 Shu Ohmura
Shu Ohmura Hideki Ohmura
Hideki Ohmura Tsuyoshi Kato3
Tsuyoshi Kato3 Hirohiko Kono
Hirohiko Kono- 1Department of Physical Science and Engineering, Nagoya Institute of Technology, Nagoya, Japan
- 2National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Japan
- 3Department of Chemistry, School of Science, The University of Tokyo, Bunkyo-ku, Japan
- 4Department of Chemistry, Graduate School of Science, Tohoku University, Sendai, Japan
We present a theoretical investigation as to how multielectron dynamics of CO are manipulated by Fourier-synthesized intense laser pulses. The pulses used are assumed to be comprised of harmonics up to the fourth order. The multiconfiguration time-dependent (TD) Hartree-Fock (MCTDHF) method, where the multielectron wavefunction
Introduction
Coherence is the succinct but vital word in modern science that features a wave of definite phase such as a laser field. The coherence of light [1] can be engraved in matter. Interaction of a coherent laser field with matter induces unique phenomena such as molecular alignment [2, 3] and high-order harmonic generation (HHG) of emission by intense near-infrared laser pulses [4]. Molecules can be aligned by intense near-infrared (IR) laser fields along a given space-fixed axis or plane, depending on the choice of light polarization through the anisotropic interaction of the electric field vector of intense laser radiation with the induced dipole moment. For a polarizable molecule, the major principal axis of the polarizability tensor is forced parallel to the polarization direction of a linearly polarized laser field. The mechanism of HHG is associated with the dynamics of an electron in field-dressed continuum states: Electrons freed by tunnel ionization (TI) [5–7] are pulled away from, pulled back near to, and recollided with parent ions within one optical cycle (the well-known three-step model [8]), while synchronized with the oscillating field. High energy photons even up to the soft x-ray range are emitted upon radiative recombination of an electron in field-driven quiver motion with the parent ion. In HHG, electronic continuum states in atoms or molecules are coherently excited with certain phases (described by a linear superposition of states), which leads to quantum interference between different electron trajectories (such as short and long trajectories) in the applied laser field [9, 10]. The HHG intensity is provided by the Fourier components of the induced dipole moment associated with individual trajectories. Coherent electronic motion in an ensemble of atoms or molecules, characterized by a definite dipole phase, is prerequisite for coherent emission of soft x-rays.
Phase coherence is the key concept in the optical control of quantum systems with high accuracy. Constructive and destructive interference between the wave packets created by a phase-locked pump-probe sequence can be controlled by varying the delay between the two pulses with interferometric precision [11, 12]. Various schemes for the control of optical phases have been devised to manipulate the wave functions or dynamics of quantum systems directly through the coherent nature of a laser field. This extensively growing research area is called coherent (or quantum) control [13–15]. Among various coherent control scenarios is utilization of a two-color phase-controlled laser field consisting of fundamental light and its harmonic light, which has been theoretically explored by Brumer and Shapiro [14]. For laser fields of moderate light intensity (below ∼1012 W/cm2), the population of a target state
An ultimate extension of this kind of methodology is Fourier synthesis of arbitrary light waveforms constructed of a fundamental frequency of light and its harmonics [32]. Sophisticated Fourier syntheses of laser fields have been reported [33–36]. Light wave engineering based on Fourier synthesis enables precise manipulation of electron motion beyond the case of single-frequency excitation; e.g., trajectory control of the HHG electrons in atoms and molecules by intense laser fields with various waveforms has been investigated theoretically [37] and achieved experimentally [38, 39]. Directionally asymmetric molecular TI induced by Fourier-synthesized four-color laser fields, consisting of fundamental, second-, third-, and fourth-harmonic light, can make possible orientation-selective molecular TI [40–43].
For a few-cycle single-frequency (one-color) pulse, carrier-envelope phase (CEP), i.e., the phase between the carrier wave and envelope peak of the pulse, also plays a significant role in electron dynamics such as TI and HHG. CEP stabilization has been achieved by the active feedback control which uses the combination of an f-to-2f interferometer to detect CEP drifts and a stereo-ATI (Above Threshold Ionization) phasemeter [44] to determine the value of CEP [45–48]. Few-cycle intense laser pulses with a stable CEP enable one to steer the electronic motion of atoms and molecules with an ultimate precision. Consequently, the HHG spectrum exhibits unique features depending on the CEP [49, 50].
Intense laser fields of light intensity above I≈1012–1013 Wcm−2 initiate large amplitude electronic motion in atoms, molecules, etc., which triggers various physical or chemical phenomena in a wide range of timescale. Such systems can then be excited to high-lying electronic states or ionized in a nonperturbative manner. A typical phenomenon is TI, which is the source of the HHG upon recombination with the parent ion, as mentioned above. TI occurs mainly in the sub-femotosecond or attosecond (1 as = 10–18 s) region, owing to a highly nonlinear optical response, when the electric field of the laser reaches its maximum values. A number of experimental and theoretical studies have been devoted to profoundly understanding the intense-field induced electron dynamics in atoms [51], molecules [52], solids [53], and biological systems [54].
Different types of theoretical approaches beyond perturbation theory have been developed to deal with nonperturbative electronic dynamics of molecules. Epoch-making is the one proposed by Keldysh [55], in which the intense-field ionization rate or probability of an atom is formulated as an electronic transition from the ground state to continuum states of an electron liberated in a driving laser electric field (Volkov states) [56]. In this approach, the detailed atomic energy structure, such as information on excited states or electron correlation, is not taken into account and the Coulomb interaction of the released electron in the laser field with the remaining ion core is neglected. In the Perelemov-Popov-Terent'ev (PPT) approach [57], the long-range Coulomb interaction is incorporated into the Keldysh approach as the first-order correction in the quasi-classical action of the electron. These types of approaches can be integrated into S-matrix theory in which the transition amplitude of a quantum process is formulated by the projection of the total wave function of the system onto the final state. Several versions of the S-matrix approach have been developed which are together known as the Keldysh-Faisal-Reiss (KFR) theory or Strong Field Approximation (SFA) [58, 59].
Ionization induced by intense fields is characterized by the Keldysh parameter
In the above theoretical approaches, only one atomic or molecular orbital is considered as the main ionizing orbital and is allowed to interact with the applied field [61–63]. This is the so-called single active electron (SAE) approximation, where the time-dependent (TD) Schrödinger equation to be solved for the least-bound one-electron is constructed by modelling an “effective potential” after the interaction with the remaining electrons, the nuclei, and the applied fields. In [64, 65], model effective potentials for the ionizing orbital of a CO molecule were constructed by semiempirically formulating the dynamic multielectron polarization effects induced by the applied field [66–68]. The TD version of the Hartree-Fock (HF) method, which is a first-principles approach, can be used to describe the time evolution of a multielectron system (beyond the above SAE approximation). In this TDHF, the multielectron wave function
Correlated multielectron dynamics such as simultaneous double ionization of atoms and molecules is currently one of the primary targets in the research field of attosecond science [50, 52, 69, 70]. Among related subjects are correlated intramolecular electron dynamics [71, 72] and channel interference in HHG [73]. To describe such dynamics properly, one has to go beyond the computationally inexpensive SAE approximation or TDHF method. The multiconfiguration time-dependent Hartree-Fock (MCTDHF) method [74–83] has been developed in the past two decades as a legitimate method for including electron correlation in dynamics. In the MCTDHF, the multielectron wave function
As the level of the theory employed is higher, numerical results obtained become more detailed and reliable; accordingly, what is acquired to unveil the intrinsic physics underlying behind the numerical results becomes more complicated. The TD orbitals in the MCTDHF method evolves under the effects of electron correlation, but it is hard to extract the information of electron correlation from the time evolution of each orbital. For example, the temporal change in MCTDHF orbitals cannot be linearly decomposed into the change due to the one-body interaction (interaction of an electron with nuclei and external fields) and that due to the two-body interaction.
