
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 11 July 2011
Sec. Systems Biology Archive
volume 2 - 2011 | https://doi.org/10.3389/fphys.2011.00032
This article is part of the Research TopicAdvances in Systems Immunology and CancerView all 11 articles
 Colin Campbell1
Colin Campbell1 Ranran Zhang2 Jeremy S. Haley3 Xin Liu4 Thomas Loughran4
Ranran Zhang2 Jeremy S. Haley3 Xin Liu4 Thomas Loughran4 Todd D. Schell3
Todd D. Schell3 Réka Albert1
Réka Albert1 Juilee Thakar1*
Juilee Thakar1*CD8+ T cells have the potential to influence the outcome of cancer pathogenesis, including complete tumor eradication or selection of malignant tumor escape variants. The Simian virus 40 large T-antigen (Tag) oncoprotein promotes tumor formation in Tag-transgenic mice and also provides multiple target determinants (sites) for responding CD8+ T cells in C57BL/6 (H-2b) mice. To understand the in vivo quantitative dynamics of CD8+ T cells after encountering Tag, we constructed a dynamic model from in vivo-generated data to simulate the interactions between Tag-expressing cells and CD8+ T cells in distinct scenarios including immunization of wild-type C57BL/6 mice and of Tag-transgenic mice that develop various tumors. In these scenarios the model successfully reproduces the dynamics of both the Tag-expressing cells and antigen-specific CD8+ T cell responses. The model predicts that the tolerance of the site-specific T cells is dependent on their apoptosis rates and that the net growth of CD8+ T cells is altered in transgenic mice. We experimentally validate both predictions. Our results indicate that site-specific CD8+ T cells have tissue-specific apoptosis rates affecting their tolerance to the tumor antigen. Moreover, the model highlights differences in apoptosis rates that contribute to compromised CD8+ T cell responses and tumor progression, knowledge of which is essential for development of cancer immunotherapy.
Tumors are masses of host cells containing both genetically unstable cancer cells and supporting host cells, including cells of the immune system. Tumor progression causes destructive pathogenesis within the host and ultimately death. Tumor antigens, particularly those that are unique to the tumor, can elicit an adaptive immune response (Qin and Blankenstein, 2000; Patel and Chiplunkar, 2009; Xu et al., 2009; Behboudi et al., 2010). In particular, CD8+ T cells (TCD8s) can eliminate continuously arising nascent transformed cells, inhibit carcinogenesis, and maintain cellular homeostasis under normal conditions; this process is known as immunosurveillance (Dunn et al., 2002; Schreiber et al., 2004). Tumors, nevertheless, can escape immunosurveillance through both antigenic loss and the promotion of immunosuppression, which leads to their progression. The immune response to cancer is often studied either by tumor implantation or by inducing autochthonous tumor formation in specific tissues in mice. Tumor development can be induced in transgenic mice by expressing oncoproteins under tissue-specific promoters. Transgenic mice which develop autochthonous tumors are especially interesting since the tumor antigens are often self antigens derived from non-mutated cellular proteins and tumor formation occurs over an extended period of time, reproducing some of the immunological roadblocks which limit effective immunotherapy.
Dynamic models of tumor–immune interactions have provided insights into the processes leading to immune response failure during tumor progression. Such models have been applied to study the effect of immunotherapeutic approaches (Day et al., 2006; Castiglione and Piccoli, 2007; Kirschner and Tsygvintsev, 2009) and to characterize the various stages of the immune response to infection and cancer, with particular focus on TCD8s (De Boer et al., 2003; Bocharov et al., 2004; Antia et al., 2005; de Pillis et al., 2005). These models highlight the importance of a variety of features of the immune response to tumors, including the density of tumor antigen, the duration of the interaction between MHCI-peptide complexes and the T cell receptor (TCR), TCD8 activation rates, immunological memory, and recruitment of precursor cells. Our study provides a unique perspective relative to previous studies, in that we focus on a comparative analysis of T cell responses in tumor-bearing versus wild-type (WT) mice.
The Simian virus 40 (SV40) large T-antigen (Tag) is a potent virus-encoded oncoprotein that can transform a variety of cell types (Ahuja et al., 2005). The oncogenic activity of Tag stems from its ability to inactivate tumor suppressor proteins (Rb and p53) as well as to initiate cell cycle progression (Butel and Lednicky, 1999). Tag can induce responses by MHC-I-restricted TCD8s, as is observed for other tumor antigens. Four unique Tag determinants recognized by TCD8s have been defined in C57BL/6 mice (sites I, II/III, IV, and V; Mylin et al., 2000; Tevethia and Schell, 2001). The TCD8 response to these four determinants forms a quantitative hierarchy in which site IV, for which the most T cells accumulate, is immunodominant followed by subdominant responses to sites I and II/III. The response to site V, however, is immunorecessive, as responding TCD8s are only detected following immunization with Tag variants lacking the three dominant determinants, or after site V-specific immunization (Tanaka et al., 1989; Fu et al., 1998). The expression of Tag as a self-antigen within Tag-transgenic mice can lead to TCD8s unresponsiveness by mechanisms promoting both central and peripheral tolerance (Tevethia and Schell, 2001).
We construct a dynamic model describing tumor progression, elimination of tumor cells, TCD8 expansion, and decay in the context of the TCD8 response to SV40 Tag. We developed this model to describe the TCD8 response in both WT C57BL/6 mice responding to immunization, where Tag-expressing cells are eliminated, and in mice that express Tag as a transgene, leading to the development of autochthonous tumors and TCD8 tolerance. For the purposes of this model we define tolerance as the absence of a functional T cell response in the presence of tumor antigen. In transgenic mice, site-specific TCD8s become tolerant at different time points (Theobald et al., 1997; Morgan et al., 1998; Colella et al., 2000; Nugent et al., 2000; Cordaro et al., 2002; Otahal et al., 2006; Fujimura et al., 2010) and this characteristic behavior is not seen when the WT mice encounter the Tag as a foreign antigen. Thus, the model parameterized to reproduce this observation gives insights into the characteristics of TCD8s that are changed during tumor development.
Our model quantitatively reproduces the TCD8 response to immunization with Tag-transformed cells in WT mice (Pretell et al., 1979; Mylin et al., 2000) and the qualitative behavior of the TCD8 response in mice bearing Tag-induced pancreatic tumors (Otahal et al., 2006), osteosarcoma (Schell et al., 2000), or brain tumors (Schell et al., 1999). The model reveals strong constraints on the proliferation rates and decay rates of TCD8s during tumor formation. Additionally, the model gives insight into how the activation, proliferation, and apoptosis rates of TCD8s impact the expansion and contraction phase during the site-specific immune response in normal mice, as well as during tissue-specific responses and development of tolerance. Our results indicate that though the inherent characteristics of the site-specific T cell clones are different, the overall TCD8 response dynamics are surprisingly similar when encountering antigen in different tissues. We predict and experimentally validate inequalities in the activation and decay rates of the TCD8 responding to unique determinants. We also theoretically predict the rate constants leading to tumor formation, the apoptosis rates of different TCD8 clones, the peaks of TCD8 activity in various tumor models and the mechanism of tolerance.
Our model describes the growth of tumor by modeling the Tag-expressing tumor/malignant cells (M) and their removal by site I-, II/III, IV-, and V-specific TCD8s (Ti; Figure 1). The basic model has five ordinary differential equations and assumes that the cells form well mixed populations. Since the dynamics after tumor clearance are not considered in the current study, the memory T cell state is not modeled. The site-specific TCD8s are initially activated against the tumor cells and are subsequently suppressed by the increasing tumor size in addition to their apoptosis. Tag-expressing cells exponentially proliferate at rate r which was estimated from the initial growth phase of the tumors (see e.g., Mallet and De Pillis, 2006). The four site-specific TCD8s are assumed to kill tumor cells (M) at the same rate b; however, b is modulated by a Michaelis–Menten function such that the rate of killing of tumor cells stabilizes when the tumor size increases above β (Figure A1 in Appendix).
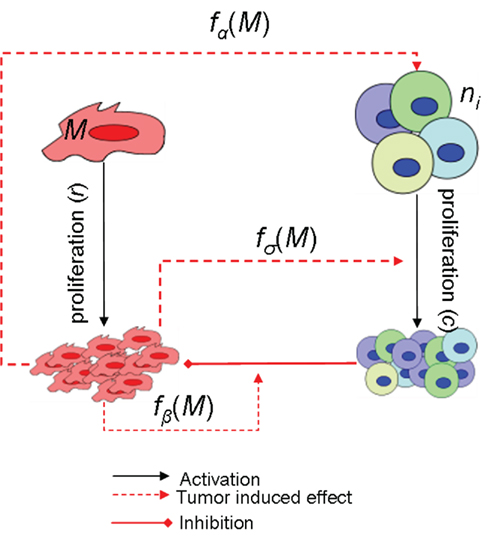
Figure 1. The interactions between Tag-expressing cells (M) and TCD8s and the parameters modeling the dynamics are depicted. Edges represent activating (black arrows), inhibiting (blunt segments), and tumor-induced (red arrows) interactions, f represents a Michaelis–Menten function.
