 Steven B. Roberts* and Mackenzie R. Gavery
Steven B. Roberts* and Mackenzie R. Gavery
- School of Aquatic and Fishery Sciences, University of Washington, Seattle, WA, USA
There is a significant amount of variation in DNA methylation characteristics across organisms. Likewise, the biological role of DNA methylation varies across taxonomic lineages. The complexity of DNA methylation patterns in invertebrates has only recently begun to be characterized in-depth. In some invertebrate species that have been examined to date, methylated DNA is found primarily within coding regions and patterning is closely associated with gene function. Here we provide a perspective on the potential role of DNA methylation in these invertebrates with a focus on how limited methylation may contribute to increased phenotypic plasticity in highly fluctuating environments. Specifically, limited methylation could facilitate a variety of transcriptional opportunities including access to alternative transcription start sites, increasing sequence mutations, exon skipping, and transient methylation.
Epigenetics refers to processes capable of inducing changes in genetic activity without altering the underlying DNA sequence (Jablonka and Lamb, 2002). Histone modifications, DNA methylation, and non-coding RNA activity (e.g., miRNA) are the most commonly described epigenetic mechanisms. DNA methylation is one of the most studied mechanisms of epigenetic regulation and refers to the addition of a methyl group to position 5 of cytosine bases. DNA methylation is presumed to be evolutionarily ancient, and, while the mark itself is prevalent across taxa, the landscape of methylation patterning is incredibly diverse.
DNA methylation has been well-studied in mammals and plants, however surprisingly little is known about this mechanism in invertebrates. Recent research characterizing DNA methylation in a handful of species is providing evidence that the absence of DNA methylation could contribute to phenotypic plasticity by increasing the number of transcriptional opportunities. Evidence of a relationship between methylation patterns and transcriptional opportunities is found primarily in studies on the mollusk, Crassostrea gigas, and the eusocial insect Apis mellifera. Here we discuss this perspective and supporting research with a particular focus on the adaptive potential this phenomenon could have on species in highly fluctuating environments. In order to provide a broad view we first outline taxonomic trends in DNA methylation patterns and describe gene-associated DNA methylation characteristics in the limited number of invertebrate species where this has been examined. Molecular mechanisms that likely contribute to phenotypic plasticity are discussed followed by a summary of fundamental questions with respect to DNA methylation in invertebrates that remain unanswered.
DNA Methylation Patterns
The relative amount of DNA methylation varies significantly across taxa. In vertebrates, ∼70–80% of cytosines in CpG dinucleotides are methylated (Bird and Taggart, 1980), a pattern referred to as global methylation. In contrast, invertebrates display a wide range of DNA methylation. In fact, two common model organisms (Drosophila melanogaster and Caenorhabditis elegans) essentially lack DNA methylation (Simpson et al., 1986; Gowher et al., 2000). Other invertebrates have an intermediate level of methylation, including sea urchins (Strongylocentrotus purpuratus; Bird et al., 1979), sea squirts (Ciona intestinalis; Simmen and Bird, 2000; Suzuki et al., 2007), honey bees (A. mellifera; Lyko et al., 2010), and oysters (C. gigas; Gavery and Roberts, 2010). Among plants, not all species studied have methylated genomes, and related species can exhibit varying degrees of methylation. For example, a global methylation pattern is observed in maize (Zea mays; Palmer et al., 2003), whereas an intermediate level, similar to that seen in invertebrates, has been reported for Arabidopsis thaliana (Zhang et al., 2006).
