

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 01 June 2012
Sec. Striated Muscle Physiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00151
This article is part of the Research Topic Muscle fatigue and muscle weakness: what we know and what we wish we did View all 10 articles
The depression in force and/or velocity associated with muscular fatigue can be the result of a failure at any level, from the initial events in the motor cortex of the brain to the formation of an actomyosin cross-bridge in the muscle cell. Since all the force and motion generated by muscle ultimately derives from the cyclical interaction of actin and myosin, researchers have focused heavily on the impact of the accumulation of intracellular metabolites [e.g., Pi, H+ and adenosine diphoshphate (ADP)] on the function these contractile proteins. At saturating Ca++ levels, elevated Pi appears to be the primary cause for the loss in maximal isometric force, while increased [H+] and possibly ADP act to slow unloaded shortening velocity in single muscle fibers, suggesting a causative role in muscular fatigue. However the precise mechanisms through which these metabolites might affect the individual function of the contractile proteins remain unclear because intact muscle is a highly complex structure. To simplify problem isolated actin and myosin have been studied in the in vitro motility assay and more recently the single molecule laser trap assay with the findings showing that both Pi and H+ alter single actomyosin function in unique ways. In addition to these new insights, we are also gaining important information about the roles played by the muscle regulatory proteins troponin (Tn) and tropomyosin (Tm) in the fatigue process. In vitro studies, suggest that both the acidosis and elevated levels of Pi can inhibit velocity and force at sub-saturating levels of Ca++ in the presence of Tn and Tm and that this inhibition can be greater than that observed in the absence of regulation. To understand the molecular basis of the role of regulatory proteins in the fatigue process researchers are taking advantage of modern molecular biological techniques to manipulate the structure and function of Tn/Tm. These efforts are beginning to reveal the relevant structures and how their functions might be altered during fatigue. Thus, it is a very exciting time to study muscle fatigue because the technological advances occurring in the fields of biophysics and molecular biology are providing researchers with the ability to directly test long held hypotheses and consequently reshaping our understanding of this age-old question.
The quest to identify the cause(s) of muscle fatigue has been a quintessential question in the field of physiology for more than 100 years. In that time our understanding of the etiology of fatigue has evolved greatly and we have a greater appreciation for the complexity of the phenomenon, recognizing that there are many potential factors that may contribute to fatigue. For example, the rate and extent of fatigue is highly dependent of the mode and intensity of contractility activity (Fitts, 1994). Based on these observations it is now clear that, in general, the factors that cause a muscle to fatigue from low intensity stimulation are distinctly different from the factors that elicit fatigue from high intensity stimulation (Fitts, 1994). This review focuses on the recent revelations with regard to fatigue resulting from short and intense bouts of contractile activity (i.e., high intensity fatigue) with a particular focus on the role of the cross-bridge cycle. This type of muscle fatigue was classically defined by a decrease in force in response to repeated intense contractile activity, however this definition has been broadened to a decrease in the expected or required power output (Fitts, 1994). This revision has two important implications; firstly it indicates that fatigue can occur at submaximal, as well as maximal, contractile intensities; and secondly that fatigue can result from a drop in force and/or the velocity of contraction. This more widely accepted definition of fatigue now defines for researchers in the field the parameters on which to focus on or identifying the underlying causes of this kind of transient loss of muscle function.
Work in the late 1970s and early 1980s used NMR spectroscopy on in vivo muscle to establish that the accumulation of metabolites, principally hydrogen ions (H+, i.e., acidosis), inorganic phosphate (Pi), and adenosine diphosphate (ADP), were correlated with the development of fatigue in response to intense bouts of contractile activity (Dawson et al., 1978). Parallel efforts using chemically skinned single muscle fibers demonstrated that elevated levels of these ions directly inhibit muscle’s ability to generate maximal isometric force and unloaded shortening velocity (Cooke et al., 1988), providing strong evidence for a causative role in fatigue. While it is clear from the skinned single muscle fiber studies that elevated levels of metabolites directly affect the force and motion generating capacity of muscle, it is still not clear how this occurs at a molecular level. More sophisticated experiments in single fibers led to hypotheses about how these ions might inhibit force and velocity at the level of a single cross-bridge including how Pi might rebind to myosin and reverse the weak to strong-binding transition (Hibberd et al., 1985; Dantzig et al., 1992). Our current understanding of the role of the cross-bridge cycle in fatigue based on muscle fiber experiments has recently been reviewed (Fitts, 2008). In the present review we examine the research at the molecular level largely incorporating in vitro findings that provide more detailed insight into the underlying mechanisms of putative agents of fatigue on actomyosin function. The big advantage of using these methods is that the behavior of a single cross-bridge can be directly observed rather than inferred from the properties of a whole muscle or even single muscle fiber where the parameters measured represent the collective action of more than a billion individual cross-bridges. Furthermore, intact muscle contains a host of proteins in addition to actin and myosin that act to modulate and regulate contractile function, making it difficult to isolate which proteins are mediating the effects of fatigue. For these reasons researchers have resorted to in vitro approaches to understand both which proteins are involved and how the function of a single cross-bridge is affected.
Great technological advances in the fields of biophysics and molecular biology are now enabling researchers to gain unprecedented insight into some of the most fundamental mechanisms underlying the loss of the force and motion generating capacities of muscle during fatigue. It is these recent efforts that will be the focus of this review. It is important to note that at the molecular level the efforts to understand muscle fatigue are often confluent with the efforts to understand the basic molecular mechanism of contraction and thus this review incorporates some literature focused on the basic mechanism of contraction as it pertains to understand fatigue.
The force and motion generated by muscle are ultimately the result of the cyclical interaction of myosin and actin in a process coupled to the hydrolysis of ATP. This process, referred to as the cross-bridge cycle, links myosin’s ATPase cycle with the mechanical events that drive force and motion. Although many of the specific details remain controversial, extensive study over many years has provided a basic model for how a myosin molecule converts the energy from ATP hydrolysis into force and motion (Holmes and Geeves, 2000). This simple working model of the cross-bridge cycle that incorporates the salient features of the cycle can be used to explore the molecular basis of fatigue (Figure 1). In this basic model, Pi-release is closely associated with myosin’s lever arm rotation, the key molecular event responsible for force and motion in muscle. This event is followed by two kinetic transitions that occur while myosin is strongly bound to actin. In the first step myosin, now strongly bound to actin, releases ADP from the active site putting actomyosin in a rigor state; subsequently, in the second step, the cross-bridge waits in a rigor state until a new ATP molecule rebinds to myosin’s active site and facilitates the dissociation from actin. While myosin is detached (or more precisely weakly bound) from actin, ATP is hydrolyzed, a biochemical event coupled to resetting of the lever arm, ensuring that the next binding event causes another productive displacement. It is important to point out that more complex models have been proposed based on recent in vitro findings including additional AM.ADP states that could be coupled to a structural change in the position of the lever arm (Capitanio et al., 2006) and strongly strongly bound pre-powerstroke states (Takagi et al., 2004) but this simple model is consistent with much of the data from in vitro experiments (Palmiter et al., 1999; Baker et al., 2002; Debold et al., 2008, 2010) and provides the best starting point for understanding the effects of elevated levels of Pi, H+, and ADP on actomyosin. In fact, years of investigations using skinned muscle fibers have led to several hypotheses regarding how elevated levels of Pi, H+ and ADP during fatigue could directly inhibit specific steps in the cross-bridge cycle (Cooke, 2007). More recent in vitro approaches using isolated proteins have attempted to address these hypotheses more directly by taking advantage of the advances in the field of biophysics (Debold et al., 2008, 2011).
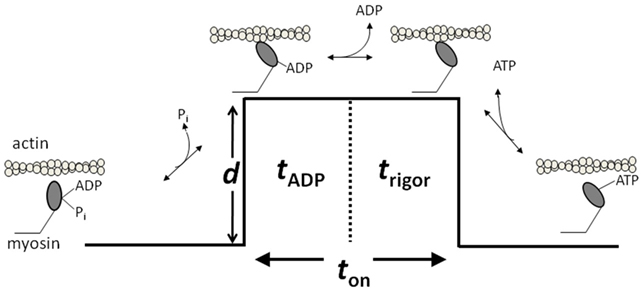
Figure 1. A model of the cross-bridge cycle depicting the biochemical mechanical events. d indicates the powerstroke that causes the displacement. ton refers to the duration of strong actomyosin strong-binding. ton is made up of tADP corresponding to the ADP-lifetime and the rigor lifetime trigor. Figure is modified from Debold et al. (2011), reprinted with permission from the American Physiological Society.
As far back as 1880 acidosis was known to increase in contracting muscle and was postulated to be directly involved in the loss of force during periods of stress (Gaskell, 1880). The idea that the build-up of acidosis was causative in fatigue gained support from observations that the decrease in muscle performance could be temporally correlated with a decrease in the intracellular pH (Dawson et al., 1978). Consistent with this hypothesis early work on isolated muscle suggested that fatiguing levels of acidosis inhibit force production (Donaldson et al., 1978; Fabiato and Fabiato, 1978; Edman and Lou, 1990; Kentish, 1991; Ricciardi et al., 1994). However, more recent work in isolated single fibers demonstrates that the effect of acidosis on force is highly temperature-dependent (Pate et al., 1995). In fact, at physiological temperatures most studies now indicate that there is little, if any, effect of acidosis on maximal isometric force (Pate et al., 1995; Knuth et al., 2006). Some authors have even suggested that acidosis may actually prevent, rather than contribute to, the loss in force during fatigue (Pedersen et al., 2004), but this remains controversial (Kristensen et al., 2005). Thus at the very least it appears that acidosis contributes little if at all to the decrease in force during fatigue, at least at saturating levels of intracellular Ca++ (see Effects of Acidosis on Muscle Activation).
