
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 07 May 2012
Sec. Vascular Physiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00128
This article is part of the Research Topic Vascular macrophages and dendritic cells View all 8 articles
 David G. Harrison1*
David G. Harrison1* Paul J. Marvar2 and Jens M. Titze1
Paul J. Marvar2 and Jens M. Titze1
Hypertension is a common disorder with uncertain etiology. In the last several years, it has become evident that components of both the innate and adaptive immune system play an essential role in hypertension. Macrophages and T cells accumulate in the perivascular fat, the heart and the kidney of hypertensive patients, and in animals with experimental hypertension. Various immunosuppressive agents lower blood pressure and prevent end-organ damage. Mice lacking lymphocytes are protected against hypertension, and adoptive transfer of T cells, but not B cells in the animals restores their blood pressure response to stimuli such as angiotensin II or high salt. Recent studies have shown that mice lacking macrophages have blunted hypertension in response to angiotensin II and that genetic deletion of macrophages markedly reduces experimental hypertension. Dendritic cells have also been implicated in this disease. Many hypertensive stimuli have triggering effects on the central nervous system and signals arising from the circumventricular organ seem to promote inflammation. Studies have suggested that central signals activate macrophages and T cells, which home to the kidney and vasculature and release cytokines, including IL-6 and IL-17, which in turn cause renal and vascular dysfunction and lead to blood pressure elevation. These recent discoveries provide a new understanding of hypertension and provide novel therapeutic opportunities for treatment of this serious disease.
Hypertension is an enormous clinical burden in Western Societies and represents a major risk factor for stroke, myocardial infarction, and heart failure. One third of the population is hypertensive, while another third has “pre-hypertension” and commonly develop overt hypertension in 2 years (Lenfant et al., 2003; Julius et al., 2006; Narayan et al., 2010). By age 70, 70% of United States citizens have hypertension. Despite the frequency of this disease, its cause in the majority of adults is unknown. In virtually all cases of adult hypertension, systemic vascular resistance is increased, suggesting a vascular cause of the disease. In keeping with this, vasodilator agents such as calcium channel antagonists and hydralazine lower blood pressure in many instances. In addition, the kidney is often implicated in hypertension. Most inherited single gene defects that cause hypertension affect the distal nephron, leading to sodium and volume retention (Lifton et al., 2001). Finally, there is ample evidence that the central nervous system is altered in hypertension. Central administration of angiotensin II promotes neuronal firing in specific centers including the subfornical organ and this increases sympathetic outflow, thereby increasing systemic blood pressure (Scroop and Lowe, 1968; Peterson et al., 2006). Increased renal sympathetic nerve traffic promotes hypertension, and renal sympathectomy lowers blood pressure in both experimental animals and humans (Dibona and Esler, 2010; Esler et al., 2010). The precise mechanisms whereby these various systems interact to cause hypertension remain unclear.
For almost half a century there has been evidence that the immune system is involved in hypertension. Inflammatory cells, including T cells and macrophages, have been observed in the kidneys of hypertensive animals and humans and have been implicated in renal damage. In the past decade, new methodology and experimental models for study of adaptive and innate immunity have been developed that have led to a new understanding of this complex disease. This new information will be covered in this review and a proposal for the role of inflammation in hypertension will be discussed.
As reflected above, organs most often implicated in hypertension are the vasculature, the kidney, and the brain. Inflammatory cells accumulate in all of these organs in the setting of hypertension, although most research to date has focused on the vasculature and the kidney. In the blood vessel, we have found that chronic angiotensin II infusion causes vascular accumulation of T cells, B cells, macrophages, and to a lesser extent, dendritic cells. Of note, these cells predominantly accumulate in the adventitia and perivascular adipose tissue (PVAT) of the larger vessels and of smaller resistance vessels such as the mesenteric arcade. Our preliminary data indicate that the degree of PVAT inflammation in hypertension is greater than in other visceral fat, and that hypertensive stimuli do not promote inflammation in the subcutaneous fat. The precise reason why inflammatory cells accumulate in the perivascular regions remains unclear, however sympathetic nerves terminate in these regions, and might contribute to signaling this inflammatory process. Inflammation in the PVAT occurs not only in hypertension, but in other cardiovascular conditions, including obesity and atherosclerosis (Hagita et al., 2011; Ohman et al., 2011). PVAT releases factors that affect vascular tone and generates chemotactic molecules that promote inflammation. An important player in accumulation of leukocytes in the PVAT is P-selectin glycoprotein ligand-1 (PSGL1), a key ligand for both P- and E-selectins on leukocytes (Russo et al., 2010). Mice lacking this have reduced accumulation of PVAT macrophages when crossed to a genetically obese background and reduced rolling of leukocytes on venular endothelium compared to animals with this ligand. Moreover, PSGL1 deficient mice have reduced serum levels of soluble E and P-selectin and MCP1. Bone marrow transplant studies have shown that this is a property of PSGL1 on the leukocytes rather than the fat or vasculature per se. In addition, perivascular venules likely express other chemokines that promote leukocyte transmigration.
There is also evidence that inflammatory cells accumulate in perivascular regions in the kidney, and in and around glomeruli. Theuer et al. (2002) have shown that there is extensive perivascular infiltration of leukocytes in the kidney and heart of double transgenic rats harboring the human renin and angiotensinogen genes. These investigators found that pyrrolidine dithiocarbamate (PDTC), an antioxidant and an inhibitor of NFκB, prevented monocyte/macrophage infiltration in these animals, and reduced expression of NFκB-dependent genes, including the intracellular cellular adhesion molecule-1 (ICAM-1) and the inducible nitric oxide synthase (iNOS). The authors also showed that PDTC markedly enhanced survival in these rats, reduced albuminuria, decreased cardiac hypertrophy, and reduced blood pressure. PDTC also reduced NFκB activation, ICAM-1, and iNOS levels in this experimental model. This study emphasized the role of NFκB-mediated inflammation in blood pressure elevation and its importance in producing end-organ damage. There is also evidence of macrophage and T cell infiltration in the glomeruli of hypertensive animals and humans. Chronic infusion of endothelin-1 causes endothelial injury, podocyte damage, and enhances glomerular leukocyte infiltration in rats, likely via endothelin A receptor activation (Saleh et al., 2010). In humans, Imakiire et al. (2007) have shown that angiotensin converting enzyme inhibitors reduce macrophage infiltration of the glomerulus.