The location of DNA methylation across the genome is also diverse among taxa. In vertebrates, the limited amount of the genome that is not methylated is often found in CpG rich gene promoter regions called CpG islands. Gene bodies are typically methylated in vertebrates, though the degree of methylation decreases in 5′ and 3′ regions. In invertebrates, tracts of methylated CpGs are interspersed with unmethylated regions across the genome, referred to as a mosaic pattern (Suzuki et al., 2007). Another example of spatial heterogeneity is the predominance of methylation in exons. This phenomenon has been observed in A. mellifera (Lyko et al., 2010), C. intestinalis (Suzuki et al., 2007), and C. gigas (Gavery and Roberts, 2010). This is in contrast to the blood fluke (Schistosoma mansoni) where methylation has been found in a highly repetitive intronic region (Geyer et al., 2011). In plants, methylation occurs predominantly on repetitive DNA elements and transposons (Zhang et al., 2006), though gene bodies are substantially methylated in some species (Zhang et al., 2006; Zilberman et al., 2006).
Gene-Associated DNA Methylation in Invertebrates
If one considers the significant diversity of methylation across taxa it seems plausible that these marks could have different functions, and potentially different mechanisms of action, across organisms and evolutionary time. Here we will focus on a functional role of DNA methylation in invertebrate species where DNA methylation patterns are associated with transcript coding regions. A discussion of the functional relationship of DNA methylation in other taxonomic systems can be found elsewhere (Regev et al., 1998; Colot and Rossignol, 1999; Hendrich and Tweedie, 2003; Suzuki and Bird, 2008; Law and Jacobsen, 2010).
In contrast to well-studied mammalian and plant systems, there are limited studies on DNA methylation in invertebrates. Some of the first evidence supporting a regulatory role of intragenic DNA methylation in invertebrates comes from in silico analyses. Initial computational analysis revealed a relationship between gene body methylation and gene function. This analysis is based on the known hyper-mutability of methylated cytosines, which readily deaminate to thymine residues (Coulondre et al., 1978). The mutation is not easily corrected by DNA repair machinery and, as a result, consistently methylated regions of DNA are depleted of CpG dinucleotides over evolutionary time (Schorderet and Gartler, 1992). Consequently, regions of DNA with a low CpG observed versus expected ratio (denoted as CpG O/E) are predicted to be methylated at the germline, whereas regions with a high CpG O/E (approaching 1.0) are predicted to be sparsely methylated. Germline methylation refers to the methylation state that is inherited.
In A. mellifera, ubiquitously expressed critical genes were predicted to be methylated at the germline, whereas caste-specific genes were predicted to lack methylation (Elango et al., 2009; Foret et al., 2009). From their study, Elango et al. (2009), hypothesized that genes predicted to be unmethylated (caste-specific) might have greater epigenetic flexibility, which allows for higher regulatory control of these inducible classes of genes via transient methylation. In a previous publication, we described a similar relationship in the Pacific oyster, C. gigas. In C. gigas, genes predicted to be hyper-methylated are ubiquitously expressed critical genes such as those involved in DNA and RNA metabolism (Gavery and Roberts, 2010). Likewise, genes predicted to be sparsely methylated (i.e., higher CpG O/E) are associated with tissue specific and inducible expression, including those involved in general immune function (e.g., cell adhesion, cell–cell signaling, and signal transduction; Gavery and Roberts, 2010). These results suggest DNA methylation has regulatory functions in genes involved in stress and environmental responses.
In order to experimentally corroborate the in silico analysis that predicts hyper-methylated genes in oysters are ubiquitously expressed critical genes, our lab has recently performed deep sequencing of the methylated portion of the C. gigas genome. Methyl-CpG binding domain protein sequencing (MBD-seq; see Li et al., 2010) was carried out followed by Gene Ontology based analysis. Our results indicate that genes involved in DNA and protein metabolism were most prevalent in the MBD-library (thus having the highest amount of methylation) and the most underrepresented genes in the library are involved in cell adhesion (Figure 1). These analyses are consistent with the results of the in silico analysis.
