


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 19 October 2011
Sec. Cardiac Electrophysiology
volume 2 - 2011 | https://doi.org/10.3389/fphys.2011.00067
This article is part of the Research Topic Autonomic regulation of the heart - emerging regulators and arrhythmogenic mechanisms View all 8 articles
Strong emotion and mental stress are now recognized as playing a significant role in severe and fatal ventricular arrhythmias. The mechanisms, although incompletely understood, include central processing at the cortical and brain stem level, the autonomic nerves and the electrophysiology of the myocardium. Each of these is usually studied separately by investigators from different disciplines. However, many are regulatory processes which incorporate interactive feedforward and feedback mechanisms. In this review we consider the whole as an integrated interactive brain–heart system.
“Died of fright” and “worried to death” are amongst many colloquial expressions that testify to the long held belief that strong emotions may precipitate sudden cardiac death. Until relatively recently such a notion has been largely confined to the realms of anecdote, however, there is now substantial and compelling evidence linking mental and emotional stress with arrhythmias and sudden death.
History is replete with anecdotal examples of an emotionally charged event followed almost immediately by the death of the person (Mauck and Hockman, 1967; Engel, 1971). An often quoted example are the words of the surgeon John Hunter, who collapsed and died following a heated board room meeting: “My life is at the mercy of any scoundrel who should put me in a passion” (Williams and Edwards, 1968). Population studies have shown an increase in the incidence of sudden death at times of heightened psychological stress such as natural disasters or wars (Trichopoulos et al., 1983; Meisel et al., 1991; Leor et al., 1996; Steinberg et al., 2004a). Studies in animal models and humans support the contention that emotion is an important factor in arrhythmogenesis, for example: laboratory studies in animals suggest that strong emotions such as anger may be a potent cause of ventricular fibrillation (Skinner and Reed, 1981; Verrier, 1990); studies in humans show that emotion may influence the ease of arrhythmia induction and termination (Lampert et al., 2000) and several studies have shown that emotion may influence ECG measures of ventricular repolarization known to be important in arrhythmogenesis (for example: Toivonen et al., 1997; James et al., 2000; Kop et al., 2004; Taggart et al., 2005; Lampert et al., 2009).
However, the mechanisms by which emotion may destabilize cardiac electrophysiology and initiate ventricular arrhythmia are at present incompletely understood. It is generally considered that both the spatial and temporal patterns of autonomic input to the heart play a key role together with altered myocardial electrophysiological parameters as a result of disease. There is now emerging evidence that central neural processing of emotions may also play an important role (Zipes and Rubart, 2006; Coote, 2007). In this review we will therefore consider the possible role of emotion and mental stress in arrhythmias and sudden death in the context of an interactive brain–heart system (Figure 1).
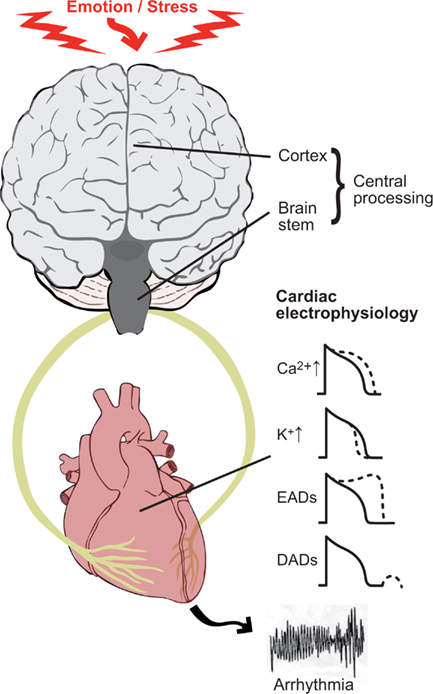
Figure 1. The theme of this review is to consider the brain and heart as an interactive unit and its role in the generation of ventricular arrhythmias. Key components are (1) the electrophysiological changes occurring in the myocardium as a result of autonomic nerve stimulation; (2) the modulatory role of the sympathetic and parasympathetic nerves which tend to oppose and balance each other (3) central neural processing of emotional input and afferent input from the heart and circulation. Sympathetic nerve stimulation acting through β-adrenergic receptors influences a number of ion channels and transporters in cardiac myocytes. These effects include an increase in inward calcium current ICa and decrease in outward potassium current Ik, thereby tending to lengthen and shorten action potential duration (and refractoriness) respectively. Sympathetic stimulation favors the formation of both early after depolarizations (EAD) and delayed after depolarizations (DAD). These effects may be proarrhythmic as a result of triggered activity and/or modulation of refractoriness.
The two components of the autonomic nervous system usually act in a reciprocal fashion on target organs in keeping with the concept of opponent processing which is generally considered to be a fundamental functioning principle of autonomic control. In general, in the ventricles, enhanced sympathetic stimulation is proarrhythmic while enhanced parasympathetic activity is protective (Schwartz, 1984; Schwartz and Zipes, 2004). Notable exceptions include the long QT3 syndrome and Brugada syndrome (BrS) where the opposite may be the case and arrhythmias are commonly induced by parasympathetic activity, although it is difficult to separate increased vagal activity from decreased sympathetic activity. The proarrhythmic potential of sympathetic stimulation has been established in a wide range of experimental models including an isolated heart preparation with autonomic nerves intact (Ng et al., 2007) and selective stimulation of sympathetic and parasympathetic nerves in vivo (Myers et al., 1974; Verrier et al., 1974; Podrid et al., 1990; Vanoli et al., 1991). Depressed vagal responsiveness was a risk factor for ventricular fibrillation during exercise induced enhanced sympathetic activity (La Rovere et al., 1998). The clinical antiarrhythmic benefits of stellate ganglion section and β-adrenergic blockade are well known (Gottlieb et al., 1998; Schwartz and De Ferrari, 2011). The balance between sympathetic and parasympathetic activity is not a simple see–saw effect as co-activation also occurs (Koizumi and Kollai, 1981; Zipes and Miyazaki, 1990; Paton et al., 2005).
Based on a wide range of asymmetric behavior of left and right cerebral hemispheres, Lane and Jennings proposed the “brain–heart laterality hypothesis” (Lane and Jennings, 1995; Lane et al., in press; Figure 2). This hypothesis maintains that lateralization of emotional processing in the cortex is conveyed ipsilaterally through the brainstem to the autonomic nerves which in turn are distributed asymmetrically in the ventricular myocardium. An increase in the naturally occurring spatial inhomogeneity of repolarization facilitates the development of re-entrant arrhythmias (Wu et al., 2004). The left and right autonomic nerves are asymmetrically distributed over the ventricles, although knowledge of the anatomy of the afferent nerves is sparse compared with knowledge on the efferent pathways. Despite an extensive extracardiac anastomotic plexus (Pauza et al., 2000, 2002; Armour and Ardell, 2004), functional autonomic asymmetry exists in response to selective autonomic nerve stimulation, with a trend for left sided neural innervation of the postero-inferior aspect and left ventricle and right sided neural innervation of the anterior aspect and right ventricle, albeit with substantial overlap (Levy et al., 1966; Yanowitz et al., 1966; Randall and Ardell, 1990). Another study in dogs showed that right and left stellate ganglion stimulation and block induced regional wall motion changes with similar geographic distribution (Schlack and Thamer, 1996). It should be underscored, however, that the interindividual differences in the left–right effects are substantial in dogs (Opthof et al., 1991). Selective stimulation of stellate ganglion nerve branches has been shown to exhibit functional regionality (Kralios et al., 1975). Myocardial infarction may result in areas of denervation some of which may exhibit hypersensitivity to circulating catecholamines. Nerves which have been interrupted by ischemia/infarction may undergo regrowth referred to as nerve sprouting, resulting in patchy areas of hyperinnervation (Chen et al., 2001; Schwartz, 2001; Chen and Chen, 2004; Zhou et al., 2004). These processes promote electrophysiological inhomogeneity as a result of inhomogeneous autonomic effects and facilitate arrhythmogenesis.
