is due to both the high concentrations of alcohol found in the portal blood (versus systemic), as well as the metabolic consequences of ethanol metabolism. Alcoholic Liver Disease (ALD) is a spectrum of disease states that includes steatosis (fatty liver), steatohepatitis, and in severe cases, fibrosis and/or cirrhosis. Steatosis, characterized by fat accumulation in hepatocytes, develops in 90% of individuals who drink more than 16 g of alcohol/day (Crabb, 1999), but resolves upon cessation of alcohol consumption (Bergheim et al., 2005). In only a minority of even heavy drinkers, steatosis progresses to steatohepatitis, which is characterized by the persistence of fatty liver accompanied by inflammation. In later stages of ALD, collagen deposition and regenerative nodules can result in the development of fibrosis and cirrhosis, respectively. Abstinence from alcohol is beneficial for patients in all stages of ALD and is necessary to prevent progression of liver injury in those with early stages of disease (Bergheim et al., 2005). Unfortunately, a high rate of recidivism among chronic alcohol abusers greatly reduces the opportunity for disease remission via abstinence. Therefore, better understanding of the mechanisms by which alcohol damages the liver may yield new pharmacologic strategies to blunt, halt, or reverse disease progression, potentially even in inveterate alcoholics.
Although alcohol exposure has been studied for decades, most work has focused on chronic exposure and our understanding of the mechanisms and effects of acute alcohol exposure are lacking. Acute alcohol exposure, commonly known as “binge drinking,” is defined by the National Survey on Drug Use and Health (NSDUH) as five or more drinks on a single occasion, within a time period of 3 h. However, acute alcohol exposure can also include a period of heavy drinking that may span several days or periods of intermittent, repeated episodes of heavy drinking. For example, in the clinical setting an episode of “binge” drinking often describes a period of alcohol consumption and intoxication that lasts upward of 2 days (Weschsler and Austin, 1998). The NSDUH in 2010 showed that more than 58 million Americans have participated in binge drinking on at least one occasion within 30 days (Substance Abuse and Mental Health Services Administration, 2011). Due to the increasing prevalence of binge drinking in people aged 18–25, greater interest has turned toward the effects of acute alcohol exposure. Although chronic alcohol abuse is the most commonly recognized source of illness caused by alcohol consumption, acute alcohol abuse is also detrimental. Many acute alcohol-related deaths are due to severe CNS depression, which slows motor coordination and respiratory rates. Acute alcohol exposure also impairs recovery from infection and trauma and delays wound healing (Radek et al., 2005, 2012). As such, acute alcohol consumption is a major underlying cause of morbidity and mortality during hospital admittance (Jones et al., 1991).
Acute Alcohol-Induced Liver Injury
The current understanding of the effects of binge drinking on liver injury is not as complete as our knowledge regarding the effects of chronic ethanol exposure. There are, however, some parallels between acute and chronic alcohol exposure. Potential mechanisms by which acute alcohol causes liver injury are discussed below.
Mechanisms of Steatosis
As mentioned above, the first and most common hepatic change caused by alcohol consumption is steatosis, or fatty liver. Fat accumulation can be both macrovesicular (having one large fat droplet per hepatocyte and lateral displacement of the nucleus) or microvesicular (many small fat droplets per hepatocyte; Ishak et al., 1991). One mechanism by which alcohol exposure causes steatosis is directly via alcohol metabolism. Concentrations of alcohol can easily reach the mM range in the portal/hepatic circulation during alcohol consumption. In the process of metabolizing ethanol to acetate, two equivalents of reduced NADH are generated per equivalent of ethanol oxidized. This metabolism robustly increases the ratio of NADH:NAD+ within the cell, which then inhibits the β-oxidation of fatty acids in the liver. Furthermore, ethanol metabolism also increases the rate of esterification of fatty acids (Ontko, 1973). The net effect is to favor fat accumulation in the hepatocytes.
While alcohol metabolism may be sufficient to cause fat accumulation, it is not the only mechanism by which steatosis develops. Blocking lipopolysaccharide (LPS) signaling (Yin et al., 2001) and pro-oxidant producing enzymes (Kono et al., 2000; McKim et al., 2003) in rodents via genetic alterations also protect against steatosis, without altering ethanol metabolism. Fat accumulation can also be stimulated by mediators that alter lipid metabolism, such as pro-inflammatory cytokines (Grunfield et al., 1990). Signaling downstream of these cytokines contributes to hepatic fat accumulation in two ways – by increasing the deposition of fatty acids, and decreasing the breakdown and secretion of lipids into the circulation. For example, TNFα increases free fatty acid release from peripheral adipocytes (Hardardottir et al., 1992), increases de novo lipid synthesis (Feingold and Grunfeld, 1987), and inhibits β-oxidation of fatty acids (Nachiappan et al., 1994). Ultimately, these changes stimulate fat accumulation in the liver by increasing fatty acid supply, and concomitantly impairs the liver’s capacity for fatty acid metabolism and secretion.
Hepatic steatosis develops acutely in the majority of individuals consuming even moderate amounts of alcohol. Steatotic changes are also seen in rodent models of binge drinking (Kaiser et al., 2009; Donohue et al., 2011). Steatosis is considered an asymptomatic disease state, which readily reverses with abstinence (Bergheim et al., 2005). Indeed, at the level of the organism, hepatic steatosis can be viewed as a protective measure, as it partitions lipids away from the blood and stores them for potential later use (van Ginneken, 2008). However, although steatosis is an inert pathology per se, it sensitizes the liver to injury caused by a second insult (Day and James, 1998). According to this “two-hit hypothesis,” alcohol consumption alone does not cause progression of ALD from steatosis to steatohepatitis, cirrhosis, or fibrosis; rather, a second risk factor or insult is required for development of later stages of disease. Such a second insult could include an inflammatory response, reactive oxygen species, or hypoxia, among others. For example, acute ethanol exposure is well known to enhance liver pathology induced by xenobiotics, such as bacterial cell wall products (e.g., lipopolysaccharide). Furthermore, acute alcohol consumption itself could provide the second hit; for example, an episode of binge drinking can trigger entry into acute alcoholic hepatitis in a chronic alcoholic (Rivara et al., 1993; Barrio et al., 2004).
