 Adele Stewart
Adele Stewart

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 13 April 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00095
This article is part of the Research Topic Autonomic regulation of the heart - emerging regulators and arrhythmogenic mechanisms View all 8 articles
It has been nearly a century since Otto Loewi discovered that acetylcholine (ACh) release from the vagus produces bradycardia and reduced cardiac contractility. It is now known that parasympathetic control of the heart is mediated by ACh stimulation of Gi/o-coupled muscarinic M2 receptors, which directly activate G protein-coupled inwardly rectifying potassium (GIRK) channels via Gβγ resulting in membrane hyperpolarization and inhibition of action potential (AP) firing. However, expression of M2R–GIRK signaling components in heterologous systems failed to recapitulate native channel gating kinetics. The missing link was identified with the discovery of regulator of G protein signaling (RGS) proteins, which act as GTPase-activating proteins to accelerate the intrinsic GTPase activity of Gα resulting in termination of Gα- and Gβγ-mediated signaling to downstream effectors. Studies in mice expressing an RGS-insensitive Gαi2 mutant (G184S) implicated endogenous RGS proteins as key regulators of parasympathetic signaling in heart. Recently, two RGS proteins have been identified as critical regulators of M2R signaling in heart. RGS6 exhibits a uniquely robust expression in heart, especially in sinoatrial (SAN) and atrioventricular nodal regions. Mice lacking RGS6 exhibit increased bradycardia and inhibition of SAN AP firing in response to CCh as well as a loss of rapid activation and deactivation kinetics and current desensitization for ACh-induced GIRK current (IKACh). Similar findings were observed in mice lacking RGS4. Thus, dysregulation in RGS protein expression or function may contribute to pathologies involving aberrant electrical activity in cardiac pacemaker cells. Moreover, RGS6 expression was found to be up-regulated in heart under certain pathological conditions, including doxorubicin treatment, which is known to cause life-threatening cardiotoxicity and atrial fibrillation in cancer patients. On the other hand, increased vagal tone may be cardioprotective in heart failure where acetylcholinesterase inhibitors and vagal stimulation have been proposed as potential therapeutics. Together, these studies identify RGS proteins, especially RGS6, as new therapeutic targets for diseases such as sick sinus syndrome or other maladies involving abnormal autonomic control of the heart.
Heart rate (HR) and stroke volume are controlled by the opposing branches of the autonomic nervous system capable of rapidly adjusting cardiac output in response to the body’s oxygen demand. Neurotransmitters released from both sympathetic and parasympathetic nerves activate members of the G protein-coupled receptor (GPCR) family, the largest family of cell surface receptors comprising over 800 genes (Fredriksson et al., 2003) and estimated to be the target of one-third of FDA approved and marketed drugs (Hopkins and Groom, 2002). In heart, these GPCRs control various cellular functions including automaticity and contractility as well as cell growth and survival. Aberrations in these cardiac GPCR signaling cascades have numerous pathophysiological consequences and represent the major contributors to cardiovascular diseases (Salazar et al., 2007). Unsurprisingly, drugs designed to interfere with autonomic control of the heart and vasculature are used in the treatment of arrhythmias, hypertension, and heart failure.
The sympathetic division increases cardiac output as a part of the “fight-or-flight” response under conditions of stress or high physical activity by both facilitating action potential (AP) initiation and conduction in the sinoatrial (SAN) and atrioventricular nodes (AVN) and increasing cardiac muscle contractility in the atrium and ventricles. These end results are primarily achieved by catecholamine-mediated activation of G protein-coupled β1 adrenergic receptors (β1AR) expressed in nodal cells and cardiac myocytes. Activated β1ARs act as guanine nucleotide exchange factors (GEFs) promoting the exchange of GDP for GTP-bound to the αs subunit of the heterotrimeric G protein complex. GTP-Gαs then stimulates adenylate cyclase-mediated production of the second messenger cyclic AMP (cAMP), activation of cAMP-dependent protein kinases, and facilitation of the pacemaker current (If) in nodal cells and an increase in L-type calcium channel current (ICa,L) via channel modification in atrial and ventricular myocytes and the SA/AV nodes.
Resting HR in humans is, however, primarily determined by the parasympathetic division due to greater vagal discharge under resting conditions compared to electrical activity from sympathetic efferents. Acetylcholine (ACh) released from the vagus nerve, the primary conduit for efferent parasympathetic nerve activity, stimulates Gαi/o-coupled muscarinic M2 receptors (M2Rs), which influence membrane excitability both through inhibition of cAMP production in direct opposition to β1AR signaling as well as through direct Gβγ-mediated activation of G protein-coupled inwardly rectifying potassium (GIRK) channels. GIRK channels represent the primary determinants of ACh-activated potassium current (IKACh) promoting potassium efflux, membrane hyperpolarization, and inhibition of AP firing in the pacemaker and electrically conducting portions of the heart (Mark and Herlitze, 2000). Studies in mice lacking GIRK4, the cardiac specific GIRK channel subunit, revealed that approximately 50% of the bradycardic response to ACh is mediated by GIRK channel activation (Wickman et al., 1998). Under normal physiological conditions the vagal nerve is activated by numerous reflex control pathways involving arterial baroreceptors, peripheral chemoreceptors, trigeminal receptors, and cardiopulmonary receptors with vagal afferents designed to sense changes in blood pressure and oxygenation and stimulate compensatory alterations in HR.
The magnitude and duration of GPCR effector responses is limited by members of the regulator of G protein signaling (RGS) family of proteins, which act as GTPase accelerating proteins (GAPs) for Gα. By stabilizing the transition state of Gα in GTP hydrolysis, RGS proteins accelerate their intrinsic GTPase activity to facilitate termination of both Gα- and Gβγ-mediated cellular signaling (Figure 1). Twenty canonical RGS proteins, divided into four subfamilies based on sequence homology and the presence and nature of additional non-RGS domains, act as functional GAPs for Gαi/o, Gαq/11, or both (Table 1). Many of these proteins are detectable at the mRNA level in SAN, AVN, atria, or ventricles (Table 1), but lack of specific antibodies with corresponding genetic knockout controls has made detection of endogenous RGS proteins difficult in vivo, making investigations into the physiological significance of RGS proteins in the heart even more challenging. In fact, only RGS2, 3, 4, 5, and 6 have been detected at the protein level in heart (Table 1). Furthermore, physiological functions have only been attributed to a handful of RGS proteins in the heart and vasculature. Among these, RGS2 is a key regulator of vascular smooth muscle cell contractility and control of blood pressure (Heximer et al., 2003; Tang et al., 2003) and both RGS2 and RGS5 have been implicated as key inhibitors of cardiac hypertrophy and fibrosis in response to pressure overload (Takimoto et al., 2009; Li et al., 2010; Zhang et al., 2011).
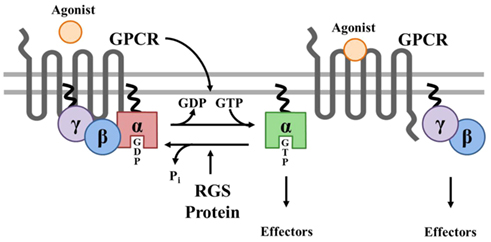
Figure 1. Canonical Regulation of GPCR signaling by RGS proteins. Agonist binding to G protein-coupled receptors (GPCRs) induces a conformational change that facilitates the exchange of GDP for GTP on the α subunit of the heterotrimeric complex. Both GTP-bound Gα in the active form and the released Gβγ dimer can then go on to stimulate a number of downstream effectors. RGS proteins are GTPase accelerating proteins (GAPs) for Gα, which function to terminate signaling through GPCRs by accelerating the intrinsic GTPase activity of Gα and promoting re-association of the heterotrimeric complex with the receptor at the cell membrane.
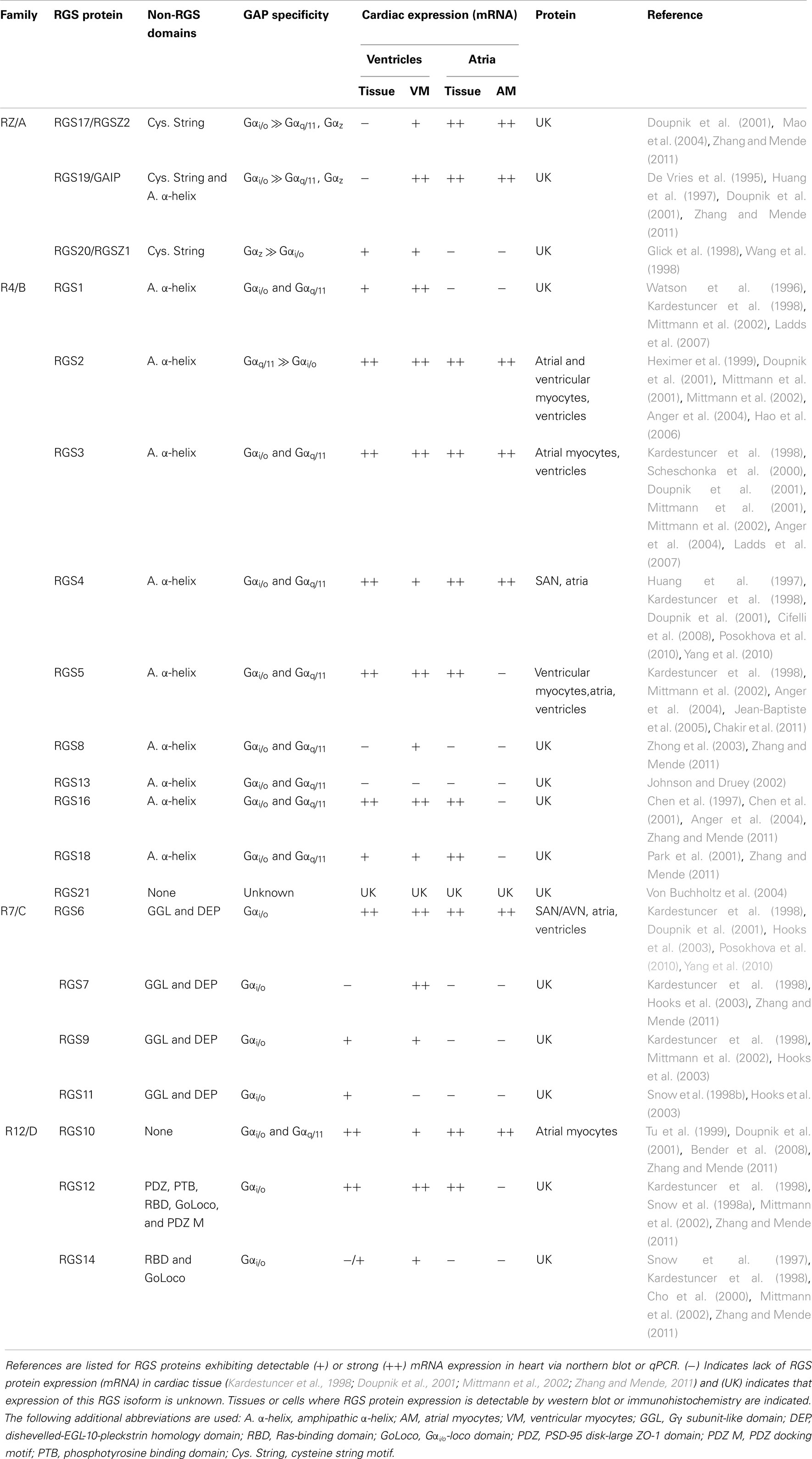
Table 1. Canonical RGS protein structure, GAP activity, and expression in cardiac tissue.