In contrast to its minimal effects on force, there is good evidence from muscle fibers that it may play a role in slowing the velocity of contraction during fatigue (Knuth et al., 2006; Karatzaferi et al., 2008). Single fiber studies demonstrate that decreasing pH from a resting value (∼7.0) to a value reached during fatigue (∼6.2) can slow unloaded shortening velocity by over 30% even near physiological temperatures (30°C). These findings suggest acidosis may have other effects on the actomyosin interaction, specifically that it might slow the step in the cross-bridge cycle that limits shortening velocity. However, since these studies were done in muscle fibers it is not clear exactly which step in the cross-bridge cycle might be affected or which of the contractile proteins are helping to mediate the effect.
Confirmation that acidosis can directly affect the actomyosin interaction was demonstrated using an in vitro motility assay (a measure analogous to unload shortening velocity), where the velocity at which isolated muscle myosin translocates fluorescently labeled actin filaments can be quantified (Debold et al., 2008). Increasing acidosis from a resting level (7.4) to fatiguing levels (6.4), near physiological temperatures (30°C), in this assay decreases actin filament velocity (Vactin) by over 65% (Debold et al., 2008), a finding qualitatively consistent with observations in single muscle fibers (Knuth et al., 2006). Interestingly, the acidosis-induced decrease in Vactin is greater than that observed in fibers, suggesting that the structure of the sacromere and/or the presence of additional contractile proteins present in intact muscle may attenuate some of the loss in unloaded shortening velocity. For example, the highly ordered arraignment of the thick filaments in muscle fibers absent in in vitro assays where myosin is randomly coated on the surface. And there is evidence that the orientation of myosin to actin affects the single molecule mechanics (Tanaka et al., 1998). Alternatively, the difference in magnitude may be related to the absence of the regulatory proteins in the in vitro assays discussed above. Here there is strong evidence for the involvement of the troponin and tropomyosin in the depressive effects of acidosis (as detailed below) and these proteins may act to attenuate the magnitude of the acidosis (Fujita and Ishiwata, 1999). It will be important and informative to reconcile this difference as we attempt to understand the potential role of acidosis in slowing velocity during fatigue.
In the simplest model (Huxley, 1990), Vactin is proportional to myosin’s unitary step size (d) and the duration of the actomyosin strong-binding (ton; i.e., Vactin = d/ton). Using this paradigm the acidosis-induced decrease in Vactin could result from either a decrease in d and/or an increase in ton. The advent of the three-bead laser trap assay (Finer et al., 1994) now provides an unprecedented means of directly determining which single molecule parameter is affected by acidosis (Figure 2). Using this assay Debold et al. (2008) found that while d was largely unaffected by acidosis at 30°C, but that ton was increased by almost threefold at low pH, which could quantitatively account for the 65% decrease in Vactin that was observed in the in vitro motility assay (Debold et al., 2008).
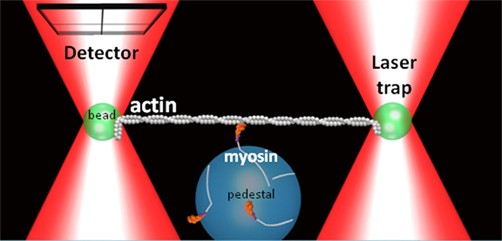
Figure 2. Three-bead laser trap assay. A schematic representation of the three-bead laser trap assay showing a single myosin molecule interacting with a single actin filament. A quadrant photodiode detector tracks the motion a trapped bead connected to the actin filament.
As illustrated in Figure 1, ton is composed of two biochemical states of myosin; the ADP bound state (AM.ADP) and the rigor state (AM). By manipulating the ATP concentration in the single molecule laser trap assay one can further delineate whether the prolongation of ton results from a slowing of the rate of ADP release or the rate of ATP-induced dissociation from rigor. The results of this series of experiments suggested that acidosis has little effect on the rate of ATP-induced dissociation of actin and myosin, but increases the duration of the ADP-bound state by over threefold (Debold et al., 2008). The latter effect can fully explain the acidosis-induced increase in ton and therefore the slowing of Vactin at a fatiguing level acidosis. Thus these findings provided some of the first direct evidence of the effects of a fatiguing level of acidosis on a single cross-bridge, suggesting that the effect can be attributed to a single biochemical transition in myosin’s cross-bridge cycle.
Now that this information is known what we wish we knew is the specific structures and mechanisms responsible for mediating this effect. Given the knowledge gained from revelation of both the full amino acid sequence and myosin’s atomic structure (Rayment et al., 1993) this task may now be a less daunting one. An interesting starting point to identify the important structures would be to take advantage of the differential sensitivity to acidosis of fast type II and slow type I muscle fibers (Metzger and Moss, 1990). Fibers expressing the fast type II, IIx, or IIb myosin heavy chain (MHC) seem to be more sensitive to a decrease in acidosis than fibers with slow type I MHC. A Comparison of all the different residues of each protein might be inefficient, however since velocity is strongly governed by the rate of ADP-release (Siemankowski et al., 1985) the structures affecting this rate present an attractive area to target in myosin’s heavy chain. The sequences for type I and each type II isoform (IIa, IIx, and IIb) are highly homologous except for the surface binding loops (Chikuni et al., 2004). And there is evidence that these binding loops can influence the overall ATPase rate, shortening velocity, and the ADP-release rate (Kurzawa-Goertz et al., 1998) therefore this may be a key area to probe the differential sensitivity to acidosis.
In addition to direct effects on the actomyosin cross-bridge cycle, acidosis is also thought to indirectly affect the actomyosin interaction by altering the Ca++ sensitivity of muscle. A decreased Ca++-sensitivity means that less force will be produced at the same level of activation, thus unlike the reduced effect of acidosis observed at saturating Ca++ levels (Pate et al., 1995) if acidosis disrupts muscle activation force will be compromised at sub-saturating Ca++ levels. This may play a particularly important role during the latter stages of fatigue, when the intracellular [Ca++] is thought to compromised, due to decreased release from the sarcoplasmic reticulum (Lee et al., 1991).
Muscle activation starts when a complex series of molecular motions following the binding of Ca++ to TnC ultimately lead to the movement of tropomyosin away from a position where it blocks the myosin-binding sites on actin. Improvements in biophysical techniques combined with new high resolution structures of the contractile proteins have revealed new details of this process (Galinska-Rakoczy et al., 2008). These findings are leading to more sophisticated hypotheses about how contraction is regulated at the molecular level and will be crucial to understanding how this process might be disrupted during fatigue. Much of these new data support a model that posits that Tm oscillates between three distinct positions on actin (McKillop and Geeves, 1993); the first being a “Blocked-state” where the myosin binding sites on actin are completely unavailable; a second, “Closed-state” where Tm only reveals the weak binding sites on actin; and a third, “Open-state” in which the sites for strong myosin-binding are available and myosin can therefore generate force and motion (Gordon et al., 2000). In this model the binding of Ca++ to TnC increases occupancy of the Closed-state, but only after myosin strongly binds to actin in the Open-state is the filament fully activated. Thus full activation of the thin filament requires both Ca++ dependent process and myosin being strongly bound to actin. We also know that this process is highly cooperative, meaning that the binding of one myosin increases the probability that neighboring myosin molecules will bind to actin. And there is evidence that the putative agents of fatigue might affect this cooperative behavior (Debold et al., 2006), which would likely show the greatest alterations under conditions where Ca++ release is compromised.
Support for the above model of activation and its cooperative properties have been provided from structural evidence showing that Tm can exist in three distinct positions on actin (Galinska-Rakoczy et al., 2008). In addition, recent single molecule laser trap experiments using actin filaments reconstituted with TnTm demonstrate that in the absence of Ca++ TnTm decrease the probability of myosin strong-binding by 100-fold, but that the binding of one myosin increases the probability of a neighboring myosin binding by more than 10-fold (Kad et al., 2005). The later result is consistent both with this “three-state model” of thin filament activation and nicely demonstrates the cooperative aspect at the single molecule level. This later technique could provide interesting insight into how acidosis might exerts its effects on during fatigue.
In addition to these studies which highlight the recent work characterizing Tm dynamics, the revelation of the crystal structure of the core domain of Tn has provided exciting details of the intra-molecular dynamics of Tn resulting from Ca++-binding to TnC (Takeda et al., 2003), and the movements that might ultimately couple to the positions of Tm on actin. The most significant finding from these structural investigations suggests that in the absence of Ca++ actin and myosin are prevented from interacting because the C-terminal portion of the inhibitory subunit of Tn (TnI) is tightly bound to actin. This constrains Tm in a position that blocks the myosin binding sites on actin. Binding of Ca++ to TnC opens up a hydrophobic patch the N-terminal lobe TnC that has an affinity for a specific helix in the C-terminal portion of TnI causing it to dissociate from actin and bind to N-terminal lobe of TnC. This dissociation of TnI from actin frees Tm to move out of way of the myosin binding sites on actin, allowing actin, and myosin to bind. These new molecular insights are providing unprecedented detail of the molecular basis of muscle activation and will therefore help us to pinpoint where activation process acidosis might exert its depressive effects.
Early work, using skinned single muscle fibers, established that acidosis decreases Ca++ sensitivity (Fabiato and Fabiato, 1978) and more recent evidence indicates this effect can be recapitulated in the in vitro motility assay where acidosis can slow the Vactin of actin filaments reconstituted with TnTm (Sata et al., 1995; VanBuren et al., 2002). To better understand the molecular basis of this effect and isolate the specific structures involved researchers have used advances in molecular biology and biophysical techniques to both manipulate the structural of the regulatory proteins and then directly assess the impact on function in vitro.
The interest in identifying the structural elements responsible for the acidosis-induced decrease in Ca++-sensitivity originally stemmed from an interest in understanding the effects of acute ischemia on cardiac function (Blanchard and Solaro, 1984; Solaro et al., 1988), a condition that shares with fatigue the rapid accumulation of metabolites. It was readily apparent that cardiac muscle was much more strongly affected by acidosis than skeletal muscle, particularly at sub-maximal levels of activation. In fact after cardiac muscle, fast skeletal muscle is the next most sensitive followed by slow skeletal, which shows the smallest decrease in Ca++-sensitivity under acidic conditions (Morimoto et al., 1999). The structural differences between skeletal isoforms may provide avenues to pinpoint the key structures and molecular motions which gives rise to these effects. Fortuitously, researchers have focused heavily on understanding the effects acidosis, often using pH levels experienced during fatigue (Fabiato and Fabiato, 1978; Solaro et al., 1988; Ball et al., 1994). Thus the findings provide important insight into the role of acidosis in the fatigue process as well.