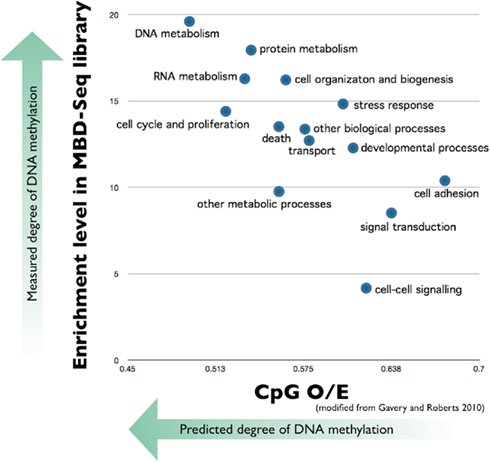
Figure 1. Predicted methylation level of C. gigas genes categorized by biological processes compared to measured level of DNA methylation. Mean CpG O/E for 10,699 C. gigas genes categorized according to Biological Process Gene Ontology (GO) Slim terms are plotted on the x-axis (modified from Gavery and Roberts, 2010). DNA methylation was empirically measured by performing MBD-seq on the SOLiD 4 platform (Applied Biosystems). Genes identified in the MBD-library were associated with respective GO terms and enrichment analysis was performed based on the entire transcriptome (Fleury et al., 2009) using DAVID (Huang et al., 2009a,b). Results indicate the most underrepresented genes in the library are involved in cell adhesion and genes involved in DNA and protein metabolism were most prevalent in the MBD-library.
Direct measurements of DNA methylation patterns in A. mellifera have also been carried out. Using bisulfite treatment coupled with high-throughput sequencing, Lyko et al. (2010) found that methylated cytosines occur primarily in exons and that methylated genes had a higher degree of conservation across species than unmethylated genes. Just as with the oyster data, these results confirmed the inverse relationship between germline methylation and CpG O/E. Other trends that arose from this analysis were that (1) methylated cytosine clusters were associated with alternatively spliced exons and (2) genes containing introns were more likely to be methylated than those lacking introns. The authors also highlighted an example where an increased level of methylation in an alternatively spliced exon in the worker bee brain was associated with an increased expression of the variant lacking the exon. Lyko et al. (2010) concluded methylation may not be functioning as an “on/off” switch but instead allowing for “fine tuning” of transcriptional control of these conserved genes.
Another characteristic of gene-associated DNA methylation in invertebrates is that genes predicted to be methylated at the germline (i.e., low CpG O/E) have less genetic diversity compared to genes lacking germline methylation (i.e., high CpG O/E). One source of evidence of this relationship comes from recent analyses in our own lab where high-throughput sequencing reads from a pooled oyster gill tissue cDNA library were mapped to the oyster transcriptome and single nucleotide polymorphisms characterized. There was as positive relationship among the mean number of polymorphisms per nucleotide and CpG O/E. This is consistent with results from Lyko et al. (2010) where they showed increased sequence conservation in low CpG O/E genes in A. mellifera.
DNA Methylation and Transcriptional Opportunities
Given the similarities in DNA methylation patterns between A. mellifera and C. gigas it is possible that the mechanism of action is conserved at some level. However, given the dramatic differences in specific life history characteristics of the species, the role of DNA methylation in bees and oysters could have diverged over evolutionary time. In the following section we will primarily focus on a putative role of DNA methylation in the oyster, however a majority of the concepts discussed are in agreement with what has been observed in other taxa. For an in-depth review of the functional role of DNA methylation in insects see Glastad et al. (2011).
Based on what we currently know concerning DNA methylation in invertebrates, we propose the absence of germline methylation facilitates random variation that contributes to phenotypic plasticity and thus could increase adaptive potential. Another way to consider this is that in species that experience a diverse range of environmental conditions, processes have evolved to increase the number of potential phenotypes in a population in order to improve the chances for an individual’s survival. This would be particularly important for estuarine species such as C. gigas, where a large number of planktonic larvae are dispersed by currents and can settle in a range of habitats. Germline methylation of genes essential for normal biological function, such as those involved in DNA and protein metabolism, essentially “protects” these genes from the inherent genome wide plasticity, as this would likely be lethal. Thus, as a result of their low methylation status, those genes involved in responding to environmental perturbation may be subject to one of several transcriptional opportunities.
