
- Institut für Neurophysiologie, Medizinische Hochschule Hannover, Hannover, Germany
ClC-K channels form a subgroup of anion channels within the ClC family of anion transport proteins. They are expressed predominantly in the kidney and in the inner ear, and are necessary for NaCl resorption in the loop of Henle and for K+ secretion by the stria vascularis. Subcellular distribution as well as the function of these channels are tightly regulated by an accessory subunit, barttin. Barttin improves the stability of ClC-K channel protein, stimulates the exit from the endoplasmic reticulum and insertion into the plasma membrane and changes its function by modifying voltage-dependent gating processes. The importance of ClC-K/barttin channels is highlighted by several genetic diseases. Dysfunctions of ClC-K channels result in Bartter syndrome, an inherited human condition characterized by impaired urinary concentration. Mutations in the gene encoding barttin, BSND, affect the urinary concentration as well as the sensory function of the inner ear. Surprisingly, there is one BSND mutation that causes deafness without affecting renal function, indicating that kidney function tolerates a reduction of anion channel activity that is not sufficient to support normal signal transduction in inner hair cells. This review summarizes recent work on molecular mechanisms, physiology, and pathophysiology of ClC-K/barttin channels.
CLC Channels and Transporters
The ClC family of anion channels and transporters is the largest known family of anion transport proteins (Jentsch, 2008). It was identified by Jentsch et al. (1990), when they cloned a chloride channel, ClC-0, from the electric organ of Torpedo. In the following years a large number of homologs was identified in a variety of organisms and shown to serve multiple physiological functions (Jentsch, 2008).
At present, the ClC family contains nine mammalian isoforms, ClC-1 to ClC-7 and ClC-K1/ClC-Ka, and ClC-K2/ClC-Kb. Four ClC isoforms were shown to function as anion channels and the remaining ones to mediate coupled antiport of anions and protons (Jentsch, 2008).
ClC-1 is exclusively expressed in adult skeletal muscle and provides the large resting anion conductance necessary for the electrical stability of muscle fibers (Steinmeyer et al., 1991a,b). Mutations in the gene encoding ClC-1, CLCN1, cause myotonia congenita (Koch et al., 1992; George et al., 1993). ClC-2 is expressed in a broad variety of cells. It appears to be important for cell excitability in the central nervous system (Kleefuss-Lie et al., 2009; Saint-Martin et al., 2009; Foldy et al., 2010; Rinke et al., 2010), as well as for anion resorption in various epithelia (Bosl et al., 2001; Catalan et al., 2004; Pena-Munzenmayer et al., 2005).
ClC-K channels form a subgroup of ClC channels expressed mainly in the kidney and the inner ear (Uchida and Sasaki, 2005; Sile et al., 2006; Jentsch, 2008). In human, two ClC channels, hClC-Ka and hClC-Kb, are expressed in distinct parts of the loop of Henle and fulfill transepithelial transport functions that are necessary for urinary concentration. Both channels are also expressed in the stria vascularis and simultaneous loss of function of both channels results in reduced K+ secretion and impaired sensory transduction.
ClC-3, ClC-4, ClC-5, ClC-6, and ClC-7 reside predominantly in intracellular compartments (Günther et al., 1998; Friedrich et al., 1999; Stobrawa et al., 2001; Wang et al., 2006; Graves et al., 2008; Neagoe et al., 2010). They function as coupled antiporters of Cl− and H+ (Picollo and Pusch, 2005; Scheel et al., 2005; Graves et al., 2008; Neagoe et al., 2010), presumably with a two Cl−: one H+ stoichiometry (Graves et al., 2008; Zifarelli and Pusch, 2009). The physiological role of these transporters is illustrated by rare human syndromes that are associated with impaired endosomal function and are caused by mutations in the genes encoding ClC-5 (CLCN5) (Lloyd et al., 1996) or ClC-7 (CLCN7) (Kornak et al., 2001). Intracellular ClCs were initially suggested to provide a shunt conductance in order to support electrogenic proton transport by V-type ATPases (Günther et al., 1998). However, work with engineered mice carrying ClC mutants that solely support passive anion transport demonstrated that this transport function is not sufficient to prevent pathological conditions found in ClC-5 and ClC-7 knock-out mice (Novarino et al., 2010; Weinert et al., 2010). Coupled anion–proton transport thus rather serves establishment of high intravesicular [Cl−].
ClC channels exhibit several functional properties that differ from other channel families. The existence of anion channels and transporters in the same family illustrates the functional similarity of ion channels and transporters. ClC proteins display a unique functional stoichiometry. They are dimeric proteins with two subunits and two independent active centers (Dutzler et al., 2002). For ClC channels, this peculiar structure gives rise to the appearance of single channel openings with two conductance states (Miller, 1982; Saviane et al., 1999; Fischer et al., 2010). Moreover, in double-barreled ClC channels two structurally distinct types of gating transitions occur, fast protopore gating and slow common gating (Miller, 1982; Saviane et al., 1999; Zuniga et al., 2004; de Santiago et al., 2005; Fischer et al., 2010). For fast gating of ClC-0, ClC-1, and ClC-2, a highly conserved glutamate at the amino-terminal end of the F-helix is important. In these ion channels, neutralization of this glutamate results in marked increases of fast gate open probability suggesting that this side chain contributes to the formation of the fast gate (Fahlke et al., 1997; Dutzler et al., 2003; Zuniga et al., 2004; de Santiago et al., 2005). For ClC-type transporters, neutralization of this “gating glutamate” abolishes coupled transport (Accardi and Miller, 2004; Picollo and Pusch, 2005; Scheel et al., 2005).
Anion Channels in the Loop of Henle of the Kidney and the Stria Vascularis of the Inner Ear
ClC-K channels are expressed predominantly in the loop of Henle of the kidney and in the stria vascularis of the inner ear (Figure 1) (Uchida, 2000; Estevez et al., 2001). In both types of epithelia anion channels with important physiological roles have been known for many years (Takeuchi et al., 1995; Wangemann et al., 1995; Uchida, 2000; Reeves et al., 2001) and later identified to be ClC-K/barttin channels.
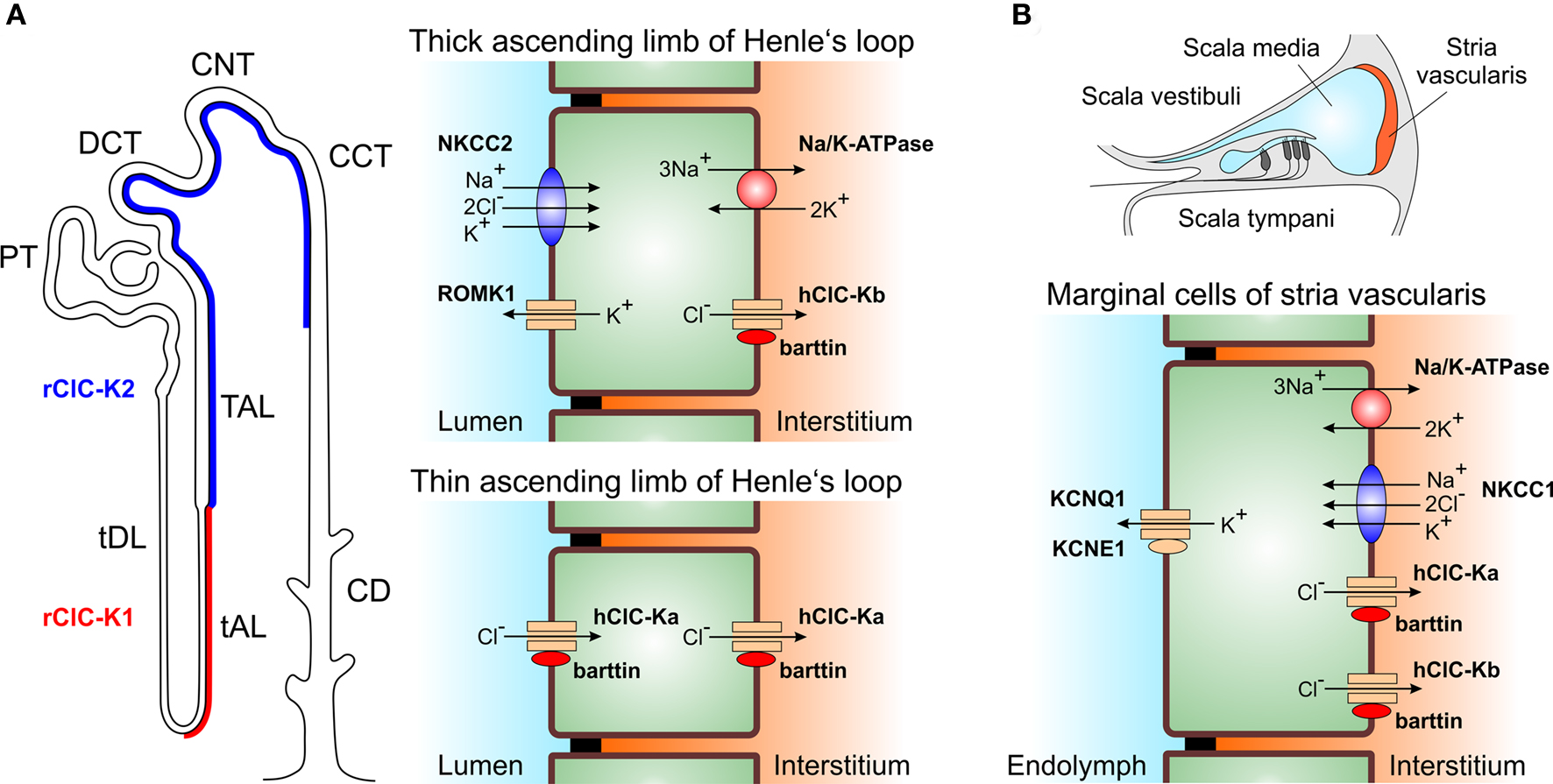
Figure 1. Physiological functions of ClC-K channels. (A) Major expression sites of ClC-K channels in the nephron. ClC-Ka/ClC-K1 permits passive transepithelial transport of chloride in the thin ascending limb of Henle. ClC-Kb/ClC-K2 is necessary for secondary-active sodium chloride reabsorption in the outer medulla and cortical region of the kidney. (PT, proximal tubule; tDL, thin descending limb of Henle’s loop; tAL, thin ascending limb of Henle’s loop; TAL, thick ascending limb of Henle’s loop; DCT, distal convoluted tubule; CNT, connecting tubule; CCT, cortical collecting tubule; CD, collecting duct). (B) Both ClC-Ka/ClC-K1 and ClCKb/ClC-K2 are expressed in the marginal cells of the stria vascularis of the inner ear and contribute to potassium secretion into the endolymph required for sensory transduction in inner hair cells.
