 Harshita Srivastava
Harshita Srivastava Ambrish Kumar Srivastava
Ambrish Kumar Srivastava
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Phys. , 05 April 2022
Sec. Chemical Physics and Physical Chemistry
Volume 10 - 2022 | https://doi.org/10.3389/fphy.2022.870205
This article is part of the Research Topic Superhalogens & Superalkalis: Exploration of Structure, Properties and Applications View all 10 articles
The activation of carbon dioxide is essential not only for global carbon balance but also for its conversion into fuel. As CO2 is highly stable, it is quite challenging to activate or reduce CO2. Recently, the ability of superalkalis to easily transfer an electron to CO2 has been proposed in several studies. The superalkalis are species possessing lower ionization energy than alkali atoms. These are hypervalent species, having an excess of electrons. Owing to this, they possess strong reducing power and cause the linear structure of CO2 to bend by transferring an electron to it. Herein, we present a comprehensive account of the single-electron reduction and activation of CO2 by various kinds of superalkalis. This review also includes a novel strategy for the capture and storage of CO2 by superalkali.
Carbon dioxide (CO2) is a colorless and odorless gas with the property of trapping greenhouse gases, which are produced due to human activities as well as natural processes. Recently, the sharp decline in CO2 levels has been noticed at the expense of the COVID-19 pandemic, which has caused a severe health emergency in the world and is not sustainable. The fueling of CO2 in our environment is mainly through the CO2 emissions from power plants and other industrial facilities, primarily waste products, and the developed economies are the leading contributors. As mentioned, it traps greenhouse gases which generally cause a change in the behavior of climate since it is a major contributor to global warming. In order to reduce its contribution to global warming, it is necessary to convert CO2 into value-added products. The best way to back-pedal climate change without using expensive methods is extricating CO2 from the air and then converting it into a useful product like fuel. These important issues were addressed by numerous techniques, which can be employed to reduce and capture CO2 by other molecules [1–7]. As CO2 is an extremely stable molecule [8, 9], it is quite challenging to convert it into usable fuel. To convert CO2 into fuel, it is needed to activate CO2 by some means or chemically reduce it by catalysts. CO2 can be reduced either electrochemically (electrical energy) or photoelectrochemically (incident light) into CO. Single-electron reduction of CO2 to CO2− was experimentally not viable due to the large energy of reorganization between linear CO2 and bent CO2− anion. Notably, the electron affinity of CO2 is negative so that CO2− is metastable. CO2− anion is stable in the 2A1 state [10] which can be treated as an activated CO2 moiety with the weaker C-O bond. The potential energy surface of the CO2− anion suggests three vibronically coupled bound states [11].
It is difficult to extract an electron from carbon dioxide because of its high ionization energy (13 eV) [12]. However, it has been revealed that there is a possibility of oxidation of CO2 using superhalogens [13], whose electron affinity overrides the halogen atoms [14]. The counterparts of superhalogens are superalkalis which bear lower ionization energy than alkali atoms [15]. Due to the stronger reducibility of superalkalis over alkali atoms, they might activate stable CO2. To investigate this, a few studies have been performed recently. In this review, we will provide an overview of how superalkalis play a significant role in the activation or reduction of CO2, which is the initial step to convert CO2 into fuel. Exploration of chemical processes used in the reduction of CO2 is of tremendous importance in various fields, like biological, environmental, and industrial processes [16]. Before we go further, let us first have a look at superalkalis.
Alkali atoms possess the lowest ionization energy (IE), ranging from 5.39 to 3.89 eV, among all the elements in the periodic table. However, superalkalis are clusters whose ionization energies are even lower than this range. These clusters were originally introduced by Gutsev and Boldyrev in 1982 using sp-block elements [15]. They proposed species like Li2F, Li3O, and Li4N as superalkalis. In the form of a superatom, these clusters impersonate the behavior of alkali atoms. There have been several studies on the design of various kinds of superalkalis [17–24]. For instance, the binuclear superalkalis including F2Li3, have been widely studied [17]. Hou et al. [18] described non-metallic binuclear cations such as F2H3+ and O2H5+. Zhao et al. [19] proposed some special superalkalis like N4Mg6Li, Al3, Mn(B3N3H6)2, B9C3H12, Al12P, and C5NH6, which were designed by using different schemes like jellium rule, 18-electron rule, Wade–Mingos rule, and Huckle’s rule, respectively. Al12P was reported to be an alkali–metal-like superatom [20, 21]. Recently, Sikorska and Gaston [22] reported the superalkali behavior of polynuclear N4Mg6M for M = Li, Na, and K. Srivastava [25] noticed that the IE of C6Li6, being lower than that of Li, makes it a closed-shell superalkali. The IE scale of these superalkalis is depicted in Figure 1.
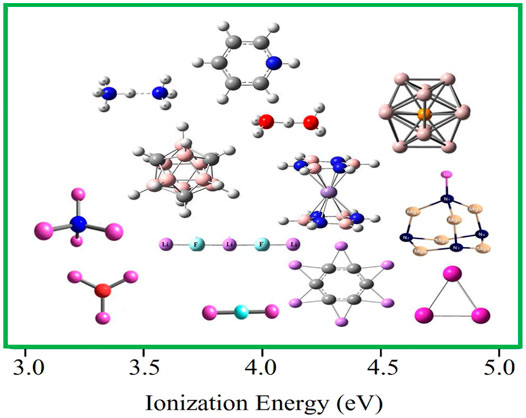
FIGURE 1. Ionization energy scale of various superalkalis is considered here. The values are taken from Refs. 17–22, 25, and 35.
Superalkalis find applications in the design of supersalts [26–28], superbases [29–31], alkaloids [32–34], and so forth. Due to their low IE, superalkalis play an important role in chemical industries as reducing agents. Here, we provide an account of how superalkalis are exploited to activate the CO2 molecule.
CO2 is known to be a highly stable molecule due to its very high IE [12], as mentioned earlier, and no positive electron affinity [36, 37]. However, the low IE of superalkalis enables them to transfer an electron to CO2, reducing it to CO2− anion and thus activating it. In Figure 2, we show the structures of CO2 and its anion, along with the charge distribution. One can see that the CO2− anion is bent, in which the bond length is increased as compared to neutral CO2 due to the negative charge.
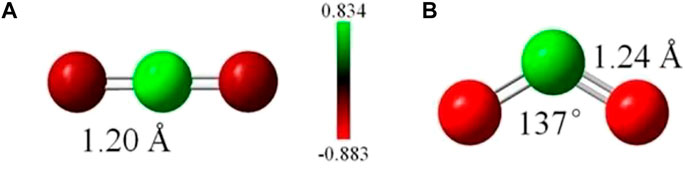
FIGURE 2. Structure and NPA charge distribution in (A) neutral CO2 and (B) CO2−anion, Ref. 19 with the permission of the Royal Society of Chemistry.
Thus, the activation of CO2 requires the following conditions to be satisfied:
1) The negative charge on CO2 moiety is close to unity.
2) The structure of the CO2 moiety is bent.
3) The bond length of CO2 moiety is increased.
It has been reported earlier [38] that CO2 would assume a bent structure when an electron is transferred to it or due to its interaction with the electrons of the metal atom. One would expect that the stable geometry of the M-CO2 complex depends upon the IE of the metal atom, M. This may lead us to infer that an atom with a smaller IE should be able to transfer an electron to CO2 more easily than one with a large IE. Later, we will discuss the interaction of CO2 with various superalkalis described earlier.