Dysregulation of Immunity and Inflammation
The two-hit hypothesis is mirrored on a molecular level by the concepts of “priming” and “sensitization” which are now considered to be fundamental to alcohol-induced liver injury (Tsukamoto et al., 2001). Here, “priming” refers to the ability of ethanol pre-exposure to cause inflammatory cells of the liver (e.g., Kupffer cells) to more robustly release pro-inflammatory cytokines in response to a second stimulus, such as LPS. Alcoholic hepatitis patients have larger amounts of circulating TNFα both basally and after stimulus (McClain and Cohen, 1989). Additionally, ethanol pre-exposure can prime Kupffer cells to LPS stimulation, resulting in enhanced TNF-α release (Enomoto et al., 1998). Ethanol can also sensitize cells, causing cell populations downstream of inflammatory cytokine signaling to respond more robustly. Patients with fatty liver are more sensitive to LPS-induced liver injury (Yang et al., 1997). This sensitization to LPS is recapitulated in rodent models of acute alcohol exposure (Bergheim et al., 2006; Beier et al., 2009).
Although some studies have shown that alcohol exposure primes macrophages to induce more inflammatory tissue damage, others have shown that acute alcohol intoxication (i.e., when alcohol is present in the system) actually impairs the immune response. For example, binge drinking increases the risk of infection (e.g., Gentilello et al., 1993; Gamble et al., 2006; Griffin et al., 2009). Furthermore, acute ethanol consumption worsens the prognosis of sepsis (Lin et al., 2009). A major mechanism by which alcohol can impair the immune response is via altering Toll-like receptor (TLR) signaling, which serve as pattern recognition receptors on macrophages and other cells of the innate immune system (Beutler, 2009). TLRs are involved in both direct cytotoxic and effector responses of macrophages, and therefore serve as key early initiators of an appropriate immune response. Importantly, ethanol intoxication has been shown to blunt the stimulation of macrophages by a number of TLR ligands, including zymosan A (TLR2) and LPS (TLR4; Pruett et al., 2004; Goral and Kovacs, 2005).
Although it may seem that these effects of acute ethanol on macrophage responsiveness are contradictory (i.e., both impairing and priming the macrophages), the timing of the inflammatory stimulus relative to ethanol exposure seems to be important. For example, acute ethanol (4–6 g/kg) blunted inflammatory liver damage caused by LPS, when LPS was injected 3 h after ethanol exposure (during intoxication), but enhanced damage if the LPS was administered 24 h after ethanol (Enomoto et al., 1998). The mechanisms by which acute ethanol causes both tolerance and priming of the innate immune system are still incompletely understood. However, it is clear that acute alcohol exposure produces a dangerous situation in which the likelihood of infection is increased during acute intoxication, but liver damage is enhanced by the inflammatory response to that infection.
Gut Permeability and Endotoxin
Even in the absence of infection, alcohol can alter the response to inflammatory stimuli. The association between increased circulating bacterial endotoxin (i.e., LPS) and liver injury has been established since the 1970s (Nolan, 2010). Circulating levels of LPS are increased in ALD patients (Bode et al., 1987; Fukui et al., 1991) as well as in experimental models of both acute (Lambert et al., 2003) and chronic (Nanji et al., 1994; Keshavarzian et al., 2001) alcohol consumption. Increased circulating LPS in response to alcohol consumption is due, at least in part, to increased gut permeability, which thereby allows translocation of LPS into the portal blood. In fact, elimination of intestinal bacteria reduces alcohol-induced liver injury via reduced circulating endotoxin levels in mice (Nanji et al., 1994). Experimental models of both chronic (Mathurin et al., 2000; Tamai et al., 2002) and acute (Tamai et al., 2000; Lambert et al., 2003) alcohol exposure showed that alcohol increased permeation of LPS. Acute alcohol consumption increased circulating levels of LPS in both healthy volunteers and alcoholics with ALD (Bode et al., 1987; Fukui et al., 1991).
Under normal conditions, the spaces between epithelial cells that make up the intestinal barrier are sealed by tight junctions. These tight junctions prevent transfer of toxic compounds, like LPS, into the circulation (Purohit et al., 2008). Changes in intestinal permeability caused by ethanol may be due in part to the disruption of apical junctional proteins in the small intestine. In vitro studies using Caco-2 enterocytes showed that alcohol can disrupt ZO-1 tight junctions, ultimately causing gaps in the paracellular space (Ma et al., 1999). Furthermore, chronic alcohol exposure reduces ZO-1 and occludin in the ileum of mice (Zhong et al., 2010). The same studies also demonstrated that the reduction in tight junction proteins was associated with oxidative stress in the intestine. Potential sources of oxidative stress in the GI tract after alcohol exposure include acetaldehyde and nitric oxide (Purohit et al., 2008). Similar mechanisms may be involved in gut permeability due to acute alcohol exposure.
Summary
Taken together, acute alcohol exposure can cause a “perfect storm” that favors inflammatory liver damage. Acute alcohol exposure enhances the risk of infection and permeability of the GI tract. Either or both mechanisms will increase the delivery of TLR ligands to macrophages in the liver. At later times after alcohol exposure, the inflammatory response of macrophages to TLR ligands is primed, and more cytotoxic cytokines (e.g., TNFα) are produced. Furthermore, steatosis in parenchymal cells sensitizes them to cytotoxic killing by the cytokines released by macrophages.