While less studied, the brain is also a site of inflammation in hypertension. Particularly important are microglial cells, which are the resident immune cells of the CNS. Microglia are derived from monocytic cells and develop properties similar to activated macrophages during inflammation (Ransohoff and Perry, 2009). Recently, Shi et al. (2010) showed that chronic angiotensin II infusion in rats activates microglial cells in the paraventricular nucleus (PVN), a site that integrates signals from the forebrain and in turn sends efferent projections to the brainstem to modulate blood pressure. The authors found that concomitant intracerebroventricular (ICV) infusion of the anti-inflammatory antibiotic minocycline not only prevents microglial activation, but also hypertension. The authors also showed that angiotensin II increases mRNA expression of the proinflammatory cytokines 1L-1β, TNFα, and IL-6 and a decrease in the anti-inflammatory cytokine IL-10. Minocycline reverses these perturbations of cytokine production. Moreover, the authors used adenoviral gene therapy to overexpress IL-10 and found that this prevents hypertension while ICV injection of IL-1β causes hypertension.
Patton and co-workers have implicated brainstem inflammation in hypertension, demonstrating a role of junctional adhesion molecule-1 (JAM-1) in hypertension (Waki et al., 2007). JAM-1 is an adhesion molecule expressed at endothelial cell junctions and involved in leukocyte binding to the endothelium. The authors found that JAM-1 is upregulated in the brainstem of spontaneously hypertensive rats (SHR), and that overexpression of JAM-1 in the nucleus tractus solitarius raises blood pressure and leads to leukocyte adhesion in the brainstem of normal rats. This paper emphasized a role of leukocyte/endothelial adhesion in central nervous system in hypertension.
As in other organs, resident immune cells in the CNS are localized in the perivascular space. In the CNS this specialized region is known as the Virchow–Robin space. This pre-lymphatic space provides a communication between the central nervous system and the peripheral immune system (Williams et al., 2001; Marin-Padilla and Knopman, 2011). Interestingly, studies using magnetic resonance imaging have shown that hypertension increases in size of the Virchow–Robins space in humans (Heier et al., 1989). The Virchow–Robin space is similarly dilated in dementia and aging, and might reflect a common pathology between these diseases and hypertension.
Cells of the monocyte/macrophage line have been identified in the kidney and the vasculature in hypertension for many years. In the SHR, Clozel et al. (1991) found that the distribution of subendothelial macrophages corresponded to endothelial function and that treatment with an angiotensin I-enzyme converting inhibitor improved endothelial function and reduced the number of vascular macrophages. Various models of hypertension, including angiotensin II infusion, lead-induced hypertension, and high salt have been shown to increase renal accumulation of macrophages (Franco et al., 2007; Rodriguez-Iturbe et al., 2008). Despite these observational studies, to date only a few studies have attempted to genetically or chemically modify the level of macrophages to examine their role in hypertension. In two studies, Schiffrin and co-workers examined hypertension in Op/Op mice, which have a deficiency of macrophage colony stimulating factor and reduced monocytes and macrophages (De Ciuceis et al., 2005; Ko et al., 2007). These mice were found to develop less vascular remodeling, endothelial dysfunction, and vascular oxidative stress as compared to wild-type mice in response to either angiotensin II or DOCA–salt challenge.
Recently Wenzel et al. employed an elegant approach to deplete macrophages in mice to examine their role in hypertension. The investigators used Cre–Lox technology to induce expression of the diphtheria toxin receptor in LysM positive cells, leading to depletion of neutrophils and macrophages in these animals (Wenzel et al., 2011). This genetic manipulation markedly blunted virtually all consequences of angiotensin II infusion, including blood pressure elevation, the induction of vascular adhesion molecules, vascular dysfunction, and superoxide production.
While the studies by Wenzel et al. (2011) and Schiffrin and co-workers have shown that macrophages play an essential role in hypertension, the precise mechanisms by which this occurs remain unclear. In all of these studies, elimination of macrophages reduced vascular oxidative stress, improved vascular function and reduced the deleterious vascular remodeling that accompanies hypertensive stimuli. A simple interpretation of these studies is that macrophages are powerful sources of ROS, which underlie all of these phenomena. It is also possible that products of macrophages, including ROS and cytokines, diffuse to the adjacent endothelial cells and vascular smooth muscle cells to alter function of these cells. This could include activation of ROS generating enzymes in these adjacent cells, stimulation of vascular smooth muscle hypertrophy and growth, promotion of vascular chemokines, cytokines and adhesion molecules, alterations of NO production, and other alterations of vascular cell signaling. Similar actions of ROS and cytokines on renal epithelial cells could also promote sodium and volume retention. These phenomena likely interact to promote hypertension. Finally, is likely that the genetic manipulations in the studies by Wenzel et al. and Schiffrin and co-workers could affect at least some populations of dendritic cells, which in addition to macrophages can present antigens involved in T cell activation, which is important in the genesis of hypertension. This is discussed in later in this review.
In addition to a deleterious role in hypertension, some subsets of macrophages are likely protective. Recently, Machnik et al. (2009) showed that salt loading increases macrophage accumulation in the subcutaneous space and that these are stimulated by the hypertonic environment in this site to produce the vascular endothelial growth factor C (VEGF-C). This leads to exuberant proliferation of subcutaneous lymphatics. This response is protective, because clodronate-mediated depletion of macrophages prevents the lymphatic proliferation and leads to hypertension in response to salt loading.
It is rather easy to understand why macrophages participate in the inflammatory response in hypertension. Macrophages are components of the innate immune system, and are poised to respond to non-specific stimuli, such as might be present in the tissue damage induced by angiotensin II, salt, or elevated pressure. Much more surprising is the concept that the adaptive immune response is involved in the genesis of hypertension. This idea is not new, but has been established in the literature for almost half a century. Studies by Grollman and co-workers showed that immunosuppressive therapy targeted toward adaptive immunity attenuates experimental hypertension caused by partial renal infarction (White and Grollman, 1964). The investigators found anti-kidney antibodies in these animals, and showed that transfer of lymph node cells from rats with renal infarction to normal rats increases blood pressure in the recipients (Okuda and Grollman, 1967). Ba et al. (1982) found that engraftment of normal thymus into SHR restores T cell function and lowers blood pressure. Bendich et al. (1981) found that treatment with anti-thymocyte serum lowers blood pressure in this model of spontaneous hypertension. The immunosuppressant cyclophosphamide also transiently lowers blood pressure in SHR (Dzielak, 1991). Moreover, in mineralocorticoid-induced hypertension, several early studies showed that T cells are also important. Immune deficient mice do not sustain this form of hypertension (Svendsen, 1976), and transfer of splenocytes from rats with DOCA–salt hypertension raises blood pressure in recipient rats (Olsen, 1980).