In our previous papers [86–90], a novel approach, i.e., a single orbital picture was established under the framework of the MCTDHF. We adopted the representation of TD natural spin-orbitals (SOs)
Directional anisotropy in the intense-field induced ionization of CO has been extensively investigated [20, 21, 26, 92] by using space-asymmetric ω+2ω two-color fields. The emission direction of C+ or O+ from CO in two-color field ionization experiments indicates that ionization is enhanced when the laser electric field
The direction of anisotropic ionization does not always agree with the prediction based solely on the shape of the HOMO. For OCS, the HOMO has a large amplitude around the C-S axis but ionization in a circularly polarized field is enhanced when the electric field turns to the direction from O to S [93]. This anisotropy is attributed to the linear Stark effect for polar molecules which increases (or reduces) the ionization potential of HOMO when
The results obtained by the conventional MO-ADK theory [61] are in agreement with the experimental result that the ionization rate of the HOMO of CO takes the maximum when
Among other relevant factors to be considered for intense field ionization are the combined contribution from multiple orbitals [101, 102], field-induced multielectron correlation effects [87], and dynamic electron polarization [64, 99]. Although the emission direction of C+ or O+ from CO in the two-color ionization experiments [20, 21, 26] suggests that the main channel of the TI in CO is the 5σ HOMO, next lower lying orbitals such as 1π HOMO-1 and 4σ HOMO-2 can contribute to the ionization yield [88, 101]. These factors also affect the HHG process. The effects of dynamic electron polarization on HHG have also been discussed in [103, 104].
In previous studies [88–90], we calculated the effective potentials for natural orbitals of CO in an intense near-IR field (λ = 760 nm, I = 1014 Wcm−2) from the MCTDHF wave function and investigated the mechanism of anisotropic ionization of CO. The analysis of the 5σ HOMO effective potential
We have so far shown how useful the effective potential approach is to unveil the intrinsic nature of multielectron dynamics [88–90]. In this paper, we examine the role of electron-electron interaction or electron correlation in CO interacting with Fourier-synthesized intense laser pulses by monitoring the temporal change in effective potentials, though the effective potential itself comes from a single-electron picture. The structure of this paper is as follows. The MCTDHF method for the calculation of multielectron dynamics is outlined in Methodology for Effective Potential, together with a brief derivation of the effective potentials for time-dependent natural orbitals. The results and discussion on the ionization and multielectron dynamics of CO in one-color to four-color phase-controlled fields are presented in Results and Discussion. Finally, conclusions about the manipulation of multielectron dynamics of CO by Fourier-synthesized pulses are given in Conclusion.
Methodology for Effective Potential
In this section, we outline the MCTDHF method developed for the simulation of multielectron dynamics of atoms and molecules. In our approach, the multielectron wave function
Outline of the Multiconfiguration Time-Dependent Hartree-Fock Method
The dynamics of an Ne-electron system is governed by the TD Schrödinger equation for the wave function
where
where
The working EOMs for
where
where the (i, j) matrix element of the 1RDM,
Up to this point,
We now show briefly how to derive the effective potential for each SO. To begin with, we present in this paragraph the EOM for
where
the time propagation of
How to Derive the Effective Potentials for Natural Orbitals
In the case of
The elements
where
We start with the natural orbitals at
The constraint that
By substituting Eq. 11 and
where
where
We designate the expectation value of
Results and Discussion
In addition to a one-color (ω) two-cycle pulse with different carrier-envelope phases [88–90], we theoretically investigated the response of CO to intense multicolor pulses; two-color (ω+2ω), three-color (ω+2ω+3ω), and four color (ω+2ω+3ω+4ω) pulses with different relative phases. We present the numerical results of ionization dynamics of a CO molecule interacting with the above four types of pulses to extract how laser field coherence affects multielectron dynamics from the spatial profiles of TD effective potentials of the 5σ HOMO natural orbital.
Applied Pulses
The C-O axis is assumed to be parallel to the polarization direction of the applied electric field
where
and otherwise
These four phases
where
TD-CASSCF Calculation
In One-Color Pulses, Two-Color Pulses, Three-Color Pulses, Four-Color Pulses, we discuss the characteristic features of the ionization of CO for one- and multi-color pulses. The effects of electron correlation is in detail examined on the basis of the TD effective potentials for the 5σ natural orbitals obtained in the MCTDHF framework. We trace the temporal change in effective potentials to investigate how distinctly electron correlation influences the electronic dynamics.
In the numerical simulations for CO in one-color pulses of λ = 760 nm [88–90], we considered 10 spatial orbitals 1σ, 2σ, 3σ, 4σ, 2×1π, 5σ HOMO, 2×2π LUMO (lowest unoccupied molecular orbital) and 6σ. There is a limit to the number of molecular orbitals (MOs) that we can handle in practical applications of the MCTDHF. The most commonly used scheme is the complete active space (CAS) method, where the orbitals used in the Slater determinants (or configuration state functions) are divided into inactive (core) and active orbitals. The two inactive spin-orbitals (SOs) with the same spatial function are singly occupied respectively in all electron configurations; all possible electron configurations are generated by distributing the other electrons among the active orbitals. This type of expansion scheme is called the time-dependent complete-active-space self-consistent-field (TD-CASSCF) method [107, 108]. Various wave-function-based multiconfigurational TD approaches to the dynamics of indistinguishable particles are compiled in a recent review [109]. We adopted the following TD-CASSCF scheme in the present study; the lower lying core SOs up to 4σ were treated to be occupied by one electron and the shapes of the orbitals are allowed to vary according to the EOMs derived for the core SOs. The high lying 6 SOs, i.e., 2×1π, 5σ, 2×2π, 6σ, were treated as active orbitals that constitute the CAS to accommodate 14−8=6 electrons (the expansion length M = 400).