Some of the initial work suggested that the differential sensitivity between cardiac and skeletal muscle might be attributable to the subtle structural differences in the isoforms of troponin (Blanchard and Solaro, 1984). A subsequent comparison of neonatal cardiac muscle, which expresses a slow skeletal isoform of TnI (ssTnI) with adult cardiac muscle, which expresses cTnI, revealed that the neonatal isoform of TnI was much less sensitive to acidosis (Solaro et al., 1988). This suggested that the differential response might be attributed to the specific regions in TnI that differ between ssTnI and cTnI. However, it is important to point out that in this experiment the isoforms of TnC and the tropomyosin binding subunit of TnT were also slightly different, preventing the authors from attributing the effect exclusively to TnI. To attempt to resolve this issue Ball et al. (1994) extracted Tn from fast skeletal muscle fibers (fsTn) and replaced the fsTnI with the cardiac isoform of TnI isoform and then incorporated the full Tn complex back into the skeletal muscle fibers. These manipulated skeletal fibers displayed the mild acidosis sensitivity characteristic of skeletal muscle fibers despite containing cardiac TnI, suggesting that TnI alone does not govern the differential response to acidosis. In a subsequent set of experiments both the fsTnI and fsTnC were replaced with cTnI and cTnC and under these conditions the fibers demonstrated this increased sensitivity to acidosis characteristic of cardiac muscle. This led the authors to conclude that the interaction between TnC and TnI is crucial for mediating the acidosis-induced depression in Ca++-sensitivity. This finding may not be surprising in light of the recent structural evidence that the interaction of these two subunits is crucial for activation (Takeda et al., 2003) but clearly more precise manipulation of the structures was required to understand this more effect more fully.
Based on the above evidence researchers have focused on indentifying the crucial structural regions within the TnI subunit that might be responsible for the differential response of cardiac and skeletal muscle. Initial experiments in this process used chimeras of ssTnI and cTnI and measured the effect on the force-pCa relationship (Day et al., 2007). By systematically reducing the number of amino acids that differed between the cardiac and skeletal isoforms researchers were able to assign a good portion of the differential sensitivity to acidosis to the carboxy-terminus region of TnI. In fact the same research group used point mutations to suggest that the differential response to acidosis was largely attributable to a difference of one amino acid at residue 164 (Day et al., 2006). In an elegant follow-up study they directly tested this hypothesis by substituting the alanine at residue 164 in cTnI for the histadine, present in ssTnI, made the cardiac muscle respond nearly identical to slow skeletal muscle, essentially protecting it against the depressive effects of acidosis (Day et al., 2006). The authors postulated that the since pKa of histadine is in the physiological range, its presence makes ssTnI better able to buffer the free protons and therefore maintain its Ca++-sensitivity under acidic conditions (Westfall and Metzger, 2007). This of course has important potential applications for the treatment of myocardial ischemia but also highlights nicely how functional biophysical assays paired with molecular biology provide exceptional insight into the molecular basis of the depressive effects of acidosis during fatigue.
Based on these experiments it is clear that the depressive effects of acidosis involve TnI, particularly in cardiac muscle, however there is also evidence that the other subunits of Tn may be involved in this decreased Ca++-sensitivity, particularly in skeletal muscle. In an effort to delineate a possible role for TnC in the acidosis-induced depression of Ca++-sensitivity, Metzger et al. (1993) replaced the TnC in skeletal muscle fibers (sTnC) with the cardiac isoform (cTnC) and found that this increased the sensitivity of the skeletal fibers to acidosis suggesting it may be more important to fatigue than TnI (Metzger et al., 1993). Based on this evidence and findings that indicate that acidosis directly reduces the affinity of Ca++ binding sites on TnC (Parsons et al., 1997) authors have suggested that high levels of H+ may temporally alter the structure of the Ca++ binding sites on TnC. In addition, the C-terminal region of TnC makes important contacts with TnI during activation (Takeda et al., 2003) and this could help explain why both TnI and TnC are implicated in mediating this depressive effect of acidosis (Ball et al., 1994). Since the effects of acidosis on TnC’s Ca++ binding affinity are significant in both cardiac and fast skeletal isoforms, but the effects mediated through TnI are more prominent in cardiac muscle, this suggests that the effects mediated through TnC may be more important for skeletal muscle fatigue. The greater potential involvement of TnC in the fatigue process could stem from the fact that the fsTnC has two low-affinity Ca++-binding sites while cTnC has just one (Gordon et al., 2000), although this is an idea that needs further exploration.
There is also evidence that Tm-binding subunit of Tn (TnT) is involved in the acidosis-induced decrease in Ca++ sensitivity in skeletal muscle (Ogut et al., 1999). In support of this notion Nosek et al. (2004) demonstrated in mice, genetically engineered to express the fast skeletal isoform of TnT in the myocardium, that there was an increased sensitivity to acidosis compared to those with the normal those with the normal cTnT. This is particularly interesting for skeletal muscle fatigue because it suggests that while fsTnI may attenuate the Ca++-sensitivity, fsTnT may actually accentuate the decrease in Ca++-sensitivity in skeletal muscle. This would imply a greater role for TnT than TnI in skeletal muscle fatigue. Since the structural differences between the cardiac and skeletal isoforms of TnT are now well known the authors concluded that the subtle charge differences in the NH-terminal domain between cardiac and fast skeletal may account for the differential response to acidosis (Nosek et al., 2004). Here a systemic approach, similar to the one for taken for TnI, could be employed to identify the structures and mechanisms responsible TnT’s role in the acidosis-sensitivity of skeletal muscle.
These investigations into the structure and function of Tn have been very important for understanding how the loss of contractility during ischemic heart disease and provide an important starting point for understanding the role in of acidosis in the decreased Ca++-sensitivity during fatigue. Interestingly, while much of the effect in cardiac muscle can be localized to a specific region in TnI the evidence from skeletal muscle suggests a greater involvement of TnC and TnT. Future work in which systematic series of structural perturbations of each Tn subunit and Tm are paired with functional assessments in an in vitro setting will more clearly delineate the role that the regulatory proteins play in the acidosis-induced decrease in Ca++-sensitivity.
One of the strongest correlates with the loss of force during fatigue is the accumulation of Pi in muscle, as demonstrated in NMR studies using in vivo muscle (Dawson et al., 1978). The associated decrease in force is believed to be due to a direct effect in the contractile proteins since in non-fatigued muscle fibers it is routinely observed that high levels of Pi reduce maximal isometric force (Cooke and Pate, 1985; Hibberd et al., 1985; Nosek et al., 1987; Cooke et al., 1988; Pate and Cooke, 1989; Debold et al., 2004, 2006).
Muscular force, at the molecular level, is dependent on the product of the unitary force of a single cross-bridge (Funi) and the duty cycle (the percentage of the ATPase cycle spent strongly bound to actin; VanBuren et al., 1995). Therefore Pi could affect either one or both of these parameters to cause a reduction in force. The prevailing model suggests that the duty cycle is reduced because the rebinding of Pi rapidly reverses the lever arm rotation and leads to detachment of myosin from actin reducing the number of strongly bound cross-bridges (Takagi et al., 2004). Strong support for this model comes from the observation that elevated levels of Pi reduce maximal isometric force in skinned muscle fibers in a concentration-dependent manner (Pate and Cooke, 1989). Furthermore, elevating Pi in a single molecule assay suggests that Pi release and myosin’s lever arm rotation are closely coupled (Baker et al., 2002). Therefore the presence of high levels of Pi would act to decrease myosin’s duty cycle with little or no effect on Funi. However, this mechanism of force production still remains controversial because isolated muscle preparations that incorporate spin-labeled probes to sensitive to myosin’s lever arm rotation do not detect a reverse shift the distribution of myosin heads from the post-powerstroke to pre-powerstroke state following exposure to high levels of Pi. This finding suggests that rather than reversing lever arm rotation and inducing detachment elevated Pi may decrease the force per cross-bridge (Baker et al., 1999). Findings such as this have led to the proposal of thermodynamic models of muscular contraction (Baker et al., 1999; Karatzaferi et al., 2004) as opposed to the more conventional molecular models (Huxley, 1957). In support of these types of models maximal isometric force is linearly decreases with the logarithm of the [Pi] (Pate and Cooke, 1989). This suggests that elevated levels of Pi during fatigue decrease force by decreasing the available energy from the hydrolysis of ATP.
In addition, to the thermodynamic information we are also gaining important atomic level insight into the internal motions of myosin that involve Pi. In fact, the effects of Pi on the cross-bridge cycle are important to understanding the molecular basis of contraction because the release of Pi from myosin is believed to be the step most closely associated to the rotation of myosin’s lever arm, the key molecular event underlying the generation of force and motion (Holmes and Geeves, 2000). In a conventional molecular model of the cross-bridge cycle Pi is thought to reduce force by rebinding to myosin in the AM.ADP state and rapidly, causing both reversal of myosin’s lever arm rotation and the dissociation from actin (Takagi et al., 2004). In isometrically contracting muscle this molecular process would lead to a decrease in the number of strongly bound cross-bridges and therefore force. In the 1980s this may have provided a complete mechanistic explanation for the effect of Pi on force, however advances in structural biology leading to the revelation of the 3-D atomic structure of myosin (Rayment et al., 1993; Figure 3) means that we can now gather much more detail about how this process might occur. For example, what internal molecular motions within myosin’s active site lead to the Pi-induced reversal of the lever arm rotation that ultimately lead to the reversal of the lever arm rotation and detachment from actin? Atomic resolution structures of myosin in different nucleotide states have provided important clues into these motions, at least from the perspective of Pi-release and lever are rotation(Holmes and Geeves, 2000). In fact, with all this new structural information, the suggestion that Pi reduces force during fatigue by simply reversing force generating step is analogous to suggesting that a clock tells time by the moving its hands. In the case of the clock you would be ignoring the intricate motions of gears and springs behind the face of the clock that precisely govern the rotation of the hands. Similarly, we now know that the reversal of force-generating step by myosin likely involves the coordinated movement of several regions within the myosin molecule.