Tag-expressing cells present four determinants I, II/III, IV, and V that are recognized by MHCI molecules. MHCI-peptide complexes are then recognized by TCR, which leads to the differentiation of naive cells and subsequently the recruitment of TCD8s to the tumor site. In the model, Tag-expressing cells induce a proportion ni of competent site-specific TCD8s at a rate proportional to c. Based on our recent experimental results (T. Schell, unpublished) a 0.3/1/4 ratio for site V/I/IV TCD8 activation from naïve T cells was used to determine ni. Since the experimental observations could be reproduced by using the same value of the proliferation rate c, it was kept the same for all site-specific TCD8s. Thus nic represents the activation rate of TCD8s from precursor cells and c represents the proliferation of TCD8s. TCD8s undergo natural death at rate wi which is different for the four determinants. The immunogenicity of the determinants is decided by the site-specific activation and decay rates. Tumor cells alter the microenvironment leading to the suppression of the TCD8 response (Ganss and Hanahan, 1998; Gajewski et al., 2006). This suppression is modeled as a negative modulation of the proliferation and activation rates in response to a higher number of tumor cells. In the case of proliferation, the effect is modeled with a Michaelis–Menten function which sets proliferation to 0 for M = 0 and saturates at c for M >> σ. The negative modulation of activation rates is modeled in the opposite way, using a “repressive” Michaelis–Menten function: for M << αi, activation occurs at a constant rate ni; for M >> αi, it asymptotically approaches 0. In the absence of spatial compartments, inhibition of the activation of new TCD8s represents the suppression of the recruitment of the TCD8s at the site of tumor which can occur by diffusible cytokines and chemokines produced by the tumor cells.
In Tag-transgenic mice both central (Faas et al., 1987; Schell et al., 1999; Colella et al., 2000; Zheng et al., 2002) and peripheral tolerance (Ye et al., 1994; Schell et al., 2000; Cordaro et al., 2002; Otahal et al., 2006) is observed in response to tumors. Tolerance is modeled by the absence of functional TCD8s even in the presence of the Tag-expressing cells. In central tolerance self-reactive T cells are deleted during development in the thymus and there is no recruitment at the site of tumors, which can be modeled by setting the value of ni to 0. During peripheral tolerance self-reactive T cells that escape to the periphery are maintained in a state of unresponsiveness or are deleted following activation. The gradual process of peripheral tolerance is simulated by the tumor-induced suppression of TCD8s and by retraining the parameter values (explained in Section “Results”).
Thus the dynamics of tumor cells (M) and TCD8s specific for site i (Ti) are given by
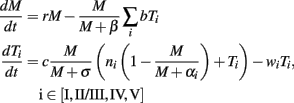
Note that though a constant source of site-specific precursor cells (constant ni) was used in the model, their effective number is not unlimited. In the WT model precursor cells stop differentiating after the clearance of Tag-expressing cells, and in the tumor models activation of new TCD8s is inhibited by large tumors (so that ≈15 TCD8s are activated for 1000 tumor cells) and has a minimal effect after the first few days.
Immunization of transgenic mice was modeled, similarly to Kirschner and Panetta (1998), by introducing a variable si that follows the clearance dynamics of Tag-transformed cells injected in WT mice by obeying the relation dsi/dt = −γSi. In the presence of immunization, the T equation becomes
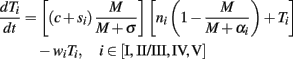
The nullcline analysis (Figures A2B–E in Appendix) is performed to study the effect of parameter values on the temporal trajectories of the tumor cells and site-specific TCD8s (Figures 2B–E). The nullcline analysis finds the equilibrium of the system when M and Ti are in a steady state (dM/dt = dT/dt = 0). Considering a single type of site-specific TCD8 for simplicity the equations become:
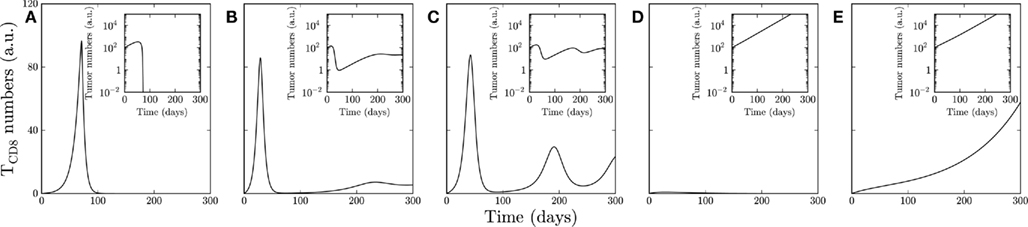
Figure 2. The response of site IV-specific TCD8s is shown in case of (A) clearance, (B) non-zero steady state, (C) oscillations, (D) tumor, and (E) uncontrolled response of Tag-expressing cells (Tag-expressing cells shown in insets). In all cases the initial condition is Tag-expressing cells = 100, site IV-specific TCD8 = 0. The parameters r = 0.03/day, n = 1.8 (proportion), b = 0.5/day, c = 0.5/day, w = 0.2/day, α = 40 cells, β = σ = 10 cells are used in all figures with the following exceptions: (A) n = 0.3 (proportion), c = 0.29/day; (B) β = σ = 50/day; (C) β = σ = 100/day; (D) c = 0.15/day; (E) c = 0.21/day.
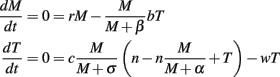
Solving these equations in terms of M and T yields
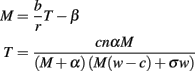
For the biologically accepted range of β (>0) in our model, M = T = 0 is an unstable steady state. That is, for small number of tumor cells (M) and TCD8s (T), the value of tumor cells will always increase in the tumor model. Additional biologically significant steady states are discussed in Section “Results.”
We used published experimental data corresponding to TCD8s derived from the spleen of WT C57BL/6 mice and three Tag-transgenic mouse strains that develop distinct tumors. Data corresponding to splenic TCD8 responses were used since they are available for all models and are representative for the systemic response to the antigen. In the case of brain tumors, T cell accumulation at the tumor site is correlated with spleen dynamics with a time lag (Ryan and Schell, 2006).
Tag-specific TCD8 cells are measured by MHC tetramer staining after encountering antigen which correlates with the number of TCD8s that produce IFNγ upon peptide-specific in vitro stimulation (Mylin et al., 2000). The published data and our own were obtained from two independent sets of experiments that were quantitatively different. In vivo model systems are often difficult to standardize. Hence, to account for lab-specific differences and to align the data, we scaled the data from Mylin et al. (2000) by a multiplicative factor that maximized the agreement.
The number of endogenous TCD8s in Tag-transgenic mice can be very low and difficult to detect. Hence induction of detectable TCD8 responses can be stimulated by immunization of mice with Tag-transformed cells expressing full-length WT Tag or Tag variants in which specific determinants have been eliminated by mutagenesis. If immunization is not sufficient to induce the T cell response, due to deletion of T cell precursors during T cell development, then splenocytes from WT mice, which contain naïve TCD8s as well as other immune cell subsets, are injected (adoptively transferred) into the Tag-transgenic mice. In such experiments, saturating amounts of splenocytes were given to achieve maximal T cell response.
All chemical reagents were purchased from Sigma-Aldrich (St Louis, MO, USA). RPMI-1640 with Glutamax and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA, USA). Benzonase® Nuclease was purchased from EMD Chemicals (San Diego, CA, USA). Annexin V Apoptosis Detection Kit, rat anti-mouse CD16/CD32 Fc block, Cytofix/Cytoperm, PermWash, fluorescein isothiocyanate (FITC), phycoerythrin (PE), allophycocyanin (APC) or APC-Cy7-labeled anti-mouse CD8α, PE-labeled anti-CD90.1, and FITC-labeled anti-5-bromo-2-deoxyuridine (BrdU) antibody were purchased from BD Biosciences (San Jose, CA, USA).
Male and female C57BL/6 (H-2b) mice (4–6 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, Maine) and routinely used between the ages of 7 and 12 weeks. SV11 (H-2b) mice express full-length SV40 T Ag under the control of the SV40 promoter (Brinster et al., 1984). Line SV11 mice were maintained by breeding hemizygous Tag transgene+ males with C57BL/6J females and transgene positive animals identified as previously described (Schell et al., 1999). TCR-IV transgenic mice expressing the TCRα and TCRβ chains specific for Tag site IV have been previously described (Tatum et al., 2008) and were maintained by breeding transgene positive males with B6.PL-Thy1a/CyJ females. All mice were maintained in the animal facility at the Pennsylvania State University College of Medicine, Hershey, PA, USA and experiments were performed under guidelines approved by the Institutional Animal Care and Use Committee.
