 Kai Liu
Kai Liu Keke Bai1†
Keke Bai1† Ying Liu
Ying Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Phys., 13 June 2022
Sec. Condensed Matter Physics
Volume 10 - 2022 | https://doi.org/10.3389/fphy.2022.856526
Antimonene is a novel two-dimensional topological semiconductor material with a strain-driven tunable electronic structure for future electronic devices, but the growth of clean antimonene is not fully understood. In this work, the growth process of antimonene on the silver substrate has been studied in detail by using the density functional theory and particle swarm optimization algorithms. The results show that, in addition to the experimental reported flat honeycomb and β-phase antimonene, α-phase antimonene was observed to be able to grow on the substrates, and the phases of antimonene were deeply dependent on the reconstructed supercells and surface alloys. It has been demonstrated that the surface alloys on the substrate play an active role in the growth of antimonene.
Antimonene, one of the novel monoelemental class of two-dimensional (2D) material, has been extensively studied from theory to experiment [1–4], since it was first reported by Zhang et al. [5] in 2015. As a member of the 2D group-V family [6], the freestanding monolayer antimonene, either puckered (α-phase) or buckled (β-phase) structure, is a semiconductor. Compared to α-phase antimonene (α-Sb), β-phase antimonene (β-Sb) with a buckled honeycomb structure similar to black phosphorus [7] is more thermodynamically stable. It was reported to have a strain-tunable energy gap and high carrier mobility [5, 7–12]. Recently, Zhao et al. [13] achieved band inversion by applying a large stretching strain on the lattice, and β-Sb was transferred from a trivial semiconductor to a nontrivial quantum spin hall insulator. Unlike β-Sb, the structure of α-Sb is a structural analog of blue phosphorus, with four atoms in the unitcell, which are arranged in a rectangular lattice in puckered shape with two distorted sublayers. It was found to be a kind of 2D topological insulator when the in-plane anisotropic strain was applied [14]. Applying the in-plane strain is a very important method to tune the electronic structure of antimonene, and how to obtain strained antimonene becomes critical.
To obtain monolayer antimonene, scientists have done experiments with different synthesis methods, including micromechanical exfoliation (MME), liquid-phase exfoliation (LPE), and molecular beam epitaxy (MBE). By applying the MME method, which is used to obtain graphene [15]. Ares et al. [16] have successfully obtained few-layer antimonene, but it is hard to obtain monolayer antimonene. Some other works have reported successes in the preparation of antimonene using the LPE method [17–19], while the samples obtained were mostly freestanding antimonene. It usually does not meet the requirements for obtaining topological properties. By using the MBE method, it may be possible to obtain strain-stretched antimonene. In 2016, Ji et al. [20] synthesized high-quality, few-layer β-Sb polygons on mica substrate through van der Waals epitaxy. Some works have reported successful epitaxial growth of β-Sb on various types of substrates, such as layered materials, transition metals, and transition metal-oxides. According to our collection, layered material substrates include PdTe2 [21], MoS2 [22], graphene [23], Bi2Se3 [24], and WSe2 [25]; transition metal substrates include Ag (111) [26–28], Cu(111) [29], and Au (111) [30] surfaces; transition metal-oxide substrates include Cu3O2 [31] and sapphire [32]. During the epitaxial growth of antimonene, the substrate played a very important role. With the lower mismatch between the selected substrates and antimonene, the lattice parameters of the grown antimonene were closer to that of the freestanding antimonene. For example, the lattice constant of antimonene on PdTe2 was 4.13Å [21], and 4.1 Å [24] on Bi2Se3. The lattice parameter of antimonene grown on transition metals was much larger than that of the freestanding antimonene. It was 4.43 Å on Cu(111) surface [29] and 5.0 Å on Ag (111) surface [26, 27]. Thus, transition metals may be good substrates for the epitaxial growth of antimonene with stress tensile.
The Density Functional Theory (DFT) based simulations have played an increasingly important role in assisting experimental efforts to synthesize and analyze two-dimensional (2D) materials in these years. The structural analysis of 2D materials is one of the main functions. In this article, the structural analysis of antimonene on Ag (111) substrate, on which antimonene could have about 20% stretching of lattice, was carried out with the DFT calculations based on the following fundamental questions: 1) How do the structures evolve with an increasing Sb coverage? 2) What is the most stable phase of antimonene on substrates? Our results demonstrated that the antimonene phase was strongly affected by the structure of the substrate and reconstructed supercell.
To find the most stable configuration of antimonene on the Ag (111) substrate, we have selected different sizes of Ag (111) supercells. Then, the surface reconstruction search under various Sb coverage using the particle swarm optimization (PSO) algorithm, which is well-designed in the software package CALYPSO [33–35], was applied. Our calculations were based on the fact that antimony atoms were sufficiently contracting with the substrate and distributed uniformly. Therefore, the evolution of the most stable structures at different coverages was able to describe the continuous deposition of Sb on the Ag (111) surface theoretically.
The coverage of antimony (CSb) on the substrate was the key parameter in this work. Usually, we describe it in terms of CSb = NSb/NSb-sheet. In this equation, the NSb-sheet would not be constant for different phases of the substrate with the same reconstructed area. Therefore, for the ease to describe CSb, we defined it as follows:
where NSb is the number of Sb atoms adsorbed on substrate, and NSub is the number of atoms in a monolayer of the substrate. For the supercells in type of
To measure the structural stability, the formation energy of Sb adsorbed onto the substrates is defined by the following formula:
where Etot is the total energy of the substrate with the Sb atoms adsorbed, Esub is the total energy of the substrate,
To the best of our knowledge, the formation of surface alloy on the substrate is crucial for the epitaxial growth of two-dimensional materials. It has been reported that when being deposited on the Ag (111) surface, the Sb atoms react with the surface and form Ag2Sb surface alloy, which is combined by a monolayer of a 1 × 1 Ag2Sb matching with a
All the PSO calculations were energetically converged with a population size of 20 and a max step of 10 (for saving compute resources, five steps for NSb = 1–4, 10 or 15 steps for NSb ≥ 5) to meet the global optimization required. Then, the possible stable surface structures with lower entropy and different structural symmetry were collected to do the next step of high precision structure optimization. After all the DFT calculations had been done, the energetically favorable structures of all the coverages were obtained to characterize the most probable structural evolution path.
