


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 17 May 2012
Sec. Integrative Physiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00139
This article is part of the Research TopicThe spatiotemporal dynamics of longevity-defining cellular processes and its modulation by genetic, dietary and pharmacological anti-aging interventionsView all 10 articles
Mitochondrial dysfunction activates intracellular signaling pathways that impact yeast longevity, and the best known of these pathways is the retrograde response. More recently, similar responses have been discerned in other systems, from invertebrates to human cells. However, the identity of the signal transducers is either unknown or apparently diverse, contrasting with the well-established signaling module of the yeast retrograde response. On the other hand, it has become equally clear that several other pathways and processes interact with the retrograde response, embedding it in a network responsive to a variety of cellular states. An examination of this network supports the notion that the master regulator NFκB aggregated a variety of mitochondria-related cellular responses at some point in evolution and has become the retrograde transcription factor. This has significant consequences for how we view some of the deficits associated with aging, such as inflammation. The support for NFκB as the retrograde response transcription factor is not only based on functional analyses. It is bolstered by the fact that NFκB can regulate Myc–Max, which is activated in human cells with dysfunctional mitochondria and impacts cellular metabolism. Myc–Max is homologous to the yeast retrograde response transcription factor Rtg1–Rtg3. Further research will be needed to disentangle the pro-aging from the anti-aging effects of NFκB. Interestingly, this is also a challenge for the complete understanding of the yeast retrograde response.
Mitochondrial dysfunction underlies the spectacular manifestations of a class of diseases known as mitochondrial encephalomyopathy. It is found in cardiac hypertrophy and neurological disorders such as Parkinson’s disease. Mitochondrial dysfunction is also a hallmark of cancer (Wallace and Fan, 2010). It would seem that the mitochondrial deficits that contribute to the above disorders would invariably prevent any cell survival. However, cells often respond to mitochondrial stress with specific responses, allowing them to survive in some cases. Thus, adaptations to loss of mitochondrial function are widespread (Jazwinski, 2012). It has become clear over the past decade that these adaptations or compensations are also evident during normal aging, and they may in part determine lifespan. The best known of these cellular responses to mitochondrial dysfunction is the yeast (Saccharomyces cerevisiae) retrograde response (Liu and Butow, 2006), which plays a role in determining replicative lifespan (Kirchman et al., 1999). However, similar responses have been described in Caenorhabditis elegans, Drosophila melanogaster, and the mouse (Jazwinski, 2012). They have even been found in human cells in tissue culture, demonstrating their pervasiveness. We review these developments here to search for common principles in the response to mitochondrial dysfunction.
The yeast retrograde signaling pathway has been elucidated in great detail (Liu and Butow, 2006). The retrograde signaling pathway cross talks with other signaling pathways in the cell. Thus, it is embedded in a mesh of signal transduction events that adapt the cell to a variety of internal and external environments. This range of interactions has recently expanded, and it is our purpose to review them here to provide clues to the signaling modules that may operate in the response of mammalian cells to mitochondrial dysfunction. This augments the bioinformatics approach we have taken earlier to identify the retrograde response transcription factors in mammalian cells (Srinivasan et al., 2010). As that analysis suggested, the “master regulator” NFκB has likely taken on in metazoans the role of the yeast retrograde transcription factor Rtg1–Rtg3. This conclusion gathers further support from the current review.
Yeast cells missing mtDNA (rho0) display a wide variety of changes in nuclear gene expression compared to rho+ cells (Epstein et al., 2001; Traven et al., 2001). The activated genes encode metabolic and stress proteins destined for the mitochondrion, the cytoplasm, and the peroxisome, and they portend a realignment of metabolism that compensates for the mitochondrial dysfunction. The loss of respiratory ability in rho0 cells eliminates the tricarboxylic acid (TCA) cycle as a source of glutamate for biosynthesis, because the activity of succinate dehydrogenase is compromised. However, the first three reactions of the TCA cycle remain intact, and this part of the TCA cycle can give rise to α-ketoglutarate, the precursor of glutamate, as long as a supply of citrate is available. This citrate is provided by the activation of the glyoxylate cycle, which utilizes oxaloacetate and acetyl-coenzyme A to generate citrate, but unlike the TCA cycle retains the carbons of acetate rather than ultimately releasing them as carbon dioxide. This central feature of the metabolic adaptation in rho0 cells is surrounded by additional niceties that allow the cell to function without an active electron transport chain. The oxidative phosphorylation that is abrogated in these cells is easily supplanted by the glycolytic production of ATP. The phenotypic changes described above are together termed the retrograde response.
The key event in retrograde signaling is the translocation of the retrograde transcription factor from the cytoplasm to the nucleus (Rothermel et al., 1995; Rothermel et al., 1997; Sekito et al., 2000). The retrograde transcription factor is a heterodimer of two basic helix-loop-helix/leucine zipper proteins, Rtg1 and Rtg3, which binds to the sequence GTCAC (R box; Liao and Butow, 1993; Jia et al., 1997). Of the two, Rtg1 is atypical for such proteins because it possesses a truncated basic domain with no apparent transcriptional activation domains. Yet, only as a heterodimer can Rtg3 bind the R box and activate transcription. A hierarchical analysis of transcription factor regulatory networks in yeast has shown that Rtg1 is in the top layer, because it is not regulated by any other transcription factor (Jothi et al., 2009). Top layer transcription factors are comparatively abundant, long-lived, and noisy in terms of expression from cell to cell. This variability may allow at least some members of a yeast clone or population to respond to one or another environmental challenge by launching a response whose precision is maximized by the tightly regulated transcription factors in lower layers.
Translocation of Rtg1–Rtg3 requires the Rtg2 protein (Sekito et al., 2000), which has no known homologs in higher organisms (Figure 1). Rtg2 promotes the dephosphorylation of Rtg3 by binding Mks1 and preventing Mks1 from forming a complex with the 14-3-3 protein Bmh1 or Bmh2, a complex which maintains Rtg3 in a hyperphosphorylated state (Sekito et al., 2000; Dilova et al., 2002; Sekito et al., 2002; Liu et al., 2003; Dilova et al., 2004). Partial phosphorylation of Rtg3 is necessary, however, to expose its nuclear localization signal and thus to render the Rtg1–Rtg3 capable of activating retrograde target genes. Mks1 is removed by ubiquitin-mediated degradation promoted by the ubiquitin ligase component Grr1 (Liu et al., 2005). Thus, Grr1 is a positive regulator of the retrograde response, while Mks1 is a negative regulator.
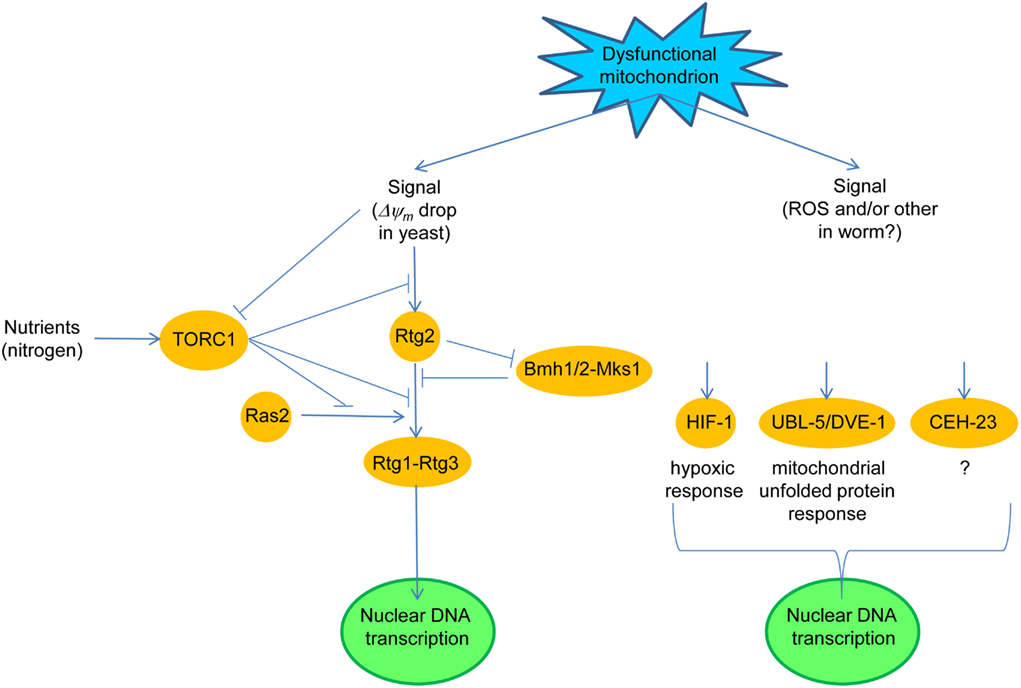
Figure 1. Dysfunctional mitochondria trigger a retrograde response in yeast and in round worms. In yeast, a drop in mitochondrial membrane potential (ΔΨm) initiates retrograde signaling through Rtg2, by preventing the Mks1-Bmh1/2 complex from inhibiting the partial dephosphorylation of Rtg3 in the Rtg1–Rtg3 retrograde transcription factor which is stimulated by Rtg2. Rtg1–Rtg3 translocates from the cytoplasm to the nucleus where it activates the expression of retrograde response target genes. The nutrient-responsive, target of rapamycin (TOR) complex 1 (TORC1) blocks the retrograde response both upstream and downstream of Rtg2. TORC1 also suppresses the stimulatory effect of Ras2 on retrograde signaling. In the worm, a signal(s) elicited by dysfunctional mitochondria activate anyone of at least three retrograde signaling pathways defined by the transcription factor which is activated. The transcription factor can be HIF-1, activated by reactive oxygen species (ROS), UBL-5 and DEV-1, or the putative transcription factor CEH-23.