Models of Acute Ethanol Exposure
The majority of research on alcoholic liver injury has investigated the effects of chronic alcohol consumption. However, clinical evidence highlighting the detrimental effects of acute alcohol consumption has spurred recent interest in experimental models of binge drinking, intermittent heavy drinking, and other acute alcohol exposures. These models include in vitro and in vivo paradigms, with the latter spanning many species including rodents, dogs, primates, and micropigs (Dolganiuc and Szabo, 2009). The majority of acute alcohol research is performed in rodent models. Such models achieve pathological states (e.g., steatosis, inflammation) that resemble the early stages of liver injury seen in humans. The use of rodents by the research community has increased since the development of a variety of knockout and other genetically modified mouse strains that allow for more convenient and specific mechanistic studies.
One of the major obstacles in rodent models of alcohol exposure is their aversion to ethanol. Rodents do not voluntarily consume alcohol at concentrations that will recapitulate liver disease found in humans. Therefore, forced bolus administration by intragastric gavage is the most common approach to modeling acute alcohol abuse (Siegmund et al., 2005). Rodent models of acute alcohol exposure often fall into one of three categories: single bolus dose models, multiple bolus dose models, and “2-hit” models. Ethanol is administered at bolus doses of up to 6 g/kg, which takes into account differences in ethanol kinetics in rodents versus humans (Gershman and Steeper, 1991; Carson and Pruett, 1996). The peak concentrations of alcohol in rodents are similar to those achievable in humans during an acute alcohol binge.
Chronic models are costly, and it is often difficult to distinguish between effects and proximate causes in chronic models of liver damage. In addition to studying acute hepatotoxicity, acute models are therefore also useful as a screening tool and/or mechanistic analysis to complement chronic studies. This approach is supported by the observation that animals exposed to acute alcohol administration develop steatosis and inflammation in a manner similar to animals in chronic ethanol studies. Further strengthening this idea of “a model of a model” is the mechanistic overlap of protective agents in both acute and chronic alcohol exposure models. For example, Enomoto et al. (2000) demonstrated that compounds known to protect against liver damage in chronic exposure models [e.g., GdCl3 (Adachi et al., 1994), antibiotics (Adachi et al., 1995), nimodipine (Iimuro et al., 1996)] were also protective against steatosis in a rat model of acute ethanol exposure. Because of their use as models of chronic alcohol exposure, acute models of alcohol exposure are particularly valuable for screening compounds that may offer therapeutic benefit, as well as for the investigation of new mechanisms downstream of therapeutic compounds. Thus far, acute alcohol exposure models have lent few false positives when used to screen for efficacy of therapeutic molecules for chronic study.
PAI-1 and Fibrin – New Mediators in Acute Alcohol-Induced Liver Injury
Plasminogen Activator Inhibitor-1
Plasminogen activator inhibitor-1 (PAI-1) is an acute phase protein that plays an important role in injury and inflammation, at least in part via regulation of fibrinolysis. PAI-1 prevents the activation of plasmin and subsequent degradation of fibrin into fibrin degradation products (FDP) by inhibiting both the urokinase-type and tissue-type plasminogen activators (uPA and tPA; Figure 1; Kruithof, 1988). A role for PAI-1 in alcohol-induced liver injury was first seen in cirrhotic patients, who had an increased PA:PAI-1 ratio (Violi et al., 1992; Hu et al., 2001), resulting in hyperfibrinolysis. In contrast, during the development of ALD, levels of circulating PAI-1 are elevated and the PA:PAI-1 is lower, favoring hypofibrinolysis (Marques-Vidal et al., 1995; Dimmitt et al., 1998; Mukamal et al., 2001). Levels of PAI-1 correlate with the degree of lipid accumulation in experimental models of alcohol-induced liver steatosis (Alessi et al., 2003), and may be used as an index of severity of later stages of ALD (Tran-Thang et al., 1989).
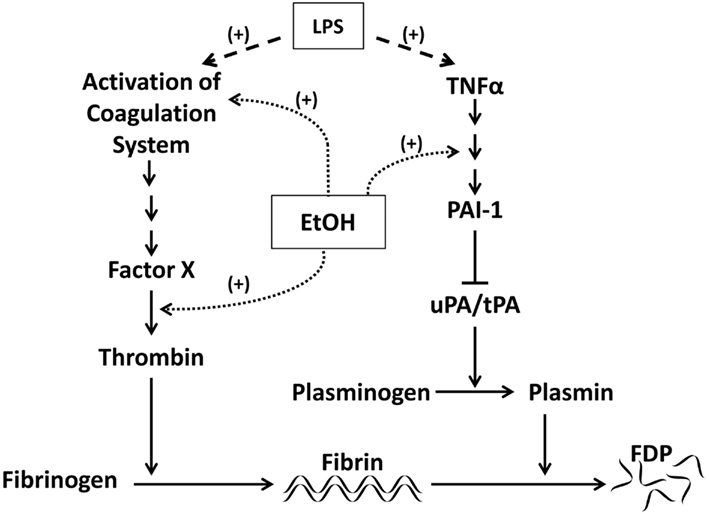
Figure 1. Effects of LPS and ethanol on PAI-1 and fibrinolysis. Activation of the coagulation cascade by LPS via thrombin results in accumulation of cross-linked fibrin. Cross-linked fibrin is degraded by plasmin. PAI-1 blocks fibrinolysis by inhibiting the plasminogen activators uPA and tPA and therefore the activation of plasmin. Both ethanol and LPS can modulate signaling downstream of the coagulation cascade, increasing fibrin deposition. Ethanol may also blunt fibrin degradation by altering signaling downstream of TNFα. LPS can have similar effects on fibrinolysis by stimulating release of TNFα.