Srivastava [35] studied the interaction of CO2 with FLi2, OLi3, and NLi4 superalkalis using the second-order Møller–Plesset perturbative (MP2) method [39] and the 6–311+G(d) basis set in the Gaussian 09 program [40]. Such interaction leads to the formation of complexes, as shown in Figure 3, and corresponding parameters can be found in Table 1. It is evident that the minimum energy of these complexes corresponds to the structure in which the interaction between CO2, and superalkalis is mediated by both the O atoms of CO2. The bond length of Li-O lies between 1.865 and 1.892 Å. The low-lying isomers of FLi2-CO2 and OLi3-CO2 are of higher energy in which CO2 interacts via a single atom, whereas in the case of NLi4-CO2, there are no competing isomers obtained.
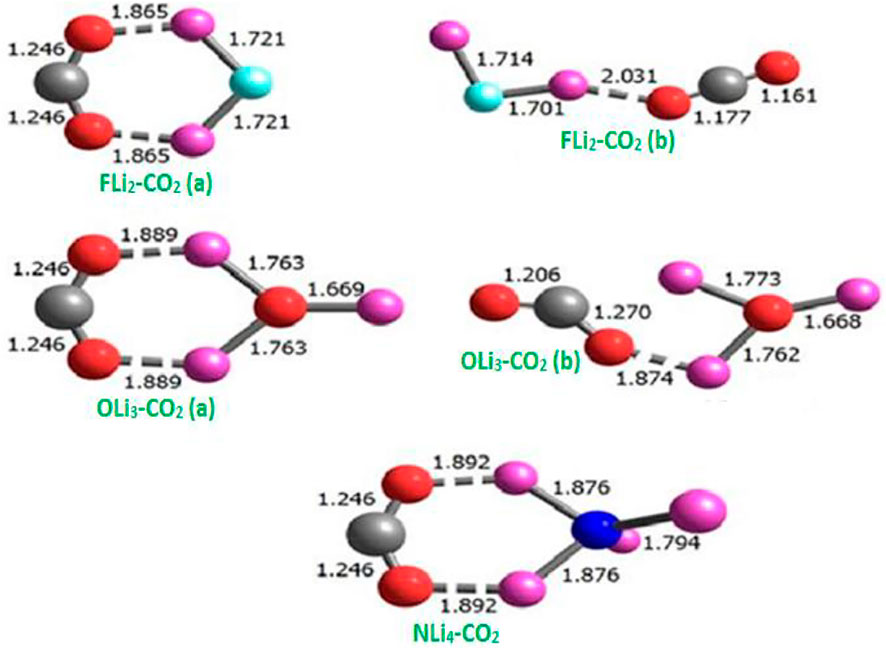
FIGURE 3. Equilibrium structures of CO2 complexes with typical superalkalis, with bond lengths in Å from Ref. 35 with the permission of Wiley.
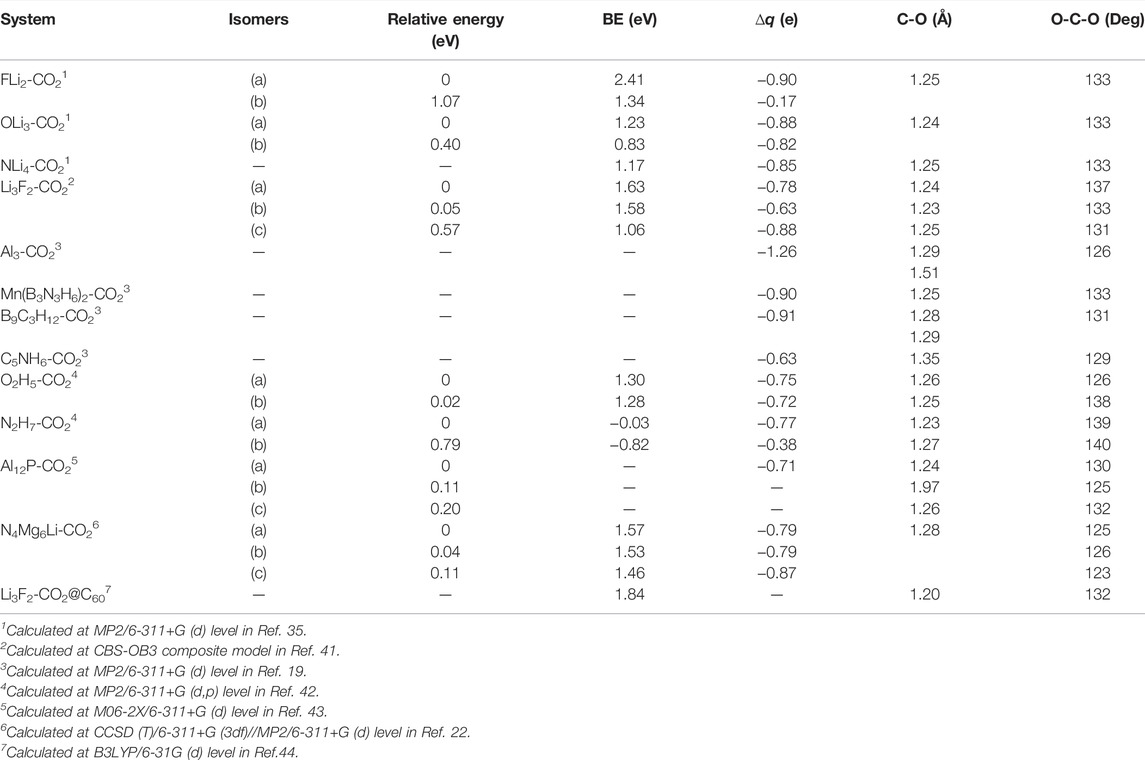
TABLE 1. Relative energy, binding energy (BE), NPA charge (∆q), bond length (C-O), and bond angle (O-C-O) of the complexes of CO2 with various superalkalis.
The binding energy (BE) of superalkali–CO2 complexes is calculated and listed in Table 1. The BE of these complexes monotonically decreases with the increase in the size of superalkalis. This can be explained on the basis of a more delocalized electron cloud that is generally associated with the larger superalkalis. The natural population analysis (NPA) [45] charges (∆q) on CO2 have also been listed. The most stable structure of superalkali–CO2 complexes takes the values of ∆q as −0.90e for FLi2, −0.88e for OLi3, and −0.85e for NLi4. In isomer (b) of FLi2-CO2, ∆q has a very small magnitude (−0.17e), which is consistent with an almost linear CO2 moiety just as in a neutral CO2 molecule. On the contrary, the ∆q in isomer (b) of OLi3-CO2 is, albeit smaller than that in its lowest energy structure (a), large enough to bend the CO2 moiety. It should be noted that the size of the superalkalis is a more important factor than their IE in CO2 activation. As per calculated binding energy and charge transfer, FLi2 is more effective for CO2 reduction. In these complexes, the CO2 moiety is bent by 133° and the bond length C-O becomes 1.246 Å which is comparable to the bond lengths of 1.237 Å and bond angle 137° in the CO2− anion, obtained at the MP2/6-311+G(d) level. This study suggests simple and catalyst-free single-electron reduction of CO2 by using typical superalkalis such as FLi2, OLi3, and NLi4.
Park and Meloni [41] reported the interaction of CO2 and superalkali species Li3F2 using the CBS-OB3 composite model [46] through the Gaussian 09 program. They obtained three isomers, two planar (a) and (b), as well as one non-planar (c), of the Li3F2-CO2 complex, as shown in Figure 4. There was no appreciable change in bond lengths between Li and F on interaction with CO2. Despite some structural changes in the superalkalis, the structure of CO2 changes from linear to bending. Therefore, it is clear that the strongly bound CO2 is activated upon interaction with the superalkali. From Table 1, the BE of Li3F2-CO2 isomers is found to be in the range of 1.06–1.63 eV (106–163 kJ/mol). The lowest BE was obtained for the isomer (c) in which one oxygen interacts with both the terminal Li atoms and the other oxygen with the central Li atom. The isomer (a) possesses greater BE and therefore stronger intermolecular interaction than isomer (b) because the electron density is more localized between the two terminal Li atoms and two oxygen atoms.