The B6/WT-19 cell line was derived previously by transformation of B6 mouse embryo fibroblasts with WT SV40 strain VA45-54 (Pretell et al., 1979; Tevethia et al., 1980). Production and characterization of the Db/Tag site I (Db/I), Kb/Tag site IV (Kb/IV), Db/influenza virus (Flu) nucleoprotein (NP) 366–374 (Db/Flu), and Kb/HSV gB 498–505 (Kb/gB) PE-conjugated tetramers were described previously (Mylin et al., 2000). For immunization, 5 × 107 live B6/WT-19 cells were injected by the intraperitoneal route. For adoptive transfer, SV11 or transgene negative mice were injected intravenously with lymphocytes derived from TCR-IV transgenic mice containing 5 × 105 clonotypic site IV-specific TCD8s. For tetramer staining, mouse spleens were harvested at the indicated time points post immunization and processed to single cell suspensions as previously described (Schell et al., 1999). Erythrocyte-depleted splenocytes were washed twice in PBS–FBS [PBS supplemented with 2% (vol/vol) FBS], resuspended at 2 × 107 cells/ml in PBS–FBS, and incubated with rat anti-mouse CD16/CD32 (33 mg/ml) for 15 min on ice. Following incubation, cells were washed once in PBS–FBS and resuspended in fluorescence-activated cell sorter (FACS) buffer [PBS–FBS supplemented with 0.1% (wt/vol) sodium azide]. Aliquots containing 2 × 106 cells were prepared and the appropriate MHC tetramer plus anti-mouse CD8α antibody were added. Alternatively, TCR-IV transgenic T cells were identified according to their surface expression of CD90.1. In this case, cells were incubated with anti-CD90.1 antibody at room temperature for 15 min as well as anti-CD8 and MHC tetramer to minimize non-specific staining. Proliferation and apoptosis analysis focused on the population of CD8+, Tetramer+, and CD90.1+ cells. Cells were then resuspended in FACS buffer and kept on ice or processed into apoptosis assay prior to flow cytometry.
2 × 106 erythrocyte-depleted and MHC tetramer- and/or anti-CD90.1-stained cells were incubated with conjugated Annexin V and 7-AAD (1:100 dilution) in 100 μl 1× Annexin V staining buffer for 15 min at room temperature in the dark. Cells were immediately assessed by flow cytometry (BD FACSCalibur or FACSCanto). At least 10000 events were collected in the live cell gate and analyzed for Annexin V and 7-AAD staining. Annexin V negative, 7-AAD positive cells were considered non-viable and excluded from further analysis.
Mice received a 1-mg dose of 1 mg/ml BrdU solution (diluted in PBS) 3 h before sacrifice by intraperitoneal injection at the indicated times post immunization. Splenocytes were stained for BrdU incorporation using a modified staining protocol (BD Biosciences). Briefly, 2 × 106 splenocytes were stained with MHC tetramers and anti-mouse CD8α as described above. Cells were then resuspended in 100 μl of Cytofix/Cytoperm (Becton Dickinson) and incubated for 30 min at room temperature. The cells were washed once with 1× PermWash, resuspended again in 100 μl of Cytofix/Cytoperm and incubated for 10 min at room temperature. Cells were washed again and resuspended in 100 μl Cytofix/Cytoperm and incubated for 5 min at room temperature. After one wash, cells were incubated at 37°C for 1 h with 20 U Benzonase nuclease in 100 μl DPBS with 1 mM MgCl2 and washed once. Cells were then stained with 5 μl of FITC-labeled anti-BrdU antibody (eBioscience) in 40 μl 1× PermWash for 20 min at room temperature. Cells were washed and then fixed with 2% paraformaldehyde in PBS and analyzed by flow cytometry as above.
We performed the Welch two-sample t-test to assess whether the ratio of the percentage of site IV-specific TCD8s proliferating in WT to the similar percentage in SV11 mice is significantly greater than the similar ratio of the apoptotic cells. Three data points were taken from the WT mice and four data points were taken from SV11 mice. To construct the two groups for the statistical test we used the percentage of proliferating and apoptotic cells in all combinations in which WT values were in the numerator.
To study the characteristics of TCD8s modulation during tumor development we developed a dynamic model of the interactions between tumor cells (M) and TCD8 (Ti) cells elicited in response to the four unique determinants of Tag. In this section we discuss the repertoire of dynamical behaviors that emerged from the model by describing the site IV-specific TCD8s in response to the growing tumor (refer to Materials and Methods and Appendix for details). Tag-expressing cells exponentially proliferate at rate r and are killed by TCD8s at a rate that grows with tumor size, until the tumor size becomes large compared to β (Figure A1 in Appendix); after which the killing rate saturates to a rate b. Site IV-specific TCD8s are activated against the tumor cells and proliferate but their differentiation is subsequently suppressed by the increasing tumor size and they also undergo natural death at rate wiv. Proliferation and activation of TCD8s is suppressed by large tumors and the suppression is modeled by Michaelis–Menten functions parameterized by σ and αi respectively.
A repertoire of dynamical behaviors emerged from the model including clearance of Tag-expressing cells, as in WT mice, or tumor formation, as in Tag-transgenic mice. If Tag-expressing cells are cleared, the T cell response contracts (Figure 2A). Tag-expressing cells and T cells can also reach homeostasis (Figure 2B), as is observed when mice are immunized prior to the development of Tag-induced pancreatic tumors (Otahal et al., 2006). In some cases, the number of TCD8 and Tag-expressing cells fluctuate for extended periods of time before reaching a steady state (Figure 2C). If the TCD8 response is incapable of controlling the proliferating Tag-expressing cells, they increase exponentially, and TCD8s either undergo tolerance, becoming unresponsive (Figure 2D), or themselves expand continuously (Figure 2E).
To understand the effect of the parameter values on the dynamic behavior we performed a nullcline analysis (refer to Materials and Methods and Appendix for details). The nullcline analysis provides the long-term outcome resulting from the trajectories of change in the concentrations of Tag-expressing cells and TCD8s. The clearance scenario (Figure 2A; Figure A2A in Appendix) shows increasing initial trajectories of Tag-expressing cells and TCD8s. The increasing efficiency of TCD8-mediated killing of tumor cells leads to first a slowing down of the increase, and later a decrease in tumor cell numbers (modeled by the value of M). When the number of Tag-expressing cells is below a certain threshold value given by the nullcline analysis, TCD8s begin decreasing and eventually Tag-expressing cells and TCD8s are depleted. In the clearance scenario the TCD8 response is maximized by setting the Michaelis–Menten constant αi, which models the effect of tumor size on the recruitment of new TCD8s, higher than the Tag-expressing cells. Though lower values of αi are used in the tumor models, increasing αi cannot clear the tumor cells because it stimulates the differentiation of effector cells from naïve T cells which is a linear process and has a limited role in the control of exponential tumor growth (Figure A1 in Appendix).
The nullcline analysis identified the salient parameters that drive the system from one behavior type to another (Figures 2B–E). Unlike in the clearance scenario in which tumor cells are cleared, a larger value of Michaelis–Menten constants β and/or σ, which model the effect of tumor size on the TCD8 activity, launch a protective TCD8 response characterized by a steady state like behavior (Figure 2B; Figure A2B in Appendix). Further increase in these Michaelis–Menten constants (β and/or σ) leads to extended oscillations of TCD8s and tumor cells (Figures 2C and 3; Figure A2C in Appendix). As expected, decreased tumor growth (lower r) and increased TCD8-mediated killing (b) (Figure 2E; Figure A2E in Appendix) pushes the system toward tumor clearance. The extreme cases when r < 0 are not relevant in the systems that lead to tumor formation. In the following sections we consider the dynamics of all the determinants in which case site-specific TCD8 apoptosis rates and inhibition by tumor cells lead to the characteristic TCD8 response against each determinant.
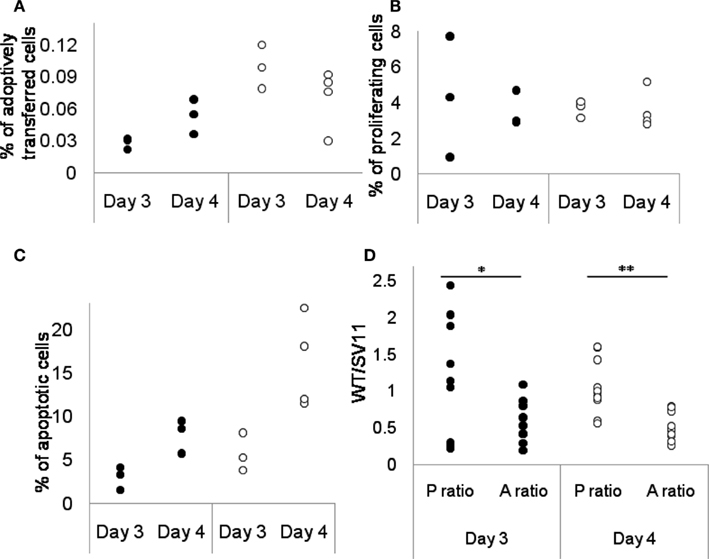
Figure 3. Site IV-specific TCD8s have different proliferation and apoptosis kinetics under WT and tumor conditions. Data was obtained 3 and 4 days after immunization using Tag-expressing cells. From (A) to (C), each circle represents data obtained from one mouse, either WT (•) or SV11 (○). (A) The percentage of adoptively transferred site IV-specific TCD8s in total splenocytes as assessed by flow cytometry. Although not statistically significant, there was an increase of site IV-specific TCD8s percentage in WT mice, but a decrease of site IV-specific TCD8s percentage in SV11. Site IV-specific TCD8s was defined as CD8+, Tetramer IV+ (indicating T cells specific for epitope IV), and CD90.1+ (indicating T cells derived from the TCR-IV mouse line). (B) The percentage of proliferating site IV-specific TCD8s in total CD8+ T cells of WT and SV11 mice after immunization as assessed by BrdU assay. The proliferation rate remained similar under WT and SV11 condition. (C) The percentage of apoptotic site IV-specific TCD8s in total CD8+ cells of WT and SV11 mice as assessed by apoptosis assay. Although not statistically significant, there is an increase of apoptotic site IV-specific TCD8s percentage in both WT and SV11, with SV11 more so than WT. (D) The WT to SV11 ratio of percentages of proliferating site IV-specific TCD8s (P ratio: proliferation ratio) and the same ratio of percentages of apoptotic site IV-specific TCD8s (A ratio: apoptosis ratio) for both time points. Each circle indicates one possible ratio between WT data and SV11 data at the same time point, either proliferation ratio (•) or apoptosis ratio (•). Proliferation ratio is higher than the apoptosis ratio at day 3 (*P < 0.032, t = 2.07) and day 4 (**P < 0.0001, t = 4.57).