While its importance in some diseases (e.g., atherosclerosis) has been established, the role of PAI-1 in ALD is still unclear. Studies using knockout mice strains, particularly PAI-1−/− mice, have begun to clarify the role of PAI-1 in lipid accumulation. Ma et al. (2004) showed that PAI-1−/− mice were protected from hepatic lipid accumulation in a model of high fat/high carbohydrate diet induced obesity. Additionally, our group has shown that both acute and chronic ethanol-induced steatosis correlate with hepatic PAI-1 expression, and that blocking PAI-1, either pharmacologically or genetically, confers protection against steatosis (Bergheim et al., 2006). In this study, TNFR1−/− mice were also protected against steatosis with concomitant blunting of PAI-1 (Bergheim et al., 2006), which suggests that the mechanism of PAI-1 induction after alcohol exposure is downstream of TNFα, possibly via MAPK signaling (Fearns and Loskutoff, 1997). PAI-1 also appears to be critical in mediating organ inflammation. For example, mice lacking PAI-1 were protected against leukocyte infiltration and tissue damage in a model of glomerulonephritis (Kitching et al., 2003). Work in this laboratory has shown that PAI-1−/− mice were nearly completely protected against inflammation caused by chronic alcohol (Arteel, 2008). How PAI-1 mediates inflammation, particularly after alcohol exposure, remains unclear. One potential mechanism is that PAI-1 mediates changes in the proteolytic activation/deactivation of cytokines important in the inflammatory response. These effects could include prevention of the activation of cytokines including transforming growth factor-β (Espevik et al., 1987; Sato et al., 1990), or stabilization of pro-inflammatory cytokines, such as the chemoattractant IL-8 (Marshall et al., 2003).
Fibrin
Another potential mechanism underlying the inflammatory effects of PAI-1 involves impaired fibrinolysis and subsequent fibrin accumulation (Holdsworth et al., 1979; Loike et al., 1995). Fibrin(ogen) is a structural component of the extracellular matrix (ECM). Upon activation of the coagulation cascade, the latent form, fibrinogen, is cleaved by thrombin into the active form, fibrin (Figure 1). Fibrin can crosslink to create clots or be degraded by plasmin into fibrinogen degradation products. In addition to its role in hemostasis, fibrin can also function as a signaling molecule via interaction with cell surface receptors. Fibrin’s role in inflammation has been highlighted by its ability to exert a myriad of pro-inflammatory effects. For example, Qi et al. (1997) showed that fibrin induces IL-8 expression in vascular endothelial cells. Fibrin matrices can regulate the migration of inflammatory cells in vitro, including IL-8-stimulated neutrophils (Loike et al., 1995). Additionally, defibrillation reduces macrophage migration in the kidney (Holdsworth et al., 1979).
Fibrin has been shown to have a direct role in experimental ALD as well. Our group has shown that fibrin deposition in hepatic sinusoids is increased by chronic ethanol consumption (Figure 2) and that fibrin accumulation correlates with inflammation in an acute-exposure model of alcohol-induced liver injury. Furthermore, blocking fibrin deposition either directly, using the thrombin inhibitor Hirudin, or indirectly, using the MEK-inhibitor U0126, protected against enhanced liver injury caused by alcohol pre-exposure (Beier et al., 2009). Blocking fibrin accumulation also protected against inflammation (Beier et al., 2009). These data implicate fibrin as an important player in enhanced inflammation and injury mediated by PAI-1 after alcohol exposure.
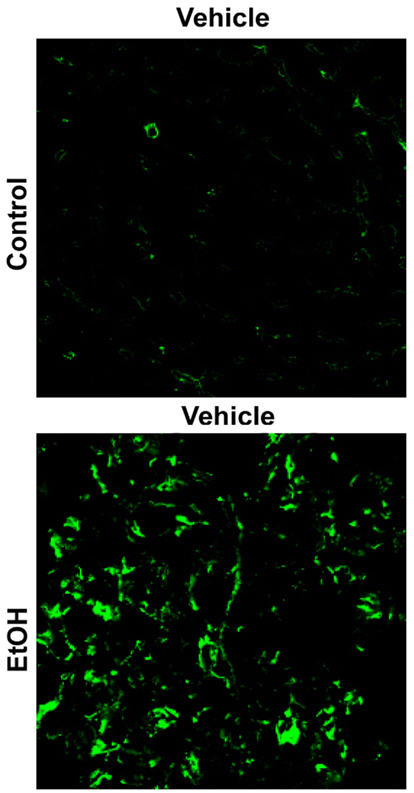
Figure 2. Chronic ethanol increases hepatic fibrin(ogen) deposition. Representative photomicrographs (40×) depicting immunofluorescent detection of fibrin(ogen) ECM are shown. Liver tissue is from mice fed either control diet or enteral ethanol diet for 4 weeks.
Integrins – Mediators of Cell-to-ECM Communication
While this group demonstrated a strong correlation between alcohol-induced fibrin deposition and hepatic inflammation and injury, the mechanism(s) by which fibrin is mediating its inflammatory effects remain unclear. One mechanism by which fibrin accumulation mediates inflammatory effects is via interaction with cell surface receptors such as integrins. Integrins are heterodimeric receptors expressed on many cell types including endothelial cells, inflammatory cells including macrophages and neutrophils, and platelets. Upon interaction with extracellular components, including surrounding matrices and cells, the beta subunit of an integrin undergoes a rapid conformational change. This change in integrin structure allows for transfer of information across the plasma membrane by activation of intracellular signaling pathways. Adapter proteins, including Integrin-Linked Kinase (ILK), are important mediators in signal transduction downstream of integrin activation and play a role in specifying which signaling cascades are activated within the cell. The integrins represent a diverse superfamily with at least 24 different heterodimers that are important in a variety of cell processes including proliferation, angiogenesis, and inflammation.
