 Wei Kang
Wei Kang Xue Du
Xue Du
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Phys. , 06 April 2023
Sec. Chemical Physics and Physical Chemistry
Volume 11 - 2023 | https://doi.org/10.3389/fphy.2023.1188513
This article is part of the Research Topic Calculation and Design of Two-dimensional Thermoelectric and Piezoelectric Materials View all 14 articles
As one of the first experimentally found and naturally stable two-dimensional (2D) ferromagnetic materials, the monolayer Cr2Ge2Te6 has garnered great interest due to its potential hires in electronics and spintronics. Yet, the Curie temperature of monolayer Cr2Ge2Te6 is lower than the ambient temperature, severely restricting the creation of valuable devices. Using the first-principle calculations, we explored how the adsorption of 3d transition metals affects the electronic and magnetic properties of the monolayer Cr2Ge2Te6 (from Sc to Zn). Our findings indicate that depending on the 3d transition metals to be adsorbed, the electronic properties of the Cr2Ge2Te6 adsorption system may be adjusted from semiconductor to metal/half-metal. We found that the adsorption of Ti and Fe leads to a transformation from semiconductor to metal. While in Cr2Ge2Te6@Sc, V, Co, Ni, and Cu, the absorption realizes the changes from semiconductor to half metal. Moreover, adsorption may modify the magnetic moment and Curie temperature of the adsorbed system to enhance the ferromagnetic stability of the monolayer Cr2Ge2Te6. Furthermore, we are able to modulate the half-metallic of Cr2Ge2Te6@Mn by means of electric fields. Hence, adsorption is a viable method for modulating the ferromagnetic half-metallic of 2D ferromagnets, paving the door for the future development of nano-electronic and spintronic devices with enhanced performance for 2D ferromagnetic materials.
Since Geim and Novoselov initially reported [1] the successful fabrication of graphene by the transparent tape exfoliation technique in 2004, there has been a continuous attempt to identify two-dimensional (2D) materials with superior characteristics. In the realm of contemporary materials research, atomic-level thickness layered van der Waals materials are suitable as a broad class of materials with excellent physical properties [2], and they are now gaining popularity among scientists. The van der Waals force interaction within the interlayer of layered 2D materials is a typical weak coupling interaction. In particular, with the discovery of boronene [3], MoS2 [4], and black phosphorus [5], single or multiple-layer 2D layered materials, the interest in 2D semiconductor layered materials, and their relative heterostructures have been intensively studied. Yet, another essential attribute of materials, magnetic properties, has been of interest and is absent from many 2D van der Waals materials. For example, graphene [6], a band-gapless, nonmagnetic 2D material, impedes its employment in spintronic devices. Hence, researchers are hopeful of discovering a series of 2D stacked magnetic materials.
Low-dimensional ferromagnetic semiconductors are urgently required to construct the next-generation of nanoscale spintronic devices. In recent years, there are some 2D half-metallic materials, for example, monolayer Mxene [7, 8] and multiferroic nanosheets ACr2S4 [9] (A = Li, Na, K Rb). The transition-metal trichalcogenides (TMTC), another class of typical layered van der Waals materials that possess both magnetic properties and a unique layered structure, have also attracted the interest of the public [10]. They are coupled between layers by weak van der Waals interactions; for instance, MnPS3 has excellent physicochemical properties and is easy to prepare [11, 12]. In addition, the materials of this structure display diverse electronic properties, some as semiconducting and others as insulating, and they also have unique magnetic properties. As a potential candidate for 2D magnetic layered van der Waals materials, it has become the focus of recent research. In particular, as a member of the family of layered van der Waals materials, crystalline Cr-based transition metal trichloride has attracted significant interest. For example, the Curie temperature of CrSiTe3 [13], one of the transition metal trisulfide materials, increases as the number of layers decreases. It is reported to be a well-known 2D ferromagnetic [14] semiconductor because both the block and monolayer exhibit different magnetic ground states. In addition, it has been reported [15] that the Curie temperature of CrI3 is 45 K. Being a multilayer van der Waals insulator, its Curie temperature drops as the number of layers reduces, and it is also regarded as an excellent 2D ferromagnetic material. In recent years, Cr2Ge2Te6 has also been selected by researchers because of its van der Waals geometry, intrinsic ferromagnetism, and higher Curie temperature than CrI3. It has been stated [16] that V. Carteaux produced the first bulk of Cr2Ge2Te6 in 1995. In 2017, a ferromagnetic monolayer, Cr2Ge2Te6, with a Curie temperature of 61 K, in experimental, was exfoliated [17]. 2D layered Cr2Ge2Te6 [18] of novel electronic, magnetic, optical, optoelectronic, piezoelectric, and thermoelectric properties are easily compatible with nanoelectronic devices. It was even reported [17] that ferromagnetism exists in the new Cr2Ge2Te6 atomic layer and that the Curie temperature can be easily regulated by modulating the applied magnetic field. This finding suggests Cr2Ge2Te6 is an ideal candidate for the study of 2D magnetic phenomena and spintronic device materials. On the other hand, the focus of research on this type of 2D layered van der Waals magnetic materials will also significantly advance our understanding of the magnetic properties of low-dimensional materials, which, when combined with their rich electronic and optical properties, will open up numerous application possibilities for 2D magnetoelectric and magneto-optical applications.
In particular, research on intrinsically magnetic semiconductors with high Curie temperatures, large band gaps, and high carrier mobility is of extraordinary significance for developing the next-generation of ultra-miniaturized, highly integrated spintronics and magneto-optics devices. Nevertheless, it is challenging to have long-range ordered ferromagnetic and semiconducting properties in 2D materials [19], thus hoping that the substrate material can be tuned by some conventional modifications to produce magnetic properties. In the past decades, many modifications, such as magnetic doping [20], nonmagnetic doping [21], or hole doping [22], have been used in an attempt to introduce long-range ordered ferromagnetic sequences in semiconductors. Functional adsorption is one of the most traditional techniques to modulate the electronic and magnetic properties of the material effectively. Many efforts have previously been devoted to studying metal atom interactions in 2D materials inducing magnetic for applications in spintronics. Particularly, graphene-like 2D materials interact with magnetic atoms to produce half metal, and this modulation has been considered as an effective and interesting means of application to spintronics. Some theoretical studies have shown that the adsorption of metal atoms on the graphene sheets significantly increases the Curie temperature up to 438 K [23]. The adsorption of transition metal atoms on the surface of black phosphorus also effectively modulates its electronic and magnetic properties [24]. It was reported that a strong orbital coupling between GaSe monolayer and transition metals was found to produce 100% spin-polarized currents [25]. A recent study also found that monolayer Cr2Ge2Te6 nanosheet interacted with alkali metals, enhancing their ferromagnetism and exhibiting half-metallic properties [26]. Therefore, the combination of 2D layered van der Waals materials, intrinsic ferromagnetism, half-metallic properties, and adsorption will likely provide a good platform for applying new low-dimensional magnetic materials in the field of spintronics.
Using first-principles calculation, we want to examine the electronic and magnetic properties of 3d transition metals adsorbed on monolayer Cr2Ge2Te6 nanosheet. We found that 3d transition metals adsorbed on the substrate Cr2Ge2Te6 significantly change the magnetic moments of the system. Except for Ti and Cr adsorbed, the Curie temperature of the adsorption system (TM = Sc, V, Mn, Fe, Co, Ni, Cu, and Zn) is significantly enhanced by a factor of two, with an enhancement of Curie temperature increasing rate over to 200%. Based on the optimized structure of the adsorbed system, we analyzed the geometry, electronic structure, spin density of states, magnetic moments, and Curie temperature of the ten-absorbed system for first-principles calculation. We found that the adsorption energy changes significantly as the 3d transition metal is adsorbed at different positions. The degree of interaction between the transition metal atoms and monolayer Cr2Ge2Te6 significantly differs at different adsorption positions. We also found that the 3d transition metals change the electronic properties of the absorbed system, especially Cr2Ge2Te6@Ti and Cr2Ge2Te6@Fe become metallic. At the same time, the Sc, V, Co, Ni, and Cu absorbed systems show half-metallic. In addition, we found that the adsorption of 3d transition metals can significantly increase the Curie temperature of the system. Therefore, the present study reveals that the monolayer Cr2Ge2Te6 can change its electronic structure and magnetic properties by adsorption of 3d transition metal atoms, thus predicting the unlimited application prospects of monolayer 2D Cr2Ge2Te6 ferromagnet in the next-generation of functional spintronic devices.