All the DFT calculations were carried out as implemented in the Vienna ab-initio simulation package (VASP) [39–41]. The electron–ion interaction was described by the projector augmented-wave potentials [42], and the exchange-correlation function was given by generalized gradient approximation parametrized by Perdew, Burke, and Ernzerhof [43, 44]. The van der Waals corrections were treated by the semi-empirical DFT-D3 method [45] in the surface reconstruction calculations. The cutoff energy was set to 400 eV for all calculations. The vacuum layer was set to be at least 15 Å to eliminate the interaction between the layers. All the structures were fully relaxed until the force on each atom was less than 0.02 eV/Å, and the energy convergence criterion was met to 10–8 eV. The substrates were built on the basis of the bulk structure of FCC Ag, and the Ag bulk lattice constant obtained by structural optimization was 4.137 Å, which was well converged in the VASP. The total number of layers in the substrate was set to three ( four for the substrate with surface alloy), and the atom positions in the bottom layer were fixed during the geometry optimization.
In this section, we have carried out the structural search for the most stable configuration with different coverage of Sb atoms on pure Ag (111) substrate. In Figure 1A, we depicted the variation of formation energies with CSb on Ag (111) substrate. There are formation energy valleys at the corresponding coverages of CSb = 2/3, 2/3, 4/9 for type
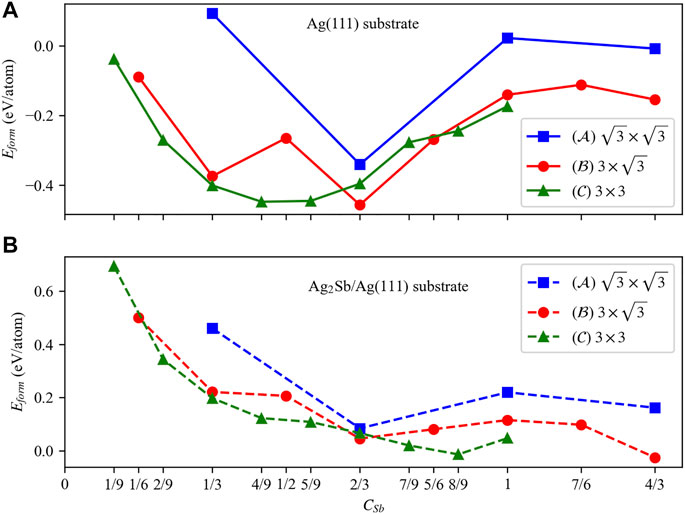
FIGURE 1. The formation energies of the Sb atoms adsorbed on different types of pure Ag (111) and Ag2Sb/Ag (111) substrates. (A) and (B) show the evolution curves of the formation energy, where blue, red, and green represent
The upper four subfigures in Figure 2 show the structures with the lowest energy at different coverage of antimony on an
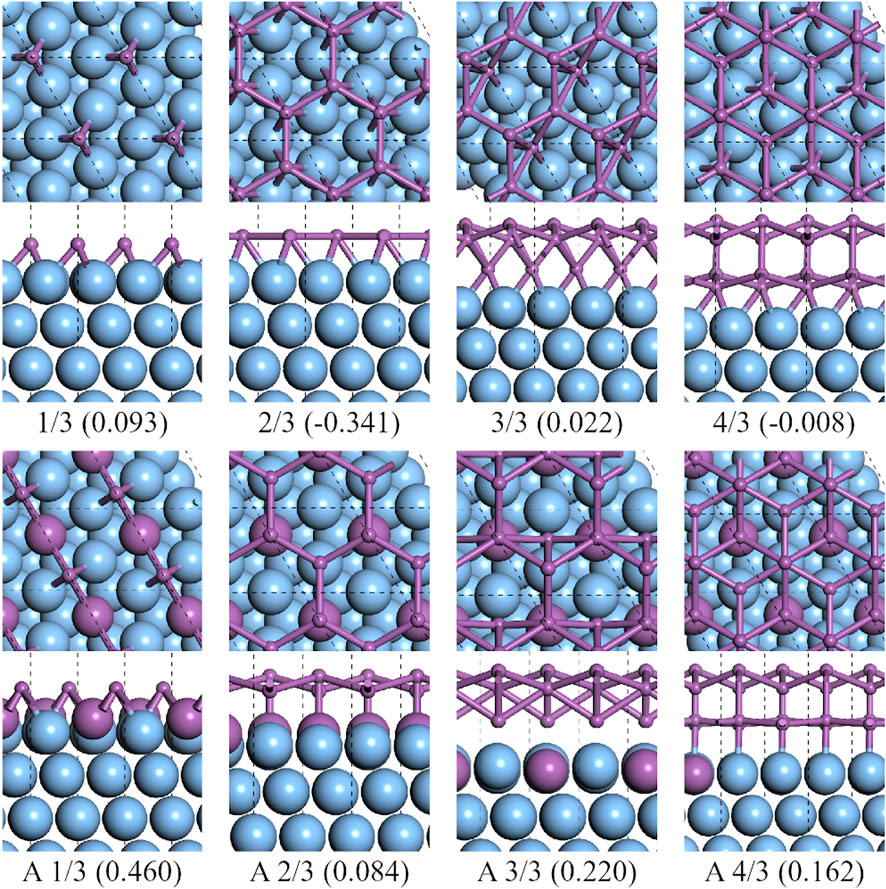
FIGURE 2. Top and side views of the lowest formation energy favorable configurations of antimony adsorbed on the
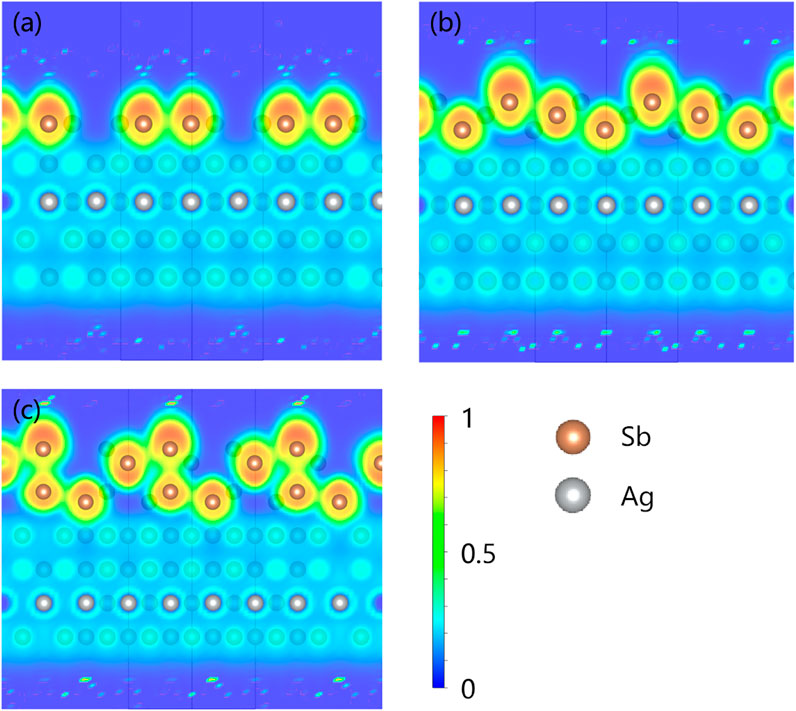
FIGURE 3. 2D contour plot of the electron localization function (ELF) of the Sb atoms on a pure Ag (111) substrate at CSb = (A) 2/3, (B) 3/3, and (C) 4/3 along the cut of the Miller index (110). The minimum ELF value is 0.0 (blue) and the maximum value is 1.0 (red). The brown and silver balls represent Sb and Ag, respectively.