Since the molecular axis is parallel to the polarization direction, cylindrical symmetry is maintained; then, the cylindrical coordinates z and ρ are convenient for the numerical grid point representation of spatial orbitals; z is chosen parallel to the molecular axis. The nuclei C is placed at
One-Color Pulses
The temporal change in induced dipole moments characterizes the overall electronic dynamics of a molecule. Shown in Figure 1A are the one-color pulse
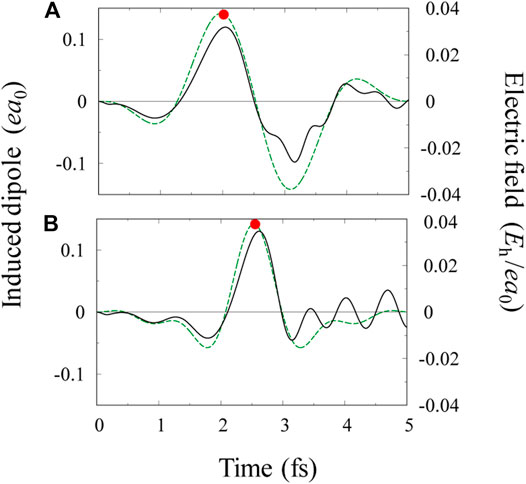
FIGURE 1. (A) Electric field profile
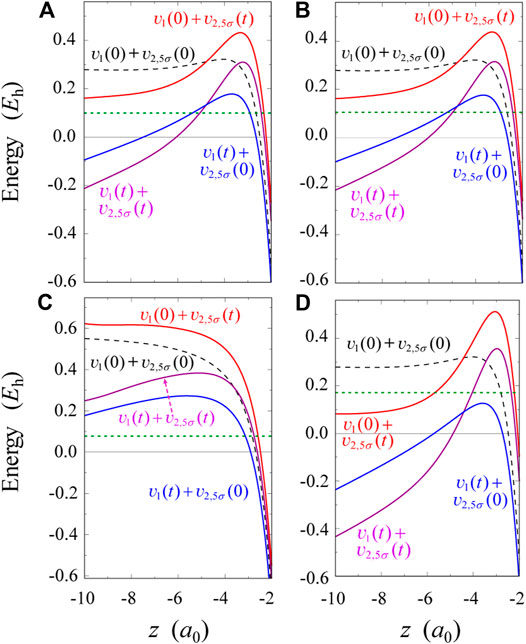
FIGURE 2. Four kinds of 5σ effective potentials plotted parallel to the z-axis at the fixed value of ρ = 0.755a0: (A) potentials obtained by TD-CASSCF at t = t1 ≈ 2 fs under the interaction with
We have numerically confirmed that ionization exclusively occurs from the 5σ orbital when the electric field points from C to O, i.e.,
The origin of anisotropic ionization of CO in near-IR fields has been argued mostly in connection with the fact that the 5σ HOMO natural orbital has a large lobe around the C atom. In what follows, we examine what role electron correlation plays in the anisotropic ionization process. We investigate the mechanism of anisotropic near-IR induced ionization of CO by tracing the TD effective potentials of natural orbitals, defined by Eq. 13, which are changing every moment. More generally, the (correlated) multielectron nature intrinsic in intense-field-induced phenomena can be extracted from the analysis of the temporal change in the effective potential of each natural orbital.
Figure 2 display different types of 5σ HOMO effective potentials
When the sign of
To quantify the role of electron correlation, we compare the effective potentials in Figure 2A with TDHF ones shown in Figure 2C. In Figure 2C,
In Four-Color Pulses, we provide a more concrete picture to understand the root of hump formation in
Two-Color Pulses
The one-color field
The largest peak in
Three-Color Pulses
Multicolor fields can be used to manipulate the modulation between the peaks of the electric field. In this subsection, we present
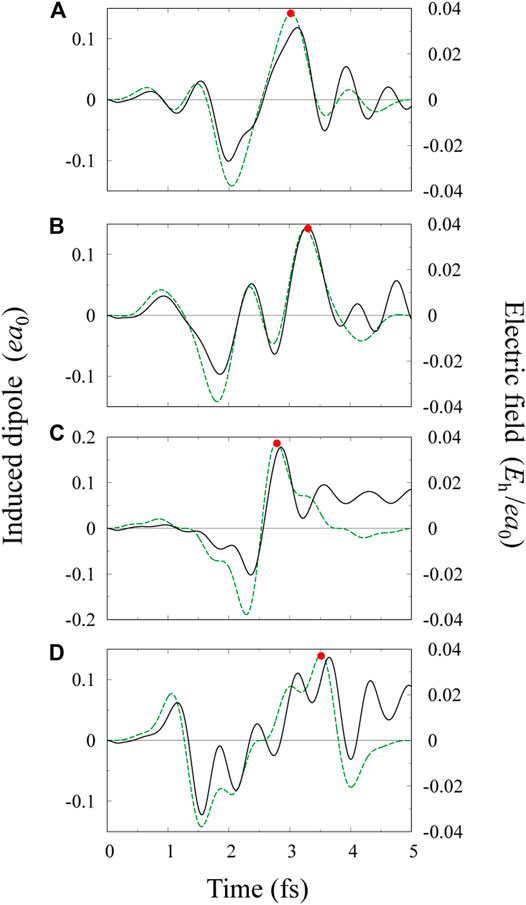
FIGURE 3. Electric field profiles for multi-color pulses (green broken lines): (A, B) three-color pulses with relative intensities
The induced dipole moment
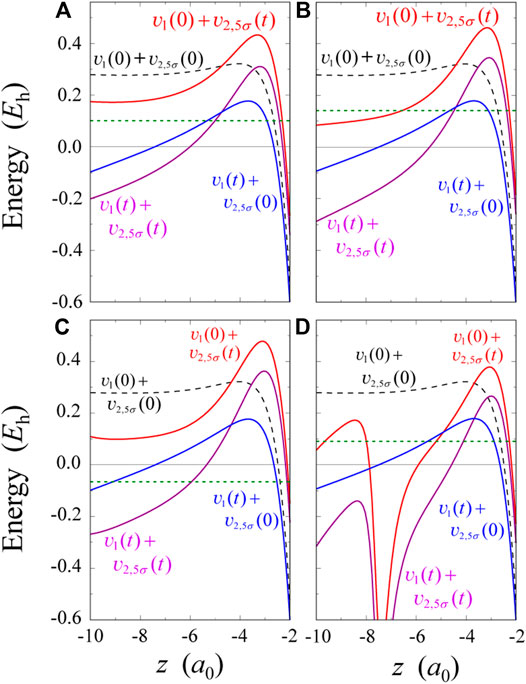
FIGURE 4. Four kinds of 5σ effective potentials plotted parallel to the z-axis at the fixed value of ρ = 0.755a0. The pulses used in four panels (A–D) correspond to those in Figure 3(A–D), respectively.