Our density functional theory (DFT) calculations were performed using Vienna ab initio Simulation Package [27, 28]. The projector augmented wave formalism (PAW) [29, 30] describes the ion-electron potential. The exchange of correlation functions during the simulations is mainly handled by the generalized gradient approximation (GGA) [31] and is parameterized by the Perdew-Burke-Ernzerhofer (PBE) parameterization. Projection-enhanced wave functions have also been used to describe the electron-ion potential [27, 32]. The kinetic cut-off energy of the plane wave basis group is set as 400 eV. We set the condition of full convergence at ion relaxation as long as the total energy is less than 10–5 eV and the force on each atom is less than 0.01 eV/Å when the lattice constant, as well as the atomic positions, will be the most stable state of complete relaxation. To eliminate the interaction between periodic boundaries, we simulated the properties of the monolayer Cr2Ge2Te6 using a 20 Å thick slab model in the z-direction. In addition, to fully characterize the van der Waals interactions between the adsorbed 3d transition metal and the monolayer Cr2Ge2Te6, a DFT-D3 correction method [33, 34] with the Grimme scheme is used to perform the whole system study calculations. We adopted the 2 × 2 supercell structure for monolayer Cr2Ge2Te6 with one transition metal. Besides, the Brillouin zone k-point mesh of 3 × 3 × 1 is used for optimizing, and a denser 8 × 8 × 1 k-point mesh is used for electronic structure computations. Here, we analyzed these results by utilizing the PBE + U functional to describe the high correlations between the electrons in the d shell. The on-site interactions
It is known from previous studies [42] that there is a large van der Waals gap in the structure of bulk Cr2Ge2Te6, and the interlayer interactions are pretty weak, so it is possible to extract 2D Cr2Ge2Te6 nanosheet by experimental peeling. Therefore, before studying the adsorption of transition metal atoms by the substrate monolayer Cr2Ge2Te6, we first investigated the structural, electronic, and magnetic properties of the monolayer Cr2Ge2Te6. Supplementary Figure S1A shows the top and side views of a monolayer Cr2Ge2Te6 supercell (2 × 2). Cr2Ge2Te6 is a hexagonal lattice structure. Each layer can be considered an octahedral CrTe6 at the edge of the hexagonal lattice and a Ge dimer in the center [43]. After structural optimization, there are 40 atoms in the supercell (2 × 2) with lattice constants of a = b = 13.65 Å, α = 120°, and a bond length of 2.78 Å between the Cr atom and the nearest neighboring Te atom. Our calculated results are in agreement with previous literature [18]. The spin-polarized band structure shown in Supplementary Figure S1B explains the electronic properties of monolayer Cr2Ge2Te6. It reveals that the monolayer Cr2Ge2Te6 is a semiconductor whose band gap in the spin-down channel is indirect, with the valence band in the Γ point being the top of the valence band (VBM) and the bottom of conduction band (CBM) located in the Κ point, and the value of band gap in the spin-down channel is 0.52 eV; In the spin-up channel, we can see that the VBM is located at Γ point, while the CBM is located between K point and M point, and its indirect band gap value is 0.31 eV, which is consistent with the results of the literature [18, 44]. Supplementary Figure S1C shows the total density of states of the monolayer Cr2Ge2Te6. We can see that near the Fermi surface, both the conduction band and the valence band are asymmetric in both spin channels, and the VBM in the spin-up channel is closer to the Fermi surface than its VBM in the spin-down energy band, while the CBM in the spin-up channel is closer to the Fermi surface than that in the spin-down channel, which also indicates that both the conduction band and the valence band edges near the Fermi surface are fully spin-polarized. Thus the band gap in the spin-up channel is smaller than that in the spin-down channel, implying that Cr2Ge2Te6 is a bipolar magnetic semiconductor [45]. In addition, the contribution of the Cr atom in the spin-up is significantly larger than that in the spin-down, while the density of states of Te atoms is in the conduction band, where the electronic density of states in the spin-up is significantly larger than that in the spin-down. In addition, the valence band edge is mainly contributed by Te atoms, and at the conduction band edge, it is contributed primarily by Cr and Te atoms [18]. The spin polarization at the Fermi surface is also caused by the interaction between Cr and Te atoms. The valence band top is occupied by the p orbital of the Te atom, while the hybridized orbital of the d orbital of the Cr atom and the p orbital of the Te atom occupies the conduction band bottom.
To confirm the magnetic ground state of the most stable monolayer Cr2Ge2Te6 nanosheet, we have calculated two different magnetic configurations for monolayer Cr2Ge2Te6 nanosheet, i.e., ferromagnetic (FM) and antiferromagnetic (AFM) states. The energy difference (∆E) is defined as the difference between the energy of AFM and FM per cell, that is ∆E = EAFM−EFM, where
Previous work has shown [18] that low-dimensional materials are sensitive to external stimuli, which helps us investigate the adsorption system’s fundamental properties. After studying the electronic properties and magnetic properties based on monolayer Cr2Ge2Te6, we selected transition metal (TM = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) atoms and monolayer Cr2Ge2Te6 nanosheet for the analytical study of the adsorption mechanism. Studying this class of transition metal adsorption will be a good guide for applying 2D magnetic materials in spintronic devices. We selected 2 × 2 supercell monolayer Cr2Ge2Te6 as substrates for the adsorbing 3d transition metals from Sc atoms to Zn atoms sequentially. To find the most favorable adsorption sites, as shown in Figures 1A–C, we considered three locations where the monolayer Cr2Ge2Te6 nanosheet adsorb the transition metal atoms. They are positions A (Cr-top), B (Te-top), and C (Ge-top), respectively. The adsorption distance (d) is the distance between 3d transition metal atoms and the substrate surface atoms, as shown in Figure 1D. To evaluate the interaction between the substrate and 3d transition metals, we calculated the adsorption energy of the adsorption system. We used the adsorption energy to describe the absorption stability with the following equation:
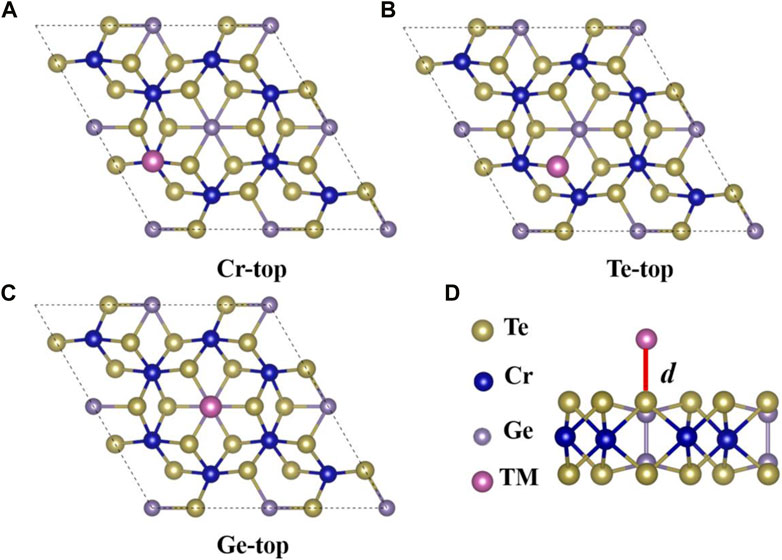
FIGURE 1. The top views of the crystalline structure of a 2 × 2 monolayer Cr2Ge2Te6, with TM atoms absorbed at three different positions labeled as (A) Cr-top, (B) Te-top, and (C) Ge-top, respectively. The dotted black lines in the graph indicate the 2 × 2 supercell. (D) The side view of the monolayer Cr2Ge2Te6 (2 × 2) nanosheet adsorbed by the transition metals, and the adsorption distance d is represented by the solid red line. The atoms of the Te, Cr Ge, and 3d transition metals are represented by the pale yellow, indigo, purple, and pink balls represent, respectively.