Target of rapamycin complex 1 is also a negative regulator of the retrograde response. TORC1 appears to act both upstream and downstream of Rtg2 (Komeili et al., 2000; Giannattasio et al., 2005; Breitkreutz et al., 2010). One of the components of TORC1 is the WD-protein Lst8, which in genetic studies was shown to act upstream and downstream of Rtg2 depending on the identity of the mutation in Lst8 which was examined (Liu et al., 2001; Chen and Kaiser, 2003). This coincides well with the fact that TORC1 impinges upon the retrograde response at multiple points. The regulation of the retrograde response by TORC1 ensures that it is not active when nutrients, such as glutamate, are plentiful.
Target of rapamycin (TOR) complex 1 is subject to negative feedback from dysfunctional mitochondria, because TORC1-mediated phosphorylation of Sch9, an AGC protein kinase, is down-regulated in rho0 cells (Kawai et al., 2011). Phosphorylated Sch9 antagonizes stress responses under the control of the Msn2–Msn4 transcription factor and promotes ribosome biogenesis (Urban et al., 2007). It also inhibits protein kinase A activity, balancing cell growth and metabolism with stress resistance (Zhang et al., 2011). This occurs because protein kinase A negatively regulates Msn2–Msn4 mediated stress responses and because it feedback inhibits its own activation, which likely prevents an exaggerated response to the feedback inhibition of TORC1 by dysfunctional mitochondria. Osmotic stress also reduces Sch9 phosphorylation by TORC1, but only transiently (Urban et al., 2007). Osmotic stress is known to recruit the Rtg1–Rtg3 transcription factor (Pastor et al., 2009). Thus, retrograde signaling responds not only to metabolic stress but to other types of stress as well.
Ras2 is a positive regulator of the retrograde response (Kirchman et al., 1999); however, it is not clear at which point in the retrograde signaling pathway Ras2 exerts its effect. Interestingly, MKS1 was originally identified as a negative regulator of the Ras2–cAMP pathway (Matsuura and Anraku, 1993). This, together with the effects of TORC1 on protein kinase A suggests that it is the Ras2–cAMP pathway that contributes to the retrograde response. However, this interpretation is complicated. Activation of the retrograde response extends yeast replicative lifespan, which is measured by the number of times an individual cell divides (Kirchman et al., 1999). Ras2 also extends replicative lifespan (Sun et al., 1994). However, it does so via a cAMP-independent pathway. Thus, it is not clear which of the Ras2 pathways impacts the retrograde response, and indeed both the cAMP-dependent and independent pathways may be involved.
Rtg2 plays multiple roles in the cell. As discussed above, it is a positive regulator of the retrograde response, by promoting dephosphorylation of Rtg3 in the cytoplasm. In addition, it has at least two other roles in the nucleus. Rtg2 is an integral component of the transcriptional co-activator SAGA-like (SLIK) complex that contains the histone acetyltransferase Gcn5 (Pray-Grant et al., 2002). SLIK is required for the induction of the retrograde response target gene CIT2, and it has been shown to bind to the CIT2 promoter. The other role Rtg2 plays in the nucleus is promotion of genome stability (Bhattacharyya et al., 2002; Borghouts et al., 2004). The mechanism by which it extends this protection is not known, except that it does not involve the participation of an intact SLIK complex (Kim et al., 2004).
The retrograde signal transducer proximal to the dysfunctional mitochondrion is Rtg2 (Liu and Butow, 2006). However, the nature of the mitochondrial signal that triggers the retrograde response has not been clear until recently. One of the candidates was the drop in membrane potential (ΔΨm) in dysfunctional mitochondria. Manipulation of ΔΨm genetically, irrespective of the presence or absence of mtDNA, has shown that loss of ΔΨm is necessary and sufficient to activate the retrograde response. However, the loss of mtDNA can augment this effect (Miceli et al., 2011). The question now becomes how this signal is read by Rtg2. A ROS scavenger does not block the signal, and it does not appear that a drop in cellular ATP levels is involved. Thus, the loss of ΔΨm itself must be relayed to Rtg2. Even though they are not part of retrograde regulation, mitochondrial ROS somehow signal increased chronological lifespan (survival in stationary phase) in yeast cells in which TORC1 signaling is attenuated (Pan et al., 2011). Thus, mitochondrial ROS can perform a signaling function in some instances in yeast.
There is a gradual loss of ΔΨm as yeasts replicatively age, which occurs without loss of mtDNA, and this is accompanied by a progressive activation of the retrograde response (Lai et al., 2002; Borghouts et al., 2004). Thus, it appears it is loss of ΔΨm that triggers the retrograde response during the yeast replicative lifespan. In fact, the activation of the retrograde response may allow yeasts to live as long as they do. Indeed, the greater the forced induction of the retrograde response at the beginning of their lifespans is the greater the lifespan extension (Jazwinski, 2000). This indicates that the retrograde response is a compensatory mechanism for mitochondrial dysfunction.
There are two other pathways that signal mitochondrial dysfunction and extend replicative lifespan that have recently been described in yeast. Mitochondrial back-signaling is activated upon deletion of the AFO1/MRPL25 gene, which encodes a protein found in mitochondrial ribosomes, and this activation extends replicative lifespan (Heeren et al., 2009). This requires an active TORC1 and the transcription factor Sfp1, which activates expression of cytoplasmic ribosomal proteins. This pathway is activated only in rho0 cells. However, this occurs during growth on glucose which represses the retrograde response in the yeast strain studied. The deletion of nuclear genes that encode components of the mitochondrial translation complex (MTC), which activates translation of mtDNA-encoded proteins, also extends yeast replicative lifespan in a Sir2-dependent manner (Caballero et al., 2011). It had been known for quite some time that interruption of mitochondrial translation with erythromycin extends yeast replicative lifespan (Holbrook and Menninger, 2002). The relationship of mitochondrial back-signaling and the MTC to the retrograde response is of interest, but it is not known at present.
The glyoxylate cycle is upregulated in C. elegans as a function of age and also in certain mutants that display an increased lifespan (Vanfleteren and De Vreese, 1995). This bears much resemblance to the yeast retrograde response. It has been shown that knockdown of respiratory chain components in this worm can extend lifespan (Dillin et al., 2002; Lee et al., 2003). Furthermore, a systematic search for lifespan extending genes has led to the conclusion that there is a retrograde response that extends worm lifespan (Cristina et al., 2009).
Recent studies have addressed the pathways that comprise the worm retrograde response (Figure 1). Knockdown of cco-1, among several respiratory chain components, extends life span, and activates the hypoxia-inducible transcription factor HIF-1 (Lee et al., 2010a). Parenthetically, activation of HIF-1 involves ceramide signaling and the hyl-2 encoded ceramide synthase, a homolog of the yeast longevity assurance gene LAG1 (Mehta et al., 2009; Menuz et al., 2009), which will have significance below. The mitochondrial signal in this cco-1 knockdown appears to be the ROS generated during mitochondrial stress, but it is not known whether this follows changes in ΔΨm. In another study, down-regulation of COX4 (cco-1) by RNAi extended lifespan and concomitantly activated the mitochondrial unfolded protein response, which recruited the transcription factors UBL-5 and DVE-1 (Durieux et al., 2011). This response was cell-non-autonomous implying the secretion of a “mitokine” by certain cells, to which other cells respond. In yet another study, an RNAi screen for reduced mitochondrial electron transport chain function identified CEH-23, a predicted transcription factor, in the longevity increase (Walter et al., 2011). In each of the above studies, the respective transcription factors were shown to be necessary and sufficient for life span extension. Thus, the mutual relationships of the “retrograde responses” uncovered in these studies is not clear at present.
This discussion of the worm retrograde response suggests a marked heterogeneity of the responsible signaling pathways, as compared to the yeast retrograde response. However, recent studies in yeast also point to more than a single, comprehensive response to mitochondrial dysfunction. The worm signaling pathways appear more disparate, and they may reflect the increased complexity of this metazoan. C. elegans does not possess an NFκB homolog, which may have evolved as a master regulator along with the appearance of more complex immune systems (Srinivasan et al., 2010). Thus, NFκB may have gathered together the capacity to respond to a variety of inputs with a variety of outputs, including the equivalent of a retrograde response as we discuss below.
