

 Naoko Yanagisawa1
Naoko Yanagisawa1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 02 April 2012
Sec. Gastrointestinal Sciences
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00077
This article is part of the Research Topic Recent advances in Pancreatology View all 10 articles
The etiopathogenesis of many autoimmune disorders has not been identified. The aim of this paper is to focus on the involvement of bacterial exposure, as an environmental factor, in the pathogenesis of autoimmune pancreatitis (AIP), which is broadly categorized as autoimmune disorders involving pancreatic lesions. Avirulent and/or commensal bacteria, which may have an important role(s) as initiating/progressing factors in the pathogenesis of autoimmune disorder AIP, will be emphasized.
Autoimmune diseases arise from an overactive immune response of the body against tissues normally present in the body. Organ-specific autoimmune disease can develop through a combination of hereditary and environmental factors that regulate adaptive immune responses to self antigens (Mills, 2011). There are well-established links between infection and autoimmune diseases. Several studies have implicated microorganisms in the environmental etiology of autoimmune disorders based on observations such as, infection with Chlamydia pneumoniae, human herpesvirus 6, and Epstein–Barr virus (EBV) are triggered or exacerbated by multiple sclerosis (Mills, 2011). The regression of autoimmune thrombocytopenia is seen after the eradication of Helicobacter pylori (Gasbarrini et al., 1998). In Guillain–Barré syndrome (GBS), amino acid similarities exist between the gangliosides of the nerve system and the lipopolysaccharides (LPSs) of Campylobacter jejuni, suggesting that sensitization by microbes may be based on autoimmunity from molecular mimicry between bacteria and the targeted system of the host (Yuki et al., 2004; Houliston et al., 2011). The link has been attributed to either molecular mimicry between pathogen-derived antigens and self antigens or non-specific activation of innate immunity leading to a breakdown in immunological tolerance and the development of self antigen-specific T cell and antibody response (Mills, 2011). A common recent theory of the cause of autoimmune diseases is that an infectious agent triggers a cycle of events, which leads to the upregulation of the host immune response to self antigens (Aoki, 1999; Tlaskalová-Hogenova et al., 2004).
Autoimmune pancreatitis (AIP) is a putative autoimmune disease and is a chronically progressing inflammatory disease of the pancreas (Park et al., 2009; Okazaki et al., 2011a). The morphological characteristics of AIP include diffuse or localized enlargement of the pancreas and irregular narrowing of the main pancreatic duct. Histologically, the disease is also associated with progressive lymphoplasmacytic infiltration, predominantly localized to the ductal structures, and varying degrees of parenchymal and acinar destruction (Okazaki et al., 2008).
There are two types of AIP that differ in their clinical features, such as the gender ratio, mean age, and associated immune-related diseases. Type 1 AIP is associated with the histological finding of lymphoplasmacytic sclerosing pancreatitis (LPSP). Its serological hallmark is an elevation in the serum levels of the IgG4 subclass of IgG (Okazaki et al., 2011a). Type 1 AIP appears to be the pancreatic manifestation of a systemic disease called IgG4-associated systemic disease (ISD) or IgG4-related sclerosing disease, affecting not only the pancreas, but also other organs including the bile duct, retroperitoneum, kidney, lymph nodes, and salivary glands (Kamisaw and Okamoto, 2006; Okazaki et al., 2011b). Type 2 AIP is a form of idiopathic chronic pancreatitis (ICP), histologically associated with granulocyte-epithelial lesions (Shimosegawa and Kanno, 2009). The pathogenesis of AIP remains unknown. Genetic associations between susceptibility to the disease and the human leukocyte antigen (HLA) DRB1*0405-DQB1*0401 haplotype (Kawa et al., 2002; Muraki et al., 2006; Ota et al., 2007), Fc receptor-like gene 3 (FCRL3; Kojima et al., 2007), and the CTLA-4 gene (Umemura et al., 2008) have been suggested.
An outstanding finding in type 1 AIP is hypergammaglobulinemia and the existence of a high serum concentration for IgG4, which has been documented in 90% of patients (Hamano et al., 2001). This occurs in parallel to an abundant IgG4 positive plasma cell infiltration in the pancreatic tissue (Aoki et al., 2005). The fibroinflammatory process characterizing AIP occurs at the pancreatic basement membranes where IgG4/IgG/complement–immune complexes are deposited (Detlefsen et al., 2010). IgG1 is believed to be an opsonizing antibody and activates the complement classical pathway, namely opsonization of a bacterium by activated complement and antibodies. Combined opsonization by both complement and antibodies considerably enhances the uptake of the bacterium by the phagocyte (Wilson et al., 2011). AIP is occasionally associated with elevated circulating immune complex levels, which are significantly linked to increased serum IgG1 and complement activation via the classical pathway (Muraki et al., 2006). IgG4 is unable to activate the classical pathway of complements, but binds IgG1, 2, and 3 and forms an immune complex by Fc–Fc interaction in patients with AIP (Kawa et al., 2008). Although the role of IgG4 in the immune response and autoimmunity has not yet been fully elucidated, it may be hypothesized that IgG4 blocks the Fc-mediated effector functions of IgG1 and dampens the inflammatory response to an as yet unidentified primary trigger of the inflammatory process in AIP (Ito et al., 2010).
To which antigens the immunoglobulins in the deposits along the basal membranes of the pancreatic ducts and acini react remains unclear and definitive disease-specific antibodies have not been identified. However, several candidates for AIP-specific autoantibodies have been reported, such as anti-lactoferrin (LF; Okazaki et al., 2000), anti-carbonic anhydrase (CA)-II (Okazaki et al., 2000), anti-CA IV (Nishimori et al., 2005), anti-pancreatic secretory trypsin inhibitor (PSTI; Asada et al., 2006), anti-amylase-alpha (Endo et al., 2009), anti-heat shock protein (HSP) 10 (Takizawa et al., 2009), and anti-plasminogen-binding protein (PBP) peptide autoantibodies (Frulloni et al., 2009). These putative AIP-specific autoantibodies give us some hints toward considering the possibility that microorganisms are involved in the pathogenesis of AIP.
In order to survive in a host, a pathogenic microbe has to follow these steps; (i) attach to the host cells for colonization, (ii) evade the host’s innate and adaptive immune defense and persist in the host, (iii) obtain iron and other nutrients, (iv) disseminate or spread within a host, and (v) produce symptoms of disease in the host (although production of symptoms is not necessary; Wilson et al., 2011). Following the above mentioned processes, it might be valid to hypothesize that microbes are the pathogenic factors of AIP, based upon the possible roles of AIP-specific antibodies.
