 Phuong Mai Dinh
Phuong Mai Dinh Marc Vincendon1
Marc Vincendon1 Eric Suraud
Eric Suraud Paul-Gerhard Reinhard
Paul-Gerhard Reinhard
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Phys. , 19 February 2020
Sec. Nuclear Physics
Volume 8 - 2020 | https://doi.org/10.3389/fphy.2020.00027
This article is part of the Research Topic Advances in Time-Dependent Methods for Nuclear Structure and Dynamics View all 16 articles
This brief review illustrates on a few typical applications fully fledged dynamical simulations of finite electronic systems (atoms, molecules, clusters) using time-dependent density-functional theory (TDDFT). It concentrates on aspects which are different from nuclear applications. These are: the correct handling of electron emission, the self-interaction correction, the enormous versatility of laser excitation to probe systems properties, and with it the exploitation of detailed observables of electron emission as photo-electron angular distributions and photo-electron spectra (PES). Finally, we demonstrate the impact of electronic dissipation putting question marks on the reliability of TDDFT simulations over long times.
Far-off equilibrium dynamics in quantum many-body systems is since numerous decades a challenging task much studied in experimental and theoretical investigations. In the realm of finite electronic systems, the availability of versatile laser pulses has given way to detailed analysis of the response of clusters and molecules [1–11]. In particular it allows one to explore the properties of electrons emitted after irradiation in correlation with the laser pulse. This has led to detailed analysis of angular- and energy-resolved electron spectra, e.g., their angular distributions and photoelectron spectra (PES) or both simultaneously as angular resolved PES (ARPES) recorded via Velocity Map Imaging techniques [12]. The theoretical description of such far-off equilibrium situations is also demanding. It requires large phase spaces and must cover vastly different time scales ranging from basic processes of excitation over collisional redistribution with subsequent relaxation to ionic motion and possible coupling to environment. They thus require dedicated fully time-dependent approaches, such as the widely used time-dependent density functional theory (TDDFT), often combined with molecular dynamics for the description of ionic motion (see e.g., [13]). This approach is both versatile and robust and allows one to simulate numerous dynamical scenarios with high degree of accuracy. The electronic part, TDDFT, is formally exactly the same as time-dependent Hartree-Fock (TDHF) in the nuclear domain [14]. The difference lies mainly in energy functionals which are constructed by very different strategies, see Dreizler and Gross [15] for electronic systems and Bender et al. [16] for nuclei.
In a typical laser irradiation, electrons immediately react to the external electro-magnetic field driving the system quickly far-off equilibrium long before ionic motion and thermal relaxation processes set on [17]. Thus, the system fully remains in the quantum regime for quite a while, as seen in recent experiments, exhibiting clear quantum and thermal pattern in photoelectron spectra [18–20]. The crucial first stage, i.e., the doorway to any photo-reaction, is predominantly quantum-mechanical electron dynamics which is (as in nuclear dynamics) well-described by TDDFT and we shall focus on this domain below.
Actual implementations of TDDFT are often based on the Time-Dependent Local Density Approximation (TDLDA) [8, 21, 22] which usually performs well at moderate excitation. However, TDLDA is plagued by a self-interaction error [23] which arises because the local approximation of Coulomb exchange spoils the subtle balance with the direct Coulomb term (which was still maintained in full Hartree-Fock calculations). This is particularly disastrous for simulation of emission properties because the ionization potentials in terms of the energy of the highest occupied single-electron orbital are underestimated. The problem is cured in ad hoc manner by augmenting TDLDA with a self-interaction correction (SIC). As this is an aspect which is less important in the nuclear domain and thus ignored, we will address it below. Another aspect which is missing in TDLDA and which becomes important in later stages of the dynamical evolution are dynamical correlations and associated dissipative features. Extensions beyond TDLDA which take dissipation into account are presently under development. We shall address also this question on one example.
The paper is organized as follows. Section 2 provides a brief summary of the formalism, TDDFT in real time with SIC and computation of observables, particularly ionization and PES. Section 3 presents a few selected application examples starting with the impact of SIC and basic features of optical responses as the latter plays a crucial role as doorway to any dynamical scenario. We then illustrate the capabilities of short laser pulses and finally discuss the impact of dissipation.