More recently we have found that RAG-1−/− mice, which lack T and B cells, are resistant to the development of hypertension in response to either chronic angiotensin II infusion, DOCA–salt challenge, or norepinephrine. Adoptive transfer of T cells, but not B cells, restores hypertension to these various challenges (Guzik et al., 2007; Marvar et al., 2010). These studies have been replicated in mice with severe combined immunodeficiency (Crowley et al., 2010). Transfer of CD4+ cells from rats with pregnancy-related hypertension (pre-eclampsia) to normal pregnant rats increases blood pressure in the recipient rats (Wallace et al., 2011). Taken together, these studies demonstrate that T cells are essential for development of many types of experimental hypertension, despite the underlying stimulus.
We have also performed several studies to understand how the central nervous system contributes to T cell activation in hypertension. It is well known that the central nervous system communicates with the immune system and plays a role in the activation of immune cells (Oke and Tracey, 2009). The spleen and lymph nodes receive sympathetic innervation and possess adrenergic receptors which modulate their activation, antigen presentation, and cytokine production in diverse ways (Marvar et al., 2011). We have performed several studies showing that central stimuli are upstream of T cell activation in hypertension. To address this question we produced lesions anterior and ventral to the 3rd ventricle (AV3V lesions) of mice. Lesions at this site disrupt signals from the subfornical organ to the hypothalamus and are known to prevent most forms of experimental hypertension. We found that AV3V lesions reduce blood pressure elevation and prevent T cell activation in response to angiotensin II (Marvar et al., 2010). Moreover, AV3V lesions prevent the vascular accumulation of leukocytes, composed largely of T cells and macrophages, in response to angiotensin II. This is a specific response to the AV3V lesioning, because norepinephrine infusion, which bypasses the effect of AV3V lesions, causes hypertension, and T cell activation after such lesions. In other studies, we increased central signaling using two approaches. In the first, we deleted the extracellular superoxide dismutase (ecSOD or SOD3) in the circumventricular organs (CVO) of mice using Cre–Lox technology (Lob et al., 2010). This increases oxidative stress in these sites, particularly in the subfornical organ, which in turn increases sympathetic outflow, causes a modest elevation in baseline blood pressure and markedly increases the hypertension caused by a low-dose of angiotensin II. Moreover, T cell activation is markedly enhanced in mice with SOD3 deleted in the CVO in response to this normally subpressor dose of angiotensin II. These studies clearly demonstrate an important link between central modulation of blood pressure and peripheral immune activation in the genesis of hypertension.
We have also examined the role of T cells in the blood pressure response to chronic psychological stress (Marvar et al., 2012). In these studies, repeated daily cage switch and restraint stress produced modest hypertension in normal mice, but not in RAG-1−/− mice. Adoptive transfer of T cells to RAG-1−/− mice restores the hypertensive response to chronic stress. Moreover in normal mice, chronic stress causes activation of circulating T cells and increases vascular accumulation of T cells. These effects are markedly enhanced by infusion of angiotensin II. These studies provide a link between chronic emotional stress, inflammation, and hypertension commonly reported in the epidemiologic literature.
In many immune responses, T regulatory (Tregs) cells provide a brake to the inflammatory process, and recent evidence suggests that is true for hypertension. These are a distinct subset of CD4+ cells characterized by expression of the forkhead transcription factor FOXP3 and the surface marker CD25, and have a critical role in self-tolerance (Sakaguchi et al., 2006). Genetic deletion of FoxP3 leads to a loss of Tregs and a severe, fatal lympho-proliferative disorder (Fontenot et al., 2003). In a recent study, Kvakan et al. (2009) showed that adoptive transfer of Tregs does not affect blood pressure elevation, but reduces cardiac inflammation, hypertrophy, and fibrosis caused by chronic angiotensin II infusion and high salt feeding. Treg adoptive transfer also reduces the percent of circulating T cells that express markers of activation and improved electrical stability. Three recent studies have shown that adoptive transfer of Tregs cells, but not T effector T cells, reduces hypertension caused by either angiotensin II or aldosterone (Barhoumi et al., 2011; Matrougui et al., 2011; Kasal et al., 2012). In the most recent of these, angiotensin II infusion was found to cause a decrease in splenic Tregs cells and to stimulate apoptosis of Tregs when administered in culture. The authors also found evidence of macrophage activation in hypertensive mice, and that adoptive transfer of Tregs reduced this. Interestingly, coronary endothelial function is impaired in angiotensin II-treated mice, and Treg adoptive transfer ameliorates this (Matrougui et al., 2011).
Additional information regarding the role of Tregs in hypertension comes from studies by Viel et al. (2010) who studied rats harboring the Dahl salt-sensitive (SS) genome with a substitution of chromosome 2 of the Brown Norway normotensive strain (SSBN2 rats). This chromosome has quantitative trait loci for hypertension and contains genes associated with both hypertension and inflammation. The authors found that these SSBN2 rats have reduced hypertension, less vascular hypertrophy, and reduced aortic inflammation compared to Dahl SS rats. The SSBN2 rats also have more aortic Tregs as evidenced by increased FoxP3b mRNA. The anti-inflammatory cytokine IL-10 both induces and is produced by Treg cells. The genetic substitution in the SSBN2 rats led to increased IL-10 production by Tregs compared to Tregs of the Dahl SS rats. These data indicate that Tregs could mitigate both blood pressure elevation and end-organ damage in the SSBN2 animals. In keeping with a protective role of IL-10, Didion and co-workers found that carotid arteries of IL-10−/− mice develop marked endothelial dysfunction when incubated with angiotensin II, while arteries of wild-type mice are resistant to this insult. Likewise, angiotensin II increases vascular superoxide in IL-10−/− mice, but not in wild-type animals (Didion et al., 2009).
These studies support the concept that Tregs cells modulate hypertension and the end-organ damage caused by stimuli such as high salt, angiotensin II, and aldosterone. This is compatible with the known role of these cells to modulate other immune responses. It is unclear if hypertensive stimuli might also lead to Tregs cell dysfunction or alter homing of these cells to areas of inflammation.