There may in fact be more than one type of mitochondrial “retrograde response” in the worm. Recent work has demonstrated that the location of the respiratory chain disruption that extends C. elegans longevity affects the associated phenotypic manifestations (Yang and Hekimi, 2010b). Evidence has also been presented that mitochondrial ROS serve a signaling function in lifespan extension, in a pathway distinct from any known lifespan extending mechanism in the worm (Yang and Hekimi, 2010a). Curiously, this novel ROS pathway may have bile acid-like mediators that perform a hormonal function (Liu et al., 2012).
A retrograde response has been characterized in D. melanogaster in a variety of RNAi strains in which anyone of several respiratory chain components were knocked down (Copeland et al., 2009). Many of these strains displayed an increase in life span. A mutant in the sbo gene involved in coenzyme Q biosynthesis also demonstrated extended lifespan (Liu et al., 2011). The identity of the signaling pathway(s) involved here in lifespan extension is not known.
In mice, reduced activity of MCLK1, involved in coenzyme Q biosynthesis and resulting in a defective electron transport chain, markedly extended life span, with no apparent tradeoff in growth or fertility (Lapointe and Hekimi, 2008). These mouse studies followed up on early work on the C. elegans clk-1 gene, mutants of which extended worm longevity. In a SURF1 knockout mouse, mitochondrial complex IV assembly is disrupted, and this also results in substantially increased longevity (Dell’Agnello et al., 2007). It is not clear which signal transduction proteins are utilized in these mouse retrograde responses, but they are likely to differ from the yeast RTG genes.
Mitochondrial respiratory defects elicit expression of nuclear genes in mammalian cells (Heddi et al., 1993). Calcium-signaling and NFκB signaling are among the potential signaling pathways that have been implicated in these “retrograde responses” (Butow and Avadhani, 2004). NFκB is a conserved master regulator that responds to a wide range of stress signals including ROS (Srinivasan et al., 2010). It is also associated with mitochondrial biogenesis. A mitochondrial stress response to aggregated ornithine decarboxylase in mammalian cells may be similar to the mitochondrial unfolded protein response in C. elegans, described above; however, it specifically involves the CHOP transcription factor and not UBL-5 and DVE-1 homologues (Zhao et al., 2002). CHOP is a target for activation by ceramide generated by the LASS6-encoded ceramide synthase in human cells, which protects the cells from endoplasmic reticulum stress-induced apoptosis (Senkal et al., 2010). This connection to ceramide signaling will become more significant in the discussion below.
Gene expression differences in rho+/rho0 pairs of three different human cell types are heterogeneous between cell types and may reflect cellular pathology (Miceli and Jazwinski, 2005). However, the gene expression changes consistent across cell types reflected an adaptation to loss of respiratory function by stimulation of glycolysis, enhanced protection from ROS, and compensation for genome instability. These gene expression changes reflect the physiologic events in yeast cells in which the retrograde response is activated. One of the genes commonly activated was c-Myc, a basic helix-loop-helix/leucine zipper transcription factor. The Myc–Max heterodimer is homologous to Rtg1–Rtg3 (Srinivasan et al., 2010). Significantly, NFκB has two binding sites in the Myc promoter, suggesting that the mammalian retrograde response may involve both NFκB and Myc (Duyao et al., 1990). NFκB would be upstream of Myc in this scenario. This is further consistent with the observation of both increased NFκB activity and elevated levels of Myc activity with age, which is suppressed by Cdk2 in a cell senescence model (Semsei et al., 1989; Campaner et al., 2010). Acute mitochondrial respiratory stress activates NFκB through a novel calcineurin-dependent pathway (Biswas et al., 2008). Stress-activated calcineurin participates in TORC2 regulation of ceramide synthase, as discussed below. This protein phosphatase also responds to the rise in cytoplasmic calcium found in mammalian rho0 cells and to which NFκB is known to respond (Butow and Avadhani, 2004). Despite these similarities between the yeast and mammalian retrograde responses, homologues of Rtg2 have not yet been found in mammals. Thus, this particular link to mitochondrial dysfunction in mammalian retrograde signaling is still missing.
There also appears to be a link between mitochondrial dysfunction in mammalian cells and cell lifespan (Passos et al., 2007). Mild mitochondrial uncoupling with dinitrophenol delays the replicative senescence of normal human diploid fibroblasts, while lowering ROS production, reducing telomere shortening, preventing the appearance of DNA repair foci in the nucleus, and inducing a variety of gene expression changes. Thus, mammalian cells show many of the molecular features of yeast retrograde signaling, and they also display the extended life span characteristic of the retrograde response. It will be important to identify the relevant signaling pathways.
General autophagy is a gene regulated process that is non-selective for removal of proteins and organelles through their degradation in the lysosome (Nakatogawa et al., 2009). This process can become selective, however. In the case of mitochondria, the Atg32 protein tags yeast mitochondria for selective removal by autophagy, in a process termed mitophagy (Kim et al., 2007; Kissova et al., 2007). This can serve to channel nutrients to starving cells. It can also allow remodeling of the cell to adapt to changing metabolic needs, as well as to remove damaged and dysfunctional organelles. The mitochondrial fission–fusion cycle plays a role in mitophagy, since deletion of DNM1 which is required for fission attenuates mitophagy without entirely eliminating it (Kanki et al., 2009). Interestingly, deletion of this gene extends yeast replicative lifespan, suggesting that some but perhaps not excessive mitophagy is consistent with long life (Scheckhuber et al., 2007).
Stationary phase mitophagy requires the AUP1 gene in yeast (Journo et al., 2009). The Aup1 protein is located in the mitochondrial intermembrane space, and it specifies a protein phosphatase. During stationary phase mitophagy, the retrograde response is activated, and this requires AUP1 (Journo et al., 2009). Furthermore, deletion of RTG3 prevents this activation of the retrograde response and mitophagy, and Aup1 affects the phosphorylation status of Rtg3. Thus, the signal generated by mitochondria appears somehow to be relayed to Rtg3 during stationary phase to elicit retrograde target gene expression and induce mitophagy. These activities are consistent with the extensive remodeling of metabolism as yeast cells adapt to survival in stationary phase.
General autophagy is inhibited by TORC1, which phosphorylates Atg13 a component of the Atg1 kinase that is essential for autophagy (Kamada et al., 2010). Deletion of the genes required for the synthesis of complex sphingolipids in yeast (IPT1 and SKN1) augments autophagy induced by nitrogen starvation (Thevissen et al., 2010). Sphingosine and ceramide are the substrates for complex sphingolipid biosynthesis, but sphingosine-1-phosphate and ceramide stimulate autophagy in mammalian cells (Lavieu et al., 2008). Together these observations suggest that a delicate balance exists in the sphingolipid biosynthetic pathway that can tip the scale from quality control through limited autophagy to full blown degradation. Thus, sphingolipid signaling has a modulatory effect on autophagy, while TORC1 may play the primary role in its regulation.
TORC2 also may regulate autophagy in yeast by stimulating ceramide synthase activity through the activation of Ypk2, another AGC protein kinase (Aronova et al., 2008). It does this in the presence of nutrients. The stimulation of ceramide synthesis would provide the substrate for complex sphingolipid synthesis and thus attenuation of autophagy. On the other hand, ceramide synthase is inhibited by the stress responsive protein phosphatase calcineurin, which is dependent on calcium/calmodulin (Aronova et al., 2008). In this way, stress would enhance autophagy. It appears that TORC2, through its effects on sphingolipid signaling is a potential modulator of autophagy.
The response to mitochondrial dysfunction may be linked to ceramide synthase activity. Ceramide synthase activity in yeast is encoded by the longevity assurance gene LAG1 and its homolog LAC1, and this enzyme is located in the endoplasmic reticulum membrane (D’Mello et al., 1994; Jiang et al., 1998; Guillas et al., 2001; Schorling et al., 2001). The LAG1/LAC1 orthologs in human, LASS1–6, encode the six human ceramide synthases, Lass1-6, which are also located in the endoplasmic reticulum (Venkataraman et al., 2002; Guillas et al., 2003; Teufel et al., 2009). A physical junction between the mitochondrion and the endoplasmic reticulum has been identified, and it has been proposed to regulate mitochondrial biology (Kornmann and Walter, 2010). This regulation could involve ceramide signaling, which plays an important role in processes such as cell cycle control and stress resistance (Dickson, 2010). Such a role would require exquisite balance. Perhaps this explains why the expression of the LAG1 ceramide synthase gene in yeast, which determines yeast replicative lifespan, is so finely tuned (Jiang et al., 2004). In mammalian cells, mitofusin 2 plays a decisive role in bridging the endoplasmic reticulum and mitochondria (de Brito and Scorrano, 2008). It thus impacts metabolism, apoptosis, and cell cycle progression. Importantly, the tethering of mitochondria to the endoplasmic reticulum is required for efficient mitochondrial calcium uptake (de Brito and Scorrano, 2008), which may impact NFκB activation in cells in mitochondrial respiratory stress as discussed earlier (Biswas et al., 2008). One of the hallmarks of mtDNA-less mammalian cells is an increase in cytoplasmic calcium levels, which leads to activation of calcineurin (Biswas et al., 1999), which would inhibit ceramide synthase and thus alter the balance of autophagy.