(i) LF: Considering the fact that LF is distributed in the ductal cells of several exocrine organs, including the pancreas, salivary gland, biliary duct, lungs, and renal tubules (Okazaki et al., 2011b), an anti-LF antibody might be induced as a consequence of the tissue damage caused by microbial infection and anti-LF might exert direct effects on these ductal cells of exocrine organs.
Iron is essential for the growth of bacteria. Most pathogenic bacteria require iron and thus there is a strong correlation between iron availability and virulence (Wilson et al., 2011). However bacteria have to cope with the fact that iron concentrations in nature are quite low. The concentration of free iron is particularly low in the host body due to the actions of host proteins, such as lactoferrin, transferring, ferritin, and heme, that bind most of the available iron. To survive in the body, bacteria must have some mechanism for acquiring this sequestered iron (Wilson et al., 2011).
LF is a glycoprotein, and a member of a transferrin family, thus belonging to those proteins capable of binding and transferring Fe3+ ions (Wilson et al., 2011). LF binds Fe3+ with an affinity and stability much higher than that of transferrin in the serum (Valenti and Antonini, 2005). LF is secreted from the exocrine glands and in specific granules of neutrophils, which are the main source of lactoferrin in blood plasma after degranulation. Neutrophils are at the front line of the innate immunity process (Wilson et al., 2011). LF binds iron released from transferrin, which prevents its further usage for bacterial proliferation. Besides iron, LF is capable of binding a large amount of other compounds and substances such as LPS, heparin, glycosaminoglycans, or other metal ions and certain DNA and RNA viruses. The concentration of LF in the blood increases during infection and inflammation (Adlerova et al., 2008). Since many bacteria synthesize and secrete small iron-chelating molecules (siderophores) that can compete with LF for insoluble Fe3+ ions, the growth-inhibiting activity of the protein is thus reversed. Iron sequestration by apo-LF, which is an iron-free form of LF, can effectively inhibit the growth of many bacterial species (Valenti and Antonini, 2005). LF has been related to the decreased survival of microorganisms in the host. LF combines with the bacterial surface through electrostatic and hydrophobic interactions, resulting in a perturbation of bacterial membranes. The molecular mechanisms of this bactericidal activity of LF, which is not related to iron withholding, appears to be similar for either Gram-negative or Gram-positive bacteria. Taking these characteristics of LF into consideration, blocking the LF function by an anti-LF antibody might be beneficial for microbes to get the essential growth element iron to survive and prolong persistent infection in the body of AIP patients. Assessing whether or not the anti-LF antibody obtained from AIP patients actually possesses the blocking function of the binding of Fe3+ ion to LF would be of interest.
(ii) HSP: Mammalian HSP10 and HSP60, also known as chaperonins 10 and 60, are mitochondrial proteins involved in protein folding. HSPs exported to the plasma membrane or released from dying cells are believed to be a source of “danger” signals, informing the innate and adaptive immune systems of tissue damage induced by various insults including infection, toxins, and cellular stress (Johnson et al., 2005; Shields et al., 2011).
It has not been clarified whether anti-HSP10 production is the cause or the consequence of AIP progression, but HSP10 consistently inhibits LPS-induced TLR4 signaling by interacting with HSP60 in the extracellular milieu and inhibits LPS-induced secretion of the pro-inflammatory cytokines, TNF-α, IL-6, and the pro-inflammatory chemokine regulated upon activation, normal T cell expressed and secreted (RANTES) and anti-inflammatory cytokine IL-10 (Johnson et al., 2005). From this point of view, it might be possible that the production of anti-HSP10 antibodies indicates the implication of bacteria in the pathogenesis of AIP.
(iii) Molecular mimicry: Homology between alpha-CA of H. pylori and human CA-II has been also reported. Guarneri et al. showed a significant homology between human CA-II and α-CA of H. pylori. Moreover, the homologous segments contained the binding motif of DRB1*0405 (Guarneri et al., 2005). Notably, the possession of the HLA-DRB1*0405-DQB1*0401 genotype confers a risk for AIP development (Ota et al., 2007). The anti-PBP peptide antibody was newly identified in patients with AIP. Frulloni et al. (2009) reported that 94% of AIP patients, but only 5% of pancreatic cancer patients, exhibit IgG antibodies to a PBP that is homologous to the human protein ubiquitin-protein ligase E3 component n-recognin 2 (UBR2), which is expressed in pancreatic acinar cells and is also homologous to the PBP of H. pylori. These data suggest that H. pylori infection may trigger AIP in genetically predisposed subjects through autoimmune responses triggered by molecular mimicry.
(iv) CAs: Immunization with CA-II or LF has been reported to induce systemic lesions such as pancreatitis, sialadenitis, cholangitis, and interstitial nephritis (Nishimori et al., 1995; Ueno et al., 1998). The distribution of CA-II and IV are both in the ductal cells of several exocrine organs, including the pancreas, salivary gland, biliary duct, lungs and renal tubules (Okazaki et al., 2011b). Nishimori et al. (2005) reported that serum antibodies to CAs I and II might be detected in patients with Sjögren’s syndrome and ICP perhaps as a consequence of the cross-reactivity of an antibody against another unknown antigen that mimics CAs I and II. Expression of CA IV is more restricted in tissues and cell types in the inflamed lesions that have been observed in patients with AIP and related diseases. It has been suggested that CA IV is more likely to function as the target antigen (Nishimori et al., 2005). CAs on the luminal surface of the ductal cells would eliminate excess acid from the ductal lumen, inhibiting spontaneous autoactivation of trypsin and other proteases (Nishimori et al., 1999), and therefore may contribute to attrition of bacterial killing by these enzymes in the pancreas.
(v) PSTI: PSTI, a 56-amino-acid peptide, is synthesized in pancreatic acinar cells, distributed in the ductal cells of several exocrine organs, including the pancreas, salivary gland, biliary duct, lungs, and renal tubules, and colocalizes with trypsinogen in zymogen granules. In addition to its protective role in acinar cells, PSTI inhibits activation of trypsinogen in the pancreatic duct (Asada et al., 2006). It might be beneficial for preventing degradation of bacterial pathogen(s). Additionally, it would be of interest to study whether or not the anti-PSTI antibody inhibits the function of PSTI.
Besides putative AIP-specific autoantibodies, other factors such as channel(s), transporters, and some of AIP associated genes also give us hints to consider the possible involvement of bacteria in the pathogenesis of AIP.
