


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 27 July 2011
Sec. Systems Biology Archive
volume 2 - 2011 | https://doi.org/10.3389/fphys.2011.00041
This article is part of the Research Topic Advances in Systems Immunology and Cancer View all 11 articles
Toll-like receptors (TLRs) are pattern recognition receptors that recognize conserved structures in pathogens, trigger innate immune responses, and prime antigen-specific adaptive immunity. Elucidation of crystal structures of TLRs interacting with their ligands such as TLR1-2 with triacylated lipopeptide, TLR2-6 with diacylated lipopeptide, TLR4–MD-2 with LPS, and TLR3 with double-stranded RNA (dsRNA) have enabled an understanding of the initiation of TLR signaling. Agonistic ligands such as LPS, dsRNA, and lipopeptides induce “m” shaped TLR dimers in which C-termini converge at the center. Such central convergence is necessary to bring the two intracellular receptor TIR domains closer together and promote their dimerization, which serves as an essential step in downstream signaling. In this review, we summarize TLR ECD structures that have been reported to date with special emphasis on ligand recognition and activation mechanism.
The Toll-like receptor (TLR) protein family plays an important role in the innate immune system by recognizing common structural patterns in diverse microbial molecules (Gay and Gangloff, 2007). TLRs are type I transmembrane glycoproteins characterized by the presence of an extracellular domain (ectodomain; ECD) containing leucine rich repeats (LRRs), which is primarily responsible for mediating ligand recognition, followed by a single transmembrane helix and an intracellular Toll-like/interleukin-1 (IL-1) receptor (TIR) domain that is responsible for downstream signaling. To date, 10 and 12 functional TLRs have been identified in humans and mice, respectively. TLR1-9 is conserved in both species; however, mouse TLR10 is not functional because of a retrovirus insertion, and TLR11-13 have been lost from the human genome (Kawai and Akira, 2010). “Toll” was first identified as a protein important in the early stages of development in Drosophila. Later, it was discovered that Toll signals to Dorsal (like mammalian NF-κB) and is involved in the coordination of antifungal and antibacterial responses (Rosetto et al., 1995; Lemaitre et al., 1996).
The TLR family can be largely divided into two subgroups, extracellular and intracellular, depending on their cellular localization. TLR1, 2, 4, 5, 6, and 10 are largely localized on the cell surface to recognize PAMPs. Conversely, TLR3, 7, 8, and 9 are localized in intracellular organelles such as endosomal/lysosomal compartments and the endoplasmic reticulum (ER). Among the TLRs, the ligand (lipopolysaccharide; LPS) of TLR4 was first identified by genetic studies (Lemaitre et al., 1996). Lipopeptides or lipoproteins are recognized by TLR2 in complex with TLR1 or 6, while viral double-stranded RNA (dsRNA) is recognized by TLR3, flagellin is recognized by TLR5, single-stranded RNA is recognized by TLR7 and 8, and host- or pathogen-derived DNA is recognized by TLR9. In addition to known pathogen/microbial derived ligands, TLR also recognizes the endogenous ligands (produced by stressed or damaged cells) and synthetic ligands listed in Table 1.
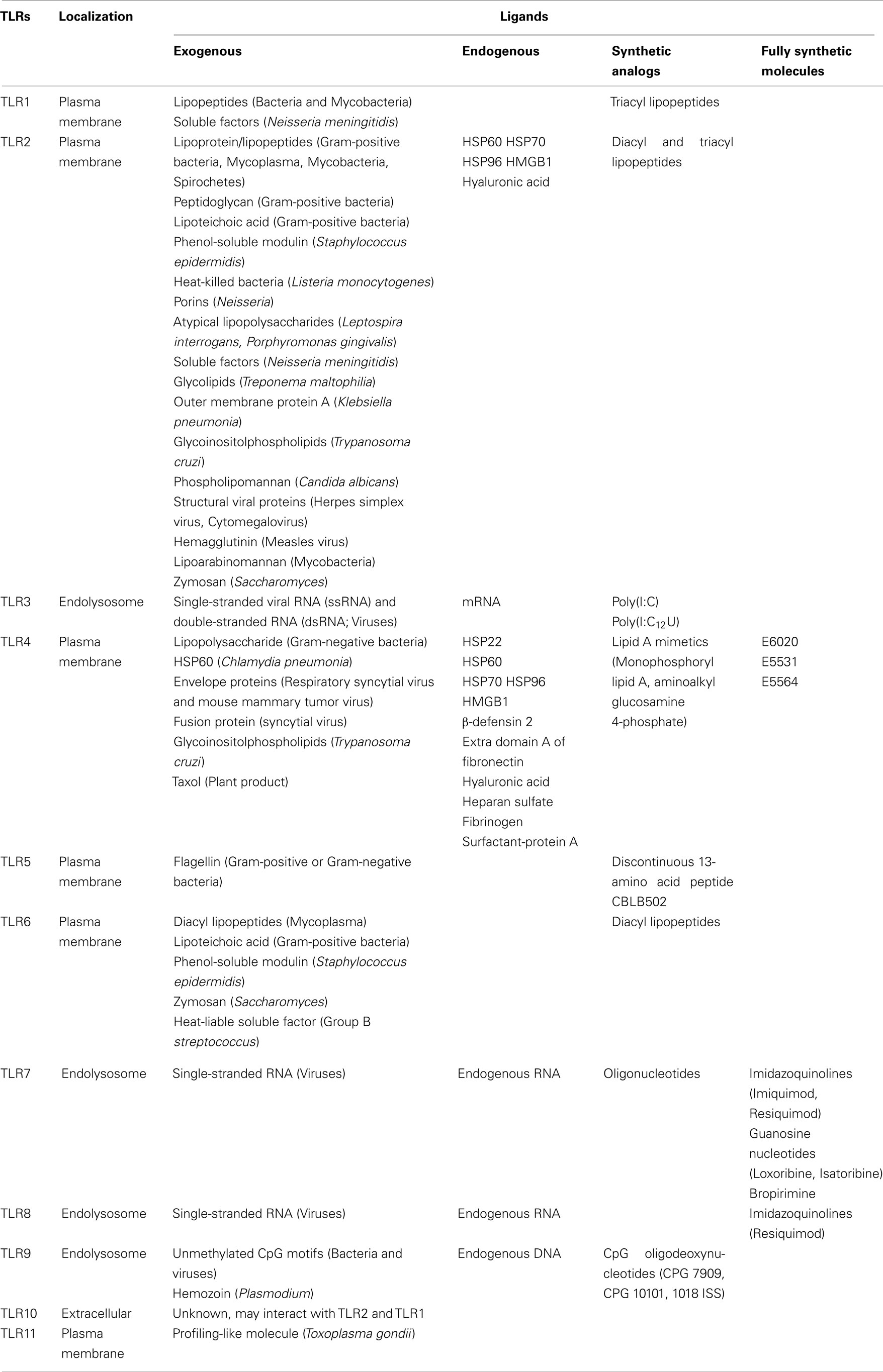
Table 1. Toll-like receptors and their principal ligands.
The common mechanism of TLR signaling is that interaction of an agonist with the ECD either induces the formation of a receptor dimer, or changes the conformation of a pre-existing dimer (Latz et al., 2007; Zhu et al., 2009) in such a way that it brings two intracellular TIR domains of the TLRs to interact physically. This simple rearrangement serves as a nucleating act for the recruitment of downstream signaling adapter proteins (Jin and Lee, 2008). Signaling cascades via the intracellular TIR domains are mediated by specific adaptor molecules such as Myd88 (Myeloid differentiation factor 88), Mal (Myd88 adaptor like), TRIF (TIR domain containing adaptor inducing interferon-β), and TRAM (TRIF related adaptor molecule). These adaptor proteins also contain TIR domains that mediate TIR–TIR interactions between TLR receptors, receptor–adaptor, and adaptor–adaptor interactions that are critical for signaling (Palsson-Mcdermott and O’Neill, 2007). In general, intracellular TIR domain of adaptor proteins are composed of approximately 160 amino acid residues and the primary sequences of TIR domains are characterized by three conserved sequence boxes designated Box 1, 2, and 3. Box 1 is considered to be the signature sequence of the family, whereas boxes 2 and 3 contain functionally important residues involved in signaling (Carpenter and O’Neill, 2009). These processes result in the formation of a large multimer complex, or “signaling platform,” that propagates downstream signaling, eventually leading to changes in the expression of several hundred primary immune response genes. However, the architecture of the TLR signaling complexes is poorly understood at this time due to a lack of reliable methods to study such interactions as well as the inherent weaknesses of individual inter- and intra-protein interactions in transitory complexes.
Structural studies of TLR–ligand complexes have been an attractive area of research that has enabled a better understanding of the structure based activation of innate immunity. Such information is essential for the development of adjuvants that specifically bind to TLR ECD and activate its signaling and also in the development of anti-inflammatory drugs that block TLR mediated signaling. To date, five TLR–ligand structures (TLR1–TLR2–Pam3CSK4, TLR2–TLR6–Pam2CSK4, TLR4–MD-2–Eritoran, TLR4–MD-2–LPS, and TLR3–dsRNA) have been determined (Jin et al., 2007; Kim et al., 2007b; Liu et al., 2008; Kang et al., 2009; Park et al., 2009). Currently, these solved atomic models can be used as templates to predict the structures of other unknown TLRs. In this review article, we discuss how similar structures of TLR ECD LRRs have evolved to bind a wide array of different ligands and their activation mechanism.