As in nuclear TDHF, the state of the electronic system is described in terms of single-particle (s.p.) Kohn-Sham (KS) wave functions φn(n = 1, …, N) from which the key ingredient in DFT, the total electronic density, is built as . The total energy is composed as Etot = Ekin + EH + Exc + Eion + Eext. The first three terms, namely kinetic energy, Coulomb Hartree energy, and exchange-correlation energy, constitute the purely electronic part and correspond to typical nuclear energy expressions. The last two terms, the ionic energy with its coupling to electrons and a possible external (laser) field, are specific to atoms/molecules. Hartree and exchange-correlation energies are approximated as functionals of ϱ: EH + Exc ≃ ETDLDA[ϱ(r, t)]. This constitutes in the dynamic domain the Time-Dependent Adiabatic Local Density Approximation (TDLDA) [15, 24, 25], the electronic analog of nuclear TDHF. Variation with respect to yields the KS equations:
The LDA potential is obtained as a functional derivative with respect to the local density : . The ionic background vion is described by pseudopotentials [26]. We use the particularly simple and efficient soft local [27] or Goedecker-type [28] pseudopotentials.
An external, coherent laser field is handled as a classical field in the long wavelength limit, adding to the mean-field Hamiltonian the potential
θ being the Heaviside function. The laser characteristics are: linear polarization (here denoted by ez), peak field strength E0 related to laser intensity (), photon frequency ℏωlas, and total pulse length Tpulse. The full width at half maximum of intensity (FWHM) is given as FWHM ≃Tpulse/3. The parameter ϕCEP, the so-called carrier-envelop phase, is the phase between the maximum of an oscillation at frequency ℏωlas and the maximum of the cos2 envelope which plays a role particularly for short pulses [29].
The KS equations introduce a self-interaction (SI) error because KS mean field vLDA employs the total density which includes also the electron on which vLDA acts. This is particularly disastrous for the long-range Coulomb term which produces thus a shifted single particle energy spectrum and, as a consequence, a wrong ionization potential (IP). To overcome the SI error, the energy-density functional is augmented by a SI correction (SIC) [30]
The price to pay is to deal with a Hamiltonian that is now non-Hermitian and state-dependent, since the variation with respect to of the SI-corrected energy explicitly produces a functional of the s.p. density ϱn instead of the total electronic density (for a broad overview of orbital-dependent functionals, see Kümmel and Kronik [31]). This difficulty is particularly severe in the time domain and requires elaborate strategies to maintain unitary time evolution [32, 33].
Still, a full solution of dynamical SIC is expensive. Fortunately, there are many situations where one can employ a drastic simplification, namely the average density SIC (ADSIC) which was proposed already in the 1930s [34], and applied since then in clusters [35]. The idea is to assume that all electrons fill about the same region of space and thus contribute about equally to the SI error. This amounts to replace Equation (3) by
where ϱσ and Nσ are the total electronic density and the total number of particles of spin σ, respectively. This is, again, a functional of the local spin-density only and thus can be treated in the same manner as any LDA scheme. This works nicely in a wide class of compact atomic/molecular systems [36] and particularly well for metal clusters. It is not applicable to situations with fragmented electron density and to bulk systems (the latter because the particle numbers Nσ grow infinite).
The KS equations are solved in real time on a 3D grid with standard techniques [13, 17, 37]. The predominantly local structure of the KS Hamiltonian allows one to use the very efficient time-splitting scheme for time propagation [38]. The Coulomb field is computed with successive over-relaxation [39] or Fast Fourier Transform techniques [37, 40]. To describe ionization dynamics, we use absorbing boundary conditions [17], which, when properly optimized [41], gently remove outgoing electron flow at the boundaries of the numerical grid. This is a crucial ingredient which is so far not much used in nuclear dynamics. But it is essential in atoms/molecules where observables from electron emission (explained below) are very important.
The analysis of electron dynamics is performed through a set of well-established observables [10, 17, 42, 43] that we briefly recall here. As the laser field couples to the electronic dipole, dipole response is the most prominent observable in many studies [10, 13, 42, 44]. We compute the dipole moment of electrons (with respect to ionic background) from the electronic density as:
Spectral properties are then obtained from the time-frequency Fourier transform after unfolding the spectrum of the exciting pulse [44]. Spectral analysis becomes particularly simple for excitation by an instantaneous dipole boost. The dipole strength becomes then and the dipole power spectrum .
For sufficiently strong excitation, electron emission becomes important. It can be analyzed at various levels of sophistication thanks to the use of absorbing boundary conditions mentioned above. Simplest is the global measure, the total ionization which can be computed from the given density as
where N is the initial electron number.