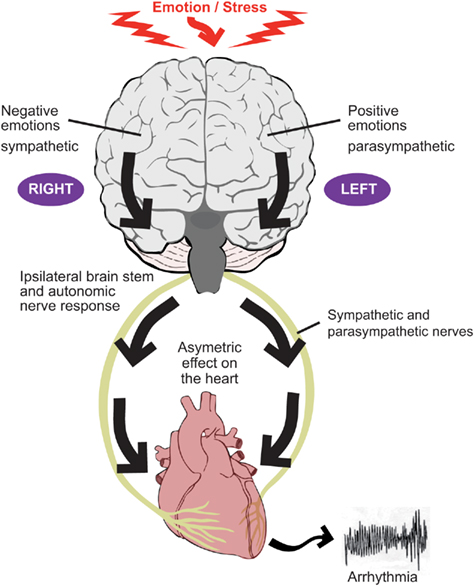
Figure 2. The two components of the autonomic nervous system, the sympathetic nerves and parasympathetic (vagus) nerves tend to act in a reciprocal manner such that, for example, an increase in sympathetic activity is accompanied by a decrease (or lesser increase) in parasympathetic activity. Evidence suggests that the right cerebral hemisphere is predominantly concerned with negative emotion and sympathetic activity, whereas the left hemisphere is predominantly concerned with positive emotion and parasympathetic activity. Autonomic nerve traffic from the brain to the heart is mainly ipsilateral between the brainstem and heart. There is also some degree of lateralization of the distribution of the right and left autonomic nerves on the heart. These considerations form the basis for the laterality hypothesis whereby central neural processes may be represented asymmetrically on the heart and thereby induce inhomogeneous repolarization and be proarrhythmic.
Several reflexes exist which regulate autonomic efferent input to the heart in response to afferent information from mechano or chemoreceptors in the myocardium, thoracic blood vessels, and lungs. The baroreflex regulates the balance of sympathetic/parasympathetic neural input to the heart in response to central pressure/volume changes leading to deformation of the aortic and carotid baroreceptors in order to maintain homeostasis. Mechanical or chemical stimulation of the postero-inferior aspect of the left ventricle may result in enhanced parasympathetic activity while stimulation of the anterior aspect may induce enhanced sympathetic activity (Malpas, 1988; Hainsworth, 1991; Longhurst et al., 2001).
Recently evidence is emerging for an afferent–efferent feedback loop interacting with higher brain centers than the vasomotor center in the medulla oblongata. In a study using PET imaging in patients with heart disease, mental, and physical stress caused a right lateralized shift in dorsal pons and midbrain activity (Critchley et al., 2005) associated with enhanced sympathetic drive to the heart [estimated from heart rate variability (HRV) measurements] and potentially proarrhythmic changes in cardiac repolarization. This was compatible with a stress-related lateralized relay between cortex and heart. We retested this notion using electroencephalography in cardiac patients during mental stress paradigms. This study measured an electrical waveform in the electroencephalogram known as the cardiac evoked potential, considered to represent afferent sensing of the heart beat. The results suggested a dynamic interplay between cortical brain regions and a feedback loop from an abnormal heart with possible implications for brain mechanisms of stress induced sudden cardiac death (Gray et al., 2007).
Sympathetic stimulation acting through β-adrenergic receptors may be proarrhythmic through a number of mechanisms (Steinberg et al., 2004b). As shown in (Figure 3), binding of an agonist to the receptor results in activation of a G-protein (Gs), the consequent activation of adenylate cyclase (AC), the consequent elevation of the intracellular level of cyclic adenosine monophosphate (cAMP), and the consequent activation of protein kinase A (PKA). The elevation of cAMP will directly activate the funny current, If: cAMP binds to funny (HCN) channels, resulting in a depolarizing shift of the If activation curve; this can result in greater activation of If at diastolic potentials (Di Francesco, 2010). HCN channels are pacemaker channels. HCN1 and HCN4 are preferentially expressed in pacemaker tissues – not only the sinoatrial node, atrioventricular node, and His-Purkinje system, but also the atrioventricular ring tissues and retroaortic node (Yanni et al., 2009). However, HCN2 and HCN4 are expressed in working myocardium (Stillitano et al., 2008). Furthermore, expression of HCN channels in working myocardium is upregulated in heart failure and atrial fibrillation (AF; Lai et al., 1999; Stillitano et al., 2008). Activation of If is potentially proarrhythmic by facilitating ectopic pacemaker activity.
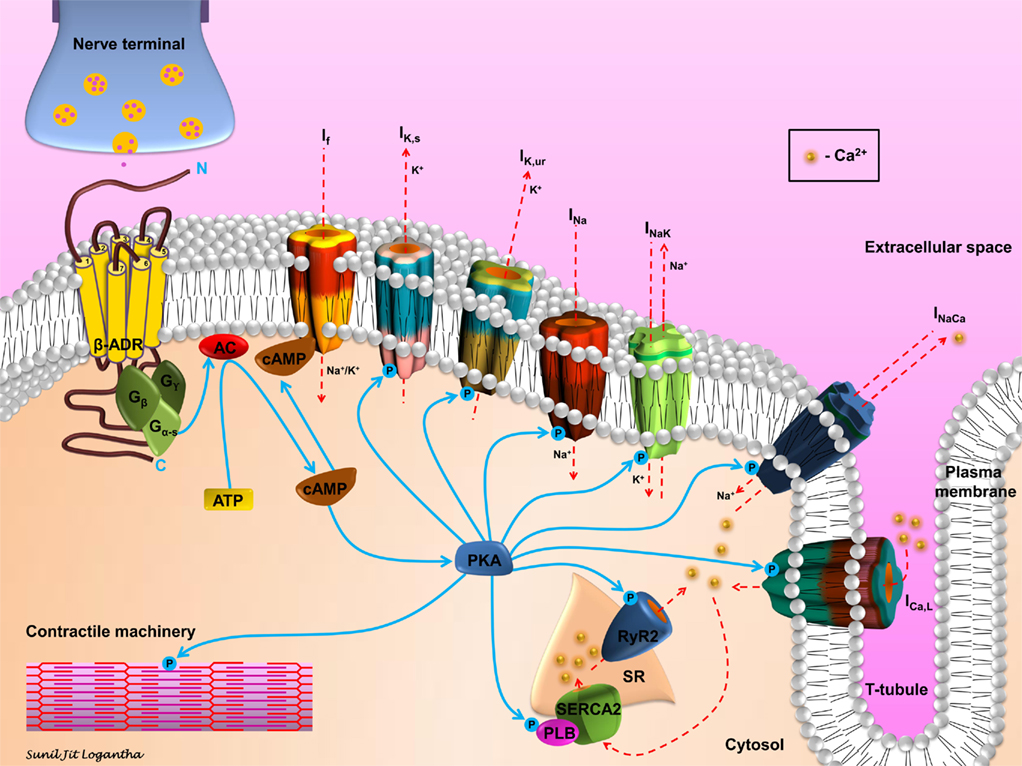
Figure 3. Schematic representation of β-adrenergic signaling in cardiomyocytes. Sympathetic stimulation and the consequent neurotransmitter release initiates the β-adrenergic signaling cascade in cardiomyocytes. Agonist binding to the β-adrenergic receptor (β-ADR) activates, via Gs, the plasma membrane bound adenylate cyclase (AC), which catalyzes the conversion of ATP to cAMP. cAMP activates If and PKA. PKA modulates, via phosphorylation, a number of cellular substrates, including ion channels, transporters, exchangers, intracellular Ca2+-handling proteins, and the contractile machinery.
However, the activation of PKA results in the phosphorylation of many other targets (Figure 3). PKA phosphorylates a range of ion channels resulting in an increase in the Na+ current (INa); (Wang et al., 2009) L-type Ca2+ current (ICa,L) and the ultra-rapid and slow delayed rectifier K+ currents (IK,ur and IK,s; Yue et al., 1999; Lei et al., 2000). PKA phosphorylation also results in an increase in Na+–Ca2+ exchange current (INaCa; Perchenet et al., 2000) and it has complex effects on Na+–K+ pump current (Ip or INaK in Figure 3; Gao et al., 1996). β-adrenergic stimulation is known to increase the amplitude of the action potential plateau, but hasten repolarization and shorten the action potential duration. The increase in ICa,L accounts for the former effect and the increase in IK,ur and IK,s the latter effect. There is strong evidence that repolarization gradients in the ventricles create a substrate for reentry. The effects of β-adrenergic stimulation on repolarization may facilitate arrhythmogenesis by enhancing the inhomogeneity of repolarization and refractoriness in the ventricles, especially in the border zone of infarcts or with regional ischemia, and so facilitate arrhythmia by reentry (Schwartz and Zipes, 2004). Recent evidence suggests that action potential duration restitution properties are also important in arrhythmogenesis; a steepening of the restitution curve is proarrhythmic (Coronel et al., 2009). In this regard it is of note that β-adrenergic stimulation has been shown to steepen the restitution curve in humans (Taggart et al., 2003).