The ECD of TLR members contains multiple blocks of LRR, which are protected by cysteine rich regions to form cap-like structures at the LRR-N- and -C-terminal ends. The C-terminal capping structure of TLRs is connected to the cytoplasmic TIR domain via a single transmembrane α helix. Individual LRR module (approximately 20–30 amino acid residues long) consists of conserved “LxxLxLxxNxL” motifs and a variable region (Figure 1A). The conserved leucine residue in these motifs can be substituted by other hydrophobic amino acids (Matsushima et al., 2007). The asparagine residues that are also present in the motif form continuous H-bonds with the backbone carbonyl group of neighboring strands throughout the entire protein, resulting in an asparagine ladder. These conserved asparagine residues are important in maintaining the overall shape of the ECD, which can also be replaced by other residues such as cysteine, threonine, or serine, which are able to form H-bonds (Kajava et al., 1995; Kobe and Deisenhofer, 1995; Bell et al., 2003). The variable “x” residues present in the motif are exposed to the solvent. Among them, only few residues are involved in ligand recognition. The “LxxLxLxxNxL” motifs located in the inner concave surfaces of the horseshoe-like structure form parallel β-strands, whereas the variable region forms a convex surface generated by α helices, β-turns, and loop structures (Figure 1A). LRR proteins are present in a very large and diverse group of proteins and have been found to be involved in a wide variety of physiological functions including immune responses, signal transduction, cell cycle regulation, enzyme regulation, and transcriptional regulation (Buchanan and Gay, 1996; Dolan et al., 2007).
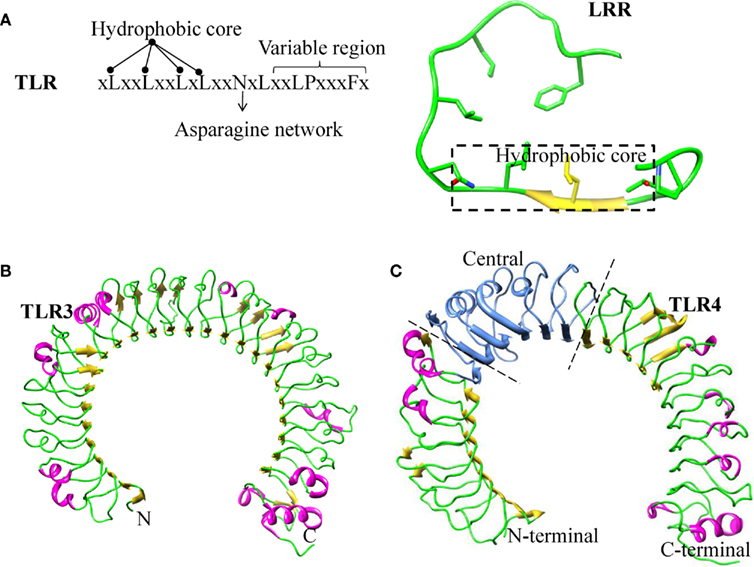
Figure 1. Structure of Leucine rich repeats. (A) LRR consensus repeats for TLR4. Residues forming the hydrophobic core, asparagine ladder and variable regions are mentioned. Secondary structure of LRR, the residues forming the hydrophobic core is highlighted in a box and the remaining portion of the LRR forming the convex surface. (B) Ribbon diagram of TLR3–ECD: LRR domain has uniform β sheet angles and a continuous asparagine network. (C) Ribbon diagram of TLR4 ECD showing the position of three sub domains: N-terminal, Central, and C-terminal.
The crystallization of some LRR proteins, including TLRs, has proven to be very difficult. This problem was overcome by the introduction of a new method known as the “hybrid LRR technique” (Jin et al., 2007; Kim et al., 2007a,b; Kang et al., 2009; Park et al., 2009). Hagfish variable lymphocyte receptors (VLRs) were chosen as fusion partners, and the TLR and VLR were fused at their conserved LxxLxLxxNxL motifs. Interestingly, the TLR–VLR hybrid demonstrated that the structure and function of the fusion proteins were not altered. Some hybrids fail to form soluble proteins due to the atomic collisions or the exposed hydrophobic core at the fusion sites. However, hybrids that produced soluble proteins formed stable heterodimers and possibly bound with ligands that were used for the crystallographic studies (Jin et al., 2007; Kim et al., 2007a,b; Kang et al., 2009; Park et al., 2009).
The LRR protein family can be classified into seven subfamilies based on their sequence and structural patterns. TLR belongs to the typical subfamily of the LRR superfamily (Kobe and Kajava, 2001; Matsushima et al., 2007). Each LRR region consists of 24 amino acid residues, possesses the conserved motif, xLxxLxxLxLxxNxLxxLPxxxFx, and displays a unique horseshoe shape structure (Figure 1B). LRR modules of TLR1, 2, 4, and 6, but not TLR3, have been shown to deviate from their conformation and length when compared with other typical members (Kim et al., 2007b; Jin and Lee, 2008; Kang et al., 2009; Park et al., 2009). These four TLRs have major structural changes in their central β-sheets; hence, their LRR domains can be divided into an N-terminal, central, and C-terminal domain, respectively (Figure 1C). The central domain of TLR1, 2, 4, 6, and 10 lacks an asparagine ladder, which is primarily responsible for the stabilization of the horseshoe-like structure. Furthermore, this broken asparagine ladder leads to unusual structural distortions. LRR modules of the central domain differ considerably in the number of residues, varying from 20 to 33. However, the LRR modules present in the majority of LRR proteins are of uniform length (Kajava et al., 1995; Kobe and Deisenhofer, 1995; Matsushima et al., 2007). LRR subfamilies with shorter LRR modules encompass loops in the convex surface, and those containing longer LRR modules have bulkier α helices. It should be noted that helices require more space than loops; therefore, subfamilies with α helices have smaller radii than those with loops that generate enough space in the convex region (Jin and Lee, 2008; Kang and Lee, 2011). This anomaly explains the structural conformation variations of TLR receptors and the ability of the receptor to bind with diverse ligands as well as co-receptors.
To date, five crystallographic structures of the TLR ECDs and their ligand complexes have been reported. Of those, four were complexed with agonistic ligands and the remaining one was complexed with a co-receptor and an antagonistic ligand. These structures provide evidence about how pattern recognition receptors (PRRs) recognize patterns present in the ligands. Additionally, these studies suggest that ECD activation mechanisms are also common among all TLR receptor family members.
Toll-like receptor-2 heterodimerizes with TLR1 or 6 to recognize multiple PAMPs of fungi, Gram-positive pathogens and mycobacteria (Kawai and Akira, 2010). TLR2 recognizes lipopeptides that are anchored to the bacterial membrane by lipid chains covalently attached to N-terminal cysteine (Hantke and Braun, 1973). Lipopeptides from Gram-negative bacteria have three lipid chains. Two of these are attached to the glycerol through an ester bond, which is in turn connected to the sulfur atom of the N-terminal cysteine. The third lipid chain is connected to the amino terminal via amide bonds. Lipopeptides from Gram-positive bacteria or mycoplasma have only two lipid chains and lack the amide-linked lipid chain (Muhlradt et al., 1997; Shibata et al., 2000). Synthetic lipopeptide analogs (Pam2CSK4, Pam3CSK4) containing a di- or tri-acylated cysteine group mimic the pro-inflammatory properties of the lipoproteins, which confirms that acylated N-terminal cysteine is the primary motif responsible for stimulating the immune response. Furthermore, TLR2 receptor also recognizes other ligands such as lipoteichoic acid, lipomannan, peptidoglycan, zymosan, and phenol-soluble modulin (Zahringer et al., 2008).
The crystal structure of TLR2 in association with TLR1 and a synthetic triacylated lipopeptide, Pam3CSK4, has been determined (Jin et al., 2007). Indeed, this is the first crystal structure of a TLR dimer resulting from the binding of agonists, which further explains the ligand-induced dimerization. In this structure, the ECD of TLR2 and 1 form an “m” shaped heterodimer, with the two N-terminals extending in the opposite direction and the C-terminals converging in the middle region (Figure 2A). Pam3CSK4 consists of three lipid chains, two of those insert into the hydrophobic pocket of TLR2 and the remaining one inserts into a narrow hydrophobic channel of TLR1 (Figure 2B). Apart from the acyl chain binding, the head groups of Pam3CSK4 also interact with TLRs 1 and 2. In particular, TLRs form H-bonds with glycerol and peptide backbone and also form hydrophobic interactions with sulfur atoms. The ligand-binding pockets of TLR1 and 2 are located at the junction of the central and C-terminal domains, indicating the importance of structural transition in the formation of ligand-binding pockets. The ligand binding in the convex surface of TLR2/1 was found to be quite unusual because most ligand-binding sites on LRR proteins that have been identified were found to be present on the concave surfaces (Kobe and Deisenhofer, 1995). The ligand bound complex of TLR1 and 2 is stabilized by non-covalent forces such as H-bonding, hydrophobic interactions and ionic interactions at the interface near the ligand-binding pocket. It is worth noting that TLR1 P315L polymorphic variation has been reported to interfere with TLR1 signaling (Omueti et al., 2007). In fact, this P315 residue is located at the TLR1/2 dimer interface, highlighting the importance of P315 in TLR1 and 2 heterodimerization. Moreover, species-specific lipoproteins response has also been observed (Grabiec et al., 2004). Lipopeptides with shorter lipid chains act as more potent activator in mouse than human TLR2. This phenomenon is mainly due to the structural variations observed in the TLR2 pocket (Jin et al., 2007).