As it is shown in Figure 4, from CSb = 1/6 to 4/6, there existed a clear pathway to form a half layer of α-Sb on
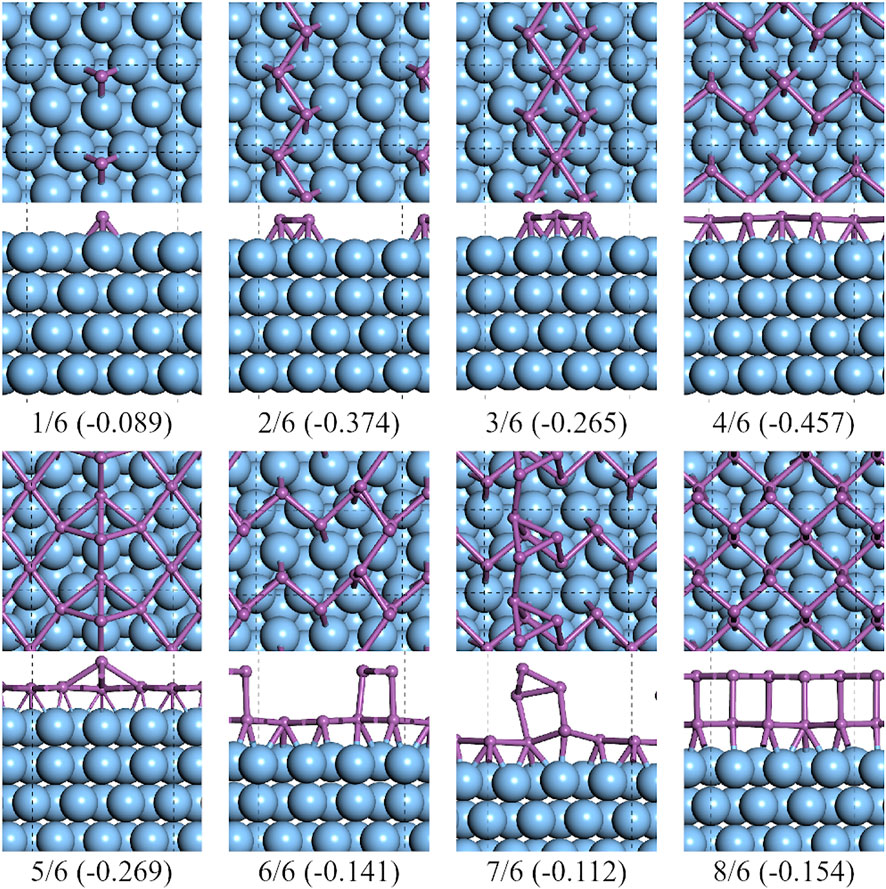
FIGURE 4. Top and side views of the lowest formation energy favorable configurations of antimony adsorbed on the
On
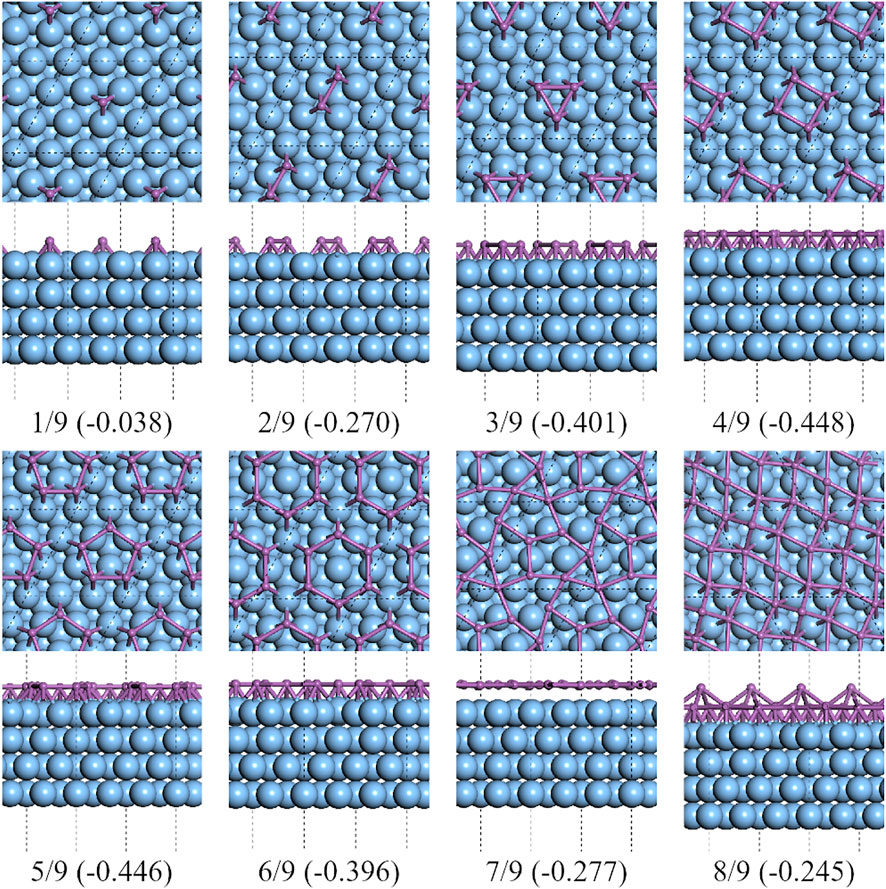
FIGURE 5. Top and side views of the lowest formation energy favorable configurations of the antimony adsorbed on 3×3 Ag (111) substrate.