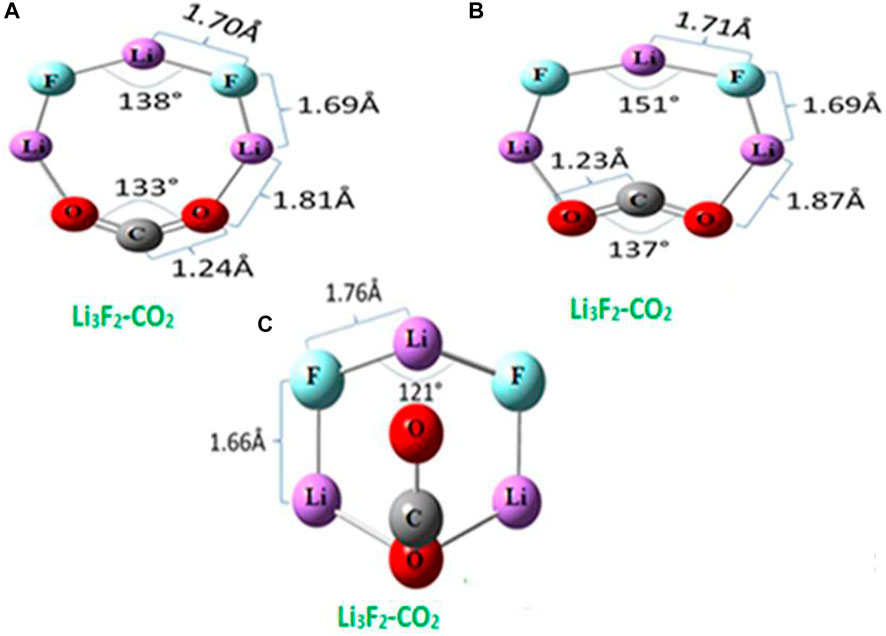
FIGURE 4. Equilibrium structures of CO2 complex with binuclear superalkali with bond lengths from Ref. 41 with the permission of the Royal Society of Chemistry.
The BE of Li3F2-CO2 complexes is comparable to or smaller than the BE of superalkali (FLi2, OLi3, NLi4)-CO2 complexes reported by Srivastava [35]. The charge transfer to CO2 in Li3F2-CO2 isomers ranges from −0.63e to −0.88e (see Table 1). Thus, the charge on the CO2 moiety along with its structure suggests that CO2 is reduced to a CO2− anion.
Zhao et al. [19] presented the rational design of superalkalis and studied the activation of CO2 by these special superalkalis using MP2/6-311+G(d) level in the Gaussian 09 program. They analyzed the interaction of CO2 with special superalkalis like Al3, Mn (B3N3H6)2, B9C3H12, and C5NH6, which leads to the complexes shown in Figure 5. The distance between the CO2 moiety and superalkali clusters has been calculated as 1.950 Å, 1.730 Å, 2.320 Å, and 1.040 Å for Al3, Mn (B3N3H6)2, B9C3H12, and C5NH6, respectively. In the case of Al3, O-C bonds extend to 1.290 and 1.510 Å, about 4.4–22% longer than those in the CO2−anion, whereas in B9C3H12, the O-C bonds are extended to 1.290 and 1.280 Å, about 3.3–4.4% longer than those in CO2−. The bond extension in Mn (B3N3H6)2 and C5NH6 is observed to be 1.250 Å which is slightly longer than that of 1.240 Å in CO2− and 1.35 Å which is about 9.3% longer than that in CO2−, respectively. The bond angle of O-C-O in Al3CO2, Mn (B3N3H6)2CO2, B9C3H12CO2, and C5NH6CO2 is 126°, 133°, 131°, and 129° making the bond bend by 8°, 3, 4, and 7% more than the corresponding value in CO2−. Thus, both the stretching of the O-C bonds and the bending of the O-C-O angle weaken the O-C bonds of CO2, making it easy to activate.
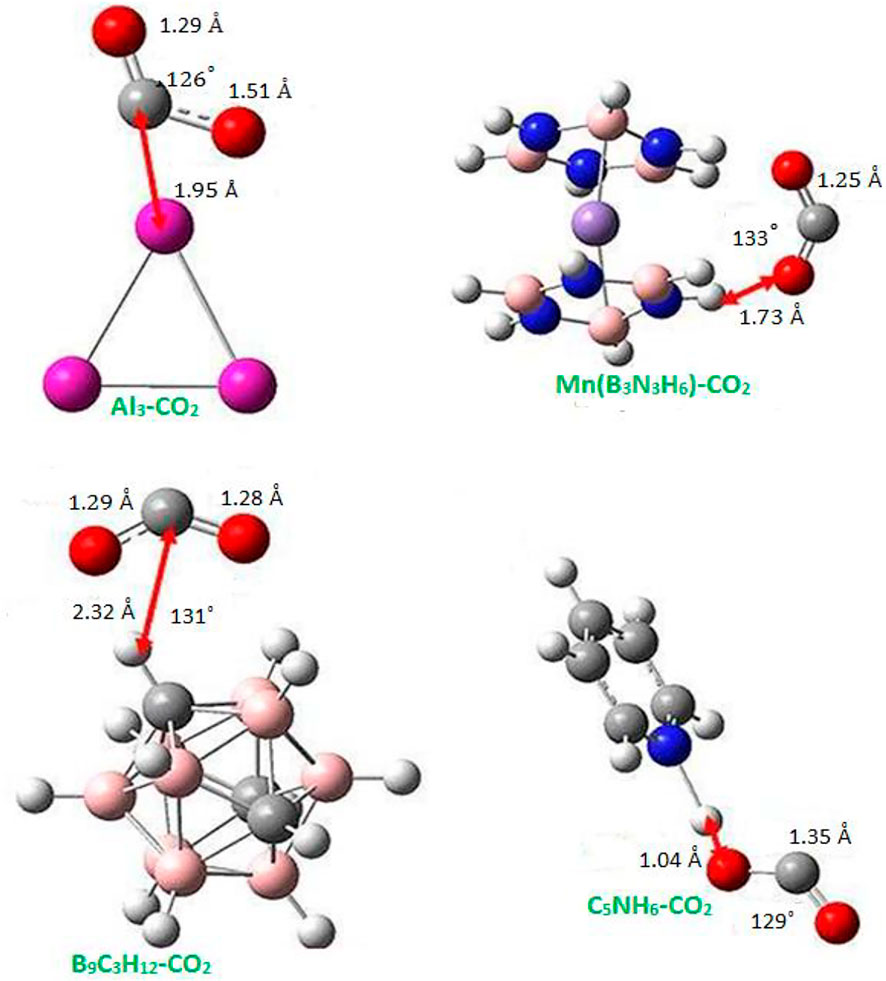
FIGURE 5. Equilibrium structures of CO2 complex with special superalkalis from Ref. 19 with the permission of the Royal Society of Chemistry.
The NPA charge has been listed in Table 1 to show how much charge is transferred to CO2. This transfer of charge results in the bending of CO2 and weakens the CO2 bond, and therefore making it easier to break. The amount of charge transferred from Al3, Mn (B3N3H6)2, B9C3H12, and C5NH6 to CO2 are −1.26e, −0.90e, −0.91e, and −0.63e, respectively. Note that this amount of charge transferred from Al3, Mn (B3N3H6)2, B9C3H12 are greater than that of noble gas (0.77e) [47] being very close to unity, whereas in the case of C5NH6, the amount of charge transferred is less. From this analysis, one may note that although the IE of Al3 is not the lowest among these four superalkalis (see Figure 1), the charge transferred is the most and it is capable of bending the CO2 molecule the most. This indicates that the quantitative nature of the activation of CO2 depends on the electronic structure and size of the superalkalis, as seen in an earlier section.