If T cells are important in hypertension, it follows that an antigen or series of antigens must be involved. It is reasonably likely that these are modifications of endogenous proteins, or so-called neoantigens, that occur in response to the stimuli leading to hypertension. Because of the known role of oxidative injury in hypertension, it is possible that these neoantigens are oxidatively modified proteins, however their identity remains unknown. The most efficient professional antigen presenting cells (APCs) are dendritic cells, but macrophages and B lymphocytes also present antigen. Little is known regarding the role of APCs in hypertension. APCs not only present antigen to the T cell receptor, but also co-stimulatory factors. Among the most important of these are the B7 ligands, which include CD80 and CD86. The B7 ligands engage CD28 on T cells during T cell receptor activation, and these two signals coordinate T cell activation. In the absence of adequate co-stimulation, T cells are either incompletely stimulated or undergo apoptosis. Recently, we have provided evidence that co-stimulation by APCs plays an important role in hypertension (Vinh et al., 2010). We found that the pharmacological agent Abatacept, which blocks the CD28/B7 interaction, prevents both angiotensin II and DOCA–salt hypertension. Moreover, the hypertension caused by angiotensin II is markedly reduced in mice lacking B7 ligands, and can be restored by bone marrow transplant with wild-type marrow. In this study, we also found that an increase in activated dendritic cells in the spleen and lymph nodes in hypertensive mice. Dendritic cell activation occurs during antigen uptake and presentation, and thus our findings suggest that dendritic cells likely are actively involved in uptake and presentation of neoantigens to T cells in hypertension.
While the above studies strongly support a role of inflammatory cells in hypertension, the precise mechanisms involved in this process remain a focus of substantial research. In the case of T cells, various hypertensive stimuli increase the percent of circulating T cells with an effector phenotype. These cells accumulate in the PVAT and in the kidney where they release cytokines that seem to have a critical role in raising blood pressure. A particularly important cytokine seems to be interleukin-17 (IL-17). TH17 cells, a unique subset of CD4+ cells, distinct from TH1 and TH2 cells, were initially thought to be the major source of this cytokine. It is now recognized that IL-17 is also made by CD8+ cells (Kondo et al., 2009), neutrophils (Li et al., 2010), and natural killer T cells (Lee et al., 2008). IL-17 contributes to a variety of human diseases including rheumatoid arthritis, inflammatory bowel disease, psoriasis, and airway inflammation (Witowski et al., 2004). We found mice lacking IL-17A (IL-17−/− mice) develop blunted hypertension during angiotensin II infusion and that the increase in superoxide production and reduction of endothelium-dependent vasodilatation observed in hypertensive wild-type mice does not occur in IL-17−/− mice (Madhur et al., 2010). In keeping with this, it has recently been reported that IL-17 can activate the NADPH oxidase, a major source of reactive oxygen species in hypertension (Pietrowski et al., 2011). We also found that the vascular accumulation of leukocytes (including other T cells) caused by angiotensin II is markedly reduced in IL-17−/− mice. This is in accord with the concept that IL-17 stimulates chemokine release and promotes chemotaxis of other inflammatory cells (Kao et al., 2005; Hartupee et al., 2007). Thus, IL-17 likely contributes to vascular pathology in hypertension not only by direct effects, but also by recruiting other inflammatory cells.
Of interest, a major driver of IL-17 expression is IL-6, which acts in concert with transforming growth factor to polarize both CD4+ and CD8+ cells toward IL-17 production. IL-6 is a multifunctional cytokine produced by many cells including T cells, B cells, fibroblasts, and endothelial cells. In vivo treatment with IL-6 causes fever, weight loss, and generalized fatigue. IL-6 stimulates the liver to produce acute phase reactants including serum amyloid A and C-reactive protein and to decrease production of albumin (Nishimoto, 2010). Antibodies to IL-6 have been used to treat a variety of human diseases, including rheumatoid arthritis, Crohn’s disease, lupus erythematosus, Casteleman’s disease, Stills disease, systemic onset juvenile onset arthritis (soJIA), and a variety of neoplasms. Both clinical observations and experimental studies have strongly implicated IL-6 in the genesis of hypertension. In the early 1990s, it was recognized that some pheochromocytomas, which cause severe hypertension, produce IL-6 (Suzuki et al., 1991). There are significant, albeit weak correlations between serum IL-6 levels and blood pressure in healthy volunteers (Chae et al., 2001; Fernandez-Real et al., 2001), and lowering blood pressure reduces serum IL-6 levels in hypertensive subjects (Vazquez-Oliva et al., 2005). A very recent study has shown that IL-6 accumulates in the kidney, and in particular the glomeruli, of patients with chronic kidney disease and hypertension, to a greater extent than in patients with CKD and no hypertension (Zhang et al., 2012). In Wystar-Kyoto rats, renal sympathetic nerve stimulation increases renal production of IL-6 (Nakamura et al., 1993). Angiotensin II stimulates the production of IL-6 by vascular smooth muscle cells via a pathway involving the AT1 receptor, increased intracellular calcium, tyrosine kinase and MAP kinase stimulation and IL-6 transcriptional activation (Funakoshi et al., 1999). Several studies have shown that IL-6 deficient mice are protected against stress-induced hypertension, angiotensin II-induced hypertension, and renal damage caused by hypertension (Lee et al., 2004; Hartupee et al., 2007; Sturgis et al., 2009; Brands et al., 2010; Zhang et al., 2012). In an elegant study, Luther et al. (2006) showed that acute angiotensin II infusion in humans increases circulating IL-6, and that this is prevented by pretreatment with spironolactone, indicating a role of aldosterone in this response.
Based on these studies, it has become clear that inflammatory cytokines such as IL-17 and IL-6 contribute to hypertension, likely both by worsening blood pressure elevation and by causing end-organ damage. Studies such as these have led to the proposal that IL-6 antagonists could be used to treat resistant hypertension (Kapoor, 2007). The precise mechanisms by which these cytokines interact remains unclear, but it is interesting to speculate that IL-6 production in the kidney or vasculature might induce T cells to produce IL-17, ultimately leading to hypertension.