To further examine the structure of antimony clusters grown on the surface of Ag (111), we calculated the various structures of 1-3 Sb4 clusters falling on the Ag (111) surface by using ab initio molecular dynamics (AIMD) embedded in the package of CP2K [46]. The substrate of Ag (111) was set to three layers with the bottom layer fixed, and the orthogonal lattice parameter was set to a = 26.327 Å, b = 25.333 Å, and c = 32.314 Å to avoid the interaction between the periodic clusters. The AIMD results showed the process of how small Sb4 clusters spread over the Ag 111) substrate into the flat ones after 5 ps at 300 K. Figure 6A plots the total energy of the systems as a function of time for three Sb4 clusters on a pure Ag (111) surface. Two of these clusters disintegrate and rapidly tile to the substrate after 0.7 ps of simulations. About 4.2 ps, the atoms in all the clusters spread out. We found that the flat tetragonal structures, which have the same structure as the CSb = 4/9 on pure Ag (111) surface, exhibit unusual stability during the simulation. To analyze the interaction between the Sb and Ag atoms, we have drawn the curve of radial distribution function (RDF), as shown in Figure 7. The curve shows that the average distance between Sb–Sb stays within 2.935 Å, and the Sb–Ag bond increases slightly with the increase of the number of Sb atoms, from about 2.815 to 2.885 Å. As the SbN cluster gets larger, the distance between SbN and the substrate gets farther, indicating that the interaction between Sb and Sb is much stronger than that between Sb and Ag. Thus, the growth of small clusters is much easier than that of Ag–Sb alloys. The results of the other two configurations are plotted in Supplementary Figure S1 and Supplementary Figure S2. We have also examined the integration curve of RDF. The average coordination number of these three systems increases from 1 to 2 with the increasing number of Sb4 clusters. It can be seen from its structural variations, where dimer, chain, and polygonal structures occupy the majority of conformations. For more visual observation of the structural changes, we exported the animations of the AIMD trajectory, as shown in the MP4 files in SI. The flat square Sb4 clusters are very stable in this simulation. This is consistent with the fact that the most stable configuration on
FIGURE 6. (A) Total energy of the system as a function of time from the AIMD simulation for three Sb4 clusters on pure Ag (111) surface at 300K and zero pressure; System structures at (B) 0 fs (initial configuration), (C) 703.5 fs (two of Sb4 clusters collapse to planar configuration), (D) 4,225 fs (all three Sb4 clusters collapse to planar configuration), and (E) 6,304.5 fs (the configuration with the lowest energy) are listed at right, respectively.
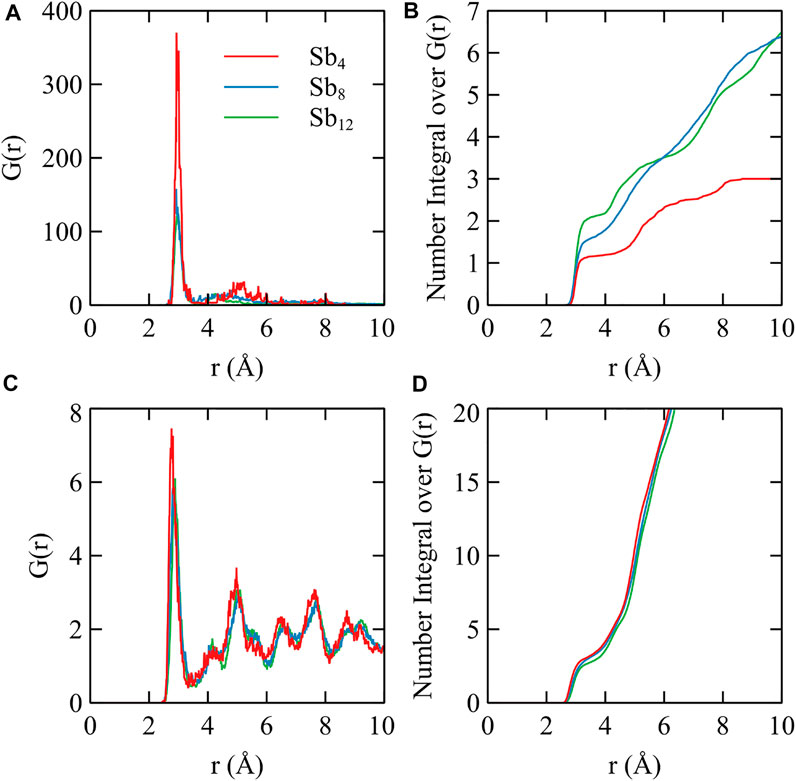
FIGURE 7. (A) and (C) are the radial distribution function for Sb–Sb and Sb–Ag, respectively. (B) and (D) are integration curve of (A) and (C). The line in red, blue, and green correspond to one, two, and three Sb4 clusters on the Ag (111) substrate, respectively.
Theoretically, the larger the substrate, the closer the results are to reality. Combining the results of these three substrates, it can be seen that at lower coverages, flat 2D clusters occupy the major part, and when the antimony atoms increase enough to spread over the substrate or even more, it is quite possible to grow the so-called β- and α-Sb structures. However, in terms of energy comparison, the most stable configuration is the flat half layer of α-Sb.
Before calculating the deposition of Sb atoms on the Ag2Sb/Ag (111) substrate, the climbing image nudged elastic band (cNEB) method [47] was applied to search the transition state to analyze the process of penetration and replacement of Sb to Ag on the Ag (111) surface. We started with modeling the Sb/Ag (111) interface by adding a single Sb atom onto 3 × 3 supercell Ag (111) surface (see Figure 8 (a), the structure diagram on the left). The adsorption energy was calculated by the formula:
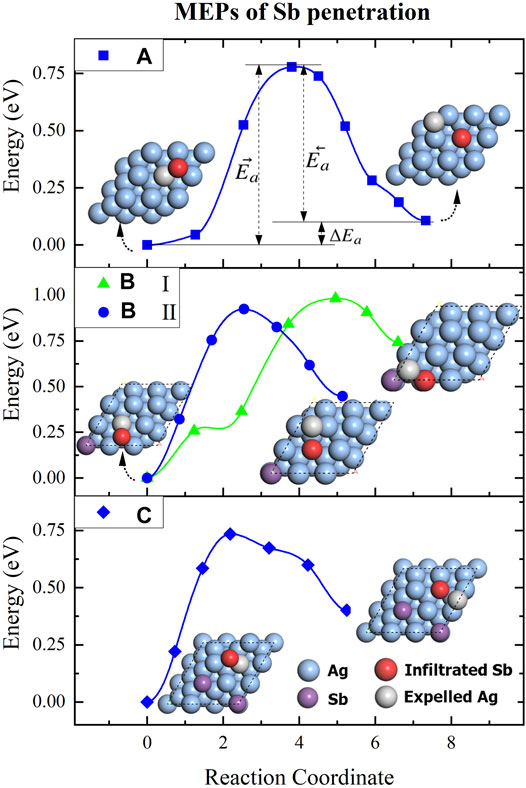
FIGURE 8. The MEPs of the Sb atoms continuously replace the Ag atoms in surface of Ag(111) substrate. (A) is the first step of one Sb atom penetrating into Ag(111) surface. B (I) and B (II) are the second step of another Sb atom penetrate into the final state of step A at neighbor and sub-neighbor sites, respectively. C is the third step of Sb atom penetrate into the final state of step B (II) at sub-neighbor site. The purple and blue balls represent the Ag and Sb atoms, the red and gray balls represent the infiltrated Sb atom and expelled Ag atoms, respectively.