Fibrin(ogen) ECM is capable of interacting with the integrin family of cell surface adhesion receptors, such as the integrin αvβ3 (Cheresh et al., 1989). The role of integrin αvβ3 in tumor angiogenesis and metastasis is well documented, and stimulation of the αvβ3 integrin has been shown to contribute to inflammation (Zhou et al., 2009). Integrin αvβ3 is strongly expressed by endothelial cells (Carloni et al., 1996), including those that line the hepatic sinusoids (HSECs), which is a region that colocalizes with fibrin accumulation after alcohol exposure (Figure 2). Integrin αvβ3 binds fibrin(ogen) via an RGD sequence on the protein (Cheresh et al., 1989), which is pharmacologically targetable with small peptide antagonists. Studies by this group show that inhibition of αvβ3 using the small peptide antagonist, CycloRGDfV, protects against enhanced LPS-induced liver injury caused by acute alcohol in a “2-hit” model of acute alcohol-induced liver injury (Figure 3) without any effects on fibrin accumulation. Additionally, protection from liver injury by CycloRGDfV was concomitant with the blunting of LPS-induced neutrophil infiltration in this model (Figure 3). These data suggest that inflammation and liver injury due to LPS after acute alcohol exposure may be due to interaction between sinusoidal fibrin and integrin αvβ3.
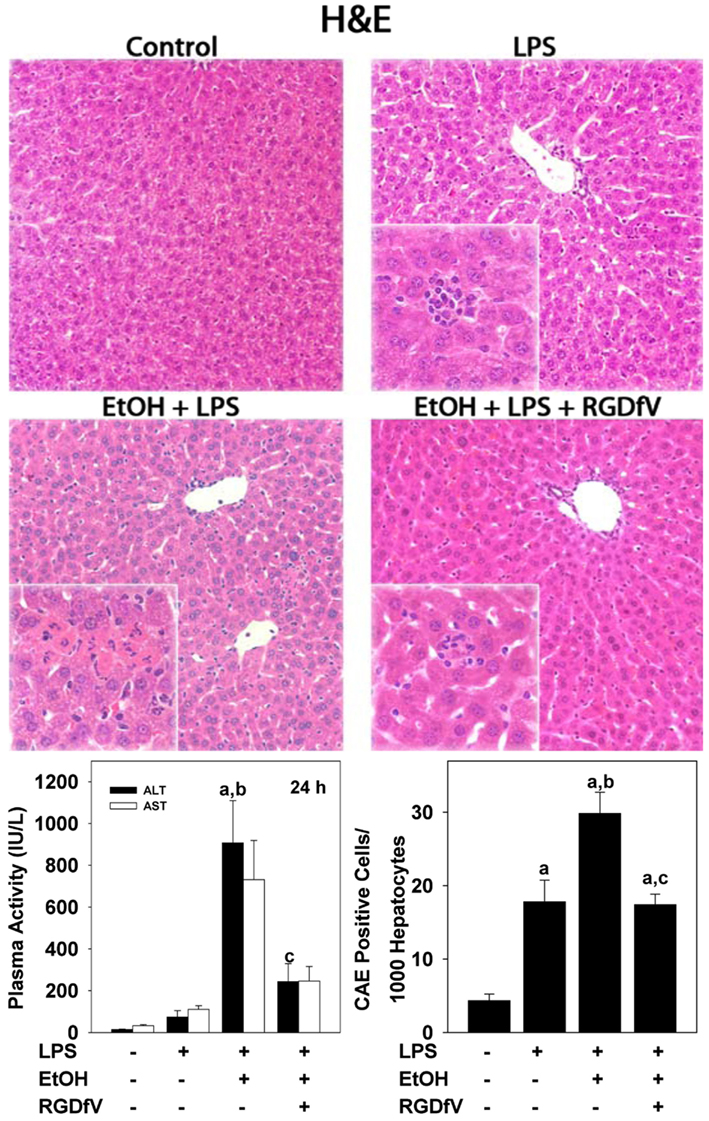
Figure 3. Inhibition of Integrin αvβ3 protects against enhanced LPS-induced liver injury caused by ethanol pre-exposure. Representative photomicrographs (100×; insets 400×) of hematoxylin and eosin staining are shown in upper panel. Circulating ALT levels and neutrophil migration (determined by CAE staining) are in the lower left and right panels, respectively. Animals were exposed to alcohol (6 g/kg i.g.) for 3 days, followed by LPS administration (10 mg/kg; i.p) 24 h after last dose of ethanol and were sacrificed 24 h after ethanol administration. Some animals received CycloRGDfV (3 mg/kg i.p.) 1 h prior to LPS.
Summary and Conclusion
A safe and effective therapy for ALD in humans is still elusive, despite significant advances in our understanding of how the disease is initiated and progresses. It is clear that alcohol binges not only can be acutely toxic to the liver, but also can contribute to the chronicity of ALD. Potential mechanisms by which acute alcohol causes damage include steatosis, dysregulated immunity and inflammation, and altered gut permeability. Recent interest in modeling acute alcohol exposure has yielded new insights into potential mechanisms of acute injury, that also may well be relevant for chronic ALD. Recent work by this group on the role of PAI-1 and fibrin metabolism in mediating acute alcohol-induced liver damage serve as an example of possible new targets that may be useful for alcohol abuse, be it acute or chronic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported, in part, by a grant from the National Institute of Alcohol Abuse and Alcoholism (AA003624). Veronica L. Massey was supported by a predoctoral (T32) fellowship from National Institute of Environmental Health Science (ES011564).
References
Adachi, Y., Bradford, B. U., Gao, W., Bojes, H. K., and Thurman, R. G. (1994). Inactivation of Kupffer cells prevents early alcohol-induced liver injury. Hepatology 20, 453–460.
Adachi, Y., Moore, L. E., Bradford, B. U., Gao, W., and Thurman, R. G. (1995). Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108, 218–224.
Alessi, M. C., Bastelica, D., Mavri, A., Morange, P., Berthet, B., Grino, M., and Juhan-Vague, I. (2003). Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Arterioscler. Thromb. Vasc. Biol. 23, 1262–1268.
Arteel, G. E. (2008). New role of plasminogen activator inhibitor-1 in alcohol-induced liver injury. J. Gastroenterol. Hepatol. 23, S54–S59.