(vi) AQP1: Aquaporin (AQP) water channels are intrinsic membrane proteins expressed most of the cell types which have high osmotic water permeability. Among them AQP1 (Aquaporin 1) is a predominant water channel expressed in the plasma membranes of human pancreatic ducts (Ko et al., 2009). Altered tissue water homeostasis may contribute to edema formation during various stress including bacterial infection (Schweitzer et al., 2007). AQP1 indeed contributes to maintain water homeostasis even in cells with plasma membrane damage following insult with bacterial infection (Schweitzer et al., 2007). Interestingly, AQP1 expression was significantly increased in plasma membranes of pancreatic ducts in AIP (Ko et al., 2009). Upregulation of AQP1 expression seen in pancreatic ducts of patients with AIP is speculated to be caused by the reduced fluid secretion from the pancreas as compensation (Ko et al., 2009).
Membranes serve as a barrier to prevent pathogenic bacteria from entering the nutrient-rich host cytosol (Radtke and O’Riordan, 2008). Aquaporins may be an integral part of infection and pathology caused by some microbial pathogens, so regulation of aquaporin expression and function is a key aspect of host–pathogen interaction (Radtke and O’Riordan, 2008). In addition, AQP1 regulates swelling of secretory vesicles, associated with pathogen-containing vacuoles (PCV). AQP1-dependent modulation of PCV was triggered by bacterial induced membrane damage and ion flux (Radtke and O’Riordan, 2008). As described above, in the pancreas of AIP patients, expression of AQP1 was increased markedly, not only to on the plasma membrane but also in the cytoplasm of pancreatic duct cells. To clarify the association between the upregulation of AQP1 in the cytoplasm and PCV would be of interest.
(vii) CFTR: It was reported that aberrant cystic fibrosis transmembrane conductance regulator (CTFR) localization in the pancreatic ducts was observed in AIP patients. The CTFR regulates overall pancreatic ductal fluid/overall ion transport across most epithelia and plays a central role in pancreatic ductal  secretion (Ko et al., 2010). Altered acidity of the pancreatic fluid may affect growth of the normal bacterial inhabitant and immunity of the upper intestinal environment.
secretion (Ko et al., 2010). Altered acidity of the pancreatic fluid may affect growth of the normal bacterial inhabitant and immunity of the upper intestinal environment.
(viii) ABCF1: The ABCF1 (ATP-binding cassette, sub-family F) gene proximal to C3-2-11 microsatellite in HLA class I regions, is thought to be one of the HLA-linked susceptibility regions for AIP (Ota et al., 2007). At this point, biological function of the putative ABCF1 protein is mostly speculative, however it is thought to be regulated by TNF-α, a prime cytokine in inflammatory reactions (Ota et al., 2007). From this point, it could be possible to suspect that bacteria would lie in the pathogenesis of AIP.
Several experimental models of AIP have been described (Table 1). As for specific induction models, CA-II, LF, and amylase-specific T cells could be listed (Nishimori et al., 1995; Uchida et al., 2002; Davidson et al., 2005). As for the microorganisms related induction group, virus-induced AIP models, such as C57BL/6 mice infected with the murine leukemia retrovirus LP-BM5, developed histological findings similar to human AIP (Suzuki et al., 1993; Watanabe et al., 2003). The spontaneous development of pancreatitis via an autoimmune mechanism in MRL/Mp mice is accelerated by the administration of polyinosinic:polycytidylic acid (poly I:C), a synthetic double-stranded RNA and TLR 3 ligand (Qu et al., 2002; Soga et al., 2009; Asada et al., 2010; Nishio et al., 2011). Sensitization occurs with not only viral components, such as double-stranded RNA poly I:C, but also bacterial LPS in interleukin (IL)-10-deficient mice (Nishio et al., 2011). TLRs play important roles in innate immunity by initiating intracellular signaling to macrophages and dendritic cells after stimulation with various antigens. The majority of known TLRs mediate the development of Th1 cell-inducing dendritic cells (Li et al., 2009). Thus, pattern-recognition receptors (PRRs) that bind pathogen-associated molecular patterns (PAMPs) may trigger an autoimmune response.
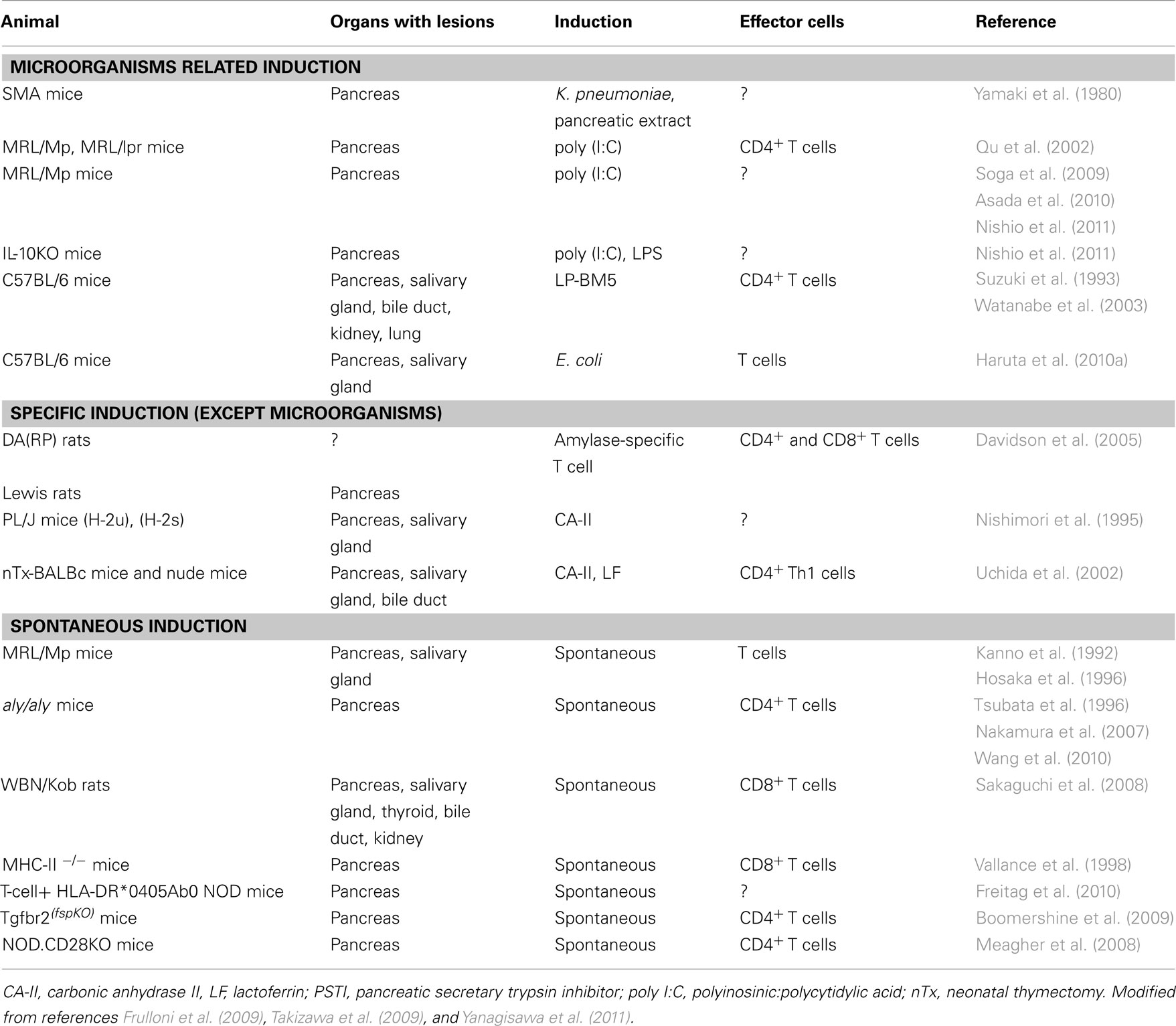
Table 1. Experimental animal models of autoimmune pancreatitis.