Next, we added the second Sb atom to the substrate while removing the Ag atom that was expelled. Since there was already a penetrated Sb atom after step (A), so we chose to calculate the MEPs for the penetration at the neighbor and subneighbor site separately, see Figure 8B. From the results, it is clear that path B (II) is more favorable than path B (I), with a lower energy of the final state and a relatively shorter reaction path. Based on this, we continued to add a 3rd Sb atom to the final state of path B (II) and made it penetrate at the subneighbor site, and the MEP of this process was plotted in Figure 8C. All the energies of MEPs are listed in Table 1, the ΔEa are all positive, which proves that Sb penetration is a heat absorption process. Such subneighbor atomic arrangement characteristics predetermine the impossibility of continuous replacement of Ag by Sb, and with Sb penetration occurring all over the surface, and an Ag2Sb alloy monolayer is formed. Till now, we have confirmed that if a surface alloy could be formed, it would be Ag2Sb.

TABLE 1. Forward energy barrier
Then, we continued to study the adsorption of Sb atoms on the Ag2Sb/Ag (111) substrate. Same as pure Ag (111) substrates, three types of substrates were constructed, and the curve of the formation energies vs CSb were plotted in Figure 1B. Overall, the formation energies on 3 × 3 substrates were generally lower than on the other two types of substrates. Unlike pure Ag (111) substrates, Sb exhibits different growth characteristics on the Ag2Sb monolayer, the small antimony clusters are no longer more stable, and the formation energy continues to decline until at coverage of 8/9. At CSb = 8/9, the Sb layer shows a 2 × 2 reconstructed β-phase structure with a minor deformation on
For A-type of Ag2Sb/Ag (111) substrates, the results of single Sb on Ag2Sb/Ag (111) shows that the Ag–Ag bridge site was the most favorable, but a monolayer of β-Sb with a layer height of 0.736 Å formed at CSb = 2/3 and Sb atoms occupy the top sites of both the Ag and Sb atoms in the substrate, see Figure 2A 2/3). At CSb = 3/3, the bilayer structure consisted of a layer of β-Sb and intercalated atoms, while the intercalated atoms were located at the Ag–Ag bridge-sites of the substrate. At CSb = 4/3, the bilayer structure consisted of two layers of β-Sb with AB stacking, the bottom layer was almost flat, and the top layer was a buckled honeycomb lattice with buckling higher the 1.022 Å. On such substrates, the flat honeycomb antimonene cannot be stabilized due to the surface alloy, but is replaced by a honeycomb structure with buckling. The interaction between the Ag and Sb atoms affects the structures of antimonene layers. It is worth mentioning that the formation energies of
The lowest formation energy on the
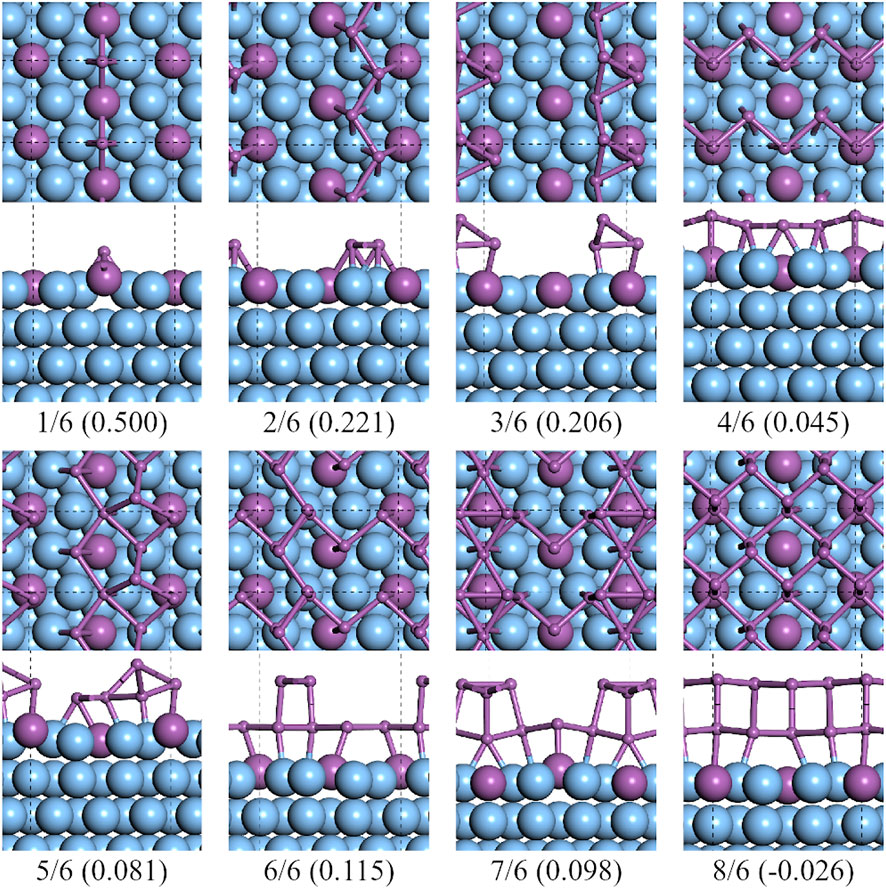
FIGURE 9. Top and side views of the lowest formation energy favorable configurations of antimony adsorbed on the
On C-type Ag2Sb/Ag (111) substrates, the lowest formation energy was at CSb = 8/9 of a reconstructed 2 × 2 supercell of β-Sb, see Figure 10. The reconstructed supercell contains eight Sb atoms, divided into two sublayers, with three of the four Sb atoms in the bottom layer at the bridge site of the Ag–Ag pair and the other Sb atom at the top site of the Sb atom in the surface alloy. This structure is highly consistent with that in Sun’s work [28]. It can be seen from the green dashed line in Figure 1 that the formation energy decreases with the increase of CSb. The existence of the alloy surface changes the potential energy surface of the Ag (111) substrate. Under its impact, the flat polygonal clusters, that appear on the pure silver substrate, are no longer the more stable configurations.