Though various parameters can affect the fate of the tumors and consequently the TCD8 response, only the net growth of TCD8s, described by the difference between the rate of activation (c) and cell death (wi), decides the tolerance behavior (unresponsiveness of the TCD8 cells). Decrease in the net growth transitions the system from a clearance state (if c − wi is positive and large) observed in WT mice to a tumor state observed in transgenic mice (if c − wi is negative; see Figure 2D; Figure A2D in Appendix). Thus to reproduce the tolerance of TCD8s observed in Tag-transgenic mice wi is assumed to be greater than c in all tumor models. Our parameter analysis (Figure A3 in Appendix) indicates a large deviation from the experimental results when the above condition on the net growth of TCD8s is not implemented.
The TCD8 response to Tag in WT mice and in transgenic mice bearing tumors provides two unique immunological environments which are modeled by assuming positive and negative net growth (c − wi) of TCD8s in WT and transgenic mice respectively. The fact that the TCD8 response is strong in WT mice but is undetectable in transgenic mice (Schell et al., 2000; Otahal et al., 2006) lends support to this assumption. The early expression of antigen in Tag-transgenic mice might lead to a lower rate of expansion (c) of TCD8s, allowing tumors to grow. Indeed, it is experimentally observed that high amounts of antigen reduce the proliferation of TCD8s during tolerance (Ganss and Hanahan, 1998).
To validate our hypothesis of the positive versus negative net growth of TCD8s in WT versus transgenic mice we experimentally assessed proliferation and apoptosis of TCD8s in both WT mice and Tag-transgenic mice bearing brain tumors (Schell et al., 1999). It is difficult to obtain the absolute value of the rate of activation (c) and the rate of apoptosis (wi) through experimentation, which renders it improper to directly compare the experimental values. However, we noticed that if the net growth is positive in WT (wWT < cWT) and negative in transgenic mice (wTumor > cTumor), it implies that for a specific TCD8 clone, the ratio of proliferation rates in WT to transgenic mice is greater than the similar ratio of apoptosis rates [(cWT/cTumor) > (wWT/wTumor)]. The converse implication is satisfied if the rates of T cell proliferation are comparable in WT and tumor-bearing mice. To test this relationship, we utilized TCD8s from TCR transgenic mouse line TCR-IV in which 90% of the TCD8s were specific for site IV (Tatum et al., 2008). Splenocytes isolated from TCR-IV transgenic mice were adoptively transferred into groups of 3∼4 WT or SV11 mice before immunization with Tag-expressing cells. The percentage of proliferating and apoptotic TCR-IV cells recovered from the recipients was assessed through in vivo BrdU incorporation assay (Figure 3B) and Annexin V apoptosis assay (Figure 3C) on days 3 and 4 after immunization, respectively. These data were subsequently used to estimate the ratio of proliferation (P ratio – cWT/cTumor) and apoptosis (A ratio – wWT/wTumor) rates of site IV-specific TCD8s in WT to transgenic mice. As shown in Figure 3D, the value of the proliferation ratio is higher than the apoptosis ratio for both time points, and the difference is statistically significant [P < 0.032 (t = 2.07) at 3 days and P < 0.0001 (t = 4.57) at day 4 after immunization]. Moreover, different trends of change in the frequency of TCR-IV cells in WT and brain tumor-bearing transgenic mice between days 3 and 4, with a slight increase in TCR-IV T cells in WT and slight decrease in TCR-IV T cells in transgenic mice (Figure 3A) suggest that the rate of apoptosis is higher than the rate of proliferation in the presence of tumors. The results suggest that the net growth in TCD8s is negative in transgenic mice, while positive in WT mice, assuming that the proliferation rate of the TCD8 clones in WT and transgenic mice is in a comparable range. Next, we discuss the dynamics of site-specific TCD8s in WT and transgenic mice.
We used our own experimental data (closed symbols in Figure 4) and the data from (Mylin et al., 2000; open symbols in Figure 4) to model the behavior of the TCD8 response to the four H-2b-restricted Tag determinants following immunization with Tag-transformed cells in WT mice. Mice were immunized with 5 × 107 SV40 Tag-transformed cells and the site-specific TCD8 response was analyzed by staining with site-specific MHC tetramers at the indicated time points. The simulations of the TCD8 activity were then fit to the experimental data (Figure 4).
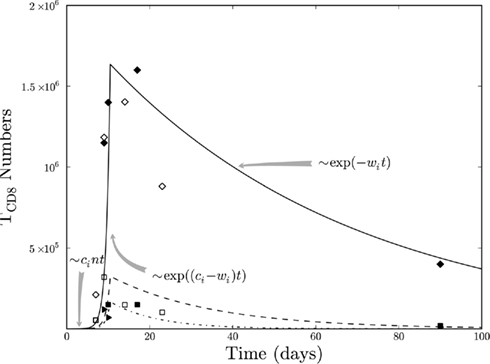
Figure 4. Simulated (lines) and experimental (symbols) TCD8s in response to the immunization of WT mice with Tag-transformed cells. The response of site I-, II/III-, IV-, and V-specific TCD8s is shown by the solid, dashed, dash-dot, and dotted lines, respectively. The gray arrows represent the terms in the mathematical model dominating the dynamics. The data from Mylin et al. (2000); empty symbols) and current study (filled symbols) representing site I (squares), II/III (triangles), and IV (diamonds) specific TCD8s is shown. The data from Mylin et al. (2000) is scaled by a multiplicative factor to minimize the variation between the two experiments. The initial conditions were, TCD8s = 0, and Tag-expressing cells (non-proliferating) = 106. The parameter values are n = 5 × 0.3/1/4 proportion for site V/I/IV respectively, b = 0.50/day, c = 1.08/day, wI = 0.04/day, wII/III = 0.09/day, wIV = 0.02/day, wV = 1.02/day, αi = 1E100 cells.
Figure 4 shows that TCD8s specific for site IV are highest in numbers followed by TCD8s specific for site I and then TCD8s specific for site II/III. The model can reproduce the observation that the addition of excess site I-specific precursor cells reverses the hierarchy so that the number of TCD8s specific to site I is higher than that of site IV-specific TCD8s (Tatum et al., 2010). This result indicates that the dominance hierarchy, at least among the three most dominant determinants, is impacted by the precursor frequency. However, the undetectable numbers of site V-specific TCD8s are not explained by a lower precursor frequency but due to their smallest net growth upon exposure to the antigen. These cells become detectable upon immunization with mutated Tag expressing only site V determinant because of the availability of tumor cells to drive their proliferation. For the fit shown in Figure 4 at the peak of the response, the hierarchy of the Tag site-specific TCD8 response is dependent on the site-specific activation of new TCD8s and the apoptosis rates. The proliferation rate (c) is kept the same for all site-specific TCD8s since it was not required to be different to reproduce the observed data in Figure 4.
The TCD8 dynamics can be characterized by an initial linear conversion of naïve cells into activated TCD8s in response to tumor cells, followed by exponential growth of the TCD8s. As a result, the number of tumor cells rapidly decreases toward 0 (not shown). In simulations, tumor cells are cleared around day 10, which coincides with the experimentally observed time after which minimal effector TCD8 are proliferating. When tumor cell numbers become negligible, the contraction phase of TCD8s begin which is dominated by exponential decay (at the rate wi). In this phase, the relationships between the rates of apoptosis (wi) of site-specific TCD8s can be estimated.
TCD8s targeting the dominant site IV and subdominant site I make up 80% of the Tag-specific TCD8 response. The model predicts that the only way to reproduce the highest and most prolonged site IV-specific TCD8 response is to have a lower apoptosis rate for site IV-specific TCD8s compared to site I-specific TCD8s (wIV < wI). To test this novel prediction of site-specific apoptosis rates, we immunized groups of three WT mice by intraperitoneal injection of C57BL/6-derived Tag-transformed cells. The percentage of apoptotic and proliferating TCD8s specific for site I and IV was assessed 9 and 14 days after immunization as estimations of the respective rates of apoptosis and proliferation. In addition, the percentage of site I-specific and site IV-specific TCD8s in splenocytes was assessed 7, 9, 14, and 23 days after immunization. Since the model estimates the relationship between the two rates of apoptosis when the activation of the TCD8s is minimal, we tested our prediction after day 14 when no significant proliferation was observed in the experiments (Figure 5A). As shown in Figure 5B, the percentage of apoptosis for site I-specific TCD8 was significantly higher than for site IV-specific TCD8s (P < 0.012). A differential cell death rate was also reflected in Figure 5C, which showed the percentage of site-specific TCD8s in splenocytes. While the percentage of site IV-specific TCD8s remained consistent after day 9, there was a drop in the percentage of site I-specific TCD8s between day 9 and day 14. Taken together, these results suggest that site I-specific TCD8s undergo cell death at a higher average rate compared to site IV-specific TCD8s, explaining the prolonged high level accumulation of site IV-specific T cells. We note that the same rates of proliferation for site I- and IV-specific TCD8s are used in the model; a parsimonious assumption which is also supported by Figure 5A. However, day 9 is at the end of the expansion phase and many parameters in addition to the proliferation rates may play a role in explaining the observations (Figures 5A,B).