In addition to the physical connection between the mitochondrion and the endoplasmic reticulum, there are other ways in which the activities in these cell compartments may be coordinated. The LAC1 gene is coordinately regulated with the multi-drug resistance family of membrane transporters in yeast (Kolaczkowski et al., 2004). These transporters are activated in rho0 cells by retrograde signaling and by mechanisms independent of the RTG genes in the presence of other mitochondrial defects (Hallstrom and Moye-Rowley, 2000; Moye-Rowley, 2005). All in all, it appears that the cellular response to mitochondrial damage in the form of the retrograde response and autophagy/mitophagy is coordinated.
The PINK1 protein kinase, together with the ubiquitin ligase Parkin, plays a crucial role in the elimination of dysfunctional mitochondria by mitophagy (Deas et al., 2011; Kawajiri et al., 2011). The mitochondrial signal that triggers this process is the loss of ΔΨm (Narendra et al., 2009). This process is defective in Parkinson’s disease, which can be caused by mutations in PARKIN, resulting in its aggregation, as well as by mutations in PINK1 (Deas et al., 2011; Kawajiri et al., 2011). PINK1 is localized on the mitochondrial outer membrane. Mitochondrial depolarization results in the translocation of Parkin to mitochondria, and this requires PINK1. One of the targets of polyubiquitination by Parkin is the voltage-dependent anion channel (VDAC1; Geisler et al., 2010). The mitochondrial outer membrane protein Miro is a PINK1 substrate, and it is degraded via Parkin-mediated polyubiquitination (Wang et al., 2011). This detaches the dysfunctional mitochondria from microtubules preventing their trafficking, and likely aiding in their removal by mitophagy. There are no PINK1 or Parkin homologues in yeast. However, Gem1, the yeast Miro homolog, is found in mitochondria-endoplasmic reticulum contact sites and regulates the association of the two organelles (Kornmann et al., 2011), which suggests that Miro degradation would also release mitochondria from the endoplasmic reticulum. It will be of interest to determine whether ceramide signaling is engaged in the processes described here, because of the potential effects of the mitochondria-endoplasmic reticulum junctions on ceramide synthase activity.
Sequestosome 1 (p62) can aggregate ubiquitylated proteins and accumulates on mitochondria that have undergone Parkin-mediated polyubiquitination (Geisler et al., 2010). It is not clear whether or not p62 is required for mitophagy. However, its role in the response to mitochondrial damage may be related to its activation of the NFκB pathway, as discussed below. The similarities and differences between yeast and human cells in the activation of some of the responses to changes in mitochondrial metabolism are highlighted in Figure 2. We have discussed those aspects related to mitophagy. We turn our attention below to the master regulator NFκB, which incorporates the roles of the retrograde transcription factor Rtg1–Rtg3 into its repertoire.
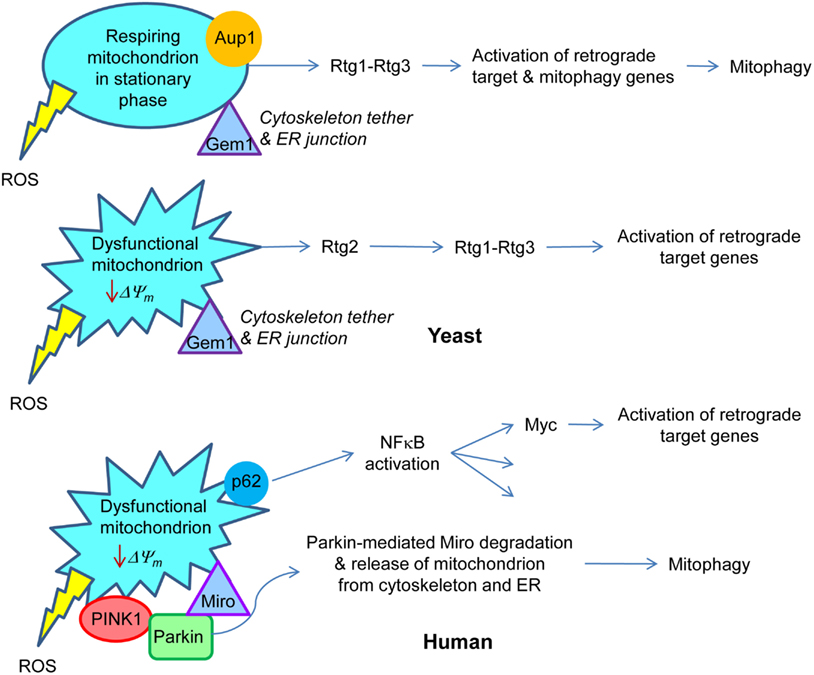
Figure 2. Retrograde signaling in yeast and human. In yeast, respiring mitochondria in non-dividing, stationary phase cells signal the retrograde response that activates both retrograde response target genes, similar to those in dividing cells, and mitophagy genes. This results in the metabolic adaptation to stationary phase. Aup1, a protein phosphatase in the intermembrane space in mitochondria, is essential for this gene induction. Rtg1–Rtg3 is the retrograde transcription factor. On the other hand, dysfunctional mitochondria in growing cells trigger the classical retrograde response with activation of retrograde response target genes. Rtg2 plays an essential role in this process. Gem1 is a Miro homolog in yeast which is important for maintaining junctions between mitochondria and the endoplasmic reticulum. By analogy with mammalian cells, it would also tether the mitochondria to the cytoskeleton. In human cells, a drop in mitochondrial membrane potential (ΔΨm) recruits Parkin by the PINK1 protein kinase to the mitochondrial membrane. Parkin mediates ubiquitylation of Miro, which releases the mitochondria from the cytoskeleton and also, presumably, from the endoplasmic reticulum. This facilitates the removal of dysfunctional mitochondria by mitophagy. Sequestosome 1 (p62) aggregates proteins polyubiquitinated by Parkin on the surface of mitochondria. p62 is known to stimulate NFκB, which among its many target genes has Myc. The Myc–Max dimer is homologous to Rtg1–Rtg3. Transcription of Myc is activated in human cells devoid of mtDNA, and Myc itself activates the transcription of metabolic genes, typical for the retrograde response. The production of reactive oxygen species (ROS) by the mitochondria may elicit responses as well.
The components of NFκB heterodimers are diverse, with roles in innate immunity, inflammation, and anti-apoptosis, and they respond to a wide diversity of stressors. NFκB forms a class of transcription factor with several members including RelA and p50, and their activation relies on IKK kinases phosphorylating inhibitory IκB proteins. Phosphorylated subunits of IκB are short-lived and are degraded by the proteasome, through the ubiquitin proteasome system (UPS) pathway. In mammalian cells, NFκB is localized at the mitochondrial membrane (Cogswell et al., 2003), and it participates with mitochondria in innate immune responses (West et al., 2011). The cluster formed by IKK, IκB, and NFκB resembles pathway connections and mediators known for the RTG genes (Figure 3). One of the constitutive mechanisms of NFκB activation is oncogenic RAS, utilizing Raf-dependent and independent MAPK signaling pathways (Norris and Baldwin, 1999). NFκB can be activated by a number of atypical mechanisms including the endoplasmic overdose response (Pahl and Baeuerle, 1997) and aging (Kriete and Mayo, 2009), making NFκB a universal stress response sensor.