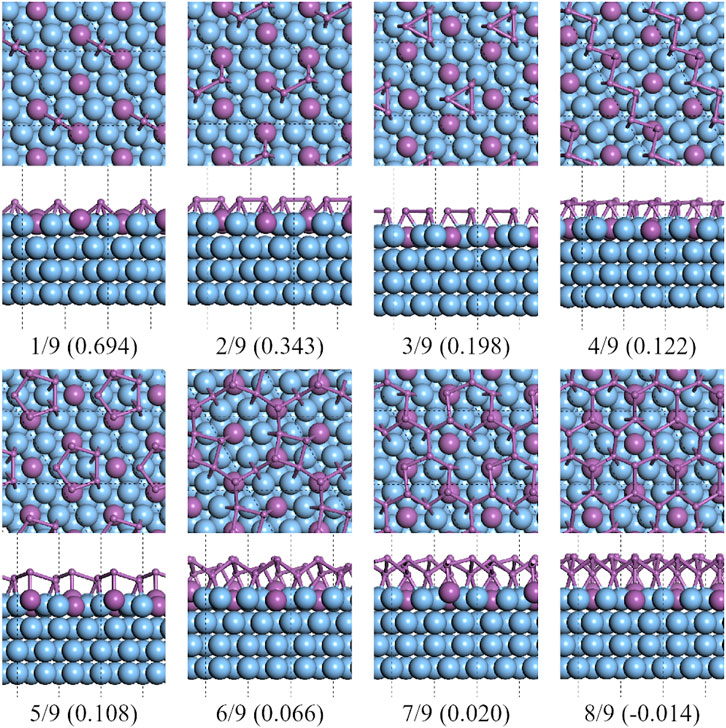
FIGURE 10. Top and side views of the lowest formation energy favorable configurations of antimony adsorbed on the 3×3 Ag2Sb/Ag (111) substrate.
From the calculation results of the abovementioned three substrates, we can see that the presence of the surface alloy changes the adsorption site. The Sb–Sb interaction is somewhat stronger than that of Sb–Ag, so the presence of Sb atoms in the surface alloy can affect the growth of antimonene, especially in the earlier stage. The energetically stable adsorption sites change from the hollow sites to the Ag–Ag bridge sites. Moreover, unlike the pure silver substrate, the Sb top site is also a more stable adsorption site.
Our calculation shows that the formation energies of β-Sb (-0.014 eV/atom) and α-Sb (-0.026 eV/atom) on the Ag2Sb/Ag (111) substrates are very close. Since both α- and β-Sb monolayers are stable in ambient conditions, we need to compare their structural stability and find their structural growth paths to determine which phrase is easier to grow on and which one is relatively stable. Therefore, we carefully examined all the structures, including the substable ones, from the half-layer to the full layer and found two evolutionary paths for each phrase on the Ag2Sb/Ag (111) substrate. In Figure 11A, the formation energies increased and then decreased with the increasing coverage, where the two relatively stable configurations were half- and full-layer α-Sb at CSb = 4/6 and 8/6, respectively. The structures from CSb = 5/6 to 8/6 were all grown based on the stable structure at CSb = 4/6. It is clear that α-Sb is grown half-layer by half-layer on this substrate. Although, there exists a small energy barrier of 0.069 eV/atom along this path. For 3 × 3 Ag2Sb/Ag (111) substrate in Figure 11B, at CSb = 4/9 and 5/9, there were quadrilateral and pentagonal rings with pucker, in which the Sb5 is a very special one, two of the five atoms are sitting on the top site of the Sb atoms in the substrate, and the other three are on the top site of the Ag atoms. This pucker structure is exactly a part of the β-Sb at CSb = 8/9. The continuously deposited Sb atoms placed in their gap positions can form the structures of CSb = 6/9 and 7/9, and the structure of CSb = 7/9 is exactly β-Sb with a Sb vacancy. The formation energy decreases with the increasing concentration of the adsorbed Sb atoms, which indicates that the deposition process is exothermic and can react autonomously. The growth of β-Sb is more advantageous under lower-temperature, while at a high-temperature, there exists a competition between the two phases. Therefore, we believe that it should be possible to observe the growth of both the structures simultaneously during a given experiment.
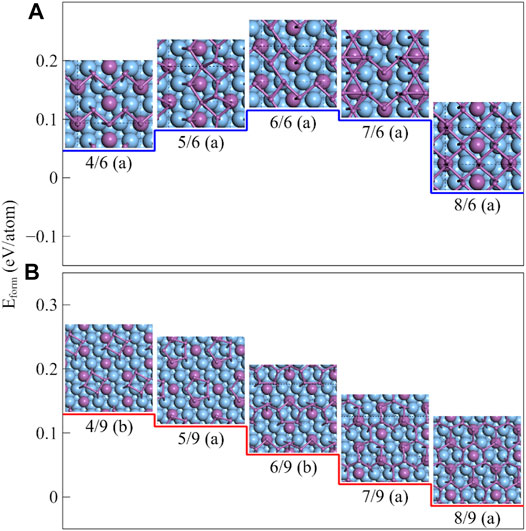
FIGURE 11. Structural evolution paths of
Considering the results of all the types of substrates together, both the size of reconstructed supercells and the surface alloy have a significant effect on growth. No matter with or without the surface alloy, the structure of antimonene on the
The existence of the surface alloy of Ag2Sb has also played an important role in the growth of antimonene. At lower coverages (CSb < 2/3), clusters of SbN (N = 3, 4, and 5), although present, are no longer the most stable structures. On the other hand, at higher coverages (CSb > 2/3), the formation energies of α- and β-Sb shift from the higher to the lowest levels with the presence of the surface alloy. This has a positive effect on the formation of a stable monolayer of α-antimonene.