Limited methylation might passively facilitate specific transcriptional opportunities including access to alternative transcription start sites, increasing sequence mutations, and exon skipping. Furthermore, there is the opportunity for these genes to be transiently methylated in somatic tissue, which could also influence transcription. Conversely, germline methylation limits transcriptional opportunities in critical genes. This theory provides an inclusive framework that suggests a suite of specific mechanisms that contribute to evolutionary success by increasing the number of phenotypes via gene-associated, random variation (Figure 2). This theory is consistent with what has been described in A. mellifera by Lyko et al. (2010) suggesting that methylation could “control which of several versions of a gene is expressed.” Furthermore, researchers have shown a relationship between DNA methylation, alternative splicing, and sequence conservation (e.g., Lyko et al., 2010; Park et al., 2011) and suggested a role for DNA methylation in influencing ecologically important traits (e.g., Angers et al., 2010).
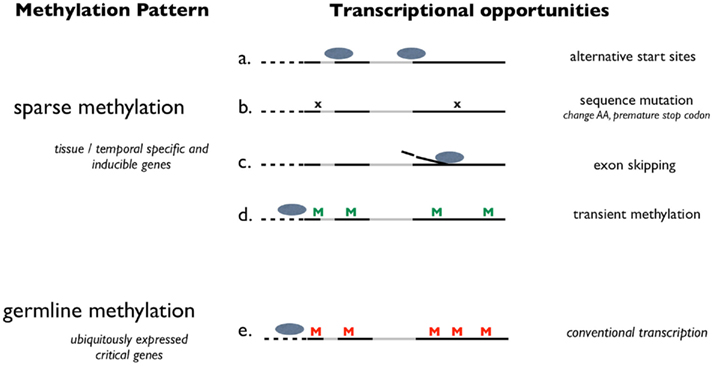
Figure 2. Schematic representation of a how DNA methylation potentially influences transcriptional activity in invertebrate species. This theory proposes the absence of germline methylation (sparse methylation) contributes to adaptive potential by allowing for multiple transcriptional opportunities. Transcriptional opportunities are diagrammed for genes with sparse methylation (a–d) and genes methylated at the germline (e). Dashed lines represent the 5′ UTR, solid lines represent exons and gray lines indicate introns. “M” designates a methylated CpG. “x” Represents a sequence mutation. Ovals represent putative promoter complexes.
The absence of DNA methylation in genes that are induced in response to changing conditions could allow for multiple transcripts indirectly by providing access to alternative promoter sites. This explanation is consistent with the ability of DNA methylation to inhibit binding of transcription factors to response elements in mammalian promoter regions (Iguchi-Ariga and Schaffner, 1989). A recent mammalian study provided direct evidence of this, revealing that intragenic methylation limits the generation of alternate gene transcripts by masking intragenic promoters (Maunakea et al., 2010). Direct evidence of DNA methylation associated with alternative transcripts is also available in invertebrates (Lyko et al., 2010).
Sequence mutation is another important source of potential phenotypic variation. Transcript variations that could be associated with function include those that contribute to an alteration in amino acid or result in a premature stop codon. There are several instances of evidence supporting an inverse relationship between methylation density and sequence variation. As described above we have characterized this relationship in the oyster using high-throughput sequence analysis and a similar pattern has been reported in honey bees (Lyko et al., 2010). Furthermore, a recent study in the jewel wasp (Nasonia vitripennis) showed high CpG O/E ratios correspond with higher substitution rates between related species for synonymous, non-synonymous, and intron sites (Park et al., 2011). In other words, there was more genetic variation in genes lacking germline methylation.
Another means by which a transcriptional variant might be produced is through exon skipping, and there is evidence to suggest methylation is associated with this phenomenon in invertebrates. In A. mellifera, the gene GB18602 has two forms (long and short), which are distinguished by a cassette-exon being skipped in the long form (Lyko et al., 2010). This exon contains a stop codon that creates a shorter, alternative transcript. The researchers went on to find numerous examples of genes where the methylated CpGs were associated with differentially spliced exons (Lyko et al., 2010). This phenomenon would be consistent with the transient (or differential) methylation that could lead to alternative transcripts under different environmental conditions.