The loop of Henle is a U-shaped tubule portion that connects the proximal and the distal tubule of the nephron (Figure 1). Its principal function is to generate a hyperosmotic interstitium using counter current mechanisms. An osmotic gradient between tubulus lumen and interstitium serves as the driving force for water resorption in the collecting duct and is thus necessary for urinary concentration. Transpithelial transport processes in the thin ascending and also in the thick ascending limb of Henle require anion fluxes through anion channels (Reeves et al., 2001).
The thin ascending limb of Henle is impermeable to water but exhibits an effective passive and electroneutral NaCl transport (Figure 1A). Measurements of apical and basolateral membrane potentials demonstrated a conductive pathway for anions on both membrane sides, whereas Na+ appears to diffuse on a paracellular pathway (Gross et al., 1975; Imai and Kokko, 1976). In the thick ascending limb cooperation of apical Na+–2Cl−–K+ co-transporters (NKCC2) and inwardly rectifying potassium channels (ROMK1) with basolateral anion channels results in electrogenic transcellular NaCl reabsorption (Greger, 1981; Greger and Schlatter, 1981, 1983; Greger et al., 1983) (Figure 1A). The thus generated transepithelial voltage provides a driving force for paracellular absorption of mono- and divalent cations.
Strial marginal cells and vestibular dark cells of the inner ear absorb K+ from the intrastrial fluid space and secrete it into the endolymph. K+ secretion is accomplished by basolateral K+ uptake by the Na+−2Cl–−K+ co-transporter (NKCC1) and Na+/K+-ATPase and apical secretion by a potassium channel consisting of KCNQ1 pore-forming and KCNE1 accessory subunits (Wangemann et al., 1995; Shen et al., 1997; Lang et al., 2007). On the basolateral membrane site, Cl– recycles through anion channels (Takeuchi et al., 1995) (Figure 1B).
Physiological Roles of CLC-K Channels
Rat ClC-K1 was cloned by Uchida et al. (1993) as the first kidney-specific ClC isoform. Subsequently, related isoforms from rat (rClC-K2) and human (hClC-Ka and hClC-Kb) were identified (Adachi et al., 1994; Kieferle et al., 1994). Whereas no functional expression of the other ClC-K isoforms could be initially achieved (Kieferle et al., 1994), rClC-K1 expression in Xenopus oocytes resulted in outwardly rectifying anion currents with Br > Cl > I conductivity sequence and a permeability sequence of Cl > Br > NO3 > I (Uchida et al., 1993; Waldegger and Jentsch, 2000). Moreover, anion currents in Xenopus oocytes heterologously expressing rClC-K1 channels were reduced by pharmacological agents that block transepithelial NaCl resorption in the thin ascending limb (Uchida et al., 1995).
rClC-K1 is upregulated in water-restricted animals (Uchida et al., 1993), and its deletion in a ClC-K1 knock-out mouse results in diabetes insipidus (Matsumura et al., 1999). rClC-K1 knock-out mice can maintain salt and water balance under normal conditions but are unable to concentrate urine under water restriction. Exogenous application of vasopressin agonists did not increase urinary osmolarity even though the integration of water channels into the collecting duct was intact, indicating that Diabetes insipidus resulted from a reduced osmotic gradient in the medulla due to impaired function of the thin ascending limb of Henle (Matsumura et al., 1999).
In situ hybridization and antibody studies (Vandewalle et al., 1997; Yoshikawa et al., 1999; Kobayashi et al., 2001) demonstrated that rClC-K1 is expressed in the thin ascending limb of Henle. RT-PCR experiments additionally detected lower amounts in the more distal segments of the nephron (Waldegger et al., 2002; Nissant et al., 2004). There are conflicting data about the subcellular distribution of rClC-K1 in the thin ascending limb of Henle. Uchida et al. (1995) demonstrated insertion into basolateral as well as into apical membranes. However, Vandewalle et al. (1997) reported exclusive staining of basolateral membranes in immunohistochemical experiments.
The distribution of rClC-K2 in the kidney is relatively widespread. Because of the high degree of sequence similarity between rClC-K1 and rClC-K2, no isoform-specific antibody has been generated so far. However, antibodies binding both rClC-K1 and rClC-K2 allowed determination of the intrarenal location of rClC-K2 in rClC-K1 knock-out mice (Kobayashi et al., 2001). This was found in the basolateral membrane of the thick ascending limb of Henle, also in the distal tubule and the cortical collecting duct (Uchida, 2000). With RT-PCR rClC-K2 was found to be highly expressed within the outer medulla and cortex but not in the thin ascending limb of Henle and other parts of the inner medulla, thus indicating a different physiological role for rClC-K2 than for rClC-K1 (Waldegger et al., 2002).
Sequence analysis was not sufficient to relate the human isoforms hClC-Ka and hClC-Kb to their mice orthologs, because homology between rClC-K1 and rClC-K2 is about 80% compared to approximately 80% homology between rClC-K1 and hClC-Ka. Kobayashi et al. (2002) generated transgenic mice harboring the green fluorescence protein driven by the human ClC-Kb gene promoter to determine the intrarenal distribution of human ClC-Kb. They observed fluorescent staining of the thick ascending limb of Henle, distal tubules, connecting tubules, and intercalated cells, demonstrating close similarity to tissue distributions of rClC-K2 (Kobayashi et al., 2002). Furthermore, genetic alterations of CLCNKB cause Bartter syndrome in human patients (Simon et al., 1997), a genetic syndrome with symptoms clearly distinct from Diabetes insipidus found in ClC-K1 knock-out mice (Matsumura et al., 1999). Taken together, these data indicate that hClC-Kb is the human homolog of rClC-K2, and hClC-Ka of rClC-K1.
Data reported to date provide compelling evidence that rClC-K1/hClC-Ka allows passive Cl- transepithelial resorption in the thin ascending limb of Henle (Figure 1A). It is a crucial component of the counter current system necessary for the establishment of the corticomedullary osmotic gradient that drives water resorption from the collecting duct. In contrast, rClC-K2/hClC-Kb is responsible for anion exit from the basolateral side of the thick ascending limb of Henle, supporting secondary-active NaCl resorption in this section of the nephron.
Immunohistochemical data demonstrated co-expression of both ClC-K channels and barttin in basolateral membranes of marginal cells in the stria vascularis (Estevez et al., 2001) (Figure 1B). Since neither human patients with CLCNKB mutations nor ClC-K1 knock-out mice exhibit any significant hearing loss, it may be deduced therefore that each isoform can act as a substitute for the lack of function of the other isoform.
The Accessory Subunit Barttin
For many ClC-K isoforms, heterologous expression did not initially result in the occurrence of functional anion channels (Kieferle et al., 1994; Waldegger and Jentsch, 2000). The identification of an accessory ClC-K channel subunit, barttin, provided a molecular basis for the lack of functional expression in these cases. Barttin is the gene product of BSND, the disease gene of a rare human disease, called Bartter syndrome type IV (Birkenhager et al., 2001). Patients with Bartter IV suffer from a severe impairment of urinary concentration ability as well as sensorineural deafness (Landau et al., 1995; Jeck et al., 2001). The clinical phenotype suggested that the disease gene might encode a subunit of a transport protein expressed in the kidney and the inner ear.
In all tested heterologous expression systems, co-expression of ClC-K channels with barttin resulted in the occurrence of functional anion channels. Barttin increases the number of ClC-K channels in the surface membrane of injected oocytes (Estevez et al., 2001; Waldegger et al., 2002), changes the subcellular distribution of ClC-K channels in transfected mammalian cells (Hayama et al., 2003; Scholl et al., 2006; Janssen et al., 2009), modifies the gating of rClC-K1 and switches human ClC-K channels from a non-conducting form into an active form (Scholl et al., 2006; Fischer et al., 2010) (Figure 2).
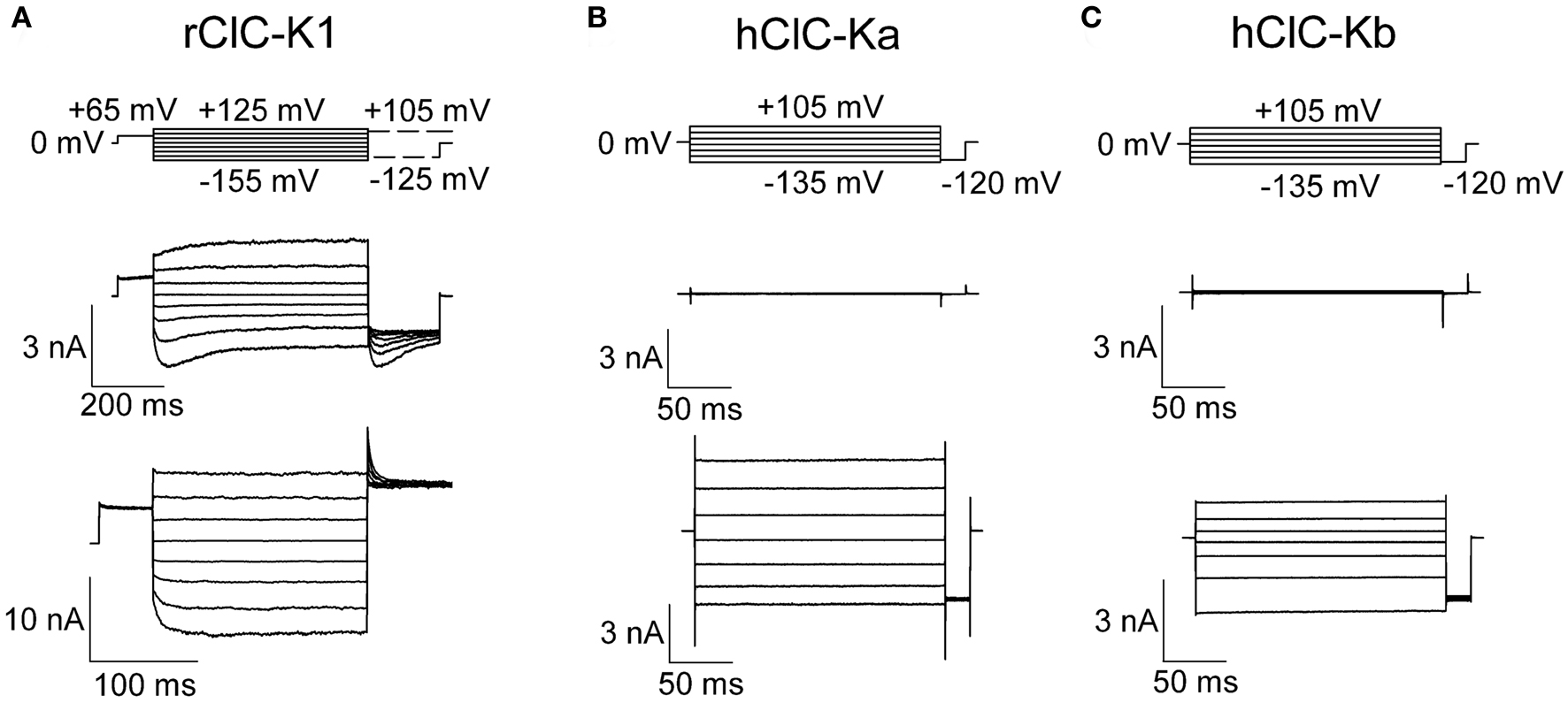
Figure 2. Representative current recordings of ClC-K channels heterologously expressed in mammalian tsA201 cells. Whole-cell patch clamp recordings of macroscopic currents of rClC-K1 (A), hClC-Ka (B), and hClC-Kb (C) in the absence (upper row) and presence (lower row) of the accessory subunit barttin. Channels were studied under nearly symmetrical chloride concentrations (pipette solution: 124 mM; bathing solution: 150 mM).