Kumar et al. [42] explored the scope of non-metallic superalkalis in the activation of CO2. They studied the interaction of CO2 with non-metallic superalkalis such as O2H5 and N2H7, employing the MP2/6-311++G (d,p) level via the Gaussian 09 program. The equilibrium structures of O2H5-CO2 and N2H7-CO2 are shown in Figure 6, and related parameters are listed in Table 1. It was noticed that in O2H5-CO2 complexes, O atoms of CO2 interact with the H-atom of superalkali, unlike in N2H7-CO2, in which the C-atom of CO2 interacts with the H-atom. This may be due to the repulsion between excess electrons of N and O atoms.
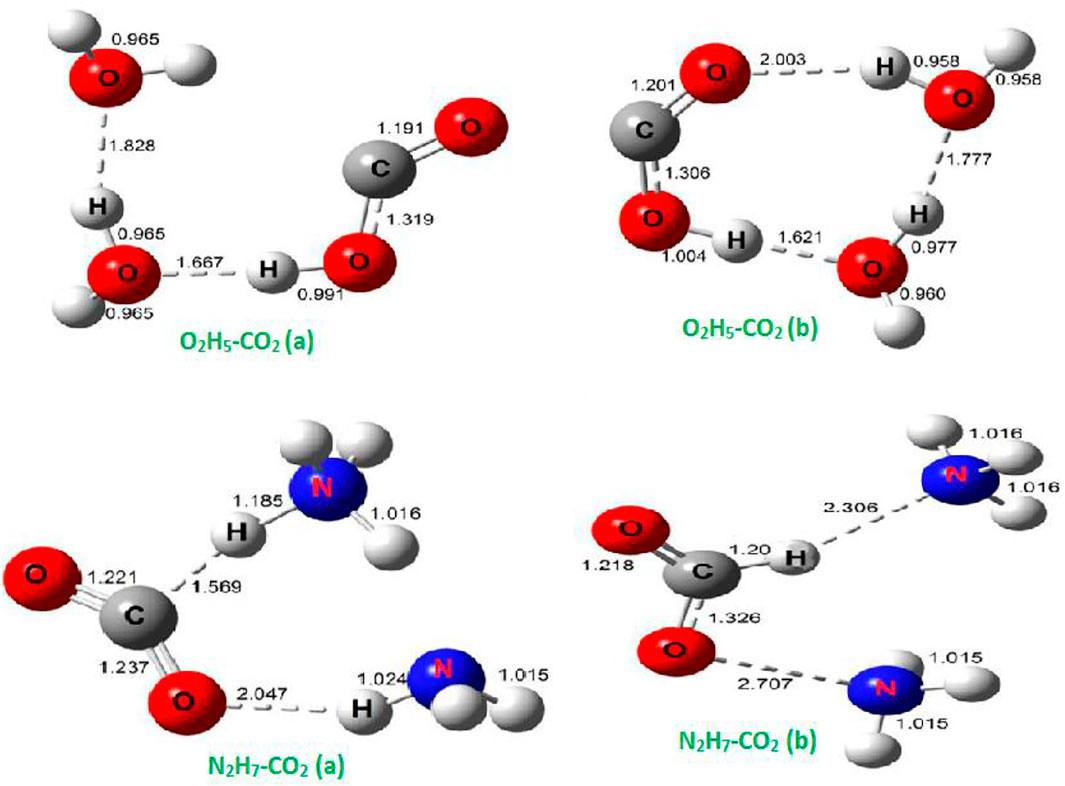
FIGURE 6. Equilibrium structures of CO2 complexes with non-metallic superalkalis from Ref. 42 with the permission of the Taylor & Francis.
The (b) isomers of O2H5-CO2 and N2H7-CO2 are 0.24 and 0.78 eV, higher in energy in which CO2 interacts through the O-atom as well as the C-atom in the N2H7-CO2 isomer. The relative stability of isomers can be explained on the basis of H-bond interactions. For instance, the bond lengths of O-H and C-H are 2.060 Å and 3.710 Å, respectively. The BE of these complexes provides relative strength through the interaction of CO2 with non-metallic superalkalis. From Table 1, the BE suggests that O2H5-CO2 isomers are stable, whereas N2H7-CO2 is slightly destabilized due to the negative value of BE.
From Table 1, the value of NPA charges of CO2 is calculated to be −0.75e for O2H5-CO2 and −0.77e for N2H7-CO2 lowest energy structures (a). Thus, the NPA charge values are very close to each other. In isomer (b) of O2H5-CO2 and N2H7-CO2, the CO2 moiety is bent, similar to that in its lowest energy structure. Therefore, the activation and the consequential reduction of CO2 can also be possible by non-metallic superalkalis such as O2H5, if not by N2H7.
The compact (quasi) icosahedral Al12X (X = Be, Al, C, and P) clusters have been employed to analyze the dissociation and absorption of small gas molecules [48–54]. Zhang et al. [43] studied the interaction of Al12P superalkali with CO2 using Minnesota density functional (M06-2X) [55] and 6-311+G(d) basis set in the Gaussian 09 program. They obtained three isomers of the Al12P-CO2 complex as shown in Figure 7. The lowest energy corresponds to the isomer (a) in which the interaction is mediated by both O atoms. The isomers (b) and (c) are found to have a high energy of 0.11 eV (2.64 kcal/mol) and 0.20 eV (4.52 kcal/mol), respectively. Obviously, the chemisorbed CO2 molecule undergoes structural changes from linear to bending in each Al12P-CO2 isomer.
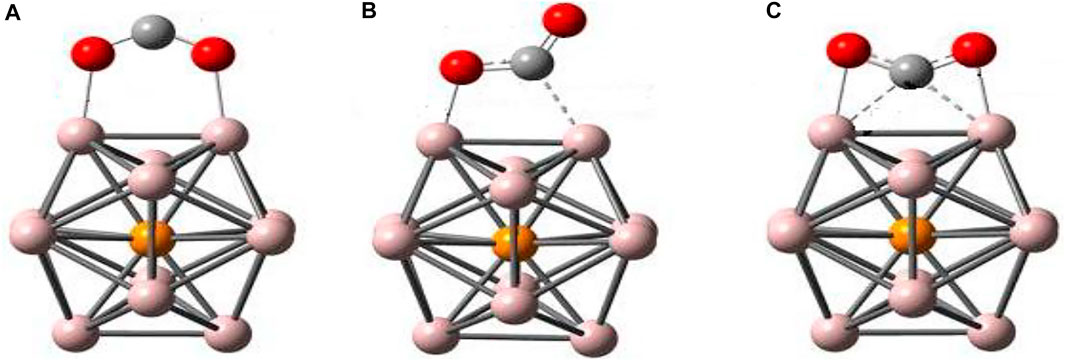
FIGURE 7. Equilibrium structures of CO2 complex with Al12P from Ref. 43 with the permission of Wiley.
They confined their analyses to the global minimum structure, that is, isomer (a). The bond distance of C-O in isomer (a) is 1.24 Å, which is found to be 7.6% larger than free CO2 (1.15 Å), consequently, weakening the C-O bond. Moreover, the variation of the C-O-C angle from 180° to 130.4° in isomer (a) advocates the change in hybridization of carbon in CO2 from sp to quasi-sp2 after activation by Al12P. Furthermore, the C-O bond is marginally larger (1.23 Å) as compared to isolated CO2−, whereas bond bending is also larger, about 4.7%, than that in isolated CO2−, which clearly supports the activation of CO2 assimilated on the Al12P cluster. The computed total NPA charge on the CO2 subunit is −0.707e, which shows the transfer of almost one electron charge from Al12P to CO2 in the complex. Thus, CO2 is successfully reduced to CO2− anion. The low IE of Al12P superatom is the main source of CO2 reduction as it facilitates the transfer of charge to CO2, which ultimately results in the contraction of the O-C-O angle and the weakening of the C-O bond of the CO2 moiety. The small activation barrier of 23 kcal/mol is calculated for the chemisorption of CO2 on Al12P to form the Al12P-CO2 complex (a), which further suggests the application of Al12P as a potential catalyst for CO2 conversion. It has been found that the Al12P complex shows high adsorption intensities in the visible region and, hence, promotes photocatalysis or photothermal catalysis of CO2 and its transformation by absorbing sunlight.