Figure 2A shows the calculated adsorption energy at three positions after complete structural relaxation. All adsorption energies at all positions are negative, except for the Cr adsorbed at the Cr-top position and Ti atoms adsorbed at the Ge-top site. The adsorption energies are negative, indicating that this reaction is exothermic, suggesting the monolayer Cr2Ge2Te6 can adsorb transition metals at three different positions, indicating that these adsorption systems are stable. Still, the interaction strength between the transition metal and the substrate differs from 3d transition metals. For the adsorption of Mn and Zn, there is no significant difference in the adsorption energies at the three different sites. Still, the adsorption energy of the Cr2Ge2Te6@Mn fluctuating between −0.55 and −0.66 eV, is larger than that of the Cr2Ge2Te6@Zn, indicating that the interaction strength in Cr2Ge2Te6@Mn is stronger than that of the Cr2Ge2Te6@Zn. For the systems adsorbed Sc, Ti, V, Cr, Fe, Co, Ni, and Cu, their adsorption energies significantly changed at different adsorption positions, implying the strength of interaction between 3d transition metals and the substrate was quite different. Similarly, from Supplementary Table S1, the adsorption distances of 3d transition metals adsorbed at different positions have significantly different variations. Cr2Ge2Te6@Sc has relatively high adsorption energy, indicating a strong interaction between the Sc atoms and the substrate. However, for the Cr2Ge2Te6@Zn, the adsorption distance fluctuates around 2.92 Å. In contrast, the adsorption energies at all three positions are small and almost negligible, indicating minimal interaction between the Zn atoms and the substrate material. On the other hand, for the adsorption systems of Ti, V, Cr, Mn, Fe, Co, Ni, and Cu, the adsorption distance and adsorption energies obviously differ significantly at three different positions. For example, for the Cr2Ge2Te6@Fe, we found that the value of the adsorption energy −1.65 eV at the Cr-top position is considerably larger than that of −0.39 eV at the Te-top position. At the same time, its adsorption distance of 1.20 Å is much larger than that of 2.79 Å at the Te-top position, indicating that the adsorption of Fe atoms at the Cr-top position is more stable than that at the Te-top position. More stable the absorption and their interactions are stronger.
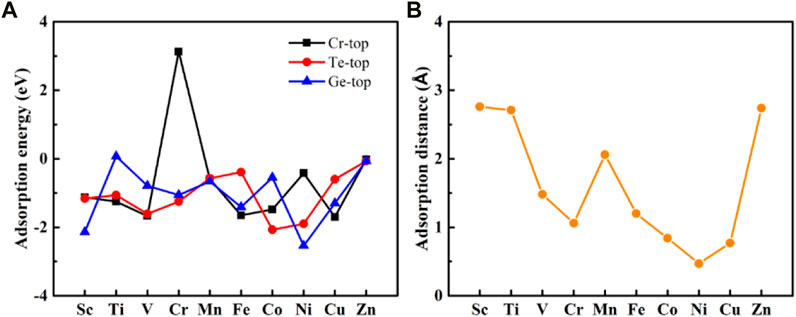
FIGURE 2. (A) The adsorption energy of monolayer Cr2Ge2Te6 for transition metals at three distinct adsorption positions: Cr-top, Te-top, and Ge-top. (B) The adsorption distance of transition metals adsorbed by monolayer Cr2Ge2Te6.
We can determine the best adsorption site based on the minimum adsorption energy after structural relaxation. In Cr2Ge2Te6@Fe, Co adsorbed at the Te-top position was significantly shifted and moved above the Cr atom, so the best adsorption site for the transition metal Co is at the Cr-top position. Similarly, we found that the Zn also moved from the Te-top site to the Ge-top site, so the best adsorption site for Zn is at the Ge-top position. Also, Cr2Ge2Te6@Ni has a similar trend, having the Te-top position as the adsorption site. As a result (shown in Supplementary Table S1, S2), three categories may be used to group the best adsorption site for the ten absorption systems. The optimum adsorption site for Ti, V, Fe, Co, and Cu is at the Cr-top position (A site); the best adsorption site for Cr and Ni is at the Te-top position (B site); and the best adsorption site for Sc, Mn, and Zn is located at the Ge-top position (C site). Figure 2B shows the adsorption distance d at the best adsorption site. The Cr2Ge2Te6@Zn has the smallest negative value of adsorption energy at −0.07 eV. As we all know, the outermost shell of the Zn atom is in an entire electron state. The Zn binds to the monolayer Cr2Ge2Te6 through weak interaction, most likely as physical adsorption. Except for Zn absorbed, the Eads values of the other absorption system are pretty large, ranging from −2.54 to −0.66 eV, indicating a strong bonding between 3d transition metals and the substrate. For Ti, V, Cr, Mn, Fe, Co, Ni, and Cu absorbed, the higher the negative values of the adsorption energy and, at the same time, the smaller their adsorption distance d, indicating strong bonding, most likely by chemical interaction. The adsorption energies of the adsorbed Sc and Ni atoms are the highest, reaching −2.14 and −2.54 eV, respectively, indicating that the bonding between Sc and Ni atoms and monolayer Cr2Ge2Te6 is the strongest. For the adsorption system, the stability of 3d transition metal adsorption: Cr2Ge2Te6@Ni > Cr2Ge2Te6@Sc > Cr2Ge2Te6@Co > Cr2Ge2Te6@Cu > Cr2Ge2Te6@V > Cr2Ge2Te6@Fe > Cr2Ge2Te6@Ti = Cr2Ge2Te6@Cr > Cr2Ge2Te6@Mn > Cr2Ge2Te6@Zn. In addition, except for Sc, Ti, and V absorbed, the trend of adsorption energy change is consistent with the trend of its adsorption distance d change. In conclusion, by adsorbing 3d transition metals at various places, we determined that the optimal adsorption sites for various transition metals are situated at various locations. In general, the greater the negative value of their adsorption energy and the shorter the adsorption distance, the easier and stronger the mutual interaction between the substrate material and the transition metals.
Figure 3 depicts the spin-polarized band structure of the adhered nanosheet. As is evident, the transition metal has a considerable influence on the electronic structure of the monolayer Cr2Ge2Te6 nanosheet of the substrate material. The half-metallic attribute of a material is that it exhibits metallic qualities and semiconductor or insulator properties for electrons with two distinct spin orientations. When the spin polarization rate is 100%, the material is half-metallic, meaning that the energy band in the spin-up channel is metallic while the energy band in the opposite spin direction is non-metallic. For Cr2Ge2Te6@Sc, V, Cr, Co, Ni, and Cu, the spin-up energy bands cross the Fermi level showing the metallic. In contrast, the spin-down energy bands have band gaps, indicating that the spin-down energy bands are semiconducting in nature, suggesting that these systems become half metallic and supply 100% of the spin-polarized current due to their spin polarizabilities 100% at the Fermi level. For the half metallic adsorption systems, their corresponding half metallic gaps (Eg) were computed. The half metallic gap is the minimum between the lowest energy of spin-up (down) conduction bands in relation to the Fermi level and the greatest energy of spin-up (down) valence bands in absolute values. And the results showed that in the Cr2Ge2Te6@Sc, V, Cr, Co, Ni, and Cu, half metallic gaps, Eg were 40.82, 121.14, 24.56, 100.78, 46.33, and 118.06 meV, indicating the half metallic of these adsorption systems is relatively stable. For the Cr2Ge2Te6@Sc (Figure 3A), the spin-down channel retains the band structure of the pristine Cr2Ge2Te6. The substrate material has acquired electrons because the spin-up energy band is heavily hybridized, and its conduction band is moved downward relative to intrinsic Cr2Ge2Te6. With Cr2G2Te6@V and Co, the spin-down exhibits a similar pattern; the spin-down energy band is visibly moved down relative to the intrinsic Cr2G2Te6, and the spin-up is evidently annihilated, showing that 3d transition metal and the substrate have a strong interaction. As shown in Figures 3B, F, the Cr2Ge2Te6@Ti and Fe have similar energy band structures, but their spin-up energy bands are more severely destroyed, indicating more intense energy band hybridization. Both the spin-up and spin-down energy bands exhibit metallic properties with conductivity, indicating the transition from semiconducting to metallic of the system. In the Cr2G2Te6@Mn (Figure 3E), although the spin-down and spin-up valence bands preserve most of the intrinsic energy bands, the conduction bands exhibit notable change, suggesting that the Mn interacts with the substrate quite significantly. Yet, both spin-up and spin-down energy bands preserve the semiconductor character, with a very narrow band gap of 0.07 eV and 0.65 eV, respectively, showing that the entire system offers the semiconducting. Cr2G2Te6@Zn (Figure 3J) preserves the intrinsic Cr2G2Te6 energy band in both the spin-down and spin-up energy bands, indicating a very weak interaction and monolayer Cr2G2Te6. It still exhibits semiconducting, with a band gap of about 0.18 eV in the spin-up and 0.48 eV in the spin-down. We also find that the energy band is slightly shifted downward, showing that Cr2G2Te6 receives electrons. Hence, monolayer Cr2G2Te6 energy bands are significantly hybridized with 3d transition metal except for Zn absorbed. As such, according to the interaction between the transition metal and the substrate material, it can be divided into four categories: half metal (Cr2Ge2Te6@Sc, Cr2Ge2Te6@V, Cr2Ge2Te6@Cr, Cr2Ge2Te6@Co, Cr2Ge2Te6@Ni, and Cr2Ge2Te6@Cu); metal (Cr2Ge2Te6@Ti and Cr2Ge2Te66@Fe); semiconductor (Cr2Ge2Te6@Mn and Cr2Ge2Te6@Zn).