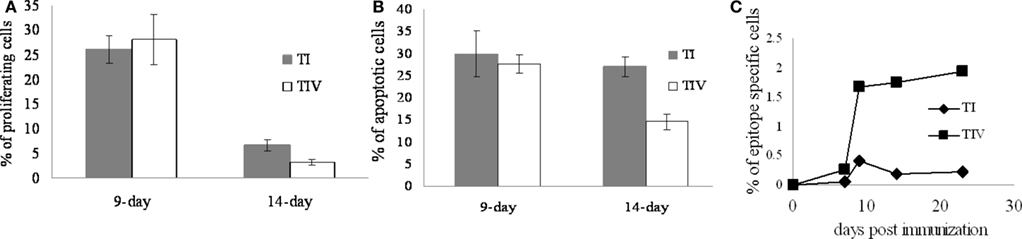
Figure 5. Site I- and site IV-specific TCD8s have different apoptosis kinetics after immunization. (A) Percentage of proliferating cells assessed by BrdU proliferation assay and (B) Annexin V positive cells indicating apoptotic cells among site I (filled) and site IV (empty) specific TCD8s. The site I-specific TCD8s undergoing apoptosis are significantly higher than site IV-specific TCD8s (*P < 0.012). (C) Percentage of site I-specific TCD8s (♦) and site IV-specific TCD8s (□) among total splenocytes 7, 9, 14, and 23 days after immunization. Comparing to site IV-specific TCD8s, there is a drop in the percentage of site I-specific TCD8s between days 9 and 14.
The three Tag-transgenic systems that autonomously develop tumors vary in the lifespan of the mice, metastasis of the tumors, responsiveness to immune-therapies and tolerance of TCD8s. The computer simulations were run to cover the duration of the life span for each transgenic mouse line. The simulated tumor phenotype reproduces the exponential tumor growth and TCD8 expansion during the early period of antigen expression, followed by an unresponsiveness of TCD8s. The tolerance onset time is different for site-specific TCD8s in different transgenic mouse lines. This information was used to parameterize the apoptosis rates (Table 1; Figure A3A in Appendix) and all the other parameters were kept the same across different tumor models. Figures 6A,B reproduce the sequential loss (tolerance onset) of site-specific TCD8s in pancreatic tumor (Otahal et al., 2006) and osteosarcoma (Schell et al., 2000) models, respectively. Tissue specific death rates for TCD8s against each site (wi) reproduce the correct tolerance onset in different transgenic mouse lines. We observed interesting regularities between the rates of apoptosis. For example, the apoptosis rate of site I-specific TCD8s is always greater than that of site IV-specific TCD8s as seen in the WT model. Thus while the specific values of the parameters can be different, the unavoidable similarities pointed out by our dynamic model can improve our understanding of the tumor growth and TCD8 response.
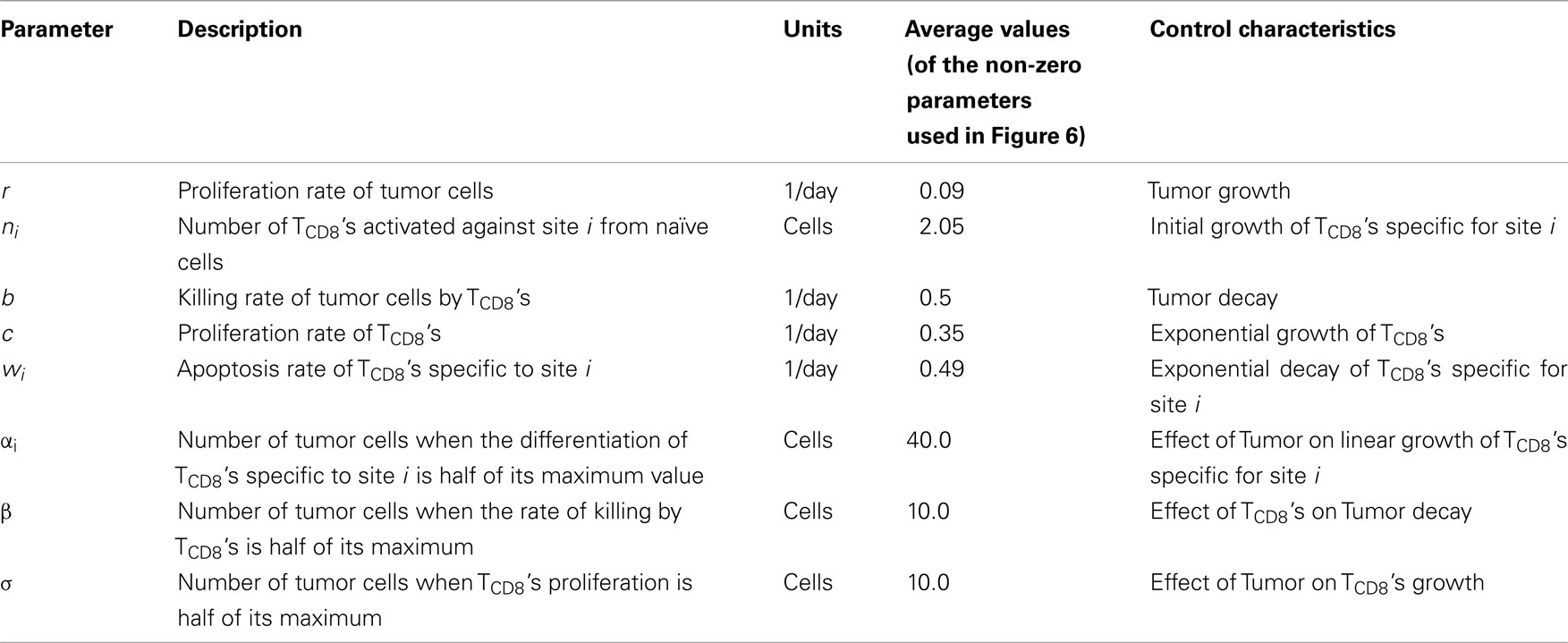
Table 1. Model parameters and description.
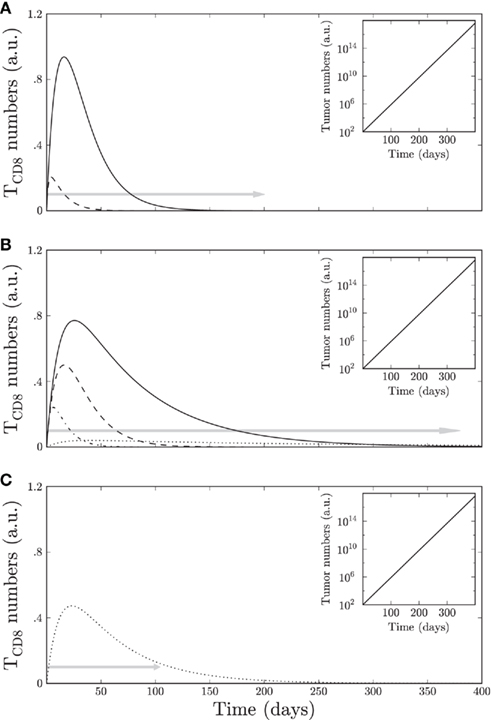
Figure 6. Simulated TCD8s response in (A) pancreatic tumors, (B) osteosarcomas, and (C) brain tumors. Y-axis depicts the number of TCD8s in arbitrary units and x-axis depicts the time in days. Site I-specific TCD8s is shown with a dashed line, TII/III with a dash-dot line, site IV-specific TCD8s with a solid line, and TV with a dot line. Light gray arrows show the average lifespan of mice. All cases show a sequential loss of TCD8 cells against different sites. The insets indicate tumor growth.
Changes in the other parameters including the growth rate of tumor cells (r) and the rate of TCD8 mediated killing of tumor cells (b) were not necessary to reproduce any of the observations, though they could affect the modeled tumor size. The unresponsiveness of TCD8s targeting particular determinants was confirmed by the absence of TCD8 activation after simulating in silico immunization with Tag-expressing cells (modeled by si; Figure 7). In the brain tumor model (Schell et al., 1999) TCD8s specific for the three most dominant Tag sites undergo central tolerance; hence we only see the TCD8s specific for the immunorecessive site V which remain responsive throughout the lifespan of these transgenic mice. However, the simulations indicate that the activity of these TCD8s is lower than in WT mice (Figure 6C).