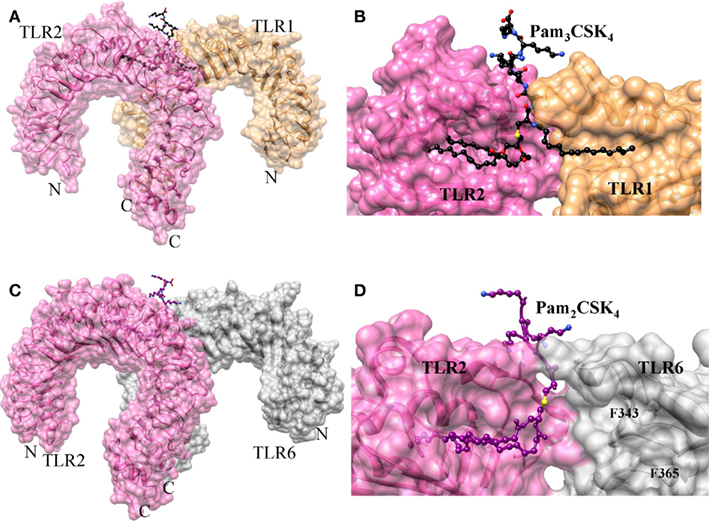
Figure 2. Structures of TLR2–TLR1/6 heterodimers induced by lipopeptides. (A) Crystal structure of TLR1/2–Pam3CSK4 complex. TLR1, TLR2, and Pam3CSK4 are colored in sandy brown, hot pink, and black, respectively. (B) Lipid-binding pocket in TLR1/2–Pam3CSK4 complex. The structures of TLRs are omitted to reveal the shape of lipid-binding pocket. (C) Crystal structure of TLR2/6–Pam2CSK4 complex. TLR2, TLR6, and Pam2CSK4 are colored in hot pink, gray, and deep magenta, respectively. (D) Lipid-binding pocket in TLR1/2–Pam2CSK4 complex. The lipid-binding channel is blocked by F343 and F365.
The crystal structure of TLR2 in association with TLR6 and a synthetic diacylated lipopeptide Pam2CSK4 has been determined (Kang et al., 2009). In this structure, the ECD of TLR2 and 6 form an “m” shaped heterodimer, with the two N-terminals extending in the opposite direction and the two C-terminal ends converging in the middle region (Figure 2C). The dimeric arrangement of TLR2/6 is similar to TLR2/1 complex. However, TLR1 and 6 contain important structural differences in their ligand-binding sites and dimerization interface. In TLR6, the side chains of two phenylalanine (F343 and F365) residues block the lipid-binding pocket, leading to a pocket that is less than half the length of the TLR1 (Figure 2D). This structural feature provides selectivity for diacylated over triacylated lipopeptides, as confirmed by the mutation studies of these phenylalanine residues to the corresponding amino acids of TLR1 that rendered TLR6 fully responsive not only to diacyl but also to triacylated lipopeptides. In the TLR2/6 complex, two-ester bound lipid chains of Pam2CSK4 are inserted into a hydrophobic pocket in TLR2 that is located between the LRR11 and 12 loops. Whereas, F319 located in the LRR11 loop of TLR6, forms an H-bond with the peptide bond of the ligand. Such an H-bond network is absent in the TLR2–TLR1–Pam3CSK4 structure. Moreover, TLR2-6 heterodimerization is primarily mediated by surface exposed residues of LRR11-14 modules. In the TLR2-1 complex, the amide bound lipid chain plays an important role in bridging the two TLRs. Although Pam2CSK4 lacks these amide bound chains, it still forms a dimer, primarily through hydrophobic and hydrophilic interactions of their surface exposed residues between the two TLRs. This area of hydrophobic interaction is 80% larger than in the TLR1/2 complex, suggesting that this surface interaction together with the H-bond between LRR11 and the ligand drives the heterodimerization of TLR6.
During the course of TLR2–TLR6–diacylated lipopeptide complex determination, TLR2 in complex with two non-peptide ligands, Streptococcus pneumonia lipoteichoic acid (pnLTA) and PE-DTPA (1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-diethylenetriaminepentaacetic acid), has been determined (Kang et al., 2009). PE-DTPA is a synthetic derivative of phospholipid in which metal coordinating DTPA is attached to the ethanolamine head group. In the monomeric TLR2–pnLTA structure, the overall horseshoe-shaped structure of TLR2 and the ligand-binding pocket remain unchanged. When compared with TLR2-6–Pam2CSK4, the position of the sugar head group of LTA of the TLR2–pnLTA complex displaces upward by ∼5.2 Å and rotated by 110 Å toward the lateral surface of the ECD. Moreover, the hydrogen donor and acceptor atoms in the sugar head group of pnLTA have a different arrangement than the lipopeptides. Hence, it is not possible to form an H-bonding network. Due to the shift, TLR1 or TLR6 cannot approach TLR2 to form heterodimers. In the TLR2–PE-DTPA structure, the acyl chain and head group arrangements are similar to those of TLR2–pnLTA. When compared with TLR2-6 lipopeptide complexes, the head group of PE-DTPA is shifted ∼4.3 Å. This structural shift primarily occurs due to a lack of proper H-bonding between the ligand head group and the TLRs, as well as to repulsion of the hydrophilic oxygen atom of the ligand, whose corresponding position in lipopeptide contains sulfur that forms a hydrophobic interaction with TLRs. These complexes (pnLTA and PE-DTPA) have little or no ability to activate TLR2 because of the structural shift in ligand head groups, which strongly suggests that the ligand/lipopeptide head group plays an important role in TLR2 activation via heterodimerization. A large proportion of TLR2 ligands are lipopeptides that can bind to the TLR2 hydrophobic pocket, but some TLR2 ligands including peptidoglycan, hyaluronic acid, teichoic acid, and zymosan do not contain this hydrophobic region (Table 1). Hence, the interaction of these ligands with TLR2 might use different binding sites. Further crystallographic or modeling studies are required to clarify the exact binding sites of non-lipid ligands and to verify whether these bindings induce the formation of similar heterodimeric structures such as TLR1-2 or TLR2-6.
Toll-like receptor-3 has been shown to recognize dsRNA produced during viral replication (Alexopoulou et al., 2001). The first TLR3 structure was identified independently by two different groups (Bell et al., 2005; Choe et al., 2005). Both groups have shown that the LRR region of TLR3 displays a heavily glycosylated horseshoe-shaped solenoid structure. Choe et al. (2005), postulated that dsRNA might bind at the convex surface because this region is a glycan-free face, which enables dsRNA to bind to the positively charged residues of the TLR ECD. However, Bell et al. (2005) suggested that the nucleotide binding site is located in the concave surface. This is likely due to the fact that during crystallization, two sulfate molecules from the crystallization medium stably bound to residues in LRRs 12 and 20, and these two LRRs contain large insertions. As the sulfate ions share the same atomic arrangement as phosphate groups, those present in the dsRNA backbone might be able to bind to one or both of the sulfate binding sites. Hence, each group prediction differs in the dsRNA binding sites and it was not clear how TLR3 specifically recognizes dsRNA and initiates signaling. However, the recently solved crystal structure of mTLR3 bound to dsRNA explains how this is accomplished (Liu et al., 2008). TLR3 ECD exists as a monomer in solution and the dimerization only occurs upon ligand binding. In the structure, dsRNA interacts with both the N- and C-terminal sites on the lateral side of the convex surface of the TLR3 ECD (Figure 3A). The N-terminal interaction sites are composed of LRRNT and LRR1-3 modules, whereas the C-terminal site is composed of LRR19-21 modules. The dsRNA in the complex retains a typical A-DNA like structure, in which the ribose phosphate backbone and the position of the grooves are the major determinants in binding (Figure 3B). The mTLR3–ECD interacts with the sugar phosphate backbones, but not with individual bases, which accounts for the lack of any particular nucleotide specificity in binding (Alexopoulou et al., 2001; Leonard et al., 2008). This feature would prevent the viruses from escaping detection by mutation (Botos et al., 2011). Moreover, the identified structure reveals the possible reasons for the inability of TLR3 to recognize dsDNA. The helical structure of dsDNA is the B form, whereas dsRNA is present in A form. The B form helical structure would not be structurally compatible with the two terminal binding sites on the TLR3–ECD. Moreover, several H-bonds were observed between TLR3–ECD and the 2′-OH groups of dsRNA that is missing in dsDNA.