Four-Color Pulses
The effects of sawtooth wave forms on multielectron dynamics can be investigated by using four-color fields. The relative amplitudes are fixed as
The induced dipole moment
Four types of 5σ effective potentials at t ≈ 2.8 fs for the pulse in Figure 3C are shown in Figure 4C. The difference between
In the present treatment, the effective potential of CO is a two-dimensional (2D) function, i.e., a function of z and ρ. We have already examined TD-CASSCF effective potentials in 2D representation for a one-color pulse [90]. In Figure 5A, we present a 2D contour plot of
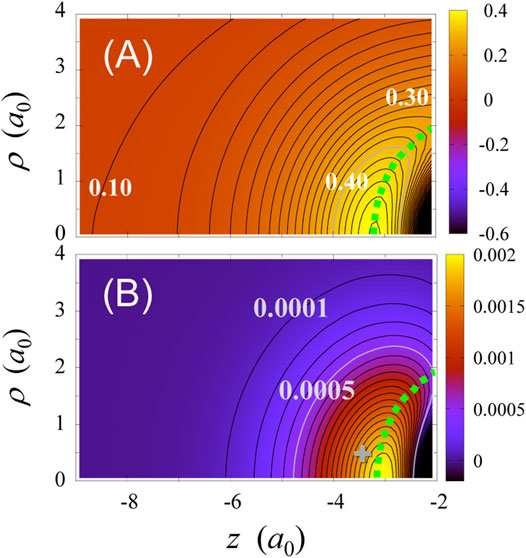
FIGURE 5. TD-CASSCF results for the pulse in Figure 3C: (A) 2D contour plot of
The change in field-induced electron-electron interaction is definitely affected by the spatial change in total electron density
The hump ridge elevates as the field strength
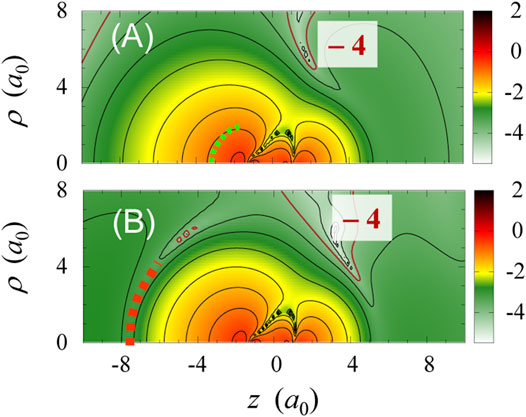
FIGURE 6. 2D contour plots of
For the pulse of Figure 3D, effective potentials at t ≈ 3.5 fs are shown in Figure 4D. The potential
An interpretation based on the present single-electron picture is that the valley in
On the Information out of High-Order Harmonic Generation Spectra
We here discuss the connection of the present results of TI with HHG. The HHG spectra of asymmetric molecules, which reflect the presence and motion of the charges in applied fields, can be utilized to investigate the mechanism of coherent control and to assess the degree of controllability. The ionization potential for a polar molecule in an intense field is a time-dependent one, as denoted by
Another feature inherent in the HHG spectra of asymmetric molecules was pointed out by Bandrauk et al. [113, 114], who theoretically investigated the role of excited states in HHG for HeH2+. They found that a transient localized state (Stark-shifted or field-dressed first excited state of HeH2+) is resonantly populated prior to ionization by laser induced electron transfer from the He2+ side to the H+ side. This field-dressed excited state is able to get back directly to the ground state with emission. A resulting new HHG channel, regarded as the quasi two-step pathway, gives birth to an intense resonance peak (<
Intermediate states prepared by tailored multi-color fields, such as represented by a “squeezed” orbital in Figure 6B, may work as such transient excited states leading to resonance HHG. A realistic attempt to confirm the existence of such resonance states and to assess the controllability of the wave function is to examine how the HHG spectrum (regarding the cutoff energy, individual peak positions and intensities, etc.) changes by varying the relative phases among multi-color fields. In line with this, it is necessary to quantify how long the intermediate states (orbitals) prepared live. We would like to take on the applications of the effective potential approach to the setup of new experimental schemes and to the search of possible results, as discussed in [90].
Conclusion
We presented the results of theoretical investigation of the multielectron dynamics of CO in intense laser fields and discussed various manipulation schemes by Fourier-synthesized coherent fields comprised of harmonics up to the fourth order. The multielectron wavefunction
We then quantified the multielectron nature such as electron correlation by using our effective potential approach: the time-dependent natural orbitals
Two-body interaction
We found that
The final point to be discussed here concerns the future extension of the present effective potential approach to chemical reactions. The strong coupling between intense fields and valence electrons dramatically distorts the potential hypersurfaces which determine the motion of the nuclei and brings about decisive changes in reaction pathways [17, 117, 118]. Kübel et al. [118] ionized H2 by a few-cycle visible pulse and prepared a wave packet on the σg state of
A feasible extension along with this line is to include the nuclear coordinates
The various effective potential approaches abovementioned would help reveal the entire picture of the quantum electronic and nuclear dynamics of molecules and help contribute to further development of coherent control of chemical reactions. Challenges are widespread ahead of the frontier of the research on Coherent Phenomena in Molecular Physics.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
SO: Computation, preparation of figures. HO: conceptual proposal based on experimental feasibility, preparation of manuscript. TK: development of theoretical framework. HK: project coordination, preparation of manuscript. All authors developed the concept of the control of multielectron dynamics by multicolor fields and contributed to the article and approved the submitted version.
Funding
This work was supported in part by JSPS KAKENHI Grant Number JP16H04091.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Prof. S. Koseki for providing us results of the electronic structure calculation of CO.
References
1. Glauber RJ. Quantum Theory of Optical Coherence: Selected Papers and Lectures. Weinheim, Germany: Wiley VCH (2007). doi:10.1002/9783527610075.ch16
2. Friedrich B, Herschbach DR. Alignment and Trapping of Molecules in Intense Laser Fields. Phys Rev Lett (1995) 74:4623. doi:10.1103/PhysRevLett.74.4623−6
3. Stapelfeldt H, Seideman T. Colloquium: Aligning Molecules with Strong Laser Pulses. Rev Mod Phys (2003) 75: 543–57. doi:10.1103/RevModPhys.75.543
4. Agostini P, Fabre F, Mainfray G, Petite G, Rahman NK. Free-free Transitions Following Six-Photon Ionization of Xenon Atoms. Phys Rev Lett (1979) 42: 1127–30. doi:10.1103/PhysRevLett.42.1127
5. Chin SL, Yergeau F, Lavigne P. Tunnel Ionisation of Xe in an Ultra-intense CO2 Laser Field (1014 Wcm−2) with Multiple Charge Creation. J Phys B: Atom Mol Phys (1985) 18:L213–5. doi:10.1088/0022-3700/18/8/001
6. Augst S, Meyerhofer DD, Strickland D, Chint SL. Laser Ionization of Noble Gases by Coulomb-Barrier Suppression. J Opt Soc Am B (1991) 8:858. doi:10.1364/josab.8.000858
7. DeWitt MJ, Levis RJ. Observing the Transition from a Multiphoton-Dominated to a Field-Mediated Ionization Process for Polyatomic Molecules in Intense Laser Fields. Phys Rev Lett (1998) 81:5101–4. doi:10.1103/PhysRevLett.81.5101