Protein kinase A also phosphorylates targets concerned with intracellular Ca2+ handling (Figure 3). Effects on ICa,L and INaCa have already been mentioned. In addition, PKA phosphorylates troponin I, thereby decreasing the Ca2+ sensitivity of the troponin complex (Ramirez-Correa and Murphy, 2007). The effect of this is to decrease the sensitivity of the contractile machinery to intracellular Ca2+, but more importantly to facilitate the release of Ca2+ from the myofilaments and in this way hasten relaxation (Ramirez-Correa and Murphy, 2007). PKA also phosphorylates phospholamban, releasing its inhibition of the sarcoplasmic reticulum (SR) Ca2+ pump, SERCA2 (Ramirez-Correa and Murphy, 2007). As a consequence, Ca2+ uptake into the SR is accelerated and this too hastens relaxation (Ramirez-Correa and Murphy, 2007). The increase in ICa,L as well as the activation of SERCA2 is also expected to result in an increase in the Ca2+ content of the SR and if “Ca2+ overload” occurs this is expected to result in an increase in spontaneous releases of Ca2+ from the SR (manifested as Ca2+ sparks and waves). Such an increase in Ca2+ sparks and waves with β-adrenergic stimulation has been observed in rat ventricular myocytes by Venetucci et al. (2006). This is important for arrhythmogenesis, because Ca2+ sparks and waves can result in activation of inward INaCa and induction of delayed after-depolarizations (DADs; Heijman et al., 2011). If suprathreshold, DADs can initiate an action potential and in this way be responsible for ectopic pacemaker activity. Finally, PKA also phosphorylates the SR Ca2+ release channel, the ryanodine receptor (RYR2; Shan et al., 2010). This facilitates SR Ca2+ release and, therefore, may be proarrhythmic.
Atrial fibrillation can be triggered by changes in autonomic tone. Coumel originally described vagally mediated AF occurring usually at night during a period of bradycardia. The atria are richly innervated by ganglionic plexi and sympatho-vagal nerve tracts. Experimental studies have shown that pulmonary vein activity responsible for triggering and potentially maintaining AF, can be increased by both vagal and sympathetic stimulation (Patterson et al., 2005). This probably operates through a number of mechanisms including shortening action potential duration (APD) by acetylcholine, increased Ca2+ transients and Ca2+ release through sympathetic stimulation resulting in increased triggered activity. This triggered activity is expressed as increased pulmonary vein ectopic firing. Clinically, stress has been shown to be associated with an increased incidence of lone AF in combination with increased caffeine consumption and a Type A personality (Pope and Smith, 1991; Suarez et al., 1997; Mattioli et al., 2008). In one study of 400 individuals presenting with a first episode of AF (Mattioli et al., 2008), recent stress, high coffee intake, and obesity were associated with greater risk of AF. Patients who developed AF after acute stress showed the highest probability of spontaneous conversion, and high espresso coffee consumption and obesity were associated with an increased risk of persistent AF. Type A individuals are more likely to be hypercholesterolemic and have high levels of circulating catecholamines with diminished mononuclear leukocyte β-adrenergic receptor function. This creates a proarrhythmic autonomic milieu in vulnerable atria particularly if there has been a degree of electrophysiological remodeling in the myocardium following prolonged episodes of AF with resultant shortening of APD and fibrosis to maintain re-entrant wavelet activity.
The next question is how does emotion engage the autonomic control system. Emotion could impact on arrhythmogenesis in several ways including altered sympathetic/parasympathetic balance, alterations in the spatial distribution of autonomic input to the heart, or by causing coronary arterial vasoconstriction and ischemia.
It is a popular belief that increased emotion/stress equates with adrenaline and hence the sympathetic nervous system. However this is an over simplification and some emotions appear to be associated with parasympathetic activity. The issue of whether different emotions are associated with different patterns of autonomic activity and different cortical representation, has been an ongoing debate since James (1884) originally proposed specificity of emotion and Cannon (1927) argued against it. Clinical studies have identified anger as the most common emotion precipitating ventricular arrhythmia (Lampert et al., 2002). For example in patients with cardioverter defibrillators (ICD), the state of anger occurred significantly more frequently in periods prior to ICD shocks than during control periods. In contrast this was not observed for the incidence of anxiety, worry, sadness, happiness, or feeling in control or being interested (Lampert et al., 2002). The greater arrhythmogenic effect of anger could be due to either engaging specific properties of the autonomic nerves, or simply due to eliciting a greater arousal response. Support for the former concept derives from studies which have demonstrated specific patterns of autonomic activity for anger and other emotions in response to emotional facial expressions (Ekman et al., 1983; Levenson et al., 1990), film clips (Christie and Friedman, 2004), and to the recall of previous emotional experiences (Rainville et al., 2006). The study by Rainville et al. (2006) incorporated Fourier analysis of the ECG RR tachogram (HRV; Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology, 1996) using the high frequency component (HF–HRV) as a measure of parasympathetic activity. They showed that anger recall was associated with an increase in heart rate but no change in HF–HRV suggesting relative dominance of sympathetic activity, as reported previously in anger by McCraty et al. (1995). In contrast fear, happiness, and sadness were associated with an increase in heart rate but a decrease in HF–HRV (parasympathetic activity) suggesting an overall decrease in parasympathetic activity, or increase in sympathetic/parasympathetic ratio. This is in keeping with a reduction in vagal tone reported in patients with panic attacks (Friedman and Thayer, 1998).
Early studies using ambulatory ECG recordings reported substantial increase in heart rate during a range of real life stressful situations, with heart rates in the region of 90–100 bpm during road car driving; in the region of 130 bpm during the more intense mental challenge of public speaking, and between 140 and 180 bpm during the extreme stresses of parachute jumping and racing car driving (Taggart et al., 1983). These observations accord with the popular belief that mental stress in associated with an increase in heart rate. However subjects undergoing dental procedures or watching violence on movies have been shown to develop heart rate slowing despite feeling highly stressed and concomitant increased circulating levels of catecholamines (Taggart et al., 1983). These observations would be in accord with a complex interplay between the sympathetic and parasympathetic limbs of the autonomic nervous systems in response to different types of emotional input. Overall there is considerable evidence supporting specificity whereby different emotions have different autonomic signatures (Levenson et al., 1990; Rainville et al., 2006).
Asymmetric autonomic neural traffic to the heart in response to emotion could be generated by several cortical upstream mechanisms.
Considerable evidence suggests that the left and right halves of the human forebrain are associated differentially with specific emotions (Craig, 2005). There is a model that ascribes positive emotions to the left and negative emotions to the right hemisphere with specific neural pathways used for particular emotions (Craig, 2005). A similar lateralization is apparent for the cortical control of cardiac activity (Oppenheimer et al., 1992) which may be related directly to predominantly sympathetic effects arising from the right hemisphere and predominantly parasympathetic effects arising from the left hemisphere (Wittling, 1995). These confluent asymmetries suggest a neuroanatomical homeostatic model of emotional asymmetry in which the right forebrain is associated mainly with sympathetic activity and the left forebrain with parasympathetic activity (Craig, 2005).
Considerable progress has been made in identifying the central circuitry of emotional processing in humans and animal models (Critchley et al., 2000; Napadow et al., 2008; Critchley, 2009; Xavier et al., 2009). For example diminished coupling bilateral amygdala, bilateral hippocampus, and Brodmann areas 9 and 45 have been shown to be associated with increased anxiety and sympathetic activation in humans supporting regulation by these regions as a control system (Mujica-Parodi et al., 2009).
As indicated earlier, studies suggesting a functional lateralization of right and left sided autonomic nerves on the heart (Yanowitz et al., 1966) combined with lateralization of emotional processing in the cortex and ipsilateral conveyance through brainstem to the autonomic nerves, forms the basis of the “brain–heart laterality hypothesis” (Lane and Jennings, 1995; Lane et al., in press). However there is undoubtedly considerable overlap and inter individual variation in this distribution as well as an extensive intercommunicating epicardial plexus. Nevertheless there is considerable evidence in support of such an hypothesis (Lane and Jennings, 1995; Lane et al., in press).
Severe or fatal ventricular arrhythmias occur most commonly in subjects with ischemic heart disease, usually coronary artery disease. Myocardial ischemia is known to play a major role in the initiation and persistence of ventricular arrhythmias (Rubart and Zipes, 2005). Mental stress may induce ischemia as a result of epicardial and/or microvascular constriction together with an increased oxygen demand (Rozanski et al., 1988; Burg et al., 1993; Gottdiener et al., 1994; Goldberg et al., 1996). Also, mental stress may aggravate the effects of ischemia on vasoconstriction superimposed on already insufficient perfusion.