When expressed in MDCK cells without barttin, rClC-K2 was reported to reside mainly in the Golgi apparatus (Hayama et al., 2003). A different subcellular distribution was described by Scholl et al. (2006) who found fluorescent protein-tagged rClC-K1 and hClC-Kb to be retained within the endoplasmic reticulum. Co-transfection with barttin promotes insertion into the surface membrane often resulting in exclusive staining of the surface membrane of transfected cells (Scholl et al., 2006). Barttin has profound effects on the glycosylation status of ClC-K channels (Janssen et al., 2009). It stimulates complex glycosylation of ClC-K as indicator of effective exit from the endoplasmic reticulum. Moreover, since complex glycosylation represents a protective mechanism against protein degradation, this barttin effect might explain the higher protein stability of ClC-K when associated with barttin (Hayama et al., 2003).
In polarized epithelia, heterologously expressed ClC-K/barttin channels are located predominantly on the basolateral membrane site. Barttin appears to be the major determinant of the polarized insertion of ClC-K/barttin channels in epithelial cells. In the absence of ClC-K, barttin inserts preferentially into the basolateral membrane of MDCK cells (Janssen et al., 2009). Co-transfection with ClC-K does not modify the subcellular distribution of barttin, and optimal co-localization of ClC-K and barttin can be observed. A naturally occurring barttin mutation (E88X) abolishes basolateral targeting of barttin and results in apical and basolateral insertion of ClC-K/barttin channels (Janssen et al., 2009). At present, the regulation of the polarized distribution of ClC-K/barttin in epithelia is not fully understood. In contrast to the almost exclusive basolateral insertion of ClC-K/barttin in heterologous expression systems, anion channels with functional properties resembling ClC-K appear to reside also in the apical membrane (Uchida et al., 1995; Uchida, 2000). One might speculate that ClC-K channels could interact with additional subunits.
Although there are hClC-Ka and hClC-Kb channels present in the surface membrane, even in the absence of barttin (Estevez et al., 2001; Scholl et al., 2006), the lack of macroscopic currents indicates that these channels are non-functional. This hypothesis was verified by experiments using heterodimeric concatamers consisting of hClC-Kb and hClC-1, a muscle-specific ClC isoform that inserts into the plasma membrane independently of barttin (Steinmeyer et al., 1991b). Although hClC-Kb reached the surface membrane at higher density as a component of such a heterodimeric channel, anion currents were undistinguishable from pure hClC-1 currents (Scholl et al., 2006). However, co-transfection of the concatamer and barttin resulted in anion currents that corresponded to the addition of hClC-1 and hClC-Kb/barttin current components. Barttin thus controls macroscopic anion currents by determining the number of ClC-K channels in the surface membrane as well as its ability to conduct anions.
Genetically altered mice that lack barttin in the kidney and in the inner ear suffer from severe dehydration and die within a few days after birth. Rickheit et al. (2008) therefore engineered inner-ear specific BSND knock-out mice. These mice suffer from deafness, however, no impairment of renal function nor any obvious alteration of cochlear morphology could be detected. Moreover, normal [K + ] in the endolymph was observed. However, the endocochlear potential was markedly decreased, thus fully explaining the auditory phenotype of the mouse model. The authors demonstrated not only lack of barttin, but also of ClC-K proteins in the inner ear, indicating the importance of barttin for ClC-K protein stability. These findings demonstrate the necessity of ClC-K/barttin channels for the endocochlear potential and fully explain sensorineural deafness in patients suffering from mutations in BSND.
Functional Properties of Rat and Human CLC-K Channels
So far, three ClC-K isoforms have been functionally studied in detail, rat ClC-K1, human ClC-Ka and ClC-Kb (Waldegger and Jentsch, 2000; Waldegger et al., 2002; Scholl et al., 2006; Riazuddin et al., 2009; Fischer et al., 2010; Zifarelli et al., 2010) (Figure 2). Rat ClC-K1 is unique within the known ClC-K isoforms due to its ability to form functional anion channels without the accessory subunit barttin (Figure 2A). The distinct isoforms share a high degree of sequence homology (83% among rat isoforms, 91% among human isoforms, Kieferle et al., 1994), and at present the basis of the unique functional properties of rClC-K1 is not understood.
Rat ClC-K1 channels exhibit pronounced voltage-dependent gating that is distinct for channels with or without barttin. In the absence of barttin, biphasic voltage-dependent gating indicates the coexistence of two processes controlling channel opening and closing. Hyperpolarizing voltage steps result in channel activation followed by slower deactivation, whereas positive voltage steps elicit current deactivation followed by slower activation. Co-expression with barttin removes depolarization-induced gating and results in monophasic gating with channel opening upon voltage step into the negative direction.
rClC-K1 is assumed to share the common architecture of ClC channels with two subunits that exhibit two independent ion conduction pathways (Miller, 1982; Dutzler et al., 2002). Hence, fast and slow gating could either individually affect protopores or represent cooperative gating steps that jointly open or close both protopores. We used noise analysis (Katz and Miledi, 1970; DeFelice, 1981) and single channel recordings to study the molecular basis of the two different gating processes (Fischer et al., 2010) (Figure 3). In double-barreled ion channels, noise may arise from common openings and closings of both protopores together, from gating of individual protopores or from a combination of both. If the individual gates of all protopores are continuously open, noise analysis will provide unitary current amplitudes that are twice the protopore current amplitude, since unitary gating events will correspond to simultaneous openings of both protopores via the common gate. Alternatively, if common gates are completely open and individual protopores switch between open and closed state, currents and variances will indicate the same unitary conductance as a single protopore.
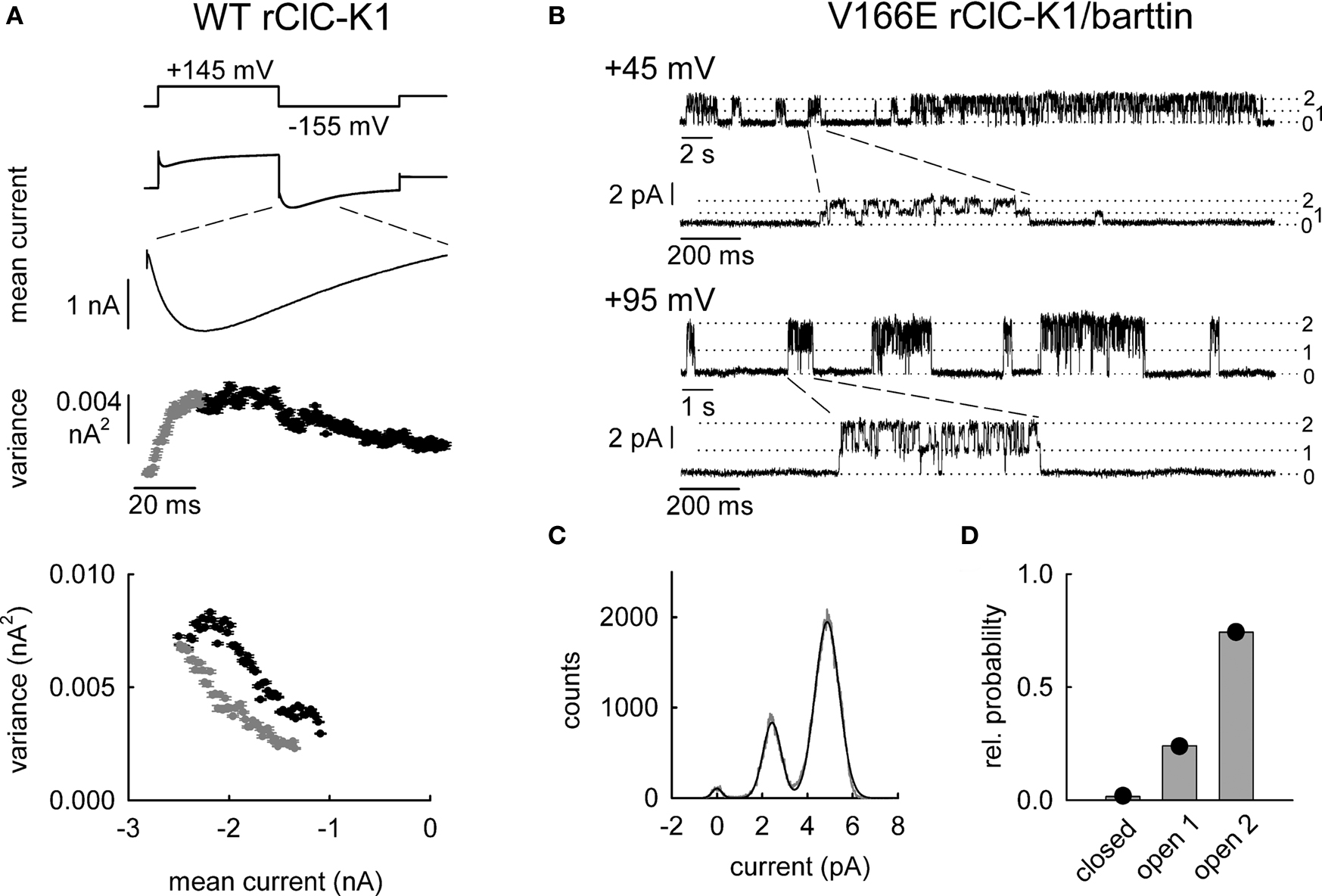
Figure 3. Analysis of the functional stoichiometry of rClC-K1 channels by non-stationary noise analysis (A) and single channel recordings (B–D). (A) Non-stationary noise analysis of WT rClC-K1 currents in the absence of barttin obtained from whole-cell patch clamp recordings of heterologously expressed channels in mammalian tsA201 cells. Mean currents (upper traces) and variances (middle trace) upon a voltage step that elicits biphasic responses result in a loop-like dependence when displayed in a plot of variances versus mean current amplitudes (lower panel). (B–D) Inside–out patch clamp recordings of mutant V166E rClC-K1 under nearly symmetrical chloride concentration (pipette solution: 150 mM; bathing solution: 124 mM). (B) Single channel recordings in presence of barttin at +45 and +95 mV show fast protopore gating with two equally spaced conductance states. (C) Amplitude histogram from bursts of registration at +95 mV. (D) Probabilities of the three current levels, closed, open 1 and open 2 at +95 mV (bars). Closed circles denote the predicted values for binomially distributed states. (modified from Fischer et al., 2010).