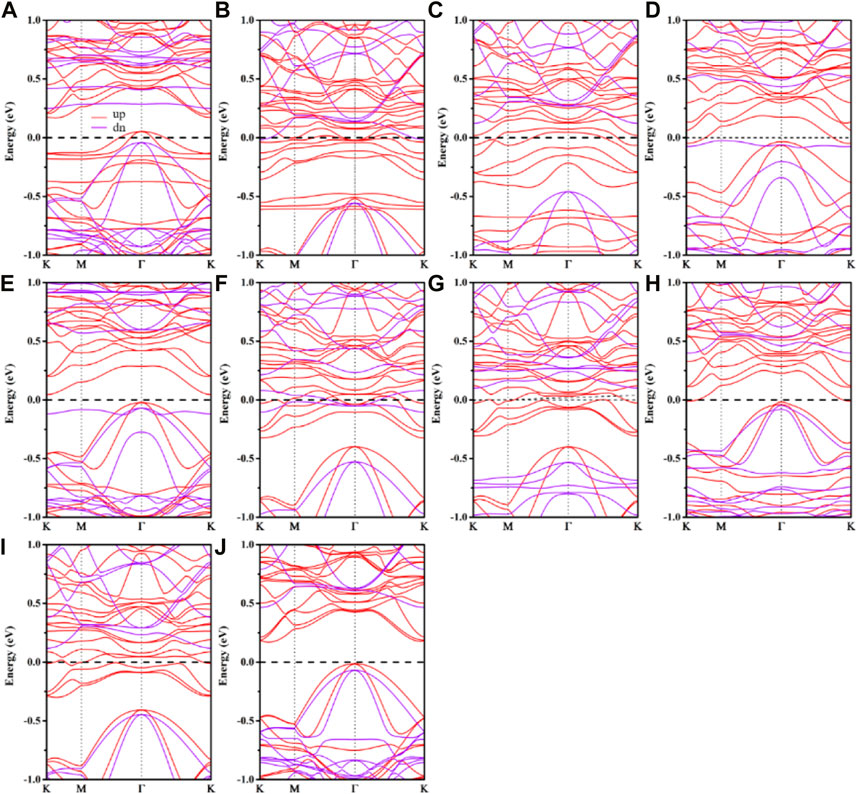
FIGURE 3. The calculated spin-polarized band structures of (A) Cr2G2Te6@Sc, (B) Cr2G2Te6@Ti, (C) Cr2G2Te6@V, (D) Cr2G2Te6@Cr, (E) Cr2G2Te6@Mn, (F) Cr2G2Te6@Fe, (G) Cr2G2Te6@Co., (H) Cr2G2Te6@Ni, (I) Cr2G2Te6@Cu, (J) Cr2G2Te6@Zn. In the band structures, the solid red lines correlate to the spin-up, while the solid purple lines correspond to the spin-down. The Fermi level is set to zero.
After that, we investigate the density of states of the adsorbed system, shown in Supplementary Figure S2. It should come as no surprise that the density of states is found to be asymmetric. In Cr2Ge2Te6@Sc, the spin-up energy band near the Fermi level comes mostly from the 3d orbitals of Sc and partially from the 3d orbitals of Cr. In Cr2Ge2Te6@V, the density of states near the Fermi energy level is predominantly provided by the 3d orbitals of V. Moreover, a portion of the density of states comes from the 3d orbitals of the Cr atom that is close as well as the p orbitals of the Ge atom. It should be noted that the formation of half metal in Cr2Ge2Te6@Co and Cu is exactly the same as in Cr2Ge2Te6@V. In contrast, the half-metallic of Cr2Ge2Te6@Ni is mostly due to the contribution of the 3d orbitals of Cr. Notably, Cr2Ge2Te6@Cr exhibits half-metallic properties, mainly coming from the d orbitals of the Cr in the substrate. Its VBM in the spin-down channel results from the hybridization of the 3d orbitals of both the substrate and the adsorbed Cr and the p orbitals of the Ge atom. In Cr2Ge2Te6@Ti and Fe, their metallic properties are mainly derived from the mutual hybridization between the Cr 3d orbitals, the Ge p orbitals, and the transition metal 3d orbitals. In both Cr2Ge2Te6@Mn and Cr2Ge2Te6@Zn, the conduction band bottom and valence band top at the Fermi plane are mainly contributed by the 3d orbitals of Cr atoms and partly by the p orbitals of Ge.
In order to get a comprehensive comprehension of the shifts in charge transfer that occur between 3d transition metals and monolayer Cr2Ge2Te6, we carried out the differential charge density, as shown in Figure 4. We discovered that, with the exception of the charge that was transferred between the Zn and the monolayer Cr2Ge2Te6, which was a very small amount, there was a large charge transfer in all of the other absorption systems. In addition, we used Bader charge [48] analysis to investigate the flow and magnitude of transferred charge between 3d transition metals and the monolayer Cr2Ge2Te6. As shown in Supplementary Table S2, there is almost no charge transfer between Zn atom and the substrate, exactly in agreement with Figure 4J, indicating that the substrate material and transition metal interact weakly, matching the bandstructure (Figure 3J) and the very small adsorption energy (−0.07 eV). The charge transferred between Co and monolayer Cr2Ge2Te6 is significantly more than that transferred in Cr2G2Te6@Zn, as shown in Figure 4G. And the loss of charge accumulates near the Co, while the accumulation of charge clearly occurs at the surface of the monolayer Cr2Ge2Te6, as well as slightly partial charge accumulation in the intermediate Cr atomic layer, indicating that strong chemical interaction between the Co atoms and monolayer Cr2Ge2Te6, and a roughly 0.21 e loss of Co by Bader analysis. Furthermore, the charge transfer that occurs between Sc, Ti, V, Mn, Fe, and Cu and monolayer Cr2Ge2Te6 is even much stronger, with charge loss occurring clearly around the 3d transition metals and charge accumulation apparently still present in the adsorbed intermediate region as well as around the surface and interlayer of monolayer Cr2Ge2Te6. This suggests that the connection between the transition metals Sc, Ti, V, Mn, Fe, and Cu atoms and monolayer Cr2Ge2Te6 is relatively robust and most likely involves chemisorption. Furthermore, in the Cr2Ge2Te6@Cr and @Ni systems, the loss of charge occurs mainly around 3d transition metals, and electron accumulation is evident not only at the surface atoms of monolayer Cr2Ge2Te6 but also more very evident on the Cr atom of the intermediate layer, suggesting equally strong hybridization in the absorbed system. In accordance with the Bader charge analysis, we conduct quantitative research to determine the magnitude of the charge that is transmitted from the transition metal to the single layer Cr2Ge2Te6. We discover that the number of transferred charges between Zn and monolayer Cr2Ge2Te6 is very low, coming in at just 0.04 e, which suggests that the interaction between Zn and the substrate is relatively weak. Strong contact exists between monolayer Cr2Ge2Te6 and the remainder of the 3d transition metals. The substrate Cr2Ge2Te6 function as an acceptor to take up the lost electrons by the 3d transition metals. Just shown in Supplementary Table S2, the number of electrons transferred from Sc, Ti, V, Cr, Mn, Fe, Co, Ni, and Cu to monolayer Cr2Ge2Te6 is 0.76, 0.65, 0.83, 0.71, 0.64, 0.40, 0.21, 0.54, and 0.39 e, respectively, indicating a significant increase in the number of charges transferred. This result lines up perfectly with the findings about the adsorption energy.