Summary
Here we have set out to provide a perspective on the functional role of DNA methylation in invertebrates. We propose that an absence of germline methylation in genes involved in responding to fluctuating conditions facilitates phenotypic variation, which could contribute to increased adaptive potential. However, there are several questions that remain to be answered. Foremost is what contributes to the proposed promiscuous transcriptional nature in certain invertebrates that acts in concert with DNA methylation to enhance phenotypic plasticity? Here we suggest that the probability of a transcriptional opportunity occurring is random, however it is also possible that an environmental stressor could have a specific effect on methylation patterns that directly impacts the physiological response. Furthermore, it is not clear what mechanism(s) are responsible for transient methylation in invertebrates or how common transient methylation occurs. Finally, it is not known if DNA methylation patterns are heritable independent of genetic inheritance. Future research efforts will certainly begin to shed light on these questions as well as test the theory proposed here. Given the evidence we have to date, what we learn about DNA methylation and epigenetics in invertebrates has the potential to considerably change how we view organismal physiology and population biology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Angers, B., Castonguay, E., and Massicotte, R. (2010). Environmentally induced phenotypes and DNA methylation: how to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 19, 1283–1295.
Bird, A. P., and Taggart, M. H. (1980). Variable patterns of total DNA and rDNA methylation in animals. Nucleic Acid Res. 8, 1485–1497.
Bird, A. P., Taggart, M. H., and Smith, B. A. (1979). Methylated and unmethylated DNA compartments in the sea urchin genome. Cell 17, 889–901.
Colot, V., and Rossignol, J. (1999). Eukaryotic DNA methylation as an evolutionary device. Bioessays 21, 402–411.
Coulondre, C., Miller, J. H., Farabaugh, P. J., and Gilbert, W. (1978). Molecular basis of base substitution hotspots in Escherichia coli. Nature 274, 775–780.
Elango, N., Hunt, B. G., Goodisman, M. A. D., and Yi, S. (2009). DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc. Natl. Acad. Sci. U.S.A. 106, 11206–11211.
Fleury, E., Huvet, A., Lelong, C., de Lorgeril, J., Boulo, V., Gueguen, Y., Bachère, E., Tanguy, A., Moraga, D., Fabioux, C., Lindeque, P., Shaw, J., Reinhardt, R., Prunet, R., Davey, G., Lapègue, S., Sauvage, C., Corporeau, C., Moal, J., Gavory, F., Wincker, P., Moreews, F., Klopp, C., Mathieu, M., Boudry, P., and Favrel, B. (2009). Generation and analysis of a 29,745 unique expressed sequence tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database: the GigasDatabase. BMC Genomics 10, 341. doi:10.1186/1471-2164-10-341
Foret, S., Kucharski, R., Pittelkow, Y., Lockett, G. A., and Maleszka, R. (2009). Epigenetic regulation of the honey bee transcriptome: unraveling the nature of methylated genes. BMC Genomics 10, 472. doi:10.1186/1471-2164-10-472
Gavery, M., and Roberts, S. B. (2010). DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genomics 11, 483. doi:10.1186/1471-2164-11-483
Geyer, K., Rodriguez-Lopez, C. M., Chalmers, I. W., Munshi, S. E., Truscott, M., Heald, J., Wlkinson, M. J., and Hoffmann, K. F. (2011). Cytosine methylation regulates oviposition in the pathogenic blood fluke Schistosoma mansoni. Nat. Commun. 2, 424.
Glastad, K. M., Hunt, B. G., Yi, S. V., and Goodisman, M. A. D. (2011). DNA methylation in insects: on the brink of the epigenomic era. Insect Mol. Biol. 20, 553–565.