As mentioned in the introduction of this chapter, there remains substantial debate about the precise roles of the central nervous system, the kidney, and the vasculature in hypertension and a clear understanding of how a stimulus like angiotensin II can coordinate dysfunction of all of these remains undefined. We propose that inflammation provides a link between these systems, and by producing dysfunction in each, leads to an elevation of blood pressure. This working hypothesis is pictured in Figure 1. Stimuli such as angiotensin II, high salt, or chronic stress activates regions of the brain such as the CVO, leading to an increase in sympathetic outflow and perhaps other signals that cause modest elevations in systemic pressure (pre-hypertension) and promote local production of cytokines. The elevations in pressure, in concert with the direct insults of angiotensin II and increased neurotransmitters such as norepinephrine lead to tissue injury, release of tissue-derived cytokines such as IL-6 and formation of neoantigens, perhaps due to oxidative modifications. APCs, including dendritic cells and macrophages are involved in presenting these neoantigens, leading to T cell activation. The activated T cells produce cytokines such as IL-17, which are critical in the hypertensive process. This inflammatory milieu, comprised of IL-17, IL-6, catecholamines, angiotensin II, and ROS promote sodium retention in the kidney and in the vasculature causes vasoconstriction and vascular remodeling. These events cause progression of pre-hypertension to overt severe hypertension.
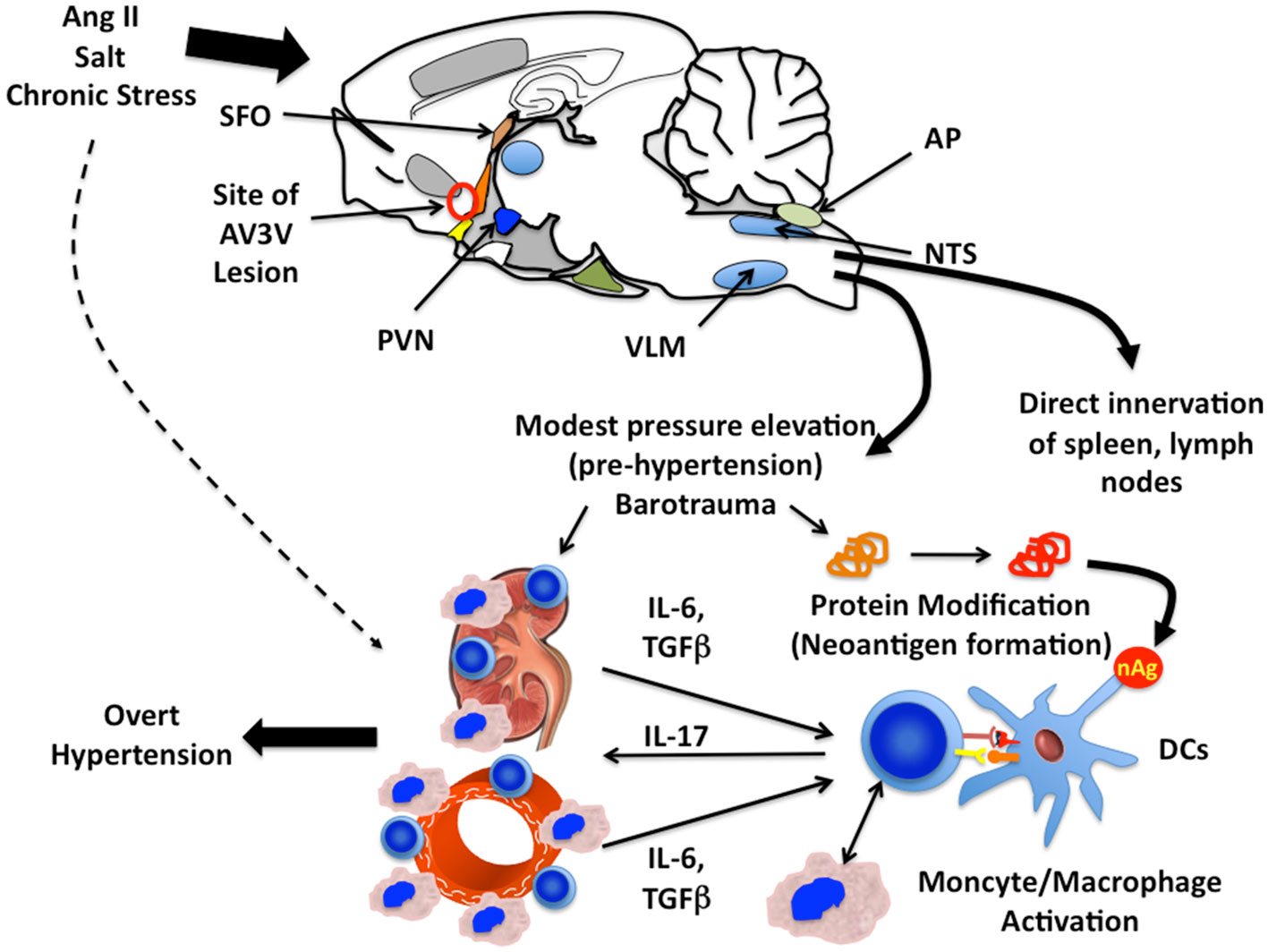
Figure 1. Proposed paradigm for inflammation and immune cell activation in hypertension. Stimuli including angiotensin II, salt, and chronic stress act on the central nervous system and increase sympathetic outflow. The circumventricular organs (CVO), including the subfornical organ (SFO), the medium preoptic eminence (MPO, orange structure) organum vasculosum lateral terminalis (OVLT, yellow structure), and the area postrema (AP) have a poorly formed blood–brain barrier and are responsive to circulating angiotensin II and sodium. These stimuli increase ROS production in the CVO, which provide input into hypothalamic centers including the paraventricular nucleus (PVN). Microglial cells are activated in this process, and increase input into brainstem centers including the ventral lateral medulla (VLM) and the nucleus tractus solitarius (NTS). These increase sympathetic outflow, which causes a modest elevation in blood pressure to levels compatible with pre-hypertension. Sympathetic stimulation also increases renal production of IL-6 and acts on T cell adrenergic receptors to modify their polarization. The elevations of pressure, direct actions and angiotensin II and catecholamines activate ROS production in the kidney and vasculature, increasing chemokine production and adhesion molecule expression. We propose that neoantigens (nAg) are formed from endogenous proteins in the kidney and vasculature, which are presented by dendritic cells to T cells. Activated T cells interact with monocyte/macrophages, promoting macrophage transformation and these leukocytes accumulate in the kidney. IL-6 and TGFβ, produced in these organs, help direct T cell IL-17 production. IL-17 and other cytokines produced by these cells promote ROS production in the vascular smooth muscle and kidney, leading to vasoconstriction, sodium retention, and ultimately severe hypertension.