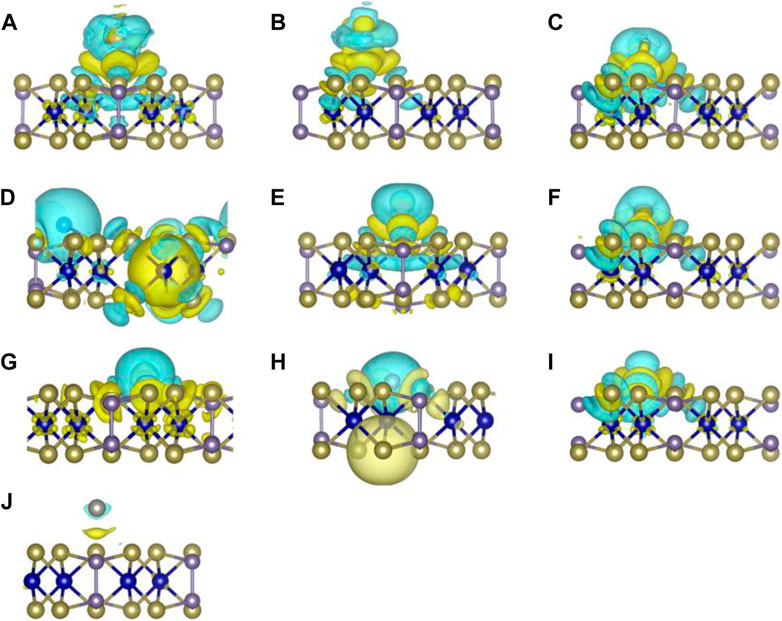
FIGURE 4. The differential charge density of (A) Cr2G2Te6@Sc, (B) Cr2G2Te6@Ti, (C) Cr2G2Te6@V, (D) Cr2G2Te6@Cr, (E) Cr2G2Te6@Mn, (F) Cr2G2Te6@Fe, (G) Cr2G2Te6@Co., (H) Cr2G2Te6@Ni, (I) Cr2G2Te6@Cu, (J) Cr2G2Te6@Zn. The isosurface value is set as 0.001 e/Å3. Charge depletion (accumulation) is represented by the blue (yellow) isosurface in the graph.
After that, we began to investigate how the adsorption of three-dimensional transition metals might affect the magnetic properties of a single layer of Cr2Ge2Te6. In order to investigate the magnetic ground state of the monolayer Cr2Ge2Te6 adsorbed with 3d transition metals, we took into account the four magnetic states that are depicted in Figures 5A–D. These states are, in order, ferromagnetic state, Néel-antiferromagnetic state, Stripy-antiferromagnetic state, and Zigzag-ferromagnetic state, respectively. As can be seen in Table 1, the ferromagnetic state is significantly lower after the adsorption of 3d transition metal compared to the other three magnetic states. This suggests that the monolayer Cr2Ge2Te6 absorbed with 3d transition metals is still relatively resistant in the ferromagnetic state. The ferromagnetic state is unaffected by the adsorption of 3d transition metals since it is an intrinsic magnetic state of the monolayer Cr2Ge2Te6. In addition, we defined the energy difference between the Néel-antiferromagnetic and ferromagnetic states as ∆E1:
where E (Neel-AFM) and
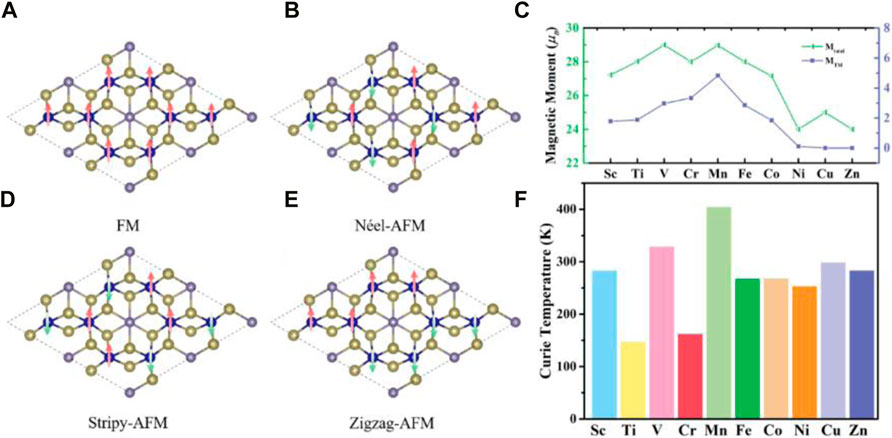
FIGURE 5. Schematics of four different magnetic structures: (A) FM, (B) Néel-AFM, (C) Stripy-AFM, and (D) Zigzag-AFM; (E) The total magnetic moment of the monolayer Cr2Ge2Te6 absorbed with transition metals in the 2 × 2 supercell and the magnetic moment of transition metals in the monolayer Cr2Ge2Te6 absorbed with transition metals. (F) The variation in the Curie temperature of monolayer Cr2Ge2Te6 absorbed by conventional metals.
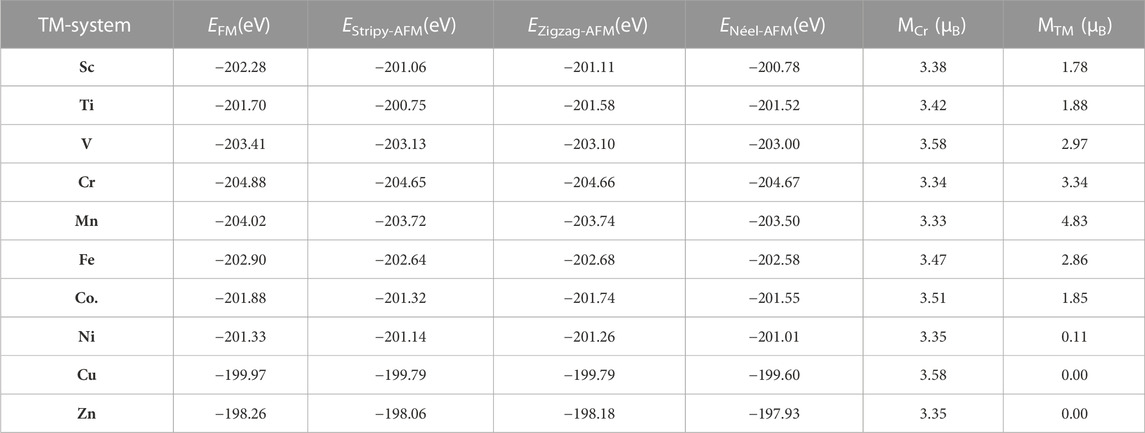
TABLE 1. The calculated total energy of the monolayer Cr2Ge2Te6 absorbed the transition metals with four different magnetic configurations, the magnetic moment of the Cr atom near the absorption site (
In the ferromagnetic state, the supercell 2 × 2 monolayer Cr2Ge2Te6 with 8 Cr atoms, is expected to have a total magnetic moment of 24.00 µB. Furthermore, we calculated the magnetic moments of Cr near the optimal adsorption site (denoted by MCr) and the magnetic moments of the transition metals in the Cr2Ge2Te6@TM adsorption system (represented by MTM) separately. The findings are presented in Figure 5E. From Table 1, we can see that when 3d transition metals are absorbed, the magnetic moments of Cr near the optimal adsorption site do not vary, mainly in the range of 3.33–3.58 µB, which is significantly higher than 3.37 µB for monolayer Cr2Ge2Te6. Among them, with the adsorption of V and Cu, the MCr reached a maximum of 3.58 µB; however, the MCr changed the least after adopting Mn, coming to 3.33 µB. On the other hand, we calculated the magnetic moments MTM of the transition metals in the Cr2Ge2Te6@TM adsorption system. And the results showed that from Sc to Zn atoms, the magnetic moments MTM were 1.78, 1.88, 2.97, 3.34, 4.83 4.83, 2.86, 1.85, 0.11, 0.00和0.00 µB. The results are consistent with the results of Supplementary Figure S3. The 3d transition metals affect the magnetic properties of the adsorption system. In addition, we also calculated the total magnetic moments for the ten absorbed systems, as shown in Figure 5E. We discovered that the total magnetic moment first goes through an increase, then slightly decreases after the Cr adsorption, and then continues to increase again after Fe adsorption, all the way to the Zn, where the total magnetic moment falls; however, the decrease in the total magnetic moment for the Ni atom adsorption is greater than that for Cu atom adsorption. As we all know, the 4s orbitals from Sc to V are full of electrons, and the outermost 3d orbitals are unfilled with the number of electrons 3.00, 4.00, and 5.00 e. The total magnetic moments of the Cr2Ge2Te6@Sc, Ti, and V systems are all integers, rising to 27.00, 28.00, and 29.00 µB in that sequence. This may be explained by the fact that the number of electrons in the outermost layer is connected to the total magnetic moments. When the Sc is absorbed, the total magnetic moments of Cr2Ge2Te6@Sc increase by 3.00 µB, which coincides with the number of 4 s, 3d orbital electrons. In addition, from Mn to Co, the 4 s orbital is full of electrons, and the outermost 3d orbital is unfilled, with 5.00, 6.00, and 7.00 e, respectively. The total magnetic moments of the adsorbed systems of Cr2Ge2Te6@Mn, Cr2Ge2Te6@Fe, and Cr2Ge2Te6@Co are also integers, decreasing linearly from a magnetic moment of 29.00 µB–27.00 µB, which is also related to the number of electrons in the outermost 3d orbitals. As we all know, the outermost 3d orbitals of Mn are arranged in 5.00 e. When Mn is adsorbed, the total magnetic moment of the Cr2Ge2Te6@Mn reaches 29.00 µB. This is precisely 5.00 µB more than the total magnetic moment of the monolayer Cr2Ge2Te6 without the adsorbed transition metal, which is 24.00 µB. This is due to the fact the outermost 3d orbitals of Mn have five unpaired electrons. Compared to 24.00 µB of the monolayer Cr2Ge2Te6, the total magnetic moment of the Cr2Ge2Te6@Fe is 28.00 µB, an increase of 4.00 µB. This can be interpreted as the Fe has 6 electrons in the outermost 3d orbitals but only 4 unpaired electrons. Compared to 29.00 µB for the Cr2Ge2Te6@Mn, the total magnetic moment of the Cr2Ge2Te6@Fe is reduced by 1.00 µB, and the total magnetic moment of the Cr2Ge2Te6@Co adsorption system is reduced to 27.00 µB, which is likely to be the same reason. Since the 3d orbital of Cu is in the full electron state and the 4 s orbital is half full with only one electron, the total magnetic moment of the Cr2Ge2Te6@Cu increases to 25.00 µB. Compared to 24.00 µB for a single layer of Cr2Ge2Te6, the total magnetic moment of the Cr2Ge2Te6@Cu increases by only 1 µB. Obviously, the magnetic moment of Zn, whose outermost 3d orbitals are in the full electron state, is zero; hence the total magnetic moment of the Cr2Ge2Te6@Zn does not change.