Two RGS proteins, RGS6 and RGS4, have recently been identified as critical negative regulators of M2R signaling in heart, functioning to set the parasympathetic “tone” by acting as the brake on vagal stimulation of the heart (Figures 2 and 3). It remains unclear, however, whether RGS6 and RGS4 are redundant inactivators of cardiac M2R signaling or if they cooperate to ensure proper parasympathetic control of HR by regulating distinct channel, GPCR, or G protein populations or by functioning in different cardiac cells. Nevertheless, dysregulation in the expression of either RGS6 or RGS4 could contribute to the loss of vagal tone observed in cardiovascular diseases. Complete and comprehensive understanding of the role of RGS proteins in regulating parasympathetic stimulation of the heart could lead to the development of novel treatment strategies for cardiac pathologies involving arrhythmias and conduction defects that result from unchecked parasympathetic signaling. Among these are pathological AV block, sick sinus syndrome, and arrhythmias (Fu et al., 2007). Considering the cardioprotective effect of increased vagal tone, these RGS proteins are also being considered as viable druggable targets in the treatment of heart failure and doxorubicin-induced cardiotoxicity.
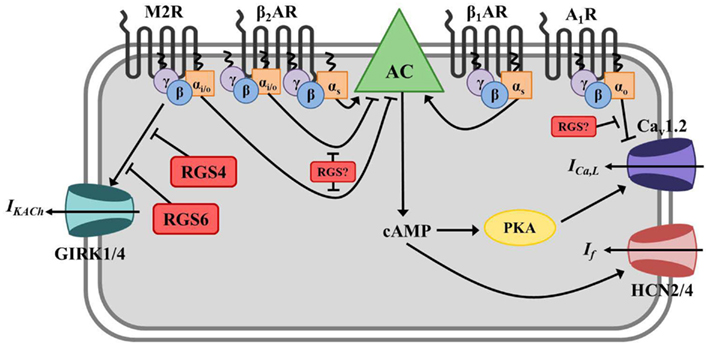
Figure 2. Regulator of G protein signaling-mediated regulation of cardiac automaticity in the sinoatrial node (SAN). In the SAN, Gs-coupled β1- or β2-adrenergic receptors (β1/2AR) stimulate adenylate cyclase (AC)-mediated production of the second messenger cyclic AMP (cAMP), activation of cAMP-dependent protein kinase (PKA), activation of hyperpolarization-activated cyclic nucleotide-gated cation (HCN) channels and L-type calcium channels (primarily Cav1.2), and induction of the pacemaker current (If) and calcium current (ICa,L). The net effect is membrane depolarization and increased nodal cell excitation. AC is inhibited by Gαi/o-coupled muscarinic M2 receptors (M2R), and activated M2Rs can also directly induce G protein-coupled inwardly rectifying potassium channel (GIRK) current (IKACh) via Gβγ resulting in membrane hyperpolarization and inhibition of nodal cell firing. β2ARs can also couple to Gαi/o in these cells and block AC-mediated cAMP production. Adenosine A1 receptors (A1R) also have a negative chronotropic effect in the SAN via Gαo-dependent inhibition of ICa,L, though the exact mechanism whereby this occurs remains unclear. RGS4 and RGS6 both function to inactivate Gαi/o activated by M2Rs and block subsequent acetylcholine-activated GIRK current (IKACh) in SAN. The specific RGS protein(s) regulating A1Rs and Gαi/o-coupled β2AR in this tissue have yet to be identified.
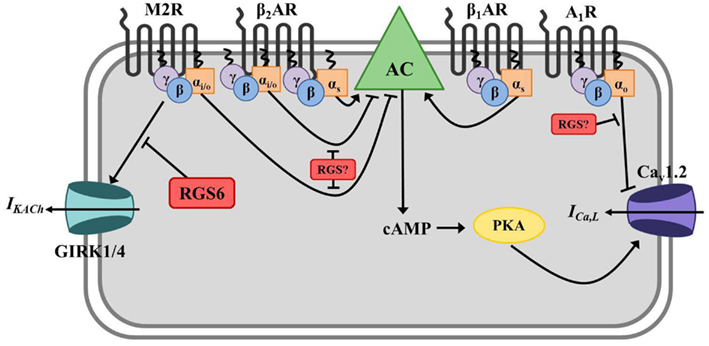
Figure 3. Regulator of G protein signaling-mediated regulation of cardiac automaticity in atrial myocytes. In atrial myocytes, Gs-coupled β1- or β2-adrenergic receptors (β1/2AR) stimulate adenylate cyclase-mediated production of the second messenger cyclic AMP (cAMP), activation of cAMP-dependent protein kinase (PKA), and induction of the calcium current (ICa,L) through L-type calcium channels (primarily Cav1.2). The net effect is membrane depolarization, increased cell excitation, and enhanced cardiac contractility. Adenylate cyclase is inhibited by Gαi/o-coupled muscarinic M2 receptors (M2R), and activated M2Rs can also directly induce G protein-coupled inwardly rectifying potassium channel (GIRK) current (IKACh) via Gβγ resulting in membrane hyperpolarization and inhibition of cell firing. β2ARs can also couple to Gαi/o in these cells and block AC-mediated cAMP production. Adenosine A1 receptors (A1R) also have a negative chronotropic effect in atrial myocytes via Gαo-dependent inhibition of ICa,L, though the exact mechanism whereby this occurs remains unclear. RGS6 functions to inactivate stimulated M2Rs and block subsequent GIRK current (IKACh) in atrial myocytes. The specific RGS protein(s) regulating A1Rs and Gαi/o-coupled β2AR in this tissue have yet to be identified.
Prior to the discovery of RGS proteins in 1995 (Dohlman and Thorner, 1997; Berman and Gilman, 1998; Ross and Wilkie, 2000), the model of M2R signaling in cardiac myocytes remained incomplete as cardiac specific GIRK channels expressed in CHO cells exhibited deactivation kinetics up to 40 times slower than those observed in native tissues (Doupnik et al., 1997). These results suggested that an additional component required for normal inactivation of these channels following M2R stimulation was missing. Expression of several members of the RGS protein family (including RGS4) along with GIRK channels and M2Rs restored the activation and deactivation kinetics of heterologously expressed GIRK channels to those observed in native atrial cells (Doupnik et al., 1997). These findings demonstrated the essential role of RGS proteins in regulating M2R signaling, but failed to identify whether this added regulation was required in vivo.
One major challenge to investigating RGS protein function in living animals is the potential for functional redundancy and compensatory changes in RGS protein expression resulting from loss of a single protein. To circumvent this issue, a series of transgenic mice were developed that express knock-in alleles of RGS-insensitive Gα mutants. In place of the endogenous protein, these mice instead express Gα with a point mutation (G184S in Gαi2) in the switch I region that blocks the interaction with RGS proteins necessary for GTPase activation (Lan et al., 1998) without affecting the intrinsic GTPase activity of Gα or its ability to bind Gβγ, GPCRs, and effectors (Fu et al., 2004). Thus, this mouse model can be used to evaluate the net regulatory actions of RGS proteins on various GPCR signaling pathways in vivo.
Using embryonic stem cell derived cardiocytes (ESDCs) expressing knock-in RGS-insensitive alleles of Gαi2 and Gαo, endogenous RGS proteins were identified as key negative regulators of the agonist-induced negative chronotropic responses to stimulation of Gαi/o-coupled M2Rs and Gαo-coupled adenosine A1 receptors (A1R). Enhancement in M2R-mediated bradycardia primarily occurred through increased GIRK current while the decreased HR induced by A1R activation occurred via a GIRK channel-independent mechanism. Mutant cells of each genotype also showed impaired β2-adrenergic receptor (β2AR)-mediated tachycardia. Because this effect was sensitive to pertussis toxin, an inhibitor of Gαi/o, these results implicated RGS proteins in regulation of Gαi/o-coupled β2ARs. In vivo, the β2AR can couple either Gαs or Gαi/o with opposing effects on HR. These results indicate that endogenous RGS proteins specifically regulate β2ARs coupled to Gαi/o such that, under basal conditions, the Gαs dependent, non-RGS regulated effect dominates (Fu et al., 2006). Thus far, only the Gαi2/M2R-dependent sensitization to ACh in the RGS-insensitive mice has been confirmed in live animals.
Because loss of RGS protein-mediated regulation of Gαi2 could influence cardiac automaticity through various mechanisms including changes in GPCR signaling in the peripheral vasculature or central nervous system, it was necessary to confirm that the enhanced bradycardia observed in this mouse model was due to intrinsic alterations in cardiac pacemaker activity. In a follow up study, enhanced M2R-mediated bradycardia and AV block were observed in isolated, perfused hearts from mice expressing RGS-insensitive Gαi2 both under basal conditions and in the presence of isoproterenol, a non-specific β-adrenergic receptor agonist used to mimic sympathetic input. Studies in these mice revealed no genotype-dependent differences in adenosine-stimulated bradycardia consistent with the previously demonstrated preferential coupling of A1R to Gαo (Fu et al., 2007). While these studies did firmly establish the necessity of RGS proteins in controlling HR downstream of M2R/Gαi2 stimulation, it failed to determine which of the numerous RGS proteins present in heart (Table 1) are responsible for this activity.
RGS4 is a member of the R4 subfamily of RGS proteins, which contain only short N- and C-terminal protein sequences in addition to their conserved RGS domain. These proteins act as non-discriminatory GAPs for both Gαi/o and Gαq/11 (Table 1). RGS4 mRNA is enriched in the heart, but there is little to no detectable RGS4 protein expression (Posokhova et al., 2010; Yang et al., 2010). The lack of correlation between RGS4 mRNA and protein levels observed (Xie et al., 2009) may be due to a destabilizing N-terminal cysteine residue whose arginylation targets RGS4 for degradation through the ubiquitin-dependent N-end rule pathway (Davydov and Varshavsky, 2000; Lee et al., 2005). Using an RGS4 knockout mouse expressing LacZ behind the endogenous RGS4 promoter, selective enrichment of RGS4 mRNA in SAN was observed. This is an indirect means to measure RGS4 protein localization and may not reflect endogenous RGS4 levels due to differences in protein regulation (e.g., by the N-end rule pathway) between RGS4 and the exogenously expressed LacZ.