It is interesting to note that there is a consistent theme of interplay between oxidative events and inflammation in this pathway. An initial step involves activation of CNS centers including the CVO. This leads to an inflammatory response in the hypothalamus, characterized by activation of microglia, which alters input to brainstem cardiovascular centers and promotes sympathetic outflow. Mechanical deformation of vascular cells promotes a cascade of signaling and transcriptional events that stimulate expression of adhesion molecules and chemokines necessary for leukocyte trafficking. Formation of neoantigens, perhaps due to oxidative modifications might lead to T cell activation. The ultimate increase in vascular superoxide is clearly T cell dependent, and is absent in RAG-1−/− mice and mice lacking IL-17A. Paradoxically, small clinical trials have failed to show a long-term benefit of commonly employed vitamin antioxidants in hypertension (Kim et al., 2002; Ward et al., 2007). These clinical studies might reflect the fact that ROS can also have beneficial effects, that scavenging all ROS might be deleterious and that these commonly employed agents are simply not very effective when administered in vivo (Widder and Harrison, 2005).
Finally, the studies described above have largely been performed in the early phases of hypertension in experimental animals. It is unclear if inflammation and immune cells contribute to established, long-standing human hypertension. Moreover, commonly employed anti-inflammatory drugs, including cyclosporine and non-steroidal inflammatory drugs (NSAIDs) have off target effects that promote, rather than prevent hypertension. Cyclosporine increases sympathetic outflow and endothelin expression in the kidney (Kon et al., 1990; Scherrer et al., 1990; Zhang et al., 2000), while NSAIDs inhibit production of vasodilator and natriuretic prostaglandins that could promote hypertension (Morrison et al., 2007). It is conceivable that agents that specifically interfere with T cell activation without off target effects could be used to treat refractory hypertension in humans, however additional studies are needed to address this issue.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by Dr. Harrison’s funding NIH R01HL039006, P01HL058000, P01HL095070, P01GM015431, R01HL105294-02 and Dr. Marvar’s funding NIH K99HL107675.
Ba, D., Takeichi, N., Kodama, T., and Kobayashi, H. (1982). Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J. Immunol. 128, 1211–1216.
Barhoumi, T., Kasal, D. A., Li, M. W., Shbat, L., Laurant, P., Neves, M. F., Paradis, P., and Schiffrin, E. L. (2011). T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 57, 469–476.
Bendich, A., Belisle, E. H., and Strausser, H. R. (1981). Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem. Biophys. Res. Commun. 99, 600–607.
Brands, M. W., Banes-Berceli, A. K., Inscho, E. W., Al-Azawi, H., Allen, A. J., and Labazi, H. (2010). Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56, 879–884.
Chae, C. U., Lee, R. T., Rifai, N., and Ridker, P. M. (2001). Blood pressure and inflammation in apparently healthy men. Hypertension 38, 399–403.
Clozel, M., Kuhn, H., Hefti, F., and Baumgartner, H. R. (1991). Endothelial dysfunction and subendothelial monocyte macrophages in hypertension. Effect of angiotensin converting enzyme inhibition. Hypertension 18, 132–141.
Crowley, S. D., Song, Y. S., Lin, E. E., Griffiths, R., Kim, H. S., and Ruiz, P. (2010). Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1089–R1097.
De Ciuceis, C., Amiri, F., Brassard, P., Endemann, D. H., Touyz, R. M., and Schiffrin, E. L. (2005). Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler. Thromb. Vasc. Biol. 25, 2106–2113.
Dibona, G. F., and Esler, M. (2010). Translational medicine: the antihypertensive effect of renal denervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R245–R253.
Didion, S. P., Kinzenbaw, D. A., Schrader, L. I., Chu, Y., and Faraci, F. M. (2009). Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension 54, 619–624.
Dzielak, D. J. (1991). Immune mechanisms in experimental and essential hypertension. Am. J. Physiol. 260, R459–R467.
Esler, M. D., Krum, H., Sobotka, P. A., Schlaich, M. P., Schmieder, R. E., and Bohm, M. (2010). Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 376, 1903–1909.
Fernandez-Real, J. M., Vayreda, M., Richart, C., Gutierrez, C., Broch, M., Vendrell, J., and Ricart, W. (2001). Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J. Clin. Endocrinol. Metab. 86, 1154–1159.
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat. Immunol. 4, 330–336.
Franco, M., Martinez, F., Quiroz, Y., Galicia, O., Bautista, R., Johnson, R. J., and Rodriguez-Iturbe, B. (2007). Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R251–R256.
Funakoshi, Y., Ichiki, T., Ito, K., and Takeshita, A. (1999). Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension 34, 118–125.
Guzik, T. J., Hoch, N. E., Brown, K. A., Mccann, L. A., Rahman, A., Dikalov, S., Goronzy, J., Weyand, C., and Harrison, D. G. (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 204, 2449–2460.
Hagita, S., Osaka, M., Shimokado, K., and Yoshida, M. (2011). Adipose inflammation initiates recruitment of leukocytes to mouse femoral artery: role of adipo-vascular axis in chronic inflammation. PLoS ONE 6, e19871. doi:10.1371/journal.pone.0019871
Hartupee, J., Liu, C., Novotny, M., Li, X., and Hamilton, T. (2007). IL-17 enhances chemokine gene expression through mRNA stabilization. J. Immunol. 179, 4135–4141.
Heier, L. A., Bauer, C. J., Schwartz, L., Zimmerman, R. D., Morgello, S., and Deck, M. D. (1989). Large Virchow-Robin spaces: MR-clinical correlation. AJNR Am. J. Neuroradiol. 10, 929–936.
Imakiire, T., Kikuchi, Y., Yamada, M., Kushiyama, T., Higashi, K., Hyodo, N., Yamamoto, K., Oda, T., Suzuki, S., and Miura, S. (2007). Effects of renin-angiotensin system blockade on macrophage infiltration in patients with hypertensive nephrosclerosis. Hypertens. Res. 30, 635–642.
Julius, S., Nesbitt, S. D., Egan, B. M., Weber, M. A., Michelson, E. L., Kaciroti, N., Black, H. R., Grimm, R. H. Jr., Messerli, F. H., Oparil, S., and Schork, M. A. (2006). Feasibility of treating prehypertension with an angiotensin-receptor blocker. N. Engl. J. Med. 354, 1685–1697.
Kao, C. Y., Huang, F., Chen, Y., Thai, P., Wachi, S., Kim, C., Tam, L., and Wu, R. (2005). Up-regulation of CC chemokine ligand 20 expression in human airway epithelium by IL-17 through a JAK-independent but MEK/NF-kappaB-dependent signaling pathway. J. Immunol. 175, 6676–6685.