On the other hand, we estimate the density of states of Cr2Ge2Te6@TM shown in Figure 6. The total magnetic of the adsorption system is greater, the more asymmetric the spin-polarized density of states. It is noticeable that the spin-polarized density of states asymmetry becomes more and more pronounced with the absorption of Sc and V, indicating that Cr2Ge2Te6@V has a greater total magnetic moment than that of Cr2Ge2Te6@Sc. Similarly, the Cr2Ge2Te6@Mn has a greater spin-polarized density of states asymmetry than the Cr2Ge2Te6@Cu system, indicating a bigger total magnetic moment. While the asymmetry of the spin-polarized density of states of Cr2Ge2Te6@Ni and Cr2Ge2Te6@Zn is significantly weaker, so the total magnetic moments of these two transition metal adsorption systems are also the smallest. In order to investigate more progressively the origin of the total magnetic moment and magnetism of Cr2Ge2Te6@TM, we also calculated the spin density distribution by the following equation:
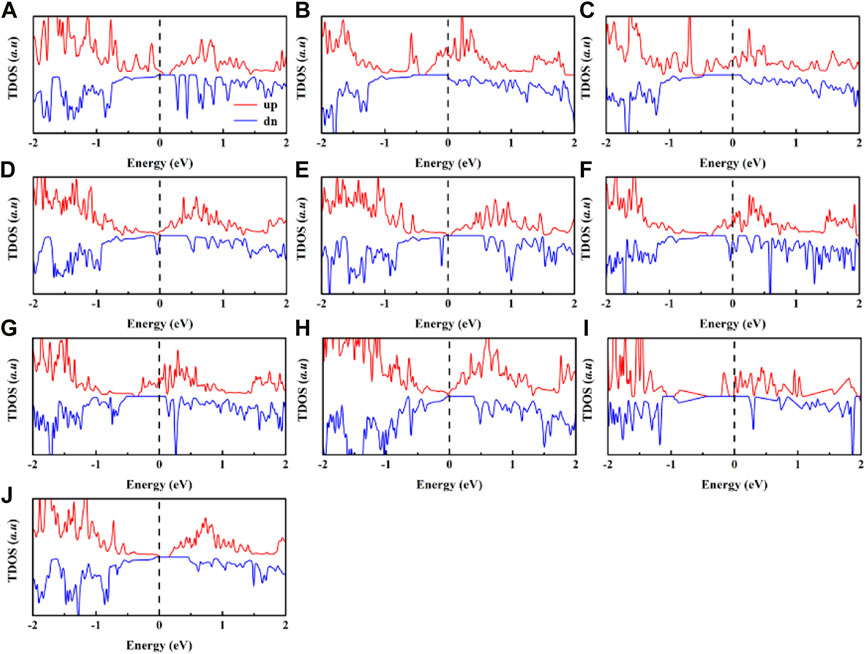
FIGURE 6. Spin-polarized total density of states in the (A) Cr2G2Te6@Sc, (B) Cr2G2Te6@Ti, (C) Cr2G2Te6@V, (D) Cr2G2Te6@Cr, (E) Cr2G2Te6@Mn, (F) Cr2G2Te6@Fe, (G) Cr2G2Te6@Co., (H) Cr2G2Te6@Ni, (I) Cr2G2Te6@Cu, (J) Cr2G2Te6@Zn. The Fermi level is set to zero.
Where
In spintronic devices, the Curie temperature influences the transition temperature from ferromagnetic to paramagnetic and the stability of the ferromagnetic sequence. Calculating the Curie temperature of each adsorption system using Monte Carlo simulation is the next step to directly evaluate whether the ferromagnetic state coupling of the adsorbent system is improved. According to the literature [49, 50], based on the energy difference calculated above and Ising model simulations, we obtain the following expression for the spin exchange parameter J,
where N represents the number of magnetic atoms, Z represents the number of nearest neighbor magnetic atoms of Cr atoms, and ∆E1 is the energy difference between the Néel-antiferromagnetic and ferromagnetic states that we calculated earlier. As shown in Figure 5F, the Curie temperatures of Cr2Ge2Te6@Ti and Cr are slightly lower, around 150 K, while the Curie temperatures of other transition metal adsorption systems typically exceed 200 K. However, compared to that of the monolayer Cr2Ge2Te6, the adsorption of 3d transition metals result in a significant increase in the Curie temperature of Cr2Ge2Te6@TM and an improvement in the ferromagnetic coupling. According to the Goodenough-Kanamori rules [165,166], the ferromagnetic state coupling of monolayer Cr2Ge2Te6 is the product of superexchange interaction and direct exchange interaction.
Since the bond length between Cr-Cr of the Cr2Ge2Te6@TM system is relatively large, about 4.10 Å and the direct exchange interaction is mainly by the direct electron hopping of two adjacent Cr atoms showing the antiferromagnetic coupling, their direct exchange interaction is relatively weak. It is well knowledge that the superexchange contact will have a greater propensity to display ferromagnetic coupling. The superexchange interaction in the adsorbed system is generated by the Te atom acting as an intermediate anion. Since the bond angle of Cr-Te-Cr is mostly around 90°, this results in a more powerful superexchange interaction. Hence, the struggle between the two exchange interactions leads to the dominance of superexchange interactions in the adsorption system, which shows ferromagnetic coupling as a consequence. In view of this, the adsorption of 3d transition metals by the monolayer Cr2Ge2Te6 leads to the enhancement of the ferromagnetic state. After that, we also define the Curie temperature increasing rate ε for the adsorption system by equation,
According to our calculation, the Curie temperature enhancement ratio of Cr2Ge2Te6@TM reaches 224%, 68%, 277%, 85%, 367%, 207%, 207%, 190%, 242%, and 224%, respectively. All adsorbed transition metals improved their Curie temperatures by a factor of 2, with the exception of Ti and Cr atoms, whose adsorption Curie temperature improvement was less than 100%, suggesting that transition metals may dramatically increase their Curie temperatures.