Barrio, E., Tome, S., Rodriguez, I., Gude, F., Sanchex-Leira, J., Perez-Becerra, E., and Gonzalez-Quintela, A. (2004). Liver disease in heavy drinkers with and without alcohol withdrawal syndrome. Alcohol. Clin. Exp. Res. 28, 131–136.
Beier, J. I., Luyendyk, J. P., Guo, L., von Montfort, C., Staunton, D. E., and Arteel, G. E. (2009). Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology 49, 1545–1553.
Bergheim, I., Guo, L., Davis, M. A., Lambert, J. C., Beier, J. I., Duveau, I., Luyendyk, J. P., Roth, R. A., and Arteel, G. E. (2006). Metformin prevents alcohol-induced liver injury in the mouse: critical role of plasminogen activator inhibitor-1. Gastroenterology 130, 2099–2112.
Bergheim, I., McClain, C. J., and Arteel, G. E. (2005). Treatment of alcoholic liver disease. Dig. Dis. 23, 275–284.
Beutler, B. (2009). Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 227, 248–263.
Bode, C. H., Kugler, V., and Bode, J. C. H. (1987). Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 4, 8–14.
Carloni, V., Romanelli, R. G., Pinzani, M., Laffi, G., and Gentilini, P. (1996). Expression and function of integrin receptors for collagen and laminin in cultured human hepatic stellate cells. Gastroenterology 110, 1127–1136.
Carson, E. J., and Pruett, S. B. (1996). Development and characterization of a binge drinking model in mice for evaluation of the immunological effects of ethanol. Alcohol. Clin. Exp. Res. 20, 132–138.
Cheresh, D. A., Berliner, S. A., Vicente, V., and Ruggeri, Z. M. (1989). Recognition of distinct adhesive sites on fibrinogen by related integrins on platelets and endothelial cells. Cell 58, 945–953.
Crabb, D. W. (1999). Pathogenesis of alcoholic liver disease: newer mechanisms of injury. Keio. J. Med. 48, 184–188.
Day, C. P., and James, O. F. (1998). Steatohepatitis: a tale of two “hits?” Gastroenterology 114, 842–845.
Dimmitt, S. B., Rakic, V., Puddey, I. B., Baker, R., Oostryck, R., Adams, M. J., Chesterman, C. N., Burke, V., and Beilin, L. J. (1998). The effects of alcohol on coagulation and fibrinolytic factors: a controlled trial. Blood Coagul. Fibrinolysis 9, 39–45.
Dolganiuc, A., and Szabo, G. (2009). In vitro and in vivo models of acute alcohol exposure. World J. Gastroenterol. 15, 1168–1177.
Donohue, T. M. Jr., Osna, N., Trambly, C., Whitaker, N., Thomes, P., Todero, S., and Davis, J. S. (2011). Early growth response-1 contributes to steatosis development after acute ethanol administration. Alcohol. Clin. Exp. Res. 36, 759–767.
Enomoto, N., Ikejima, K., Bradford, B. U., Rivera, C. A., Kono, H., Brenner, D. A., and Thurman, R. G. (1998). Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology 115, 443–451.
Enomoto, N., Ikejima, K., Yamashina, S., Enomoto, A., Nishiura, T., Nishimura, T., Brenner, D. A., Schemmer, P., Bradford, B. U., Rivera, C. A., Zhong, Z., and Thurman, R. G. (2000). Kupffer cell-derived prostaglandin E(2) is involved in alcohol-induced fat accumulation in rat liver. Am. J. Physiol. 279, G100–G106.
Espevik, T., Figari, I. S., Shalaby, M. R., Lackides, G. A., Lewis, G. D., Shepard, H. M., and Palladino, M. A. Jr. (1987). Inhibition of cytokine production by cyclosporin A and transforming growth factor beta. J. Exp. Med. 166, 571–576.
Fearns, C., and Loskutoff, D. J. (1997). Induction of plasminogen activator inhibitor 1 gene expression in murine liver by lipopolysaccharide. Cellular localization and role of endogenous tumor necrosis factor-alpha. Am. J. Pathol. 150, 579–590.
Feingold, K. R., and Grunfeld, C. (1987). Tumor necrosis factor-alpha stimulates hepatic lipogenesis in the rat in vivo. J. Clin. Invest. 80, 184–190.
Fukui, H., Brauner, B., Bode, J. C., and Bode, C. (1991). Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J. Hepatol. 12, 162–169.
Gamble, L., Mason, C. M., and Nelson, S. (2006). The effects of alcohol on immunity and bacterial infection in the lung. Med. Mal. Infect. 36, 72–77.
Gentilello, L. M., Cobean, R. A., Walker, A. P., Moore, E. E., Wertz, M. J., and Dellinger, E. P. (1993). Acute ethanol intoxication increases the risk of infection following penetrating abdominal trauma. J. Trauma. 35, 669–674.
Gershman, H., and Steeper, J. (1991). Rate of clearance of ethanol from the blood of intoxicated patients in the emergency department. J. Emerg. Med. 9, 307–311.
Goral, J., and Kovacs, E. J. (2005). In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J. Immunol. 174, 456–463.
Griffin, R., Poe, A. M., Cross, J. M., Rue, L. W., and McGwin, G. (2009). The association between blood alcohol level and infectious complications among burn patients. J. Burn Care Rehabil. 30, 395–399.
Grunfield, C., Soued, M., Adi, S., Moser, A. H., Dinarello, C. A., and Feingold, K. R. (1990). Evidence for two classes of cytokines that stimulate hepatic lipogenesis: relationships among tumor necrosis factor, interleukin-1 and interferon-alpha. Endocrinology 127, 46–54.
Hardardottir, I., Doerrler, W., Feingold, K. R., and Grunfeld, C. (1992). Cytokines stimulate lipolysis and decrease lipoprotein lipase activity in cultured fat cells by a prostaglandin independent mechanism. Biochem. Biophys. Res. Commun. 186, 237–243.