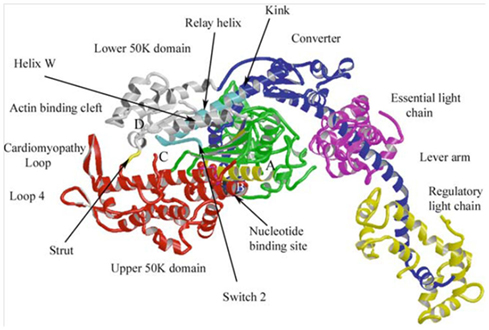
Figure 3. Atomic structure of a myosin molecule A ribbon representation of the head portion myosin (S1). The heavy chain is colored in blue, gray, green, and red, denoting different regions of the protein. The light chains are colored in yellow (Essential) and pink (Regulatory). The upper portion of the motor domain is separated by a large cleft (actin-binding cleft), the opening and closing of which modulates actin affinity. Subtle changes in the nucleotide-binding site are amplified in the converter region resulting in a large rotation of the lever arm. Figure from Geeves and Holmes (2005), reprinted with permission from the publisher.
In recent years systematic and careful experimental designs have providing important insights into the basic intra-molecular motions deep within myosin that link subtle changes in the ATP binding site to large conformational changes in the converter region and lever arm that are associated with force generation (Holmes and Geeves, 2000). Since myosin is quite a large protein, there must be pathways of communication that allow distant regions of the protein coordinate the biochemical and structural to ensure energy is not wasted. Most importantly, strong-binding of myosin to actin must be precisely timed and coordinated with rotation of the lever arm to effectively displace an actin filament and/or generate force. Both of these events are also coupled to the release of Pi from myosin’s active site, this mechanochemical coupling is thought to ensure efficient use of the energy from the hydrolysis of ATP (Holmes and Geeves, 2000). Coordination of these events is believed to be initiated through the opening and closing of a large cleft in the motor domain, involving a strut that spans the cleft (Figure 3). This large cleft, which reaches from the actin-binding domain deep into active site, closes upon strong-binding to actin, a movement coupled to subtle rearrangements within the nucleotide binding pocket and to the release of Pi. Through a series of linkages and switches these subtle motions in the active site are communicated and amplified by the converter domain to cause the lever arm to rotate and generate the ∼10 nm displacement of an actin filament (Holmes, 2005). The events of rebinding are not yet known in the same intricate detail, but it is generally assumed that the reaction is reversible and the rebinding of Pi during fatigue reverses this sequence of events to reduce force. Thus these basic findings have the potential to provide unprecedented molecular detail of the fatigue within muscle’s molecular motor and it will be interesting to follow the developments as the number of structures and the resolution increases.
It is clear these and similar structural data are providing important insights into the molecular basis of muscle contraction and indirectly insight into the process of fatigue, however it is important to point out that structures are only “snapshots in time.” Therefore they do not provide a full characterization of all the motions within the protein through time. What we wish we knew, and could see directly, is how Pi rebinds to the active site of myosin and how this event is linked to conformational changes in the actin-binding site, converter, and lever-arm in real-time. Given the time scales of these events this will be difficult but recent developments in molecular dynamics simulations may soon provide important insights into these events in the near future (Cecchini et al., 2010).
Despite extensive work on the effects of Pi on force and the wealth of knowledge of structural states there are still effects of Pi on contractile function that are not easily explained with existing models. For example, the effects of Pi on shortening velocity are much more equivocal, and seemingly more complex, than the effects on force. In skinned single muscle fibers high levels of Pi most often have little effect on unloaded shortening velocity (Cooke et al., 1988), however altering the conditions slightly can cause Pi to elicit a decrease (Pate and Cooke, 1989) or even a minor increase in velocity (Pate and Cooke, 1989). Similar observations have been made in in vitro motility experiments where Pi can either increase or decrease Vactin depending on the experimental conditions (Debold et al., 2011). In both preparations the effects of Pi are highly dependent on the ATP concentration, at high ATP Pi can cause a slight increases in Vactin (Homsher et al., 1992) while at μM ATP levels it slows Vactin (Warshaw et al., 1991). This issue has recently been re-examined and the data suggest that the opposing effects result from the ability of Pi to rebind to the either AM.ADP or rigor states (see Figure 1) of myosin (Debold et al., 2011). In this variation of the simple detachment-limited model (Figure 1), Pi rebinding to the AM.ADP state facilitates dissociation from a post-powerstroke state and therefore limits duration of strong-actin binding and increases velocity by ∼5%. However the lifetime of the AM.ADP state is very short in skeletal muscle myosin and the affinity rather low so this sequence is not readily observed under normal conditions. By contrast the when Pi rebinds to the rigor state it prevents ATP from binding, prolonging the strongly bound state (i.e., ton) and therefore slows velocity. Simulations governed by this mechanism were able to accurately capture both the Pi-induced increase in velocity at ATP and the depression in velocity at low ATP (Debold et al., 2011). These findings were then extended to more fully understand the effect during fatigue-like conditions by manipulating the pH as well as the Pi levels (Debold et al., 2011). In this experiment it was demonstrated that the Pi-induced increase in Vactin was significantly enhanced at low pH. For example, at pH 6.5, the addition of 30 mM Pi caused Vactin to almost double (Figure 4). The effect was still significant but smaller in magnitude in a less acidic pH of 6.8 and recently a similar effect was reported under even more severe fatigue-like conditions (Greenberg et al., 2010). Taken together these findings strongly suggest the effects of Pi on velocity are dependent on the pH. Debold et al. (2011) interpreted the findings using the same modified simple model described above. The simulations using the model suggested that the changes in ton were due to either acidosis prolonging the AM.ADP state or by Pi shortening the lifetime of the AM.ADP state. Since our earlier work demonstrated that pH prolongs the AM.ADP state this suggests that there is more time for Pi to rebind to actomyosin, making the increase in Vactin more pronounced at low pH, providing a kinetic basis for the pH dependence of the effect (Debold et al., 2011).
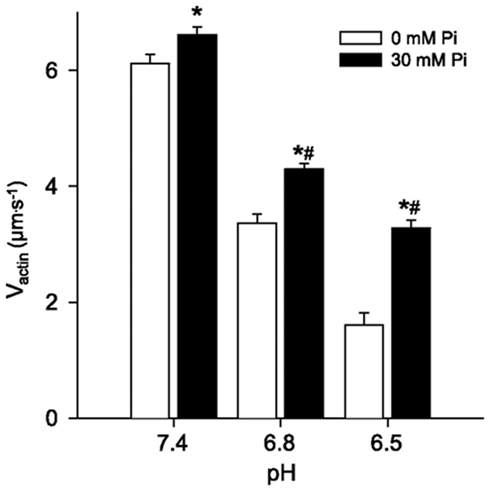
Figure 4. The effect of Pi at low pH on Vactin. In vitro motility actin filament velocities from chicken skeletal myosin and actin under various levels of pH and Pi. Values represent mean ± SEM for Vactin at each pH and Pi level. Data were analyzed using a two-way ANOVA, * indicates significantly (p < 0.05) different from 0 mM Pi and # indicates a significant interaction, meaning that the increase in Vactin in response to Pi was greater at both pH 6.8 and 6.5 than at 7.4. Figure from Debold et al. (2011), reprinted with permission from the American Physiological Society.
These findings have relevance to fatigue because they suggest that the depressive effects of acidosis on Vactin may be offset by the effects of Pi. However, since pH also slows myosin’s overall ATPase rate (Stone and Prevost, 1973) these ions would likely act additively to reduce force in muscle and thus power. This hypothesis is supported by the observation that in a loaded in vitro motility assay a fatigue-like milieu (pH 6.2, 30 mM Pi, 0.3 mM ADP) reduced myosin’s force generating capacity at a given velocity and translated into profound reduction in the peak power generating capacity of myosin (Figure 5). Therefore a Pi-induced increase in shortening velocity may come at the expense of force generation and ultimately power at the molecular level. Here we wish we knew how these ionic changes might affect force and power in vitro and with recent advances in the laser trap assay (Debold et al., 2005) this is something that could be answered in the near future.
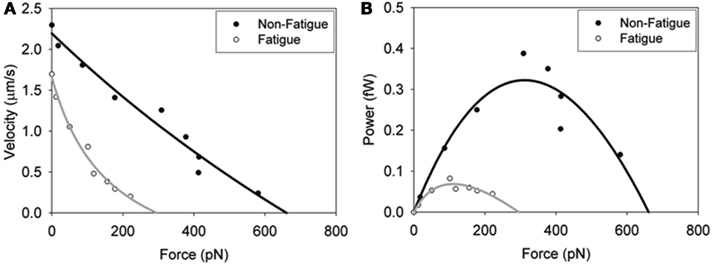
Figure 5. In vitro force–velocity (A) and force–power (B) relationships. Fatigue was simulated by exposing the myosin pH 6.2, 30 mM Pi and 0.3 mM ADP and the effects on skeletal muscle myosin’s force–velocity relationship determined. Load was applied by adding increasing amounts of an actin-binding protein (alpha-actinin). Data were fit with the Hill force–velocity equation (Hill, 1938) with the black line normal conditions and the gray line fatigue-like conditions. The force–power relationships (B) were derived by calculating the product of force and velocity and fit to the Hill force–power equation (Hill, 1938). Figure reprinted from Greenberg and Moore (2010) with permission from John Wiley and Sons.
The effects of fatiguing levels of Pi, like acidosis, typically decrease the Ca++-sensitivity of force in single muscle fibers (Millar and Homsher, 1990, 1992; Walker et al., 1992; Palmer and Kentish, 1994; Metzger, 1996). Furthermore, recent work suggests that this effect this is more pronounced near physiological temperatures vs lower temperatures (Debold et al., 2006; Figure 6). This effect may be particularly relevant for fatigue because it means that during the later stages of fatigue, when intracellular Ca++ is reduced (Westerblad et al., 1991), increases in Pi would have a more dramatic depressive effect on isometric force. Despite being a well-established phenomenon, the molecular mechanisms underlying this Pi-induced depression of Ca++-sensitivity effect remain poorly understood.