In addition, we study how the electric field modulates the electrical characteristics of the adsorption system. According to earlier research, the Cr2Ge2Te6@Mn system is a semiconductor; thus, we choose this system as an example and apply a positive electric field, that is, an electric field directed from the Cr2Ge2Te6 to the adsorbed atoms. We discovered that the application of an additional positive electric field has a considerable influence on the electronic properties of the Cr2Ge2Te6@Mn, particularly the spin-polarized electronic states of the monolayer Cr2Ge2Te6. The calculated projected spin-up and spin-down band structures of the Cr2Ge2Te6@Mn adsorption system under 0.10 and 0.60 V/Å electric fields are shown in Figures 7A, B, respectively. After applying the electric field, the spin-up energy band extended all the way to the Fermi level, whereas the spin-down energy band remained semiconducting. This caused a spin-polarization current with a spin-polarization rate of 100% at the Fermi level. In addition, compared to the energy band of the intrinsic monolayer Cr2Ge2Te6, the conduction band in the spin-up channel of Cr2Ge2Te6@Mn moves increasingly downward with an increasing positive electric field, suggesting that as its magnitude increases, the electric field increasingly impacted the conduction band in the spin-up channel. Consequently, the applied positive electric field transforms Cr2Ge2Te6@Mn from a semiconductor to a half-metal, suggesting that the applied electric field has a distinct benefit in managing the electronic and magnetic properties of the absorbed system. In other words, as seen in Figure 7C, the applied positive electric field causes more charges initially localized in a certain energy band to shift in relative positions. Once the energy band at these positions crosses the Fermi energy level under the influence of the electric field, the materials also transform into a half-metal, i.e., one spin-up electronic state moves downward across the Fermi plane, while the electric field has little effect on the other spin-down electronic state, thus achieving a modulation of the electronic and magnetic properties of the absorbed system.
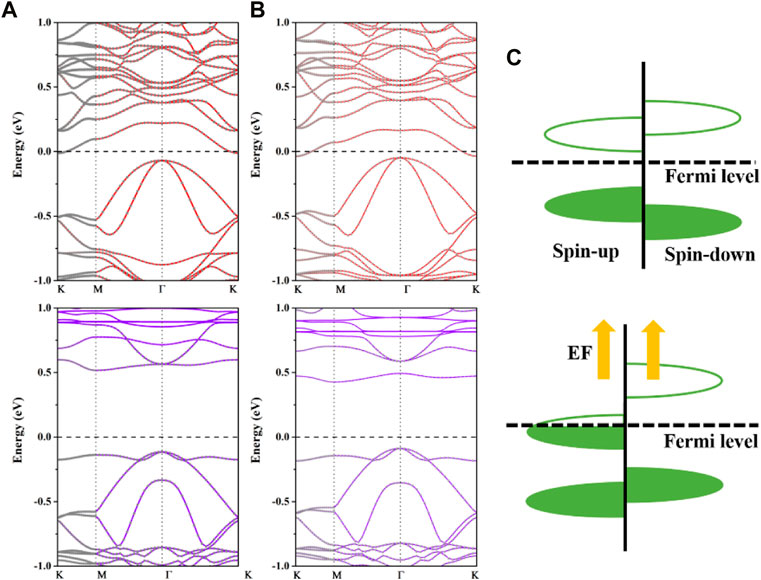
FIGURE 7. The calculated projected spin-up and spin-down band structures of Cr2Ge2Te6@Mn with electric fields of (A) 0.1 and (B) 0.6 V/Å. In the band structures, the solid red lines correlate to the spin-up, while the solid purple lines correspond to the spin-down. The solid grey circles in the spin-up and spin-down band structures correspond to the contribution from the Cr2Ge2Te6 layer. The Fermi level is set to zero. (C) A schematic illustration of the electric field-induced transition from a ferromagnetic semiconductor to a half metal.
In conclusion, based on first-principles calculation, the electronic and magnetic properties of the monolayer Cr2Ge2Te6 absorbed with 3d transition metal atoms have been investigated. Based on the calculated absorption energy, equilibrium absorption distance, and charge redistribution, the monolayer Cr2Ge2Te6 absorbed Zn via a weak interaction. At the same time, the rest of the 3d transition metals are fascinated with the substrate via a strong interaction. Our results show that Cr2Ge2Te6@TM can possess different electronic properties. We found that the adsorption of Ti and Fe leads to a transformation from semiconductor to metal. While in Cr2Ge2Te6@Sc, V, Co, Ni, and Cu, one of their spin energy bands appears to be metallic, the other spin energy band remains semiconductor, so the absorption realizes the changes from semiconductor to half metal. Furthermore, the ferromagnetic coupling is the most stable state in Cr2Ge2Te6@TM, and their respective Curie temperatures are greatly enhanced by adsorption, demonstrating that adsorption may effectively boost the ferromagnetic state of monolayer Cr2Ge2Te6. This relates to the outcome of the competition between direct exchange and superexchange. This research demonstrates a feasible method for not only modifying the electrical characteristics of the monolayer Cr2Ge2Te6 but also enhancing their ferromagnetic stability. We are certain that this method can be experimentally achieved in the near future, which will considerably ease the deployment of CGT in nanoelectronic and spintronic devices.
The original contribution presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
WK completed the computational analysis related to the paper and wrote the manuscript. XD and others helped me to check the grammatical problems of the manuscript and the corrections of the related icons.
This work was supported by the Natural Science Foundation of Chongqing (Grant No. cstc2020jcyj-msxmX0686), the Fundamental Research Funds for the Central Universities (2020CDJ-LHZZ-073), the National Natural Science Foundation of China (11674042), and the Thousand Youth Talents Program of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ZW declared a shared affiliation with the author(s) to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2023.1188513/full#supplementary-material
1. Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, et al. Electric field effect in atomically thin carbon films. Science (2004) 306:666–9. doi:10.1126/science.1102896
3. Mannix AJ, Zhang Z, Guisinger NP, Yakobson BI, Hersam MC. Borophene as a prototype for synthetic 2D materials development. Nat Nanotechnol (2018) 13:444–50. doi:10.1038/s41565-018-0157-4
4. Manzeli S, Ovchinnikov D, Pasquier D, Yazyev OV, Kis A. 2D transition metal dichalcogenides. Nat Rev Mater (2017) 2:17033. doi:10.1038/natrevmats.2017.33
5. Kong X, Liu Q, Zhang C, Peng Z, Chen Q. Elemental two-dimensional nanosheets beyond graphene. Chem Soc Rev (2017) 46:2127–57. doi:10.1039/c6cs00937a
7. Liu J, Huang CC, Wang Y, Yao Q, Wang LY, Li DF. A promising robust intrinsic half-metallic MXene nanosheet Cr2CuC2 with high Curie temperature. Phys E Low-dimens Syst Nanostructures (2022) 143:115276. doi:10.1016/j.physe.2022.115276
8. Huang C-C, Wang Y, Yao Q, Li D-F, Liu J. MXene monolayer Mn2ZnN2: A promising robust intrinsic half-metallic nanosheet. J Phys Condens Matter (2022) 34:105301. doi:10.1088/1361-648x/ac3cb3
9. Liu J, Wang Y, Wang LY, Yao Q, Huang CC, Huang HY, et al. Several promising non-vdW multiferroic half-metallic nanosheets ACr2S4 (A = Li, Na, K, Rb): The first-principles researches. Eur Phys J Plus (2023) 138:224. doi:10.1140/epjp/s13360-023-03789-6
10. Sivadas N, Daniels MW, Swendsen RH, Okamoto S, Xiao D. Magnetic ground state of semiconducting transition-metal trichalcogenide monolayers. Phys Rev B (2015) 91:235425. doi:10.1103/physrevb.91.235425
11. Wang K, Ren K, Cheng Y, Chen S, Zhang G. The impacts of molecular adsorption on antiferromagnetic MnPS3 monolayers: Enhanced magnetic anisotropy and intralayer dzyaloshinskii–moriya interaction. Mater Horiz (2022) 9:2384–92. doi:10.1039/d2mh00462c
12. Long G, Henck H, Gibertini M, Dumcenco D, Wang Z, Taniguchi T, et al. Persistence of magnetism in atomically thin MnPS3 crystals. Nano Lett (2020) 20:2452–9. –9. doi:10.1021/acs.nanolett.9b05165