Recently, Sikorska and Gaston [22] explored new superalkali species, N4Mg6M (M = Li, Na, K) by performing the MP2/6-311+G(d) and single-point CCSD(T)/6-311+G (3df) calculations Ref. 56 in the Gaussian 09 program. They studied the catalytic behavior of N4Mg6Li, N4Mg6Na, and N4Mg6K for CO2 activation. For the sake of brevity, we will discuss the interaction of CO2 with N4Mg6Li superalkali. CO2 interacts with N4Mg6M and the resultant N4Mg6M-CO2 complexes are displayed in Figure 8 for M = Li. In all isomers, the interaction between CO2 and N4Mg6M takes place via the C-N bond of 1.452–1.454 Å, which supports the evolution of a single bond between carbon and nitrogen.
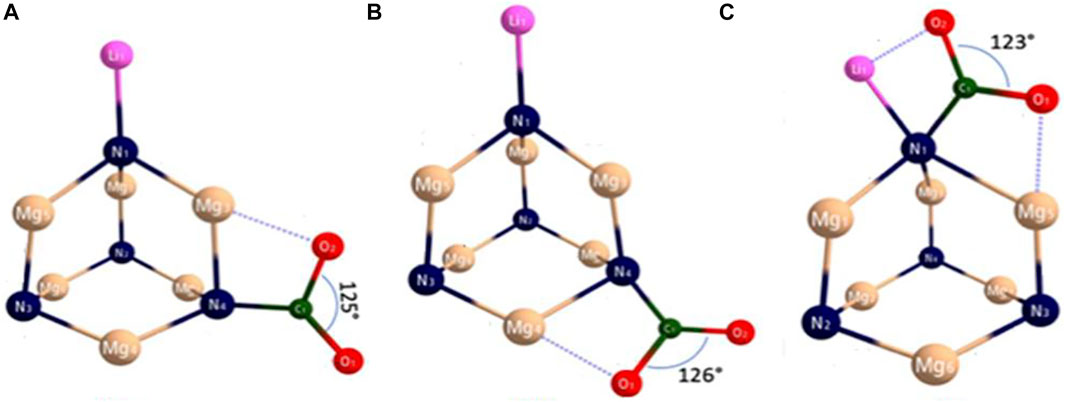
FIGURE 8. Equilibrium structures of CO2 complex with N4Mg6Li from Ref. 22 with the permission of the American Institute of Physics.
The isomers (b) and (c) are found to be 0.04 eV (0.97 kcal/mol) and 0.11 eV (2.48 kcal/mol) higher in energy than the lowest energy isomer (a) in the case of N4Mg6Li-CO2. The binding energy of these isomers lies in the range of 1.46–1.57 eV (see Table 1). The C-O bond lengths, being in the range of 1.223–1.224 Å and 1.335–1.336 Å are indeed 8% larger than that of the CO2 anion. Furthermore, the bending of the angle O-C-O, 123–126°, was 9% more than the angle of isolated CO2−. The NPA charge transferred from N4Mg6M superalkalis to CO2 varies from −0.799e to −0.806e. Ionization energy plays a major role in the activation process of CO2, as the amount of transferred charge increases with the decrease in ionization energy because it is effortless to transfer charge from species with low ionization energy. Therefore, the extension of the bond distance of C-O along with the bending of angle O-C-O results in the weakening of C-O bonds of CO2. Thus, the newly designed N4Mg6M superalkalis could be used as a catalyst for CO2 activation.
The interaction of CO2 with the binuclear Li3F2 superalkali was reported by Park and Meloni as discussed in an earlier section. Recently, Meloni et al. [44] investigated the interaction of CO2 with Li3F2 inside fullerene (C60) by using B3LYP [57, 58] with the 6-31G (d) basis set in the Gaussian 09 program. They noticed two important features. When CO2 is encapsulated within C60, it gets destabilized as its binding energy is −147 kJ/mol. The Li3F2 inside C60 assumes trigonal bipyramidal (D3h) geometry with a binding energy of 119 kJ/mol, unlike the free Li3F2 linear cluster (see Figure 1). Thus, C60 strongly interacts with Li3F2 and there is no reduction of C60.
The unforeseen result has been noticed on insertion of CO2 inside Li3F2(D3h)@C60 endofullerene, as displayed in Figure 9. On the inspection of the geometry inside the C60, it was found that CO2 has been activated by making a ∠OCO 132° and that the bond length of C-O has been increased to 1.20 Å. The activation of CO2 has been attained by the transfer of F atom from Li3F2 to CO2, due to the F-C interaction with the bond distance of 1.38 Å. Thus, the endo-reaction simulates a non-planar (trigonal pyramidal) FCO2 interacting with the FLi3-like species (also shown in Figure 9). There have been several studies [59, 60] in which the structures and interactions between species are greatly modified by encapsulation within C60.
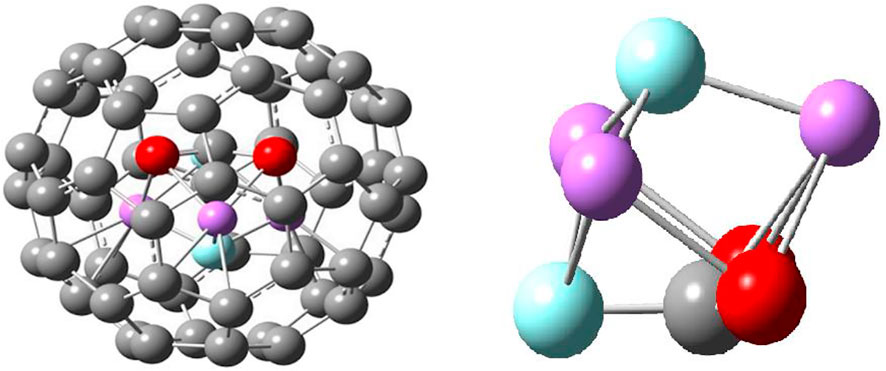
FIGURE 9. Equilibrium structures of [Li3F2-CO2]@C60 endofullerene and Li3F2-CO2 inside C60 from Ref. 44.
Thus, the strong reducing power enables superalkalis to reduce CO2 as well as several other molecules [61, 62], So far, it has been found that the superalkalis are restricted to activate one CO2 molecule per unit, that is, only one CO2 molecule is reduced by a superalkali. This may limit the capture and storage of superalkalis for practical applications. Srivastava [63] reported that a single molecule of hexalithiobenzene (C6Li6) is not only capable of reducing but also capturing up to six CO2 molecules sequentially using the ωB97xD functional [64] and 6-311+G(d) basis set in the Gaussian 09 program. The planar C6Li6 molecule has equal ring bond lengths of 1.418 Å, whose IE is reported to be lower than that of Li, thereby characterizing it as a superalkali molecule [25]. Note that planar star-like C6Li6 has been previously studied by several groups [65–67]. The sequential interaction of CO2 molecules with C6Li6 results in the C6Li6-nCO2 complexes as displayed in Figure 10.