As for specific induction, thymectomized mice immunized with CA-II or LF develop a pathology that closely resembles AIP under a regulatory T (Treg) cell-depleted background (Uchida et al., 2002). However, T cells specific for CA-II and LF were unable to induce pancreatitis in the adoptive transfer of an amylase-specific rat model (Davidson et al., 2005), suggesting that autoantibodies against these enzymes in AIP represent a late consequence of tissue destruction.
As for the spontaneous induction group, recently, the spontaneous development of AIP in CD4+ T cell-competent HLA-DR*0405 transgenic Ab0 NOD mice (Freitag et al., 2010), Treg-deficient backgrounds in neonatally thymectomized mice (Uchida et al., 2002), NOD.CD28 knockout mice (Meagher et al., 2008), WBN/Kob rats (Sakaguchi et al., 2008), and Tgfbr2fsp knockout mice (Boomershine et al., 2009) have demonstrated genetic polymorphisms of the effector cells in the etiologies of AIP. However, because these mice are genetically engineered they may not completely reflect the onset of human diseases.
Instances of where microbial antigens might underlie the pathogenesis of IgG4-related disease have been reported (Akitake et al., 2010; Watanabe et al., 2012). We previously reported that when C57BL/6 mice were inoculated intraperitoneally (i.p.) with heat-killed E. coli weekly for 8 weeks, marked cellular infiltration with fibrosis was observed in the exocrine pancreas accompanied by a high serum gamma globulin level and the production of autoantibodies against CA-II and LF. Bacterial infection apparently triggered AIP-like pathological alterations in mice that strikingly resembled AIP in humans (Haruta et al., 2010a).
C57BL/6 mice inoculated weekly with E. coli for 8 weeks were utilized as donors, and the spleens were intravenously transferred to RAG2−/− mice. The pancreas in the recipient RAG2−/− mice showed cellular infiltration in the exocrine pancreas, especially around the pancreatic ducts, indicating that the E. coli-inoculated mouse spleen cells possess the ability to reproduce pathological alterations in the pancreas of naïve mice. Similarly, when the spleen cells of donor S. intermedius-inoculated mice were transferred to RAG2−/− mice, primary biliary cirrhosis (PBC)-like cholangitis in the liver was induced, similar to that seen in the donor (Haruta et al., 2010b). The AIP-like inflammatory region in the pancreases of recipient mice with spleen cells transferred from E. coli-inoculated mice (Haruta et al., 2010a), showed that most of the cellular infiltrates in the target organs were CD3-positive, indicating that these cells in both models originated from the donor mice.
The criteria for determining whether a condition may be considered to be autoimmune, according to Witebsky’s postulates with modern revision by Rose and Bona (1993), include (i) indirect evidence based on the reproduction of the autoimmune disease in experimental animals, (ii) direct evidence of the transfer of pathogenic antibodies, or (iii) pathogenic T cells and indirect evidence of the isolation of autoantibodies or autoreactive T cells. Several lines of evidence have suggested that repeated E. coli-inoculated AIP-like inflammation in the C57BL/6 mice pancreas is possibly of autoimmune origin.
It is worth mentioning that long before the concept of AIP was proposed as a definite disease entity, Yamaki et al. (1980) had established inflammation of the exocrine pancreas using syngeneic pancreatic antigens. Repeated injection of pancreatic extract from syngeneic mice mixed with capsular polysaccharide (CPS) of the Klebsiella pneumoniae type 1 Kasuya strain (CPS-K), as an adjuvant, caused pathological alteration in the pancreas, consistent with infiltration by lymphocytes, plasma cells and mononuclear cells, degradation and lysis of the acinar cells, and destruction of the lobular architecture, replacement of fatty tissue and fibrous connective tissue and anti-pancreas antibody production. No histological changes were observed in tissues other than the pancreas and either CPS-K or the pancreatic extract alone were insufficient to induce inflammation in the pancreas. K. pneumoniae is one of the commonly found bacteria in Enterobacteriaceae (Yamaki et al., 1980), the natural habitat of the intestinal tract, and it is an opportunistic bacterium. The CPS is a voluminous outer layer and is involved in protection against complement deposition that occurs mainly in the inhibition of macrophage phagocytosis (Evrard et al., 2010). It is of great interest that inflammation had been induced only when the syngeneic pancreatic antigen and bacterial component both existed, which suggest the possibility that the bacterial component may exhibit an adjuvant effect to the antigen.
It is currently believed that the microbiota is essential for the development of a functional immune system and the gut microbiota has a far-reaching influence on the immune system, beyond the gastrointestinal tract, i.e., within and outside the gut. In other words, gut microbiota has a greater influence on the systemic immune system than previously anticipated (Kranich et al., 2011). From the aspect of gut microbiota and autoimmunity, several animal models have been reported. For instance, it was reported that the relationship between certain harmful species of gut bacteria affect T cell populations in the periphery and thereby control the development of autoimmune arthritis. Segmented filamentous bacteria could promote experimental autoimmune encephalomyelitis (Kranich et al., 2011).