Figure 3A shows noise analysis on rClC-K1 current responses to a voltage protocol that results in fast activation and consecutive slow deactivation in the absence of barttin. A plot of current variances versus the corresponding mean current amplitude shows a loop-like dependence deviating from the parabolic relationship observed for other types of ion channels (Sigworth, 1980; Alvarez et al., 2002). Currents during the rising phase immediately after the voltage step produce lower variances than corresponding anion currents during the decaying phase (Figure 3A). This behavior demonstrates the existence of two gating processes with different unitary currents (Fischer et al., 2010). The slower process exhibits unitary current amplitudes that are approximately two-fold larger than for the fast process. This finding establishes that individual and cooperative gating processes exist in rClC-K1 and that fast gating opens and closes individual protopores and slow gating mediates cooperative processes. Co-expression of barttin results in the disappearance of slow gating of rClC-K1 and in monophasic time dependences of rClC-K1/barttin currents (Figure 2A). Currents activate upon membrane hyperpolarization, and deactivate at steps to positive voltages. Gating of rClC-K1/barttin is comparable to fast gating of rClC-K1 in the absence of barttin (Fischer et al., 2010).
ClC-K channels are unique among mammalian ClC channels in the absence of the “gating” glutamate that provides fast opening and closing of individual protopores. rClC-K1 nevertheless exhibits fast protopore gating. However, a point mutation V166E that reconstitutes the lacking glutamate does not install additional gating processes (Fischer et al., 2010). It causes profound alterations of rClC-K1 channel gating that resembles an apparent inversion of the voltage dependence of channel gating (Waldegger and Jentsch, 2000; Scholl et al., 2006). Separation of fast and slow gating demonstrated that V166E rClC-K1 without barttin displays only two distinct gating processes: fast protopore gating that is activated by membrane depolarization and slow common gating induced by membrane hyperpolarization (Fischer et al., 2010). An artificial “gating” glutamate in rClC-K1 modifies the voltage dependence of fast and slow gating conferred by another structural domains.
Single channel recordings of V166E rClC-K1/barttin provided definitive proof that fast gating affects exclusively the individual protopores (Figure 3B). After a rundown process following patch excision we observed long-lasting closures, interrupted by bursts of channel openings. In the bursts, rapid transitions between one closed and two conductance states occurred. The two conductance states were equally spaced and open probabilities were binomially distributed (Figures 3C,D). This behavior resembles the binomial burst behavior of ClC-0 (Miller, 1982), and demonstrates that there is voltage-dependent protopore gating in V166E rClC-K1/barttin channels. The open probability of wild type rClC-K1/barttin channels turned out to be too low to permit a similar analysis for WT channels.
The functional properties of hClC-Ka and hClC-Kb are different from rClC-K1 (Figure 2). hClC-Ka and hClC-Kb channels are not active in the absence of barttin (Kieferle et al., 1994; Waldegger and Jentsch, 2000; Scholl et al., 2006) but require the accessory subunit to become anion conducting (Figures 2B,C). hClC-Ka/ClC-Kb/barttin channels exhibit time-independent currents upon both positive and negative voltage steps (Scholl et al., 2006; Janssen et al., 2009). In contrast to hClC-Kb/barttin that exhibits bidirectional rectification, macroscopic hClC-Ka/barttin conductances decrease at very negative potentials, indicating a reduced open probability of hClC-Ka/barttin channels. Gating produces high frequency noise production at very negative potentials, indicating short closed and open states of the channels. Stationary noise analysis allowed determination of unitary current amplitudes as well as absolute open probabilities (Riazuddin et al., 2009). hClC-Ka/barttin channels exhibit a linear current–voltage relationship above −100 mV with unitary conductance of 26 pS. Absolute open probabilities are 1 at voltages positive to 0 mV and decay following a Boltzmann distribution with an inflection point at around −200 mV.
A surprising observation is the clear kinetic difference between ClC-K channels expressed in mammalian cells (Scholl et al., 2006; Janssen et al., 2009; Riazuddin et al., 2009; Sile et al., 2009) and Xenopus oocytes (Estevez et al., 2001; Waldegger et al., 2002; Embark et al., 2004; Liantonio et al., 2008; Martinez and Maduke, 2008; Zifarelli et al., 2010). When expressed in oocytes, both hClC-Ka/barttin and hClC-Kb/barttin show time- and voltage-dependent gating. Non-stationary noise analysis on excised patches containing hClC-Ka/barttin channels exhibits low open probability at 0 mV that increases at negative potentials without indication for saturation (Zifarelli et al., 2010). At present, the reason for this difference is not clear. One might speculate that unidentified subunits in mammalian cells or amphibian oocytes might be the basis of this difference. Alternatively, certain intracellular signal cascades that modify gating of ClC-K/barttin might exist only in one of the two expression systems.
Both rat ClC-K1/barttin and human ClC-Ka/barttin and ClC-Kb/barttin channels are modified by the composition of the external medium. They are blocked upon reduction of external pH (Uchida et al., 1995; Estevez et al., 2001; Waldegger et al., 2002). ClC-K channels resemble ClC-2 in its external pH dependence (Niemeyer et al., 2009) but differ from the muscle-type ClC isoforms, ClC-0 and ClC-1, that are activated by external protons (Hanke and Miller, 1983; Fahlke et al., 1996; Rychkov et al., 1996).
rClC-K1/barttin and both human hClC-K/barttin channels are unique among ClC isoforms in their sensitivity to external Ca2+ (Uchida et al., 1995; Estevez et al., 2001; Waldegger et al., 2002; Martinez and Maduke, 2008; Gradogna et al., 2010). Increasing external [Ca2+] from 0 to 5 mM increases ClC-Kb/barttin current approximately two-fold in Xenopus oocytes (Martinez and Maduke, 2008). Even more pronounced effects were found for rClC-K1 without barttin (Waldegger et al., 2002) or ClC-Ka/barttin (Martinez and Maduke, 2008; Gradogna et al., 2010). The physiological importance of the Ca2+-dependence of renal chloride channels is unclear. Increased external [Ca2+] will stimulate NaCl absorption by the thick ascending limb and thus Ca2+ reabsorption.
Many ClC channels display small unitary current amplitudes. At −160 mV, single channel amplitudes of ClC-1 and ClC-2 are about 280 fA (Pusch et al., 1994; Hebeisen and Fahlke, 2005) and 400 fA (Weinreich and Jentsch, 2001; Garcia-Olivares et al., 2008), respectively. Rat ClC-K1 and human ClC-Ka channels displayed voltage-independent single channel conductances of 33 pS (rClC-K1/barttin) and 26 pS (hClC-Ka/barttin) and are thus, to date, the ClC channels with the largest unitary conductance. In WT rClC-K1, single channel current measurements revealed unchanged amplitudes with and without barttin. The substitution of valine by glutamic acid (V166E) reduces the single channel conductance to 10 pS in the absence and 22 pS in the presence of barttin (Scholl et al., 2006; Fischer et al., 2010). Thus, barttin increases unitary current amplitudes of V166E rClC-K1, but leaves it unaltered in WT rClC-K1.
For rClC-K2/barttin or hClC-Kb/barttin unitary conductances have not yet been investigated in heterologous expression systems. However, Lourdel et al. (2003) and Nissant et al. (2004) investigated single chloride channels in the basolateral membrane of the murine distal convolute tubule and observed channels with low conductance of ∼9 pS as well as a permeability sequence and pH dependence that was comparable to macroscopic currents of rClC-K2/barttin and hClC-Kb/barttin in heterologous expression systems.
Structural Determinants of Barttin Function
The predicted transmembrane topology of barttin consists of two transmembrane helices encompassing the amino acids between 9 and 54 and a large cytoplasmic carboxy-terminus. Using various truncation mutants Scholl et al. (2006) demonstrated that key functions of barttin are mediated by the transmembrane core and a short stretch of amino acids carboxy-terminal to the second transmembrane helix. While the transmembrane core of barttin is sufficient for ClC-K channel exit from the endoplasmic reticulum and incorporation into the surface membrane, an additional stretch of 15–17 amino acids (I72X barttin) is necessary to turn hClC-Kb into a conductive anion channel (Figure 4). Separate regions of barttin thus confer the two functional properties of barttin, activation of ClC-K/barttin channels and promotion of membrane insertion.