Though RGS4-null animals exhibited no difference in basal HR compared to their wild-type counterparts, these mice show enhanced bradycardia in response to systemic administration of the M2R agonist carbachol (CCh). In anesthetized animals, where parasympathetic tone is increased, RGS4-null mice did show reduced basal HR. These results are consistent with the fact that sympathetic drive dominates in conscious rodents as opposed to the greater parasympathetic stimulation of heart under basal conditions observed in humans. Knockout animals also displayed a larger change in HR upon administration of atropine, a M2R antagonist, suggesting these mice exhibit increased vagal tone. These results were replicated in isolated perfused hearts indicting that the enhanced bradycardia resulting from RGS4 loss is due to intrinsic alterations in cardiac automaticity (Cifelli et al., 2008).
SAN cells isolated from RGS4-deficient animals also showed increased sensitivity to CCh-induced inhibition of AP firing and changes in maximum diastolic potential (MDP). The correlation between inhibition of AP firing and MDP implicated enhanced GIRK current as a possible determinant of the enhanced M2R signaling RGS4-null cells. Sinoatrial myocytes isolated from RGS4 knockout mice also exhibited a loss of rapid M2R-activated GIRK current activation and deactivation kinetics as well as a loss of current desensitization with no difference in the magnitude or dose-dependency of the response (Cifelli et al., 2008). The increased time course of receptor deactivation and desensitization are consistent with a model whereby RGS4, acting as a GAP for Gαi/o-coupled M2Rs, functions to inactivate Gβγ-induced IKACh. Indeed, it was previously established that endogenous RGS proteins are required for proper activation and deactivation kinetics of agonist-induced GIRK currents (Lambert et al., 2010). RGS4 was shown to mediate these effects in the SAN, but RGS4 mRNA was not detected in atrial and ventricular cardiomyocytes indicating lack of RGS4 involvement in M2R signaling in these cell types (Cifelli et al., 2008). RGS4 appears to also mediate agonist and voltage-dependent GIRK channel relaxation (Fujita et al., 2000; Inanobe et al., 2001; Ishii et al., 2001), a process involving relief of channel pore blockade by intracellular cations and polyamines at hyperpolarizing potentials and increasing agonist concentration. As these experiments were performed in heterologous expression systems or isolated cells, the in vivo significance of this latter effect remains unclear. Nevertheless, the phenotype of enhanced parasympathetic tone in mice expressing RGS-insensitive Gα mutants may be due, in part, to loss of RGS4-mediated regulation of M2Rs in the SAN.
RGS6 is a member of the R7 subfamily of RGS proteins characterized by a distinct three domain structure. The conserved RGS domain confers their GAP activity directed specifically toward Gαi/o subunits while the Disheveled-EGL-10-Pleckstrin (DEP) homology domain and Gγ subunit-like (GGL) domain facilitate binding of R7 family members to two accessory proteins: R7 family binding protein (R7BP), required for membrane targeting, and Gβ5, an atypical G protein required for R7 family protein stability, respectively (Posner et al., 1999; Anderson et al., 2009). Unlike RGS4, RGS6 expression is detectable at appreciable levels in the SAN, AVN, atria, and ventricles of mouse hearts via PCR, western blot, and immunohistochemical staining (Posokhova et al., 2010; Yang et al., 2010). RGS6 is also highly enriched in the SAN and AVN along with cardiac GIRK channel subunit GIRK1 (Posokhova et al., 2010; Yang et al., 2010). Similar to the phenotype observed in RGS4-deficient mice, loss of RGS6 was associated with exaggerated bradycardia in response to CCh in conscious and anesthetized mice and isolated perfused hearts (Yang et al., 2010). Isolated perfused hearts from RGS6−/− mice also exhibited significant CCh-induced AV block as evidenced by dramatic prolongation of the PR interval on electrocardiogram tracings.
Consistent with this phenotype of enhanced parasympathetic stimulation of heart, CCh-induced inhibition of spontaneous AP firing was exaggerated in SAN pacemaker cells isolated from RGS6−/− animals (Yang et al., 2010). Like RGS4, RGS6 also appears to be required for CCh-induced GIRK channel gating kinetics. In atrial myocytes from wild-type mice, application of CCh elicited rapid IKACh that showed significant desensitization over time, followed by rapid deactivation upon removal of agonist. Conversely, cells isolated from RGS6−/− mice exhibited a significant reduction in the time course of activation and deactivation, as well as the extent of IKACh desensitization (Posokhova et al., 2010; Yang et al., 2010). The observation that RGS6 is required for desensitization and rapid activation and deactivation of IKACh in atrial myocytes and SAN cells is consistent with its role as a GAP for Gi/o. Indeed, such defects in GIRK desensitization and deactivation in RGS6-deficient atrial cells likely underlie the exaggerated bradycardia response to CCh in RGS6−/− mice and isolated hearts. Therefore, like RGS4, RGS6 is also essential for modulation of M2R signaling in heart and loss of either protein recapitulates the phenotype of enhanced parasympathetic tone observed in mice expressing RGS-insensitive Gαi2. The phenotypes of the various mouse models used to evaluate the role of RGS proteins in modulating autonomic control of heart are summarized in Table 2.
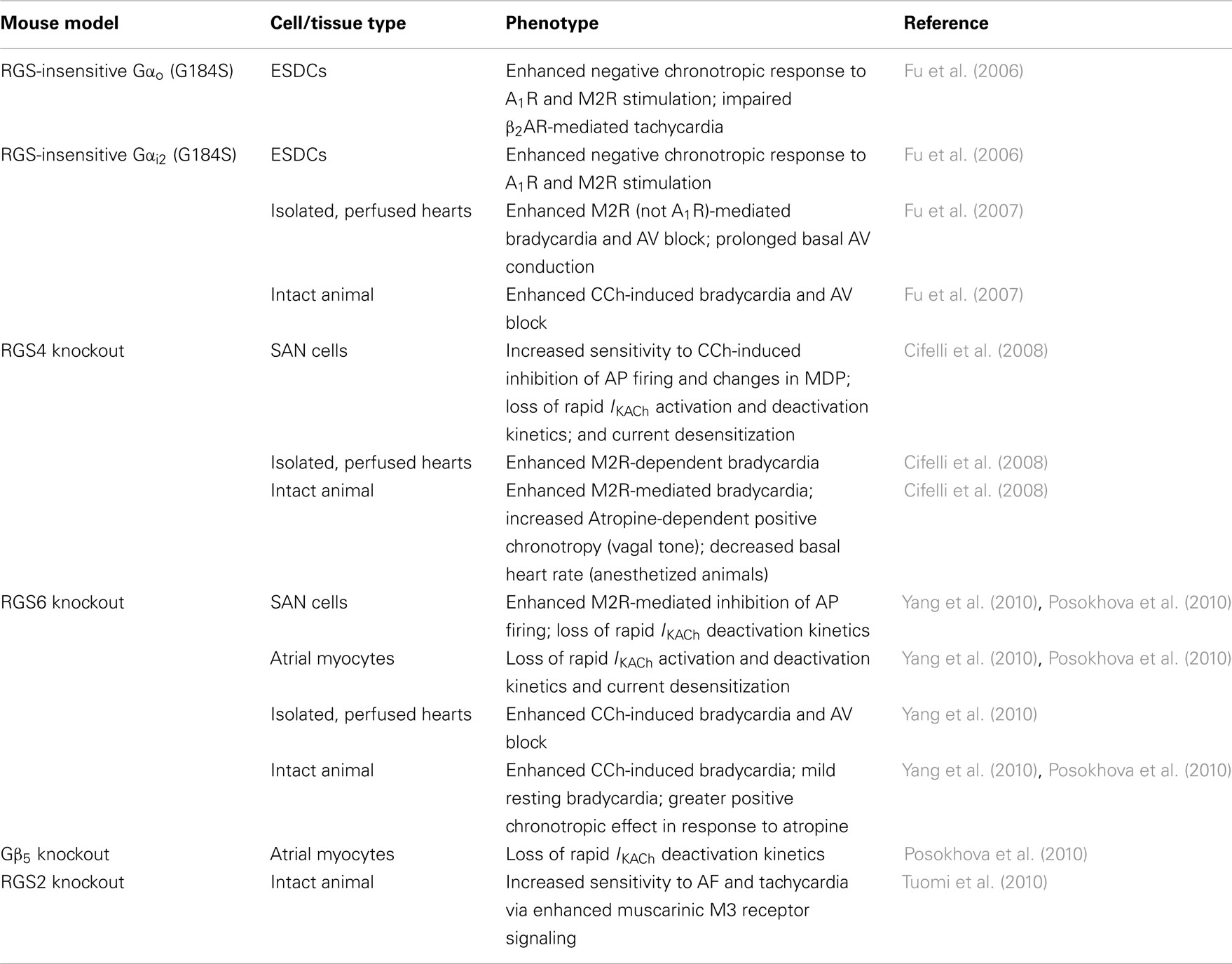
Table 2. Mouse models implicating RGS proteins as key modulators of cardiac automaticity and their associated phenotypes.
That fact that both RGS4 and RGS6 knockout animals exhibit similar phenotypes of enhanced M2R signaling imply that, while each of these proteins may function in a similar signaling cascade, they fail to completely compensate for each other’s loss. A number of hypotheses can be posed to explain these observations. It is possible that RGS4 and RGS6 regulate distinct receptor sub-populations, act as GAPs for different G proteins (e.g., Gαi vs. Gαo), or function to regulate M2Rs in separate cell types. Indeed, RGS6 is known to bind Gαo with higher affinity than Gαi isoforms in vitro (Hooks et al., 2003) implying it may selectively regulate M2Rs coupled to Gαo. The observed lack of RGS4 expression outside of the SAN implies that RGS6 may be the primary negative regulator of M2R-mediated GIRK channel activation in AVN and extra-nodal tissues. In transfected cells expressing exogenous proteins, RGS4 appears to form a complex with the M2R and GIRK channel subunit GIRK1, though this interaction has not been confirmed in vivo (Jaen and Doupnik, 2006). Conversely, RGS6 fails to directly bind GIRK1 in vitro or in vivo (Posokhova et al., 2010; Yang et al., 2010), but does interact with GIRK4 in a heterologous expression system (Posokhova et al., 2010). These results suggest RGS4 and RGS6 may selectively regulate distinct GIRK channel subunits in heart, but the physiological significance of these observations remains unclear. Clearly, the generation of double knockout mice is required to facilitate investigations into the redundant, additive, or synergistic function of RGS4 and RGS6 in regulating parasympathetic stimulation of heart.
Many of the other RGS proteins expressed at the mRNA level in heart are capable GAPs for Gαi/o (Table 1) and their role in regulating cardiac automaticity remains unknown. A few RGS proteins are emerging as potential additional players in the regulation of cardiac automaticity. For example, the enhanced susceptibility to atrial fibrillation (AF) in RGS2−/− mice suggests involvement of additional RGS players in heart, though RGS2 is a selective GAP for Gαq/11 (Heximer et al., 1999) and likely mediates these effects by regulating Gαq-coupled muscarinic M3 receptors (Tuomi et al., 2010). There is also evidence for crosstalk between β1AR signaling and M2R signaling in heart as isoproterenol, a β-adrenoreceptor agonist, induces a slowing of IKACh deactivation kinetics, a process abolished by knockdown of RGS10 in isolated atrial myocytes (Bender et al., 2008). These results implicate RGS10 as a downstream target of the β1AR in heart and suggest RGS10 might also function to regulate M2R signaling, though this effect has yet to be confirmed in vivo. Delineation of the distinct functions of each of the RGS proteins in controlling cardiac automaticity is essential to the design and utilization of targeted therapeutics aimed at modulating RGS protein function in heart. GPCR signaling pathways involved in cardiac automaticity and known to be regulated by endogenous RGS proteins in the SAN and atrial myocytes are summarized in Figures 2 and 3, respectively.