Extensive experimental work has demonstrated that inadequate oxygenated blood flow delivery to the heart muscle rapidly induces a series of metabolic and electrophysiological changes (Janse and Wit, 1989) which subsequently may destabilize the regular heart rhythm by a number of different mechanisms, but primarily reentry following initiating extrasystoles due to “injury current” flowing from the normal to the ischemic myocardium (Janse et al., 1980; Janse and Wit, 1989). Both exercise and emotion are well known to induce ischemia in susceptible individuals often manifest as the development of anginal chest pain. However, most episodes of mental stress induced ischemia are symptomatically silent (Rozanski et al., 1988; Gottdiener et al., 1994). In the original study by Deanfield et al. (1984) 12 of 16 patients with coronary artery disease had a regional myocardial perfusion defect (rubidium-82 myocardial PET imaging) during a mental stress test (mental arithmetic) indicative of ischemia, but only six patients had ST depression in the ECG and only four patients had anginal chest pain. This pattern has since been corroborated in many other laboratories giving rise to the use of the term mental stress induced “silent ischemia.” Overall the substantial proportion of patients who develop ischemia during exercise and also develop ischemia during mental stress is between 30 and 50%. It should be emphasized that this is the behavior of patients with myocardial disease and not normal individuals. In these patients, during everyday activities mental activities are as potent a cause of ischemia as exercise, particularly anger (Gabbay et al., 1996).
Studies in animal models demonstrate the importance of the combination of ischemia with mental stress in the development of fatal arrhythmias. In canines, the combination of mental stress with ischemia induced with a coronary artery snare, was a potent cause of ventricular fibrillation (Verrier, 1990). These studies also showed that vulnerability was prevented by beta blockade or stellectomy and reduced by vagal activity (Verrier, 1990). For example, in the studies by Skinner and Reed (1981) in pigs the combination of mental stress was a much more potent initiator of ventricular fibrillation (VF) than either stress or ischemia alone. In another study in canines the combination of a behavioral challenge with ischemia resulted in a much greater increase in precordial T wave alternans than either the stress or ischemia separately. Atrial pacing to 180 did not increase T wave alternans, and beta blockade with metoprolol greatly reduced it (Kovach et al., 2001).
Anger has been shown to be the commonest emotion prior to the onset of ventricular arrhythmia. It is not clear whether this relates just to the intensity or to differences in the sympathetic response to anger and other emotions.
In humans, anger as well as other stresses have been shown to both increase plasma catecholamines (Lampert et al., 2005), and decrease vagal activity (Bernardi et al., 2000). Creating an anger like state in a canine model induced an increase in heart rate, mean blood pressure, and plasma catecholamines, together with a decrease in the stimulation threshold for repetitive programmed extrasystoles (Verrier and Hagelstad, 1987). In another canine model a fear paradigm was created resulting in an elevated heart rate and mean blood pressure. The anger like state was associated with a predominant rise in plasma noradrenaline whereas the fear like state was associated with a predominant rise in adrenaline (Verrier, 1990).
Episodes of anger have been shown to promote greater increases in sympathetic activity compared with other emotions (McCraty et al., 1995; Rainville et al., 2006). Several studies from the laboratory of Lampert and colleagues along with others have characterized the effects of anger on arrhythmogenesis in humans. As already mentioned anger was shown to be the commonest emotion prior to ventricular arrhythmia onset in patients with ICDs. Other mood states were not significantly correlated (Lampert et al., 2002). Anger induced T wave alternans in laboratory testing in patients with ICDs was found to be predictive of future arrhythmic events suggesting that emotion induced repolarization instability is one mechanism linking emotion and sudden death (Lampert et al., 2009). The induction of anger in patients with coronary artery disease during laboratory mental stress tests increases the likelihood of developing T wave alternans (Kovach et al., 2001; Kop et al., 2004). Anger triggered ventricular arrhythmias in patients with ICDs are more likely to be pause dependent and polymorphic suggesting that anger may create a substrate for more disorganized rhythms (Stopper et al., 2007). Even anger recall in patients mainly with cardioverter defibrillators (CAD) during non-invasive EP studies can make ventricular arrhythmias easier to induce and more difficult to terminate (Lampert et al., 2000).
At the brain level, there is an emerging literature relating the gene–environment interactions determining the neural patterning responsible for the stress response. Heritability accounts for 30–40% of the variance contributing to risk for mood and anxiety disorders as well as post-traumatic stress disorder (Katz and McGuffin, 1993). Childhood exposure to abuse and other early life adverse events increases risk for the later development of these conditions. Recent research from a number of areas suggests that childhood experiences in combination with genetic factors appear to contribute to alterations in biologically based stress response systems. For example, a persistent finding in patients with depression is the elevation of corticotrophin-releasing hormone (CRH) and arginine-vasopressin neurons in the paraventricular nucleus (PVN) in comparison to control subjects (Raadsheer et al., 1994; Purba et al., 1995). Recently, experiments conducted in a rat model to examine CRH function in the amygdala may provide insight into these findings (Keen-Rhinehart et al., 2009). Genetically modified lentiviral vectors were introduced into the central nucleus of the amygdala (CeA) resulting in overexpression of CRH and arginine-vasopressin within the CeA as well as the PVN. Overexpression of CRH in these structures was accompanied physiologically by decreased glucocorticoid negative feedback, and behaviorally by increased anxiety-like behavior (acoustic startle test) and depressive-like behavior (forced swimming test). These data suggest that unrestrained CRH synthesis in the CeA may produce dysregulation of the hypothalamic–pituitary–adrenal axis, which is associated with many of the behavioral, physiological, and reproductive consequences associated with stress-related disorders. Furthermore, gene association studies associate certain CRHR1 polymorphisms with depression and suicidality (Licinio et al., 2004; Liu et al., 2006; Wasserman et al., 2008; Utge et al., 2010). This indicates that there are genetic factors influencing the neurobiology of behavioral and emotional responses including myocardial electrophysiological responses to emotional stress.
The revolution in molecular genetics has elucidated the pathophysiological role of specific cardiac ion channel currents in sudden arrhythmic death through the detailed evaluation of a number of inherited arrhythmia syndromes including Long QT, BrS, and catecholaminergic polymorphic ventricular tachycardia (CPVT). Each of these conditions illustrate how abnormalities in the electrical milieu at the ion channel level can influence the myocardial response to anger and mental stress (Figure 4).
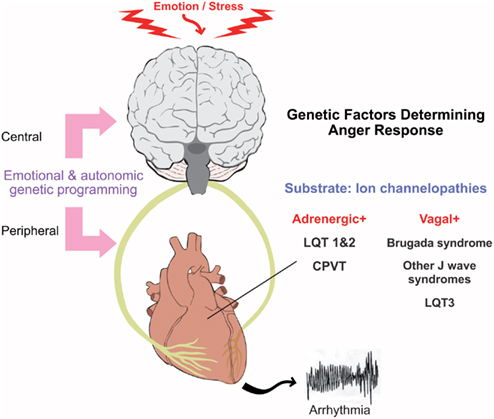
Figure 4. Genetic predisposition to emotion and stress induced arrhythmias.
The congenital long QT syndrome (LQTS) is an inherited disorder characterized by prolongation of the QT interval and susceptibility to ventricular arrhythmias and sudden death (Ackerman and Clapham, 1997; Sauer et al., 2007). It has a prevalence of 1 in 5000–6000 mainly arising through mutations in a number of cardiac ion channel genes (Morita et al., 2008).
In LQT1 and LQT2, mutations in the IK,s and IK,r channel complexes respectively reduce the currents leading to prolongation of phase 3 of the ventricular action potential. Overall prolongation of repolarization time leads to QT prolongation on the surface ECG. Lethal ventricular arrhythmia in long QT is thought to arise through a combination of triggered beats due to early after depolarizations and large spatial gradients in repolarization. These create the optimal conditions for the development of functional block and reentry giving rise to Torsade de Pointes, polymorphic ventricular tachycardia, and ventricular fibrillation. In LQT3, a gain of function mutation in SCN5A leads to a situation in which a small amount of current persists during the plateau phase and thereby prolongs the action potential and the QT interval.
The triggers for cardiac arrest or syncope seem to track with specific LQT subtypes (Schwartz et al., 2001). In LQT1 exercise accounted for 68% of events but only 15% of events in LQT2. However, emotional stimuli predominated in LQT2 (51%) but less so in LQT1 (28%). Sleep and rest without arousal acted as a trigger in 55% of LQT3 cases.
An association between stressful life conditions and arrhythmic events has been demonstrated recently in LQT subjects (Hintsa et al., 2010). Symptomatic patients with LQTS had experienced more stressful life conditions and the level of vital exhaustion as a measure of chronic stress was more than three times higher among patients with LQTS with arrhythmic events than in asymptomatic LQTS mutation carriers.