Holdsworth, S. R., Thomson, N. M., Glasgow, E. F., and Atkins, R. C. (1979). The effect of defibrination on macrophage participation in rabbit nephrotoxic nephritis: studies using glomerular culture and electronmicroscopy. Clin. Exp. Immunol. 37, 38–43.
Hu, K. Q., Yu, A. S., Tiyyagura, L., Redeker, A. G., and Reynolds, T. B. (2001). Hyperfibrinolytic activity in hospitalized cirrhotic patients in a referral liver unit. Am. J. Gastroenterol. 96, 1581–1586.
Iimuro, Y., Ikejima, K., Rose, M. L., Bradford, B. U., and Thurman, R. G. (1996). Nimodipine, a dihydropyridine-type calcium channel blocker, prevents alcoholic hepatitis due to chronic intragastric ethanol exposure in the rat. Hepatology 24, 391–397.
Ishak, K. G., Zimmerman, H. J., and Ray, M. B. (1991). Alcoholic liver disease: pathologic, pathogenetic and clinical aspects. Alcohol. Clin. Exp. Res. 15, 45–66.
Jones, J. D., Barber, B., Engrav, L., and Heimback, D. (1991). Alcohol use and burn injury. J. Burn Care Rehabil. 12, 148–152.
Kaiser, J. P., Beier, J. I., Zhang, J., David, H. J., von Montfort, C., Guo, L., Zheng, Y., Monia, B. P., Bhatnagar, A., and Arteel, G. E. (2009). PKCepsilon plays a causal role in acute ethanol-induced steatosis. Arch. Biochem. Biophys. 482, 104–111.
Keshavarzian, A., Choudhary, S., Holems, E. W., Yong, S., Banan, A., Jakate, S., and Fields, J. Z. (2001). Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J. Pharmacol. Exp. Ther. 299, 442–448.
Kitching, A. R., Kong, Y. Z., Huang, X. R., Davenport, P., Edgtton, K. L., Carmeliet, P., Holdsworth, S. R., and Tipping, P. G. (2003). Plasminogen activator inhibitor-1 is a significant determinant of renal injury in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 14, 1487–1495.
Kono, H., Rusyn, I., Yin, M., Gabele, E., Yamashina, S., Dikalova, A., Kadiiska, M. B., Connor, H. D., Mason, R. P., Segal, B. H., Bradford, B. U., Holland, S. M., and Thurman, R. G. (2000). NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Invest. 106, 867–872.
Lambert, J. C., Zhou, Z., Wang, L., Song, Z., McClain, C. J., and Kang, Y. J. (2003). Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J. Pharmacol. Exp. Ther. 305, 880–886.
Lin, J. N., Tsai, Y. S., Lai, C. H., Chen, Y. H., Tsai, S. S., Lin, H. L., Huang, C. K., and Lin, H. H. (2009). Risk factors for mortality of bacteremic patients in the emergency department. Acad. Emerg. Med. 16, 749–755.
Loike, J. D., el Khoury, J., Cao, L., Richards, C. P., Rascoff, H., Mandeville, J. T., Maxfield, F. R., and Silverstein, S. C. (1995). Fibrin regulates neutrophil migration in response to interleukin 8, leukotriene B4, tumor necrosis factor, and formyl-methionyl-leucyl-phenylalanine. J. Exp. Med. 181, 1763–1772.
Ma, L. J., Mao, S. L., Taylor, K. L., Kanjanabuch, T., Guan, Y., Zhang, Y., Brown, N. J., Swift, L. L., McGuinness, O. P., Wasserman, D. H., Vaughan, D. E., and Fogo, A. B. (2004). Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 53, 336–346.
Ma, T. Y., Nguyen, D., Bui, V., Nguyen, H., and Hoa, N. (1999). Ethanol Modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 276, G965–G974.
Marques-Vidal, P., Cambou, J. P., Nicaud, V., Luc, G., Evans, A., Arveiler, D., Bingham, A., and Cambien, F. (1995). Cardiovascular risk factors and alcohol consumption in France and Northern Ireland. Atherosclerosis 115, 225–232.
Marshall, L. J., Ramdin, L. S., Brooks, T., DPhil, P. C., and Shute, J. K. (2003). Plasminogen activator inhibitor-1 supports IL-8-mediated neutrophil transendothelial migration by inhibition of the constitutive shedding of endothelial IL-8/heparan sulfate/syndecan-1 complexes. J. Immunol. 171, 2057–2065.
Mathurin, P., Deng, Q. G., Keshavarzian, A., Choudhary, S., Holmes, E. W., and Tsukamoto, H. (2000). Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 32, 1008–1017.
McClain, C. J., and Cohen, D. A. (1989). Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology 9, 349–351.
McKim, S. E., Gabele, E., Isayama, F., Lambert, J. C., Tucker, L. M., Wheeler, M. D., Connor, H. D., Mason, R. P., Doll, M. A., Hein, D. W., and Arteel, G. E. (2003). Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology 125, 1834–1844.
Mukamal, K. J., Jadhav, P. P., D’Agostino, R. B., Massaro, J. M., Mittleman, M. A., Lipinska, I., Sutherland, P. A., Matheney, T., Levy, D., Wilson, P. W., Ellison, R. C., Silbershatz, H., Muller, J. E., and Tofler, G. H. (2001). Alcohol consumption and hemostatic factors: analysis of the Framingham offspring cohort. Circulation 104, 1367–1373.
Nachiappan, V., Curtiss, D., Corkey, B. E., and Kilpatrick, L. (1994). Cytokines inhibit fatty acid oxidation in isolated rat hepatocytes: synergy among TNF, IL-6 and IL-1. Shock 1, 123–129.
Nanji, A. A., Khettry, U., and Sadrzadeh, S. M. H. (1994). Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver disease. Proc. Soc. Exp. Biol. Med. 205, 243–247.
Nolan, J. P. (2010). The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology 52, 1829–1835.
Ontko, J. A. (1973). Effects of ethanol on the metabolism of free fatty acids in isolated liver cells. J. Lipid Res. 14, 78–86.