We propose a hypothetical pathogenesis of bacteria-induced AIP. Under normal conditions, commensal bacteria are not pathogenic and may exist in the host from early life. However, additional factor(s) such as genetic factors including the haplotype of class II antigen of the major histocompatibility complex (MHC) and so on would be synchronized with the existence of microorganisms, and the progression switch to generate the status of AIP would be turned on. During the initiation phase, infection with microorganisms possessing PAMPs and/or non-pathogenic microorganisms associated molecular patterns (MAMPs) trigger and upregulate the innate immune system. In this step, we are especially focusing on the commensal bacteria. Inflammatory T cells are induced by molecules derived from pathogens or commensal microorganisms, as well as by endogenous stress-induced self molecules. These respective MAMPs and damage-associated molecular patterns (DAMPs) induce inflammatory T cells either indirectly, through the induction of pro-inflammatory cytokine production by binding to pattern recognition receptors (PRRs) on innate immune cells, or directly, by binding to pathogen-derived peptide specific receptors on T cells (Mills, 2011).
Second, the progressive phase features the persistence of this PAMPs/MAMPs attack, stimulation by molecular-mimicking antigen, perhaps released as DAMPs, exposure to or stimulation from commensal flora possessing the same antigenic epitope, and/or non-specific activation of innate immunity. These slowly progressive steps lead to a breakdown in immunological tolerance and the development of self antigen-specific T cell and antibody responses (Mills, 2011) and eventually establish the autoimmune diseases. Throughout these processes, TLRs might participate as the key member of PRRs involved in driving autoimmune inflammation (Mills, 2011; Figure 1). Taking these considerations together, the pancreas is the field of the specific inflammation which would be triggered by the interaction with microorganisms, and a provider of the autoantigen to induce the production of autoantibodies. These sequential processes would be orchestrated to constitute the status of AIP.
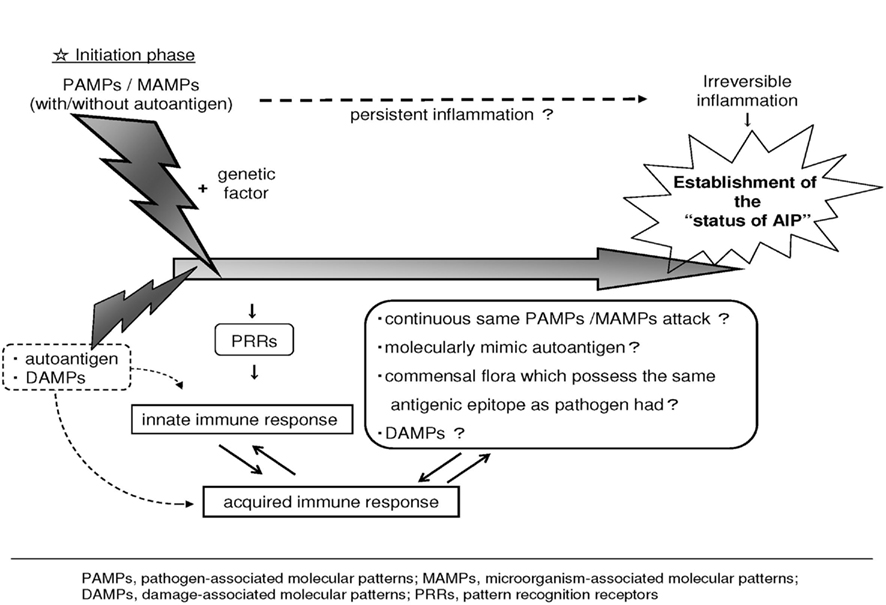
Figure 1. Hypothetical pathogenesis of AIP. Under normal condition, commensal bacteria are not pathogenic and may exist in the host from early life. However, additional factor(s) such as genetic factors would be synchronized with the existence of microorganisms, and the progression switch to generate the status of AIP would be turned on. During the initiation phase, PAMPs and/or non-pathogenic MAMPs trigger and upregulate the innate immune system. These respective PAMPs/MAMPs and DAMPs induce inflammatory response PRRs. Second, the progressive phase features the persistence of this PAMPs/MAMPs attack or stimulation by a molecular-mimicking antigen, perhaps released as DAMPs and/or exposure to or stimulation from commensal flora possessing the same antigenic epitope that the initial pathogen possessed, thereby upregulating the host immune response to the target antigen. These slowly progressive steps eventually lead to the development of autoimmune diseases. Modified from previous reports (Haruta et al., 2010a; Yanagisawa et al., 2011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was financially supported in part by the Grant-in-Aid for Scientific Research (C23590522 to Ikuko Haruta) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and The Takako Satake Scientific Award (2011) and The 28th Itoe Okamoto Scientific Award (2011; to Naoko Yanagisawa), Shiseikai, Tokyo Women’s Medical University, Tokyo, Japan.
AIP, autoimmune pancreatitis; ANAs, antinuclear antibodies; CA-II, carbonic anhydrase II; CPS, capsular polysaccharide; CTLA-4, cytotoxic T-lymphocyte antigen-4; DAMPs, damage-associated molecular patterns; FCRL3, Fc receptor-like gene 3; GBS, Guillain–Barré syndrome; GEL, granulocyte-epithelial lesions; HLA, human leukocyte antigen; HSP, heat shock protein; IDCP, idiopathic duct-centric pancreatitis; LF, lactoferrin; LPS, lipopolysaccharide; LPSP, lymphoplasmacytic sclerosing pancreatitis; MAMPs, microorganism-associated molecular patterns; PAMP, pathogen-associated molecular patterns; PBC, primary biliary cirrhosis; PBP, plasminogen-binding protein; poly I:C, polyinosinic:polycytidylic acid; PRR, pattern-recognition receptor; Treg, regulatory T cell; TGF, transforming growth factor; TLR, toll-like receptor; UBR2, ubiquitin-protein ligase E3 component N-recognin 2.
Adlerova, L., Bartoskova, A., and Faldyna, M. (2008). Lactoferrin: a review. Vet. Med. (Praha) 9, 457–468.
Akitake, R., Watanabe, T., Zaima, C., Uza, N., Ida, H., Tada, S., Nishida, N., and Chiba, T. (2010). Possible involvement of T helper type 2 responses to Toll-like receptor ligands in IgG4-related sclerosing disease. Gut 59, 542–545.
Aoki, S., Nakazawa, T., Ohara, H., Sano, H., Nakao, H., Joh, T., Murase, T., Eimoto, T., and Itoh, M. (2005). Immunohistochemical study of autoimmune pancreatitis using anti-IgG4 antibody and patients’ sera. Histopathology 47, 147–158.
Asada, M., Nishio, A., Akamatsu, T., Tanaka, J., Saga, K., Kido, M., Watanabe, N., Uchida, K., Fukui, T., Okazaki, K., and Chiba, T. (2010). Analysis of humoral immune response in experimental autoimmune pancreatitis in mice. Pancreas 39, 224–231.