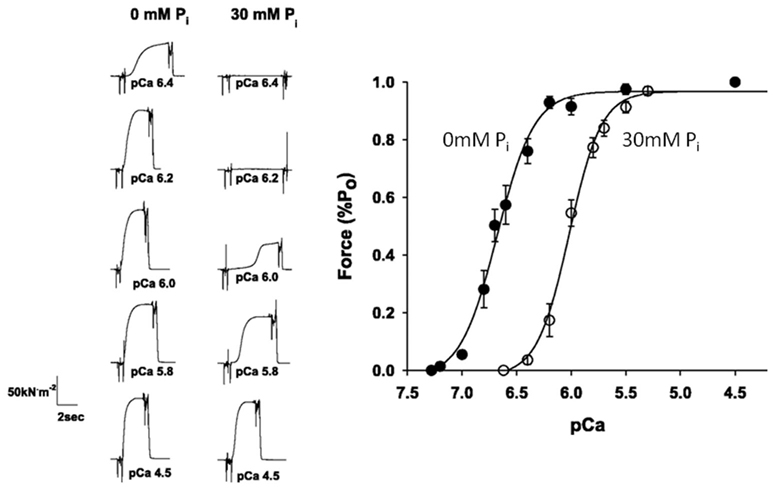
Figure 6. Effect of Pi on the force–pCa relationship. Skinned single fiber muscle forces measured at increasing concentrations of free Ca++ (displayed in pCa, −log units) in the presence and absence of 30 mM Pi (A). The forces are normalized to the maximum reached at saturating pCa (4.5) and fit with the Hill plot (B). The leftward shift in the curve indicates that elevated Pi makes the fibers less sensitive to Ca++. Figure is modified from Debold et al. (2006), reprinted with permission from the American Physiological Society.
In contrast to the putative effects of acidosis on muscle activation, most evidence indicates that Pi does not disrupt the function of Tn, including Ca++ binding to TnC (Palmer and Kentish, 1994). This suggests that the effect is mediated through a distinctly different mechanism than the acidosis-induced depression in Ca++-sensitivity (Fabiato and Fabiato, 1978). Rather most evidence suggests that Pi’s strong depressive effect on the force–pCa relationship is likely mediated through Pi’s effect on a strongly bound actomyosin cross-bridge (Millar and Homsher, 1990, 1992; Walker et al., 1992; Palmer and Kentish, 1994; Metzger, 1996; Debold et al., 2006). Strong-binding of myosin to actin is required for full activation of the thin filament (McKillop and Geeves, 1993) therefore at high concentrations, Pi rebinding to myosin is thought to cause the reversal of the force generating step (Takagi et al., 2004). This is believed to have several effects on the level of thin filament activation; first the stabilization of Tm in the Open state (i.e., full activation) requires myosin strong binding (Gordon et al., 2000) thus this effect would be reversed by elevated Pi. Secondly, strong-binding also contributes to the cooperative binding of myosin through propagation of the motions of Tm on actin (Gordon et al., 2000) from one regulatory unit (7Actin monomers, 1Tn, 1Tm) to its nearest neighbors, here a Pi-induced decrease in strong-binding would be expected to reduce this effect and thus activation. Another source of cooperative activation that is also dependent on strong-binding is the force-induced increase in TnC’s affinity for Ca++ (Gordon et al., 2000), an increase in Pi would reduce this effect, leading to a muscle that is less sensitive to Ca++ all of these contributing to a reduced sensitivity to Ca++ in a force–pCa relationship.
While the above findings provide plausible mechanisms for the rightward shift in the force–pCa relationship there are some findings that suggest our understanding is incomplete. For example, elevated levels of Pi can increase the steepness of the force–pCa curve suggesting an increase rather than a decrease in cooperativity, even in the face of decreasing Ca++-sensitivity as evidenced by a decrease in the pCa50 (Millar and Homsher, 1990). The increased cooperativity seems inconsistent with the mechanism outlined above and suggests that a cross-bridge to which Pi rebinds behaves differently than a myosin molecule in a state prior to Pi-release. In addition, the effects of the velocity–pCa relationship have been equivocal with the earliest work demonstrating that Pi decreases Ca++sensitivity of Vactin (Sata et al., 1995), yet more recent work suggests that elevated Pi seems to have little if any impact on the velocity–pCa relationship (Gorga et al., 2003). This resolution of these issues will be crucial to understanding how this important ion mediates its potent effects on fatigue. Here it might be informative to directly observe the effects on Pi on the behavior of a single actomyosin cross-bridge by using a regulated thin filament in the laser trap assay (Kad et al., 2005) both under unloaded and loaded conditions.
At rest, ADP is typically maintained quite low (∼10 μM) inside the cell due to the creatine kinase reaction strongly favoring the formation of ATP. However, with intense contractile activity the concentration of creatine phosphate rapidly decreases and creatine reciprocally increases. These changes shift the equilibrium of the reaction so that when creatine phosphate is completely depleted intracellular levels of ADP can be as high as 1 mM (Nagesser et al., 1993). However others have argued that this might significantly overestimate the values reached during fatigue in in vivo muscle (Cooke, 2007). This controversy aside it is clear that even 0.5 mM ADP can significantly affect both the force and velocity generating capacity of myosin in vitro (Greenberg et al., 2010).
Observations from skinned single muscle fibers consistently reveal that mM levels of ADP slow unloaded shortening velocity (Cooke and Pate, 1985; Metzger, 1996). ADP has a similar effect on Vactin measured in a motility assay, suggesting that ADP slows velocity by directly inhibiting myosin function (Greenberg et al., 2010). In contrast high levels of ADP have the opposite effect on fiber force, consistently causing small but significant increases maximal isometric force (Cooke and Pate, 1985; Hoar et al., 1987; Metzger, 1996; Fukuda et al., 2000).
The opposing effects on force and velocity can be explained at the molecular level by assuming that ADP competes with ATP for binding to myosin’s active site. In a simple detachment model (Figure 1), when levels are high ADP can readily rebind to myosin’s empty nucleotide-binding site in the rigor state resulting in the reformation of the AM.ADP state. This state is a strongly bound post-powerstroke state, therefore high ADP acts to prolong ton, while having no effect on d, and therefore slows velocity (according to V = d/ton). This molecular concept is supported by observations from the single molecule laser trap assay where mM ADP levels have been shown to prolong the strongly bound lifetime of a single actomyosin bond without affecting d (Baker et al., 2002). Prolongation of ton, while slowing velocity, would also increase the number of strongly bound cross-bridges at any given moment. Therefore, since force is directly dependent on the number of attached cross-bridges this would act to increase maximal isometric force in muscle. These data and accompanying interpretations provide a simple, molecular level explanation for the slowing of velocity but increased force observed with increasing ADP levels.
While these data provide important information, maximal isometric force and unloaded shortening velocity only represent two endpoints on the force–velocity relationship continuum. Therefore to fully understand if ADP contributes to the loss of muscular power during fatigue it may be more informative to characterize its effects on myosin’s load-dependent velocity. This was recently accomplished using a modified motility assay where myosin worked against the load of an actin-binding protein (Greenberg et al., 2010). The results revealed that elevated levels of ADP attenuated load-induced decrease in Vactin that characterizes muscle and myosin. This implies that elevated ADP levels may have little effect on myosin’s power-generating capacity and thus might play only a small or even protective role in in vivo fatigue. Here, examining the effect of ADP the load dependence of a single actomyosin interaction, a technique recently used to characterize the load dependence of smooth muscle muscle (Veigel et al., 2003). These are related techniques may provide even more crucial insight into both the mechanism of fatigue and the role of ADP-release in contraction.
As detailed in the previous section the potential role of ADP in fatigue has been strongly questioned due to the debate over the concentrations achieved during high intensity exercise (Cooke, 2007). However the highest estimates suggest the ADP can reach 1 mM and this level of ADP can have a significant impact on the Ca++-sensitivity both in muscle fibers and in vitro (Greenberg et al., 2010). In skinned single muscle fibers elevated levels of ADP slow unloaded shortening velocity but enhance maximal isometric force and it also increases the sensitivity to Ca++ evidenced by a leftward-shift in the force–pCa relationship (Hoar et al., 1987). This might seem beneficial during fatigue since it could potentially offset a loss in force caused by other metabolites, however ADP has the opposite effect on contraction velocity, slowing unloaded shortening velocity at both maximal and submaximal Ca++ concentrations (Metzger, 1996). Thus the ADP-induced slowing of velocity might offset an increase in force and as a result power may ultimately be compromised.
The ADP-induced slowing of Vactin is thought to be mediated by a direct effect on myosin since the observations from the in vitro motility assay using reconstituted thin filaments (actin + Tn + Tm) also show ADP slows actin filament velocity (Gorga et al., 2003). In these in vitro investigations, ADP levels of only 0.5 mM can significantly decreases Vactin at both maximal and submaximal Ca++ levels, and this effect is concentration dependent with 5 mM ADP causing Vactin to decrease by ∼80% at maximal Ca++ (Gorga et al., 2003; Figure 7).
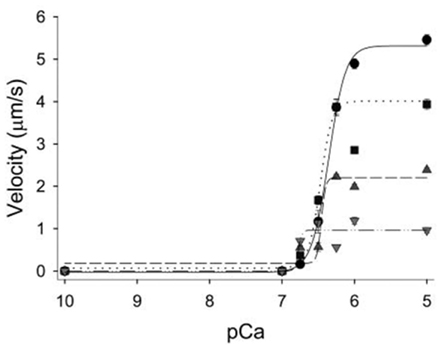
Figure 7. Effect of ADP on the velocity–pCa relationship in vitro. Regulated actin filament velocities plotted as a function of increasing ADP concentrations. The ADP levels were increased from 0.5 mM ADP (squares, dotted line), to 2 mM (triangles up, dashed regression), and 5 mM (triangles down, dash-dot regression) and compared to control experiments with no added ADP (circles, solid regression). Elevated ADP significantly decreased maximal velocity but increased the pCa50 of the thin filament in a concentration-dependent manner. Figure modified from Gorga et al., 2003, reprinted with permission from Cell Press.