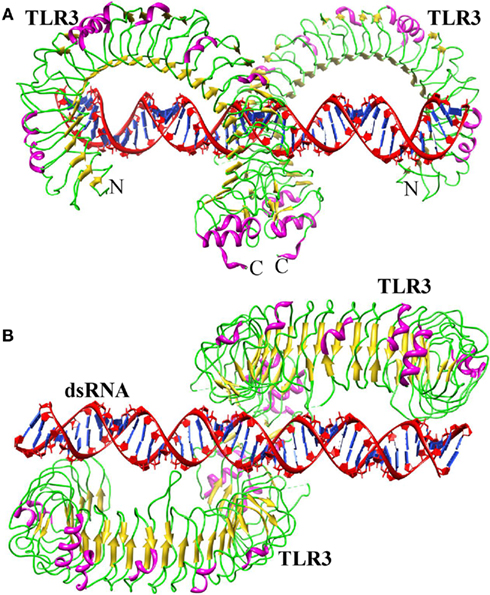
Figure 3. Structure of TLR3–dsRNA. (A) Ribbon structure of TLR3 dimer (colored according to the secondary structure: helix-pink; sheet-yellow; loop-green) bound with dsRNA (red). (B) Top view.
The TLR3–TLR3 interaction site located near the LRRCT occupies only a small portion, demonstrating that ligand–protein interaction as the major driving force behind TLR3 dimerization. The ligand interaction sites (two TLR3 ECD N-terminal regions) are separated by about 120 Å, thus showing why only 40–50 base pairs are sufficient for the stabilized binding of dsRNA to TLR3 (Leonard et al., 2008). However, there have also been study reports of dsRNA of substantially less than 40 base pairs being able to initiate TLR signaling (Kariko et al., 2004; Kleinman et al., 2008). This raises the possibility that the N-terminal interaction site is not essential for efficient TLR3 signal induction in some experimental conditions. Moreover, mutation studies have identified functional amino acid residues in three different regions (N-terminal, C-terminal, and dimerization region) of the TLR3 ECD. H539E and N541A mutation in the C-terminal, H39A/E and H60A/E mutation in the N-terminal region, and D648A, T679A, and P680L in the dimerization region leads to a loss of TLR3 activity (Bell et al., 2006; Ranjith-Kumar et al., 2007; Fukuda et al., 2008; Wang et al., 2010). Although hydrophobic interactions play a crucial role in binding of lipopeptides to TLR1/2 and 2/6, the TLR3 interaction with dsRNA mainly involves electrostatic interactions and H-bonds. Despite these differences in ligand interactions, the ligand-induced dimers of TLR3, TLR2-6, and TLR1-2 adopt a similar fold, the “m” shaped dimer, in which the two C-termini of the TLR ECDs are in proximity, thereby bringing the two TIR domains together on the cytoplasmic side and providing a scaffold for the recruitment of adaptor proteins and subsequent initiation of further downstream signaling.
A crystal structure of human TLR4–MD-2 complex binding with an antagonist (Eritoran) has been described (Kim et al., 2007b). Unlike other TLRs that recognize ligands directly, TLR4 does not directly interact with ligands. Alternatively, TLR4 forms a stable 1:1 heterodimer with MD-2 and uses the hydrophobic pocket in MD-2 to interact with the LPS of Gram-positive bacteria (Shimazu et al., 1999). Two accessory proteins such as lipid-binding protein (LBP) and CD14, whose main function is to extract LPS from the bacterial membrane and transferring it efficiently into MD-2. The general structure of bacterial LPS consists of a hydrophobic lipid A domain, an oligosaccharide core and a distal polysaccharide (the O antigen; Bryant et al., 2010). Lipid A moiety alone is sufficient to activate innate immune responses. Lipid A consists of a diglucosamine diphosphate head group that is substituted with a variable number of acyl chains, ranging from four to eight. In general, lipid A moieties consisting of hexa acylated lipid chain and two phosphate groups are powerful immune stimulators, whereas Lipid A with five acyl chains have ∼100-fold less activity. Several synthetic derivatives of lipid A have been developed as candidate drugs against sepsis and septic shock syndrome. Eritoran or E5564 is a synthetic molecule derived from the lipid A component of non-pathogenic LPS of Rhodobacter sphaeroides. This compound contains only four acyl chains and acts as a strong antagonist of TLR4–MD-2 complex and is currently in Phase III clinical trial (Mullarkey et al., 2003; Rossignol and Lynn, 2005).
Toll-like receptor-4 ECD has 22 LRRs capped by LRRNT and LRRCT at its N- and C-termini, respectively. MD-2 has a cup fold like structure and is composed of antiparallel β sheets forming a large hydrophobic core, with the surface area of ∼1000 Å that is able to bind with ligand. The opening of the pocket is lined with positively charged residues and three disulfide bridges that stabilize the cup-like structure. It should be noted that MD-2 does not have either a transmembrane or an intracellular domain; hence it is not able to transmit the signals. Recent TLR4 and MD-2 complex clearly indicated that only one-third of MD-2 is involved in TLR4 binding, the remaining part is available for the interaction with ligands (Kim et al., 2007b; Park et al., 2009). The MD-2 binding site of TLR4 can be divided into two chemically and evolutionary distinct areas, termed as A and B patches. The A patch is provided by the N-terminal domain of TLR4, which is mainly comprised of negatively charged amino acids. The B patch is located in the central domain that is predominantly comprised of positively charged residues. The TLR4 binding surface of MD-2 shows a clear charge complementarity to the TLR4 surface (Figure 4E). In the crystal structure, four acyl chains of Eritoran occupy approximately 90% of the solvent accessible volume of the MD-2 pocket. Of those, two acyl chains are in the fully extended conformation within the binding pocket, while the remaining two acyl chains are bent in the middle (Figure 4A). The diglucosamine backbone is fully exposed to the solvent and the phosphate groups make ionic contacts with positively charged residues at the surface of the pocket. Additionally, there is no direct interaction between Eritoran and TLR4 (Kim et al., 2007b). Indeed, this is very similar to the recently identified structure of MD-2 in complex with the lipid IVA (Figure 4B; Ohto et al., 2007). Lipid IVA, or compound 406, is an intermediate in LPS biosynthesis, which contains four lipid chains with lengths and structures that differ from the Eritoran. Lipid IVA acts as an antagonist of human TLR4–MD-2, but behaves as an agonist of mouse TLR4–MD-2 (Means et al., 2000). Despite the significant structural differences seen between lipid IVA and Eritoran, their binding modes are similar. The structural superimposition of TLR4–MD-2–Eritoran and MD-2–lipid IVA have shown that lipid chains of different lengths are accommodated in the MD-2 pocket with only a 2-Å shift in the glucosamine backbone (Kim et al., 2007b). In both lipid IVA–MD-2 and Eritoran-TLR4–MD-2 structures, the ligands did not induce any conformational changes in the receptors, thereby demonstrating that these molecules are antagonists.
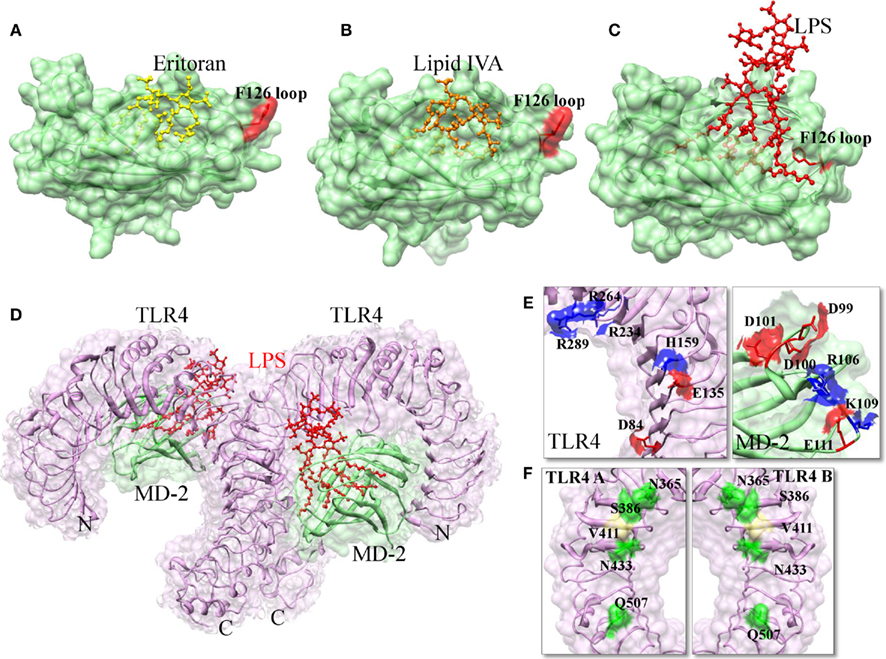
Figure 4. The interactions of agonist and antagonistic ligands in TLR4–MD-2 complex. (A) When TLR4–MD-2 binds to Eritoran, the F126 loop is exposed to the solvent area. (B) When MD-2 binds to lipid IVA, the F126 loop is exposed to the solvent area. (C) When TLR4–MD-2 binds to LPS; the F126 loop forms hydrophobic interactions with lipid chains and the second TLR4. This interaction causes a structural shift in the F126 loop, which enables the correct positioning of the R2 lipid chain to interact with the second TLR4 as well as TLR4 dimerization to occur. (D) Structure of TLR4–MD-2–LPS complex. TLR4, MD-2, and LPS are colored in magenta, light green, and red, respectively. (E) TLR4–MD-2 dimer interface formed by electrostatic interaction. Positive and negative charged residues are marked in blue and red color, respectively. (F) TLR4 homodimer interface. Hydrophilic and hydrophobic residues are colored in green and khaki, respectively.