Investigations into regulatory mechanisms controlling RGS4 and RGS6 activity in heart might further elucidate the importance of RGS4 and RGS6 in both physiological and pathophysiological regulation of parasympathetic stimulation of heart and the identification of novel means to target their activity. In addition to regulation of RGS4 expression via the N-end rule pathway, additional post-translational modifications control RGS4 stability, and subcellular localization. Palmitoylation of RGS4 appears to protect the protein from N-end rule-mediated degradation (Wang et al., 2010), but the significance of these two opposing processes in regulating RGS4 expression and localization in heart remains unclear. It is possible that inhibition or activation of key enzymes involved in this proteolytic cascade could allow for increases or decreases in RGS4 expression, respectively, and might be valid pharmacologic means to manipulate RGS4 levels and activity. Atrial natriuretic peptide induces a PKG-dependent RGS4 phosphorylation and membrane translocation in astrocytes that enhances RGS4 activity, but the relevance of this effect in heart has yet to be determined (Pedram et al., 2000). The calcium sensor calmodulin, involved in numerous cardiac pathologies (Zhang et al., 2004), has also been shown to bind directly to RGS4 relieving phosphatidylinositol 3,4,5-trisphosphate (PIP3)-mediated inhibition of RGS4 GAP activity (Popov et al., 2000). Such a mechanism might represent a means to inhibit IKACh in the SAN in order to enhance cellular excitability and ensure AP generation in response to increases in intracellular calcium.
Gβ5 is an atypical Gβ subunit in that, instead of interacting with Gγ, Gβ5 interacts exclusively with R7 family RGS proteins including RGS6 in vivo (Snow et al., 1999). This interaction is essential for the stability, expression, and function of R7 RGS proteins as confirmed by co-expression studies and the discovery that genetic deletion of Gβ5 in mice resulted in a loss of all R7 family members (Posner et al., 1999; Kovoor et al., 2000; Chen et al., 2003). Conversely, deletion of RGS9, the only R7 RGS protein in photoreceptors, caused a complete loss of Gβ5 (Chen et al., 2003) indicating a co-stabilization relationship between Gβ5 and R7 RGS proteins. The almost complete loss of Gβ5 expression observed in the atria of RGS6−/− mice (Posokhova et al., 2010; Yang et al., 2010) identifies RGS6 as the predominant R7 RGS protein expressed in cardiac tissue. R7BP protein expression is not detectable in heart (Posokhova et al., 2010; Yang et al., 2010), but low levels of mRNA expression have been reported in aorta (Drenan et al., 2005; Martemyanov et al., 2005). Indeed, complex formation between RGS6 and Gβ5 but not R7BP is detectable in atrial tissue (Posokhova et al., 2010; Yang et al., 2010). Gβ5 appears to also be necessary for normal GIRK channel gating kinetics in response to M2R stimulation as atrial myocytes isolated from Gβ5−/− mice exhibit a similar loss of current deactivation as that observed in mice lacking RGS6 (Posokhova et al., 2010). Thus, targeting of Gβ5 expression might represent an alternate means to control RGS6 activity in heart. RGS6 mRNA is also subjected to complex alternative splicing in brain yielding 36 distinct isoforms of the protein (Chatterjee et al., 2003), a number of which are detectable in heart (Doupnik et al., 2001; Fisher et al., 2008). This variation influences the ability of the individual isoforms to associate with Gβ5 and R7BP and, as a result, impacts their stability and subcellular localization. The differential role of the various RGS6 splice forms in regulation of cardiac GPCRs has not been explored as studies investigating the function of RGS6 in heart were performed in a global knockout mouse lacking expression of all RGS6 isoforms.
Thus far the specific RGS proteins (RGS4 and RGS6) involved in regulating Gαi/o-coupled M2Rs have been identified. The enhanced bradycardia in mice lacking either of these RGS proteins appears to be determined, at least in part, by enhanced M2R-mediated IKACh. Ventricular myocytes isolated from mice expressing RGS-insensitive Gαi2 also exhibit sensitization to CCh-induced inhibition of contractility implicating endogenous RGS proteins as key regulators of this process (Waterson et al., 2011). Due to its expression in atria and ventricles, RGS6 might also regulate this inotropic response to M2R activation, though this effect has not been directly tested. Studies in cells expressing RGS-insensitive knock-in Gα alleles also implicated endogenous RGS proteins as key negative regulators of both the Gαo-coupled A1R and β2AR signaling through Gαi/o. To date, the consequences of loss of RGS-mediated regulation of cardiac GPCRs on cAMP levels and PKA-dependent modulation of If and ICa,L remains unexplored. As the cardioprotective effects of β2AR stimulation occur via Gαi/o-dependent mechanisms (Xiao, 2001; Patterson et al., 2004), identification of the specific RGS protein(s) responsible for negative regulation of this signaling cascade might allow for the development of targeted therapeutics aimed at specifically enhancing the protective, Gαi/o-mediated effect without simultaneous activation of Gαs.
Both conscious RGS6−/− and anesthetized RGS4-null mice display mild resting bradycardia and a greater positive chronotropic response to administration of the muscarinic antagonist atropine implying these mice exhibit increased vagal tone (Cifelli et al., 2008; Posokhova et al., 2010). Thus far, enhanced vagal tone in these knockout mice has only been assessed using muscarinic cholinergic receptor blockade. Information gleaned from this method is limited and fails to assess the full physiological significance of enhanced vagal tone in these animal models. Fluctuations in parasympathetic nerve activity under resting conditions are the primary determinant of HR variability and such measures provide an additional index of the magnitude of parasympathetic modulation of HR (Chapleau and Sabharwal, 2011). ECG recordings in mice under basal conditions compared to recording upon administration of autonomic blockers can be used to assess sympathovagal balance, which would be expected to be skewed toward the parasympathetic division in mice lacking RGS6 or RGS4. The vagus is activated by multiple reflex pathways including the arterial baroreceptors and peripheral chemoreceptors to slow HR in response to increases in blood pressure or decreased blood oxygen, respectively (Chapleau and Sabharwal, 2011). RGS null mice may exhibit exaggerated bradycardia upon administration of a potent vasoconstrictor or under conditions of hypoxia, hypercapnia, or acidosis. Such measurements would give a more accurate approximation of the physiologically relevant regulation of cardiac M2Rs by RGS proteins without the administration of exogenous pharmacological agents.
In humans, RGS6 may indeed be essential for proper control of parasympathetic stimulation of heart as a single nucleotide polymorphism in the RGS6 gene is associated with deficits in HR recovery after exercise (Vasan et al., 2007). During periods of intense physical exercise, the sympathetic nervous system is activated with a corresponding inhibition of the parasympathetic division. Upon exercise termination, reactivation of vagal efferents is required for the rapid, exponential decline in HR (Chapleau and Sabharwal, 2011). Unsurprisingly, muscarinic receptor blockade compromises HR recovery after exercise (Coote and Bothams, 2001). Post-exercise HR recovery is used as a diagnostic and prognostic tool clinically as it has been shown to be predictive of morbidity and mortality in chronic heart failure and other cardiovascular diseases with faster recovery, indicative of increased vagal tone, associated with better health and lower mortality risk (Thayer and Lane, 2007; Tang et al., 2009). Indeed, shorter HR recovery is predictive of overall mortality independent of cardiovascular risk factors (Cole et al., 1999). Based on its essential role in regulating M2R signaling in heart, loss of RGS6 and the consequent increased vagal tone may promote faster HR recovery in mice. Such a result would implicate RGS6 as a novel target for therapeutic intervention in human patients with failing cardiovascular health.
Given their essential role in regulating parasympathetic activation in heart, alterations in RGS4 or RGS6 expression might provide symptomatic relief in patients with cardiac diseases involving impaired or excessive vagal activity. Amongst these disorders are sick sinus syndrome and AV block (Fu et al., 2007). Excessive vagal stimulation leads directly to AV block, an effect that is primarily dependent on increased GIRK current in mice (Drici et al., 2000; Hardouin et al., 2002) but can also result from inhibition of ICa,L by maternal auto-antibodies in cases of congenital heart block in humans (Garcia et al., 1994). Activation of RGS6 or RGS4 activity in heart would be expected to dampen parasympathetic stimulation and relieve these symptoms.
Atrial fibrillation is also known to lead to sinoatrial nodal dysfunction in humans and rodents (Sparks et al., 1999; Hadian et al., 2002; Hocini et al., 2003). Often GIRK currents are constitutively active in human patients with chronic AF (Dobrev et al., 2005) and a polymorphism in the Gβ3 subunit of the heterotrimeric G protein complex resulting in reduced GIRK current is linked to decreased risk of AF in humans (Schreieck et al., 2004). Loss of M2R-mediated GIRK channel activation in mouse models, either through genetic ablation of cardiac specific GIRK channel GIRK4 or reduced membrane targeting of Gβγ, also resulted in reduced HR variability and risk for AF (Kovoor et al., 2001; Gehrmann et al., 2002). Clearly the extent of M2R-mediated GIRK current is a critical determinant of AF risk. Thus, activation of RGS protein-mediated negative regulation of cardiac IKACh might be a viable means of treating AF in the clinic.
Heart failure can lead to diseases of the cardiac conduction system such as sick sinus syndrome and AVN dysfunction, which may result from both cardiac damage as well as negatively chronotropic pharmacological interventions including the use of β-blockers and digoxin (Monfredi et al., 2010). Impairment of the baroreceptor reflex is also associated with increased mortality in heart failure (Mortara et al., 1997; Schwartz and De Ferrari, 2011). Emerging evidence suggests that increased parasympathetic tone may be protective in heart failure. Donepezil, an acetylcholinesterase inhibitor used to enhance cholinergic signaling in patients with Alzheimer’s disease, can produce adverse cardiovascular events involving bradycardia and AV block (Hernandez et al., 2009) presumably due to enhanced ACh-mediated M2R signaling in heart. Nevertheless, Donepezil treatment regimens also reduce the risk for heart failure related mortality in human patients (Sato et al., 2010), suggesting that under certain pathological conditions, enhanced parasympathetic stimulation of the heart may be cardioprotective. These results have been recapitulated in rodent models of heart failure following aortocaval shunt placement (Handa et al., 2009) and during the acute and chronic phases post-myocardial infarction (Okazaki et al., 2010; Arikawa et al., 2011). The effects are likely due to increased parasympathetic modulation of heart as an increase in HR variability, a common measure of parasympathetic tone, was observed in Donepezil-treated animals (Okazaki et al., 2010). Similar observations have also been found using vagal nerve stimulation protocols in humans (Schwartz and De Ferrari, 2009) and rodents (Li et al., 2004).