13. Lin M-W, Zhuang HL, Yan J, Ward TZ, Puretzky AA, Rouleau CM, et al. Ultrathin nanosheets of CrSiTe3: A semiconducting two-dimensional ferromagnetic material. J Mater Chem C (2016) 4:315–22. –22. doi:10.1039/c5tc03463a
14. Casto LD, Clune AJ, Yokosuk MO, Musfeldt JL, Williams TJ, Zhuang HL, et al. Strong spin-lattice coupling in CrSiTe3. APL Mater (2015) 3:041515. doi:10.1063/1.4914134
15. Huang B, Clark G, Navarro-Moratalla E, Klein DR, Cheng R, Seyler KL, et al. Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature (2017) 546:270–3. doi:10.1038/nature22391
16. Carteaux V, Brunet D, Ouvrard G, Andre G. Crystallographic, magnetic and electronic structures of a new layered ferromagnetic compound Cr2Ge2Te6. J Phys Condens Matter (1995) 7:69–87. doi:10.1088/0953-8984/7/1/008
17. Gong C, Li L, Li Z, Ji H, Stern A, Xia Y, et al. Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature (2017) 546:265–9. doi:10.1038/nature22060
18. Li X, Yang J. CrXTe3 (X = Si, Ge) nanosheets: Two dimensional intrinsic ferromagnetic semiconductors. J Mater Chem C (2014) 2:7071. –6. doi:10.1039/c4tc01193g
19. Wolf SA, Awschalom DD, Buhrman RA, Daughton JM, von Molnár S, Roukes ML, et al. Spintronics: A spin-based electronics vision for the future. Science (2001) 294:1488–95. doi:10.1126/science.1065389
20. Tu NT, Hai PN, Anh LD, Tanaka M. High-temperature ferromagnetism in heavily Fe-doped ferromagnetic semiconductor (Ga,Fe)Sb. Appl Phys Lett (2016) 108:192401. doi:10.1063/1.4948692
21. Yazyev OV, Helm L. Defect-induced magnetism in graphene. Phys Rev B (2007) 75:125408. doi:10.1103/physrevb.75.125408
22. Červenka J, Katsnelson MI, Flipse CFJ. Room-temperature ferromagnetism in graphite driven by two-dimensional networks of point defects. Nat Phys (2009) 5:840–4. –4. doi:10.1038/nphys1399
23. Naji S, Belhaj A, Labrim H, Bhihi M, Benyoussef A, El Kenz A. Adsorption of Co and Ni on graphene with a double hexagonal symmetry: Electronic and magnetic properties. J Phys Chem C (2014) 118:4924–9. –9. doi:10.1021/jp407820a
24. Wang H, Zhu S, Fan F, Li Z, Wu H. Structure and magnetism of Mn, Fe, or Co adatoms on monolayer and bilayer black phosphorus. J Magn Magn Mater (2016) 401:706–10. –10. doi:10.1016/j.jmmm.2015.10.131
25. Lu Y, Ke C, Fu M, Lin W, Zhang C, Chen T, et al. Magnetic modification of GaSe monolayer by absorption of single Fe atom. RSC Adv (2017) 7:4285–90. –90. doi:10.1039/c6ra27309b
26. Ilyas A, Wu H, Usman T, Khan SA, Deng R. The induction of half-metallicity and enhanced ferromagnetism in a Cr2Ge2Te6 monolayer via electron doping and alkali metal adsorption. J Mater Chem C (2021) 9:5952–60. doi:10.1039/d1tc00006c
27. Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B (1996) 54:11169–86. doi:10.1103/physrevb.54.11169
28. Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci (1996) 6:15–50. doi:10.1016/0927-0256(96)00008-0
29. Blöchl PE. Projector augmented-wave method. Phys Rev B (1994) 50:17953–79. doi:10.1103/physrevb.50.17953
30. Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B (1999) 59:1758–75. doi:10.1103/physrevb.59.1758
31. Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett (1996) 77:3865–8. –8. doi:10.1103/physrevlett.77.3865
32. Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B (1976) 13:5188–92. doi:10.1103/physrevb.13.5188
33. Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys (2010) 132:154104. doi:10.1063/1.3382344
34. Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem (2006) 27:1787–99. doi:10.1002/jcc.20495
35. Jiang P, Wang C, Chen D, Zhong Z, Yuan Z, Lu Z-Y, et al. Stacking tunable interlayer magnetism in bilayer CrI3. Phys Rev B (2019) 99:144401. doi:10.1103/physrevb.99.144401
36. Tang C, Zhang L, Sanvito S, Du A. Electric-controlled half-metallicity in magnetic van der Waals heterobilayer. J Mater Chem C (2020) 8:7034–40. doi:10.1039/d0tc01541e
37. Chen S, Huang C, Sun H, Ding J, Jena P, Kan E. Boosting the Curie temperature of two-dimensional semiconducting CrI3 monolayer through van der Waals heterostructures. J Phys Chem C (2019) 123:17987–93. doi:10.1021/acs.jpcc.9b04631
38. Liu M, Chen Q, Huang Y, Cao C, He Y. A first-principles study of transition metal doped arsenene. Superlattices Microstruct (2016) 100:131–41. doi:10.1016/j.spmi.2016.09.014
39. Zhang J, Zhao B, Yao Y, Yang Z. Robust quantum anomalous Hall effect in graphene-based van der Waals heterostructures. Phys Rev B (2015) 92:165418. doi:10.1103/physrevb.92.165418
40. Liechtenstein AI, Anisimov VI, Zaanen J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys Rev B (1995) 52:R5467–70. doi:10.1103/physrevb.52.r5467
41. Tang C, Zhang L, Du A. Tunable magnetic anisotropy in 2D magnets via molecular adsorption. J Mater Chem C (2020) 8:14948–53. doi:10.1039/d0tc04049e
42. Xing W, Chen Y, Odenthal PM, Zhang X, Yuan W, Su T, et al. Electric field effect in multilayer Cr2Ge2Te6: A ferromagnetic 2D material. arXiv (2017) 4:024009. doi:10.48550/arXiv.1704.08862
43. Wang H, Qi J, Qian X. Electrically tunable high Curie temperature two-dimensional ferromagnetism in van der Waals layered crystals. Appl Phys Lett (2020) 117:083102. doi:10.1063/5.0014865
44. Tang X, Fan D, Guo L, Tan H, Wang S, Lu X, et al. Achieving higher thermoelectric performance for p-type Cr2Ge2Te6 via optimizing doping. Appl Phys Lett (2018) 113:263902. doi:10.1063/1.5064787
45. Li X, Wu X, Li Z, Yang J, Hou JG. Bipolar magnetic semiconductors: A new class of spintronics materials. Nanoscale (2012) 4:5680. –5. doi:10.1039/c2nr31743e
46. Kang S, Kang S, Yu J. Effect of coulomb interactions on the electronic and magnetic properties of two-dimensional CrSiTe3 and CrGeTe3 materials. J Electron Mater (2019) 48:1441–5. –5. doi:10.1007/s11664-018-6601-2
47. Suzuki M, Gao B, Koshiishi K, Nakata S, Hagiwara K, Lin C, et al. Coulomb-interaction effect on the two-dimensional electronic structure of the van der Waals ferromagnet Cr2Ge2Te6. Phys Rev B (2019) 99:161401. doi:10.1103/physrevb.99.161401
48. Clarendon Richard FWB. A theory of molecules: Atoms in molecules. A quantum theory. Science (1991) 5012:1566–7.
49. Naji S, Belhaj A, Labrim H, Bhihi M, Benyoussef A, El Kenz A. Electronic and magnetic properties of iron adsorption on graphene with double hexagonal geometry. Int J Quan Chem. (2014) 114:463–7. –7. doi:10.1002/qua.24592
Keywords: monolayer Cr2Ge2Te6, 3d transition metal, electronic properties, ferromagnetism, half-metal, curie temperature
Citation: Kang W, Du X, Wang J, Ye Z, Zhao J, Wang W, Wang Y, Wang L and Liu X (2023) Two-dimensional half-metallicity in transition metal atoms decorated Cr2Ge2Te6. Front. Phys. 11:1188513. doi: 10.3389/fphy.2023.1188513
Received: 17 March 2023; Accepted: 28 March 2023;
Published: 06 April 2023.
Edited by:
Guangzhao Wang, Yangtze Normal University, ChinaReviewed by:
H.N. Dong, Chengdu Normal University, ChinaCopyright © 2023 Kang, Du, Wang, Ye, Zhao, Wang, Wang, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Kang, MjgzOTYzNDIzQHFxLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.