The much anticipated TLR4–MD-2–LPS complex has recently been solved (Park et al., 2009). The authors demonstrated that TLR4 and MD-2 proteins associate with each other without LPS, but the dimerization of the TLR4–MD-2 complex with another TLR4–MD-2 occurs only via binding of LPS. The receptor multimer is composed of two copies of the TLR4–MD-2–LPS complex arranged in a symmetrical fashion (Figure 4D). In the crystal structure, five of the six lipid chains of LPS bind to this pocket, while the remaining lipid chain that is exposed on the surface of MD-2 forms hydrophobic interactions (F440, F463, and L444) with the second TLR4 (Figure 4C). Mutation of the F440 and F463 interface residues disrupt TLR4 dimerization and its signaling (Resman et al., 2009). The binding of LPS induces localized conformational changes in MD-2, primarily on the F126 loop region, which leads to the hydrophilic residues in the F126 loop and R90 residues of MD-2 form H-bonds and ionic interactions with the second TLR4, further stabilizing the complex. In addition to the above major interaction, TLR4 makes an additional contribution to dimerization by directly interacting with second TLR4 (Figure 4F). The previously solved MD-2 bound to the Eritoran and lipid IVA structures revealed that F126 of MD-2 was exposed to the solvent, thereby showing no conformational changes and hence MD-2 complex was unable to induce TLR4 dimerization. Park et al. (2009) clearly demonstrated that structural changes that mainly occurred at the F126 loop of MD-2 following LPS simulation are necessary for the dimer formation and subsequent initiation of downstream signaling. Mutation studies of the F126 residue of MD-2 supports this finding. The mutation of F126 did not affect LPS binding; however, it abolished the ability of the TLR4–MD-2 heterodimer to form the activated heterotetramer, suggesting that these residues form part of the dimerization region (Kobayashi et al., 2006; Kim et al., 2007b). Moreover, LPS contain two phosphate groups that are important for forming ionic interactions with positively charged residues on both TLR4 and MD-2. Comparison of LPS bound MD-2 with Eritoran–MD-2 indicates that the additional two lipid chains in LPS displace the phosphorylated glucosamine backbone upward by 5 Å toward the solvent area, which allows the phosphate groups to associate with the second TLR4 (Park et al., 2009). In addition to the displacement, the glucosamine backbones are also rotated by 180°, interchanging the phosphate groups. It should be noted that there is a general rule for TLR signaling (based on the structural and biochemical studies); specifically, TLR agonists induce TLR dimerization, whereas antagonists are likely to interfere with dimerization (Brodsky and Medzhitov, 2007).
Crystallographic studies have provided almost 50% of the mammalian TLR structures (TLR1, 2, 3, 4, and 6), which have provided a basis for the understanding of agonistic induced TLR activation and antagonistic mediated TLR inhibition. Each TLR member recognizes “n” number of ligands starting from the microbes, and each ligand has its own unique properties. From this review, we come to know that the binding sites of these ligands cannot be similar in all TLRs. For example, TLR4 recognizes various ligands (Table 1), but the binding site of those ligands are not the same as LPS in TLR4–MD-2 complex. X-ray crystallographic studies have revealed that there are only a limited number of TLR ECD interactions with ligands. The identification of all ligand interactions with each TLR member (listed in Table 1) using X-ray crystallographic studies have proven to be very difficult. Hence, we have to rely on molecular modeling studies along with biochemical validation, to gain further insights into these interactions.
To date, approximately 20 molecular modeling studies have investigated on TLR signaling. These studies include: (i) prediction of TLR ECD using available TLR crystal structures as a template and identification of its possible ligand-binding region. (ii) Structural basis identification of positive and negative regulators in TLR signaling and (iii) Identification of the interaction between the TIR domain and its adaptor molecules, which provides structural insights into the mechanism responsible for TLR mediated downstream activation or inhibition.
The first modeling study reported the structures of the mouse (m) and human (h) TLR4 ECD. These structures were generated using the first solved hTLR3 structure as a template (Kubarenko et al., 2007). Their target–template alignment showed that N-terminal and C-terminal domains aligned with the template, but the central domain did not align well. Hence, the alignment of this portion was conducted individually by matching LRRs in hTLR3. These sub domains (N-terminal, C-terminal, and individual LRRs) were manually assembled and subjected to MD simulation. Their analysis revealed that the central domain of TLR4 ECD (LRR9-13) is hypervariable across human and mouse. It should be noted that the ECDs of TLR7 and 9 are cleaved in the endolysosome to recognize ligands, and this cleaved form is necessary for Myd88 activation (Kawai and Akira, 2010; Basith et al., 2011b). Wei et al. (2009) generated structural models of cleaving ligand-binding domains of TLR7, 8, and 9. Based on comparison of the structures, they have identified potential ligand-binding sites as well as possible configurations of the receptor–ligand complexes. Conversely, Kubarenko et al. (2010) modeled full length ECD structures of TLR7, 8, and 9. Structural comparison of these ECDs revealed that the insertion mainly takes place in the TLR9 loop regions (LRR2, 5, and 8), which contains primarily cysteine and few proline residues (Kubarenko et al., 2010). Finally, the loop insertion residues have been quantified through biochemical studies and identified the functional role of these residues (C98, C110, P183, C184, C265, C268, and P269) in TLR9 signaling. The first modeling report to show the ligand binding to the TLR ECD is TLR5, whose concave surface interacts with flagellin and the biochemical studies provided that D296 and D367 of TLR5 are necessary for mediating this interaction (Andersen-Nissen et al., 2007).
Recently, the LRRML and TollML tools were designed to identify appropriate templates for each LRR and the functional annotation of TLR primary sequences, respectively (Wei et al., 2008; Gong et al., 2011). LRRML, the program produces the alignment for each LRR along with templates that were subsequently used for homology modeling of LRR proteins. Generally, one or more full length protein has been used as a template for modeling. However, due to variations in the LRR numbers among TLRs, sequences with low similarity between the target and full length template are usually not sufficient for homology modeling. The LRRML tool was developed to address this issue. This tool currently contains 1261 individual LRRs (obtained from 112 PDB structures) that serve as a local template for each target. As a test case, the developers modeled the structure of the mouse TLR3 ECD and excluded the LRRs of the mouse/human TLR3 ECD from the LRRML dataset. The final 26-line multiple alignments were generated by 25 template sequences and the target sequences were used for modeling. Superimposition of the modeled TLR3 structure with the actual TLR3 crystal structure revealed an RMSD value of 1.9 Å, confirming the reliability of modeling studies. This method has since been used to predict series of human TLR5-10 and mouse 11–13 (Wei et al., 2010). These models can be used to conduct ligand docking studies or design mutagenesis experiments to investigate the TLR–ligand-binding mechanism. Recent studies by our group have shown that the Pam3CSK4 might be the ligand for the TLR2/10 complex and Pam2CSK4 might activate TLR10/6 and TLR10 homodimer. The predicted TLR10 complexes are similar to the available TLR1 family complexes. However, the binding orientation of TLR10 homodimer was different due to the presence of negatively charged surface near LRR11-14, that defined the specific binding pocket (Govindaraj et al., 2010). This has been the first study to suggest the possible ligands for TLR10. Our predictions were also confirmed by the recent biochemical studies by showing that chimeric receptors [TLR10 ECD and endodomain (TIR) TLR1] along with TLR2 recognize triacylated lipopeptides (Guan et al., 2010).
It is well known that lipid IVA acts as an agonist or antagonist for TLR4–MD-2 complex, depending upon the species. To identify the species specificity, Walsh et al. (2008) conducted modeling studies and identified differences in primary sequences among the species (mouse, cat, horse, and human). Mouse, cat and horse species were able to induce signaling in response to lipid IVA, whereas human species were not able to induce signaling, primarily due to the conservative substitution. However, this reason alone cannot be expected to have a large influence on the overall structure of the protein. Furthermore, they identified significant differences in the local charge distribution on the surfaces of MD-2 and TLR4 from different species, which suggests that electrostatic forces also govern the pharmacology of lipid IVA, further leading to the transduction of TLR4 signaling. In general, the assembly of active TLR4 complexes is a stepwise process, with initial TLR4–MD-2 complex formation being induced by the binding of lipid IVA, further promoting the subsequent homodimerization of receptor ECDs. In the modeled complex structure, LRR 15–17 modules were found to participate in the main dimerization interface of TLR4. Their predicted modeling and mutagenesis data were remarkably accurate when the LPS bound TLR4–MD-2 crystal structure was released (Park et al., 2009).