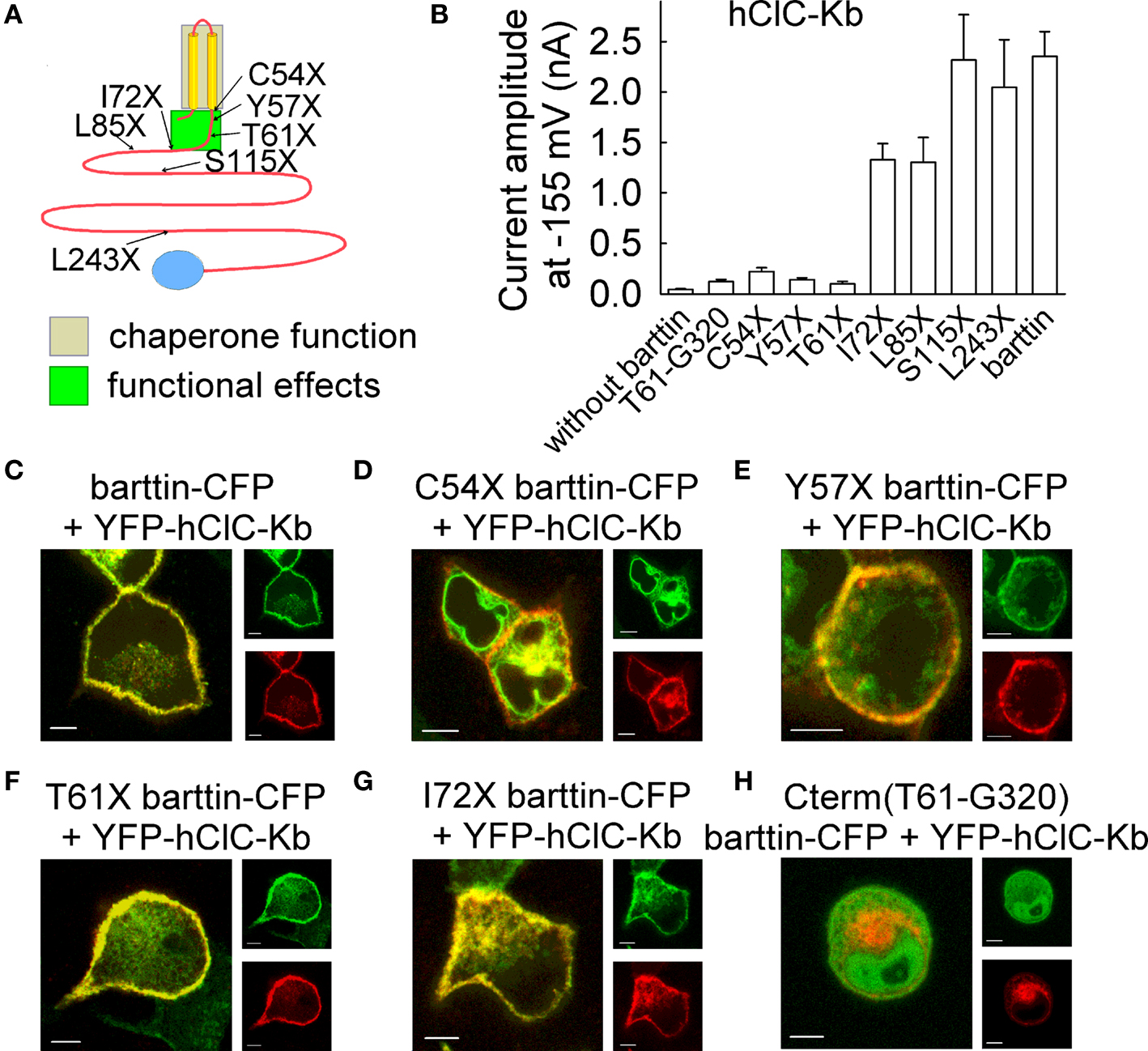
Figure 4. Truncation mutants of barttin affect subcellular distribution and functional properties of hClC-Kb channels. (A) Localization of tested carboxy-terminal truncations that result in functional deficits of barttin. Chaperone function of barttin is assigned to the two transmembrane domains. (B) Mean isochronal current amplitudes of hClC-Kb determined 2 ms after a voltage step to −155 mV on cells co-expressing WT or various truncated versions of barttin. (C–H) Confocal images of MDCK cells co-expressing YFP-hClC-Kb (red) and various mutants of barttin-CFP (green). Scale bars: 5 μm. (modified from Scholl et al., 2006, Proc. Natl. Acad. Sci. U. S. A 103, 11411–11416, copyright (2006) National Academy of Sciences, U.S.A).
In contrast to initial suggestions the carboxy-terminus is not involved in ClC-K/barttin interaction. In co-expression experiments of an isolated carboxy-terminal fragment and hClC-Kb no ClC-Kb/barttin currents were observed (Figure 4B). Expression of a fluorescent carboxy-terminal fusion protein results in fluorescent staining of the whole cell, since the protein is soluble and small enough to diffuse through nuclear pores into the cell nucleus (Figure 4H). In co-expression experiments with hClC-Kb, no redistribution of the carboxy-terminal fusion protein could be observed. Thus, the carboxy-terminus of barttin does not associate with ClC-K channel protein.
Tajima et al. (2007) studied interacting domains of barttin and ClC-K by co-expression of barttin and rClC-K2 fragments and co-immunoprecipitation experiments. They found that barttin interacts with the B and the J helix of rClC-K2 and suggested that barttin binds to ClC-K in a four-barttin to two-ClC-K subunit stoichiometry. Since it appears possible that transmembrane helices expose different binding domains in isolation than in the context of a full channel, this conclusion awaits further substantiation using other experimental approaches. Moreover, direct approaches are required to determine the subunit stoichiometry.
Disease-Causing Mutations in CLC-K and Barttin
Bartter syndrome is a group of genetic diseases characterized by impaired urinary concentration, polyuria, polydipsia, hypokalemia, alkalosis, elevated plasma renin and aldosterone, and normal blood pressure. Based on differences in the clinical phenotype, five different types of Bartter syndrome can be distinguished. For each Bartter syndrome, a distinct disease gene has been identified. In all cases, Bartter genes encode components of the NaCl reabsorption machinery in the thick ascending limb of Henle (Figure 1). Bartter syndrome type I and type II are caused by mutations in the genes encoding NKCC2 and ROMK1, respectively. CLCNKB was identified as underlying disease gene for Bartter syndrome type III (Simon et al., 1997). Type V is related to gain of function mutations of a calcium-sensing receptor that is activated by increased extracellular calcium and inhibits NKCC2 and ROMK1 in a G protein-coupled fashion (Hebert, 2003; Seyberth, 2008).
The majority of disease-causing mutations in CLCNKB induced a loss-of-function, resulting either in greatly reduced or absent macroscopic anion currents in heterologous expression systems. One mutation, R538P (Konrad et al., 2000), caused a change in the calcium sensitivity of hClC-Kb (Martinez and Maduke, 2008). However, it is not clear how such a change in calcium sensitivity might affect the kidney’s ability to concentrate urine.
There are also naturally occurring CLCNKA and CLCNKB mutations that result in a gain-of-function of renal and inner ear anion channels. Jeck et al. (2004) studied polymorphic mutations in the CLCNKB gene and found that a polymorphism predicting T481S is associated with hypertension as well as reduced hearing thresholds (Frey et al., 2006). This polymorphism was later found to be associated with hypertension in a South Australian population (Milton et al., 2006), but not in another study performed by a Swedish group (Fava et al., 2007). Hypertension is not only linked to CLCNKB, but also to CLCNKA. CLCNKA polymorphisms were associated with increased arterial pressure in an Italian population (Barlassina et al., 2007). Unfortunately, the functional consequences of these polymorphisms have not been reported so far.
Insertion of T481S had pronounced functional effects on ClC-Kb in heterologous expression system. In Xenopus oocytes (Jeck et al., 2004) and in mammalian cells (Sile et al., 2009) T481S ClC-Kb is active even in the absence of barttin. In oocytes, but not in mammalian cells, T481S increased ClC-Kb/barttin currents. These results indicate that T481 plays a crucial role in the functional switch between non-conducting and active ClC-Kb by barttin (Scholl et al., 2006). Furthermore, it underlines the physiological importance of the functional regulation of ClC-K channels by the accessory subunit barttin.
Bartter syndrome type IV is a rare subtype of the Bartter syndromes that results in both severe renal salt-wasting and sensorineural deafness. Fetal polyuria causes maternal polyhydramnios (Landau et al., 1995; Jeck et al., 2001), and postnatally, salt-wasting and polyuria occur as consequences of impaired tubular sodium chloride reabsorption. In some patients, renal failure may occur at a young age. Since mutations in the BSND gene (Birkenhager et al., 2001) trigger a combined dysfunction of ClC-Ka and ClC-Kb (Estevez et al., 2001), the identification of digenic mutations in CLCNKA and CLCNKb with comparable symptoms to monogenic barttin mutations was not surprising (Schlingmann et al., 2004; Nozu et al., 2008).
So far, seven BSND mutations were found in patients with Bartter syndrome IV that allow expression of a substantial portion of barttin, R8L (Birkenhager et al., 2001), R8W (Birkenhager et al., 2001), G10S (Birkenhager et al., 2001; Shalev et al., 2003), G47R (Garcia-Nieto et al., 2006), Q32X (Kitanaka et al., 2006), and E88X (Ozlu et al., 2006). Additionally, a mutation predicting I12T was identified in four large families with non-syndromic deafness (Riazuddin et al., 2009).
R8L, R8W, G10S, and Q32X result in loss-of-function of ClC-K/barttin channels expressed in mammalian cells (Janssen et al., 2009). This finding fully explains deafness and renal salt-wasting in affected patients. Q32X abolished surface membrane insertion of ClC-K/barttin and G47R decreased the number of ClC-K/barttin channels. However, in contrast to Q32X, G47R did not prevent activation of hClC-Kb/barttin channels, but rather appears only to reduce the binding affinity for ClC-K channels. G47R was reported in patients with an adult onset of renal symptoms, and the less pronounced renal phenotype correlates aptly to the preserved function of G47R barttin.
E88X does not modify the function of ClC-Kb/barttin channels, but abolishes the preferential insertion of ClC-K/barttin channels into the basolateral membrane. This finding demonstrates the importance of subcellular targeting for normal physiology. Increased apical anion conductance in the thick ascending limb or in the stria vascularis causes significant impairment of epithelial transport processes and results in deafness and reduced urinary concentration.
Heterologous co-expression of mutant I12T barttin and ClC-K channels in mammalian cells demonstrate full activation of the channels but reduced insertion into the surface membrane. I12T leaves unitary current amplitudes and absolute open probabilities of ClC-K/barttin unaffected and only impairs late steps in the intracellular trafficking pathway (Riazuddin et al., 2009).
These data provide an accurate correlation between barttin dysfunction and severity of the clinical phenotype (Figure 5). Only mutations that abolish barttin expression, such as Q32X (Janssen et al., 2009), or mutations that insert early stop codons (Birkenhager et al., 2001) are associated with a severe course and early deterioration of renal function (Birkenhager et al., 2001; Jeck et al., 2001). In contrast, renal function was preserved in all patients carrying barttin mutations that do not prevent insertion into the surface membrane. Mutations that result only in minor alteration of ClC-K/barttin function (G47R) or do not affect the subcellular distribution of barttin (G10S) were found to have the most benign clinical course.