A study of QT and RR intervals during the stages of sleep in LQT1 and LQT2 patients revealed that women with LQT2 developed significant prolongations in QT interval and RR shortening during the transition from NREM (autonomically stable) to REM (autonomically unstable) sleep which were not observed in LQT1 patients or males with LQT2 (Lanfranchi et al., 2010). This is clinically important since it is recognized that 49% of LQT2 patients die during sleep/rest or have arrhythmic events that are triggered by unexpected acoustic stimuli during rest or sleep (Schwartz et al., 2001). This demonstration of large fluctuations in QT and RR intervals during sleep gives an indication that the autonomic instability and IK,r mutations conspire to induce ventricular arrhythmia in this population. This study also suggests that the abnormal IK,r channel characteristics in LQT2 may not be the only explanation for sudden death and that differences in autonomic patterning are critical. This could be interpreted that the mutation creates the substrate of the arrhythmia, but the autonomic nervous system is a modulating factor that leads to the fatal triggers.
The BrS is characterized by a triad of right bundle branch block (RBBB), ST elevation in the right precordial leads and lethal ventricular arrhythmia and is one of the important causes of sudden cardiac death in the young, accounting for about 20% of all cases (Wilde et al., 2002). The molecular basis remains uncertain as ion channel mutations have only been identified in a maximum of 30% of cases (Grant, 2005) and the role of structural aberrations is increasingly recognized (Coronel et al., 2005; Hoogendijk et al., 2010). This is quite remarkable when it is considered that the absence of structural abnormalities has been defined as a key factor in the diagnosis of BrS (Wilde et al., 2002). In BrS dynamic changes in J point elevation and ventricular arrhythmia are induced by increased vagal tone (Nakazawa et al., 2003). These authors reported that high vagal tone and low sympathetic tone are specific properties of symptomatic BrS by analysis of HRV using Holter ECG recordings. Dynamic changes in J point elevation are more prominent at night particularly in patients with previous VF. The precise relationship between vagal tone and pro-arrhythmia is subject to debate. Increased vagal tone is thought to reduce the Ca2+ transient during phase 2 of the action potential resulting in increased transmural dispersion of repolarization. Then during phase 2 parts of the myocardium may have shorter action potentials. When current can flow between closely apposed areas with longer and shorter action potentials, triggered activity erroneously labeled as phase 2 reentry may ensue giving rise to arrhythmias. Equally increased autonomic tone may exaggerate apical–basal APD gradients promoting increased tissue heterogeneities in conduction and repolarization leading to wave break and VF. There is debate on whether BrS should be considered a conduction or repolarization disease (Wilde et al., 2010).
Catecholaminergic polymorphic ventricular tachycardia is a condition which causes ventricular arrhythmia in situations of heightened adrenergic drive, specifically anger and exercise (Coumel et al., 1978; Leenhardt et al., 1995). The patient, usually a child or young adult, develops bidirectional ventricular tachycardia or polymorphic ventricular tachycardia under conditions of exercise or acute stress implicating an adrenergically triggered mechanism. The molecular deficit lies in the intracellular cycling of sarcomeric Ca2+ (Lahat et al., 2004; Györke, 2008; Cerrone et al., 2009). Although it is generally agreed that CPVT mutations lead to increased Ca2+ leak in the cytoplasm, the precise molecular, and biophysical mechanisms remain to be fully resolved. Three principle hypotheses have been proposed (i) enhanced store overload induced Ca2+ release (ii) hyperactivity of the RyR2 receptor (iii) FKB12.6 RyR2 binding protein disruption – under conditions of adrenergic stress this complex is disrupted due to PKA phosphorylation and prevents the RyR 2 channel from remaining closed in diastole.
Ample evidence implicates anger together with other emotions and mental stress in playing a significant role in arrhythmias and sudden death. The mechanisms involved embrace neuroscience, physiology of the autonomic nervous system and cardiac electrophysiology which are usually investigated and reported by investigators from different disciplines. Growing evidence favors specific cortical representation of emotions in concert with autonomic reflexes and the molecular physiology of the myocardium. Feedback mechanisms from heart to brain probably play a significant modulatory role, particularly in pathological conditions. Such a highly interdependent schema could be regarded as a control system underlining the importance of an interdisciplinary approach to this field.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ackerman, M. J., and Clapham, D. E. (1997). Ion channels – basic science and clinical disease N. Engl. J. Med. 336, 1575–1586.
Armour, J. A., and Arndell, J. L.. (2004). Basic and clinical neurocardiology. Oxford, UK: Oxford University Press.
Bernardi, L., Wdowczyk-Szulc, J., Valenti, C., Castoldi, S., Passino, C., Spadacine, G., and Sleight, P. (2000). Effects of controlled breathing, mental activity, and mental stress with or without verbalisation on heart rate variability J. Am. Coll. Cardiol. 35, 1462–1469.
Burg, M. M., Soufer, J. D., Kerns, R. D., and Zaret, B. L. (1993). Role of behavioural and psychological factors in mental stress induced silent left ventricular dysfunction in coronary artery disease J. Am. Coll. Cardiol. 22, 440–448.
Cannon, W. B. (1927). The James-Lange theory of emotions: a critical examination and an alternative theory Am. J. Psychol. 39, 106–124.
Cerrone, M., Napolitano, C., and Priori, S. G. (2009). Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated with impaired Ca2+ regulation Heart Rhythm 11, 1652–1659.
Chen, L. S., and Chen, P. S. (2004). “Nerve sprouting and cardiac arrhythmias,” in Cardiac Electrophysiology: From Cell to Bedside, eds D. P. Zipes and J. Jalife (Philadelphia, PA: W. B. Saunders), 299–305.
Chen, P. S., Chen, L. S., Cao, J. M., Sharifi, B., Karagueuzian, H. S., and Fishbein, M. S. (2001). Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden death Cardiovasc. Res. 50, 409–416.
Christie, I. C., and Friedman, B. H. (2004). Autonomic specificity of discrete emotion and dimensions of affective space: a multivariate approach Int. J. Psychophysiol. 51, 143–153.
Coote, J. H. (2007). Landmarks in understanding the central nervous control of the cardiovascular system Exp. Physiol. 92, 3–18.
Coronel, R., Casini, S., Koopman, T. T., Wilms-Schopman, F. J., Verderk, A. O., de Groot, J. R., Bhuiyan, Z., Bezzina, C. R., Veldkamp, M. W., Linnenbank, A. C., van der Wall, A. C., Tan, H. L., Brugada, P., Wilde, A. A., and de Bakker, J. M. (2005). Right venrtricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study Circulation 112, 2769–2777.
Coronel, R., Wilms-Schopman, F. J., Opthof, T., and Janse, M. J. (2009). Dispersion of repolarization and arrhythmogenesis Heart Rhythm 6, 537–543.
Coumel, P., Fidelle, J., Lucet, V., Attuel, P., and Bouvrain, Y. (1978). Catecholamine-induced severe ventricular arrhythmias with Adam-Stokes in children: report of four cases Br. Heart J. 40(Suppl.), 28–37.
Craig, A. D. (2005). Forebrain emotional asymmetry: a neuroanatomical basis? Trends Cogn. Sci. 9, 566–571.
Critchley, H. D. (2009). Psychophysiology of neural, cognitive and affective integration: fMRI and autonomic indicants Int. J. Psychophysiol. 73, 88–94.
Critchley, H. D., Corfield, D. R., and Chandler, M. P. (2000). Cerebral correlates of autonomic cardiovascular arousal: a functional neuroimaging investigation J. Physiol. (Lond.) 523, 259–270.
Critchley, H. D., Taggart, P., Sutton, P. M., Holdright, D. R., Batchvarov, V., Hnatkova, K., Malik, M., and Dolan, D. (2005). Mental stress and sudden cardiac death: asymmetric midbrain activation as a possible linking mechanism Brain 128, 75–85.
Deanfield, J. E., Shea, M., Kensett, M., Horlock, P., Wilson, R. A., de Landsheere, C. M., and Selwyn, A. P. (1984). Silent myocardial ischaemia due to mental stress Lancet 2, 1001–1005.
Di Francesco, D. (2010). The role of the funny current in pacemaker activity Circ. Res. 106, 434–446.
Ekman, P., Levenson, R. W., and Friesen, W. V. (1983). Autonomic nervous system activity distinguishes among emotions Science 221, 1208–1210.
Engel, G. L. (1971). Sudden and rapid death during psychological stress Ann. Intern. Med. 74, 771–782.
Friedman, B. H., and Thayer, J. F. (1998). Autonomic balance revisited: panic anxiety and heart rate variability J. Psychosom. Res. 44, 133–151.
Gabbay, R. H., Krantz, D. S., Kop, E. J., Hedges, S. M., Klein, J., Gottdiener, J. S., and Rozanski, A. (1996). Triggers of myocardial ischemia during daily life in patients with coronary artery disease: physical and mental activities, anger and smoking J. Am. Coll. Cardiol. 27, 585–592.