African swine fever viruses (ASFV) encode a novel protein (pI329L) that has been shown to inhibit TLR3 signaling pathway. Modeling studies have shown that pI329L structural arrangement is similar to TLRs (Henriques et al., 2011). However, the difference observed in ECD of pI329L, which is shorter than the TLR. This protein forms a heterodimer with TLR3, thus acting like a decoy receptor, demonstrating that viral protein hinders the TLR3 homodimerization, and thereby inhibiting the TRIF mediated pathways. A recent study showed that the pentameric B subunit of type IIb Escherichia coli enterotoxin (LT-IIb-B5), a non-lipidated protein ligand, activates TLR2/1 signaling pathways. Molecular modeling along with mutagenesis studies showed that the upper pore of LT-IIb-B5 (M69E, A70D, L73E, and S74D) defines an interactive surface for binding with the concave surface of the TLR2/1 central domain (Liang et al., 2009). Unlike TLR2–TLR1–triacylated lipopeptide complex, non-lipidated ligands cannot fit into the small hydrophobic channel; however, these ligands can engage in TLR surface interactions via specific residues.
Toll-like receptor ECD activation leads to TIR dimerization of TLRs, which creates specific scaffold for the binding of adaptor proteins such as Myd88, Mal, TRIF, and TRAM. This assembly of the TIR complexes activates the downstream signaling pathways, leading to the expression of pro-inflammatory cytokines, antiviral response and also in the initiation of adaptive immunity. To date, five mammalian TIR structures have been reported (TLR1, TLR2, TLR10, IL-1RAPL, and Myd88; Xu et al., 2000; Tao et al., 2002; Khan et al., 2004; Nyman et al., 2008; Ohnishi et al., 2009). All these TIR domains, containing alternative β strands and α helices are arranged as a central five stranded parallel β sheets surrounded by α helices. The TIR domains of TLR1 and TLR2 exist as a monomer in the crystal. Conversely, TLR10 TIR domain without the extracellular and transmembrane regions behaves as a monomer in solution, but it forms a homodimer in the crystal asymmetric unit. This structure has been used to represent the signaling dimer of TIRs. In the TLR10 TIR dimer interface, BB-loop connecting the βB strand and the αB helix, and the death domain (DD) loop connecting the βD strand and the αD helix, have been reported to be important for the downstream signaling. Moreover, part of the BB-loop exposed to the surface is essential for the binding of the adaptor proteins during signal transduction (Nyman et al., 2008).
On the basis of TLR10 TIR structure, TLR4 TIR homodimer has been modeled by computational studies and identified two symmetrically related interfaces that are potentially capable of binding to adaptors, Mal and TRAM (Figure 5; Nunez Miguel et al., 2007). It is of worth noting that TLR4 TIR P681H polymorphism variation has been reported to abolish signal in response to LPS. In fact, this P681 located at the BB-loop, highlights its importance in TIR dimerization. Moreover, this model indicates that two adaptors could bind simultaneously to the TLR4 TIR dimer. Another important question raised by this study is whether adaptors binding is mutually exclusive, that is whether a single activated receptor complex recruits either Mal or TRAM, but not both simultaneously. Kagan et al. (2008) suggested that TLR4 signaling via Mal–Myd88 occurs at the plasma membrane and the signaling via TRAM–TRIF might be endosomal.
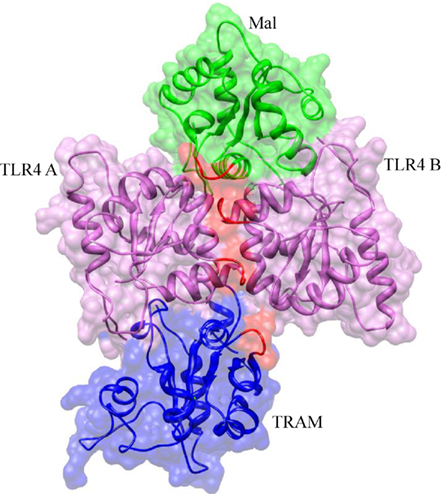
Figure 5. Molecular model of MAL and TRAM TIR domains bridged to the activated TLR4 TIR domains. The BB-loops in each TIR domain are highlighted in red. MAL and TRAM proteins are both predicted to bind to the TLR4 homodimer interface. It is probable that binding of MAL or TRAM protein is mutually exclusive, with the former binding to activated receptors at the cell surface and the latter in endosomes.
The crystal structures of bacterial (Chan et al., 2010) and the plant TIR domains (Chan et al., 2009) are highly homologous to those of mammalian TIRs. In bacterial TIR domain, the dimerization interface involves DD loop but not the BB-loop (important for TLR10 dimer). Chan et al. (2009) suggest that the BB-loop is not important for the homotypic interactions but may have a defined role in the heterotypic interactions with Mal or Myd88 TIR domains. Moreover, the available TIR structures lack the region immediately following their transmembrane segment, further making it hard to predict their exact orientation. Myd88 contains an N-terminal DD that is separated from the C-terminal TIR domain by a short linker sequence. After binding of Myd88 TIR domain to TLR TIR domains, Myd88 DD can interact with DD members of IRAK family, to activate their downstream signaling cascades. Recently, Lin et al. (2010) identified the complex crystal structures formed by DD of Myd88, IRAK4, and IRAK2. This complex structure known as Myddosome, consists of left handed helical structure in the order of 6 Myd88, 4 IRAK4, and 4 IRAK2 DDs. Like TIRs, DD are small globular proteins but have an anti parallel α helical fold rather than α–β structure. The dimerization of TLR TIR dimer recruits two Myd88 TIR domains then the larger myddosome superhelix could possibly bridge several activated receptor dimer in the network (Gay et al., 2011). Polymorphism of S34Y and R98C in the human DD, interfere with the myddosome assembly and may contribute susceptibility to infection (George et al., 2011).
Single immunoglobulin interleukin-1 receptor TIR domain (SIGIRR) and ST2L, belong to the TIR/IL-1R superfamily, which act as a negative regulator of Myd88-dependent TLR signaling. Specifically, this family attenuates the recruitment of Myd88 adaptors to the receptors via its intracellular TIR domain. Thus, these molecules are highly important for the treatment of autoimmune diseases caused by TLRs. Gong et al. (2010) proposed a residue detailed structural framework of SIGIRR inhibiting the TLR4 and 7 signaling pathways. In their multimer complex, SIGIRR exerts its inhibitory effect by blocking the molecular interface of TLR4, TLR7, and Myd88 adaptors, mainly via its BB-loop region. Our group proposed a structural framework of ST2L inhibiting the TLR4, TLR2/1, and TLR2/6 signaling pathways (Basith et al., 2011a). Apart from this, our group identified the structure based modulation of IκB family proteins. These proteins are structurally similar that are activated by TLR signaling and it has specific role in the cytoplasm and the nucleus by interacting with different subunits NF-κB dimer. Although the structures are similar, the binding specificities of these proteins remain unknown. The modeling studies have identified that variation in charged surfaces among the IκB proteins and also differences in the flexible residual position might be the chief factor for the IκB protein binding specificities (Manavalan et al., 2010, 2011).
In the past few years, there has been tremendous progress in the study of interaction of TLRs with their ligands and activators. Herein, we have discussed recent structural information regarding the TLR family and its proposed activation and inhibition mechanisms. Recent crystallographic studies of TLR1/2, 2/6, 4, and 3 have provided an explanation for in vivo, in vitro, and clinical observations. The solved structures have demonstrated that TLR exists as a monomer in solution and that dimerization takes place only upon ligand binding. Conversely, TLR8 and 9 exist as preformed dimers that subsequently change the conformation upon ligand binding. The solved (TLR1, 2, 6, 3, and 4) and modeled TLR ECD structures appear to have a common fold that belong to a well known LRR family with repeated LRR modules. Sequence and structural analyses indicate that TLRs present in the extracellular membrane (TLR1, 2, 4, 6, and 10) belong to a three-domain subfamily that binds to hydrophobic ligands such as lipoprotein, LTA and LPS. Conversely, TLRs present in the endolysosome (TLR3, 7, 8 and 9) belong to a single domain family that interacts with hydrophilic proteins or nucleic acids. This ligand-induced dimerization leads to the juxtamembrane sequences at the C-terminal ECDs coming into close proximity. These sequences are then transmitted across the transmembrane, resulting in reorientation or homodimerization between the receptor TIR domains. The homodimeric receptor TIR domains provide specific molecular surfaces for the recruitment of adaptor TIR domains. Although the structures of the TLRs are similar, the binding pocket and electrostatic surfaces are not conserved among these receptors. These variations are mandatory for the discrimination of the ligand specificity in each TLR family member. For example, triacylated lipopeptides bind to the hydrophobic binding pocket of TLR1/2; however, LT-IIb-B5 protein binds to the same receptor on another surface rather than the hydrophobic pocket. This is primarily due to the patterns present in the ligands with different properties (lipids and proteins), which causes the binding site of ligands to vary among all TLRs according to the surface and cavity provided by the receptors.