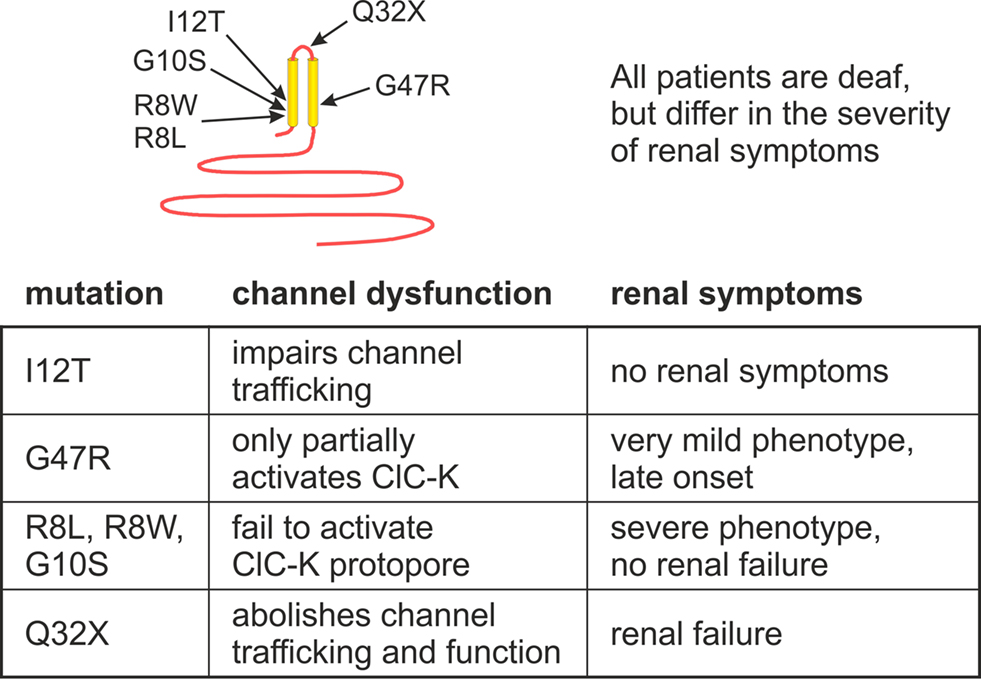
Figure 5. Disease-causing barttin mutations result in deafness and varying severity of renal symptoms depending on the type of functional deficits of ClC-K/barttin channels.
I12T is the only BSND mutation that almost exclusively impairs inner ear function (Riazuddin et al., 2009). The clinical phenotype and the alteration of channel function allowed insights into different roles of anion conductances in renal and inner ear epithelia. Reduction of ClC-K/barttin anion urinary by impaired trafficking results in deafness but not in a decreased concentration ability. The inner ear seems to be more sensitive than the kidney to a reduction in chloride conductances. Moreover, basolateral chloride flux by ClC-K/barttin appears to be a rate-limiting step in K+ secretion in the endolymph.
Pharmacology of CLC-K/Barttin Channels
ClC-K channels are promising candidates for the treatment of various human diseases. Block of ClC-Ka/barttin and ClC-Kb/barttin will impair the counter current mechanism in the loop of Henle and thus water resorption by the collecting duct. Since basolateral anion currents appear to be a rate-limiting step in K+ secretion by the stria vascularis of the inner ear (Riazuddin et al., 2009), stimulation of ClC-K/barttin currents will reduce hearing thresholds (Frey et al., 2006) and potentially improve certain hearing disorders. Moreover, ClC-K and barttin dysfunction in Bartter syndrome or in certain forms of non-syndromic deafness requires pharmacological restoration of ClC-K/barttin channels.
The coexistence of ClC-K/barttin channels in the kidney and the inner ear makes isoform-specific medication necessary. The vast majority of patients will not tolerate antihypertensive treatment causing deafness as side effect. The group of Conte-Camerino has focused on the generation of such drugs in recent years (Liantonio et al., 2004, 2006, 2008; Picollo et al., 2004). Based on a comparison of existing chemical compounds and detailed biophysical analysis of the blocking mechanisms (Picollo et al., 2007; Zifarelli et al., 2010), they produced high affinity blockers for ClC-Ka as well as for ClC-Kb by modifying activating compounds (Liantonio et al., 2008). It thus appears possible to create novel ligands with optimized properties concerning biophysical effects, isoform specificity and affinity to treat the different clinical phenotypes. Such rationally designed compounds carry great promises for future pharmacotherapy.
Open Questions and Outlook
The last two decades have provided much progress in our understanding of renal and inner ear chloride channels. Pore-forming and accessory subunits have been identified and their dysfunction in certain genetic diseases clarified. As in every ion channel, ClC-K/barttin channels are tightly regulated by intracellular signal cascades and many of these regulatory pathways are already known (Estevez et al., 2001; Embark et al., 2004; Schmidt et al., 2007; Bergler et al., 2008). Surprisingly, much basic information is still missing. There is still uncertainty as to the number of barttin subunits within a ClC-K/barttin complex. Although the interaction phase of both proteins has been defined (Tajima et al., 2007), it is still unclear how barttin exerts the variety of functions on ClC-K proteins (Scholl et al., 2006).
Many accessory subunits fulfill functions in conjunction with the modification of channel properties (Hidalgo and Neely, 2007). Such additional functions have not yet been identified for barttin. They might provide an explanation for the difference in the clinical course of Bartter IV patients with missense and non-sense mutations (Figure 5) and explain some of the clinical symptoms in this disease that cannot be directly explained by dysfunctional epithelial anion channels.
Improved understanding of molecular function as well as progress in pharmacotherapy carries the hope for novel therapeutic strategies to treat diseases associated with ClC-K/barttin channel dysfunction. Some of the clinical conditions are serious and debilitating, and such progress is therefore urgently needed.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Drs Gabriel Stölting and Daniel Wojchiechowski for helpful discussions. Our work is supported by the DFG (Fa 301/10-1).
References
Accardi, A., and Miller, C. (2004). Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature 427, 803–807.
Adachi, S., Uchida, S., Ito, H., Hata, M., Hiroe, M., Marumo, F., and Sasaki, S. (1994). Two isoforms of a chloride channel predominantly expressed in thick ascending limb of Henle’s loop and collecting ducts of rat kidney. J. Biol. Chem. 269, 17677–17683.
Alvarez, O., Gonzalez, C., and Latorre, R. (2002). Counting channels: a tutorial guide on ion channel fluctuation analysis. Adv. Physiol Educ. 26, 327–341.
Barlassina, C., Dal, F. C., Lanzani, C., Manunta, P., Guffanti, G., Ruello, A., Bianchi, G., Del, V. L., Macciardi, F., and Cusi, D. (2007). Common genetic variants and haplotypes in renal CLCNKA gene are associated to salt-sensitive hypertension. Hum. Mol. Genet. 16, 1630–1638.
Bergler, T., Stoelcker, B., Jeblick, R., Reinhold, S. W., Wolf, K., Riegger, G. A., and Kramer, B. K. (2008). High osmolality induces the kidney-specific chloride channel ClC-K1 by a serum and glucocorticoid-inducible kinase 1 MAPK pathway. Kidney Int. 74, 1170–1177.
Birkenhager, R., Otto, E., Schurmann, M. J., Vollmer, M., Ruf, E. M., Maier-Lutz, I., Beekmann, F., Fekete, A., Omran, H., Feldmann, Milford, D. V., Jeck, N., Konrad, M., Landau, D., Knoers, N. V., Antignac, C., Sudbrak, R., Kispert, A., and Hildebrandt, F. (2001). Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 29, 310–314.
Bosl, M. R., Stein, V., Hubner, C., Zdebik, A. A., Jordt, S. E., Mukhopadhyay, A. K., Davidoff, M. S., Holstein, A. F., and Jentsch, T. J. (2001). Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl- channel disruption. EMBO J. 20, 1289–1299.
Catalan, M., Niemeyer, M. I., Cid, L. P., and Sepulveda, F. V. (2004). Basolateral ClC-2 chloride channels in surface colon epithelium: regulation by a direct effect of intracellular chloride. Gastroenterology 126, 1104–1114.
de Santiago, J. A., Nehrke, K., and Arreola, J. (2005). Quantitative analysis of the voltage-dependent gating of mouse parotid ClC-2 chloride channel. J. Gen. Physiol. 126, 591–603.
Dutzler, R., Campbell, E. D., Cadene, M., Chait, M. B., and MacKinnon, R. (2002). X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 415, 287–294.
Dutzler, R., Campbell, E. D., and MacKinnon, R. (2003). Gating the selectivity filter in ClC chloride channels. Science 300, 108–112.
Embark, H. M., Bohmer, C., Palmada, M., Rajamanickam, J., Wyatt, A. W., Wallisch, S., Capasso, G., Waldegger, P., Seyberth, H. W., Waldegger, S., and Lang, F. (2004). Regulation of CLC-Ka/barttin by the ubiquitin ligase Nedd4-2 and the serum- and glucocorticoid-dependent kinases. Kidney Int. 66, 1918–1925.
Estevez, R., Boettger, T., Stein, V., Birkenhager, R., Otto, E., Hildebrandt, F., and Jentsch, T. J. (2001). Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature 414, 558–561.
Fahlke, Ch., Rosenbohm, A., Mitrovic, N., George, A. L. Jr., and Rüdel, R. (1996). Mechanism of voltage-dependent gating in skeletal muscle chloride channels. Biophys. J. 71, 695–706.
Fahlke, Ch., Yu, H. T., Beck, C. L., Rhodes, T. H., and George, A. L. Jr. (1997). Pore-forming segments in voltage-gated chloride channels. Nature 390, 529–532.
Fava, C., Montagnana, M., Almgren, P., Rosberg, L., Guidi, G. C., Berglund, G., and Melander, O. (2007). The functional variant of the CLC-Kb channel T481S is not associated with blood pressure or hypertension in Swedes. J. Hypertens. 25, 111–116.
Fischer, M., Janssen, A. G., and Fahlke, Ch. (2010). Barttin activates ClC-K channel function by modulating gating. J. Am. Soc. Nephrol. 21, 1281–1289.
Foldy, C., Lee, S. H., Morgan, R. J., and Soltesz, I. (2010). Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat. Neurosci. 13, 1047–1049.
Frey, A., Lampert, A., Waldegger, S., Jeck, N., Waldegger, P., Artunc, F., Seebohm, G., Lang, U. E., Kupka, S., Pfister, M., Hoppe, J., Gerloff, C., Schaeffeler, E., Schwab, M., and Lang, F. (2006). Influence of gain of function epithelial chloride channel ClC-Kb mutation on hearing thresholds. Hear. Res. 214, 68–75.
Friedrich, T., Breiderhoff, T., and Jentsch, T. J. (1999). Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 274, 896–902.