8. Corkum PB. Plasma Perspective on Strong Field Multiphoton Ionization. Phys Rev Lett (1993) 71:1994. 10.1103/PhysRevLett.71.1994
9. Lewenstein M, Salières P, L’Huillier A. Phase of the Atomic Polarization in High-Order Harmonic Generation. Phys Rev A (1995) 52: 4747–54. doi:10.1103/PhysRevA.52.4747
10. Becker W, Grasbon F, Kopold R, Milošević DB, Paulus GG, Walther H. Above-threshold Ionization: From Classical Features to Quantum Effects. Adv At Mol Opt Phys (2002) 48: 35–98. doi:10.1016/S1049-250X(02)80006-4
11. Scherer NF, Carlson RJ, Matro A, Du M, Ruggiero AJ, Romero‐Rochin V, et al. Fluorescence‐detected Wave Packet Interferometry: Time Resolved Molecular Spectroscopy with Sequences of Femtosecond Phase‐Locked Pulses. J Chem Phys (1991) 95: 1487–511. doi:10.1063/1.461064
12. Katsuki H, Chiba H, Girard B, Meier C, Ohmori K. Visualizing Picometric Quantum Ripples of Ultrafast Wave-Packet Interference. Science (2006) 311: 1589–92. doi:10.1126/science.1121240
13. Rice SA, Zhao M. Optical Control of Molecular Dynamics. New York: Wiley (2000). doi:10.1021/ja015259b
14. Shapiro M, Brumer P. Principles of the Quantum Control of Molecular Processes. 2nd ed. New York: Wiley (2011). doi:10.1002/9783527639700
15. Gordon RJ, Zhu L, Seideman T. Coherent Control of Chemical Reactions. Acc Chem Res (1999) 32:1007–16. doi:10.1021/ar970119l
16. Endo T, Fujise H, Kawachi Y, Ishihara A, Matsuda A, Fushitani M, et al. Selective Bond Breaking of CO2 in Phase-Locked Two-Color Intense Laser Fields: Laser Field Intensity Dependence. Phys Chem Chem Phys (2017) 19: 3550–6. doi:10.1039/c6cp07471e
17. Sato Y, Kono H, Koseki S, Fujimura Y. Description of Molecular Dynamics in Intense Laser Fields by the Time-Dependent Adiabatic State Approach: Application to Simultaneous Two-Bond Dissociation of CO2 and its Control. J Am Chem Soc (2003) 125:8019–31. doi:10.1021/ja0344819
18. Schafer KJ, Kulander KC. Phase-dependent Effects in Multiphoton Ionization Induced by a Laser Field and its Second Harmonic. Phys Rev A (1992) 45: 8026–33. doi:10.1103/PhysRevA.45.8026
19. Schumacher DW, Weihe F, Muller HG, Bucksbaum PH. Phase Dependence of Intense Field Ionization: A Study Using Two Colors. Phys Rev Lett (1994) 73: 1344–7. doi:10.1103/PhysRevLett.73.1344
20. De S, Znakovskaya I, Ray D, Anis F, Johnson NG, Bocharova IA, et al. Field-free Orientation of CO Molecules by Femtosecond Two-Color Laser Fields. Phys Rev Lett (2009) 103:153002. doi:10.1103/PhysRevLett.103.153002
21. Li H, Ray D, De S, Znakovskaya I, Cao W, Laurent G, et al. Orientation Dependence of the Ionization of CO and NO in an Intense Femtosecond Two-Color Laser Field. Phys Rev A (2011) 84:043429. doi:10.1103/PhysRevA.84.043429
22. Kaziannis S, Kotsina N, Kosmidis C. Interaction of Toluene with Two-Color Asymmetric Laser Fields: Controlling the Directional Emission of Molecular Hydrogen Fragments. J Chem Phys (2014) 141:104319. doi:10.1063/1.4895097
23. Ohmura H, Saito N, Tachiya M. Selective Ionization of Oriented Nonpolar Molecules with Asymmetric Structure by Phase-Controlled Two-Color Laser Fields. Phys Rev Lett (2006) 96:173001. doi:10.1103/PhysRevLett.96.173001
24. Ohmura H, Tachiya M. Robust Quantum Control of Molecular Tunneling Ionization in the Space Domain by Phase-Controlled Laser Fields. Phys Rev A (2008) 77:023408. doi:10.1103/physreva.77.023408
25. Ohmura H, Saito N, Nonaka H, Ichimura S. Dissociative Ionization of a Large Molecule Studied by Intense Phase-Controlled Laser Fields. Phys Rev A (2008) 77:053405. doi:10.1103/PhysRevA.77.053405
26. Ohmura H, Saito N, Morishita T. Quantum Control of Molecular Tunneling Ionization in the Spatiotemporal Domain. Phys Rev A (2011) 83:063407. doi:10.1103/PhysRevA.83.063407
27. Ohmura H, Saito N, Morishita T. Molecular Tunneling Ionization of the Carbonyl Sulfide Molecule by Double-Frequency Phase-Controlled Laser Fields. Phys Rev A (2014) 89:013405. doi:10.1103/PhysRevA.89.013405
28. Kitzler M, Lezius M. Spatial Control of Recollision Wave Packets with Attosecond Precision. Phys Rev Lett (2005) 95:253001. doi:10.1103/PhysRevLett.95.253001
29. Kim IJ, Kim CM, Kim HT, Lee GH, Lee YS, Park JY, et al. Highly Efficient High-Harmonic Generation in an Orthogonally Polarized Two-Color Laser Field. Phys Rev Lett (2005) 94:243901. doi:10.1103/PhysRevLett.94.243901
30. Mashiko H, Gilbertson S, Li C, Khan SD, Shakya MM, Moon E, et al. Double Optical Gating of High-Order Harmonic Generation with Carrier-Envelope Phase Stabilized Lasers. Phys Rev Lett (2008) 100:103906. doi:10.1103/PhysRevLett.100.103906
31. Brugnera L, Hoffmann DJ, Siegel T, Frank F, Zaïr A, Tisch JWG, et al. Trajectory Selection in High Harmonic Generation by Controlling the Phase between Orthogonal Two-Color Fields. Phys Rev Lett (2011) 107:153902. doi:10.1103/PhysRevLett.107.153902
32. Hänsch TW. A Proposed Sub-femtosecond Pulse Synthesizer Using Separate Phase-Locked Laser Oscillators. Opt Commum (1990) 80:71. doi:10.1016/0030-4018(90)90509-R
33. Shverdin MY, Walker DR, Yavuz DD, Yin GY, Harris SE. Generation of a Single-Cycle Optical Pulse. Phys Rev Lett (2005) 94:033904. doi:10.1103/PhysRevLett.94.033904
34. Yoshitomi D, Kobayashi Y, Torizuka K. Characterization of Fourier-Synthesized Optical Waveforms from Optically Phase-Locked Femtosecond Multicolor Pulses. Opt Lett (2008) 33:2925. doi:10.1364/OL.33.002925
35. Chan H-S, Hsieh Z-M, Liang W-H, Kung AH, Lee C-K, Lai C-J, et al. Synthesis and Measurement of Ultrafast Waveforms from Five Discrete Optical Harmonics. Science (2011) 331:1165–8. doi:10.1126/science.1198397
36. Yoshii K, Kiran Anthony J, Katsuragawa M. The Simplest Route to Generating a Train of Attosecond Pulses. Light Sci Appl (2013) 2:e58. doi:10.1038/lsa.2013.14
37. Chipperfield LE, Robinson JS, Tisch JWG, Marangos JP. Ideal Waveform to Generate the Maximum Possible Electron Recollision Energy for Any Given Oscillation Period. Phys Rev Lett (2009) 102:063003. doi:10.1103/PhysRevLett.102.063003
38. Wei P, Miao J, Zeng Z, Li C, Ge X, Li R, et al. Selective Enhancement of a Single Harmonic Emission in a Driving Laser Field with Subcycle Waveform Control. Phys Rev Lett (2013) 110:233903. doi:10.1103/PhysRevLett.110.233903