Asada, M., Nishio, A., Uchida, K., Kido, M., Ueno, S., Uza, N., Kiriya, K., Inoue, S., Kitamura, H., Ohashi, S., Tamaki, H., Fukui, T., Matsuura, M., Kawasaki, K., Nishi, T., Watanabe, N., Nakase, H., Chiba, T., and Okazaki, K. (2006). Identification of a novel autoantibody against pancreatic secretory trypsin inhibitor in patients with autoimmune pancreatitis. Pancreas 33, 20–26.
Boomershine, C. S., Chamberlain, A., Kendall, P., Afshar-Sharif, A. R., Huang, H., Washington, M. K., Lawson, W. E., Thomas, J. W., Blackwell, T. S., and Bhowmick, N. A. (2009). Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut 58, 1267–1274.
Davidson, T. S., Longnecker, D. S., and Hickey, W. F. (2005). An experimental model of autoimmune pancreatitis in the rat Am. J. Pathol. 166, 729–736.
Detlefsen, S., Brasen, J. H., Zamboni, G., Capelli, P., and Kloppel, G. (2010). Deposition of complement C3c, immunoglobulin (Ig)G4 and IgG at the basement membrane of pancreatic ducts and acini in autoimmune pancreatitis. Histopathology 57, 825–835.
Endo, T., Takizawa, S., Tanaka, S., Takahashi, M., Fujii, H., Kamisawa, T., and Kobayashi, T. (2009). Amylase alpha-2A autoantibodies: novel marker of autoimmune pancreatitis and fulminant type 1 diabetes. Diabetes 58, 732–737.
Evrard, B., Balestrino, D., Dosgilbert, A., Bouya-Gachancard, J.-L. J., Charbonnel, N., Forestier, C., and Tridon, A. (2010). Roles of capsule and lipopolysaccharide O antigen in infections of human monocyte-derived dendritic cells and Klebsiella pneumoniae. Infect. Immun. 78, 210–219.
Freitag, T. L., Cham, C., Sung, H. H., Beihack, G. F., Durinovic-Bello, I., Pastel, S. D., Bronson, R. T., Schuppan, D., and Sonderstrup, G. (2010). Human risk allele HLA-DRB1*0405 predisposes class II transgenic Ab0 NOD mice to autoimmune pancreatitis. Gastroenterology 139, 281–291.
Frulloni, L., Lunardi, C., Simone, R., Dolcino, M., Scattoni, C., Falconi, M., Benini, L., Vantini, I., Corrocher, R., and Puccetti, A. (2009). Identification of a novel antibody associated with autoimmune pancreatitis. N. Eng. J. Med. 361, 2135–2142.
Gasbarrini, A., Franceschi, F., Tartaglione, R., Landolfi, R., Pola, P., and Gasbarrini, G. (1998). Regression of autoimmune thrombocytopenia after eradication of Helicobacter pylori. Lancet 352, 878.
Guarneri, F., Guarneri, C., and Benvenga, S. (2005). Helicobacter pylori and autoimmune pancreatitis: role of carbonic anhydrase via molecular mimicry? J. Cell. Mol. Med. 9, 741–744.
Hamano, H., Kawa, S., Horiuchi, A., Unno, H., Furuya, N., Akamatsu, T., Fukushima, M., Nikaido, T., Nakayama, K., Usuda, N., and Kiyosawa, K. (2001). High serum IgG4 concentrations in patients with sclerosing pancreatitis. N. Eng. J. Med. 344, 732–738.
Haruta, I., Yanagisawa, N., Kawamura, S., Furukawa, T., Shimizu, K., Kato, H., Kobayashi, M., Shiratori, K., and Yagi, J. (2010a). A mouse model of autoimmune pancreatitis with salivary gland involvement triggered by innate immunity via persistent exposure to avirulent bacteria. Lab. Invest. 90, 1757–1769.
Haruta, I., Kikuchi, K., Hashimoto, E., Makamura, M., Miyakawa, H., Hirota, K., Shibata, N., Kato, H., Arimura, Y., Kato, Y., Uchiyama, T., Nagamune, H., Kobayashi, M., Miyake, Y., Shiratori, K., and Yagi, J. (2010b). Long-term bacterial exposure can trigger nonsuppurative destructive cholangitis associated with multifocal epithelial inflammation. Lab. Invest. 90, 577–588.
Hosaka, N., Nose, M., Kyogoku, M., Nagata, N., Miyashima, S., Good, R. A., and Ikehara, S. (1996). Thymus transplantation, a critical factor for correction of autoimmune disease in aging MRL/+ mice. Proc. Natl. Acad. Sci. U.S.A. 93, 8558–8562.
Houliston, R. S., Vinogradov, E., Dzieciatkowska Jianjun, M., Li, J., St. Michael, F., Karwaski, M. F., Brochu, D., Jarrell, H. D., Parker, C. T., Yuki, N., Mandrell, R. E., and Gilbert, M. (2011). Lipooligosaccharide of Campylobacter jejuni: similarity with multiple types of mammalian glycans beyond gangliosides. J. Biol. Chem. 286, 12361–12370.
Ito, T., Kitahara, K., Umemura, T., Ota, M., Shimozuru, Y., Kawa, S., and Bahram, S. (2010). A novel heterophilic antibody interaction involves IgG4. Scand. J. Immunol. 71, 109–114.
Johnson, B. J., Le, T. T., Dobbin, C. A., Banovic, T., Howard, C. B., Flores, F. M., Vanags, D., Naylor, D. J., Hill, G. R., and Suhrbier, A. (2005). Heat shock protein 10 inhibits lipopolysaccharide-induced inflammatory mediator production. J. Biol. Chem. 280, 4037–4047.
Kamisaw, T., and Okamoto, A. (2006). Autoimmune pancreatitis: proposal of IgG4-related sclerosing disease. J. Gastroenterol. 41, 613–625.
Kanno, H., Nose, M., Itoh, J., Taniguchi, Y., and Kyogoku, M. (1992). Spontaneous development of pancreatitis in the MRL/Mp strain of mice in autoimmune mechanism. Clin. Exp. Immunol. 89, 68–73.
Kawa, S., Kitahara, K., Hamano, H., Ozaki, Y., Arakura, N., Yoshizawa, K., Umemura, T., Ota, M., Mizoguchi, S., Shimozuru, Y., and Bahram, S. (2008). A novel immunoglobulin-immunoglobulin interaction in autoimmunity. PLoS ONE 3, e1637. doi:10.1371/journal.pone.0001637
Kawa, S., Ota, M., Yoshizawa, K., Horiuchi, A., Hamano, H., Ochi, Y., Nakayama, K., Tokutake, Y., Katsuyama, Y., Saito, S., Hasebe, O., and Kiyasawa, K. (2002). HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology 122, 1264–1269.