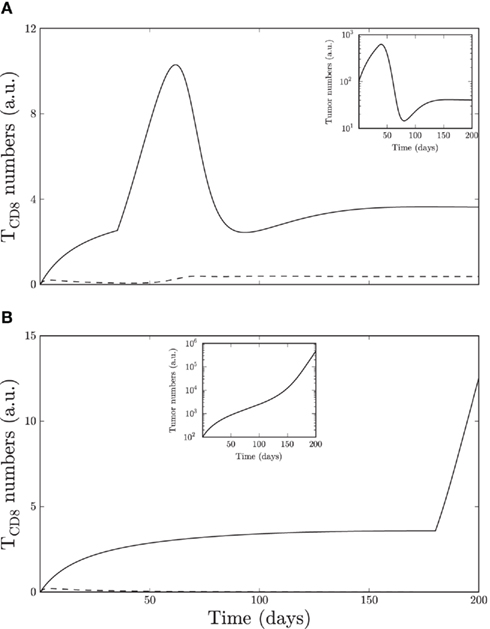
Figure 7. The pancreatic tumor shown in Figure 6 is here modified (b = 3.5, β = 100) and subjected to in silico site IV immunizations of equal strength on (A) day 35 and (B) day 180. Insets (A,B) show tumor cells on a log scale. Site I-specific TCD8s is shown with a dashed line and site IV-specific TCD8s with a solid line (TII/III and TV have negligible values). The early immunization results in a steady state of tumor cells and site IV-specific TCD8s, whereas the later immunization fails to significantly influence tumor growth.
Next, we used our model to predict the peak of TCD8 accumulation, since tumor treatment is most effective when active TCD8s are high. The tumor as well as WT models predict that site I TCD8s peak earlier than site IV TCD8s (Mylin et al., 2000). In osteosarcomas, site I-specific TCD8s reach higher numbers than in pancreatic tumors (Figure 6B). The peaks in TCD8 accumulation in the osteosarcoma model occur at later time points (at 18 days for TCD8I and 24 days for TCD8IV) as compared to the peaks of site I-specific TCD8s (4th day) and site IV-specific TCD8s (7th day) in the pancreatic model (compare Figures 6A,B). The brain tumor model (Figure 6C) predicts an earlier peak of site V-specific TCD8s compared to the osteosarcoma model (days 23 and 30, respectively) since TCD8s targeting the dominant sites are absent, resulting in increased availability of antigen. Thus our model can detect the differences in the timing of the peaks in different models.
In conclusion, the tumor models give insight into the tissue specificity, for example revealing that osteosarcomas elicit a stronger response as compared to pancreatic tumors. All tumors inhibit activation of naïve cells and reduce the net growth of TCD8 cells but differences in the apoptosis rates of the recruited TCD8s is a critical factor in determining a tissue-specific response.
Tumors can be controlled by various immune therapies including immunization with tumor antigen and the adoptive transfer of immune cells (Hersey, 2010; Moschella et al., 2010; van den Broek et al., 2010). To gain insight into the mechanisms of tumor control we simulated a known case of control of pancreatic tumors upon immunization (Otahal et al., 2006). In this study early immunization on day 35 can prevent pancreatic tumor appearance whereas immunization after day 180 cannot prevent tumor formation. Our simulations reproduce this behavior. Simulated immunization leads to a sharp increase in site IV-specific TCD8s above the endogenous levels, followed by a decrease that correlates with a drop in tumor cells. Experimentally it is expected that the TCD8 response upon immunization surpasses the response generated against the endogenous tumor. Thus the model could simulate the effect of early and late immunization in case of pancreatic tumors.
However, in transgenic mice Tag-expressing cells persist in lower numbers in case of early immunization so that the pancreatic functions are not disrupted. To reproduce this observation the rate of TCD8 proliferation, the maximal rate of TCD8 mediated killing of tumor cells, and the threshold number of tumor cells that inhibit killing have to be increased. After increasing these parameters the dose of immunization could be decreased and the control of tumors with early immunization (Figure 7A) but not late immunization (Figure 7B) was reproduced in the pancreatic tumors (Figure 7, inset). Thus our model suggests that exposure to external antigen in transgenic mice facilitates the detection of tumor cells by TCD8s and the immunogenicity of Tag.
The current work sheds light upon mechanisms that determine TCD8 tolerance onset and the characteristics leading to the site-specific TCD8s response. Both the dynamic model and the experimental data show that the apoptosis rates of TCD8 clones are context dependent and that the response in WT C57BL/6 mice is quantitatively stronger than the response in Tag-transgenic mice. This weaker response in Tag-transgenic mice aids the establishment of tolerance in the presence of progressing tumors. We hypothesize that the weaker response is driven by the immediate encounter of the peripheral T cells to Tag expressed on the tumor leading to undetectable TCD8 numbers in the transgenic mice. Figure 3 supports this hypothesis by showing a higher apoptotic population in tumor-bearing mice. In fact, in SV11 mice transferred cells encounter tumor antigen earlier than Tag from the cells used for immunization, suggesting that an early encounter with the endogenous tumor antigen also limits response to the exogenous antigen (Ryan and Schell, 2006). In the case of natural tumors, it is possible that the net growth of TCD8s varies over the course of tumor progression. The accumulation of responsive TCD8s can be enhanced by using anti-CD40 or anti-CTLA-4 antibodies (Otahal et al., 2007; Ryan et al., 2008). The tissue-specific differences in the peaks of the TCD8 response will affect the time at which immune therapy will be most effective. Differential expression of negative regulators of the receptors such as PD-L1 and Tim3 ligand within tumors of distinct tissues might lead to tissue-specific apoptosis.
Though TCD8 tolerance is often observed, particularly in the setting of cancer or transgene expression, the mechanisms leading to tolerance are not clear. Factors that have received considerable attention are the density of peptide/MHC-I complexes, the affinity of TCR for peptide/MHC complexes and the avidity of the interaction between T cells and antigen-presenting cells (Abbas et al., 2004). These factors affect the activation of TCD8s and their effects on the TCD8 response are modeled indirectly by assuming high-dose inhibition, antigen-induced cell death (Kabelitz et al., 1993), and immune suppression. While we also include saturation of TCD8s in response to large tumors (Graw and Regoes, 2009), we compare the rate of activation and apoptosis in distinct mice in which tolerance is either observed or not. Although experimentally it is difficult to measure the densities of peptide-MHCI complexes and affinities of TCR, our approach allows us to establish relationships between different parameters and test them experimentally. Moreover, our model suggests that the differentiation of naïve T cells is also affected during tolerance. Various evidence such as the disruption of MHCI-peptide and TCR complex (Nagaraj et al., 2007), TCD8 exhaustion (Moskophidis et al., 1993) and tumor-induced TCD8 suppressive microenvironment (Lee et al., 1999; Khong and Restifo, 2002) support the inhibition of the differentiation from naïve T cells during tolerance. Studies of the TCD8 response to the Tag determinants suggest that tolerance is related to immunodominance since TCD8 specific for the immunorecessive determinant (site V) are the least sensitive to tolerance even in the highly tolerogenic brain tumor model (Schell et al., 2000). In this context, it is interesting that subdominant site I-specific TCD8s undergo tolerance earlier than immunodominant site IV-specific TCD8s, suggesting that higher levels of site I/MHC versus site IV/MHC complexes may be achieved in vivo during tumor progression. We assume that the dependence of the T cell response on the number of tumor cells is described by Michaelis–Menten kinetics which is a commonly used functional form to model saturating response at high doses in biological systems. The support for such behavior comes from observations in chronic infections and cancers which limit the activation of the immune responses even when the antigen is not cleared (Kabelitz et al., 1993; Wigginton and Kirschner, 2001). We note that the Michaelis–Menten function is a special case of a Hill function which may also be a good choice but it has an additional unknown parameter as compared to the Michaelis–Menten function.
Simulations in WT mice suggest that the differential activities of site-specific TCD8s are driven by the site-specific rates of apoptosis. The rate of apoptosis was estimated in the contraction phase when Tag-expressing cells are cleared. At earlier time points the percentages observed in the experiments (Figures 3C and 6B) are the outcome of the dynamics modeled by the rate of activation (n), proliferation (c), and apoptosis (wi). In the absence of experimental estimates the rate constants used in our model represent an average rate of apoptosis over time. Hence we validate model-predicted inequalities rather than attempting to estimate the exact values of apoptosis rates. Though free decay is a commonly made assumption in dynamic models, antigen concentrations and the duration of antigen exposure can affect the apoptosis rate of the TCD8 cells which can be included in a future extension of the current model (Porter and Harty, 2006). On the contrary, the accurate prediction of the rate of proliferation (c) was not possible (Figure 4) due to the sparse data (Trinchieri et al., 1976) and various parameters affecting the expansion phase including TCD8 apoptosis (wi; Figure 5B), differentiation from naïve T cells (ni), and the proliferation of TCD8s (c). The use of similar proliferation rates for TCD8s specific to site I and IV is supported by the experimental data (Figure 5A). We maintain the same rate of proliferation for all site-specific TCD8s, a simplification which we believe is valid for the dominant sites. The rules may be different for site V since the addition of more site V-specific precursors does not overcome the weak response to site V (Otahal et al., 2005). However, estimating the true proliferation rate of site V-specific TCD8s is challenging since these cells remain below the threshold of detection following immunization of WT mice with Tag. Overall the data support a mechanism in which differences in the rate of apoptosis explain the prolonged high level accumulation of site IV-specific TCD8s cells relative to TCD8s responding to the subdominant determinants.