The exact mechanism underlying the cardioprotective effect of enhanced vagal tone remains unclear, though it is believed to involve the anti-apoptotic effects of PI3K/Akt/HIF-1α signaling through atropine-sensitive muscarinic ACh receptors (Kakinuma et al., 2005). Such receptors can couple either Gαi/o or Gαq, which activates or inhibits Akt signaling, respectively, suggesting the cardioprotective effects of vagal stimulation may occur through Gαi/o-coupled M2Rs known to be regulated by RGS6 and RGS4 in heart. Thus, the RGS6 and RGS4 knockout mice, both of which exhibit a phenotype of enhanced parasympathetic tone, would be expected to be protected from heart failure induced cardiac damage and mortality. Identification of the specific receptor signaling pathways involved would facilitate investigations into the role of RGS proteins in modulating these processes and their potential as therapeutic targets in the treatment of heart failure. Loss of RGS-mediated regulation of Gαi2 also protects isolated, perfused hearts against ischemic injury including reduction in infarct size and improved recovery of contractile function. Interestingly, inhibition of mitochondrial and sarcolemmal ATP-dependent potassium channels and not the PI3K/Akt/GSK-3β and MEK/ERK signaling cascades blocked the cardioprotective phenotype in these animals. This study points to an entirely novel, anti-apoptotic pathway regulated by RGS proteins in the heart (Waterson et al., 2011). Thus, inhibition of cardiac RGS proteins might represent a viable means to enhance the protective effects of endogenous GPCR agonists such as opioids, bradykinin, and adenosine released from the heart during periods of ischemia (Cohen et al., 2000).
RGS4 mRNA and protein levels are up-regulated in the left ventricular myocardium of human patients with dilated and ischemic cardiomyopathy (Mittmann et al., 2002) and end-stage heart failure (Owen et al., 2001) suggesting that, despite the absence of its expression in atria and ventricles under basal conditions, RGS4 might be an important regulator of GPCR signaling in heart under specific pathological conditions. Exogenous RGS4 expression in ventricular myocytes also suppressed endothelin-1 dependent activation of phospholipase C and the resulting increase in myocyte contractility. This function, if it occurs in vivo, is in direct opposition to the effects of RGS4 on M2R signaling implying that RGS4 may have opposing effects on cardiac inotropy and chronotropy depending on the physiological context (Mittmann et al., 2002). To complicate matters even more, transgenic overexpression of RGS4 in murine ventricular cardiomyocytes exacerbates heart damage and mortality due to heart failure after transverse aortic constriction but transiently improved cardiac hypertrophy (Rogers et al., 1999). RGS4-mediated protection from cardiac hypertrophy and contractile dysfunction was also observed in mice overexpressing Gαq (Rogers et al., 2001). Further investigation into the effect of RGS4 knockout on heart failure progression in mouse models might shed some light on these seemingly paradoxical observations. Indeed, a majority of the studies in mice were performed in transgenic animals overexpressing exogenous RGS4. As RGS4 expression is not detectable in appreciable levels in the myocardium under basal conditions (Posokhova et al., 2010; Yang et al., 2010), the physiological relevance of these findings is questionable. Due to the promiscuous regulation of both Gαi/o and Gαq/11-dependent signaling pathways by RGS4 and the oft times opposing functions of these GPCR cascades, RGS6, which exclusively interacts with Gαi/o, might be a more specific target to enhance parasympathetic signaling in heart. Pathological upregulation of RGS6 in failing myocardium of human patients and mouse models has yet to be explored.
Doxorubicin (Dox) is an anthracycline that is among the most effective and widely used chemotherapeutic drugs to treat human cancers including leukemia, lymphoma, multiple myeloma, and breast cancer (Carter, 1975). However, the major obstacle restricting clinical utility of anthracyclines is their dose-dependent, life-threatening cardiotoxicity (Gianni et al., 2008). Dox induces acute cardiotoxicity, occurring immediately or weeks after treatment cessation and commonly presenting as sinus tachycardia that suggests autonomic dysfunction (Bristow et al., 1978; Lipshultz et al., 2008); and chronic cardiotoxicity, which usually develops in the first year following treatment and presents as left ventricular dysfunction and chronic heart failure (Bristow et al., 1978; Praga et al., 1979; Singal and Iliskovic, 1998).
The mechanism of Dox-induced cardiotoxicity is not fully understood. However, Dox-induced production of reactive oxygen species (ROS) has been implicated as critical for subsequent cardiac myocyte apoptosis and heart damage (Li and Singal, 2000; Zhou et al., 2001; Pacher et al., 2003). The chemotherapeutic efficacy of Dox relies on its ability to kill cancer cells by activating the ATM–p53 apoptosis pathways (Evan, 1997; Canman et al., 1998; Khanna et al., 1998; Bakkenist and Kastan, 2003; Iliakis et al., 2003), which is also responsible for Dox-induced cardiotoxicity as inhibition of components of this pathway protects against Dox-induced heart injury in mice (Liu et al., 2004; Shizukuda et al., 2005; Yoshida et al., 2009; Zhu et al., 2009). RGS6 plays an essential role in mediating activation of ATM and p53 induced by Dox in a ROS-dependent manner (Huang et al., 2011), implicating RGS6, whose expression is enriched in heart (Yang et al., 2010), as a key mediator of Dox-induced cardiotoxicity. Consistently, loss of RGS6 reduces apoptosis of heart cells and consequent heart damage in Dox-treated mice (Huang et al., 2011).
RGS6 has been shown to be induced by Dox in MEFs and cancer cells (Huang et al., 2011). As described above, RGS6 plays a critical role in regulating parasympathetic vagal control in the heart. Thus, this early induction of RGS6 by Dox may be involved in Dox-induced acute AF by interfering with the normal gating kinetics of GIRK current. Evaluation of RGS6 protein levels and IKACh properties in SAN cells isolated from WT and RGS6−/− mice treated with Dox is required to further test this hypothesis.
RGS6 is also required for Dox-induced ROS production. Loss of RGS6 impairs Dox-induced ROS generation in MEFs (Huang et al., 2011). It has been recently reported that Dox induces a ROS- and CaMKII-dependent Ca2+ leakage from sarcoplasmic reticulum (SR; Sag et al., 2011). Normal heart rhythm is initiated and regulated by oscillatory SR Ca2+ release (Ca2+ clock) in SAN cells, which by rhythmically activating Na+/Ca2+ exchanger inward current, prompts membrane ion channels to fire rhythmic APs (Maltsev and Lakatta, 2007). Therefore, RGS6 may promote Dox-induced AF by mediating Dox-induced ROS generation and consequent SR Ca2+ leakage disturbing the normal rhythm of intracellular Ca2+ clock in SAN cells. Interestingly, the ability of RGS6 to mediate ROS generation is not dependent on its interaction with G proteins (Huang et al., 2011; Maity et al., 2011), suggesting a unique role of RGS6, but not other RGS proteins, in controlling Dox-induced autonomic dysfunction. The possibility that RGS6 induction by Dox might underlie cardiac pathologies resulting from anthracycline chemotherapy is intriguing, but remains an untested hypothesis requiring verification from multiple laboratories for final confirmation.
Due to their role in regulation of GPCR signaling pathways in the cardiovascular system in both physiological and pathophysiological contexts, RGS proteins have emerged as potential therapeutic targets in the treatment of various heart-related diseases. While GPCR agonists and antagonists have been used successfully in the clinic for years, additional therapeutic benefit could be derived from the additive or singular use of compounds designed to enhance or inhibit RGS protein function. In diseases involving loss of parasympathetic tone or over-activation of sympathetic nerves, inhibition, or activation of either RGS4 or RGS6 would be expected to enhance or block signaling resulting from endogenous release of vagal ACh and subsequent slowing of HR, respectively. In the case of pathological upregulation of RGS6 and RGS4 that occur in Dox-induced cardiotoxicity and heart failure, respectively, RGS protein inhibition might help to mitigate heart damage.
Small-molecule inhibitors of RGS4 have been developed. The first, CCG-4986 binds irreversibly to RGS4 and results in covalent modification and permanent, allosteric inhibition of the protein making it less than ideal for clinical use and in vivo investigation into RGS4 function (Roman et al., 2010). A more recent compound has been identified with similar but reversible effects (Blazer et al., 2010). It appears that some degree of selectivity can be achieved despite the similarity in protein sequence between members of the various RGS protein subfamilies. Targeting regulatory mechanisms might also represent viable means to manipulate the expression of specific RGS proteins. In addition to their clinical use, such compounds might also have utility in investigating the physiological and pathophysiological functions of RGS proteins, singularly, or for multiple proteins, for which there exists no genetic knockout model or those whose loss, alone or in combination, results in embryonic or post-natal lethality.
Pioneering work done in mice expressing RGS-insensitive Gα alleles identified RGS proteins as key modulators of cardiac automaticity. Further advances in transgenic mouse technology allowed for the identification of two specific RGS proteins (RGS4 and RGS6) responsible for negatively regulating M2R signaling in pacemaker cells and conducting portions of the heart. Nevertheless, many avenues relating to the physiological function of endogenous RGS proteins in controlling cardiac automaticity remain unexplored. Of the 10 RGS proteins expressed in the myocardium, only a handful have been ascribed specific functions some of which appear functionally redundant. Additional regulatory mechanisms controlling RGS protein function further complicates matters but also allows for the opportunity to identify additional means to manipulate RGS protein expression for some therapeutic benefit. As dysregulation in autonomic control of the heart contributes to numerous cardiac pathologies, elucidation of GPCR signaling cascades controlling automaticity and regulated by endogenous RGS proteins has the potential to facilitate the identification of novel treatments for a number of cardiovascular diseases, which remain the leading cause of death in the United States and worldwide. Among these are chronotropic diseases including sick sinus syndrome, pathological AV block and AF, heart failure and ischemic heart injury, and doxorubicin-induced cardiotoxicity.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
A. α-helix, amphipathic α-helix; AC, adenylate cyclase; ACh, acetylcholine; AF, atrial fibrillation; A1R, adenosine A1 receptor; AP, action potential; β1AR, β1 adrenergic receptor; β2AR, β2-adrenergic receptor; cAMP, cyclic AMP; Cys.string, cysteine string motif; DEP/DHEX, dishevelled-EGL-10-pleckstrin homology domain/DEP helical extension, Egl-10, and pleckstrin homology domain; Dox, Doxorubicin; ESDC, embryonic stem cell derived cardiocytes; GAP, GTPase accelerating protein; GEF, guanine nucleotide exchange factor; GGL, Gγ subunit-like domain; GIRK channel, G protein-activated inwardly rectifying potassium channel; GoLoco, Gαi/o-loco domain; GPCR, G protein-coupled receptor; HCN, hyperpolarization-activated cyclic nucleotide-gated cation channels; HR, heart rate; ICa,L, L-type calcium channel current; If, pacemaker current; IKACh, acetylcholine-induced GIRK current; MDP, maximum diastolic potential; MEF, mouse embryonic fibroblast; M2R, muscarinic M2 receptor; PCR, polymerase chain reaction; PDZ, PSD-95 Disk-large ZO-1 domain; PDZ M, PDZ docking motif; PKA, protein kinase A; PTB, phosphotyrosine binding domain; RBD, Ras-binding domain; RGS, Regulator of G protein signaling; RGS4, Regulator of G protein signaling 4; RGS6, Regulator of G Protein Signaling 6; R7BP, R7 family RGS binding protein; ROS, reactive oxygen species.