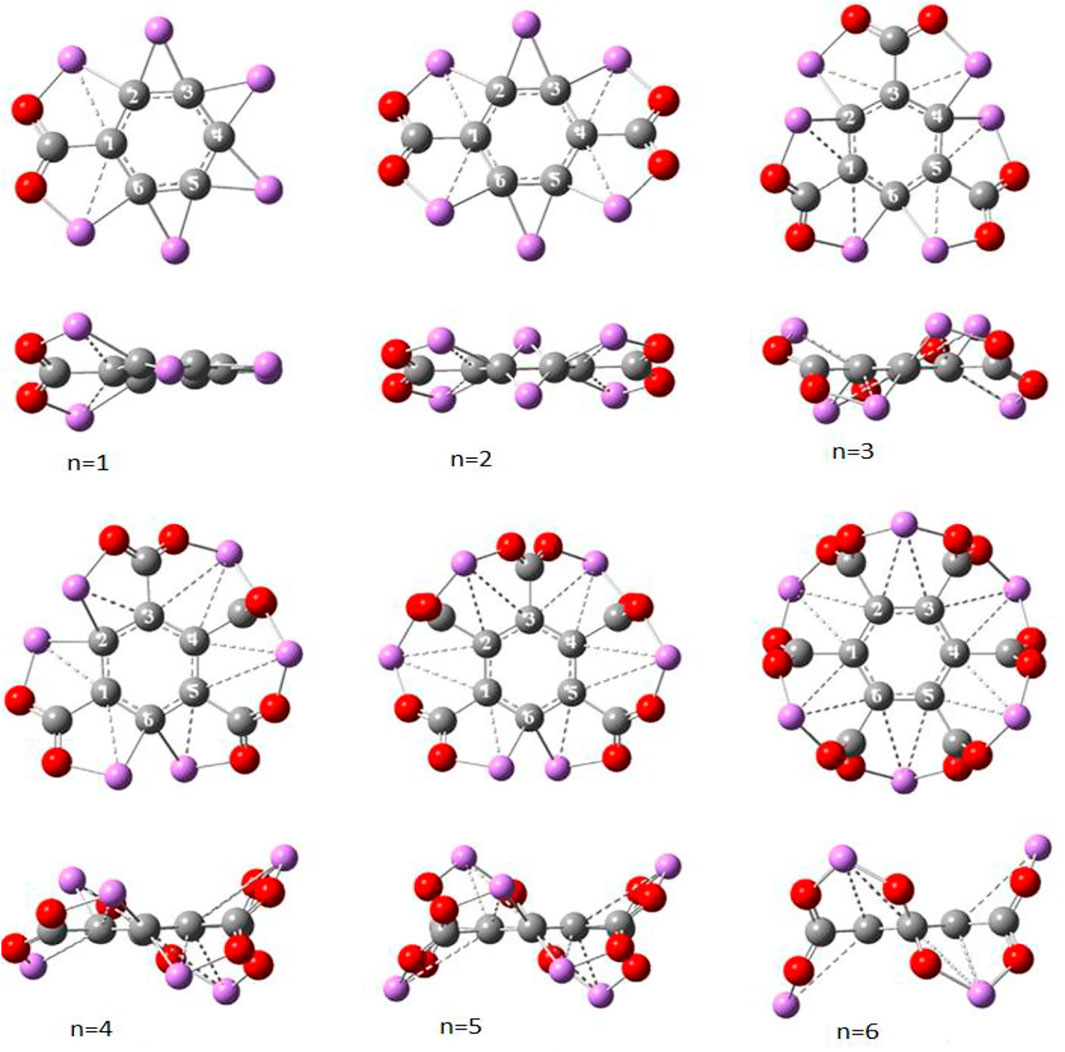
FIGURE 10. Optimized structure of C6Li6-nCO2 complexes (front and side views) for n = 1–6 from Ref. 63 with the permission of Wiley.
The interaction between CO2 and C6Li6 is mediated by one C-C and two Li-O bonds in these complexes. The C6Li6 ring moiety in C6Li6-nCO2is deviated from planarity due to the out-of-plane displacement of Li atoms because of the Coulomb-repulsion between neighboring Li-atoms for n = 1, 2, 4, and 5. However, both C6Li6-3CO2 and C6Li6-6CO2 possess the perfect planar ring moiety, having equal bond lengths of 1.42 Å and 1.40 Å, respectively. The CO2 moiety in C6Li6-CO2 has a bond length and an angle of 1.26 Å and 122.5°, respectively. With an increase in the number of CO2, the average bond length and the average bond angle of CO2 decrease and increase continuously up to 1.25 Å and 125.2°, respectively, for C6Li6-6CO2.
The activation of CO2 leads to an increase in the bond length and a decrease in the bond angle by bending. Table 2 lists the NBO charge (∆q) located at CO2 moieties, adsorption (binding) energy (Ead) per CO2 molecule, and consecutive adsorption energy (ΔEad). It is clear that the Δq of C6Li6-nCO2 becomes −0.83e, −0.81e, −0.79e, and −0.77e for n = 1–3, n = 4, n = 5, and n = 6, respectively. This suggests that C6Li6 can be employed in the activation of all CO2 molecules and, consequently, their adsorption. One can note that the Ead values are fairly large, ranging from 3.18 to 2.79 eV per CO2, which predicts the stability of these C6Li6-nCO2 complexes. It is to be noticed that the adsorption of molecules is not feasible with negative ΔEad values, whereas sequential adsorption becomes feasible with positive ΔEad values. Thus, the study suggests that C6Li6 is not only capable of activation but also effective in the sequential adsorption of six CO2 molecules.
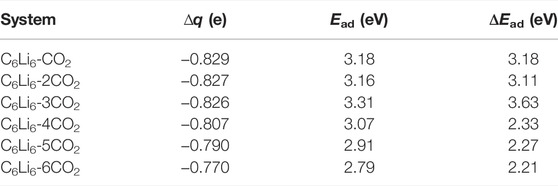
TABLE 2. ωB97xD/6-311+G (d) calculated NBO charge on CO2 (Δq), adsorption energy per CO2 (Ead), and consecutive adsorption energy (ΔEad) for C6Li6-nCO2 complexes taken from Ref. 63.
In summary, we have discussed the activation of CO2 using various superalkalis. CO2 is said to be activated when the charge is transferred to CO2 from superalkali clusters, which ultimately results in the transformation of the linear structure of CO2 to the bent structure of CO2−. Based on quantum chemical methods, CO2 is successfully reduced to CO2− by using typical superalkalis (FLi2, OLi3, and NLi4), binuclear superalkali (Li3F2), special superalkalis (Al3, Mn(B3N3H6)2, B9C3H12, C5NH6), polynuclear superalkalis (Al12P, N4Mg6M), and non-metallic superalkalis (O2H5 and N2H7). It was noticed that the amount of charge transfer depends on the electronic structure, size, and ionization energy of superalkalis. The activation of CO2 by Li3F2 inside C60 fullerene has also been discussed. It was also revealed that the C6Li6 molecule is not only capable of activating CO2 but also capturing up to six CO2 molecules. These results suggest that the superalkalis might be used as efficient catalysts for CO2 activation. Thus, this activated CO2 ion can be converted into fuel, such as methanol [68] via hydrogenation reaction.