More detailed observables are Photo-Electron Spectra (PES), obtained from measuring the distribution of kinetic energies of the emitted electrons, or their angular distribution. Measuring both simultaneously yields Angular-Resolved PES (ARPES) for which often valuable experimental data exist to compare with [43]. The strategy to compute PES consists in defining a set of “measuring points” near the absorbing boundaries, and to record the time evolution of s.p. wave functions at these points. The information thus gathered can be post-processed in two ways. Simple time integration of yields the amount of ionization at this measuring point. This together with properly accounting for the solid angle associated with the vicinity of the measuring point direction yields the angular distribution of emitted electrons [42, 45]. The PES is obtained by Fourier transforming the s.p. wave functions at the measuring point from time to frequency and identifying the kinetic energy as . This delivers the PES from s.p. state i for emission in direction of the measuring point . The total PES is then obtained by summing up the contributions from each s.p. state
This simple Fourier transform applies to weak and moderate fields. For stronger field one still needs a phase correction which is explained in detail in [46].
We illustrate the importance of SIC for TDDFT on the example of the Na5 cluster irradiated by a femtosecond (fs) laser pulse with FWHM of 10 fs, intensity I = 2.2 × 1011 W/cm2, and frequency ωlas = 10.9 eV, well above ionization threshold (ionization potential IP = 4.1 eV) and far away from dominant eigenfrequencies of the system. We use the simple ADSIC approximation (which is found to be appropriate for Na systems [35, 36]) and compare it to LDA. Figure 1 collects the results. The top right part of the figure shows the (planar) ionic configuration. The lower right panel compares the sequence of s.p. energies for LDA and ADSIC. The latter produces a global down-shift of the s.p. levels as compared to those from LDA (by 1.2 eV for the HOMO) due to the enhanced Coulomb attraction (self-interaction neutralizes the asymptotic Coulomb field). The IP can be checked independently by comparing with the difference of energies of Na5 and (Koopman's theorem) and ADSIC matches this energy difference perfectly while LDA fails badly. As a consequence, the s.p. spectrum from ADSIC carries the correct information on the ionization threshold.
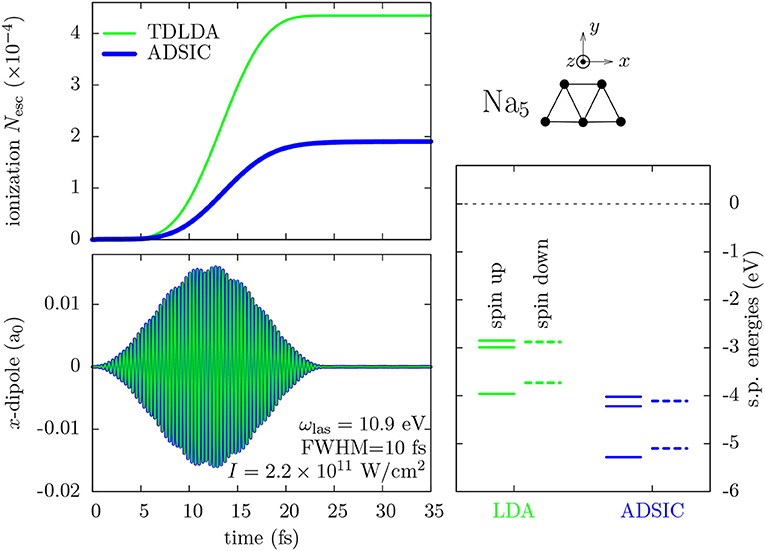
Figure 1. Comparison of (TD)LDA and ADSIC for the Na5 cluster. (Top right) Sketch of ionic configuration of Na5. (Bottom right) Single particle (s.p.) energies for spins up (full lines) and down (dashes). (Left) Time evolution of total ionization Nesc (top) and of the electronic dipole in the x direction (bottom) after irradiation by a laser pulse polarized along x with ωlas = 10.9 eV, Tpulse = 24 fs, and I = 2.2 × 1011 W/cm2.
The left panels show the dynamical response of the system to the laser pulse. The left lower panel displays the time evolution of the electronic dipole moment along laser polarization (x axis). There is almost no difference between LDA and ADSIC for the dipole signal. This is also found for the optical absorption strength (not shown here) which is no surprise because the optical absorption is another way to look at the dipole response. There is no urgent need to employ SIC when computing these quantities. This is plausible because the dipole signal is a global signal deduced from the density and density is, by construction, well-described in DFT.
The left upper panel shows the time evolution of the total ionization Nesc (see Equation 6). This signal reveals a dramatic difference between ADSIC and LDA. This is no surprise as ADSIC provides a larger IP than LDA (and altogether, more deeply bound levels, see bottom right panel). The reason is that, in TDDFT, ionization is mediated via the actual s.p. energies such that it is crucial to put the s.p. energies right within DFT.