Gowher, H., Leismann, O., and Jeltsch, A. (2000). DNA of Drosophila melanogaster contains 5-methylcytosine. EMBO J. 19, 6918–6923.
Hendrich, B., and Tweedie, S. (2003). The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 19, 269–277.
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13.
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57.
Iguchi-Ariga, S. M., and Schaffner, W. (1989). CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 3, 612–619.
Jablonka, E., and Lamb, M. J. (2002). Epigenetic Inheritance and Evolution: The Lamarckian Dimension. Oxford: Oxford University Press.
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220.
Li, N., Ye, M., Li, Y., Yan, Z., Butcher, L. M., Sun, J., Han, X., Chen, Q., Zhang, X., and Wang, J. (2010). Whole genome DNA methylation analysis based on high throughput sequencing technology. Methods 52, 203–212.
Lyko, F., Foret, S., Kucharski, R., Wolf, S., Falckenhayn, C., and Maleszka, R. (2010). The honeybee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8, e1000506. doi:10.1371/journal.pbio.1000506
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D’Souza, C., Fouse, S. D., Johnson, B. E., Hong, C., Nielsen, C., Zhao, Y., Turecki, G., Delaney, A., Varhol, R., Thiessen, N., Shchors, K., Heine, V. M., Rowitch, D. H., Xing, X., Fiore, C., Schillebeeckx, M., Jones, S. J. M., Haussler, D., Marra, M. A., Hirst, M., Wang, T., and Costello, J. F. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257.
Palmer, L. E., Rabinowicz, P. D., O’Shaughnessy, A. L., Balija, V. S., Nascimento, L. U., Dike, S., de la Bastide, M., Martienssen, R. A., and McCombie, W. R. (2003). Maize genome sequencing by methylation filtration. Science 302, 2115–2117.
Park, J., Peng, Z., Zeng, J., Elango, N., Park, T., Wheeler, D., Werren, J. H., and Yi, S. V. (2011). Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes. Mol. Biol. Evol. 28, 3345–3354.
Regev, A., Lamb, M. J., and Jablonka, E. (1998). The role of DNA methylation in invertebrates: developmental regulation or genome defense? Mol. Biol. Evol. 15, 880–891.
Schorderet, D. F., and Gartler, S. M. (1992). Analysis of CpG suppression in methylated and nonmethylated species. Proc. Natl. Acad. Sci. U.S.A. 89, 957–961.
Simmen, M. W., and Bird, A. P. (2000). Sequence analysis of transposable elements in the sea squirt, Ciona intestinalis. Mol. Biol. Evol. 17, 1685–1693.
Simpson, V. J., Johnson, T. E., and Hammen, R. F. (1986). Caenorhabditis elegans DNA does not contain 5-methylcytosine at any time during development or aging. Nucleic Acids Res. 14, 6711–6719.
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476.
Suzuki, M. M., Kerr, A. R. W., De Sousa, D., and Bird, A. (2007). CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 17, 625–631.
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S. W., Chen, H., Henderson, I. R., Shinn, P., Pellegrini, M., Jacobsen, S. E., and Ecker, J. R. (2006). Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 126, 1189–1201.
Keywords: epigenetic, methylation, oyster, plasticity, adaptation
Citation: Roberts SB and Gavery MR (2012) Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physio. 2:116. doi: 10.3389/fphys.2011.00116
Received: 16 November 2011;
Paper pending published: 30 November 2011;
Accepted: 14 December 2011;
Published online: 02 January 2012.
Edited by:
John S. Terblanche, Stellenbosch University, South AfricaReviewed by:
Jesper Givskov Sørensen, Aarhus University, DenmarkKatherine A. Mitchell, Université Catholique de Louvain, Belgium
Leigh Boardman, Stellenbosch University, South Africa
Copyright: © 2012 Roberts and Gavery. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Steven B. Roberts, School of Aquatic and Fishery Sciences, University of Washington, Seattle, 98105 WA, USA. e-mail:c3IzMjBAdS53YXNoaW5ndG9uLmVkdQ==