HS: literature survey, data collection, writing draft. AKS: conceptualization, supervision, editing, and finalizing the draft.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or any claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mirzaei S, Shamiri A, Aroua MK. Simulation of Aqueous Blend of Monoethanolamine and Glycerol for Carbon Dioxide Capture from Flue Gas. Energy Fuels (2016) 30(11):9540–53. doi:10.1021/acs.energyfuels.6b01230
2. Andreoni W, Pietrucci F. CO2 Capture in Amine Solutions: Modelling and Simulations with Non-empirical Methods. J Phys Condens Matter (2016) 28(50):503003. doi:10.1088/0953-8984/28/50/503003
3. Yeh JT, Resnik KP, Rygle K, Pennline HW. Semi-batch Absorption and Regeneration Studies for CO2 Capture by Aqueous Ammonia. Fuel Process Tech (2005) 86(14-15):1533–46. doi:10.1016/j.fuproc.2005.01.015
4. Kim HR, Yoon T-U, Kim S-I, An J, Bae Y-S, Lee CY. Beyond Pristine MOFs: Carbon Dioxide Capture by Metal-Organic Frameworks (MOFs)-Derived Porous Carbon Materials. RSC Adv (2017) 7:1266–70. doi:10.1039/c6ra26824b
5. Jiang J-X, Su F, Trewin A, Wood CD, Campbell NL, Niu H, et al. Conjugated Microporous Poly(aryleneethynylene) Networks. Angew Chem Int Ed (2008) 47(7):1167. doi:10.1002/anie.200890021
6. Plaza MG, García S, Rubiera F, Pis JJ, Pevida C. Post-combustion CO2 Capture with a Commercial Activated Carbon: Comparison of Different Regeneration Strategies. Chem Eng J (2010) 163(1-2):41–7. doi:10.1016/j.cej.2010.07.030
7. Wang L, Zhao J, Zhou Z, Zhang SB, Chen Z. First-principles Study of Molecular Hydrogen Dissociation on Doped Al12X (X = B, Al, C, Si, P, Mg, and Ca) Clusters. J Comput Chem (2009) 30(15):2509–14. doi:10.1002/jcc.21239
8. Centi G, Perathoner S. Opportunities and Prospects in the Chemical Recycling of Carbon Dioxide to Fuels. Catal Today (2009) 148(3-4):191–205. doi:10.1016/j.cattod.2009.07.075
9. Qiao J, Liu Y, Hong F, Zhang J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem Soc Rev (2014) 43(2):631–75. doi:10.1039/c3cs60323g
10. Schröder D, Schalley CA, Harvey JN, Schwarz H. On the Formation of the Carbon Dioxide Anion Radical CO2− · in the Gas Phase. Int J Mass Spectrom (1999) 185-187:25–35. doi:10.1016/s1387-3806(98)14042-3
11. Sommerfeld T, Meyer H-D, Cederbaum LS. Potential Energy Surface of the CO2? Anion. Phys Chem Chem Phys (2004) 6(1):42. doi:10.1039/b312005h
12. Wang L-S, Reutt JE, Lee YT, Shirley DA. High Resolution UV Photoelectron Spectroscopy of CO+2, COS+ and CS+2 Using Supersonic Molecular Beams. J Electron Spectrosc Relat Phenomena (1988) 47:167–86. doi:10.1016/0368-2048(88)85010-2
13. Czapla M, Skurski P. Oxidizing CO2 with Superhalogens. Phys Chem Chem Phys (2017) 19(7):5435–40. doi:10.1039/c6cp08043j
14. Gutsev GL, Boldyrev AI. DVM-xα Calculations on the Ionization Potentials of MXk+1− Complex Anions and the Electron Affinities of MXk+1 "superhalogens". Chem Phys (1981) 56(3):277–83. doi:10.1016/0301-0104(81)80150-4
15. Gutsev GL, Boldyrev AI. DVM Xα Calculations on the Electronic Structure of "superalkali" Cations. Chem Phys Lett (1982) 92:262–6. doi:10.1016/0009-2614(82)80272-8
16. Weber JM. The Interaction of Negative Charge with Carbon Dioxide - Insight into Solvation, Speciation and Reductive Activation from Cluster Studies. Int Rev Phys Chem (2014) 33(4):489–519. doi:10.1080/0144235x.2014.969554
17. Tong J, Li Y, Wu D, Li Z-R, Huang X-R. Ab Initio Investigation on a New Class of Binuclear Superalkali Cations M2Li2k+1+ (F2Li3+, O2Li5+, N2Li7+, and C2Li9+). J Phys Chem A (2011) 115(10):2041–6. doi:10.1021/jp110417z
18. Hou N, Li Y, Wu D, Li Z-R. Do nonmetallic Superalkali Cations Exist? Chem Phys Lett (2013) 575:32–5. doi:10.1016/j.cplett.2013.05.014
19. Zhao T, Wang Q, Jena P. Rational Design of Super-alkalis and Their Role in CO2activation. Nanoscale (2017) 9:4891–7. doi:10.1039/c7nr00227k
20. Molina B, Soto JR, Castro JJ. Stability and Nonadiabatic Effects of the Endohedral Clusters X@Al12 (X = B, C, N, Al, Si, P) with 39, 40, and 41 Valence Electrons. J Phys Chem C (2012) 116(16):9290–9. doi:10.1021/jp3004135
21. Akutsu M, Koyasu K, Atobe J, Hosoya N, Miyajima K, Mitsui M, et al. Experimental and Theoretical Characterization of Aluminum-Based Binary Superatoms of Al12X and Their Cluster Salts. J Phys Chem A (2006) 110(44):12073–6. doi:10.1021/jp065161p
22. Sikorska C, Gaston N. N4Mg6M (M = Li, Na, K) Superalkalis for CO2 Activation. J Chem Phys (2020) 153(14):144301. doi:10.1063/5.0025545
23. Sun W-M, Li Y, Wu D, Li Z-R. Designing Aromatic Superatoms. J Phys Chem C (2013) 117(46):24618–24. doi:10.1021/jp408810e
24. Giri S, Reddy GN, Jena P. Organo-Zintl Clusters [P7R4]: A New Class of Superalkalis. J Phys Chem Lett (2016) 7(5):800–5. doi:10.1021/acs.jpclett.5b02892
25. Srivastava AK. Organic Superalkalis with Closed-Shell Structure and Aromaticity. Mol Phys (2018) 116(12):1642–9. doi:10.1080/00268976.2018.1438678
26. Yang H, Li Y, Wu D, Li Z-R. Structural Properties and Nonlinear Optical Responses of Superatom Compounds BF4 -M (M = Li, FLi2 , OLi3 , NLi4 ). Int J Quan Chem. (2012) 112:770–8. doi:10.1002/qua.23053
27. Li Y, Wu D, Li Z-R. Compounds of Superatom Clusters: Preferred Structures and Significant Nonlinear Optical Properties of the BLi6-X (X = F, LiF2, BeF3, BF4) Motifs. Inorg Chem (2008) 47(21):9773–8. doi:10.1021/ic800184z
28. Giri S, Behera S, Jena P. Superalkalis and Superhalogens as Building Blocks of Supersalts. J Phys Chem A (2014) 118(3):638–45. doi:10.1021/jp4115095
29. Srivastava AK, Misra N. Superalkali-hydroxides as strong Bases and Superbases. New J Chem (2015) 39(9):6787–90. doi:10.1039/c5nj01259g
30. Srivastava AK, Misra N. OLi3O− Anion: Designing the Strongest Base to Date Using OLi3 Superalkali. Chem Phys Lett (2016) 648:152–5. doi:10.1016/j.cplett.2016.02.010
31. Winfough M, Meloni G. Ab Initio analysis on Potential Superbases of Several Hyperlithiated Species: Li3F2O and Li3F2OHn (N = 1, 2). Dalton Trans (2017) 47(1):159–68. doi:10.1039/c7dt03579a
32. Chen W, Li Z-R, Wu D, Li Y, Li R-Y, Sun C-C. Inverse Sodium Hydride: Density Functional Theory Study of the Large Nonlinear Optical Properties. J Phys Chem A (2005) 109(12):2920–4. doi:10.1021/jp044541c
33. Sun W-M, Fan L-T, Li Y, Liu J-Y, Wu D, Li Z-R. On the Potential Application of Superalkali Clusters in Designing Novel Alkalides with Large Nonlinear Optical Properties. Inorg Chem (2014) 53(12):6170–8. doi:10.1021/ic500655s
34. Srivastava AK, Misra N. Competition between Alkalide Characteristics and Nonlinear Optical Properties in OLi3MLi3O (M = Li, Na, and K) Complexes. Int J Quan Chem. (2017) 117(3):208–12. doi:10.1021/ic500655s10.1002/qua.25313
35. Srivastava AK. Single- and Double-Electron Reductions of CO2 by Using Superalkalis: An Ab Initio Study. Int J Quan Chem. (2018) 118:e25598. doi:10.1002/qua.25598
36. Compton RN, Reinhardt PW, Cooper CD. Collisional Ionization of Na, K, and Cs by CO2, COS, and CS2: Molecular Electron Affinities. J Chem Phys (1975) 63(9):3821. doi:10.1063/1.431875
37. Knapp M, Echt O, Kreisle D, Märk TD, Recknagel E. Formation of long-lived CO2−, N2O−, and their dimer anions, by electron attachment to van der waals clusters. Chem Phys Lett (1986) 126(3):225–31. doi:10.1016/s0009-2614(86)80074-4
38. Gutsev GL, Bartlett RJ, Compton RN. Electron Affinities of CO2, OCS, and CS2. J Chem Phys (1998) 108:6756–62. doi:10.1063/1.476091
39. Møller C, Plesset MS. Note on an Approximation Treatment for Many-Electron Systems. Phys Rev (1934) 46(7):618–22. doi:10.1103/physrev.46.618
40. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09, Revision C02. Wallingford, CT: Gaussian, Inc. (2009).