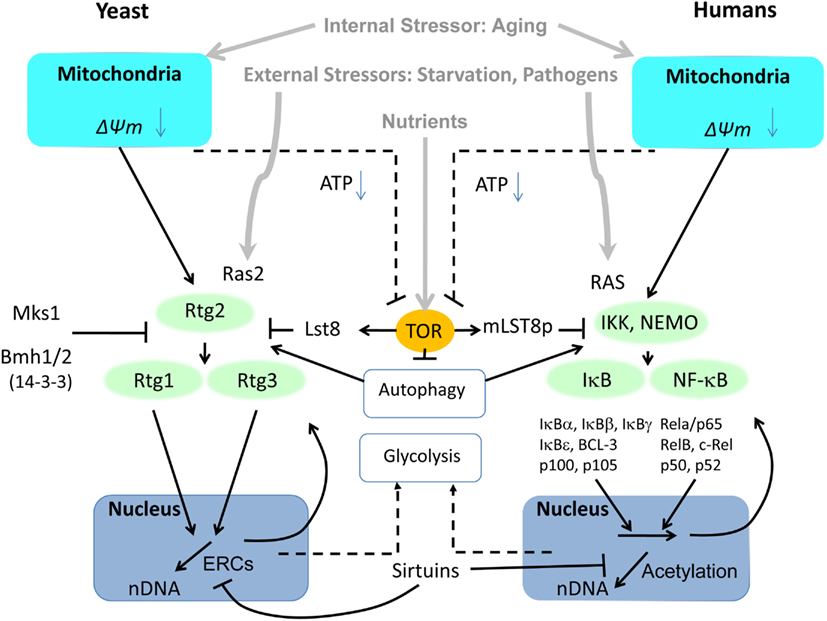
Figure 3. Side-by-side comparison of the retrograde response controlled by the heterodimeric transcription factors Rtg1–Rtg3 in yeast and NFκB in mammalian cells. In contrast to the three Rtg proteins in yeast, NFκB activators, inhibitors and transcription factors have evolved into a wide spectrum of subunits to elicit specific response patterns to a variety of stressors. Common to both pathways is their activation by mitochondrial dysfunction involving reactive oxygen species (ROS) and changes in mitochondrial membrane potential (ΔΨm). Another common activator that responds to external stressors is RAS (Ras2 in yeast). Furthermore, both pathways are modulated by TOR through LST8. LST8 dissociates from mTOR under stress, although it is not known whether this is true of TOR in yeast. Regulation of autophagy by TOR impinges on both pathways, as well. The adaptive response of both pathways to mitochondrial dysfunction includes upregulation of glycolysis to compensate for energy deficiency.
We have discussed above the similarities of the RTG genes and NFκB in metabolic regulation. One modulator of the NFκB pathway is mTOR. Inhibition of mTOR, involving separation of TOR from LST8, activates autophagy (Diaz-Troya et al., 2008). While activation of NFκB in some cases may depend on the degradation of IκB inhibitory proteins by autophagy independent of the proteasome (Jia et al., 2012), the role of mTOR and autophagy in NFκB activity is complex and involves other pathways including protein kinase B (Akt; Dan et al., 2008) and HSP90 (Qing et al., 2007). A related conundrum is the finding that IKK complexes can activate autophagy (Criollo et al., 2010, 2012). In a study involving treatment of B-cell lymphoma cells, bortezomib led to proteasome down-regulation and accumulation of polyubiquitinated proteins (Jia et al., 2012). Hereby, the autophagy adapter protein Sequestosome 1 (p62) recruits LC3-II and ubiquitinated proteins including IκBα for subsequent transport and degradation to autophagosomes. p62 can promote tumorigenesis by activating ROS scavenging via the NFκB pathway (Moscat and Diaz-Meco, 2009). It also associates with TRAF6, and in this way regulates NFκB signaling in bone homeostasis (Moscat and Diaz-Meco, 2009). p62 interacts with atypical protein kinase C proteins, such as PKCζ (Moscat and Diaz-Meco, 2009), which control NFκB (Duran et al., 2003) as well as inflammatory cytokine production in adipocytes (Lee et al., 2010b). This can lead to insulin resistance even in the absence of obesity, which has implications for aging. p62 also could be relevant for TNF-α activation of NFκB. Recent studies show that p62 interacts with Raptor and is required for TORC1 activation by amino acids, in turn downregulating autophagy (Duran et al., 2011). Thus, the regulation of autophagy and NFκB-mediated inflammation are related.
NFκB activity is controlled by ceramide signaling in some contexts. LPS-stimulated prostaglandin E2 synthesis in macrophages is mediated by the upregulation of COX2 transcription by ceramide (Wu et al., 2003). The transcription factor responsible is NFκB, whose activation is greater in old mice than in young ones. This effect is due to greater degradation of IκB in macrophages from old animals. Constitutively activating NFκB in p65 knock-in mice results in aberrant systemic inflammation involving TNFα signaling and signs of premature aging (Dong et al., 2010).
A key feature of the yeast retrograde response is activation of the glyoxylate cycle, which allows the truncated TCA cycle to serve as a source of biosynthetic intermediates (Figure 4). Tumor cells often possess mutations in mitochondrial components, which disable the electron transport chain (Mullen et al., 2012). Such cells can grow readily by generating ATP through glycolysis. However, they need a source of biosynthetic intermediates. It has recently been shown that this source is the reductive carboxylation of α-ketoglutarate derived from glutamine, which yields both acetyl-coenzyme A and the four-carbon TCA cycle intermediates that are used in various biosyntheses (Mullen et al., 2012). The key reductive carboxylation is catalyzed by isocitrate dehydrogenase-1 (IDH1) in the cytoplasm (Figure 4), although some synthesis may occur in mitochondria. The same situation occurs in normal cells under hypoxic conditions, and HIF-1α and HIF-2α both appear to play a role in the metabolic switch (Metallo et al., 2012). It is not known whether ceramide regulates these transcription factors as it does HIF-1 in C. elegans (Mehta et al., 2009; Menuz et al., 2009). If it does, the parallel between the effects of mitochondrial dysfunction on metabolism in human and in yeast and worms would be striking.
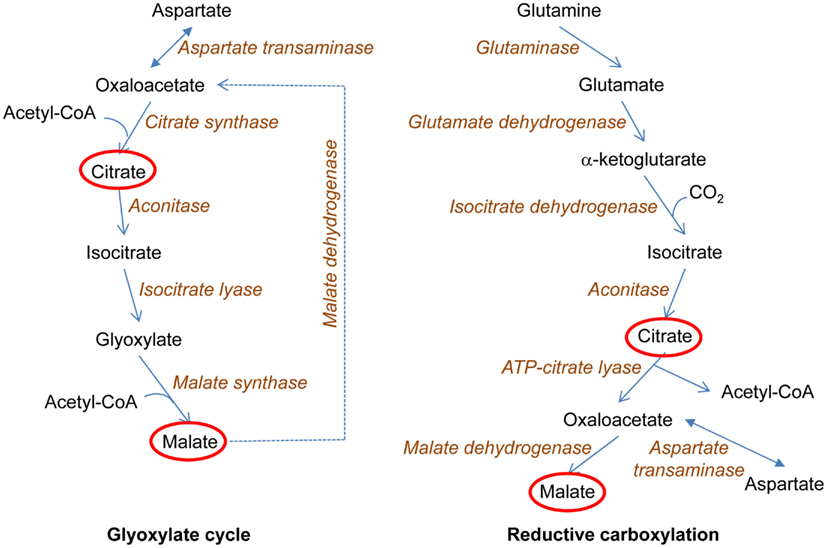
Figure 4. Metabolic adaptations to loss of biosynthetic intermediate production by a truncated tricarboxylic acid (TCA) cycle. The loss of the electron transport chain interrupts the TCA cycle at the succinate dehydrogenase reaction. This prevents the utilization of the TCA cycle for production of biosynthetic intermediates. In yeast, the glyoxylate cycle is induced. This allows acetyl-coenzyme A (acetyl-CoA) to be used for the synthesis of the TCA cycle metabolites citrate and malate, in reactions that conserve the two carbons of acetate. This, in turn, allows the first three reactions of the TCA cycle to proceed with the synthesis of α-ketoglutarate, which can be converted to glutamate, the ultimate source of nitrogen in biosynthesis (not shown here). In human cells, a related metabolic adaptation occurs. This adaptation is the reductive carboxylation of α-ketoglutarate to yield isocitrate, which in turn is a source of TCA cycle intermediates citrate and malate at the same time generating acetyl-CoA for lipid biosynthesis. The ultimate source of α-ketoglutarate in these reactions is glutamine, which allows the use of glucose for production of energy in glycolysis as well as for biosynthetic reactions. In both yeast and human, TCA cycle metabolites are used as macromolecular precursors.
There are additional hypoxia-driven changes that lead to reductive glutamine metabolism. HIF-2α promotes Myc transcription (Gordan et al., 2007). This enhances glutamine catabolism, among others by activating glutaminase expression (Gao et al., 2009). Furthermore, Myc upregulates genes involved in glycolysis, promoting the generation of lactate and ATP by substrate level phosphorylation (Collier et al., 2003), a response important in rho0 mammalian cells (Miceli and Jazwinski, 2005).
The retrograde response in yeast and related pathways in higher organisms share the common adaptive function of supporting cellular survival. Activated by external perturbations like heat and osmotic shock, bacterial pathogens, UV radiation, starvation, and related mitochondrial dysfunction, they protect the cell transiently. Environmental challenges that perturb mitochondrial function are equally likely to be encountered across species, from yeast to mammalian cells. It comes therefore as no surprise that many responses exhibit similar features as shown here for RTG and NFκB stress responses. The decisive difference between the aging process and acute environmental perturbations, regardless of the biological system, is a rather slow accumulation of damage and dysfunction in the former. Although mechanisms like the retrograde response compensate for mitochondrial deficiencies and extend lifespan, they cannot escape aging in the long term. The retrograde response was not likely selected as an anti-aging mechanism, as it operates as a double-edged sword. In yeast, its activation extends lifespan, but it also reduces genome stability which can ultimately contribute to cell demise. In mammalian cells, NFκB, optimized under evolutionary pressure to respond to acute challenges like infections, promotes inflammatory disease states when chronically activated in aging.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported in part by grant AG006168 from the National Institutes of Health (S. Michal Jazwinski) and by the Wallace H. Coulter Foundation (Andres Kriete).