It is essential that we continue to develop a thorough and detailed understanding of the structural or molecular interactions of the ligands listed in Table 1 with their corresponding TLR family members. Such studies facilitate the rational design of receptor agonists and antagonists, leading to potential improvements in the treatment of diseases. However, there are still many important unanswered questions about TLR signaling. For example, the conformation of the transmembrane spanning segment once the TLR ECDs are activated is not known. The process leading to the recruitment of adaptor proteins following TLR activation is also not clear. Furthermore, it is not known if other ligands bind to the receptors in the same orientation and induce similar “m” shaped dimerization as seen in crystal structures. TLR4 receptor activation requires a co-receptor such as MD-2, but further work is needed to determine if this mechanism holds true for TLR4 designed agonists and if these synthetic agonists also need a co-receptor to bind with TLR receptor. The recent advances that have been made in structure–function analyses should allow many of these questions to be resolved in the near future.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Basic Science Research Program through the NRF of Korea funded by the MEST (2010-0016256). This work was also partly supported by a grant from the Korea Food and Drug Administration (10182KFDA992) and the Priority Research Centers Program (NRF 2010-0028294).
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738.
Andersen-Nissen, E., Smith, K. D., Bonneau, R., Strong, R. K., and Aderem, A. (2007). A conserved surface on Toll-like receptor 5 recognizes bacterial flagellin. J. Exp. Med. 204, 393–403.
Basith, S., Manavalan, B., Govindaraj, R. G., and Choi, S. (2011a). In silico approach to inhibition of signaling pathways of toll-like receptors 2 and 4 by ST2L. PLoS ONE (in press).
Basith, S., Manavalan, B., Lee, G., Kim, S. G., and Choi, S. (2011b). Toll-like receptor modulators: a patent review (2006–2010). Expert. Opin. Ther. Pat. 21, 927–944.
Bell, J. K., Askins, J., Hall, P. R., Davies, D. R., and Segal, D. M. (2006). The dsRNA binding site of human Toll-like receptor 3. Proc. Natl. Acad. Sci. U.S.A. 103, 8792–8797.
Bell, J. K., Botos, I., Hall, P. R., Askins, J., Shiloach, J., Segal, D. M., and Davies, D. R. (2005). The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc. Natl. Acad. Sci. U.S.A. 102, 10976–10980.
Bell, J. K., Mullen, G. E., Leifer, C. A., Mazzoni, A., Davies, D. R., and Segal, D. M. (2003). Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 24, 528–533.
Botos, I., Segal, D. M., and Davies, D. R. (2011). The structural biology of Toll-like receptors. Structure 19, 447–459.
Bryant, C. E., Spring, D. R., Gangloff, M., and Gay, N. J. (2010). The molecular basis of the host response to lipopolysaccharide. Nat. Rev. Microbiol. 8, 8–14.
Buchanan, S. G., and Gay, N. J. (1996). Structural and functional diversity in the leucine-rich repeat family of proteins. Prog. Biophys. Mol. Biol. 65, 1–44.
Carpenter, S., and O’Neill, L. A. (2009). Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem. J. 422, 1–10.
Chan, S. L., Low, L. Y., Hsu, S., Li, S., Liu, T., Santelli, E., Le Negrate, G., Reed, J. C., Woods, V. L. Jr., and Pascual, J. (2009). Molecular mimicry in innate immunity: crystal structure of a bacterial TIR domain. J. Biol. Chem. 284, 21386–21392.
Chan, S. L., Mukasa, T., Santelli, E., Low, L. Y., and Pascual, J. (2010). The crystal structure of a TIR domain from Arabidopsis thaliana reveals a conserved helical region unique to plants. Protein Sci. 19, 155–161.
Choe, J., Kelker, M. S., and Wilson, I. A. (2005). Crystal structure of human Toll-like receptor 3 (TLR3) ectodomain. Science 309, 581–585.
Dolan, J., Walshe, K., Alsbury, S., Hokamp, K., O’Keeffe, S., Okafuji, T., Miller, S. F., Tear, G., and Mitchell, K. J. (2007). The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genomics 8, 320.
Fukuda, K., Watanabe, T., Tokisue, T., Tsujita, T., Nishikawa, S., Hasegawa, T., Seya, T., and Matsumoto, M. (2008). Modulation of double-stranded RNA recognition by the N-terminal histidine-rich region of the human Toll-like receptor 3. J. Biol. Chem. 283, 22787–22794.
Gay, N. J., and Gangloff, M. (2007). Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165.
Gay, N. J., Gangloff, M., and O’Neill, L. A. (2011). What the myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 32, 104–109.
George, J., Motshwene, P. G., Wang, H., Kubarenko, A. V., Rautanen, A., Mills, T. C., Hill, A. V., Gay, N. J., and Weber, A. N. (2011). Two human MYD88 variants, S34Y and R98C, interfere with MyD88-IRAK4-myddosome assembly. J. Biol. Chem. 286, 1341–1353.
Gong, J., Wei, T., Stark, R. W., Jamitzky, F., Heckl, W. M., Anders, H. J., Lech, M., and Rossle, S. C. (2010). Inhibition of Toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: a computational approach. J. Struct. Biol. 169, 323–330.
Gong, J., Wei, T., Zhang, N., Jamitzky, F., Heckl, W. M., Rossle, S. C., and Stark, R. W. (2011). TollML: a database of Toll-like receptor structural motifs. J. Mol. Model. 16, 1283–1289.
Govindaraj, R. G., Manavalan, B., Lee, G., and Choi, S. (2010). Molecular modeling-based evaluation of hTLR10 and identification of potential ligands in Toll-like receptor signaling. PLoS ONE 5, e12713.
Grabiec, A., Meng, G., Fichte, S., Bessler, W., Wagner, H., and Kirschning, C. J. (2004). Human but not murine Toll-like receptor 2 discriminates between tri-palmitoylated and tri-lauroylated peptides. J. Biol. Chem. 279, 48004–48012.
Guan, Y., Ranoa, D. R., Jiang, S., Mutha, S. K., Li, X., Baudry, J., and Tapping, R. I. (2010). Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J. Immunol. 184, 5094–5103.
Hantke, K., and Braun, V. (1973). Covalent binding of lipid to protein. Diglyceride and amide-linked fatty acid at the N-terminal end of the murein-lipoprotein of the Escherichia coli outer membrane. Eur. J.Biochem. 34, 284–296.
Henriques, E. S., Brito, R. M., Soares, H., Ventura, S., De Oliveira, V. L., and Parkhouse, R. M. (2011). Modeling of the Toll-like receptor 3 and a putative Toll-like receptor 3 antagonist encoded by the African swine fever virus. Protein Sci. 20, 247–255.
Jin, M. S., Kim, S. E., Heo, J. Y., Lee, M. E., Kim, H. M., Paik, S. G., Lee, H., and Lee, J. O. (2007). Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130, 1071–1082.
Jin, M. S., and Lee, J. O. (2008). Structures of the Toll-like receptor family and its ligand complexes. Immunity 29, 182–191.
Kagan, J. C., Su, T., Horng, T., Chow, A., Akira, S., and Medzhitov, R. (2008). TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 9, 361–368.
Kajava, A. V., Vassart, G., and Wodak, S. J. (1995). Modeling of the three-dimensional structure of proteins with the typical leucine-rich repeats. Structure 3, 867–877.
Kang, J. Y., and Lee, J. O. (2011). Structural biology of the Toll-like receptor family. Annu. Rev. Biochem. 80, 917–941.
Kang, J. Y., Nan, X., Jin, M. S., Youn, S. J., Ryu, Y. H., Mah, S., Han, S. H., Lee, H., Paik, S. G., and Lee, J. O. (2009). Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31, 873–884.
Kariko, K., Bhuyan, P., Capodici, J., and Weissman, D. (2004). Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through Toll-like receptor 3. J. Immunol. 172, 6545–6549.
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384.
Khan, J. A., Brint, E. K., O’Neill, L. A., and Tong, L. (2004). Crystal structure of the Toll/interleukin-1 receptor domain of human IL-1RAPL. J. Biol. Chem. 279, 31664–31670.
Kim, H. M., Oh, S. C., Lim, K. J., Kasamatsu, J., Heo, J. Y., Park, B. S., Lee, H., Yoo, O. J., Kasahara, M., and Lee, J. O. (2007a). Structural diversity of the hagfish variable lymphocyte receptors. J. Biol. Chem. 282, 6726–6732.
Kim, H. M., Park, B. S., Kim, J. I., Kim, S. E., Lee, J., Oh, S. C., Enkhbayar, P., Matsushima, N., Lee, H., Yoo, O. J., and Lee, J. O. (2007b). Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917.
Kleinman, M. E., Yamada, K., Takeda, A., Chandrasekaran, V., Nozaki, M., Baffi, J. Z., Albuquerque, R. J., Yamasaki, S., Itaya, M., Pan, Y., Appukuttan, B., Gibbs, D., Yang, Z., Kariko, K., Ambati, B. K., Wilgus, T. A., Dipietro, L. A., Sakurai, E., Zhang, K., Smith, J. R., Taylor, E. W., and Ambati, J. (2008). Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 452, 591–597.