Garcia-Nieto, V., Flores, C., Luis-Yanes, M. I., Gallego, E., Villar, J., and Claverie-Martin, F. (2006). Mutation G47R in the BSND gene causes Bartter syndrome with deafness in two Spanish families. Pediatr. Nephrol. 21, 643–648.
Garcia-Olivares, J., Alekov, A., Boroumand, M. R., Begemann, B., Hidalgo, P., and Fahlke, C. (2008). Gating of human ClC-2 chloride channels and regulation by carboxy-terminal domains. J. Physiol. (London) 586, 5325–5336.
George, A. L., Crackower, M. A., Abdalla, J. A., Hudson, A. J., and Ebers, G. C. (1993). Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenita). Nat. Genet. 3, 305–310.
Gradogna, A., Babini, E., Picollo, A., and Pusch, M. (2010). A regulatory calcium-binding site at the subunit interface of CLC-K kidney chloride channels. J. Gen. Physiol. 136, 311–323.
Graves, A. R., Curran, P. K., Smith, C. L., and Mindell, J. A. (2008). The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 453, 788–792.
Greger, R. (1981). Chloride reabsorption in the rabbit cortical thick ascending limb of the loop of Henle. A sodium dependent process. Pflugers Arch. 390, 38–43.
Greger, R., and Schlatter, E. (1981). Presence of luminal K + , a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch. 392, 92–94.
Greger, R., and Schlatter, E. (1983). Properties of the lumen membrane of the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch. 396, 315–324.
Greger, R., Schlatter, E., and Lang, F. (1983). Evidence for electroneutral sodium chloride cotransport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch. 396, 308–314.
Gross, J. B., Imai, M., and Kokko, J. P. (1975). A functional comparison of the cortical collecting tubule and the distal convoluted tubule. J. Clin. Invest. 55, 1284–1294.
Günther, W., Lüchow, A., Cluzeaud, F., Vandewalle, A., and Jentsch, T. J. (1998). ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. U.S.A. 95, 8075–8080.
Hanke, W., and Miller, C. (1983). Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82, 25–45.
Hayama, A., Rai, T., Sasaki, S., and Uchida, S. (2003). Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem. Cell Biol. 119, 485–493.
Hebeisen, S., and Fahlke, Ch. (2005). Carboxy-terminal truncations modify the outer pore vestibule of muscle chloride channels. Biophys. J. 89, 1710–1720.
Hidalgo, P., and Neely, A. (2007). Multiplicity of protein interactions and functions of the voltage-gated calcium channel beta-subunit. Cell Calcium 42, 398–396.
Imai, M., and Kokko, J. P. (1976). Mechanism of sodium and chloride transport in the thin ascending limb of Henle. J. Clin. Invest. 58, 1054–1060.
Janssen, A. G., Scholl, U., Domeyer, C., Nothmann, D., Leinenweber, A., and Fahlke, Ch. (2009). Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J. Am. Soc. Nephrol. 20, 145–153.
Jeck, N., Reinalter, S. C., Henne, T., Marg, W., Mallmann, R., Pasel, K., Vollmer, M., Klaus, G., Leonhardt, A., Seyberth, H. W., and Konrad, M. (2001). Hypokalemic salt-losing tubulopathy with chronic renal failure and sensorineural deafness. Pediatrics 108, E5.
Jeck, N., Waldegger, P., Doroszewicz, J., Seyberth, H., and Waldegger, S. (2004). A common sequence variation of the CLCNKB gene strongly activates ClC-Kb chloride channel activity. Kidney Int. 65, 190–197.
Jentsch, T. J. (2008). CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev. Biochem. Mol. Biol. 43, 3–36.
Jentsch, T. J., Steinmeyer, K., and Schwarz, G. (1990). Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature 348, 510–514.
Kieferle, S., Fong, P., Bens, M., Vandewalle, A., and Jentsch, T. J. (1994). Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc. Natl. Acad. Sci. U.S.A. 91, 6943–6947.
Kitanaka, S., Sato, U., Maruyama, K., and Igarashi, T. (2006). A compound heterozygous mutation in the BSND gene detected in Bartter syndrome type IV. Pediatr. Nephrol. 21, 190–193.
Kleefuss-Lie, A., Friedl, W., Cichon, S., Haug, K., Warnstedt, M., Alekov, A., Sander, T., Ramirez, A., Poser, B., Maljevic, S., Hebeisen, S., Kubisch, C., Rebstock, J., Horvath, S., Hallmann, K., Dullinger, J. S., Rau, B., Haverkamp, F., Beyenburg, S., Schulz, H., Janz, D., Giese, B., Muller-Newen, G., Propping, P., Elger, C. E., Fahlke, Ch., and Lerche, H. (2009). CLCN2 variants in idiopathic generalized epilepsy. Nat Genet. 41, 954–955.
Kobayashi, K., Uchida, S., Mizutani, S., Sasaki, S., and Marumo, F. (2001). Intrarenal and cellular localization of ClC-K2 protein in the mouse kidney. J. Am. Soc. Nephrol. 12, 1327–1334.
Kobayashi, K., Uchida, S., Okamura, H. O., Marumo, F., and Sasaki, S. (2002). Human CLC-KB gene promoter drives the EGFP expression in the specific distal nephron segments and inner ear. J. Am. Soc. Nephrol. 13, 1992–1998.
Koch, M. C., Steinmeyer, K., Lorenz, C., Ricker, K., Wolf, F., Otto, M., Zoll, B., Lehmann-Horn, F., Grzeschik, K. H., and Jentsch, T. J. (1992). The skeletal muscle chloride channel in dominant and recessive human myotonia. Science 257, 797–800.
Konrad, M., Vollmer, M., Lemmink, H., van den Heuvel, L., Jeck, N., Vargas-Poussou, R., Lakings, A., Ruf, R., Deschenes, G., Antignac, C., Guay-Woodford, L., Knoers, N., Seyberth, H. W., Feldmann, D., and Hildebrandt, F. (2000). Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J. Am. Soc. Nephrol. 11, 1449–1459.
Kornak, U., Kasper, D., Boesl, M. R., Kaiser, E., Schweizer, M., Schulz, A., Friedrich, W., Delling, G., and Jentsch, T. J. (2001). Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104, 205–215.
Landau, D., Shalev, H., Ohaly, M., and Carmi, R. (1995). Infantile variant of Bartter syndrome and sensorineural deafness: a new autosomal recessive disorder. Am. J. Med. Genet. 59, 454–459.
Lang, F., Vallon, V., Knipper, M., and Wangemann, P. (2007). Functional significance of channels and transporters expressed in the inner ear and kidney. Am. J. Physiol. Cell Physiol. 293, C1187–C1208.
Liantonio, A., Picollo, A., Babini, E., Carbonara, G., Fracchiolla, G., Loiodice, F., Tortorella, V., Pusch, M., and Camerino, D. C. (2006). Activation and inhibition of kidney CLC-K chloride channels by fenamates. Mol. Pharmacol. 69, 165–173.
Liantonio, A., Picollo, A., Carbonara, G., Fracchiolla, G., Tortorella, P., Loiodice, F., Laghezza, A., Babini, E., Zifarelli, G., Pusch, M., and Camerino, D. C. (2008). Molecular switch for CLC-K Cl- channel block/activation: optimal pharmacophoric requirements towards high-affinity ligands. Proc. Natl. Acad. Sci. U.S.A. 105, 1369–1373.
Liantonio, A., Pusch, M., Picollo, A., Guida, P., De, L. A., Pierno, S., Fracchiolla, G., Loiodice, F., Tortorella, P., and Conte, C. D. (2004). Investigations of pharmacologic properties of the renal CLC-K1 chloride channel co-expressed with barttin by the use of 2-(p-Chlorophenoxy)propionic acid derivatives and other structurally unrelated chloride channels blockers. J. Am. Soc. Nephrol. 15, 13–20.
Lloyd, S. E., Pearce, S. H. S., Fisher, S. E., Steinmeyer, K., Schwappach, B., Scheinman, S. J., Harding, B., Alessandra, B., Devota, M., Goodyear, P., Rigden, S. P. A., Wrong, O., Jentsch, T. J., Craig, I. W., and Thakker, R. V. (1996). A common molecular basis for three inherited kidney stone diseases. Nature 379, 445–449.
Lourdel, S., Paulais, M., Marvao, P., Nissant, A., and Teulon, J. (2003). A chloride channel at the basolateral membrane of the distal-convoluted tubule: a candidate ClC-K channel. J. Gen. Physiol. 121, 287–300.
Martinez, G. Q., and Maduke, M. (2008). A cytoplasmic domain mutation in ClC-Kb affects long-distance communication across the membrane. PLoS One 3, e2746. doi: 10.1371/journal.pone.0002746.
Matsumura, Y., Uchida, S., Kondo, Y., Miyazaki, H., Ko, S. B., Hayama, A., Morimoto, Liu, W., Arisawa, M., Sasaki, S., and Marumo, F. (1999). Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 21, 95–98.
Miller, C. (1982). Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 299, 401–411.
Milton, A. G., Jannes, J., Hamilton-Bruce, M. A., and Koblar, S. A. (2006). Activating mutation of the renal epithelial chloride channel ClC-Kb predisposing to hypertension. Hypertension 47, e12–e13.
Neagoe, I., Stauber, T., Fidzinski, P., Bergsdorf, E. Y., and Jentsch, T. J. (2010). The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 285, 21689–21697.
Niemeyer, M. I., Cid, L. P., Yusef, Y. R., Briones, R., and Sepulveda, F. V. (2009). Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Physiol. 587, 1387–1400.
Nissant, A., Lourdel, S., Baillet, S., Paulais, M., Marvao, P., Teulon, J., and Imbert-Teboul, M. (2004). Heterogeneous distribution of chloride channels along the distal convoluted tubule probed by single-cell RT-PCR and patch clamp. Am. J. Physiol. Renal Physiol. 287, F1233–F1243.
Novarino, G., Weinert, S., Rickheit, G., and Jentsch, T. J. (2010). Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 328, 1398–1401.
Nozu, K., Inagaki, T., Fu, X. J., Nozu, Y., Kaito, H., Kanda, K., Sekine, T., Igarashi, T., Nakanishi, K., Yoshikawa, N., Iijima, K., and Matsuo, M. (2008). Molecular analysis of digenic inheritance in Bartter syndrome with sensorineural deafness. J. Med. Genet. 45, 182–186.
Ozlu, F., Yapicioglu, H., Satar, M., Narli, N., Ozcan, K., Buyukcelik, M., Konrad, M., and Demirhan, O. (2006). Barttin mutations in antenatal Bartter syndrome with sensorineural deafness. Pediatr. Nephrol. 21, 1056–1057.