Pruett, S. B., Zheng, Q., Fan, R., Matthews, K., and Schwab, C. (2004). Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol 33, 147–155.
Purohit, V., Bode, J. C., Bode, C., Brenner, D. A., Choudhry, M. A., Hamilton, F., Kang, Y. J., Keshavarzian, A., Rao, R., Sartor, R. B., Swanson, C., and Turner, J. R. (2008). Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 42, 349–361.
Qi, J., Goralnick, S., and Kreutzer, D. L. (1997). Fibrin regulation of interleukin-8 gene expression in human vascular endothelial cells. Blood 90, 3595–3602.
Radek, K. A., Kovacs, E. J., and Dipietro, L. A. (2012). Matrix proteolytic activity during wound healing: modulation by acute ethanol exposure. Alcohol. Clin. Exp. Res. 31, 1045–1052.
Radek, K. A., Matthies, A. M., Burns, A. L., Heinrich, S. A., Kovacs, E. J., and Dipietro, L. A. (2005). Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am. J. Physiol. Heart Circ. Physiol. 289, 1084–1090.
Rivara, F. P., Jurkovich, G. J., Gurney, J. G., Seguin, D., Fligner, C. L., Ries, R., Raisys, V. A., and Copass, M. (1993). The magnitude of acute and chronic alcohol abuse in trauma patients. Arch. Surg. 128, 907–912.
Sato, Y., Tsuboi, R., Lyons, R., Moses, H., and Rifkin, D. B. (1990). Characterization of the activation of latent TGF-beta by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self-regulating system. J. Cell Biol. 111, 757–763.
Siegmund, S. V., Haas, S., and Singer, M. V. (2005). Animal models and their results in gastrointestinal alcohol research. Dig. Dis. 23, 181–194.
Substance Abuse and Mental Health Services Administration. (2011). Results from the 2010 National Survey on Drug Use and Health: Summary of National Findings. Rockville, MD: Substance Abuse and Mental Health Services Administration.
Tamai, H., Horie, Y., Kato, S., Yokoyama, H., and Ishii, H. (2002). Long-term ethanol feeding enhances susceptibility of the liver to orally administered lipopolysaccharides in rats. Alcohol. Clin. Exp. Res. 26, 75S–80S.
Tamai, H., Kato, S., Horie, Y., Ohki, E., Yokoyama, H., and Ishii, H. (2000). Effect of acute ethanol administration on the intestinal absorption of endotoxin in rats. Alcohol. Clin. Exp. Res. 24, 390–394.
Tran-Thang, C., Fasel-Felley, J., Pralong, G., Hofstetter, J. R., Bachmann, F., and Kruithof, E. K. (1989). Plasminogen activators and plasminogen activator inhibitors in liver deficiencies caused by chronic alcoholism or infectious hepatitis. Thromb. Haemost. 62, 651–653.
Tsukamoto, H., Takei, Y., McClain, C. J., Joshi-Barve, S., Hill, D., Schmidt, J., Deaciuc, I., Barve, S., Colell, A., Garcia-Ruiz, C., Kaplowitz, N., Fernandez-Checa, J. C., Yokoyama, H., Okamura, Y., Nakamura, Y., Ishii, H., Chawla, R. K., Barve, S., Joshi-Barve, S., Watson, W. H., Nelson, W. J., Lin, M., Ohata, M., Motomura, K., Enomoto, K., Ikejima, K., Kitamura, T., Oide, H., Hirose, M., Bradford, B. U., Rivera, C. A., Kono, H., Peter, S., Yamashina, S., Konno, A., Ishikawa, H., Shimizu, H., Sato, N., and Thurman, R. (2001). How is the liver primed or sensitized for alcoholic liver disease? Alcohol. Clin. Exp. Res. 25, 171S–181S.
U. S. Department of Health and Human Services. (2000). 10th Special Report to the U. S. Congress on Alcohol and Research.
van Ginneken, V. J. (2008). Liver fattening during feast and famine: an evolutionary paradox. Med. Hypotheses 70, 924–928.
Violi, F., Ferro, D., Basili, S., Quintarelli, C., Saliola, M., Alessandri, C., Cordova, C., and Balsano, F. (1992). Hyperfibrinolysis increases the risk of gastrointestinal hemorrhage in patients with advanced cirrhosis. Hepatology 15, 672–676.
Weschsler, H., and Austin, S. B. (1998). Binge drinking: the five/four measure. J. Stud. Alcohol 59, 122–124.
Yang, S. Q., Lin, H. Z., Lane, M. D., Clemens, M., and Diehl, A. M. (1997). Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. U.S.A. 94, 2557–2562.
Yin, M., Bradford, B. U., Wheeler, M. D., Uesugi, T., Froh, M., Goyert, S. M., and Thurman, R. G. (2001). Reduced early alcohol-induced liver injury in cd14-deficient mice. J. Immunol. 166, 4737–4742.
Zhong, W., McClain, C. J., Cave, M., Kang, Y. J., and Zhou, Z. (2010). The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am. J. Physiol. 298, G625–G633.
Keywords: alcohol, acute, hepatic, inflammation, steatosis
Citation: Massey VL and Arteel GE (2012) Acute alcohol-induced liver injury. Front. Physio. 3:193. doi: 10.3389/fphys.2012.00193
Received: 16 April 2012; Accepted: 22 May 2012;
Published online: 12 June 2012.
Edited by:
Ali Canbay, University Hospital Essen, GermanyReviewed by:
Wing-Kin Syn, Foundation for Liver Research, UKChristian Rust, University of Munich, Germany
Copyright: © 2012 Massey and Arteel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gavin E. Arteel, Department of Pharmacology and Toxicology, University of Louisville Health Sciences Center, 505 South Hancock Street, CTRB Room 506, Louisville, KY 40292, USA. e-mail:Z2F2aW4uYXJ0ZWVsQGxvdWlzdmlsbGUuZWR1