Note that the minimum value of the required coverage to form a monolayer are 2/3, 2/3, and 7/9 for the substrates of
In summary, we have simulated the growth process of antimonene on the Ag (111) substrate with and without Ag2Sb surface alloy using the DFT and PSO methods. According to the results, in addition to the experimental reported flat honeycomb and β-phase antimonene, α-phase antimonene had also been observed to grow on the substrates and showed a highly stable topological structure with low puckering. In the comparison of the silver substrates with and without the surface alloys, it can be seen that the presence of surface alloys does not hinder the growth of antimonene, but rather promotes the stabilization of antimonene. On the substrates with Ag2Sb surface alloy, both α-Sb and β-Sb have been observed to grow. The core of phase modulation engineering lies in the control of the relaxation rate of the antimony deposited atoms. Our work provides a more comprehensive theoretical insight into the growth of antimonene on silver substrates, complementing the possible omissions in experiments that may provide some basis for antimonene synthesis on such kind of metal substrate.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
LK, BK, and WJ contributed to conception and design of the study. LK carried out the computations and organized the database. BK and WJ performed the statistical analysis. SJ and LY contributed to the computational methods. All authors contributed to manuscript revision and read and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 12174084, 11904075, and 11874139), the National Postdoctoral Program for Innovative Talents (Grant No. BX20190104), the Scientific and Technological Research Foundation of Hebei Province (Grant No. ZD2021065), and the Key Program of Natural Science Foundation of Hebei Province (Grant No. A2021205024).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2022.856526/full#supplementary-material
1. Ares P, Palacios JJ, Abellán G, Gómez-Herrero J, Zamora F. Recent Progress on Antimonene: A New Bidimensional Material. Adv Mater (2018) 30:1703771. doi:10.1002/adma.201703771
2. Zhang S, Guo S, Chen Z, Wang Y, Gao H, Gómez-Herrero J, et al. Recent Progress in 2D Group-VA Semiconductors: From Theory to experiment. Chem Soc Rev (2018) 47:982–1021. doi:10.1039/c7cs00125h
3. Wang X, Song J, Qu J. Antimonene: From Experimental Preparation to Practical Application. Angew Chem Int Ed (2019) 58:1574–84. doi:10.1002/anie.201808302
4. Xue C-L, Li S-C. Recent Progress on Antimonene: From Theoretical Calculation to Epitaxial Growth. Jpn J Appl Phys (2021) 60:SE0805. doi:10.35848/1347-4065/abf74e
5. Zhang S, Yan Z, Li Y, Chen Z, Zeng H. Atomically Thin Arsenene and Antimonene: Semimetal-Semiconductor and Indirect-Direct Band-Gap Transitions. Angew Chem Int Ed (2015) 54:3112–5. doi:10.1002/anie.201411246
6. Meng Y-X, Zhao Y-F, Li S-C. Research Progress of Puckered Honeycomb Monolayers. Acta Phys Sin (2021) 70:148101. doi:10.7498/aps.70.20210638
7. Wang G, Pandey R, Karna SP. Atomically Thin Group V Elemental Films: Theoretical Investigations of Antimonene Allotropes. ACS Appl Mater Inter (2015) 7:11490–6. doi:10.1021/acsami.5b02441
8. Wang Y, Ding Y. Electronic Structure and Carrier Mobilities of Arsenene and Antimonene Nanoribbons: A First-Principle Study. Nanoscale Res Lett (2015) 10:254. doi:10.1186/s11671-015-0955-7
9. Aktürk OÜ, Özçelik VO, Ciraci S. Single-layer Crystalline Phases of Antimony: Antimonenes. Phys Rev B (2015) 91:235446. doi:10.1103/physrevb.91.235446
10. Wang S, Wang W, Zhao G. Thermal Transport Properties of Antimonene: An Ab Initio Study. Phys Chem Phys (2016) 18:31217–22. doi:10.1039/c6cp06088a
11. Zhao M, Zhang X, Li L. Strain-driven Band Inversion and Topological Aspects in Antimonene. Sci Rep (2015) 5:16108. doi:10.1038/srep16108
12. Kripalani DR, Kistanov AA, Cai Y, Xue M, Zhou K. Strain Engineering of Antimonene by a First-Principles Study: Mechanical and Electronic Properties. Phys Rev B (2018) 98:085410. doi:10.1103/physrevb.98.085410
13. Zhao A, Wang B. Two-dimensional Graphene-like Xenes as Potential Topological Materials. APL Mater (2020) 8:030701. doi:10.1063/1.5135984
14. Cheung C-H, Fuh H-R, Hsu M-C, Lin Y-C, Chang C-R. Spin Orbit Coupling Gap and Indirect Gap in Strain-Tuned Topological Insulator-Antimonene. Nanoscale Res Lett (2016) 11:459. doi:10.1186/s11671-016-1666-4
15. Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, et al. Electric Field Effect in Atomically Thin Carbon Films. Science (2004) 306:666–9. doi:10.1126/science.1102896
16. Ares P, Aguilar-Galindo F, Rodríguez-San-Miguel D, Aldave DA, Díaz-Tendero S, Alcamí M, et al. Mechanical Isolation of Highly Stable Antimonene under Ambient Conditions. Adv Mater (2016) 28:6332–6. doi:10.1002/adma.201602128
17. Gibaja C, Rodriguez-San-Miguel D, Ares P, Gómez-Herrero J, Varela M, Gillen R, et al. Few-Layer Antimonene by Liquid-phase Exfoliation. Angew Chem Int Ed (2016) 55:14345–9. doi:10.1002/anie.201605298
18. Wang X, He J, Zhou B, Zhang Y, Wu J, Hu R, et al. Bandgap‐Tunable Preparation of Smooth and Large Two‐Dimensional Antimonene. Angew Chem (2018) 130:8804–9. doi:10.1002/ange.201804886
19. Gibaja C, Assebban M, Torres I, Fickert M, Sanchis-Gual R, Brotons I, et al. Liquid Phase Exfoliation of Antimonene: Systematic Optimization, Characterization and Electrocatalytic Properties. J Mater Chem A (2019) 7:22475–86. doi:10.1039/c9ta06072c
20. Ji J, Song X, Liu J, Yan Z, Huo C, Zhang S, et al. Two-dimensional antimonene single crystals grown by van der Waals epitaxy. Nat Commun (2016) 7:13352. doi:10.1038/ncomms13352