41. Park H, Meloni G. Reduction of Carbon Dioxide with a Superalkali. Dalton Trans (2017) 46:11942–9. doi:10.1039/c7dt02331f
42. Kumar R, Kumar A, Srivastava AK, Misra N. Ab Initio investigations on the Interaction of CO2 and Non-metallic Superalkalis: Structure, Stability and Electronic Properties. Mol Phys (2021) 119(6):e1841311. doi:10.1080/00268976.2020.1841311
43. Zhang XL, Zhang L, Ye YL, Li XH, Ni BL, Li Y, et al. On the Role of Alkali‐Metal‐Like Superatom Al 12 P in Reduction and Conversion of Carbon Dioxide. Chem Eur J (2020) 27(3):1039–45. doi:10.1002/chem.202003733
44. Meloni G, Giustini A, Park H. CO2 Activation within a Superalkali-Doped Fullerene. Front Chem (2021) 9:712960. doi:10.3389/fchem.2021.712960
45. Reed AE, Weinstock RB, Weinhold F. Natural Population Analysis. J Chem Phys (1985) 83(2):735–46. doi:10.1063/1.449486
46. Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA. A Complete Basis Set Model Chemistry. VI. Use of Density Functional Geometries and Frequencies. J Chem Phys (1999) 110(6):2822–7. doi:10.1063/1.477924
47. Zhang X, Liu G, Meiwes‐Broer KH, Ganteför G, Bowen K. CO 2 Activation and Hydrogenation by PtH N − Cluster Anions. Angew Chem Int Ed (2016) 55(33):9644–7. doi:10.1002/ange.20160430810.1002/anie.201604308
48. Wang Y, LeVan MD. Adsorption Equilibrium of Carbon Dioxide and Water Vapor on Zeolites 5A and 13X and Silica Gel: Pure Components. J Chem Eng Data (2009) 54(10):2839–44. doi:10.1021/je800900a
49. Henry DJ, Yarovsky I. Dissociative Adsorption of Hydrogen Molecule on Aluminum Clusters: Effect of Charge and Doping. J Phys Chem A (2009) 113:2565–71. doi:10.1021/jp809619q
50. Lu QL, Chen LL, Wan JG, Wang GH. First Principles Studies on the Interaction of O2 with X@Al12 (X = Al−, P+, C, Si) Clusters. J Comput Chem (2010) 31(15):2804–9. doi:10.1002/jcc.21573
51. Zhao J-Y, Zhao F-Q, Xu S-Y, Ju X-H. DFT Studies on Doping Effect of Al12X: Adsorption and Dissociation of H2O on Al12X Clusters. J Phys Chem A (2013) 117(10):2213–22. doi:10.1021/jp309422p
52. Zhao J-Y, Zhang Y, Zhao F-Q, Ju X-H. Adsorption of Carbon Dioxide on Al12X Clusters Studied by Density Functional Theory: Effect of Charge and Doping. J Phys Chem A (2013) 117:12519–28. doi:10.1021/jp405934w
53. Zhao J-Y, Zhao F-Q, Xu S-Y, Ju X-H. Theoretical Study of the Geometries and Decomposition Energies of CO2 on Al12X: Doping Effect of Al12X. J Mol Graphics Model (2014) 48:9–17. doi:10.1016/j.jmgm.2013.11.002
54. Chakraborty D, Chattaraj PK. Reactions Involving Some Gas Molecules through Sequestration on Al12 Be Cluster: An Electron Density Based Study. J Comput Chem (2017) 39(10):535–45. doi:10.1002/jcc.25092
55. Zhao Y, Truhlar DG. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor Chem Account (2008) 120(1-3):215–41. doi:10.1007/s00214-007-0310-x
56. Watts JD, Gauss J, Bartlett RJ. Coupled‐cluster Methods with Noniterative Triple Excitations for Restricted Open‐shell Hartree-Fock and Other General Single Determinant Reference Functions. Energies and Analytical Gradients. J Chem Phys (1993) 98(11):8718–33. doi:10.1063/1.464480
57. Becke AD. Density-functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys Rev A (1988) 38:3098–100. doi:10.1103/physreva.38.30910.1103/physreva.38.3098
58. Lee C, Yang W, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys Rev B (1988) 37:785–9. doi:10.1103/physrevb.37.785
59. Ramachandran CN, Sathyamurthy N. Water Clusters in a Confined Nonpolar Environment. Chem Phys Lett (2005) 410(4-6):348–51. doi:10.1016/j.cplett.2005.04.113
60. Srivastava AK, Pandey SK, Misra N. (CH3Br⋯NH3)@C60: The Effect of Nanoconfinement on Halogen Bonding. Chem Phys Lett (2016) 662:240–3. doi:10.1016/j.cplett.2016.09.036
61. Srivastava AK. Reduction of Nitrogen Oxides (NO ) by Superalkalis. Chem Phys Lett (2018) 695:205–10. doi:10.1016/j.cplett.2018.02.029
62. Srivastava AK. DFT and QTAIM Studies on the Reduction of Carbon Monoxide by Superalkalis. J Mol Graphics Model (2021) 102:107765. doi:10.1016/j.jmgm.2020.107765
63. Srivastava AK. CO 2 ‐activation and Enhanced Capture by C 6 Li 6 : A Density Functional Approach. Int J Quan Chem. (2019) 119:e25904. doi:10.1002/qua.25904
64. Chai J-D, Head-Gordon M. Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys Chem Chem Phys (2008) 10(44):6615–20. doi:10.1039/b810189b
65. Xie Y, Schaefer HF. Hexalithiobenzene: a D6h Equilibrium Geometry with Six Lithium Atoms in Bridging Positions. Chem Phys Lett (1991) 179(5-6):563–7. doi:10.1016/0009-2614(91)87104-j
66. Smith BJ. Hexalithiobenzene: beauty Is in the Eye of the Beholder. Chem Phys Lett (1993) 207(4-6):403–6. doi:10.1016/0009-2614(93)89021-9
67. Bachrach SM, Miller JV. Structures and Relative Energies of Polylithiated Benzenes. J Org Chem (2002) 67(21):7389–98. doi:10.1021/jo02592010.1021/jo025920
Keywords: CO2 activation, CO2 reduction, charge transfer, superalkalis, theoretical studies
Citation: Srivastava H and Srivastava AK (2022) Superalkalis for the Activation of Carbon Dioxide: A Review. Front. Phys. 10:870205. doi: 10.3389/fphy.2022.870205
Received: 06 February 2022; Accepted: 08 March 2022;
Published: 05 April 2022.
Edited by:
Moyuan Cao, Tianjin University, ChinaCopyright © 2022 Srivastava and Srivastava. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ambrish Kumar Srivastava, YW1icmlzaHBoeXNpY3NAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.