39. Haessler S, Balčiunas T, Fan G, Andriukaitis G, Pugžlys A, Baltuška A, et al. Optimization of Quantum Trajectories Driven by Strong-Field Waveforms. Phys Rev X (2014) 4:021028. doi:10.1103/PhysRevX.4.021028
40. Ohmura H, Saito N. Quantum Control of a Molecular Ionization Process by Using Fourier-Synthesized Laser Fields. Phys Rev A (2015) 92:053408. doi:10.1103/PhysRevA.92.053408
41. Yoshida T, Saito N, Ohmura H. Robust Generation of Fourier-Synthesized Laser Fields and Their Estimation of the Optical Phase by Using Quantum Control of Molecular Tunneling Ionization. J Phys B: Mol Opt Phys (2018) 51:065601. doi:10.1088/1361-6455/aaac20
42. Ohmura H, Yoshida T, Saito N. Four-mode Multi-Selection in the Dual Phase Control of a Molecular Ionization Induced by Fourier-Synthesized Laser Fields. Appl Phys Lett (2019) 114:054101. doi:10.1063/1.5082578
43. Ohmura H, Saito N. Sub-Optical-Cycle Attosecond Control of Molecular Ionization by Using Fourier-Synthesized Laser Fields. Phys Rev A (2020) 101:043419. doi:10.1103/PhysRevA.101.043419
44. Paulus GG, Lindner F, Walther H, Baltuška A, Goulielmakis E, Lezius M, et al. Measurement of the Phase of Few-Cycle Laser Pulses. Phys Rev Lett (2003) 91:253004. doi:10.1103/PhysRevLett.91.253004
45. Apolonski A, Poppe A, Tempea G, Spielmann C, Udem T, Holzwarth R, et al. Controlling the Phase Evolution of Few-Cycle Light Pulses. Phys Rev Lett (2000) 85:740–3. doi:10.1103/PhysRevLett.85.740
46. Jones DJ, Diddams SA, Ranka JK, Stentz A, Windeler RS, Hall JL, et al. Carrier-Envelope Phase Control of Femtosecond Mode-Locked Lasers and Direct Optical Frequency Synthesis. Science (2000) 288:635–9. doi:10.1126/science.288.5466.635
47. Baltuska A, Fuji T, Kobayashi T. Controlling the Carrier-Envelope Phase of Ultrashort Light Pulses with Optical Parametric Amplifiers. Phys Rev Lett (2002) 88:133901. doi:10.1103/PhysRevLett.88.13390110.1103
48. Kakehata M, Takada H, Kobayashi Y, Torizuka K, Takamiya H, Nishijima K, et al. Carrier-Envelope-Phase Stabilized Chirped-Pulse Amplification System Scalable to Higher Pulse Energies. Opt Express (2004) 12:2070–80. doi:10.1364/OPEX.12.002070
49. Baltuška A, Udem T, Uiberacker M, Hentschel M, Goulielmakis E, Gohle C, et al. Attosecond Control of Electronic Processes by Intense Light Fields. Nature (2003) 421:611–5. doi:10.1038/nature01414
50. Krausz F, Ivanov M. Attosecond Physics. Rev Mod Phys (2009) 81: 163–234. doi:10.1103/RevModPhys.81.163
51. Joachain CJ, Kylstra NJ, Potvliege RM. Atoms in Intense Laser Fields. Cambridge: Cambridge University Press (2011). doi:10.1017/CBO9780511993459
52. Nisoli M, Decleva P, Calegari F, Palacios A, Martín F. Attosecond Electron Dynamics in Molecules. Chem Rev (2017) 117: 10760–825. doi:10.1021/acs.chemrev.7b00226
53. Schiffrin A, Paasch-Colberg T, Karpowicz N, Apalkov V, Gerster D, Mühlbrandt S, et al. Optical-Field-Induced Current in Dielectrics. Nature (2013) 493: 70–4. doi:10.1038/nature11567
54. Vasa P, Mathur D. Ultrafast Biophotonics. Cham, Switzerland: Springer (2016). doi:10.1007/978-3-319-39614-9
55. Keldysh LV. Ionization in the Field of a Strong Electromagnetic Wave. Sov Phys JETP (1964) 20: 1307–14.
56. Wolkov DM. Über eine Klasse von Lösungen der Diracschen Gleichung. Z Phys (1935) 94: 250–60. doi:10.1007/BF01331022
57. Perelemov AM, Popov VS, Terent'ev MV. Ionization of Atoms in an Alternating Electric Filed. Soviet Phys JETP (1966) 23: 924–34.
58. Faisal FHM. Multiple Absorption of Laser Photons by Atoms. J Phys B: Mol Phys (1973) 6:L89–L92. doi:10.1088/0022-3700/6/4/011
59. Reiss HR. Effect of an Intense Electromagnetic Field on a Weakly Bound System. Phys Rev A (1980) 22: 1786–813. doi:10.1103/PhysRevA.22.1786
60. Ammosov MV, Delone NB, Krainov VP. Tunnel Ionization of Complex Atoms and of Atomic Ions in an Alternating Electromagnetic Field. Soviet Phys JETP (1986) 64: 1191–4.
61. Tong XM, Zhao Z, Lin CD. Theory of Molecular Tunneling Ionization. Phys Rev A (2002) 66:033402. doi:10.1103/PhysRevA.66.033402
62. Abu-samha M, Madsen LB. Single-Active-Electron Potentials for Molecules in Intense Laser Fields. Phys Rev A (2010) 81:033416. doi:10.1103/PhysRevA.81.033416
63. Awasthi M, Saenz A. Breakdown of the Single-Active-Electron Approximation for One-Photon Ionization of the B1Σu+ State of H2 Exposed to Intense Laser Fields. Phys Rev A (2010) 81:063406. doi:10.1103/PhysRevA.81.063406
64. Zhang B, Yuan J, Zhao Z. Dynamic Core Polarization in Strong-Field Ionization of CO Molecules. Phys Rev Lett (2013) 111:163001. doi:10.1103/PhysRevLett.111.163001
65. Abu-samha M, Madsen LB. Multielectron Effects in Strong-Field Ionization of the Oriented OCS Molecule. Phys Rev A (2020) 102:013433. doi:10.1103/PhysRevA.102.063111
66. Zhao Z, Brabec T. Tunnel Ionization in Complex Systems. J Mod Opt (2007) 54: 981–97. doi:10.1080/09500340601043413
67. Hoang V-H, Zhao S-F, Le V-H, Le A-T. Influence of Permanent Dipole and Dynamic Core-Electron Polarization on Tunneling Ionization of Polar Molecules. Phys Rev A (2017) 95:023407. doi:10.1103/physreva.95.023407
68. Kang H-P, Xu S-P, Wang Y-L, Yu S-G, Zhao X-Y, Hao X-L, et al. Polarization Effects in Above-Threshold Ionization with a Mid-infrared Strong Laser Field. J Phys B: Mol Opt Phys (2018) 51:105601. doi:10.1088/1361-6455/aabce0
69. Kling MF, Vrakking MJJ. Attosecond Electron Dynamics. Annu Rev Phys Chem (2008) 59: 463–92. doi:10.1146/annurev.physchem.59.032607.093532
70. Vrakking MJJ, Lepine F. Attosecond Molecular Dynamics. London: Royal Society of Chemistry (2018). doi:10.1039/9781788012669
71. Harumiya K, Kawata I, Kono H, Fujimura Y. Exact Two-Electron Wave Packet Dynamics of H2 in an Intense Laser Field: Formation of Localized Ionic States H+H−. J Chem Phys (2000) 113: 8953–60. doi:10.1063/1.1319348
72. Harumiya K, Kono H, Fujimura Y, Kawata I, Bandrauk AD. Intense Laser-Field Ionization of H2 Enhanced by Two-Electron Dynamics. Phys Rev A (2002) 66:043403. doi:10.1103/PhysRevA.66.043403
73. Smirnova O, Mairesse Y, Patchkovskii S, Dudovich N, Villeneuve D, Corkum P, et al. High Harmonic Interferometry of Multi-Electron Dynamics in Molecules. Nature (2009) 460: 972–7. doi:10.1038/nature08253
74. Zanghellini J, Kitzler M, Fabian C, Brabec SA. An MCTDHF Approach to Multielectron Dynamics in Laser Fields. Laser Phys (2003) 13: 1064–8.