Kapoor, S. (2007). Interleukin-6 antagonists for the management of hypertension. Hypertension 49, e18; discussion e19.
Kasal, D. A., Barhoumi, T., Li, M. W., Yamamoto, N., Zdanovich, E., Rehman, A., Neves, M. F., Laurant, P., Paradis, P., and Schiffrin, E. L. (2012). T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension 59, 324–330.
Kim, M. K., Sasaki, S., Sasazuki, S., Okubo, S., Hayashi, M., and Tsugane, S. (2002). Lack of long-term effect of vitamin C supplementation on blood pressure. Hypertension 40, 797–803.
Ko, E. A., Amiri, F., Pandey, N. R., Javeshghani, D., Leibovitz, E., Touyz, R. M., and Schiffrin, E. L. (2007). Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 292, H1789–H1795.
Kon, V., Sugiura, M., Inagami, T., Harvie, B. R., Ichikawa, I., and Hoover, R. L. (1990). Role of endothelin in cyclosporine-induced glomerular dysfunction. Kidney Int. 37, 1487–1491.
Kondo, T., Takata, H., Matsuki, F., and Takiguchi, M. (2009). Cutting edge: phenotypic characterization and differentiation of human CD8+ T cells producing IL-17. J. Immunol. 182, 1794–1798.
Kvakan, H., Kleinewietfeld, M., Qadri, F., Park, J. K., Fischer, R., Schwarz, I., Rahn, H. P., Plehm, R., Wellner, M., Elitok, S., Gratze, P., Dechend, R., Luft, F. C., and Muller, D. N. (2009). Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119, 2904–2912.
Lee, D. L., Leite, R., Fleming, C., Pollock, J. S., Webb, R. C., and Brands, M. W. (2004). Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension 44, 259–263.
Lee, K. A., Kang, M. H., Lee, Y. S., Kim, Y. J., Kim, D. H., Ko, H. J., and Kang, C. Y. (2008). A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell. Immunol. 251, 50–55.
Lenfant, C., Chobanian, A. V., Jones, D. W., and Roccella, E. J. (2003). Seventh report of the Joint National Committee on the Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7): resetting the hypertension sails. Hypertension 41, 1178–1179.
Li, L., Huang, L., Vergis, A. L., Ye, H., Bajwa, A., Narayan, V., Strieter, R. M., Rosin, D. L., and Okusa, M. D. (2010). IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J. Clin. Invest. 120, 331–342.
Lifton, R. P., Gharavi, A. G., and Geller, D. S. (2001). Molecular mechanisms of human hypertension. Cell 104, 545–556.
Lob, H. E., Marvar, P. J., Guzik, T. J., Sharma, S., Mccann, L. A., Weyand, C., Gordon, F. J., and Harrison, D. G. (2010). Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 55, 277–283.
Luther, J. M., Gainer, J. V., Murphey, L. J., Yu, C., Vaughan, D. E., Morrow, J. D., and Brown, N. J. (2006). Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 48, 1050–1057.
Machnik, A., Neuhofer, W., Jantsch, J., Dahlmann, A., Tammela, T., Machura, K., Park, J. K., Beck, F. X., Muller, D. N., Derer, W., Goss, J., Ziomber, A., Dietsch, P., Wagner, H., Van Rooijen, N., Kurtz, A., Hilgers, K. F., Alitalo, K., Eckardt, K. U., Luft, F. C., Kerjaschki, D., and Titze, J. (2009). Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 15, 545–552.
Madhur, M. S., Lob, H. E., Mccann, L. A., Iwakura, Y., Blinder, Y., Guzik, T. J., and Harrison, D. G. (2010). Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55, 500–507.
Marin-Padilla, M., and Knopman, D. S. (2011). Developmental aspects of the intracerebral microvasculature and perivascular spaces: insights into brain response to late-life diseases. J. Neuropathol. Exp. Neurol. 70, 1060–1069.
Marvar, P. J., Lob, H., Vinh, A., Zarreen, F., and Harrison, D. G. (2011). The central nervous system and inflammation in hypertension. Curr. Opin. Pharmacol. 11, 156–161.
Marvar, P. J., Thabet, S. R., Guzik, T. J., Lob, H. E., Mccann, L. A., Weyand, C., Gordon, F. J., and Harrison, D. G. (2010). Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ. Res. 107, 263–270.
Marvar, P. J., Vinh, A., Thabet, S., Lob, H. E., Geem, D., Ressler, K. J., and Harrison, D. G. (2012). T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol. Psychiatry 71, 774–782.
Matrougui, K., Abd Elmageed, Z., Kassan, M., Choi, S., Nair, D., Gonzalez-Villalobos, R. A., Chentoufi, A. A., Kadowitz, P., Belmadani, S., and Partyka, M. (2011). Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am. J. Pathol. 178, 434–441.
Morrison, A., Ramey, D. R., Van Adelsberg, J., and Watson, D. J. (2007). Systematic review of trials of the effect of continued use of oral non-selective NSAIDs on blood pressure and hypertension. Curr. Med. Res. Opin. 23, 2395–2404.
Nakamura, A., Kohsaka, T., and Johns, E. J. (1993). Differential regulation of interleukin-6 production in the kidney by the renal sympathetic nerves in normal and spontaneously hypertensive rats. J. Hypertens. 11, 491–497.
Narayan, K. M., Ali, M. K., and Koplan, J. P. (2010). Global noncommunicable diseases – where worlds meet. N. Engl. J. Med. 363, 1196–1198.
Nishimoto, N. (2010). Interleukin-6 as a therapeutic target in candidate inflammatory diseases. Clin. Pharmacol. Ther. 87, 483–487.
Ohman, M. K., Luo, W., Wang, H., Guo, C., Abdallah, W., Russo, H. M., and Eitzman, D. T. (2011). Perivascular visceral adipose tissue induces atherosclerosis in apolipoprotein E deficient mice. Atherosclerosis 219, 33–39.
Oke, S. L., and Tracey, K. J. (2009). The inflammatory reflex and the role of complementary and alternative medical therapies. Ann. N. Y. Acad. Sci. 1172, 172–180.
Okuda, T., and Grollman, A. (1967). Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex. Rep. Biol. Med. 25, 257–264.
Olsen, F. (1980). Transfer of arterial hypertension by splenic cells from DOCA-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol. Microbiol. Scand. C 88, 1–5.
Peterson, J. R., Sharma, R. V., and Davisson, R. L. (2006). Reactive oxygen species in the neuropathogenesis of hypertension. Curr. Hypertens. Rep. 8, 232–241.