Anderson, G. R., Posokhova, E., and Martemyanov, K. A. (2009). The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem. Biophys. 54, 33–46.
Anger, T., Zhang, W., and Mende, U. (2004). Differential contribution of GTPase activation and effector antagonism to the inhibitory effect of RGS proteins on Gq-mediated signaling in vivo. J. Biol. Chem. 279, 3906–3915.
Arikawa, M., Kakinuma, Y., Handa, T., Yamasaki, F., and Sato, T. (2011). Donepezil, anti-Alzheimer’s disease drug, prevents cardiac rupture during acute phase of myocardial infarction in mice. PLoS ONE 6, e20629. doi:10.1371/journal.pone.0020629
Bakkenist, C. J., and Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506.
Bender, K., Nasrollahzadeh, P., Timpert, M., Liu, B., Pott, L., and Kienitz, M. C. (2008). A role for RGS10 in beta-adrenergic modulation of G-protein-activated K+ (GIRK) channel current in rat atrial myocytes. J. Physiol. (Lond.) 586, 2049–2060.
Berman, D. M., and Gilman, A. G. (1998). Mammalian RGS proteins: barbarians at the gate. J. Biol. Chem. 273, 1269–1272.
Blazer, L. L., Roman, D. L., Chung, A., Larsen, M. J., Greedy, B. M., Husbands, S. M., and Neubig, R. R. (2010). Reversible, allosteric small-molecule inhibitors of regulator of G protein signaling proteins. Mol. Pharmacol. 78, 524–533.
Bristow, M. R., Billingham, M. E., Mason, J. W., and Daniels, J. R. (1978). Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat. Rep. 62, 873–879.
Canman, C. E., Lim, D. S., Cimprich, K. A., Taya, Y., Tamai, K., Sakaguchi, K., Appella, E., Kastan, M. B., and Siliciano, J. D. (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679.
Chakir, K., Zhu, W., Tsang, S., Woo, A. Y., Yang, D., Wang, X., Zeng, X., Rhee, M. H., Mende, U., Koitabashi, N., Takimoto, E., Blumer, K. J., Lakatta, E. G., Kass, D. A., and Xiao, R. P. (2011). RGS2 is a primary terminator of beta-adrenergic receptor-mediated G(i) signaling. J. Mol. Cell. Cardiol. 50, 1000–1007.
Chapleau, M. W., and Sabharwal, R. (2011). Methods of assessing vagus nerve activity and reflexes. Heart Fail. Rev. 16, 109–127.
Chatterjee, T. K., Liu, Z., and Fisher, R. A. (2003). Human RGS6 gene structure, complex alternative splicing, and role of N terminus and G protein gamma-subunit-like (GGL) domain in subcellular localization of RGS6 splice variants. J. Biol. Chem. 278, 30261–30271.
Chen, C., Wang, H., Fong, C. W., and Lin, S. C. (2001). Multiple phosphorylation sites in RGS16 differentially modulate its GAP activity. FEBS Lett. 504, 16–22.
Chen, C., Zheng, B., Han, J., and Lin, S. C. (1997). Characterization of a novel mammalian RGS protein that binds to Galpha proteins and inhibits pheromone signaling in yeast. J. Biol. Chem. 272, 8679–8685.
Chen, C. K., Eversole-Cire, P., Zhang, H., Mancino, V., Chen, Y. J., He, W., Wensel, T. G., and Simon, M. I. (2003). Instability of GGL domain-containing RGS proteins in mice lacking the G protein beta-subunit Gbeta5. Proc. Natl. Acad. Sci. U.S.A. 100, 6604–6609.
Cho, H., Kozasa, T., Takekoshi, K., De Gunzburg, J., and Kehrl, J. H. (2000). RGS14, a GTPase-activating protein for Gialpha, attenuates Gialpha- and G13alpha-mediated signaling pathways. Mol. Pharmacol. 58, 569–576.
Cifelli, C., Rose, R. A., Zhang, H., Voigtlaender-Bolz, J., Bolz, S. S., Backx, P. H., and Heximer, S. P. (2008). RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535.
Cohen, M. V., Baines, C. P., and Downey, J. M. (2000). Ischemic preconditioning: from adenosine receptor to KATP channel. Annu. Rev. Physiol. 62, 79–109.
Cole, C. R., Blackstone, E. H., Pashkow, F. J., Snader, C. E., and Lauer, M. S. (1999). Heart-rate recovery immediately after exercise as a predictor of mortality. N. Engl. J. Med. 341, 1351–1357.
Coote, J. H., and Bothams, V. F. (2001). Cardiac vagal control before, during and after exercise. Exp. Physiol. 86, 811–815.
Davydov, I. V., and Varshavsky, A. (2000). RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J. Biol. Chem. 275, 22931–22941.
De Vries, L., Mousli, M., Wurmser, A., and Farquhar, M. G. (1995). GAIP, a protein that specifically interacts with the trimeric G protein Galpha i3, is a member of a protein family with a highly conserved core domain. Proc. Natl. Acad. Sci. U.S.A. 92, 11916–11920.
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., Knaut, M., and Ravens, U. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706.
Dohlman, H. G., and Thorner, J. (1997). RGS proteins and signaling by heterotrimeric G proteins. J. Biol. Chem. 272, 3871–3874.
Doupnik, C. A., Davidson, N., Lester, H. A., and Kofuji, P. (1997). RGS proteins reconstitute the rapid gating kinetics of gbetagamma-activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U.S.A. 94, 10461–10466.
Doupnik, C. A., Xu, T., and Shinaman, J. M. (2001). Profile of RGS expression in single rat atrial myocytes. Biochim. Biophys. Acta 1522, 97–107.
Drenan, R. M., Doupnik, C. A., Boyle, M. P., Muglia, L. J., Huettner, J. E., Linder, M. E., and Blumer, K. J. (2005). Palmitoylation regulates plasma membrane-nuclear shuttling of R7BP, a novel membrane anchor for the RGS7 family. J. Cell Biol. 169, 623–633.
Drici, M. D., Diochot, S., Terrenoire, C., Romey, G., and Lazdunski, M. (2000). The bee venom peptide tertiapin underlines the role of I(KACh) in acetylcholine-induced atrioventricular blocks. Br. J. Pharmacol. 131, 569–577.
Fisher, R. A., Singh, M., Anderson, M. E., and Twait, E. (2008). Expression of multiple splice variant forms of RGS6 in mouse ventricular myocytes. FASEB J. 22, Ib311.
Fredriksson, R., Lagerstrom, M. C., Lundin, L. G., and Schioth, H. B. (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272.
Fu, Y., Huang, X., Piao, L., Lopatin, A. N., and Neubig, R. R. (2007). Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am. J. Physiol. Heart Circ. Physiol. 292, H2532–H2539.
Fu, Y., Huang, X., Zhong, H., Mortensen, R. M., D’Alecy, L. G., and Neubig, R. R. (2006). Endogenous RGS proteins and Galpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ. Res. 98, 659–666.
Fu, Y., Zhong, H., Nanamori, M., Mortensen, R. M., Huang, X., Lan, K., and Neubig, R. R. (2004). RGS-insensitive G-protein mutations to study the role of endogenous RGS proteins. Meth. Enzymol. 389, 229–243.
Fujita, S., Inanobe, A., Chachin, M., Aizawa, Y., and Kurachi, Y. (2000). A regulator of G protein signalling (RGS) protein confers agonist-dependent relaxation gating to a G protein-gated K+ channel. J. Physiol. (Lond.) 526(Pt 2), 341–347.
Garcia, S., Nascimento, J. H., Bonfa, E., Levy, R., Oliveira, S. F., Tavares, A. V., and De Carvalho, A. C. (1994). Cellular mechanism of the conduction abnormalities induced by serum from anti-Ro/SSA-positive patients in rabbit hearts. J. Clin. Invest. 93, 718–724.
Gehrmann, J., Meister, M., Maguire, C. T., Martins, D. C., Hammer, P. E., Neer, E. J., Berul, C. I., and Mende, U. (2002). Impaired parasympathetic heart rate control in mice with a reduction of functional G protein betagamma-subunits. Am. J. Physiol. Heart Circ. Physiol. 282, H445–H456.
Gianni, L., Herman, E. H., Lipshultz, S. E., Minotti, G., Sarvazyan, N., and Sawyer, D. B. (2008). Anthracycline cardiotoxicity: from bench to bedside. J. Clin. Oncol. 26, 3777–3784.
Glick, J. L., Meigs, T. E., Miron, A., and Casey, P. J. (1998). RGSZ1, a Gz-selective regulator of G protein signaling whose action is sensitive to the phosphorylation state of Gzalpha. J. Biol. Chem. 273, 26008–26013.
Hadian, D., Zipes, D. P., Olgin, J. E., and Miller, J. M. (2002). Short-term rapid atrial pacing produces electrical remodeling of sinus node function in humans. J. Cardiovasc. Electrophysiol. 13, 584–586.
Handa, T., Katare, R. G., Kakinuma, Y., Arikawa, M., Ando, M., Sasaguri, S., Yamasaki, F., and Sato, T. (2009). Anti-Alzheimer’s drug, donepezil, markedly improves long-term survival after chronic heart failure in mice. J. Card. Fail. 15, 805–811.
Hao, J., Michalek, C., Zhang, W., Zhu, M., Xu, X., and Mende, U. (2006). Regulation of cardiomyocyte signaling by RGS proteins: differential selectivity towards G proteins and susceptibility to regulation. J. Mol. Cell. Cardiol. 41, 51–61.
Hardouin, S. N., Richmond, K. N., Zimmerman, A., Hamilton, S. E., Feigl, E. O., and Nathanson, N. M. (2002). Altered cardiovascular responses in mice lacking the M(1) muscarinic acetylcholine receptor. J. Pharmacol. Exp. Ther. 301, 129–137.
Hernandez, R. K., Farwell, W., Cantor, M. D., and Lawler, E. V. (2009). Cholinesterase inhibitors and incidence of bradycardia in patients with dementia in the veterans affairs new England healthcare system. J. Am. Geriatr. Soc. 57, 1997–2003.
Heximer, S. P., Knutsen, R. H., Sun, X., Kaltenbronn, K. M., Rhee, M. H., Peng, N., Oliveira-Dos-Santos, A., Penninger, J. M., Muslin, A. J., Steinberg, T. H., Wyss, J. M., Mecham, R. P., and Blumer, K. J. (2003). Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Invest. 111, 445–452.
Heximer, S. P., Srinivasa, S. P., Bernstein, L. S., Bernard, J. L., Linder, M. E., Hepler, J. R., and Blumer, K. J. (1999). G protein selectivity is a determinant of RGS2 function. J. Biol. Chem. 274, 34253–34259.