75. Kato T, Kono H. Time-Dependent Multiconfiguration Theory for Electronic Dynamics of Molecules in an Intense Laser Field. Chem Phys Lett (2004) 392: 533–40. doi:10.1016/j.cplett.2004.05.106
76. Kato T, Kono H. Time-Dependent Multiconfiguration Theory for Electronic Dynamics of Molecules in Intense Laser Fields: A Description in Terms of Numerical Orbital Functions. J Chem Phys (2008) 128:184102. doi:10.1063/1.2912066
77. Nest M, Klamroth T. Correlated Many-electron Dynamics: Application to Inelastic Electron Scattering at a Metal Film. Phys Rev A (2005) 72:012710. doi:10.1103/PhysRevA.72.012710
78. Jordan G, Caillat J, Ede C, Scrinzi A. Strong Field Ionization of Linear Molecules: a Correlated Three-Dimensional Calculation. J Phys B: Mol Opt Phys (2006) 39:S341–S347. doi:10.1088/0953-4075/39/13/S07
79. Nest M, Padmanaban R, Saalfrank P. Time-Dependent Approach to Electronically Excited States of Molecules with the Multiconfiguration Time-Dependent Hartree-Fock Method. J Chem Phys (2007) 126:214106. doi:10.1063/1.2743007
80. Sukiasyan S, McDonald C, Destefani C, Ivanov MY, Brabec T. Multielectron Correlation in High-Harmonic Generation: A 2D Model Analysis. Phys Rev Lett (2009) 102:223002. doi:10.1103/PhysRevLett.102.223002
81. Nguyen-Dang T-T, Peters M, Wang S-M, Dion F. Toward Ab-Initio Simulations of Multiple Ionization Processes in Intense Laser Field. Chem Phys (2009) 366: 71–84. doi:10.1016/j.chemphys.2009.09.007
82. Redkin P, Ganeev R. Simulation of Resonant High-Order Harmonic Generation in a Three-Dimensional Fullerenelike System by Means of a Multiconfigurational Time-Dependent Hartree-Fock Approach. Phys Rev A (2010) 81:063825.
83. Hochstuhl D, Bauch S, Bonitz M. Multiconfigurational Time-Dependent Hartree-Fock Calculations for Photoionization of One-Dimensional Helium. J Phys Conf Ser (2010) 220(10):012019. doi:10.1088/1742-6596/220/1/012019
84. Dirac PAM. The Principles of Quantum Mechanics, International Series of Monographs on Physics. 4th ed. 27. Oxford: Oxford Science (1958).
86. Kato T, Kono H. Time-Dependent Multiconfiguration Theory for Ultrafast Electronic Dynamics of Molecules in an Intense Laser Field: Electron Correlation and Energy Redistribution Among Natural Orbitals. Chem Phys (2009) 366: 46–53. doi:10.1016/j.chemphys.2009.09.017
87. Ohmura S, Kono H, Oyamada T, Kato T, Nakai K, Koseki S. Characterization of Multielectron Dynamics in Molecules: A Multiconfiguration Time-Dependent Hartree-Fock Picture. J Chem Phys (2014) 141:114105. doi:10.1063/1.4894505
88. Ohmura S, Kato T, Oyamada T, Koseki S, Ohmura H, Kono H. A Single-Electron Picture Based on the Multiconfiguration Time-Dependent Hartree-Fock Method: Application to the Anisotropic Ionization and Subsequent High-Harmonic Generation of the CO Molecule. J Phys B: Mol Opt Phys (2018) 51:034001. doi:10.1088/1361-6455/aa9e45
89. Kato T, Yamanouchi K, Kono H. Attosecond Molecular Dynamics. London: Royal Society of Chemistry (2018). p. 139–81. doi:10.1039/9781788012669
90. Ohmura S, Kato T, Ohmura H, Koseki S, Kono H. Analysis of the Multielectron Dynamics in Intense Laser-Induced Ionization of CO by the Time-Dependent Effective Potentials for Natural Orbitals. J Phys B: Mol Opt Phys (2020) 53:184001. doi:10.1088/1361-6455/ab9f0e
91. Löwdin P-O. Quantum Theory of Many-particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Phys Rev (1955) 97: 1474–89. doi:10.1103/PhysRev.97.1474
92. Kraus PM, Baykusheva D, Wörner HJ. Two-pulse Orientation Dynamics and High-Harmonic Spectroscopy of Strongly-Oriented Molecules. J Phys B: Mol Opt Phys (2014) 47:124030. doi:10.1088/0953-4075/47/12/124030
93. Holmegaard L, Hansen JL, Kalhøj L, Louise Kragh S, Stapelfeldt H, Filsinger F, et al. Photoelectron Angular Distributions from Strong-Field Ionization of Oriented Molecules. Nat Phys. (2010) 6: 428–32. doi:10.1038/nphys1666
94. Majety VP, Scrinzi A. Static Field Ionization Rates for Multi-Electron Atoms and Small Molecules. J Phys B: Mol Opt Phys (2015) 48:245603. doi:10.1088/0953-4075/48/24/245603
95. SakemiMinemoto YS, Minemoto S, Sakai H. Orientation Dependence in Multichannel Dissociative Ionization of OCS Molecules. Phys Rev A (2017) 96:011401. doi:10.1103/PhysRevA.96.011401
96. Johansen R, Bay KG, Christensen L, Thøgersen J, Dimitrovski D, Madsen LB, et al. Alignment-dependent Strong-Field Ionization Yields of Carbonyl Sulfide Molecules Induced by Mid-infrared Laser Pulses. J Phys B: Mol Opt Phys (2016) 49:205601. doi:10.1088/0953-4075/49/20/2056010.1088/0953-4075/49/20/205601
97. Sándor P, Sissay A, Mauger F, Abanador PM, Gorman TT, Scarborough TD, et al. Angle Dependence of Strong-Field Single and Double Ionization of Carbonyl Sulfide. Phys Rev A (2018) 98:043425. doi:10.1103/PhysRevA.98.043425
98. Madsen LB, Jensen F, Tolstikhin OI, Morishita T. Structure Factors for Tunneling Ionization Rates of Molecules. Phys Rev A (2013) 87:013406. doi:10.1103/PhysRevA.87.013406
99. Śpiewanowski MD, Madsen LB. Alignment- and Orientation-dependent Strong-Field Ionization of Molecules: Field-Induced Orbital Distortion Effects. Phys Rev A (2015) 91:043406. doi:10.1103/PhysRevA.91.043406