Pena-Munzenmayer, G., Catalan, M., Cornejo, I., Figueroa, C. D., Melvin, J. E., Niemeyer, M. I., Cid, L. P., and Sepulveda, F. V. (2005). Basolateral localization of native ClC-2 chloride channels in absorptive intestinal epithelial cells and basolateral sorting encoded by a CBS-2 domain di-leucine motif. J. Cell Sci. 118, 4243–4252.
Picollo, A., Liantonio, A., Babini, E., Camerino, D. C., and Pusch, M. (2007). Mechanism of interaction of niflumic acid with heterologously expressed kidney CLC-K chloride channels. J. Membr. Biol. 216, 73–82.
Picollo, A., Liantonio, A., Didonna, M. P., Elia, L., Camerino, D., and Pusch, M. (2004). Molecular determinants of differential pore blocking of kidney CLC-K chloride channels. EMBO Rep. 5, 584–589.
Picollo, A., and Pusch, M. (2005). Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436, 420–423.
Pusch, M., Steinmeyer, K., and Jentsch, T. J. (1994). Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 66, 149–152.
Reeves, W. B., Winters, C. J., and Andreoli, T. E. (2001). Chloride channels in the loop of Henle. Annu. Rev. Physiol. 63, 631–645.
Riazuddin, S., Anwar, S., Fischer, M., Ahmed, Z. M., Khan, S. Y., Janssen, A. G., Zafar, A. U., Scholl, U., Husnain, T., Belyantseva, I. A., Friedman, P. L., Riazuddin, S., Friedman, T. B., and Fahlke, Ch. (2009). Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am. J. Hum. Genet. 85, 273–280.
Rickheit, G., Maier, H., Strenzke, N., Andreescu, C. E., De Zeeuw, C. I., Muenscher, A., Zdebik, A. A., and Jentsch, T. J. (2008). Endocochlear potential depends on Cl- channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 27, 2907–2917.
Rinke, I., Artmann, J., and Stein, V. (2010). ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J. Neurosci. 30, 4776–4786.
Rychkov, G. Y., Pusch, M., Astill, D. S. J., Roberts, M. L., Jentsch, T. J., and Bretag, A. H. (1996). Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. (London) 497, 423–435.
Saint-Martin, C., Gauvain, G., Teodorescu, G., Gourfinkel-An, I., Fedirko, E., Weber, Y. G., Maljevic, S., Ernst, J. P., Garcia-Olivares, J., Fahlke, Ch., Nabbout, R., LeGuern, E., Lerche, H., Poncer, J. C., and Depienne, C. (2009). Two novel CLCN2 mutations accelerating chloride channel deactivation are associated with idiopathic generalized epilepsy. Hum. Mutat. 30, 397–405.
Saviane, C., Conti, F., and Pusch, M. (1999). The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive Myotonia. J. Gen. Physiol. 113, 457–468.
Scheel, O., Zdebik, A. A., Lourdel, S., and Jentsch, T. J. (2005). Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436, 424–427.
Schlingmann, K. P., Konrad, M., Jeck, N., Waldegger, P., Reinalter, S. C., Holder, M., Seyberth, H. W., and Waldegger, S. (2004). Salt wasting and deafness resulting from mutations in two chloride channels. N. Engl. J. Med. 350, 1314–1319.
Schmidt, C., Hocherl, K., Schweda, F., and Bucher, M. (2007). Proinflammatory cytokines cause down-regulation of renal chloride entry pathways during sepsis. Crit. Care Med. 35, 2110–2119.
Scholl, U., Hebeisen, S., Janssen, A. G., Muller-Newen, G., Alekov, A., and Fahlke, Ch. (2006). Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. U.S.A. 103, 11411–11416.
Seyberth, H. W. (2008). An improved terminology and classification of Bartter-like syndromes. Nat. Clin. Pract. Nephrol. 4, 560–567.
Shalev, H., Ohali, M., Kachko, L., and Landau, D. (2003). The neonatal variant of Bartter syndrome and deafness: preservation of renal function. Pediatrics 112, 628–633.
Shen, Z. J., Marcus, D. C., Sunose, H., Chiba, T., and Wangemann, P. (1997). IsK channel in strial marginal cells – Voltage-dependence, ion- selectivity, inhibition by 293B and sensitivity to clofilium. Audit. Neurosci. 3, 215–230.
Sigworth, F. J. (1980). The variance of sodium current fluctuations at the node of Ranvier. J. Physiol. (London) 307, 97–129.
Sile, S., Vanoye, C. G., and George, A. L. Jr. (2006). Molecular physiology of renal ClC chloride channels/transporters. Curr. Opin. Nephrol. Hypertens. 15, 511–516.
Sile, S., Velez, D. R., Gillani, N. B., Narsia, T., Moore, J. H., George, A. L. Jr., Vanoye, C. G., and Williams, S. M. (2009). CLCNKB-T481S and essential hypertension in a Ghanaian population. J. Hypertens. 27, 298–304.
Simon, D. B., Bindra, R. S., Mansfield, T. A., Nilson-Williams, C., Mendonca, E., Stone, R., Schurman, S., Nayir, A., Alpay, H., Bakkaloglu, A., Rodriguez-Soriano, J., Morales, J. M., Sanjad, S. A., Taylor, C. M., Pilz, D., Brem, A., Trachtman, H., Griswold, W., Richard, G. A., John, E., and Lifton, R. P. (1997). Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 17, 171–178.
Steinmeyer, K., Klocke, R., Ortland, C., Gronemeier, M., Jockusch, H., Grunder, S., and Jentsch, T. J. (1991a). Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 354, 304–308.
Steinmeyer, K., Ortland, C., and Jentsch, T. J. (1991b). Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 354, 301–304.
Stobrawa, S. M., Breiderhoff, T., Takamori, S., Engel, D., Schweizer, M., Zdebik, A. A., Bosl, M. R., Ruether, K., Jahn, H., Draguhn, A., Jahn, R., and Jentsch, T. J. (2001). Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29, 185–196.
Tajima, M., Hayama, A., Rai, T., Sasaki, S., and Uchida, S. (2007). Barttin binds to the outer lateral surface of the ClC-K2 chloride channel. Biochem. Biophys. Res. Commun. 362, 858–864.
Takeuchi, S., Ando, M., Kozakura, K., Saito, H., and Irimajiri, A. (1995). Ion channels in basolateral membrane of marginal cells dissociated from gerbil stria vascularis. Hear. Res. 83, 89–100.
Uchida, S. (2000). In vivo role of CLC chloride channels in the kidney. Am. J. Physiol. Renal, Fluid Electrolyte Physiol. 279, F802–F803.
Uchida, S., and Sasaki, S. (2005). Function of chloride channels in the kidney. Annu. Rev. Physiol. 67, 759–778.
Uchida, S., Sasaki, S., Furukawa, T., Hiraoka, M., Imai, T., Hirata, Y., and Marumo, F. (1993). Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J. Biol. Chem. 268, 3821–3824.
Uchida, S., Sasaki, S., Nitta, K., Uchida, K., Horita, S., Nihei, H., and Marumo, F. (1995). Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J. Clin. Invest. 95, 104–113.
Vandewalle, A., Cluzeaud, F., Bens, M., Kieferle, S., Steinmeyer, K., and Jentsch, T. J. (1997). Localization and induction by dehydration of ClC-K chloride channels in the rat kidney. Am. J. Physiol. Renal Physiol. 272, F678–F688.
Waldegger, S., Jeck, N., Barth, P., Peters, M., Vitzthum, H., Wolf, K., Kurtz, A., Konrad, M., and Seyberth, H. W. (2002). Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 444, 411–418.
Waldegger, S., and Jentsch, T. J. (2000). Functional and structural analysis of ClC-K chloride channels involved in renal disease. J. Biol. Chem. 275, 24527–24533.
Wang, X. Q., Deriy, L. V., Foss, S., Huang, P., Lamb, F. S., Kaetzel, M. A., Bindokas, V., Marks, J. D., and Nelson, D. J. (2006). CLC-3 channels modulate excitatory synaptic transmission in hippocampal neurons. Neuron 52, 321–333.
Wangemann, P., Liu, J., and Marcus, D. C. (1995). Ion transport mechanisms responsible for K+ secretion and the transepithelial voltage across marginal cells of stria vascularis in vitro. Hear. Res. 84, 19–29.
Weinert, S., Jabs, S., Supanchart, C., Schweizer, M., Gimber, N., Richter, M., Rademann, J., Stauber, T., Kornak, U., and Jentsch, T. J. (2010). Lysosomal pathology and osteopetrosis upon loss of H + -driven lysosomal Cl- accumulation. Science 328, 1401–1403.
Weinreich, F., and Jentsch, T. J. (2001). Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem. 276, 2347–2353.
Yoshikawa, M., Uchida, S., Yamauchi, A., Miyai, A., Tanaka, Y., Sasaki, S., and Marumo, F. (1999). Localization of rat CLC-K2 chloride channel mRNA in the kidney. Am. J. Physiol. 276, F552–F558.
Zifarelli, G., Liantonio, A., Gradogna, A., Picollo, A., Gramegna, G., De, B. M., Murgia, A. R., Babini, E., Camerino, D. C., and Pusch, M. (2010). Identification of sites responsible for the potentiating effect of niflumic acid on ClC-Ka kidney chloride channels. Br. J Pharmacol. 160, 1652–1661.
Zifarelli, G., and Pusch, M. (2009). Conversion of the 2 Cl-/1 H+ antiporter ClC-5 in a NO3-/H+ antiporter by a single point mutation. EMBO J. 28, 175–182.
Keywords: ClC channels, barttin, epithelial transport, loop of Henle, stria vascularis
Citation: Fahlke C and Fischer M (2010) Physiology and pathophysiology of ClC-K/barttin channels. Front. Physio. 1:155. doi: 10.3389/fphys.2010.00155
Received: 09 September 2010;
Paper pending published: 28 September 2010;
Accepted: 09 November 2010;
Published online: 26 November 2010.
Edited by:
Ali Mobasheri, The University of Nottingham, UKReviewed by:
Jorge Arreola, Universidad Autónoma de San Luis Potosi, MexicoJonathan Lippiat, University of Leeds, UK
Alfred L. George, Vanderbilt University, USA
Copyright: © 2010 Fahlke and Fischer. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Christoph Fahlke, Institut für Neurophysiologie, Medizinische Hochschule Hannover, Carl-Neuberg-Str. 1, 30625 Hannover, Germany. e-mail: fahlke.christoph@mh-hannover.de