Ko, S. B., Mizuno, N., Yatabe, Y., Yoshikawa, T., Ashiguro, H., Yamamoto, A., Azuma, S., Naruse, S., Yamao, K., Muallem, S., and Goto, H. (2010). Corticosteroids correct aberrant CFTR localization in the duct and regenerate acinar cells in autoimmune pancreatitis. Gastroenterology 138, 1988–1996.
Ko, S. B. H., Mizuno, N., Yatabe, Y., Yoshikawa, T., Ishiguro, H., Yamamoto, A., Azuma, S., Naruse, S., Yamano, K., Muallem, S., and Goto, H. (2009). Aquaporin 1 water channel is overexpressed in the plasma membranes of pancreatic ducts in patients with autoimmune pancreatitis. J. Med. Invest. 56, 318–321.
Kojima, M., Sipos, B., Klapper, W., Frahm, O., Knuth, H. C., Yanagisawa, A., Zamboni, G., Morohoshi, T., and Kloppel, G. (2007). Autoimmune pancreatitis: frequency, IgG4 expression, and clonality of T and B cells. Am. J. Surg. Pathol. 31, 521–528.
Kranich, J., Maslowski, K. M., and Macky, C. R. (2011). Commensal flora and regulation of inflammatory and autoimmune responses. Semin. Immunol. 23, 139–145.
Li, M., Zhou, Y., Feng, G., and Su, S. B. (2009). The critical role of Toll-like receptor signaling pathways in the induction and progression of autoimmune diseases. Curr. Mol. Med. 9, 365–374.
Meagher, C., Tang, Q., Fife, B. T., Bour-Jordan, H., Wu, J., Pardoux, C., Bi, M., Melli, K., and Bluestone, J. A. (2008). Spontaneous development of a pancreatic exocrine disease in CD28-deficient NOD mice. J. Immunol. 180, 7793–7803.
Mills, K. H. (2011). TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822.
Muraki, T., Hamano, H., Ochi, Y., Komatsu, K., Komiyama, Y., Arakawa, N., Yoshizawa, K., Ota, M., Kawa, S., and Kiyosawa, K. (2006). Autoimmune pancreatitis and complement activation system. Pancreas 32, 16–21.
Nakamura, Y., Yi, S. Q., Terayama, H., Naito, M., Li, J., Moriyama, H., Tsuchida, A., and Itoh, M. (2007). Sequential histopathology of pancreatic tissues in aly/aly mice. Cells Tissues Organs (Print) 86, 204–209.
Nishimori, I., Bratanova, T., Toshkov, I., Caffrey, T., Mogaki, M., Shibata, Y., and Hollingsworth, M. A. (1995). Induction of experimental autoimmune sialadenitis by immunization of PL/J mice with carbonic anhydrase II. J. Immunol. 154, 4865–4873.
Nishimori, I., Fujikawa-Adachi, K., Onishi, S., and Hollingsworth, M. M. (1999). Carbonic anhydrase in human pancreas: hypothesis for the pathophysiological roles of CA isozymes. Ann. N. Y. Acad. Sci. 880, 5–16.
Nishimori, I., Miyaji, E., Morimoto, K., Nagao, K., Kamada, M., and Onishi, S. (2005). Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis, Gut 54, 274–281.
Nishio, A., Asada, M., Uchida, K., Fukui, T., Chiba, T., and Okazaki, K. (2011). The role of innate immunity in the pathogenesis of experimental autoimmune pancreatitis in mice. Pancreas 40, 95–102.
Okazaki, K., Uchida, K., and Fukui, T. (2008). Recent advances in autoimmune pancreatitis: concept, diagnosis, and pathogenesis. J. Gastroenterol. 43, 409–418.
Okazaki, K., Uchida, K., Miyoshi, H., Ikeura, T., Takaoka, M., and Nishio, A. (2011a). Recent concepts of autoimmune pancreatitis and IgG4-related disease. Clin. Rev. Allergy Immunol. 41, 126–138.
Okazaki, K., Uchida, K., Koyabu, M., Miyoshi, H., and Takaoka, M. (2011b). Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J. Gastroenterol. 46, 277–288.
Okazaki, K., Uchida, K., Ohana, M., Nakase, H., Uose, S., Inai, M., Matsushima, Y., Katamura, K., Ohmori, K., and chiba, T. (2000). Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 118, 573–581.
Ota, M., Katsuyama, Y., Hamano, H., Umemura, T., Kimura, A., Yoshizawa, K., Kiyosawa, K., Fukushima, H., Bahram, S., Inoko, H., and Kawa, S. (2007). Two critical genes (HLA-DRB1 and ABCF1)in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics 59, 45–52.
Park, D. H., Kim, M. H., and Chari, S. T. (2009). Recent advances in autoimmune pancreatitis. Gut 58, 1680–1689.
Qu, W. M., Miyazaki, T., Terada, M., Okada, K., Mori, S., Kanno, H., and Nose, M. (2002). A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic:polycytidylic acid. Clin. Exp. Immunol. 129, 27–34.
Radtke, A., and O’Riordan, M. X. D. (2008). Homeostatic maintenance of pathogen-containing vacuoles requires TBK1-dependent regulation of aquaporin-1. Cell. Microbiol. 10, 2197–2207.
Rose, N. R., and Bona, C. (1993). Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol. Today 14, 426–430.
Sakaguchi, Y., Inaba, M., Tsuda, M., Quan, M., Ando, Y., Uchida, K., Okazaki, K., and Ikehara, S. (2008). The Wistar Bonn Kobori rat, a unique animal model for autoimmune pancreatitis with extrapancreatic exocrinopathy. Clin. Exp. Immunol. 152, 1–12.
Schweitzer, K., Li, E., Sidhaye, T., Leitch, V., Kuznetsov, S., and King, L. S. (2007). Accumulation of aquaporin-1 during hemolysin-induced necrotic cell death. Cell. Mol. Biol. Lett. 13, 195–211.
Shields, A. M., Thompson, S. J., Panayi, G. S., and Corrigall, V. M. (2011). Pro-resolution immunological networks: binding immunoglobulin protein, and other resolution-associated molecular patterns. Reumatology. doi:10.1093/rheumatology/ker412. [Epub ahead of print].
Shimosegawa, T., and Kanno, A. (2009). Autoimmune pancreatitis in Japan: overview and perspective. J. Gastroenterol. 44, 503–517.