The current model is an outcome of a step-wise process to reproduce the TCD8 response in WT and Tag-transgenic mice. As a first step we modeled all the immune processes that are hypothesized to be important in TCD8 activation. Next, we reduced the model based on what is required to reproduce the known experimental observations, for example separately modeling pMHC complexes on the antigen-presenting cells did not lead to an improvement of the model but increased the number of unknown parameters (Handel and Antia, 2008). We do not explicitly include naïve TCD8s in the model, assuming that they are not a rate limiting factor in the tumor-induced T cell interactions. However, in the biologically accepted range of parameters (Figure A3 in Appendix) the effective value of the rate of differentiation of precursor cells is never unlimited in our model. Many mathematical models have assumed a constant number of precursor cells and in those models estimates of the effective rate of thymic production vary within 25% of the nic values used in our model. The mathematical formulations modeling TCD8 response to tumors generally incorporate higher values of the source for TCD8s compared to the models of TCD8 response to viruses. This could be because the immune response is measured following immunization which usually inflates the response. We also would like to note that the value of the rate of proliferation is also in the range of estimations by other studies (De Boer et al., 2003; Bocharov et al., 2004).
In the current model we did not explore the possibility of introducing competition between TCD8 clones because recent results indicate that in WT mice competition between the endogenous TCD8s responding to sites I and IV does not play a significant role in limiting the magnitude of the T cell response (Tatum et al., 2010). We did previously observe that the presence of the dominant Tag determinants can limit the response to site V (Mylin et al., 2000). The absence of a detectable endogenous site V-specific TCD8 response upon immunization with Tag-expressing cells makes it difficult to make assumptions about the interactions between physiological levels of TCD8s specific for dominant and recessive epitopes. Though we cannot rule out the possibility of competitive interactions, non-competitive interactions mediated by weak engagement of TCR with site V-MHCI complexes can also drive the immunorecessive response as modeled in the current study. In conclusion, the mathematical model presented here is one of the few attempts to characterize in vivo TCD8 responses to known autochthonous tumors and it systematically analyzes the expansion and contraction phases during the TCD8 response to a known tumor antigen.
In the future, this model could be expanded by including competitive interactions between site-specific clones for different antigens and by separately modeling the reactivation of memory T cells in response to the antigen (Camus and Galon, 2010). Modeling of naïve T cells as a separate entity will also allow us to study the effects of adoptive transfers, which are currently under clinical investigation for several cancer types. One could also incorporate immune cells such as T regulatory cells which are implicated in inducing tolerance and model the recovery of responsive TCD8 cells (Sharabi and Ghera, 2010). Moreover, the case of uncontrolled T cell and Tag-expressing cell growth is similar to autoimmune response and though it is not a focus of the current study, the model can be used to study the relationship between tumor and autoimmunity since tolerance (Schuetz et al., 2010) and dysregulation of immune responses (Reeves et al., 2009) are implicated in both diseases. While opportunities exist to build on this basic model, relevant in vivo data are needed to inform and parameterize the expansion. Moreover, standardization of experimental techniques will be useful since the observations are not only affected by personal and lab-specific factors but also by the mice strains used. We show here that our model not only provides novel predictions that can be experimentally validated but also gives important insights based on sparse data. Models such as ours will be increasingly developed and used to provide novel predictions and biological understanding of the complex interaction between the immune system and cancer.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was partially funded by research grant R01-CA-025000 from the National Cancer Institute/National Institutes of Health (to Todd D. Schell). We would like to thank Alan M. Watson and Dr. Angela M. Tatum for their advice, Aijun Liao for her assistance with experiments, and Nate Sheaffer and Dr. David Stanford for help with acquisition and analysis of flow cytometry data. Juilee Thakar is thankful to the Cancer Research Institute for a postdoctoral fellowship.
Ethics statement: All animal studies were performed under active protocols approved by the Pennsylvania State University Institutional Animal Care and Use Committee.
Abbas, A. K., Lohr, J., Knoechel, B., and Nagabhushanam, V. (2004). T cell tolerance and autoimmunity. Autoimmun. Rev. 3, 471–475.
Ahuja, D., Saenz-Robles, M. T., and Pipas, J. M. (2005). SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24, 7729–7745.
Antia, R., Ganusov, V. V., and Ahmed, R. (2005). The role of models in understanding CD8+ T-cell memory. Nat. Rev. Immunol. 5, 101–111.
Behboudi, S., Boswell, S., and Williams, R.. (2010). Cell-mediated immune responses to alpha-fetoprotein and other antigens in hepatocellular carcinoma. Liver Int. 30, 521–526.
Bocharov, G., Ludewig, B., Bertoletti, A., Klenerman, P., Junt, T., Krebs, P., Luzyanina, T., Fraser, C., and Anderson, R. M. (2004). Underwhelming the immune response: effect of slow virus growth on CD8+-T-lymphocyte responses. J. Virol. 78, 2247–2254.
Brinster, R. L., Chen, H. Y., Messing, A., van Dyke, T., Levine, A. J., and Palmiter, R. D. (1984). Transgenic mice harboring SV40 T-antigen genes develop characteristic brain tumors. Cell 37, 367.
Butel, J. S., and Lednicky, J. A.. (1999). Cell and molecular biology of simian virus 40: implications for human infections and disease. J. Natl. Cancer Inst. 91, 119–134.
Camus, M., and Galon, J.. (2010). Memory T-cell responses and survival in human cancer: remember to stay alive. Adv. Exp. Med. Biol. 684, 166–177.
Castiglione, F., and Piccoli, B.. (2007). Cancer immunotherapy, mathematical modeling and optimal control. J. Theor. Biol. 247, 723–732.
Colella, T. A., Bullock, T. N., Russell, L. B., Mullins, D. W., Overwijk, W. W., Pierce, R. A., Restifo, N. P., and Engelhard, V. H. (2000). Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J. Exp. Med. 191, 1221–1232.
Cordaro, T. A., de Visser, K. E., Tirion, F. H., Schumacher, T. N., and Kruisbeek, A. M. (2002). Can the low-avidity self-specific T cell repertoire be exploited for tumor rejection? J. Immunol. 168, 651–660.
Day, J., Rubin, J., Vodovotz, Y., Chow, C. C., Reynolds, A., and Clermont, G. (2006). A reduced mathematical model of the acute inflammatory response. II. Capturing scenarios of repeated endotoxin administration. J. Theor. Biol. 242, 237–256.
De Boer, R. J., Homann, D., and Perelson, A. S. (2003). Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 171, 3928–3935.
de Pillis, L. G., Radunskaya, A. E., and Wiseman, C. L. (2005). A validated mathematical model of cell-mediated immune response to tumor growth. Cancer Res. 65, 7950–7958.
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998.
Faas, S. J., Pan, S., Pinkert, C. A., Brinster, R. L., and Knowles, B. B. (1987). Simian virus 40 (SV40)-transgenic mice that develop tumors are specifically tolerant to SV40 T antigen. J. Exp. Med. 165, 417–427.
Fu, T. M., Mylin, L. M., Schell, T. D., Bacik, I., Russ, G., Yewdell, J. W., Bennink, J. R., and Tevethia, S. S. (1998). An endoplasmic reticulum-targeting signal sequence enhances the immunogenicity of an immunorecessive simian virus 40 large T antigen cytotoxic T-lymphocyte epitope. J. Virol. 72, 1469–1481.
Fujimura, T., Mahnke, K., and Enk, A. H. (2010). Myeloid derived suppressor cells and their role in tolerance induction in cancer. J. Dermatol. Sci. 59, 1–6.
Gajewski, T. F., Meng, Y., Blank, C., Brown, I., Kacha, A., Kline, J., and Harlin, H. (2006). Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev. 213, 131–145.
Ganss, R., and Hanahan, D. (1998). Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 58, 4673–4681.
Graw, F., and Regoes, R. R. (2009). Investigating CTL mediated killing with a 3D cellular automaton. PLoS Comput. Biol. 5, e1000466. doi: 10.1371/journal.pcbi.1000466
Handel, A., and Antia, R. (2008). A simple mathematical model helps to explain the immunodominance of CD8 T cells in influenza A virus infections. J. Virol. 82, 7768–7772.
Kabelitz, D., Pohl, T., and Pechhold, K. (1993). Activation-induced cell death (apoptosis) of mature peripheral T lymphocytes. Immunol. Today 14, 338–339.
Khong, H. T., and Restifo, N. P. (2002). Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 3, 999–1005.
Kirschner, D., and Panetta, J. C. (1998). Modeling immunotherapy of the tumor–immune interaction. J. Math. Biol. 37, 235–252.
Kirschner, D., and Tsygvintsev, A. (2009). On the global dynamics of a model for tumor immunotherapy. Math. Biosci. Eng. 6, 573–583.