Hocini, M., Sanders, P., Deisenhofer, I., Jais, P., Hsu, L. F., Scavee, C., Weerasoriya, R., Raybaud, F., Macle, L., Shah, D. C., Garrigue, S., Le Metayer, P., Clementy, J., and Haissaguerre, M. (2003). Reverse remodeling of sinus node function after catheter ablation of atrial fibrillation in patients with prolonged sinus pauses. Circulation 108, 1172–1175.
Hooks, S. B., Waldo, G. L., Corbitt, J., Bodor, E. T., Krumins, A. M., and Harden, T. K. (2003). RGS6, RGS7, RGS9, and RGS11 stimulate GTPase activity of Gi family G-proteins with differential selectivity and maximal activity. J. Biol. Chem. 278, 10087–10093.
Huang, C., Hepler, J. R., Gilman, A. G., and Mumby, S. M. (1997). Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 94, 6159–6163.
Huang, J., Yang, J., Maity, B., Mayuzumi, D., and Fisher, R. A. (2011). Regulator of G protein signaling 6 mediates doxorubicin-induced ATM and p53 activation by a reactive oxygen species-dependent mechanism. Cancer Res. 71, 6310–6319.
Iliakis, G., Wang, Y., Guan, J., and Wang, H. (2003). DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 22, 5834–5847.
Inanobe, A., Fujita, S., Makino, Y., Matsushita, K., Ishii, M., Chachin, M., and Kurachi, Y. (2001). Interaction between the RGS domain of RGS4 with G protein alpha subunits mediates the voltage-dependent relaxation of the G protein-gated potassium channel. J. Physiol. (Lond.) 535, 133–143.
Ishii, M., Inanobe, A., Fujita, S., Makino, Y., Hosoya, Y., and Kurachi, Y. (2001). Ca(2+) elevation evoked by membrane depolarization regulates G protein cycle via RGS proteins in the heart. Circ. Res. 89, 1045–1050.
Jaen, C., and Doupnik, C. A. (2006). RGS3 and RGS4 differentially associate with G protein-coupled receptor-Kir3 channel signaling complexes revealing two modes of RGS modulation. Precoupling and collision coupling. J. Biol. Chem. 281, 34549–34560.
Jean-Baptiste, G., Li, X., Yang, Z., Heubach, J., Gaudio, S., Khoury, C., Ravens, U., and Greenwood, M. T. (2005). Beta adrenergic receptor-mediated atrial specific up-regulation of RGS5. Life Sci. 76, 1533–1545.
Johnson, E. N., and Druey, K. M. (2002). Functional characterization of the G protein regulator RGS13. J. Biol. Chem. 277, 16768–16774.
Kakinuma, Y., Ando, M., Kuwabara, M., Katare, R. G., Okudela, K., Kobayashi, M., and Sato, T. (2005). Acetylcholine from vagal stimulation protects cardiomyocytes against ischemia and hypoxia involving additive non-hypoxic induction of HIF-1alpha. FEBS Lett. 579, 2111–2118.
Kardestuncer, T., Wu, H., Lim, A. L., and Neer, E. J. (1998). Cardiac myocytes express mRNA for ten RGS proteins: changes in RGS mRNA expression in ventricular myocytes and cultured atria. FEBS Lett. 438, 285–288.
Khanna, K. K., Keating, K. E., Kozlov, S., Scott, S., Gatei, M., Hobson, K., Taya, Y., Gabrielli, B., Chan, D., Lees-Miller, S. P., and Lavin, M. F. (1998). ATM associates with and phosphorylates p53: mapping the region of interaction. Nat. Genet. 20, 398–400.
Kovoor, A., Chen, C. K., He, W., Wensel, T. G., Simon, M. I., and Lester, H. A. (2000). Co-expression of Gbeta5 enhances the function of two Ggamma subunit-like domain-containing regulators of G protein signaling proteins. J. Biol. Chem. 275, 3397–3402.
Kovoor, P., Wickman, K., Maguire, C. T., Pu, W., Gehrmann, J., Berul, C. I., and Clapham, D. E. (2001). Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143.
Ladds, G., Goddard, A., Hill, C., Thornton, S., and Davey, J. (2007). Differential effects of RGS proteins on G alpha(q) and G alpha(11) activity. Cell. Signal. 19, 103–113.
Lambert, N. A., Johnston, C. A., Cappell, S. D., Kuravi, S., Kimple, A. J., Willard, F. S., and Siderovski, D. P. (2010). Regulators of G-protein signaling accelerate GPCR signaling kinetics and govern sensitivity solely by accelerating GTPase activity. Proc. Natl. Acad. Sci. U.S.A. 107, 7066–7071.
Lan, K. L., Sarvazyan, N. A., Taussig, R., Mackenzie, R. G., Dibello, P. R., Dohlman, H. G., and Neubig, R. R. (1998). A point mutation in Galphao and Galphai1 blocks interaction with regulator of G protein signaling proteins. J. Biol. Chem. 273, 12794–12797.
Lee, M. J., Tasaki, T., Moroi, K., An, J. Y., Kimura, S., Davydov, I. V., and Kwon, Y. T. (2005). RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 15030–15035.
Li, H., He, C., Feng, J., Zhang, Y., Tang, Q., Bian, Z., Bai, X., Zhou, H., Jiang, H., Heximer, S. P., Qin, M., Huang, H., Liu, P. P., and Huang, C. (2010). Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc. Natl. Acad. Sci. U.S.A. 107, 13818–13823.
Li, M., Zheng, C., Sato, T., Kawada, T., Sugimachi, M., and Sunagawa, K. (2004). Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation 109, 120–124.
Li, T., and Singal, P. K. (2000). Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation 102, 2105–2110.
Lipshultz, S. E., Alvarez, J. A., and Scully, R. E. (2008). Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart 94, 525–533.
Liu, X., Chua, C. C., Gao, J., Chen, Z., Landy, C. L., Hamdy, R., and Chua, B. H. (2004). Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am. J. Physiol. Heart Circ. Physiol. 286, H933–H939.
Maity, B., Yang, J., Huang, J., Askeland, R. W., Bera, S., and Fisher, R. A. (2011). Regulator of G protein signaling 6 (RGS6) induces apoptosis via a mitochondrial-dependent pathway not involving its GTPase-activating protein activity. J. Biol. Chem. 286, 1409–1419.
Maltsev, V. A., and Lakatta, E. G. (2007). Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ. 16, 335–348.
Mao, H., Zhao, Q., Daigle, M., Ghahremani, M. H., Chidiac, P., and Albert, P. R. (2004). RGS17/RGSZ2, a novel regulator of Gi/o, Gz, and Gq signaling. J. Biol. Chem. 279, 26314–26322.
Mark, M. D., and Herlitze, S. (2000). G-protein mediated gating of inward-rectifier K+ channels. Eur. J. Biochem. 267, 5830–5836.
Martemyanov, K. A., Yoo, P. J., Skiba, N. P., and Arshavsky, V. Y. (2005). R7BP, a novel neuronal protein interacting with RGS proteins of the R7 family. J. Biol. Chem. 280, 5133–5136.
Mittmann, C., Chung, C. H., Hoppner, G., Michalek, C., Nose, M., Schuler, C., Schuh, A., Eschenhagen, T., Weil, J., Pieske, B., Hirt, S., and Wieland, T. (2002). Expression of ten RGS proteins in human myocardium: functional characterization of an upregulation of RGS4 in heart failure. Cardiovasc. Res. 55, 778–786.
Mittmann, C., Schuler, C., Chung, C. H., Hoppner, G., Nose, M., Kehrl, J. H., and Wieland, T. (2001). Evidence for a short form of RGS3 preferentially expressed in the human heart. Naunyn Schmiedebergs Arch. Pharmacol. 363, 456–463.
Monfredi, O., Dobrzynski, H., Mondal, T., Boyett, M. R., and Morris, G. M. (2010). The anatomy and physiology of the sinoatrial node – a contemporary review. Pacing. Clin. Electrophysiol. 33, 1392–1406.
Mortara, A., La Rovere, M. T., Pinna, G. D., Prpa, A., Maestri, R., Febo, O., Pozzoli, M., Opasich, C., and Tavazzi, L. (1997). Arterial baroreflex modulation of heart rate in chronic heart failure: clinical and hemodynamic correlates and prognostic implications. Circulation 96, 3450–3458.
Okazaki, Y., Zheng, C., Li, M., and Sugimachi, M. (2010). Effect of the cholinesterase inhibitor donepezil on cardiac remodeling and autonomic balance in rats with heart failure. J. Physiol. Sci. 60, 67–74.
Owen, V. J., Burton, P. B., Mullen, A. J., Birks, E. J., Barton, P., and Yacoub, M. H. (2001). Expression of RGS3, RGS4 and Gi alpha 2 in acutely failing donor hearts and end-stage heart failure. Eur. Heart J. 22, 1015–1020.
Pacher, P., Liaudet, L., Bai, P., Mabley, J. G., Kaminski, P. M., Virag, L., Deb, A., Szabo, E., Ungvari, Z., Wolin, M. S., Groves, J. T., and Szabo, C. (2003). Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin-induced cardiac dysfunction. Circulation 107, 896–904.
Park, I. K., Klug, C. A., Li, K., Jerabek, L., Li, L., Nanamori, M., Neubig, R. R., Hood, L., Weissman, I. L., and Clarke, M. F. (2001). Molecular cloning and characterization of a novel regulator of G-protein signaling from mouse hematopoietic stem cells. J. Biol. Chem. 276, 915–923.
Patterson, A. J., Zhu, W., Chow, A., Agrawal, R., Kosek, J., Xiao, R. P., and Kobilka, B. (2004). Protecting the myocardium: a role for the beta2 adrenergic receptor in the heart. Crit. Care Med. 32, 1041–1048.
Pedram, A., Razandi, M., Kehrl, J., and Levin, E. R. (2000). Natriuretic peptides inhibit G protein activation. Mediation through cross-talk between cyclic GMP-dependent protein kinase and regulators of G protein-signaling proteins. J. Biol. Chem. 275, 7365–7372.
Popov, S. G., Krishna, U. M., Falck, J. R., and Wilkie, T. M. (2000). Ca2+/Calmodulin reverses phosphatidylinositol 3,4, 5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J. Biol. Chem. 275, 18962–18968.
Posner, B. A., Gilman, A. G., and Harris, B. A. (1999). Regulators of G protein signaling 6 and 7. Purification of complexes with gbeta5 and assessment of their effects on g protein-mediated signaling pathways. J. Biol. Chem. 274, 31087–31093.
Posokhova, E., Wydeven, N., Allen, K. L., Wickman, K., and Martemyanov, K. A. (2010). RGS6/G{beta}5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ. Res. 107, 1350–1354.
Praga, C., Beretta, G., Vigo, P. L., Lenaz, G. R., Pollini, C., Bonadonna, G., Canetta, R., Castellani, R., Villa, E., Gallagher, C. G., Von Melchner, H., Hayat, M., Ribaud, P., De Wasch, G., Mattsson, W., Heinz, R., Waldner, R., Kolaric, K., Buehner, R., Ten Bokkel-Huyninck, W., Perevodchikova, N. I., Manziuk, L. A., Senn, H. J., and Mayr, A. C. (1979). Adriamycin cardiotoxicity: a survey of 1273 patients. Cancer Treat. Rep. 63, 827–834.