21. Wu X, Shao Y, Liu H, Feng Z, Wang Y-L, Sun J-T, et al. Epitaxial Growth and Air-Stability of Monolayer Antimonene on PdTe2. Adv Mater (2017) 29:1605407. doi:10.1002/adma.201605407
22. Chen H-A, Sun H, Wu C-R, Wang Y-X, Lee P-H, Pao C-W, et al. Single-Crystal Antimonene Films Prepared by Molecular Beam Epitaxy: Selective Growth and Contact Resistance Reduction of the 2D Material Heterostructure. ACS Appl Mater Inter (2018) 10:15058–64. doi:10.1021/acsami.8b02394
23. Sun X, Lu Z, Xiang Y, Wang Y, Shi J, Wang G-C, et al. Van der Waals Epitaxy of Antimony Islands, Sheets, and Thin Films on Single-Crystalline Graphene. ACS Nano (2018) 12:6100–8. doi:10.1021/acsnano.8b02374
24. Flammini R, Colonna S, Hogan C, Mahatha SK, Papagno M, Barla A, et al. Evidence Of β-Antimonene at the Sb/Bi2Se3 Interface. Nanotechnology (2018) 29:065704. doi:10.1088/1361-6528/aaa2c4
25. Zhang Y-W, Li J-Y, Wu C-H, Chang C-Y, Chang S-W, Shih M-H, et al. Tungsten Diselenide Top-Gate Transistors with Multilayer Antimonene Electrodes: Gate Stacks and Epitaxially Grown 2D Material Heterostructures. Sci Rep (2020) 10:5967. doi:10.1038/s41598-020-63098-1
26. Mao Y-H, Zhang L-F, Wang H-L, Shan H, Zhai X-F, Hu Z-P, et al. Epitaxial Growth of Highly Strained Antimonene on Ag(111). Front Phys (2018) 13:138106. doi:10.1007/s11467-018-0757-3
27. Shao Y, Liu Z-L, Cheng C, Wu X, Liu H, Liu C, et al. Epitaxial Growth of Flat Antimonene Monolayer: A New Honeycomb Analogue of Graphene. Nano Lett (2018) 18:2133–9. doi:10.1021/acs.nanolett.8b00429
28. Sun S, Yang T, Luo YZ, Gou J, Huang Y, Gu C, et al. Realization of a Buckled Antimonene Monolayer on Ag(111) via Surface Engineering. J Phys Chem Lett (2020) 11:8976–82. doi:10.1021/acs.jpclett.0c02637
29. Niu T, Zhou W, Zhou D, Hu X, Zhang S, Zhang K, et al. Modulating Epitaxial Atomic Structure of Antimonene through Interface Design. Adv Mater (2019) 31:1902606. doi:10.1002/adma.201902606
30. Zhou D, Si N, Jiang B, Song X, Huang H, Ji Q, et al. Interfacial Effects on the Growth of Atomically Thin Film: Group VA Elements on Au(111). Adv Mater Inter (2019) 6:1901050. doi:10.1002/admi.201901050
31. Niu T, Meng Q, Zhou D, Si N, Zhai S, Hao X, et al. Large‐Scale Synthesis of Strain‐Tunable Semiconducting Antimonene on Copper Oxide. Adv Mater (2020) 32:1906873. doi:10.1002/adma.201906873
32. Gu M, Li C, Ding Y, Zhang K, Xia S, Wang Y, et al. Direct Growth of Antimonene on C-Plane Sapphire by Molecular Beam Epitaxy. Appl Sci (2020) 10:639. doi:10.3390/app10020639
33. Wang Y, Lv J, Zhu L, Ma Y. Crystal Structure Prediction via Particle-Swarm Optimization. Phys Rev B (2010) 82:094116. doi:10.1103/PhysRevB.82.094116
34. Wang Y, Lv J, Zhu L, Ma Y. CALYPSO: A Method for crystal Structure Prediction. Comput Phys Commun (2012) 183:2063–70. doi:10.1016/j.cpc.2012.05.008
35. Lu S, Wang Y, Liu H, Miao M-s., Ma Y. Self-assembled Ultrathin Nanotubes on diamond (100) Surface. Nat Commun (2014) 5:3666. doi:10.1038/ncomms4666
36. Oppo S, Fiorentini V, Scheffler M. Theory of Adsorption and Surfactant Effect of Sb on Ag(111). Phys Rev Lett (1993) 71:2437–40. doi:10.1103/PhysRevLett.71.2437
37. Soares EA, Bittencourt C, Nascimento VB, de Carvalho VE, de Castilho CMC, McConville CF, et al. Structure Determination of Ag(111) (3×3) R30°−Sb by Low-Energy Electron Diffraction. Phys Rev B (2000) 61:13983–7. doi:10.1103/PhysRevB.61.13983
38. Quinn PD, Brown D, Woodruff DP, Bailey P, Noakes TCQ. Structural Study of the Adsorption of Sb on Ag( 111 ) Using Medium Energy Ion Scattering. Surf Sci (2002) 511:43–56. doi:10.1016/S0039-6028(02)01488-7
39. Kresse G, Hafner J. Ab Initiomolecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semiconductor Transition in Germanium. Phys Rev B (1994) 49:14251–69. doi:10.1103/PhysRevB.49.14251
40. Kresse G, Furthmüller J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput Mater Sci (1996) 6:15–50. doi:10.1016/0927-0256(96)00008-0
41. Kresse G, Furthmüller J. Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys Rev B (1996) 54:11169–86. doi:10.1103/PhysRevB.54.11169
42. Blöchl PE. Projector Augmented-Wave Method. Phys Rev B (1994) 50:17953–79. doi:10.1103/PhysRevB.50.17953
43. Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, et al. Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation. Phys Rev B (1992) 46:6671–87. doi:10.1103/PhysRevB.46.6671
44. Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys Rev Lett (1996) 77:3865–8. doi:10.1103/PhysRevLett.77.3865
45. Grimme S, Antony J, Ehrlich S, Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J Chem Phys (2010) 132:154104. doi:10.1063/1.3382344
46. Kühne TD, Iannuzzi M, Del Ben M, Rybkin VV, Seewald P, Stein F, et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package - Quickstep: Efficient and Accurate Electronic Structure Calculations. J Chem Phys (2020) 152:194103. doi:10.1063/5.0007045
Keywords: antimonene (Sb), density funcational theory, Ag(111), epitaxal growth, PSO (partial swarm optimization)
Citation: Liu K, Bai K, Wang J, Song J and Liu Y (2022) Phase-Dependent Epitaxy for Antimonene Growth on Silver Substrate. Front. Phys. 10:856526. doi: 10.3389/fphy.2022.856526
Received: 17 January 2022; Accepted: 28 April 2022;
Published: 13 June 2022.
Edited by:
Zhi-Gang Chen, University of Southern Queensland, AustraliaReviewed by:
Souraya Goumri-Said, Alfaisal University, Saudi ArabiaCopyright © 2022 Liu, Bai, Wang, Song and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Wang, andhbmdAaGVidHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.