Optical response is the most prominent observable for dynamical properties of a system. Its role in nuclear physics and in molecular or cluster physics is similarly important. The conceptually simplest way is to compute it by spectral analysis of a time-dependent simulation (see section 2.3). Traditionally, it has mostly been computed by linearized TDDFT which complicates coding, but simplifies calculations, particularly if a system has certain symmetries (see e.g., [47–49]). The pathway through spectral analysis is competitive in fully 3D calculations and it is more flexible as it also allows one to explore the transition to the non-linear regime [50]. We use that here.
Figure 2 shows optical absorption strengths for a nucleus, 208Pb, and three different electronic systems of different bond types, a metal cluster Na40, a covalent molecule H2O, and the noble gas Ar atom. The three electronic systems have significantly different spectra. For Ar and H2O, the strength is heavily fragmented over many dipole states, most of them having predominantly the structure of one-particle-one-hole excitations. Quite different is the metallic Na40, with one dominating and almost exhaustive peak, the highly collective Mie plasmon [54, 55]. The nuclear spectrum looks very similar to the cluster case. It is dominated by the a strong collective mode, the giant dipole resonance [56]. The similarity is not surprising as both, the metallic electron cloud and the nucleons, have the same bulk limit, namely a Fermi liquid [57].
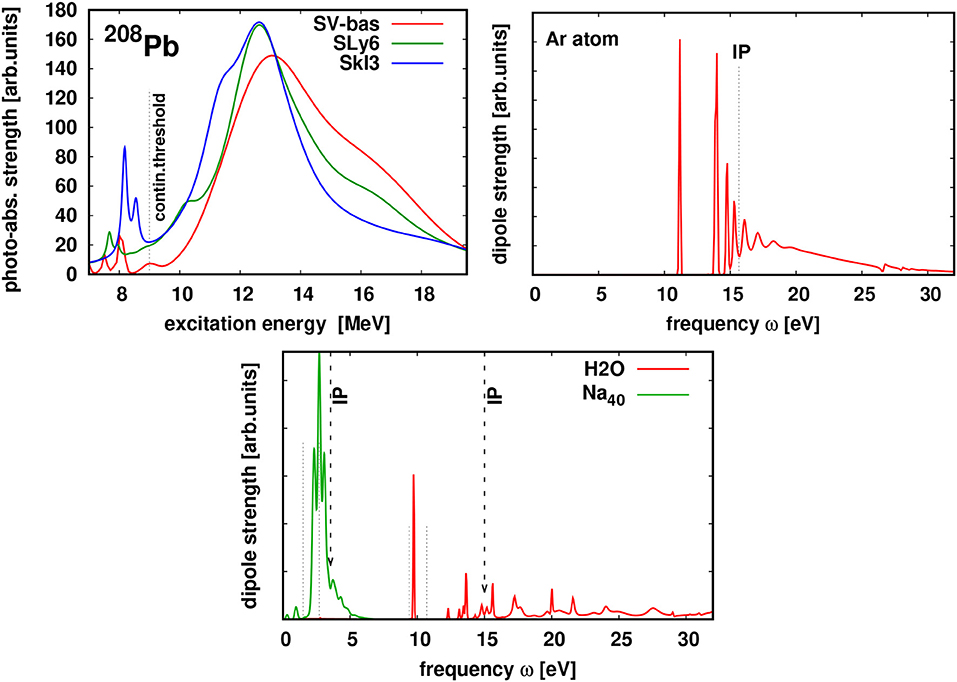
Figure 2. Optical absorption spectra for three electronic systems as indicated and the nucleus 208Pb. The electronic systems are computed with TDLDA + ADSIC as explained in section 2. The nucleus is computed with nuclear TDHF using the code of [37] (for details see Reinhard et al. [50]) and three different Skyrme parameterizations as indicated, SkI3 from [51], SLy6 from [52], and SV-bas from [53]. The vertical dashed lines indicate the particle continuum thresholds in each system, the electronic ionization potential (IP) and the neutron threshold in the nuclear case.
The nuclear dipole resonance and the cluster's plasmon resonance both have some width. However, the mechanisms producing these widths is different. The nuclear dipole resonance lies in the nucleon continuum and a large part of the width stems from limited lifetime due to nucleon emission. The remaining part is due to dissipative processes from nucleon-nucleon collisions. Both processes also play some role in metal clusters. But here the dominant broadening mechanism is given by thermal fluctuations of the underlying ionic configuration [58, 59].