In the model of the cross-bridge cycle, described above (Figure 1), elevated ADP is thought to rebind to myosin in the rigor state, pushing the cross-bridge back into the AM.ADP state. This scenario presumably prolongs the duration of the strongly bound state causing velocity to slow, but it would increase activation of the thin filament via strong-binding activation. This model nicely accounts for the increase in force and Ca++-sensitivity via an increased number of strongly bound cross-bridges and also the decrease in velocity due to the same increase in ton. Therefore while the increase in force due to elevated ADP might attenuate the effects of other metabolites (Hoar et al., 1987), any benefit to muscle performance would be offset by the much more pronounced decrease in velocity (Gorga et al., 2003). Thus this evidence implies a small but potentially significant role for elevated levels of ADP in fatigue.
Reactive oxygen species (ROS) and nitric oxide (NO) are somewhat newer putative agents of fatigue, but an accumulating body of literature is beginning to indicate that they may play a significant role in the loss of muscular force and velocity during fatigue (Smith and Reid, 2006). ROS are oxygen containing molecules, such as superoxide (O2−), that have unpaired electrons making them highly reactive and thus capable of damaging membranes and structurally modifying various contractile proteins. The potential structural modifications include the formation of disulfide bridges between neighboring cysteine residues as well as modifications of methionine residues. Many species of ROS and NO have now been demonstrated to accumulate inside contracting muscle cells during intense activity. In addition, there is also evidence that they may be playing a causative role in the fatigue process (Smith and Reid, 2006). However, the effects are concentration dependent in a complex manner. When levels of ROS are low, as might occur at rest or even in the early stages of fatigue, they appear to enhance force production in isolated muscle (Reid et al., 1993). In contrast, at higher concentrations (as might occur in the later stages of fatigue) certain species, such as H2O2, begin to inhibit contractile function (Andrade et al., 1998). Thus while small amounts of ROS may actually improve contractile function, the negative impact of higher concentrations suggests they can still play a causative role in the fatigue process.
Several groups are now beginning to explore the molecular basis of the effects of ROS and NO species on contractile function in vitro. Snook et al. (2008), recently set out to the probe the molecular basis of the effects of the ROS peroxynitrite (ONOO−) using an in vitro motility assay, exposing both myosin and actin to increasing concentrations of ONOO− and measuring its effects myosin’s ability to move actin and generate force. At concentrations greater than 10 μM, ONOO− caused significant reductions in Vactin, suggesting that this species and concentration negatively can negatively affect actomyosin function. They complemented these functional assessments with biochemical experiments to determine the nature of these structural changes, and found that ONOO− caused an increase in tyrosine nitration of both actin and myosin, with myosin showing the strongest relationship between tyrosine nitration and the decrease in Vactin (Snook et al., 2008). Interestingly, exposing myosin to the same level of ONOO− actually increased its force-generating capacity, an observation that agrees with findings from intact muscle and skinned fibers (Allen et al., 2008). This suggests that this species might attenuate the loss of force during fatigue. It is interesting that the opposite effects on velocity and force are similar to the effects of ADP on myosin, suggesting that ONOO− may slow the same step in the cross-bridge cycle, ADP-release. In terms of fatigue, however, this makes interpretation of the role of ONOO− in the fatigue process more difficult because it has opposite effects on force and velocity. The implications for its role in the fatigue process may depend on the relative magnitude of the effect force vs velocity and ultimately on power generation. This will have to be delineated by assessing the effect of ONOO− on the force–velocity and force–power relationships of myosin in vitro. It should also be noted that the concentrations used in this and similar in vitro studies were well above the levels believed to be reached in vivo during fatigue, further complicating the interpretation of such studies (Allen et al., 2008).
Some of the strongest evidence for the role of ROS in fatigue comes from the observations that the progression of fatigue can be greatly slowed (Moopanar and Allen, 2005). In addition, in muscle that has been severely fatigued exposure to a ROS scavenger can readily reverse much of the loss in force generating capacity (Moopanar and Allen, 2006). The reversibility of these changes suggests they are only transient structural modifications but the specific nature of these modifications, such as the proteins modified and the nature of the structural changes are not yet clear.
Moopanar and Allen studied the effects of fatiguing levels of ROS on single fiber contractile properties and observed that this level caused a significant depression in force. Measures of intracellular Ca++ enabled them to attribute much of the depressive effect of ROS to an alteration in the Ca++-sensitivity, suggesting the involvement of TnTm. In support of this notion, Snook et al. (2008) recently presented evidence that ROS can directly affect Tn/Tm function slowing velocity in an in vitro motility assay (Snook et al., 2008). In this experiment when both the myosin and the regulated filaments (Actin + Tn + Tm) were pretreated with peroxynitrite ONOO− the effects on Vactin were more pronounced than that observed when myosin alone was treated, suggesting in addition to myosin Tn/Tm are modified and functional altered by ONOO−. Interestingly, the depressive effects using reconstituted thin filaments (Snook et al., 2008) were less pronounced than in muscle fibers (Moopanar and Allen, 2005) suggesting that other as yet unidentified factors may be contributing in in vivo muscle.
Thus the role of ROS in fatigue is beginning to be explored but future efforts will have to determine whether the ultimate effects on muscular performance in an in vivo setting are beneficial or detrimental. Other key issues to resolve related to ROS and fatigue include the determination of the actual concentration of ROS reached during fatigue and whether the structural alterations induced by ROS are readily reversed in a live cell where antioxidants and other natural reducing agents exist to reverse the changes to structure and function.
In addition, to the transient changes induced by ROS and NO species there may also be longer lasting structural modifications to the contractile proteins that occur during fatigue that in some cases may be secondary to ROS damage. For example, when diaphragm strips are repeatedly contracted under hypoxic conditions there is a loss of both TnI and TnC from the muscle (de Paula et al., 2001). The authors speculated that the loss might be due to cleavage and degradation of TnI and TnC during the fatigue and that these structural changes contributed to the loss in force. Furthermore, much of the loss in force was reversed when full-length exogenous TnI and TnC was added back into the fatigued fibers (de Paula et al., 2001), adding further support that the cleavage of these proteins is responsible for the reduction in force. While they could not identify what caused the cleavage the authors speculated that it may stem from increased ROS production directly damaging the proteins. This hypothesis could be tested by repeating the experiments in the presence of a reducing agent to determine if it prevents the degradation of TnTm and the associated loss in force. This kind of cleavage and degradation of the regulatory proteins is similar to the response observed in cardiac muscle following a transient ischemia/reperfusion injury (a.k.a. myocardial stunning; Gao et al., 1996). This very well-characterized phenomenon results in a progressive cleavage of TnI which is dependent on the severity of the injury, and is linked to a loss in Ca++-sensitivity (McDonough et al., 1999). Thus a similar phenomenon may be occurring during hypoxic fatigue, although more investigation will be needed to confirm that this effect occurs during fatigue in vivo.
Phosphorylation of contractile proteins can also be classed as a transient structural modification, as it is a covalent modification of a protein. One particular phosphorylation event related to fatigue that has been the focus of considerable effort is the contraction-induced phosphorylation of myosin’s regulatory light chain (RLC). Phosphorylation of myosin’s RLC increases with repeated contractions and thus is high during fatigue from intense contractile activity. These observations led Karatzaferi et al. (2008) to examine the effects of Pi and H+ on single fiber contractile properties at high and low levels of RLC phosphorylation. Interestingly, they found that at physiological temperatures, elevated levels of Pi and H+ had their greatest depressive effect on single fiber force and velocity when the RLC was highly phosphorylated as they suggested would be the case during fatigue (Karatzaferi et al., 2008). However knocking out the light-chain phosphorylating kinase (MLCK) in a mouse model does not seem to prevent or even attenuate the decrease in tetanic force with fatigue, suggesting little or no role for this phosphorylation event in fatigue. Although the temperatures of the two studies were different (30 vs 25°C) could help explain a portion of the discrepancy, the power of the later in vivo work strongly suggests a limited or non-existent role for MLC phosphorylation in fatigue. A similar conclusion was reached by Greenberg et al. (2010) based findings from an in vitro motility assay where elevated levels of H+, Pi and ADP had a similar depressive effect on the phosphorylated and dephosphorylated myosin. Therefore while it is clear phosphorylation of MLC influences tetanic force it is not clear that it has a significant role in fatigue.
In addition to the RLC of myosin there are a many other potential phosphorylation sites on other contractile proteins, thus this may not be the only phosphorylation of contractile proteins associated with fatigue. One particularly interesting phosphorylation linked to oxidative stress in skeletal muscle, and thus possibly fatigue, is the phosphorylation of Tm (Houle et al., 2003). In response to elevated levels of H2O2 Houle et al. (2003) were able to detect an increase in the phosphorylation of Tm. In subsequent experiments it was demonstrated that in vitro phosphorylation of Tm can greatly increase the force and velocity of actin reconstituted with Tm in a laser trap assay (Rao et al., 2009). Interestingly, the results suggested that the effect was due to an increase in the size of the cooperative unit of the thin filament, implying that phosphorylation modifies the behavior of Tm on actin to promote strong actomyosin binding and ultimately activation of the thin filament. While such a change in Tm phosphorylation might occur during fatigue the increase force and velocity observed in vitro suggests this effect might help to prevent the loss of muscle function during fatigue rather that exacerbate it. However, more work will need to be done to definitely determine the role of this and other contraction dependent changes in phosphorylation in fatigue in vivo. In related work in the future it will be crucial to be able to link the structural modifications to the function changes. This will likely take a systemic approach to both locate each modification and assess its impact on the function of various contractile proteins in an in vitro setting.