Pietrowski, E., Bender, B., Huppert, J., White, R., Luhmann, H. J., and Kuhlmann, C. R. (2011). Pro-inflammatory effects of interleukin-17A on vascular smooth muscle cells involve NAD(P)H- oxidase derived reactive oxygen species. J. Vasc. Res. 48, 52–58.
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145.
Rodriguez-Iturbe, B., Vaziri, N. D., and Johnson, R. J. (2008). Inflammation, angiotensin II, and hypertension. Hypertension 52, e135.
Russo, H. M., Wickenheiser, K. J., Luo, W., Ohman, M. K., Franchi, L., Wright, A. P., Bodary, P. F., Nunez, G., and Eitzman, D. T. (2010). P-selectin glycoprotein ligand-1 regulates adhesive properties of the endothelium and leukocyte trafficking into adipose tissue. Circ. Res. 107, 388–397.
Sakaguchi, S., Ono, M., Setoguchi, R., Yagi, H., Hori, S., Fehervari, Z., Shimizu, J., Takahashi, T., and Nomura, T. (2006). Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 212, 8–27.
Saleh, M. A., Boesen, E. I., Pollock, J. S., Savin, V. J., and Pollock, D. M. (2010). Endothelin-1 increases glomerular permeability and inflammation independent of blood pressure in the rat. Hypertension 56, 942–949.
Scherrer, U., Vissing, S. F., Morgan, B. J., Rollins, J. A., Tindall, R. S., Ring, S., Hanson, P., Mohanty, P. K., and Victor, R. G. (1990). Cyclosporine-induced sympathetic activation and hypertension after heart transplantation. N. Engl. J. Med. 323, 693–699.
Scroop, G. C., and Lowe, R. D. (1968). Central pressor effect of angiotensin mediated by the parasympathetic nervous system. Nature 220, 1331–1332.
Shi, P., Diez-Freire, C., Jun, J. Y., Qi, Y., Katovich, M. J., Li, Q., Sriramula, S., Francis, J., Sumners, C., and Raizada, M. K. (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56, 297–303.
Sturgis, L. C., Cannon, J. G., Schreihofer, D. A., and Brands, M. W. (2009). The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1742–R1748.
Suzuki, K., Miyashita, A., Inoue, Y., Iki, S., Enomoto, H., Takahashi, Y., and Takemura, T. (1991). Interleukin-6-producing pheochromocytoma. Acta Haematol. 85, 217–219.
Svendsen, U. G. (1976). Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol. Microbiol. Scand. A 84, 523–528.
Theuer, J., Dechend, R., Muller, D. N., Park, J. K., Fiebeler, A., Barta, P., Ganten, D., Haller, H., Dietz, R., and Luft, F. C. (2002). Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc. Disord. 2, 3. doi:10.1186/1471-2261-2-3
Vazquez-Oliva, G., Fernandez-Real, J. M., Zamora, A., Vilaseca, M., and Badimon, L. (2005). Lowering of blood pressure leads to decreased circulating interleukin-6 in hypertensive subjects. J. Hum. Hypertens. 19, 457–462.
Viel, E. C., Lemarie, C. A., Benkirane, K., Paradis, P., and Schiffrin, E. L. (2010). Immune regulation and vascular inflammation in genetic hypertension. Am. J. Physiol. Heart Circ. Physiol. 298, H938–H944.
Vinh, A., Chen, W., Blinder, Y., Weiss, D., Taylor, W. R., Goronzy, J. J., Weyand, C. M., Harrison, D. G., and Guzik, T. J. (2010). Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122, 2529–2537.
Waki, H., Liu, B., Miyake, M., Katahira, K., Murphy, D., Kasparov, S., and Paton, J. F. (2007). Junctional adhesion molecule-1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension 49, 1321–1327.
Wallace, K., Richards, S., Dhillon, P., Weimer, A., Edholm, E. S., Bengten, E., Wilson, M., Martin, J. N. Jr., and Lamarca, B. (2011). CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension 57, 949–955.
Ward, N. C., Wu, J. H., Clarke, M. W., Puddey, I. B., Burke, V., Croft, K. D., and Hodgson, J. M. (2007). The effect of vitamin E on blood pressure in individuals with type 2 diabetes: a randomized, double-blind, placebo-controlled trial. J. Hypertens. 25, 227–234.
Wenzel, P., Knorr, M., Kossmann, S., Stratmann, J., Hausding, M., Schuhmacher, S., Karbach, S. H., Schwenk, M., Yogev, N., Schulz, E., Oelze, M., Grabbe, S., Jonuleit, H., Becker, C., Daiber, A., Waisman, A., and Munzel, T. (2011). Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation 124, 1370–1381.
White, F. N., and Grollman, A. (1964). Autoimmune factors associated with infarction of the kidney. Nephron 204, 93–102.
Widder, J. D., and Harrison, D. G. (2005). Can vitamin E prevent cardiovascular events and cancer? Nat. Clin. Pract. Cardiovasc. Med. 2, 510–511.
Williams, K., Alvarez, X., and Lackner, A. A. (2001). Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia 36, 156–164.
Witowski, J., Ksiazek, K., and Jorres, A. (2004). Interleukin-17: a mediator of inflammatory responses. Cell. Mol. Life Sci. 61, 567–579.
Zhang, W., Li, J. L., Hosaka, M., Janz, R., Shelton, J. M., Albright, G. M., Richardson, J. A., Sudhof, T. C., and Victor, R. G. (2000). Cyclosporine A-induced hypertension involves synapsin in renal sensory nerve endings. Proc. Natl. Acad. Sci. U.S.A. 97, 9765–9770.
Keywords: blood pressure, macrophages, interleukin 17, interleukin 6, superoxide, sympathetic nerves, T cells, dendritic cells
Citation: Harrison DG, Marvar PJ and Titze JM (2012) Vascular inflammatory cells in hypertension. Front. Physio. 3:128. doi: 10.3389/fphys.2012.00128
Received: 07 January 2012; Paper pending published: 11 February 2012;
Accepted: 16 April 2012; Published online: 07 May 2012.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Zsolt Bagi, University of Oxford, UKCopyright: © 2012 Harrison, Marvar and Titze. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David G. Harrison, Division of Clinical Pharmacology, Department of Medicine, Vanderbilt University, Room 536 Robinson Research Building, Nashville, TN 37232-6602, USA. e-mail:ZGF2aWQuZy5oYXJyaXNvbkB2YW5kZXJiaWx0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.