Soga, Y., Komori, H., Miyazaki, T., Arita, N., Terada, M., Kamada, K., Tanaka, Y., Fujino, T., Hiasa, Y., Matsuura, B., Onji, M., and Nose, M. (2009). Toll-like receptor 3 signaling induces chronic pancreatitis through the Fas/Fas ligand-mediated cytotoxicity. Tohoku J. Exp. Med. 217, 175–184.
Suzuki, K., Makino, M., Okada, Y., Kinoshita, J., Yui, R., Kanazawa, H., Asakura, H., Fujiwara, M., Mizouchi, T., and Komura, K. (1993). Exocrinopathy resembling Sjogren’s syndrome induced by a murine retrovirus. Lab. Invest. 69, 430–435.
Takizawa, S., Endo, T., Wanjia, X., Tanaka, S., Takahashi, M., and Kobayashi, T. (2009). HSP 10 is a new autoantigen in both autoimmune pancreatitis and fulminant type 1 diabetes. Biochem. Biophys. Res. Commun. 386, 192–196.
Tlaskalová-Hogenova, H., Štepánková, R., Hudcovic, T., Tuckova, L., Tucková, L., Cukrowska, B., Lodinová-Zádníková, R., Kozáková, H., Rossmann, P., Bártová, J., Sokol, D., Funda, D. P., Borovská, D., Reháková, Z., Sinkora, J., Hofman, J., Drastich, P., and Kokesová, A. (2004). Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol. Lett. 93, 97–108.
Tsubata, R., Tsubata, T., Hiai, H., Shinkura, R., Matsuura, R., Sumida, T., Miyawaki, S., Ishida, H., Kumagai, S., Nakao, K., and Honjo, T. (1996). Autoimmune disease of exocrine organs in immunodeficient alymphoplasia mice: a spontaneous model for Sjogren’s syndrome. Eur. J. Immunol. 26, 2742–2748.
Uchida, K., Okazaki, K., Nishi, T., Uose, S., Nakase, H., Ohana, M., Matsushima, Y., Omori, K., and Chiba, T. (2002). Experimental immune-mediated pancreatitis in neonatally thymectomized mice immunized with carbonic anhydrase II and lactoferrin. Lab. Invest. 82, 411–424.
Ueno, Y., Ishii, M., Takahashi, S., Igarashi, T., Toyota, T., and LaRusso, N. F. (1998). Different susceptibility of mice to immune-mediated cholangitis induced by immunization with carbonic anhydrase II. Lab. Invest. 78, 629–637.
Umemura, T., Ota, M., Hamano, H., Katsuyama, Y., Muraki, T., Arakura, N., and Kiyosawa, K. (2008). Association of autoimmune pancreatitis with cytotoxic T-lymphocyte antigen 4 gene polymorphisms in Japanese patients. Am. J. Gastroenterol. 103, 588–594.
Valenti, P., and Antonini, G. (2005). Lactoferrin: an important host defence against microbial and vial attack. Cell. Mol. Life Sci. 62, 2576–2587.
Vallance, B. A., Hewlett, B. R., Snider, D. P., and Collins, S. M. (1998). T cell-mediated exocrine pancreatic damage in major histocompatibility complex class II-deficient mice. Gastroenterology 115, 978–987.
Wang, H. X., Yi, S. Q., Li, J., Terayama, H., Naito, M., Hirai, S., Qu, N., and Itoh, M. (2010). Effects of splenectomy on spontaneously chronic pancreatitis in aly/aly mice. Clin. Dev. Immunol. 614890, 1–8.
Watanabe, S., Suzuki, K., Kawauchi, Y., Yamagiwa, S., Yoneyama, H., Kawachi, H., Okada, Y., Shimizu, F., Asakura, H., and Aoyagi, Y. (2003). Kinetic analysis of the development of pancreatic lesions in mice infected with a murine retrovirus. Clin. Immunol. 109, 212–223.
Watanabe, T., Yamashita, K., Fujikawa, S., Sakurai, T., Kudo, M., Shiokawa, M., Kodama, Y., Uchida, K., Okazaki, K., and Chiba, T. (2012). Activation of Toll-like receptors and NOD-like receptors is involved in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 64, 914–924.
Wilson, B. A., Salyers, A. A., Whitt, D. D., and Winkler, M. E. (eds). (2011). Bacterial Pathogenesis. A Molecular Approach, 3rd Edn. Washington DC: ASM press, 38–42, 193–224.
Yamaki, K., Ohta, M., Nakashima, I., Noda, A., Asai, J., and Kato, N. (1980). Microbial adjuvant and autoimmunity. IV. Production of lesions in the exocrine pancreas of mice by repeated injection of syngeneic pancreatic extract together with the capsular polysaccharide of Klebsiella pneumoniae. Microbiol. Immunol. 24, 945–956.
Yanagisawa, N., Haruta, I., Kikuchi, K., Shibata, N., and Yagi, J. (2011). Are dysregulated inflammatory responses to commensal bacteria involved in the pathogenesis of hepatobiliary-pancreatic autoimmune disease? An analysis using mice models of primary biliary cirrhosis and autoimmune pancreatitis. ISRN Gastroenterol. 2011, 513514.
Yuki, N., Susuki, K., Koga, M., Nishimoto, Y., Odaka, M., Hirata, K., Taguchi, K., Miyatake, T., Furukawa, K., Kobata, T., and Yamada, M. (2004). Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barre syndrome. Proc. Natl. Acad. Sci. U.S.A. 101, 11404–11409.
Keywords: autoimmune pancreatitis, bacteria, innate immunity, autoimmune disease, commensal flora
Citation: Haruta I, Shimizu K, Yanagisawa N, Shiratori K and Yagi J (2012) Commensal flora, is it an unwelcomed companion as a triggering factor of autoimmune pancreatitis? Front. Physio. 3:77. doi: 10.3389/fphys.2012.00077
Received: 21 February 2012; Paper pending published: 26 February 2012;
Accepted: 16 March 2012; Published online: 02 April 2012.
Edited by:
Atsushi Masamune, Tohoku University Graduate School of Medicine, JapanReviewed by:
Akira Andoh, Shiga University of Medical Science, JapanCopyright: © 2012 Haruta, Shimizu, Yanagisawa, Shiratori and Yagi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kyoko Shimizu, Department of Gastroenterology and Medicine, Tokyo Women’s Medical University, 8-1 Kawada-cho, Shinjuku-ku, Tokyo 162-8666, Japan. e-mail:a3lva29AaWdlLnR3bXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.