Aronova, S., Wedaman, K., Aronov, P. A., Fontes, K., Ramos, K., Hammock, B. D., and Powers, T. (2008). Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 7, 148–158.
Bhattacharyya, S., Rolfsmeier, M. L., Dixon, M. J., Wagoner, K., and Lahue, R. S. (2002). Identification of RTG2 as a modifier gene for CTG*CAG repeat instability in Saccharomyces cerevisiae. Genetics 162, 579–589.
Biswas, G., Adebanjo, O. A., Freedman, B. D., Anandatheerthavarada, H. K., Vijayasarathy, C., Zaidi, M., Kotlikoff, M., and Avadhani, N. G. (1999). Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 18, 522–533.
Biswas, G., Tang, W., Sondheimer, N., Guha, M., Bansal, S., and Avadhani, N. G. (2008). A distinctive physiological role for IκBβ in the propagation of mitochondrial respiratory stress signaling. J. Biol. Chem. 283, 12586–12594.
Borghouts, C., Benguria, A., Wawryn, J., and Jazwinski, S. M. (2004). Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics 166, 765–777.
Breitkreutz, A., Choi, H., Sharom, J. R., Boucher, L., Neduva, V., Larsen, B., Lin, Z. Y., Breitkreutz, B. J., Stark, C., Liu, G., Ahn, J., Dewar-Darch, D., Reguly, T., Tang, X., Almeida, R., Qin, Z. S., Pawson, T., Gingras, A. C., Nesvizhskii, A. I., and Tyers, M. (2010). A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046.
Butow, R. A., and Avadhani, N. G. (2004). Mitochondrial signaling: the retrograde response. Mol. Cell 14, 1–15.
Caballero, A., Ugidos, A., Liu, B., Oling, D., Kvint, K., Hao, X., Mignat, C., Nachin, L., Molin, M., and Nystrom, T. (2011). Absence of mitochondrial translation control proteins extends life span by activating sirtuin-dependent silencing. Mol. Cell 42, 390–400.
Campaner, S., Doni, M., Hydbring, P., Verrecchia, A., Bianchi, L., Sardella, D., Schleker, T., Perna, D., Tronnersjo, S., Murga, M., Fernandez-Capetillo, O., Barbacid, M., Larsson, L. G., and Amati, B. (2010). Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 12, 54–59.
Chen, E. J., and Kaiser, C. A. (2003). LST8 negatively regulates amino acid biosynthesis as a component of the TOR pathway. J. Cell Biol. 161, 333–347.
Cogswell, P. C., Kashatus, D. F., Keifer, J. A., Guttridge, D. C., Reuther, J. Y., Bristow, C., Roy, S., Nicholson, D. W., and Baldwin, A. S. Jr. (2003). NF-κB and IκBα are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-κB. J. Biol. Chem. 278, 2963–2968.
Collier, J. J., Doan, T. T., Daniels, M. C., Schurr, J. R., Kolls, J. K., and Scott, D. K. (2003). c-Myc is required for the glucose-mediated induction of metabolic enzyme genes. J. Biol. Chem. 278, 6588–6595.
Copeland, J. M., Cho, J., Lo, T. Jr., Hur, J. H., Bahadorani, S., Arabyan, T., Rabie, J., Soh, J., and Walker, D. W. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598.
Criollo, A., Chereau, F., Malik, S. A., Niso-Santano, M., Marino, G., Galluzzi, L., Maiuri, M. C., Baud, V., and Kroemer, G. (2012). Autophagy is required for the activation of NFκB. Cell Cycle 11, 194–199.
Criollo, A., Senovilla, L., Authier, H., Maiuri, M. C., Morselli, E., Vitale, I., Kepp, O., Tasdemir, E., Galluzzi, L., Shen, S., Tailler, M., Delahaye, N., Tesniere, A., De Stefano, D., Younes, A. B., Harper, F., Pierron, G., Lavandero, S., Zitvogel, L., Israel, A., Baud, V., and Kroemer, G. (2010). The IKK complex contributes to the induction of autophagy. EMBO J. 29, 619–631.
Cristina, D., Cary, M., Lunceford, A., Clarke, C., and Kenyon, C. (2009). A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 5, e1000450. doi: 10.1371/journal.pgen.1000450
D’Mello N, P., Childress, A. M., Franklin, D. S., Kale, S. P., Pinswasdi, C., and Jazwinski, S. M. (1994). Cloning and characterization of LAG1, a longevity-assurance gene in yeast. J. Biol. Chem. 269, 15451–15459.
Dan, H. C., Cooper, M. J., Cogswell, P. C., Duncan, J. A., Ting, J. P., and Baldwin, A. S. (2008). Akt-dependent regulation of NF-κB is controlled by mTOR and raptor in association with IKK. Genes Dev. 22, 1490–1500.
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610.
Deas, E., Wood, N. W., and Plun-Favreau, H. (2011). Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim. Biophys. Acta 1813, 623–633.
Dell’Agnello, C., Leo, S., Agostino, A., Szabadkai, G., Tiveron, C., Zulian, A., Prelle, A., Roubertoux, P., Rizzuto, R., and Zeviani, M. (2007). Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431–444.
Diaz-Troya, S., Florencio, F. J., and Crespo, J. L. (2008). Target of rapamycin and LST8 proteins associate with membranes from the endoplasmic reticulum in the unicellular green alga Chlamydomonas reinhardtii. Eukaryot. Cell 7, 212–222.
Dickson, R. C. (2010). Roles for sphingolipids in Saccharomyces cerevisiae. Adv. Exp. Med. Biol. 688, 217–231.
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., Kamath, R. S., Ahringer, J., and Kenyon, C. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401.
Dilova, I., Aronova, S., Chen, J. C., and Powers, T. (2004). Tor signaling and nutrient-based signals converge on Mks1p phosphorylation to regulate expression of Rtg1.Rtg3p-dependent target genes. J. Biol. Chem. 279, 46527–46535.
Dilova, I., Chen, C. Y., and Powers, T. (2002). Mks1 in concert with TOR signaling negatively regulates RTG target gene expression in S. cerevisiae. Curr. Biol. 12, 389–395.
Dong, J., Jimi, E., Zeiss, C., Hayden, M. S., and Ghosh, S. (2010). Constitutively active NF-κB triggers systemic TNFα-dependent inflammation and localized TNFα-independent inflammatory disease. Genes Dev. 24, 1709–1717.
Duran, A., Amanchy, R., Linares, J. F., Joshi, J., Abu-Baker, S., Porollo, A., Hansen, M., Moscat, J., and Diaz-Meco, M. T. (2011). p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 44, 134–146.
Duran, A., Diaz-Meco, M. T., and Moscat, J. (2003). Essential role of RelA Ser311 phosphorylation by ζPKC in NF-κB transcriptional activation. EMBO J. 22, 3910–3918.
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91.
Duyao, M. P., Kessler, D. J., Spicer, D. B., and Sonenshein, G. E. (1990). Binding of NF-κB-like factors to regulatory sequences of the c-myc gene. Curr. Top. Microbiol. Immunol. 166, 211–220.
Epstein, C. B., Waddle, J. A., Hale, W. T., Dave, V., Thornton, J., Macatee, T. L., Garner, H. R., and Butow, R. A. (2001). Genome-wide responses to mitochondrial dysfunction. Mol. Biol. Cell 12, 297–308.
Gao, P., Tchernyshyov, I., Chang, T. C., Lee, Y. S., Kita, K., Ochi, T., Zeller, K. I., De Marzo, A. M., Van Eyk, J. E., Mendell, J. T., and Dang, C. V. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765.
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., and Springer, W. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131.
Giannattasio, S., Liu, Z., Thornton, J., and Butow, R. A. (2005). Retrograde response to mitochondrial dysfunction is separable from TOR1/2 regulation of retrograde gene expression. J. Biol. Chem. 280, 42528–42535.
Gordan, J. D., Bertout, J. A., Hu, C. J., Diehl, J. A., and Simon, M. C. (2007). HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 11, 335–347.
Guillas, I., Jiang, J. C., Vionnet, C., Roubaty, C., Uldry, D., Chuard, R., Wang, J., Jazwinski, S. M., and Conzelmann, A. (2003). Human homologues of LAG1 reconstitute acyl-CoA-dependent ceramide synthesis in yeast. J. Biol. Chem. 278, 37083–37091.