100. Dimitrovski D, Martiny CPJ, Madsen LB. Strong-field Ionization of Polar Molecules: Stark-Shift-Corrected Strong-Field Approximation. Phys Rev A (2010) 82:053404. doi:10.1103/PhysRevA.82.053404
101. Wu J, Schmidt LPH, Kunitski M, Meckel M, Voss S, Sann H, et al. Multiorbital Tunneling Ionization of the CO Molecule. Phys Rev Lett (2012) 108:183001. doi:10.1103/PhysRevLett.108.183001
102. Akagi H, Otobe T, Itakura R. Deformation of an Inner Valence Molecular Orbital in Ethanol by an Intense Laser Field. Sci Adv (2019) 5:eaaw1885. doi:10.1126/sciadv.aaw1885
103. Le C-T, Hoang V-H, Tran L-P, Le V-H. Effect of the Dynamic Core-Electron Polarization of CO Molecules on High-Order Harmonic Generation. Phys Rev A (2018) 97:043405. doi:10.1103/PhysRevA.97.043405
104. Zhang B, Yuan J, Zhao Z. Dynamic Orbitals in High-Order Harmonic Generation from CO Molecules. Phys Rev A (2014) 90:035402. doi:10.1103/PhysRevA.90.035402
105. Pernal K, Gritsenko O, Baerends EJ. Time-Dependent Density-Matrix-Functional Theory. Phys Rev A (2007) 75:012506. doi:10.1103/PhysRevA.75.012506
106. Benavides-Rivero CL, Marques MAL. On the Time Evolution of Fermionic Occupation Numbers. J Chem Phys (2019) 151:044112. doi:10.1063/1.5109009
107. Sato T, Ishikawa KL. Time-Dependent Complete-Active-Space Self-Consistent-Field Method for Multielectron Dynamics in Intense Laser Fields. Phys Rev A (2013) 88:023402. doi:10.1103/PhysRevA.88.023402
108. Miyagi H, Madsen LB. Time-Dependent Restricted-Active-Space Self-Consistent-Field Theory for Laser-Driven Many-electron Dynamics. Phys Rev A (2013) 87:062511. doi:10.1103/PhysRevA.87.062511
109. Lode AUJ, Lévêque C, Madsen LB. Colloquium: Multiconfigurational Time-Dependent Hartree Approaches for Indistinguishable Particles. Rev Mod Phys (2020) 92:011001. doi:10.1103/RevModPhys.92.011001
110. Nielsen ES, Jo/rgensen P, Oddershede J. Transition Moments and Dynamic Polarizabilities in a Second Order Polarization Propagator Approach. J Chem Phys (1980) 73: 6238–46. doi:10.1063/1.440119
111. Uiberacker M, Uphues T, Schultze M, Verhoef AJ, Yakovlev V, Kling MF, et al. Attosecond Real-Time Observation of Electron Tunnelling in Atoms. Nature (2007) 446: 627–32. doi:10.1038/nature05648
112. Kamta GL, Bandrauk ADAD. Phase Dependence of Enhanced Ionization in Asymmetric Molecules. Phys Rev Lett (2005) 94:203003. doi:10.1103/PhysRevLett.94.203003
113. Bian X-B, Bandrauk AD. Nonadiabatic Molecular High-Order Harmonic Generation from Polar Molecules: Spectral Redshift. Phys Rev A (2011) 83(R):041403. doi:10.1103/PhysRevA.83.041403
114. Bian X-B, Bandrauk AD. Multichannel Molecular High-Order Harmonic Generation from Asymmetric Diatomic Molecules. Phys Rev Lett (2010) 105:093903. doi:10.1103/PhysRevLett.105.093903
115. Etches A, Madsen LB. Extending the Strong-Field Approximation of High-Order Harmonic Generation to Polar Molecules: Gating Mechanisms and Extension of the Harmonic Cutoff. J Phys B: Mol Opt Phys (2010) 43:155602. doi:10.1088/0953-4075/43/15/155602
116. Strelkov V. Role of Autoionizing State in Resonant High-Order Harmonic Generation and Attosecond Pulse Production. Phys Rev Lett (2010) 104:123901. doi:10.1103/PhysRevLett.104.123901
117. Kono H, Sato Y, Kanno M, Nakai K, Kato T. Theoretical Investigations of the Electronic and Nuclear Dynamics of Molecules in Intense Laser Fields: Quantum Mechanical Wave Packet Approaches. Bcsj (2006) 79:196–227. doi:10.1246/bcsj.79.196
118. Kübel M, Spanner M, Dube Z, Naumov AY, Chelkowski S, Bandrauk AD, et al. Probing Multiphoton Light-Induced Molecular Potentials. Nat Commun (2020) 11:2596. doi:10.1038/s41467-020-16422-2
119. Kato T, Yamanouchi K. Time-Dependent Multiconfiguration Theory for Describing Molecular Dynamics in Diatomic-like Molecules. J Chem Phys (2009) 131:164118. doi:10.1063/1.3249967
120. Ide Y, Kato T, Yamanouchi K. Non-Born-Oppenheimer Molecular Wave Functions of H2 by Extended Multi-Configuration Time-Dependent Hartree-Fock Method. Chem Phys Lett (2014) 595-596:180–4. doi:10.1016/j.cplett.2014.01.055
121. Kato T, Ide Y, Yamanouchi K. Molecular Wave Function and Effective Adiabatic Potentials Calculated by Extended Multi-Configuration Time-Dependent Hartree-Fock Method. AIP Conf Proceed (2015) 1702:090024. doi:10.1063/1.4938832
122. Azumi T, Matsuzaki K. What Does the Term “Vibronic Coupling” Mean? Photochem Photobiol (1977) 25:315–26. doi:10.1111/j.1751-1097.1977.tb06918.x
Keywords: muliticonfiguration time-dependent Hartree-Fock theory, natural orbital dynamics, time-dependent effective potential, multielectron dynamics, tunnel ionization, intense laser pulse, multicolor superposition
Citation: Ohmura S, Ohmura H, Kato T and Kono H (2021) Manipulation of Multielectron Dynamics of Molecules by Fourier-Synthesized Intense Laser Pulses: Effective Potential Analysis of CO. Front. Phys. 9:677671. doi: 10.3389/fphy.2021.677671
Received: 08 March 2021; Accepted: 28 May 2021;
Published: 08 July 2021.
Edited by:
Robert Gordon, University of Illinois at Chicago, United StatesReviewed by:
Andre Bandrauk, Université de Sherbrooke, CanadaArkaprabha Konar, Kent State University, United States
Copyright © 2021 Ohmura, Ohmura, Kato and Kono. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hirohiko Kono, hirohiko.kono.d6@tohoku.ac.jp