Gao, J., Mathias, R. T., Cohen, I. S., Shi, J., and Baldo, G. J. (1996). The effects of β-stimulation on the Na+-K+ pump current-voltage relationship in guinea-pig ventricular myocytes J. Physiol. (Lond.) 494(Pt 3), 697–708.
Goldberg, A. D., Becker, L. C., Bonsall, R., Cohen, J. D., Ketterer, M. W., Kaufman, P. G., Krantz, D. S., Light, K. C., McMahon, R. P., Noreuil, T., Pepine, C. J., Raczynski, J., Stone, P. H., Strother, D., Taylor, H., and Sheps, D. S. (1996). Ischemic, haemodynamic and neurohumoral responses to mental and exercise stress Circulation 94, 2402–2409.
Gottdiener, J. S., Krantz, D. S., Howell, R. H., Hecht, G. M., Klein, J., Falconer, J. J., and Rozanski, A. (1994). Induction of silent myocardial ischemia with mental stress testing: relation to the triggers of ischemia during daily life activities and to ischemia functional severity J. Am. Coll. Cardiol. 24, 1645–1651.
Gottlieb, S. S., McCarter, R. J., and Vogel, R. A. (1998). Effect of beta blockade on mortality among high-risk and low risk patients after myocardial infarction N. Engl. J. Med. 339, 489–497.
Grant, A. O. (2005). Electrophysiological basis and genetics of Brugada syndrome J. Cardiovasc. Electrophysiol. 16(Suppl. 1), S3–S7.
Gray, M. A., Taggart, P., Sutton, P. M., Groves, D., Holdright, D. R., Bradbury, D., Brull, D., and Critchley, H. D. (2007). A cortical potential reflecting cardiac function Proc. Natl. Acad. Sci. U.S.A. 104, 6818–6823.
Györke, S. (2008). Molecular basis of catecholaminergic polymorphic ventricular tachycardia Heart Rhythm 6, 123–129.
Heijman, J., Volders, P. G., Westra, R. L., and Rudy, Y. (2011). Local control of β-adrenergic stimulation: effects on ventricular myocyte electrophysiology and Ca2+-transient J. Mol. Cell. Cardiol. 50, 863–871.
Hintsa, T., Puttonen, S., Toivonen, L., Kontula, K., Swan, H., and Keltikangas-Jarvinen, L. (2010). A history of stressful life events, prolonged mental stress and arrhythmic events in inherited long QT syndrome Heart 96, 1281–1286.
Hoogendijk, M. G., Opthof, T., Postema, P. G., Wilde, A., de Bakker, J. M., and Coronel, R. (2010). The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Circ. Arrhythm. Electrophysiol. 3, 283–290.
James, P. R., Taggart, P., McNally, S. T., Newman, S. P., Sporton, S. C., and Hardman, S. M. C. (2000). Acute psychological stress and the propensity to ventricular arrhythmias Eur. Heart J. 21, 1023–1028.
Janse, M. J., Van Capelle, F. J. L., Morsink, H., Kleber, A. G., Wilms-Schopman, F., Cardinal, R., Naumann dÀlnoncourt, C., and Durrer, D. (1980). Flow of injury current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts Circ. Res. 47, 151–165.
Janse, M. J., and Wit, A. L. (1989). Electrophysiological mechanisms resulting from myocardial ischemia and infarction Physiol. Rev. 69, 1049–1168.
Katz, R., and McGuffin, P. (1993). The genetics of affective disorders Prog. Exp. Pers. Psychopathol. Res. 16, 200–221.
Keen-Rhinehart, E., Michopoulos, V., Toufexis, D. J., Martin, E. I., Nair, H., Ressler, K. J., Davies, M., Owens, M. J., Nemeroff, C. B., and Wilson, M. E. (2009). Continuous expression of corticotropin-releasing factor in the central nucleus of the amygdala emulates the dysregulation of the stress and reproductive axes Mol. Psychiatry 14, 37–50.
Koizumi, K., and Kollai, M. (1981). Control of reciprocal and non reciprocal action of vagal and sympathetic efferents: study of centrally induced reactions J. Auton. Nerv. Syst. 3, 483–501.
Kop, W. J., Krantz, D. S., Nearing, B. D., Gottdiener, J. S., Quigley, J. F., O‘Callahan, M., DelNegro, A. A., Friehling, T. D., Karasik, P., Suchday, S., Levine, J., and Verrier, R. L. (2004). Effects of acute mental stress and exercise on T-wave alternans in patients with implantable cardioverter defibrillators and controls Circulation 109, 1864–1869.
Kovach, J. A., Nearing, B. D., and Verrier, R. L. (2001). Anger like behavioural state potentiates myocardial ischemia induced T-wave alternans in canines J. Am. Coll. Cardiol. 37, 1719–1725.
Kralios, F. A., Martin, L., Burgess, M. J., and Millar, K. (1975). Local ventricular repolarization changes due to sympathetic nerve-branch stimulation Am. J. Physiol. 228, 1621–1626.
La Rovere, M. T., Bigger, J. T., Marcus, F. I., Mortara, A., and Schwartz, P. J. (1998). Baroreflex sensitivity and heart rate variability in prediction of total cardiac mortality after myocardial infarction Lancet 351, 478–484.
Lahat, H., Pras, E., and Eldar, M. (2004). A missense mutation in CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel Ann. Med. 36(Suppl. 1), 87–91.
Lai, L. P., Su, M. J., Lin, J. L., Tsai, C. H., Lin, F. Y., Chen, Y. S., Hwang, J. J., Huang, S. K., Tseng, Y. Z., and Lien, W. P. (1999). Measurement of funny current (If) channel mRNA in human atrial tissue: correlation with left atrial filling pressure and atrial fibrillation J. Cardiovasc. Electrophysiol. 10, 947–953.
Lampert, R., Jain, D., Burg, M. M., Batsford, W. P., and McPherson, C. A. (2000). Destabilising effects of mental stress on ventricular arrhythmias in patients with implantable cardio-verter defibrillators Circulation 101, 158–164.
Lampert, R., Joska, T., Burg, M. M., Batsford, W. P., McPherson, C. A., and Jain, D. (2002). Emotional and physical precipitants of ventricular arrhythmias Circulation 106, 1800–1805.
Lampert, R., Shusterman, V., Burg, M., McPherson, C., Batsford, W., Goldberg, A., and Soufer, R. (2009). Anger induced T wave alternans predicts future ventricular arrhythmias in patients with implantable cardioverter defibrillators J. Am. Coll. Cardiol. 53, 774–778.
Lampert, R., Shusterman, V., Burg, M. M., Jee, F. A., Earley, C., Goldberg, A., McPherson, C. A., Batsford, W. P., and Soufer, R. (2005). Effects of psychological stress on repolarization and relationship to autonomic and haemodynamic factors J. Cardiovasc. Electrophysiol. 16, 372–377.
Lane, R. D., Critchley, H. D., and Taggart, P. (in press). “Asymmetric autonomic innervation,” in Handbook of Cardiovascular Behavioural Medicine, eds S. Waldstein, W. Kop and L. Katzel (New York: Springer).
Lane, R. D., and Jennings, J. R. (1995). “Hemispheric asymmetry, autonomic asymmetry and the problem of sudden death,” in Brain Asymmetry, eds R. J. Davidson and K. Hugdahl (Cambridge, MA: The MIT Press), 271–304.
Lanfranchi, P. A., Ackerman, M. J., Kara, T., Shamsuzzaman, A. S., Wolk, R., Jurak, P., Amin, R., and Somers, V. K. (2010). Gene-specific paradoxical QT responses during rapid eye movement sleep in women with congenital long QT syndrome Heart Rhythm 7, 1067–1074.
Leenhardt, A., Lucet, V., Denjoy, I., Grau, F., Ngoc, D. D., and Coumel, P. (1995). Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients Circulation 91, 1512–1519.
Lei, M., Brown, H. F., and Terrar, D. A. (2000). Modulation of delayed rectifier potassium current, iK, by isoprenaline in rabbit isolated pacemaker cells Exp. Physiol. 85, 7–35.
Leor, J., Poole, W. K., and Kloner, R. A. (1996). Sudden cardiac death triggered by an earthquake N. Engl. J. Med. 334, 413–419.
Levenson, R. W., Ekman, P., and Friesen, W. V. (1990). Voluntary facial action generates emotion-specific autonomic nervous system activity Psychophysiology 27, 363–384.
Levy, M. N., Ng, M. D., and Zieske, H. (1966). Functional distribution of the peripheral sympathetic pathways Circ. Res. 14, 650–661.
Licinio, J., O’Kirwan, F., Irizarry, K., Merriman, B., Thakur, S., Jepson, R., Lake, S., Tantisira, K. G., Weiss, S. T., and Wong, M. L. (2004). Association of a corticotropin-releasing hormone receptor 1 haplotype and antidepressant treatment response in Mexican-Americans Mol. Psychiatry 9, 1075–1082.