The cause of fatigue has been a fundamental question of science for many years and the cause of muscle fatigue specifically has been studied for more than 100 years. While understanding the limits of human performance drove much of the early efforts in this field it is now clear that identifying the molecular causes will have important clinical relevance to conditions such as chronic heart failure and ischemic heart disease where the effects of fatigue can be severe enough to limit the daily tasks required for independent living (Poole-Wilson and Buller, 1988). Many of the originally proposed mechanisms, established decades ago, are now able to be directly tested and challenged with the recent technological developments in molecular and structural biology as well as single molecule biophysics. These recent findings have confirmed some long-held theories but others have been sharply challenged by the first direct evidence. And maybe more importantly, the new findings are presenting the field with a new set of questions about the nature of the mechanism of fatigue, forcing us to think at an even more basic level about the process of fatigue. Thus it is a particularly exciting time to be studying this question as the rapid pace of technological advances will now allow the field to push our knowledge even further in the very near future.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Preparation of this manuscript was supported, in part, by a grant from the American Heart Association to Edward P. Debold (AHA Grant No. 09SDG2100039).
Allen, D. G., Lamb, G. D., and Westerblad, H. (2008). Skeletal muscle fatigue: cellular mechanisms. Physiol. Rev. 88, 287–332.
Andrade, F. H., Reid, M. B., Allen, D. G., and Westerblad, H. (1998). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 509 (Pt 2), 565–575.
Baker, J. E., Brosseau, C., Joel, P. B., and Warshaw, D. M. (2002). The biochemical kinetics underlying actin movement generated by one and many skeletal muscle myosin molecules. Biophys. J. 82, 2134–2147.
Baker, J. E., LaConte, L. E., Brust-Mascher, I., and Thomas, D. D. (1999). Mechanochemical coupling in spin-labeled, active, isometric muscle. Biophys. J. 77, 2657–2664.
Ball, K. L., Johnson, M. D., and Solaro, R. J. (1994). Isoform specific interactions of troponin I and troponin C determine pH sensitivity of myofibrillar Ca2+ activation. Biochemistry 33, 8464–8471.
Blanchard, E. M., and Solaro, R. J. (1984). Inhibition of the activation and troponin calcium binding of dog cardiac myofibrils by acidic pH. Circ. Res. 55, 382–391.
Capitanio, M., Canepari, M., Cacciafesta, P., Lombardi, V., Cicchi, R., Maffei, M., Pavone, F. S., and Bottinelli, R. (2006). Two independent mechanical events in the interaction cycle of skeletal muscle myosin with actin. Proc. Natl. Acad. Sci. U.S.A. 103, 87–92.
Cecchini, M., Alexeev, Y., and Karplus, M. (2010). Pi release from myosin: a simulation analysis of possible pathways. Structure 18, 458–470.
Chikuni, K., Muroya, S., and Nakajima, I. (2004). Myosin heavy chain isoforms expressed in bovine skeletal muscles. Meat Sci. 67, 87–94.
Cooke, R. (2007). Modulation of the actomyosin interaction during fatigue of skeletal muscle. Muscle Nerve 36, 756–777.
Cooke, R., Franks, K., Luciani, G. B., and Pate, E. (1988). The inhibition of rabbit skeletal muscle contraction by hydrogen ions and phosphate. J. Physiol. 395, 77–97.
Cooke, R., and Pate, E. (1985). The effects of ADP and phosphate on the contraction of muscle fibers. Biophys. J. 48, 789–798.
Dantzig, J. A., Goldman, Y. E., Millar, N. C., Lacktis, J., and Homsher, E. (1992). Reversal of the cross-bridge force-generating transition by photogeneration of phosphate in rabbit psoas muscle fibres. J. Physiol. 451, 247–278.
Dawson, M. J., Gadian, D. G., and Wilkie, D. R. (1978). Muscular fatigue investigated by phosphorus nuclear magnetic resonance. Nature 274, 861–866.
Day, S. M., Westfall, M. V., Fomicheva, E. V., Hoyer, K., Yasuda, S., La Cross, N. C., D’Alecy, L. G., Ingwall, J. S., and Metzger, J. M. (2006). Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat. Med. 12, 181–189.
Day, S. M., Westfall, M. V., and Metzger, J. M. (2007). Tuning cardiac performance in ischemic heart disease and failure by modulating myofilament function. J. Mol. Med. 85, 911–921.
Debold, E. P., Beck, S. E., and Warshaw, D. M. (2008). The effect of low pH on single skeletal muscle myosin mechanics and kinetics. Am. J. Physiol. Cell Physiol. 295, C173–C179.
Debold, E. P., Dave, H., and Fitts, R. H. (2004). Fiber type and temperature dependence of inorganic phosphate: implications for fatigue. Am. J. Physiol. Cell Physiol. 287, C673–C681.
fillskip0ptDebold, E. P., Patlak, J. B., and Warshaw, D. M. (2005). Slip sliding away: Load-dependence of velocity generated by skeletal muscle myosin molecules in the laser trap. Biophys. J. 89, L34–L36.
Debold, E. P., Romatowski, J., and Fitts, R. H. (2006). The depressive effect of P-i on the force-pCa relationship in skinned single muscle fibers is temperature dependent. Am. J. Physiol. 290, C1041–C1050.
Debold, E. P., Saber, W., Cheema, Y., Bookwalter, C. S., Trybus, K. M., Warshaw, D. M., and VanBuren, P. (2010). Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vitro. J. Mol. Cell. Cardiol. 48, 286–292.
Debold, E. P., Turner, M., Stout, J. C., and Walcott, S. (2011). Phosphate enhances myosin-powered actin filament velocity under acidic conditions in a motility assay. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R1401–R1408.
de Paula, B. M., van Leyen, S. A., Brotto, L. S., Jin, J. P., Nosek, C. M., and Nosek, T. M. (2001). Hypoxia/fatigue-induced degradation of troponin I and troponin C: new insights into physiologic muscle fatigue. Pflugers Arch. 442, 738–744.
Donaldson, S. K., Hermansen, L., and Bolles, L. (1978). Differential, direct effects of H+ on Ca2+-activated force of skinned fibers from the soleus, cardiac and adductor magnus muscles of rabbits. Pflugers Arch. 376, 55–65.
Edman, K. A., and Lou, F. (1990). Changes in force and stiffness induced by fatigue and intracellular acidification in frog muscle fibres. J. Physiol. 424, 133–149.
Fabiato, A., and Fabiato, F. (1978). Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J. Physiol. 276, 233–255.
Finer, J. T., Simmons, R. M., and Spudich, J. A. (1994). Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature 368, 113–119.
Fitts, R. H. (2008). The cross-bridge cycle and skeletal muscle fatigue. J. Appl. Physiol. 104, 551–558.
Fujita, H., and Ishiwata, S. (1999). Tropomyosin modulates pH dependence of isometric tension. Biophys. J. 77, 1540–1546.
Fukuda, N., Kajiwara, H., Ishiwata, S., and Kurihara, S. (2000). Effects of MgADP on length dependence of tension generation in skinned rat cardiac muscle. Circ. Res. 86, E1–E6.
Galinska-Rakoczy, A., Engel, P., Xu, C., Jung, H., Craig, R., Tobacman, L. S., and Lehman, W. (2008). Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J. Mol. Biol. 379, 929–935.
Gao, W. D., Liu, Y., Mellgren, R., and Marban, E. (1996). Intrinsic myofilament alterations underlying the decreased contractility of stunned myocardium. A consequence of Ca2+-dependent proteolysis? Circ. Res. 78, 455–465.
Geeves, M. A., and Holmes, K. C. (2005). The molecular mechanism of muscle contraction. Adv. Protein Chem. 71, 161–193.
Gordon, A. M., Homsher, E., and Regnier, M. (2000). Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924.
Gorga, J. A., Fishbaugher, D. E., and VanBuren, P. (2003). Activation of the calcium-regulated thin filament by myosin strong binding. Biophys. J. 85, 2484–2491.
Greenberg, M. J., Mealy, T. R., Jones, M., Szczesna-Cordary, D., and Moore, J. R. (2010). The direct molecular effects of fatigue and myosin regulatory light chain phosphorylation on the actomyosin contractile apparatus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R989–R996.
Greenberg, M. J., and Moore, J. R. (2010). The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton 67, 273–285.
Hibberd, M. G., Dantzig, J. A., Trentham, D. R., and Goldman, Y. E. (1985). Phosphate release and force generation in skeletal muscle fibers. Science 228, 1317–1319.
Hill, A. V. (1938). The heat of shortening and the dynamic constants of muscle. Proc. R. Soc. Lond. B Biol. Sci. 126, 136–195.
Hoar, P. E., Mahoney, C. W., and Kerrick, W. G. (1987). MgADP-increases maximum tension and Ca2+ sensitivity in skinned rabbit soleus fibers. Pflugers Arch. 410, 30–36.
Holmes, K. C. (2005). The molecular basis of cross-bridge function. Adv. Exp. Med. Biol. 565, 13–22.
Holmes, K. C., and Geeves, M. A. (2000). The structural basis of muscle contraction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355, 419–431.
Homsher, E., Wang, F., and Sellers, J. R. (1992). Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. Am. J. Physiol. 262, C714–C723.
Houle, F., Rousseau, S., Morrice, N., Luc, M., Mongrain, S., Turner, C. E., Tanaka, S., Moreau, P., and Huot, J. (2003). Extracellular signal-regulated kinase mediates phosphorylation of tropomyosin-1 to promote cytoskeleton remodeling in response to oxidative stress: impact on membrane blebbing. Mol. Biol. Cell 14, 1418–1432.
Huxley, A. F. (1957). Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7, 255–318.
Huxley, H. E. (1990). Sliding filaments and molecular motile systems. J. Biol. Chem. 265, 8347–8350.
Kad, N. M., Kim, S., Warshaw, D. M., VanBuren, P., and Baker, J. E. (2005). Single-myosin crossbridge interactions with actin filaments regulated by troponin-tropomyosin. Proc. Natl. Acad. Sci. U.S.A. 102, 16990–16995.
Karatzaferi, C., Chinn, M. K., and Cooke, R. (2004). The force exerted by a muscle cross-bridge depends directly on the strength of the actomyosin bond. Biophys. J. 87, 2532–2544.
Karatzaferi, C., Franks-Skiba, K., and Cooke, R. (2008). The inhibition of shortening velocity of skinned skeletal muscle fibers in conditions that mimic fatigue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R948–R955.