Guillas, I., Kirchman, P. A., Chuard, R., Pfefferli, M., Jiang, J. C., Jazwinski, S. M., and Conzelmann, A. (2001). C26-CoA-dependent ceramide synthesis of Saccharomyces cerevisiae is operated by Lag1p and Lac1p. EMBO J. 20, 2655–2665.
Hallstrom, T. C., and Moye-Rowley, W. S. (2000). Multiple signals from dysfunctional mitochondria activate the pleiotropic drug resistance pathway in Saccharomyces cerevisiae. J. Biol. Chem. 275, 37347–37356.
Heddi, A., Lestienne, P., Wallace, D. C., and Stepien, G. (1993). Mitochondrial DNA expression in mitochondrial myopathies and coordinated expression of nuclear genes involved in ATP production. J. Biol. Chem. 268, 12156–12163.
Heeren, G., Rinnerthaler, M., Laun, P., Von Seyerl, P., Kossler, S., Klinger, H., Hager, M., Bogengruber, E., Jarolim, S., Simon-Nobbe, B., Schuller, C., Carmona-Gutierrez, D., Breitenbach-Koller, L., Muck, C., Jansen-Durr, P., Criollo, A., Kroemer, G., Madeo, F., and Breitenbach, M. (2009). The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1. Aging (Albany NY) 1, 622–636.
Holbrook, M. A., and Menninger, J. R. (2002). Erythromycin slows aging of Saccharomyces cerevisiae. J. Gerontol. A Biol. Sci. Med. Sci. 57, B29–B36.
Jazwinski, S. M. (2000). Metabolic control and gene dysregulation in yeast aging. Ann. N. Y. Acad. Sci. 908, 21–30.
Jazwinski, S. M. (2012). The retrograde response: when mitochondrial quality control is not enough. Biochim. Bsiophys. Acta. PMID: 22374136. [Epub ahead of print].
Jia, L., Gopinathan, G., Sukumar, J. T., and Gribben, J. G. (2012). Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS ONE 7, e32584. doi:10.1371/journal.pone.0032584
Jia, Y., Rothermel, B., Thornton, J., and Butow, R. A. (1997). A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol. Cell. Biol. 17, 1110–1117.
Jiang, J. C., Kirchman, P. A., Allen, M., and Jazwinski, S. M. (2004). Suppressor analysis points to the subtle role of the LAG1 ceramide synthase gene in determining yeast longevity. Exp. Gerontol. 39, 999–1009.
Jiang, J. C., Kirchman, P. A., Zagulski, M., Hunt, J., and Jazwinski, S. M. (1998). Homologs of the yeast longevity gene LAG1 in Caenorhabditis elegans and human. Genome Res. 8, 1259–1272.
Jothi, R., Balaji, S., Wuster, A., Grochow, J. A., Gsponer, J., Przytycka, T. M., Aravind, L., and Babu, M. M. (2009). Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol. Syst. Biol. 5, 294.
Journo, D., Mor, A., and Abeliovich, H. (2009). Aup1-mediated regulation of Rtg3 during mitophagy. J. Biol. Chem. 284, 35885–35895.
Kamada, Y., Yoshino, K., Kondo, C., Kawamata, T., Oshiro, N., Yonezawa, K., and Ohsumi, Y. (2010). Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 30, 1049–1058.
Kanki, T., Wang, K., Baba, M., Bartholomew, C. R., Lynch-Day, M. A., Du, Z., Geng, J., Mao, K., Yang, Z., Yen, W. L., and Klionsky, D. J. (2009). A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol. Biol. Cell 20, 4730–4738.
Kawai, S., Urban, J., Piccolis, M., Panchaud, N., De Virgilio, C., and Loewith, R. (2011). Mitochondrial genomic dysfunction causes dephosphorylation of Sch9 in the yeast Saccharomyces cerevisiae. Eukaryot. Cell 10, 1367–1369.
Kawajiri, S., Saiki, S., Sato, S., and Hattori, N. (2011). Genetic mutations and functions of PINK1. Trends Pharmacol. Sci. 32, 573–580.
Kim, I., Rodriguez-Enriquez, S., and Lemasters, J. J. (2007). Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 462, 245–253.
Kim, S., Ohkuni, K., Couplan, E., and Jazwinski, S. M. (2004). The histone acetyltransferase GCN5 modulates the retrograde response and genome stability determining yeast longevity. Biogerontology 5, 305–316.
Kirchman, P. A., Kim, S., Lai, C. Y., and Jazwinski, S. M. (1999). Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 152, 179–190.
Kissova, I., Salin, B., Schaeffer, J., Bhatia, S., Manon, S., and Camougrand, N. (2007). Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3, 329–336.
Kolaczkowski, M., Kolaczkowska, A., Gaigg, B., Schneiter, R., and Moye-Rowley, W. S. (2004). Differential regulation of ceramide synthase components LAC1 and LAG1 in Saccharomyces cerevisiae. Eukaryot. Cell 3, 880–892.
Komeili, A., Wedaman, K. P., O’Shea, E. K., and Powers, T. (2000). Mechanism of metabolic control. Target of rapamycin signaling links nitrogen quality to the activity of the Rtg1 and Rtg3 transcription factors. J. Cell Biol. 151, 863–878.
Kornmann, B., Osman, C., and Walter, P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. U.S.A. 108, 14151–14156.
Kornmann, B., and Walter, P. (2010). ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J. Cell Sci. 123, 1389–1393.
Kriete, A., and Mayo, K. L. (2009). Atypical pathways of NF-κB activation and aging. Exp. Gerontol. 44, 250–255.
Lai, C. Y., Jaruga, E., Borghouts, C., and Jazwinski, S. M. (2002). A mutation in the ATP2 gene abrogates the age asymmetry between mother and daughter cells of the yeast Saccharomyces cerevisiae. Genetics 162, 73–87.
Lapointe, J., and Hekimi, S. (2008). Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J. Biol. Chem. 283, 26217–26227.
Lavieu, G., Scarlatti, F., Sala, G., Carpentier, S., Levade, T., Ghidoni, R., Botti, J., and Codogno, P. (2008). Sphingolipids in macroautophagy. Methods Mol. Biol. 445, 159–173.
Lee, S. J., Hwang, A. B., and Kenyon, C. (2010a). Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136.
Lee, S. J., Kim, J. Y., Nogueiras, R., Linares, J. F., Perez-Tilve, D., Jung, D. Y., Ko, H. J., Hofmann, S. M., Drew, A., Leitges, M., Kim, J. K., Tschop, M. H., Diaz-Meco, M. T., and Moscat, J. (2010b). PKCζ-regulated inflammation in the nonhematopoietic compartment is critical for obesity-induced glucose intolerance. Cell Metab. 12, 65–77.
Lee, S. S., Lee, R. Y., Fraser, A. G., Kamath, R. S., Ahringer, J., and Ruvkun, G. (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40–48.
Liao, X., and Butow, R. A. (1993). RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 72, 61–71.
Liu, J., Wu, Q., He, D., Ma, T., Du, L., Dui, W., Guo, X., and Jiao, R. (2011). Drosophila sbo regulates lifespan through its function in the synthesis of coenzyme Q in vivo. J. Genet. Genomics 38, 225–234.
Liu, J. L., Desjardins, D., Branicky, R., Agellon, L. B., and Hekimi, S. (2012). Mitochondrial oxidative stress alters a pathway in Caenorhabditis elegans strongly resembling that of bile acid biosynthesis and secretion in vertebrates. PLoS Genet. 8, e1002553. doi:10.1371/journal.pgen.1002553
Liu, Z., and Butow, R. A. (2006). Mitochondrial retrograde signaling. Annu. Rev. Genet. 40, 159–185.
Liu, Z., Sekito, T., Epstein, C. B., and Butow, R. A. (2001). RTG-dependent mitochondria to nucleus signaling is negatively regulated by the seven WD-repeat protein Lst8p. EMBO J. 20, 7209–7219.
Liu, Z., Sekito, T., Spirek, M., Thornton, J., and Butow, R. A. (2003). Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Mol. Cell 12, 401–411.
Liu, Z., Spirek, M., Thornton, J., and Butow, R. A. (2005). A novel degron-mediated degradation of the RTG pathway regulator, Mks1p, by SCFGrr1. Mol. Biol. Cell 16, 4893–4904.
Matsuura, A., and Anraku, Y. (1993). Characterization of the MKS1 gene, a new negative regulator of the Ras-cyclic AMP pathway in Saccharomyces cerevisiae. Mol. Gen. Genet. 238, 6–16.
Mehta, R., Steinkraus, K. A., Sutphin, G. L., Ramos, F. J., Shamieh, L. S., Huh, A., Davis, C., Chandler-Brown, D., and Kaeberlein, M. (2009). Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 324, 1196–1198.