Liu, Z., Zhu, F., Wang, G., Xiao, Z., Wang, H., Tang, J., Wang, X., Qiu, D., Liu, W., Cao, Z., and Li, W. (2006). Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression Neurosci. Lett. 404, 358–362.
Longhurst, J. C., Tjen-A-Looi, S., and Fu, L.-W. (2001). Cardiac sympathetic afferent activation provoked by myocardial ischemia and reperfusion: mechanisms and reflexes Ann. N. Y. Acad. Sci. 940, 74–95.
Mattioli, A. V., Bonatti, S., Zennaro, M., Melotti, R., and Mattioli, G. (2008). Effect of coffee consumption, lifestyle and acute life stress in the development of acute lone atrial fibrillation J. Cardiovasc. Med. (Hagerstown) 9, 794–798.
Mauck, H. P., and Hockman, C. H. (1967). Central nervous system mechanisms mediating cardiac rate and rhythm Am. Heart J. 74, 106–109.
McCraty, R., Atkinson, M., Tiller, W. A., Rein, G., and Watkins, A. D. (1995). The effects of emotions on short-term power spectrum analysis of heart rate variability Am. J. Cardiol. 76, 1089–1093.
Meisel, S. R., Kutz, I., Dayan, K. I., Pauzner, H., Chetbourg, I., Arbel, Y., and David, D. (1991). Effect of Iraqi missile war on incidence of acute myocardial infarction and sudden death in Israeli civilians Lancet 338, 660–661.
Mujica-Parodi, L. R., Korgaonkar, M., Ravindranath, B., Greenberg, T., Tomasi, D., Wagshul, M., Ardekani, B., Guilfoyle, D., Khan, S., Zhong, Y., Chon, K., and Malaspina, D. (2009). Limbic dysregulation is associated with lowered heart rate variability and increased trait anxiety in healthy adults Hum. Brain Mapp. 30, 47–58.
Myers, R. W., Pearlman, A. S., Hyman, R. M., Goldstein, R. A., Kent, K. M., Goldstein, R. E., and Epstein, S. E. (1974). Beneficial effects of vagal stimulation and bradycardia during experimental acute myocardial ischaemia Circulation 49, 943–950.
Nakazawa, K., Sakurai, T., Takagi, A., Kishi, R., Osada, K., Nanke, T., Miyake, F., Matsumoto, N., and Kobayashi, S. (2003). Autonomic imbalance as a property of symptomatic Brugada syndrome Circ. J. 67, 511–514.
Napadow, V., Dhond, R., Conti, G., Makris, N., Brown, E. N., and Barberi, R. (2008). Brain correlates of autonomic modulation: combining heart rate variability with fmri Neuroimage 42, 169–177.
Ng, G. A., Brack, K. E., Patel, V. H., and Coote, J. H. (2007). Autonomic modulation of electrical restitution, alternans and ventricular fibrillation in the isolated heart Cardiavasc. Res. 73, 750–760.
Oppenheimer, S. M., Gelb, A. W., Girvin, J. P., and Hachinski, V. C. (1992). Cardiovascular effects of human insular cortex stimulation Neurology 42, 1727–1732.
Opthof, T., Misier, A. R., Coronel, R., Vermeulen, J. T., Verberne, H. J., Frank, R. G., Moulijn, A. C., van Capelle, F. J., and Janse, M. J. (1991). Dispersion of refractoriness in canine ventricular myocardium. Effects of sympathetic stimulation Circ. Res. 68, 1204–1215.
Paton, J. F. R., Boscan, P., Pickering, A. E., and Nalivaiko, E. (2005). The yin and yang of cardiac autonomic control: vago-sympathetic interactions revisited Brain Res. Rev. 49, 555–565.
Patterson, E., Po, S. S., Scherlag, B. J., and Lazzara, R. (2005). Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation Heart Rhythm 2, 624–631.
Pauza, D. H., Skripka, V., and Pauziene, N. (2000). Morphology, distribution and variability of the epicardiac neural ganglionated subplexuses in the human heart Anat. Rec. 259, 353–382.
Pauza, D. H., Skripka, V., and Pauziene, N. (2002). Morphology of the intrinsic cardiac nervous system in the dog: a whole-mount study employing histochemical stainingwith acetylcholinesterase Cells Tissues Organs 172, 297–320.
Perchenet, L., Hinde, A. K., Patel, K. C., Hancox, J. C., and Levi, A. J. (2000). Stimulation of Na/Ca exchange by the β-adrenergic/protein kinase A pathway in guinea-pig ventricular myocytes at 37 degrees C Pflugers Arch. 439, 822–828.
Podrid, P. J., Fuchs, T., and Cardinal, R. (1990). Role of the sympathetic nervous system in genesis of ventricular arrhythmia Circulation 82(Suppl. 1), 1103–1113.
Pope, M. L., and Smith, T. W. (1991). Cortisol excretion in high and low cynically hostile men Psychosom. Med. 53, 386–392.
Purba, J. S., Raadsheer, F. C., Hofman, M. A., Ravid, R., Polman, C. H., Kamphorst, W., and Swaab, D. F. (1995). Increased number of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of patients with multiple sclerosis Neuroendocrinology 62, 62–70.
Raadsheer, F. C., Hoogendijk, W. J., Stam, F. C., Tilders, F. J., and Swaab, D. F. (1994). Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients Neuroendocrinology 6, 436–444
Rainville, P., Bechara, A., Naqvi, N., and Damasio, A. R. (2006). Basic emotions are associated with distinct patterns of cardiorespiratory activity Int. J. Psychophysiol. 61, 5–18.
Ramirez-Correa, G. A., and Murphy, A. M. (2007). Is phospholamban or troponin I the “prima donna” in beta-adrenergic induced lusitropy? Circ. Res. 101, 326–327.
Randall, W. C., and Ardell, J. L. (1990). “Nervous control of the heart: anatomy and pathophysiology,” in Cardiac Electrophysiology: From Cell to Bedside, eds D. P. Zipes and J. Jalife (Philadelphia: W. B. Saunders), 291–299.
Rozanski, A., Bairey, C. N., Krantz, D. S., Friedman, J., Ressler, K. J., Morell, M., Hilton-Chalfen, S., Hestrin, L., Bietendorf, J., and Berman, D. S. (1988). Mental stress and the induction of silent myocardial ischemia in patients with coronary artery disease N. Engl. J. Med. 318, 1005–1012.
Rubart, M., and Zipes, D. P. (2005). Mechanisms of sudden cardiac death J. Clin. Invest. 115, 2305–2315.
Sauer, A. J., Moss, A. J., McNitt, S., Peterson, D. R., Zareba, W., Robinson, J. L., Qi, M., Goldenberg, I., Hobbs, J. B., Ackerman, M. L., Benhorin, J., Hall, W. J., Kaufman, E. S., Locati, E. H., Napolitano, C., Prioi, S. G., Schwartz, P. J., Towbin, J. A., Vincent, G. M., and Zhang, L. (2007). Long QT syndrome in adults J. Am. Coll. Cardiol. 49, 329–337.
Schlack, W., and Thamer, V. (1996). Unilateral changes of sympathetic tone to the heart impair left ventricular function Acta Anaesthesiol. Scand. 40, 262–271.
Schwartz, P. J. (1984). The rationale and the role of left stellectomy for the prevention of malignant arrhythmias Ann. N. Y. Acad. Sci. 427, 199–221.
Schwartz, P. J. (2001). QT prolongation, sudden death, and sympathetic imbalance: the pendulum swings J. Cardiovasc. Electrophysiol. 12, 1074–1077.
Schwartz, P. J., and De Ferrari, G. M. (2011). Sympathetic-parasympathetic interaction in health and disease:abnormalities and relevance in heart failure Heart Fail. Rev. 16, 101–107.
Schwartz, P. J., Priori, S. G., Spazzolini, C., Moss, A. J., Vincent, G. M., Napolitano, C., Denjoy, I., Guicheney, P., Breithardt, G., Keating, M. T., Beggs, A. H., Brink, P., Wilde, A. A., Toivonen, L., Zareba, W., Robinson, J. L., Timothy, K. W., Corfield, V., Wattanasirichaigoon, D., Corbett, C., Haverkamp, W., Schulze-Bahr, E., Lehmann, M. H., Schwartz, K., Coumel, P., and Bloise, R. (2001). Genotype-phenotype correlation in the long QT syndrome (gene-specific triggers for life threatening arrhythmias) Circulation 103, 89–95.