Rogers, J. H., Tamirisa, P., Kovacs, A., Weinheimer, C., Courtois, M., Blumer, K. J., Kelly, D. P., and Muslin, A. J. (1999). RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J. Clin. Invest. 104, 567–576.
Rogers, J. H., Tsirka, A., Kovacs, A., Blumer, K. J., Dorn, G. W. II, and Muslin, A. J. (2001). RGS4 reduces contractile dysfunction and hypertrophic gene induction in Galpha q overexpressing mice. J. Mol. Cell. Cardiol. 33, 209–218.
Roman, D. L., Blazer, L. L., Monroy, C. A., and Neubig, R. R. (2010). Allosteric inhibition of the regulator of G protein signaling-Galpha protein-protein interaction by CCG-4986. Mol. Pharmacol. 78, 360–365.
Ross, E. M., and Wilkie, T. M. (2000). GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins [review]. Annu. Rev. Biochem. 69, 795–827.
Sag, C. M., Kohler, A. C., Anderson, M. E., Backs, J., and Maier, L. S. (2011). CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. J. Mol. Cell. Cardiol. 51, 749–759.
Salazar, N. C., Chen, J., and Rockman, H. A. (2007). Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta 1768, 1006–1018.
Sato, K., Urbano, R., Yu, C., Yamasaki, F., Sato, T., Jordan, J., Robertson, D., and Diedrich, A. (2010). The effect of donepezil treatment on cardiovascular mortality. Clin. Pharmacol. Ther. 88, 335–338.
Scheschonka, A., Dessauer, C. W., Sinnarajah, S., Chidiac, P., Shi, C. S., and Kehrl, J. H. (2000). RGS3 is a GTPase-activating protein for g(ialpha) and g(qalpha) and a potent inhibitor of signaling by GTPase-deficient forms of g(qalpha) and g(11alpha). Mol. Pharmacol. 58, 719–728.
Schreieck, J., Dostal, S., Von Beckerath, N., Wacker, A., Flory, M., Weyerbrock, S., Koch, W., Schomig, A., and Schmitt, C. (2004). C825T polymorphism of the G-protein beta3 subunit gene and atrial fibrillation: association of the TT genotype with a reduced risk for atrial fibrillation. Am. Heart J. 148, 545–550.
Schwartz, P. J., and De Ferrari, G. M. (2009). Vagal stimulation for heart failure: background and first in-man study. Heart Rhythm 6, S76–S81.
Schwartz, P. J., and De Ferrari, G. M. (2011). Sympathetic-parasympathetic interaction in health and disease: abnormalities and relevance in heart failure. Heart Fail. Rev. 16, 101–107.
Shizukuda, Y., Matoba, S., Mian, O. Y., Nguyen, T., and Hwang, P. M. (2005). Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol. Cell. Biochem. 273, 25–32.
Singal, P. K., and Iliskovic, N. (1998). Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 339, 900–905.
Snow, B. E., Antonio, L., Suggs, S., Gutstein, H. B., and Siderovski, D. P. (1997). Molecular cloning and expression analysis of rat Rgs12 and Rgs14. Biochem. Biophys. Res. Commun. 233, 770–777.
Snow, B. E., Betts, L., Mangion, J., Sondek, J., and Siderovski, D. P. (1999). Fidelity of G protein beta-subunit association by the G protein gamma- subunit-like domains of RGS6, RGS7, and RGS11. Proc. Natl. Acad. Sci. U.S.A. 96, 6489–6494.
Snow, B. E., Hall, R. A., Krumins, A. M., Brothers, G. M., Bouchard, D., Brothers, C. A., Chung, S., Mangion, J., Gilman, A. G., Lefkowitz, R. J., and Siderovski, D. P. (1998a). GTPase activating specificity of RGS12 and binding specificity of an alternatively spliced PDZ (PSD-95/Dlg/ZO-1) domain. J. Biol. Chem. 273, 17749–17755.
Snow, B. E., Krumins, A. M., Brothers, G. M., Lee, S. F., Wall, M. A., Chung, S., Mangion, J., Arya, S., Gilman, A. G., and Siderovski, D. P. (1998b). A G protein gamma subunit-like domain shared between RGS11 and other RGS proteins specifies binding to Gbeta5 subunits. Proc. Natl. Acad. Sci. U.S.A. 95, 13307–13312.
Sparks, P. B., Mond, H. G., Vohra, J. K., Jayaprakash, S., and Kalman, J. M. (1999). Electrical remodeling of the atria following loss of atrioventricular synchrony: a long-term study in humans. Circulation 100, 1894–1900.
Takimoto, E., Koitabashi, N., Hsu, S., Ketner, E. A., Zhang, M., Nagayama, T., Bedja, D., Gabrielson, K. L., Blanton, R., Siderovski, D. P., Mendelsohn, M. E., and Kass, D. A. (2009). Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J. Clin. Invest. 119, 408–420.
Tang, K. M., Wang, G. R., Lu, P., Karas, R. H., Aronovitz, M., Heximer, S. P., Kaltenbronn, K. M., Blumer, K. J., Siderovski, D. P., Zhu, Y., and Mendelsohn, M. E. (2003). Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 9, 1506–1512.
Tang, Y. D., Dewland, T. A., Wencker, D., and Katz, S. D. (2009). Post-exercise heart rate recovery independently predicts mortality risk in patients with chronic heart failure. J. Card. Fail. 15, 850–855.
Thayer, J. F., and Lane, R. D. (2007). The role of vagal function in the risk for cardiovascular disease and mortality. Biol. Psychol. 74, 224–242.
Tu, Y., Popov, S., Slaughter, C., and Ross, E. M. (1999). Palmitoylation of a conserved cysteine in the regulator of G protein signaling (RGS) domain modulates the GTPase-activating activity of RGS4 and RGS10. J. Biol. Chem. 274, 38260–38267.
Tuomi, J. M., Chidiac, P., and Jones, D. L. (2010). Evidence for enhanced M3 muscarinic receptor function and sensitivity to atrial arrhythmia in the RGS2-deficient mouse. Am. J. Physiol. Heart Circ. Physiol. 298, H554–H561.
Vasan, R. S., Larson, M. G., Aragam, J., Wang, T. J., Mitchell, G. F., Kathiresan, S., Newton-Cheh, C., Vita, J. A., Keyes, M. J., O’donnell, C. J., Levy, D., and Benjamin, E. J. (2007). Genome-wide association of echocardiographic dimensions, brachial artery endothelial function and treadmill exercise responses in the Framingham Heart Study. BMC Med. Genet. 8(Suppl. 1), S2. doi:10.1186/1471-2350-8-S1-S2
Von Buchholtz, L., Elischer, A., Tareilus, E., Gouka, R., Kaiser, C., Breer, H., and Conzelmann, S. (2004). RGS21 is a novel regulator of G protein signalling selectively expressed in subpopulations of taste bud cells. Eur. J. Neurosci. 19, 1535–1544.
Wang, J., Ducret, A., Tu, Y., Kozasa, T., Aebersold, R., and Ross, E. M. (1998). RGSZ1, a Gz-selective RGS protein in brain. Structure, membrane association, regulation by Galphaz phosphorylation, and relationship to a Gz gtpase-activating protein subfamily. J. Biol. Chem. 273, 26014–26025.
Wang, J., Xie, Y., Wolff, D. W., Abel, P. W., and Tu, Y. (2010). DHHC protein-dependent palmitoylation protects regulator of G-protein signaling 4 from proteasome degradation. FEBS Lett. 584, 4570–4574.
Waterson, R. E., Thompson, C. G., Mabe, N. W., Kaur, K., Talbot, J. N., Neubig, R. R., and Rorabaugh, B. R. (2011). Galpha(i2)-mediated protection from ischaemic injury is modulated by endogenous RGS proteins in the mouse heart. Cardiovasc. Res. 91, 45–52.
Watson, N., Linder, M. E., Druey, K. M., Kehrl, J. H., and Blumer, K. J. (1996). RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature 383, 172–175.
Wickman, K., Nemec, J., Gendler, S. J., and Clapham, D. E. (1998). Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114.
Xiao, R. P. (2001). Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci. STKE 2001, re15.
Xie, Y., Wolff, D. W., Wei, T., Wang, B., Deng, C., Kirui, J. K., Jiang, H., Qin, J., Abel, P. W., and Tu, Y. (2009). Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 69, 5743–5751.
Yang, J., Huang, J., Maity, B., Gao, Z., Lorca, R. A., Gudmundsson, H., Li, J., Stewart, A., Swaminathan, P. D., Ibeawuchi, S. R., Shepherd, A., Chen, C. K., Kutschke, W., Mohler, P. J., Mohapatra, D. P., Anderson, M. E., and Fisher, R. A. (2010). RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 107, 1345–1349.
Yoshida, M., Shiojima, I., Ikeda, H., and Komuro, I. (2009). Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J. Mol. Cell. Cardiol. 47, 698–705.
Zhang, P., and Mende, U. (2011). Regulators of G-protein signaling in the heart and their potential as therapeutic targets. Circ. Res. 109, 320–333.
Zhang, P., Su, J., King, M. E., Maldonado, A. E., Park, C., and Mende, U. (2011). Regulator of G protein signaling 2 is a functionally important negative regulator of angiotensin II-induced cardiac fibroblast responses. Am. J. Physiol. Heart Circ. Physiol. 301, H147–H156.
Zhang, T., Miyamoto, S., and Brown, J. H. (2004). Cardiomyocyte calcium and calcium/calmodulin-dependent protein kinase II: friends or foes? Recent Prog. Horm. Res. 59, 141–168.
Zhong, H., Wade, S. M., Woolf, P. J., Linderman, J. J., Traynor, J. R., and Neubig, R. R. (2003). A spatial focusing model for G protein signals. Regulator of G protein signaling (RGS) protien-mediated kinetic scaffolding. J. Biol. Chem. 278, 7278–7284.
Zhou, S., Starkov, A., Froberg, M. K., Leino, R. L., and Wallace, K. B. (2001). Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 61, 771–777.
Keywords: automaticity, RGS proteins, vagal nerve, parasympathetic nervous system, muscarinic M2 receptor, heart disease, G protein-coupled receptor signaling
Citation: Stewart A, Huang J and Fisher RA (2012) RGS proteins in heart: brakes on the vagus. Front. Physio. 3:95. doi: 10.3389/fphys.2012.00095
Received: 29 December 2011; Paper pending published: 02 February 2012;
Accepted: 27 March 2012; Published online: 13 April 2012.
Edited by:
Craig Doupnik, University of South Florida College of Medicine, USACopyright: © 2012 Stewart, Huang and Fisher. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Rory A. Fisher, Department of Pharmacology, Carver College of Medicine, University of Iowa, 51 Newton Road, BSB 2-512, Iowa City, IA 52242, USA. e-mail:cm9yeS1maXNoZXJAdWlvd2EuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.