Kobayashi, M., Saitoh, S., Tanimura, N., Takahashi, K., Kawasaki, K., Nishijima, M., Fujimoto, Y., Fukase, K., Akashi-Takamura, S., and Miyake, K. (2006). Regulatory roles for MD-2 and TLR4 in ligand-induced receptor clustering. J. Immunol. 176, 6211–6218.
Kobe, B., and Deisenhofer, J. (1995). A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature 374, 183–186.
Kobe, B., and Kajava, A. V. (2001). The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 11, 725–732.
Kubarenko, A., Frank, M., and Weber, A. N. (2007). Structure-function relationships of Toll-like receptor domains through homology modelling and molecular dynamics. Biochem. Soc. Trans. 35, 1515–1518.
Kubarenko, A. V., Ranjan, S., Colak, E., George, J., Frank, M., and Weber, A. N. (2010). Comprehensive modeling and functional analysis of Toll-like receptor ligand-recognition domains. Protein Sci. 19, 558–569.
Latz, E., Verma, A., Visintin, A., Gong, M., Sirois, C. M., Klein, D. C., Monks, B. G., Mcknight, C. J., Lamphier, M. S., Duprex, W. P., Espevik, T., and Golenbock, D. T. (2007). Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat. Immunol. 8, 772–779.
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983.
Leonard, J. N., Ghirlando, R., Askins, J., Bell, J. K., Margulies, D. H., Davies, D. R., and Segal, D. M. (2008). The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. U.S.A. 105, 258–263.
Liang, S., Hosur, K. B., Lu, S., Nawar, H. F., Weber, B. R., Tapping, R. I., Connell, T. D., and Hajishengallis, G. (2009). Mapping of a microbial protein domain involved in binding and activation of the TLR2/TLR1 heterodimer. J. Immunol. 182, 2978–2985.
Lin, S. C., Lo, Y. C., and Wu, H. (2010). Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890.
Liu, L., Botos, I., Wang, Y., Leonard, J. N., Shiloach, J., Segal, D. M., and Davies, D. R. (2008). Structural basis of Toll-like receptor 3 signaling with double-stranded RNA. Science 320, 379–381.
Manavalan, B., Basith, S., Choi, Y. M., Lee, G., and Choi, S. (2010). Structure-function relationship of cytoplasmic and nuclear IkappaB proteins: an in silico analysis. PLoS ONE 5, e15782.
Manavalan, B., Govindaraj, R., Lee, G., and Choi, S. (2011). Molecular modeling-based evaluation of dual function of IkappaBzeta ankyrin repeat domain in Toll-like receptor signaling. J. Mol. Recognit. 24, 597–607.
Matsushima, N., Tanaka, T., Enkhbayar, P., Mikami, T., Taga, M., Yamada, K., and Kuroki, Y. (2007). Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate Toll-like receptors. BMC Genomics 8, 124.
Means, T. K., Golenbock, D. T., and Fenton, M. J. (2000). The biology of Toll-like receptors. Cytokine Growth Factor Rev. 11, 219–232.
Muhlradt, P. F., Kiess, M., Meyer, H., Sussmuth, R., and Jung, G. (1997). Isolation, structure elucidation, and synthesis of a macrophage stimulatory lipopeptide from Mycoplasma fermentans acting at picomolar concentration. J. Exp. Med. 185, 1951–1958.
Mullarkey, M., Rose, J. R., Bristol, J., Kawata, T., Kimura, A., Kobayashi, S., Przetak, M., Chow, J., Gusovsky, F., Christ, W. J., and Rossignol, D. P. (2003). Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J. Pharmacol. Exp. Ther. 304, 1093–1102.
Nunez Miguel, R., Wong, J., Westoll, J. F., Brooks, H. J., O’Neill, L. A., Gay, N. J., Bryant, C. E., and Monie, T. P. (2007). A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE 2, e788.
Nyman, T., Stenmark, P., Flodin, S., Johansson, I., Hammarstrom, M., and Nordlund, P. (2008). The crystal structure of the human Toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J. Biol. Chem. 283, 11861–11865.
Ohnishi, H., Tochio, H., Kato, Z., Orii, K. E., Li, A., Kimura, T., Hiroaki, H., Kondo, N., and Shirakawa, M. (2009). Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 10260–10265.
Ohto, U., Fukase, K., Miyake, K., and Satow, Y. (2007). Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316, 1632–1634.
Omueti, K. O., Mazur, D. J., Thompson, K. S., Lyle, E. A., and Tapping, R. I. (2007). The polymorphism P315L of human Toll-like receptor 1 impairs innate immune sensing of microbial cell wall components. J. Immunol. 178, 6387–6394.
Palsson-Mcdermott, E. M., and O’Neill, L. A. (2007). Building an immune system from nine domains. Biochem. Soc. Trans. 35, 1437–1444.
Park, B. S., Song, D. H., Kim, H. M., Choi, B. S., Lee, H., and Lee, J. O. (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195.
Ranjith-Kumar, C. T., Miller, W., Xiong, J., Russell, W. K., Lamb, R., Santos, J., Duffy, K. E., Cleveland, L., Park, M., Bhardwaj, K., Wu, Z., Russell, D. H., Sarisky, R. T., Mbow, M. L., and Kao, C. C. (2007). Biochemical and functional analyses of the human Toll-like receptor 3 ectodomain. J. Biol. Chem. 282, 7668–7678.
Resman, N., Vasl, J., Oblak, A., Pristovsek, P., Gioannini, T. L., Weiss, J. P., and Jerala, R. (2009). Essential roles of hydrophobic residues in both MD-2 and Toll-like receptor 4 in activation by endotoxin. J. Biol. Chem. 284, 15052–15060.
Rosetto, M., Engstrom, Y., Baldari, C. T., Telford, J. L., and Hultmark, D. (1995). Signals from the IL-1 receptor homolog, Toll, can activate an immune response in a Drosophila hemocyte cell line. Biochem. Biophys. Res. Commun. 209, 111–116.
Rossignol, D. P., and Lynn, M. (2005). TLR4 antagonists for endotoxemia and beyond. Curr. Opin. Investig. Drugs 6, 496–502.
Shibata, K., Hasebe, A., Into, T., Yamada, M., and Watanabe, T. (2000). The N-terminal lipopeptide of a 44-kDa membrane-bound lipoprotein of Mycoplasma salivarium is responsible for the expression of intercellular adhesion molecule-1 on the cell surface of normal human gingival fibroblasts. J. Immunol. 165, 6538–6544.
Shimazu, R., Akashi, S., Ogata, H., Nagai, Y., Fukudome, K., Miyake, K., and Kimoto, M. (1999). MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189, 1777–1782.
Tao, X., Xu, Y., Zheng, Y., Beg, A. A., and Tong, L. (2002). An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochem. Biophys. Res. Commun. 299, 216–221.
Walsh, C., Gangloff, M., Monie, T., Smyth, T., Wei, B., Mckinley, T. J., Maskell, D., Gay, N., and Bryant, C. (2008). Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J. Immunol. 181, 1245–1254.
Wang, Y., Liu, L., Davies, D. R., and Segal, D. M. (2010). Dimerization of Toll-like receptor 3 (TLR3) is required for ligand binding. J. Biol. Chem. 285, 36836–36841.
Wei, T., Gong, J., Jamitzky, F., Heckl, W. M., Stark, R. W., and Rossle, S. C. (2008). LRRML: a conformational database and an XML description of leucine-rich repeats (LRRs). BMC Struct. Biol. 8, 47.
Wei, T., Gong, J., Jamitzky, F., Heckl, W. M., Stark, R. W., and Rossle, S. C. (2009). Homology modeling of human Toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci. 18, 1684–1691.
Wei, T., Gong, J., Rossle, S. C., Jamitzky, F., Heckl, W. M., and Stark, R. W. (2010). A leucine-rich repeat assembly approach for homology modeling of the human TLR5-10 and mouse TLR11-13 ectodomains. J. Mol. Model. 17, 27–36.
Xu, Y., Tao, X., Shen, B., Horng, T., Medzhitov, R., Manley, J. L., and Tong, L. (2000). Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408, 111–115.
Zahringer, U., Lindner, B., Inamura, S., Heine, H., and Alexander, C. (2008). TLR2 – promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213, 205–224.
Keywords: innate immunity, ligand, myeloid differentiation factor 88, Toll-like receptor
Citation: Manavalan B, Basith S and Choi S (2011) Similar structures but different roles – an updated perspective on TLR structures. Front. Physio. 2:41. doi: 10.3389/fphys.2011.00041
Received: 26 April 2011;
Paper pending published: 06 June 2011;
Accepted: 11 July 2011;
Published online: 27 July 2011.
Edited by:
Masa Tsuchiya, Keio University, JapanReviewed by:
Vladimir N. Uversky, University of South Florida, USACopyright: © 2011 Manavalan, Basith and Choi. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Sangdun Choi, Department of Molecular Science and Technology, Ajou University, Suwon 443-749, South Korea. e-mail:c2FuZ2R1bmNob2lAYWpvdS5hYy5rcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.