Schwartz, P. J., and Zipes, D. P. (2004). “Autonomic modulation of cardiac arrhythmias,” in Cardiac Electrophysiology. From Cell to Bedside, 4th Edn, eds D. P. Zipes and J. Jalife (Philadelphia: W. B. Saunders), 300–314.
Shan, J., Kushnir, A., Betzenhauser, M. J., Reiken, S., Li, j., Lenhart, S. E., Lindegger, N., Mongillo, M., Mohler, P. J., and Marks, A. R. (2010). Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice J. Clin. Invest. 120, 4388–4398.
Skinner, J. E., and Reed, J. C. (1981). Blockade of fronto-cortical brainstem pathway prevents ventricular fibrillation of ischemic heart Am. J. Physiol. 249, H156–H163.
Steinberg, J. S., Arshad, A., Kowalski, M., Kukar, A., Suma, V., Vloka, M., Eklert, F., Herweg, B., Donelly, J., Phillip, J., Reed, G., and Rozanski, A. (2004a). Increased incidence of life threatening arrhythmias in implantable defibrillator patients after the World Trade Centre attack J. Am. Coll. Cardiol. 44, 1261–1264.
Steinberg, S. F., Robinson, R. B., and Rosen, M. R. (2004b). “Molecular and cellular bases of beta-adrenergic and alpha-adrenergic modulation of cardiac rhythm,” in Cardiac Electrophysiology. From Cell to Bedside, 4th Edn, eds D. P. Zipes and J. Jalife (Philadelphia: W. B. Saunders), 291–298.
Stillitano, F., Lonardo, G., Zicha, S., Varro, A., Cerbai, E., Mugelli, A., and Nattel, S. (2008). Molecular basis of funny current (If) in normal and failing human heart J. Mol. Cell. Cardiol. 45, 289–299.
Stopper, M., Joska, T., Burg, M. M., Batsford, W. P., McPherson, C. A., Jain, D., and Lampert, R. (2007). Electrophysiologic characteristics of anger–triggered arrhythmias Heart Rhythm 4, 268–273.
Suarez, E. C., Shiller, A. D., Kuhn, C. M., Schanberg, S. M., Williams, R. B. Jr., and Zimmerman, E. A. (1997). The relationship between hostility and beta adrenergic receptor physiology in healthy males Psychosom. Med. 59, 481–487.
Taggart, P., Carruthers, M., and Somerville, W. (1983). Some effects of emotion on the normal and abnormal heart Curr. Probl. Cardiol. 12, 1–29.
Taggart, P., Sutton, P., Chalabi, Z., Boyett, M. R., Simon, R., Elliott, D., and Gill, J. S. (2003). Effect of adrenergic stimulation on action potential duration restitution in humans Circulation 107, 285–289.
Taggart, P., Sutton, P., Redfern, C., Batchvarov, V. N., Hnatkova, K., Malik, M., James, U., and Joseph, A. (2005). The effect of mental stress on the non-dipolar components of the T wave: modulation by hypnosis Psychosom. Med. 67, 376–383.
Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. (1996). Heart rate variability standards of measurement, physiological interpretation, and clinical use Circulation 93, 1043–1065.
Toivonen, L., Helenius, K., and Vitasalo, M. (1997). Electrocardiographic repolarization during stress from awakening on alarm call J. Am. Coll. Cardiol. 30, 774–777.
Trichopoulos, D., Katsoutanni, K., Zavitsanos, X., Tronou, A., and Dalla-Vorgia, P. (1983). Psychological stress and fatal heart attack: the Athens earthquake natural experiment Lancet 1, 441–443.
Utge, S., Soronen, P., Partonen, T., Loukola, A., Kronholm, E., Pirkola, S., Nyman, E., Porkka-Heiskanen, T., and Paunio, T. (2010). A population-based association study of candidate genes for depression and sleep disturbance Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 468–476.
Vanoli, E., DeFerrari, G. M., Stramba-Badiale, M., Hull, S. S. Jr., Foreman, R. D., and Schwartz, P. J. (1991). Vagal stimulation and prevention of sudden cardiac death in conscious dogs with healed myocardial infarction Circ. Res. 68, 1471–1481.
Venetucci, L. A., Trafford, A. W., Diaz, M. E., O’Neill, S. C., and Eisner, D. A. (2006). Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes Circ. Res. 98, 1299–1305.
Verrier, R., Thompson, P., and Lown, B. (1974). Ventricular vulnerability during sympathetic stimulation: role of heart rate and blood pressure Cardiovasc. Res. 8, 602–610.
Verrier, R. L. (1990). “Behavioural stress, myocardial ischemia, and arrhythmias,” in Cardiac Electrophysiology: From Cell to Bedside, eds D. P. Zipes and J. Jalife (Philadelphia: W. B. Saundes), 343–352.
Verrier, R. L., and Hagelstad, E. L. (1987). Delayed myocardial ischemia induced by anger Circulation 75, 249–254.
Wang, H. W., Yang, Z. F., Zhang, Y., Yang, J. M., Liu, Y. M., and Li, C. Z. (2009). Beta-receptor activation increases sodium current in guinea pig heart Acta Pharmacol. Sin. 30, 1115–1122.
Wasserman, D., Sokolowski, M., Rozanov, V., and Wasserman, J. (2008). The CRHR1 gene: a marker for suicidality in depressed males exposed to low stress Genes Brain Behav. 7, 14–19.
Wilde, A. A., Antzelevitch, C., Borggrefe, M., Brugada, J., Brugada, R., Brugada, P., Corrado, D., Hauer, R. N., Kass, R. S., Nademanee, K., Priori, S. G., and Towbin, J. A. (2002). Proposed diagnostic criteria for the Brugada syndrome: consensus report Circulation 106, 2514–2519.
Wilde, A. A., Postema, P. G., DiDiego, J. M., Viskin, S., Morita, H., Fish, J. M., and Antzelevitch, C. (2010). The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization J. Mol. Cell. Cardiol. 49, 543–553.
Wittling, W. (1995). “Brain asymmetry in the control of autonomic-physiologic activity,” in Brain Asymmetry, eds R. J. Davidson and K. J. Hugdahl (Cambridge, MA: The MIT Press), 305–357.
Wu, J., Wu, J., and Zipes, D. P. (2004). “Mechanisms of initiation of ventricular tachyarrhythmias,” in Cardiac Electrophysiology; From Cell to Bedside, 4th Edn, eds D. P. Zipes and J. Jalife (Philadelphia: Saunders), 380–389.
Xavier, C. H., Nalivaiko, E., Beig, M. I., Menezes, G. B., Cara, D. C., Campagnole-Santos, M. J., and Fontes, M. A. P. (2009). Functional asymmetry in the descending cardiovascular pathways from dorsomedial hypothalamic nucleus Neuroscience 164, 1360–1368.
Yanni, J., Boyett, M. R., Anderson, R. H., and Dobrzynski, H. (2009). The extent of the specialized atrioventricular ring tissues Heart Rhythm 6, 672–680.
Yanowitz, F., Preston, J. B., and Abildskov, J. A. (1966). Functional distribution of right and left stellate innervation to the ventricles: production of neurogenic electrocardiographic changes by unilateral alteration of sympathetic tone Circ. Res. 28, 416–428.
Yue, L., Feng, J., Wang, Z., and Nattel, S. (1999). Adrenergic control of the ultra rapid delayed rectifier current in canine atrial myocytes J. Physiol. (Lond.) 516(Pt 2), 385–398.
Zhou, S., Chen, L. S., Miyanchi, Y., Miyachi, M., Kar, S., Kongavari, S., Fishbein, M. C., Sharifi, B., and Chen, P. S. (2004). Mechanisms of cardiac nerve sprouting after MI in dogs Circ. Res. 95, 76–83.
Zipes, D. P., and Miyazaki, T. (1990). “The autonomic nervous system and the heart: basis for understanding interactions and effects on arrhythmia development,” in Cardiac Electrophysiology: From Cell to Bedside, eds D. P. Zipes and J. Jalife (Philadelphia: W. B. Saunders), 312–330.
Keywords: anger, emotion, brain–heart system, mental stress
Citation: Taggart P, Boyett MR, Logantha SJRJ and Lambiase PD (2011) Anger, emotion, and arrhythmias: from brain to heart. Front. Physio. 2:67. doi: 10.3389/fphys.2011.00067
Received: 09 March 2011; Accepted: 14 September 2011;
Published online: 19 October 2011.
Edited by:
Tobias Opthof, Academic Medical Center, NetherlandsCopyright: © 2011 Taggart, Boyett, Logantha and Lambiase. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Peter Taggart, The Heart Hospital, 16-18 Westmoreland Street, London WIG8PH, UK. e-mail:cGV0ZXIudGFnZ2FydEB1Y2xoLm5ocy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.