Kentish, J. C. (1991). Combined inhibitory actions of acidosis and phosphate on maximum force production in rat skinned cardiac muscle. Pflugers Arch. 419, 310–318.
Knuth, S. T., Dave, H., Peters, J. R., and Fitts, R. H. (2006). Low cell pH depresses peak power in rat skeletal muscle fibres at both 30 degrees C and 15 degrees C: implications for muscle fatigue. J. Physiol. 575, 887–899.
Kristensen, M., Albertsen, J., Rentsch, M., and Juel, C. (2005). Lactate and force production in skeletal muscle. J. Physiol. 562, 521–526.
Kurzawa-Goertz, S. E., Perreault-Micale, C. L., Trybus, K. M., Szent-Gyorgyi, A. G., and Geeves, M. A. (1998). Loop I can modulate ADP affinity, ATPase activity, and motility of different scallop myosins. Transient kinetic analysis of S1 isoforms. Biochemistry 37, 7517–7525.
Lee, J. A., Westerblad, H., and Allen, D. G. (1991). Changes in tetanic and resting [Ca2+]i during fatigue and recovery of single muscle fibres from Xenopus laevis. J. Physiol. 433, 307–326.
McDonough, J. L., Arrell, D. K., and Van Eyk, J. E. (1999). Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ. Res. 84, 9–20.
McKillop, D. F., and Geeves, M. A. (1993). Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65, 693–701.
Metzger, J. M. (1996). Effects of phosphate and ADP on shortening velocity during maximal and submaximal calcium activation of the thin filament in skeletal muscle fibers. Biophys. J. 70, 409–417.
Metzger, J. M., and Moss, R. L. (1990). Effects of tension and stiffness due to reduced pH in mammalian fast- and slow-twitch skinned skeletal muscle fibres. J. Physiol. 428, 737–750.
Metzger, J. M., Parmacek, M. S., Barr, E., Pasyk, K., Lin, W. I., Cochrane, K. L., Field, L. J., and Leiden, J. M. (1993). Skeletal troponin C reduces contractile sensitivity to acidosis in cardiac myocytes from transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 90, 9036–9040.
Millar, N. C., and Homsher, E. (1990). The effect of phosphate and calcium on force generation in glycerinated rabbit skeletal muscle fibers. A steady-state and transient kinetic study. J. Biol. Chem. 265, 20234–20240.
Millar, N. C., and Homsher, E. (1992). Kinetics of force generation and phosphate release in skinned rabbit soleus muscle fibers. Am. J. Physiol. 262, C1239–C1245.
Moopanar, T. R., and Allen, D. G. (2005). Reactive oxygen species reduce myofibrillar Ca2 + sensitivity in fatiguing mouse skeletal muscle at 37 degrees C. J. Physiol. 564, 189–199.
Moopanar, T. R., and Allen, D. G. (2006). The activity-induced reduction of myofibrillar Ca2 + sensitivity in mouse skeletal muscle is reversed by dithiothreitol. J. Physiol. 571, 191–200.
Morimoto, S., Harada, K., and Ohtsuki, I. (1999). Roles of troponin isoforms in pH dependence of contraction in rabbit fast and slow skeletal and cardiac muscles. J. Biochem. 126, 121–129.
Nagesser, A. S., van der Laarse, W. J., and Elzinga, G. (1993). ATP formation and ATP hydrolysis during fatiguing, intermittent stimulation of different types of single muscle fibres from Xenopus laevis. J. Muscle Res. Cell. Motil. 14, 608–618.
Nosek, T. M., Brotto, M. A., and Jin, J. P. (2004). Troponin T isoforms alter the tolerance of transgenic mouse cardiac muscle to acidosis. Arch. Biochem. Biophys. 430, 178–184.
Nosek, T. M., Fender, K. Y., and Godt, R. E. (1987). It is diprotonated inorganic phosphate that depresses force in skinned skeletal muscle fibers. Science 236, 191–193.
Ogut, O., Granzier, H., and Jin, J. P. (1999). Acidic and basic troponin T isoforms in mature fast-twitch skeletal muscle and effect on contractility. Am. J. Physiol. 276, C1162–C1170.
Palmer, S., and Kentish, J. C. (1994). The role of troponin C in modulating the Ca2+ sensitivity of mammalian skinned cardiac and skeletal muscle fibres. J. Physiol. 480 (Pt 1), 45–60.
Palmiter, K. A., Tyska, M. J., Dupuis, D. E., Alpert, N. R., and Warshaw, D. M. (1999). Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. J. Physiol. 519 (Pt 3), 669–678.
Parsons, B., Szczesna, D., Zhao, J., Van, S. G., Kerrick, W. G., Putkey, J. A., and Potter, J. D. (1997). The effect of pH on the Ca2+ affinity of the Ca2+ regulatory sites of skeletal and cardiac troponin C in skinned muscle fibres. J. Muscle Res. Cell. Motil. 18, 599–609.
Pate, E., Bhimani, M., Franks-Skiba, K., and Cooke, R. (1995). Reduced effect of pH on skinned rabbit psoas muscle mechanics at high temperatures: implications for fatigue. J. Physiol. 486 (Pt 3), 689–694.
Pate, E., and Cooke, R. (1989). Addition of phosphate to active muscle fibers probes actomyosin states within the powerstroke. Pflugers Arch. 414, 73–81.
Pedersen, T. H., Nielsen, O. B., Lamb, G. D., and Stephenson, D. G. (2004). Intracellular acidosis enhances the excitability of working muscle. Science 305, 1144–1147.
Poole-Wilson, P. A., and Buller, N. P. (1988). Causes of symptoms in chronic congestive heart failure and implications for treatment. Am. J. Cardiol. 62, 31A–34A.
Rao, V. S., Marongelli, E. N., and Guilford, W. H. (2009). Phosphorylation of tropomyosin extends cooperative binding of myosin beyond a single regulatory unit. Cell Motil. Cytoskeleton 66, 10–23.
Rayment, I., Rypniewski, W. R., Schmidt-Base, K., Smith, R., Tomchick, D. R., Benning, M. M., Winkelmann, D. A., Wesenberg, G., and Holden, H. M. (1993). Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261, 50–58.
Reid, M. B., Khawli, F. A., and Moody, M. R. (1993). Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 75, 1081–1087.
Ricciardi, L., Bottinelli, R., Canepari, M., and Reggiani, C. (1994). Effects of acidosis on maximum shortening velocity and force-velocity relation of skinned rat cardiac muscle. J. Mol. Cell. Cardiol. 26, 601–607.
Sata, M., Sugiura, S., Yamashita, H., Fujita, H., Momomura, S., and Serizawa, T. (1995). MCI-154 increases Ca2+ sensitivity of reconstituted thin filament. A study using a novel in vitro motility assay technique. Circ. Res. 76, 626–633.
Siemankowski, R. F., Wiseman, M. O., and White, H. D. (1985). ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc. Natl. Acad. Sci. U.S.A. 82, 658–662.
Smith, M. A., and Reid, M. B. (2006). Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir. Physiol. Neurobiol. 151, 229–241.
Snook, J. H., Li, J., Helmke, B. P., and Guilford, W. H. (2008). Peroxynitrite inhibits myofibrillar protein function in an in vitro assay of motility. Free Radic. Biol. Med. 44, 14–23.
Solaro, R. J., Lee, J. A., Kentish, J. C., and Allen, D. G. (1988). Effects of acidosis on ventricular muscle from adult and neonatal rats. Circ. Res. 63, 779–787.
Stone, D., and Prevost, S. (1973). Characterization of modified myosin at low ionic strength. Enzymatic and spin-label studies. Biochemistry 12, 4206–4211.
Takagi, Y., Shuman, H., and Goldman, Y. E. (2004). Coupling between phosphate release and force generation in muscle actomyosin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1913–1920.
Takeda, S., Yamashita, A., Maeda, K., and Maeda, Y. (2003). Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41.
Tanaka, H., Ishijima, A., Honda, M., Saito, K., and Yanagida, T. (1998). Orientation dependence of displacements by a single one-headed myosin relative to the actin filament. Biophys. J. 75, 1886–1894.
VanBuren, P., Alix, S. L., Gorga, J. A., Begin, K. J., LeWinter, M. M., and Alpert, N. R. (2002). Cardiac troponin T isoforms demonstrate similar effects on mechanical performance in a regulated contractile system. Am. J. Physiol. Heart Circ. Physiol. 282, H1665–H1671.
VanBuren, P., Guilford, W. H., Kennedy, G., Wu, J., and Warshaw, D. M. (1995). Smooth muscle myosin: a high force-generating molecular motor. Biophys. J. 68, 256S–258S.
Veigel, C., Molloy, J. E., Schmitz, S., and Kendrick-Jones, J. (2003). Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat. Cell Biol. 5, 980–986.
Walker, J. W., Lu, Z., and Moss, R. L. (1992). Effects of Ca2+ on the kinetics of phosphate release in skeletal muscle. J. Biol. Chem. 267, 2459–2466.
Warshaw, D. M., Desrosiers, J. M., Work, S. S., and Trybus, K. M. (1991). Effects of MgATP, MgADP, and Pi on actin movement by smooth muscle myosin. J. Biol. Chem. 266, 24339–24343.
Westerblad, H., Lee, J. A., Lannergren, J., and Allen, D. G. (1991). Cellular mechanisms of fatigue in skeletal muscle. Am. J. Physiol. 261, C195–C209.
Keywords: muscle, fatigue, myosin, actin, phosphate, acidosis, troponin, tropomyosin
Citation: Debold EP (2012) Recent insights into muscle fatigue at the cross-bridge level. Front. Physio. 3:151. doi: 10.3389/fphys.2012.00151
Received: 18 January 2012; Accepted: 02 May 2012;
Published online: 01 June 2012.
Edited by:
Christina Karatzaferi, University of Thessaly, GreeceReviewed by:
Marieke Johanna Bloemink, University of Kent, UKCopyright: © 2012 Debold. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Edward P. Debold, Department of Kinesiology, University of Massachusetts, 158 Totman Building, Amherst, MA 01003, USA. e-mail:ZWRlYm9sZEBraW4udW1hc3MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.