Lee, P. P., Yee, C., Savage, P. A., Fong, L., Brockstedt, D., Weber, J. S., Johnson, D., Swetter, S., Thompson, J., Greenberg, P. D., Roederer, M., and Davis, M. M. (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685.
Mallet, D. G., and De Pillis, L. G. (2006). A cellular automata model of tumor–immune system interactions. J. Theor. Biol. 239, 334–350.
Morgan, D. J., Kreuwel, H. T., Fleck, S., Levitsky, H. I., Pardoll, D. M., and Sherman, L. A. (1998). Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 160, 643–651.
Moschella, F., Proietti, E., Capone, I., and Belardelli, F. (2010). Combination strategies for enhancing the efficacy of immunotherapy in cancer patients. Ann. N. Y. Acad. Sci. 1194, 169–178.
Moskophidis, D., Lechner, F., Pircher, H., and Zinkernagel, R. M. (1993). Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761.
Mylin, L. M., Schell, T. D., Roberts, D., Epler, M., Boesteanu, A., Collins, E. J., Frelinger, J. A., Joyce, S., and Tevethia, S. S. (2000). Quantitation of CD8(+) T-lymphocyte responses to multiple epitopes from simian virus 40 (SV40) large T antigen in C57BL/6 mice immunized with SV40, SV40 T-antigen-transformed cells, or vaccinia virus recombinants expressing full-length T antigen or epitope minigenes. J. Virol. 74, 6922–6934.
Nagaraj, S., Gupta, K., Pisarev, V., Kinarsky, L., Sherman, S., Kang, L., Herber, D. L., Schneck, J., and Gabrilovich, D. I. (2007). Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 13, 828–835.
Nugent, C. T., Morgan, D. J., Biggs, J. A., Ko, A., Pilip, I. M., Pamer, E. G., and Sherman, L. A. (2000). Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. J. Immunol. 164, 191–200.
Otahal, P., Hutchinson, S. C., Mylin, L. M., Tevethia, M. J., Tevethia, S. S., and Schell, T. D. (2005). Inefficient cross-presentation limits the CD8+ T cell response to a subdominant tumor antigen epitope. J. Immunol. 175, 700–712.
Otahal, P., Knowles, B. B., Tevethia, S. S., and Schell, T. D. (2007). Anti-CD40 conditioning enhances the T(CD8) response to a highly tolerogenic epitope and subsequent immunotherapy of simian virus 40 T antigen-induced pancreatic tumors. J. Immunol. 179, 6686–6695.
Otahal, P., Schell, T. D., Hutchinson, S. C., Knowles, B. B., and Tevethia, S. S. (2006). Early immunization induces persistent tumor-infiltrating CD8+ T cells against an immunodominant epitope and promotes lifelong control of pancreatic tumor progression in SV40 tumor antigen transgenic mice. J. Immunol. 177, 3089–3099.
Patel, S., and Chiplunkar, S.. (2009). Host immune responses to cervical cancer. Curr. Opin. Obstet. Gynecol. 21, 54–59.
Porter, B. B., and Harty, J. T.. (2006). The onset of CD8+-T-cell contraction is influenced by the peak of Listeria monocytogenes infection and antigen display. Infect. Immun. 74, 1528–1536.
Pretell, J., Greenfield, R. S., and Tevethia, S. S. (1979). Biology of simian virus 40 (SV40) transplantation antigen (TrAg). V. In vitro demonstration of SV40 TrAg in SV40 infected nonpermissive mouse cells by the lymphocyte mediated cytotoxicity assay. Virology 97, 32–41.
Qin, Z., and Blankenstein, T.. (2000). CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 12, 677–686.
Reeves, W. H., Lee, P. Y., Weinstein, J. S., Satoh, M., and Lu, L. (2009). Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 30, 455–464.
Ryan, C. M., and Schell, T. D.. (2006). Accumulation of CD8+ T cells in advanced-stage tumors and delay of disease progression following secondary immunization against an immunorecessive epitope. J. Immunol. 177, 255–267.
Ryan, C. M., Staveley-O’Carroll, K., and Schell, T. D. (2008). Combined anti-CD40 conditioning and well-timed immunization prolongs CD8+ T cell accumulation and control of established brain tumors. J. Immunother. 31, 906–920.
Schell, T. D., Knowles, B. B., and Tevethia, S. S. (2000). Sequential loss of cytotoxic T lymphocyte responses to simian virus 40 large T antigen epitopes in T antigen transgenic mice developing osteosarcomas. Cancer Res. 60, 3002–3012.
Schell, T. D., Mylin, L. M., Georgoff, I., Teresky, A. K., Levine, A. J., and Tevethia, S. S. (1999). Cytotoxic T-lymphocyte epitope immunodominance in the control of choroid plexus tumors in simian virus 40 large T antigen transgenic mice. J. Virol. 73, 5981–5993.
Schreiber, R. D., Old, L. J., Hayday, A. C., and Smyth, M. J. (2004). A cancer immunosurveillance controversy. Nat. Immunol. 5, 4–5.
Schuetz, C., Niehues, T., Friedrich, W., and Schwarz, K. (2010). Autoimmunity, autoinflammation and lymphoma in combined immunodeficiency (CID). Autoimmun. Rev. 9, 477–482.
Sharabi, A., and Ghera, N. H.. (2010). Breaking tolerance in a mouse model of multiple myeloma by chemoimmunotherapy. Adv. Cancer Res. 107, 1–37.
Tanaka, Y., Anderson, R. W., Maloy, W. L., and Tevethia, S. S. (1989). Localization of an immunorecessive epitope on SV40 T antigen by H-2Db-restricted cytotoxic T-lymphocyte clones and a synthetic peptide. Virology 171, 205–213.
Tatum, A. M., Mylin, L. M., Bender, S. J., Fischer, M. A., Vigliotti, B. A., Tevethia, M. J., Tevethia, S. S., and Schell, T. D. (2008). CD8+ T cells targeting a single immunodominant epitope are sufficient for elimination of established SV40 T antigen-induced brain tumors. J. Immunol. 181, 4406–4417.
Tatum, A. M., Watson, A. M., and Schell, T. D. (2010). Direct presentation regulates the magnitude of the CD8+ T cell response to cell-associated antigen through prolonged T cell proliferation. J. Immunol. 185, 2763–2772.
Tevethia, S. S., Greenfield, R. S., Flyer, D. C., and Tevethia, M. J. (1980). SV40 transplantation antigen: relationship to SV40-specific proteins. Cold Spring Harb. Symp. Quant. Biol. 44(Pt. 1), 235–242.
Tevethia, S. S., and Schell, T. D.. (2001). “The immune response to SV40, JCV and BKV,” in Human Polyomaviruses: Molecular and Clinical Perspectives, eds K. Khalili, and G. L. Stoner (New York: Wiley), 585–610.
Theobald, M., Biggs, J., Hernández, J., Lustgarten, J., Labadie, C., and Sherman, L. A. (1997). Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J. Exp. Med. 185, 833–841.
Trinchieri, G., Aden, D. P., and Knowles, B. B. (1976). Cell-mediated cytotoxicity to SV40-specific tumour-associated antigens. Nature 261, 312–314.
van den Broek, M., von Boehmer, L., and Knuth, A. (2010). Developments in cancer immunotherapy. Dig. Dis. 28, 51–56.
Wigginton, J. E., and Kirschner, D.. (2001). A model to predict cell-mediated immune regulatory mechanisms during human infection with Mycobacterium tuberculosis. J. Immunol. 166, 1951–1967.
Xu, Q., Liu, G., Yuan, X., Xu, M., Wang, H., Ji, J., Konda, B., Black, K. L., and Yu, J. S. (2009). Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 27, 1734–1740.
Ye, X., McCarrick, J., Jewett, L., and Knowles, B. B. (1994). Timely immunization subverts the development of peripheral nonresponsiveness and suppresses tumor development in simian virus 40 tumor antigen-transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 91, 3916–3920.
Zheng, X., Gao, J. X., Zhang, H., Geiger, T. L., Liu, Y., and Zheng, P. (2002). Clonal deletion of simian virus 40 large T antigen-specific T cells in the transgenic adenocarcinoma of mouse prostate mice: an important role for clonal deletion in shaping the repertoire of T cells specific for antigens overexpressed in solid tumors. J. Immunol. 169, 4761–4769.
Keywords: CD8+ T cells, T-antigen, tumor, dynamic model, apoptosis and proliferation rates
Citation: Campbell C, Zhang R, Haley JS, Liu X, Loughran T, Schell TD, Albert R and Thakar J (2011) Why do CD8+ T cells become indifferent to tumors: a dynamic modeling approach. Front. Physio. 2:32. doi: 10.3389/fphys.2011.00032
Received: 23 March 2011;
Accepted: 18 June 2011;
Published online: 11 July 2011.
Edited by:
Kumar Selvarajoo, Keio University, JapanReviewed by:
Gabor Balazsi, The University of Texas MD Anderson Cancer Center, USACopyright: © 2011 Campbell, Zhang, Haley, Liu, Loughran, Schell, Albert and Thakar. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Juilee Thakar, Department of Pathology, Yale University School of Medicine, 300 George Street, Suite 505, New Haven, CT 06511, USA. e-mail:anVpbGVlLnRoYWthckB5YWxlLmVkdSw=anRoYWthckBwaHlzLnBzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.