Menuz, V., Howell, K. S., Gentina, S., Epstein, S., Riezman, I., Fornallaz-Mulhauser, M., Hengartner, M. O., Gomez, M., Riezman, H., and Martinou, J. C. (2009). Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science 324, 381–384.
Metallo, C. M., Gameiro, P. A., Bell, E. L., Mattaini, K. R., Yang, J., Hiller, K., Jewell, C. M., Johnson, Z. R., Irvine, D. J., Guarente, L., Kelleher, J. K., Vander Heiden, M. G., Iliopoulos, O., and Stephanopoulos, G. (2012). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384.
Miceli, M. V., and Jazwinski, S. M. (2005). Common and cell type-specific responses of human cells to mitochondrial dysfunction. Exp. Cell Res. 302, 270–280.
Miceli, M. V., Jiang, J. C., Tiwari, A., Rodriguez-Quinones, J. F., and Jazwinski, S. M. (2011). Loss of mitochondrial membrane potential triggers the retrograde response extending yeast replicative lifespan. Front. Genet. 2:102. doi:10.3389/fgene.2011.00102
Moscat, J., and Diaz-Meco, M. T. (2009). p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137, 1001–1004.
Moye-Rowley, W. S. (2005). Retrograde regulation of multidrug resistance in Saccharomyces cerevisiae. Gene 354, 15–21.
Mullen, A. R., Wheaton, W. W., Jin, E. S., Chen, P. H., Sullivan, L. B., Cheng, T., Yang, Y., Linehan, W. M., Chandel, N. S., and Deberardinis, R. J. (2012). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388.
Nakatogawa, H., Suzuki, K., Kamada, Y., and Ohsumi, Y. (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10, 458–467.
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2009). Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5, 706–708.
Norris, J. L., and Baldwin, A. S. Jr. (1999). Oncogenic Ras enhances NF-κB transcriptional activity through Raf-dependent and Raf-independent mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 274, 13841–13846.
Pahl, H. L., and Baeuerle, P. A. (1997). The ER-overload response: activation of NF-κB. Trends Biochem. Sci. 22, 63–67.
Pan, Y., Schroeder, E. A., Ocampo, A., Barrientos, A., and Shadel, G. S. (2011). Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 13, 668–678.
Passos, J. F., Saretzki, G., Ahmed, S., Nelson, G., Richter, T., Peters, H., Wappler, I., Birket, M. J., Harold, G., Schaeuble, K., Birch-Machin, M. A., Kirkwood, T. B., and Von Zglinicki, T. (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5, e110. doi:10.1371/journal.pbio.0050110
Pastor, M. M., Proft, M., and Pascual-Ahuir, A. (2009). Mitochondrial function is an inducible determinant of osmotic stress adaptation in yeast. J. Biol. Chem. 284, 30307–30317.
Pray-Grant, M. G., Schieltz, D., Mcmahon, S. J., Wood, J. M., Kennedy, E. L., Cook, R. G., Workman, J. L., Yates, J. R. III, and Grant, P. A. (2002). The novel SLIK histone acetyltransferase complex functions in the yeast retrograde response pathway. Mol. Cell. Biol. 22, 8774–8786.
Qing, G., Yan, P., Qu, Z., Liu, H., and Xiao, G. (2007). Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-κB-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 17, 520–530.
Rothermel, B. A., Shyjan, A. W., Etheredge, J. L., and Butow, R. A. (1995). Transactivation by Rtg1p, a basic helix-loop-helix protein that functions in communication between mitochondria and the nucleus in yeast. J. Biol. Chem. 270, 29476–29482.
Rothermel, B. A., Thornton, J. L., and Butow, R. A. (1997). Rtg3p, a basic helix-loop-helix/leucine zipper protein that functions in mitochondrial-induced changes in gene expression, contains independent activation domains. J. Biol. Chem. 272, 19801–19807.
Scheckhuber, C. Q., Erjavec, N., Tinazli, A., Hamann, A., Nystrom, T., and Osiewacz, H. D. (2007). Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat. Cell Biol. 9, 99–105.
Schorling, S., Vallee, B., Barz, W. P., Riezman, H., and Oesterhelt, D. (2001). Lag1p and Lac1p are essential for the Acyl-CoA-dependent ceramide synthase reaction in Saccharomyces cerevisae. Mol. Biol. Cell 12, 3417–3427.
Sekito, T., Liu, Z., Thornton, J., and Butow, R. A. (2002). RTG-dependent mitochondria-to-nucleus signaling is regulated by MKS1 and is linked to formation of yeast prion [URE3]. Mol. Biol. Cell 13, 795–804.
Sekito, T., Thornton, J., and Butow, R. A. (2000). Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol. Biol. Cell 11, 2103–2115.
Semsei, I., Ma, S. Y., and Cutler, R. G. (1989). Tissue and age specific expression of the myc proto-oncogene family throughout the life span of the C57BL/6J mouse strain. Oncogene 4, 465–471.
Senkal, C. E., Ponnusamy, S., Bielawski, J., Hannun, Y. A., and Ogretmen, B. (2010). Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 24, 296–308.
Srinivasan, V., Kriete, A., Sacan, A., and Jazwinski, S. M. (2010). Comparing the yeast retrograde response and NF-κB stress responses: implications for aging. Aging Cell 9, 933–941.
Sun, J., Kale, S. P., Childress, A. M., Pinswasdi, C., and Jazwinski, S. M. (1994). Divergent roles of RAS1 and RAS2 in yeast longevity. J. Biol. Chem. 269, 18638–18645.
Teufel, A., Maass, T., Galle, P. R., and Malik, N. (2009). The longevity assurance homologue of yeast lag1 (Lass) gene family (review). Int. J. Mol. Med. 23, 135–140.
Thevissen, K., Yen, W. L., Carmona-Gutierrez, D., Idkowiak-Baldys, J., Aerts, A. M., Francois, I. E., Madeo, F., Klionsky, D. J., Hannun, Y. A., and Cammue, B. P. (2010). Skn1 and Ipt1 negatively regulate autophagy in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 303, 163–168.
Traven, A., Wong, J. M., Xu, D., Sopta, M., and Ingles, C. J. (2001). Interorganellar communication. Altered nuclear gene expression profiles in a yeast mitochondrial DNA mutant. J. Biol. Chem. 276, 4020–4027.
Urban, J., Soulard, A., Huber, A., Lippman, S., Mukhopadhyay, D., Deloche, O., Wanke, V., Anrather, D., Ammerer, G., Riezman, H., Broach, J. R., De Virgilio, C., Hall, M. N., and Loewith, R. (2007). Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674.
Vanfleteren, J. R., and De Vreese, A. (1995). The gerontogenes age-1 and daf-2 determine metabolic rate potential in aging Caenorhabditis elegans. FASEB J. 9, 1355–1361.
Venkataraman, K., Riebeling, C., Bodennec, J., Riezman, H., Allegood, J. C., Sullards, M. C., Merrill, A. H. Jr., and Futerman, A. H. (2002). Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J. Biol. Chem. 277, 35642–35649.
Wallace, D. C., and Fan, W. (2010). Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10, 12–31.
Walter, L., Baruah, A., Chang, H. W., Pace, H. M., and Lee, S. S. (2011). The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS Biol. 9, e1001084. doi:10.1371/journal.pbio.1001084
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., Rice, S., Steen, J., Lavoie, M. J., and Schwarz, T. L. (2011). PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906.
West, A. P., Shadel, G. S., and Ghosh, S. (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402.
Wu, D., Marko, M., Claycombe, K., Paulson, K. E., and Meydani, S. N. (2003). Ceramide-induced and age-associated increase in macrophage COX-2 expression is mediated through up-regulation of NF-κB activity. J. Biol. Chem. 278, 10983–10992.
Yang, W., and Hekimi, S. (2010a). A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 8, e1000556. doi:10.1371/journal.pbio.1000556
Yang, W., and Hekimi, S. (2010b). Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 9, 433–447.
Zhang, A., Shen, Y., Gao, W., and Dong, J. (2011). Role of Sch9 in regulating Ras-cAMP signal pathway in Saccharomyces cerevisiae. FEBS Lett. 585, 3026–3032.
Keywords: retrograde response, RTG genes, NFκB, metabolism, stress, mitophagy, Saccharomyces cerevisiae, replicative lifespan
Citation: Jazwinski SM and Kriete A (2012) The yeast retrograde response as a model of intracellular signaling of mitochondrial dysfunction. Front. Physio. 3:139. doi: 10.3389/fphys.2012.00139
Received: 30 March 2012; Accepted: 26 April 2012;
Published online: 17 May 2012.
Edited by:
Vladimir Titorenko, Concordia University, CanadaReviewed by:
Christopher Baines, University of Missouri–Columbia, USACopyright: © 2012 Jazwinski and Kriete. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: S. Michal Jazwinski, Department of Medicine, Tulane University Health Sciences Center, 1430 Tulane Avenue, SL-12, New Orleans, LA 70112, USA. e-mail:c2phendpbnNAdHVsYW5lLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.