Laser pulses offer a unique and extremely versatile tool for dedicated probing and switching of electronic systems. Coherent pulses in keV and MeV regime, as required for nuclear experiments, appear at the horizon [60–62], but deliver not yet sufficient field strengths to attain the most interesting multi-photon regime. This day will come and thus it is worth having a look at electronic applications and see what experiments with coherent pulses can reveal.
A great deal of information is gained when combining laser excitations with detailed analysis of the emitted electrons. The top panels of Figure 3 show as an example ARPES produced from the Ar atom (left) and the cluster (right) after irradiation by a few-cycle laser pulse. Cuts along a fixed angle represent the PES and cuts along a fixed kinetic energy the angular distribution. The PES decrease with increasing Ekin because more photons have to cooperate to supply the higher emitted energy. And yet, one finds a plateau around 20 eV which is typical for above-threshold ionization spectra [63, 64]. The two different systems deliver different patterns, demonstrating that system properties form the signal which, in turn, can be used to analyze a system by ARPES studies.
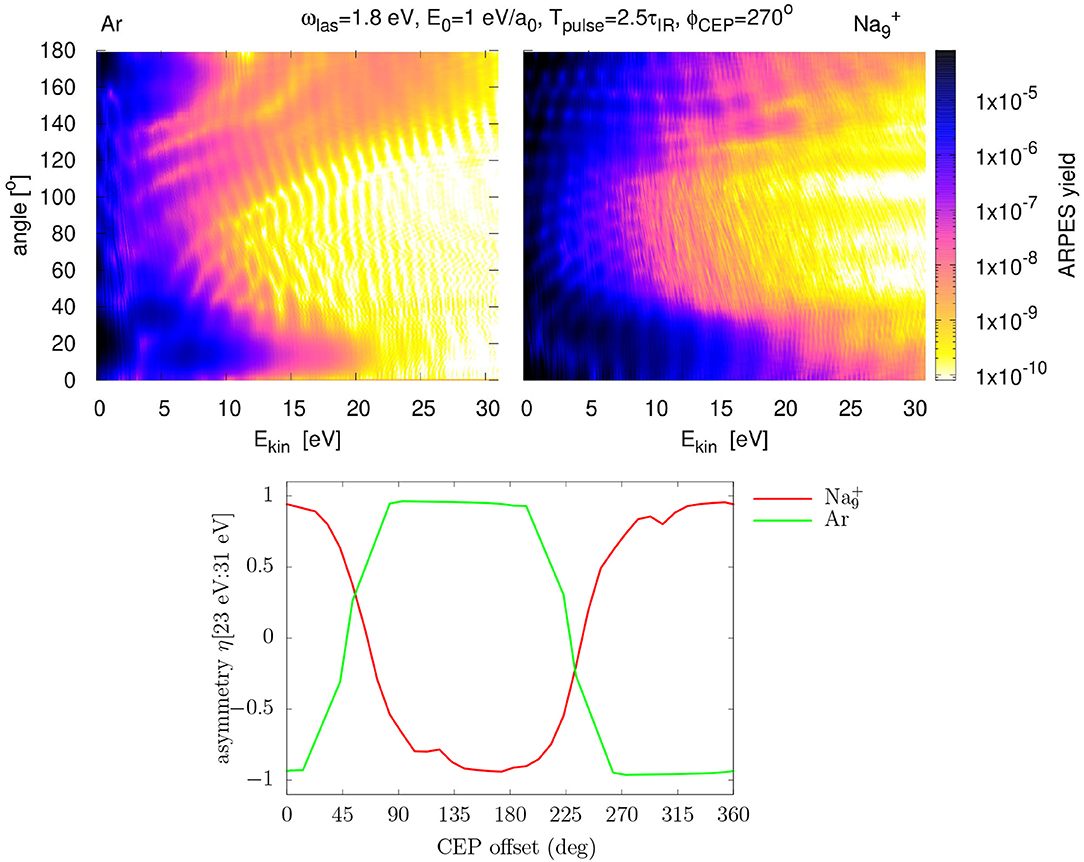
Figure 3. Ar atom and cluster excited by a laser with ωlas = 1.8 eV, I = 6 × 1013 W/cm2, pulse length Tpulse = 6 fs (corresponding to 2.5 optical cycles), and . Upper panels: ARPES yield in the plane of kinetic energy Ekin and emission angle θ, for Ar (left) and (right). Lower panel: asymmetry parameter η(Ekin) integrated on the Ekin interval [23eV:31eV].
One of the interesting features in ARPES from very short pulses is the asymmetry of the angular distributions which appears in various energy bins for both systems. Indeed, in such laser pulses, the carrier-envelop phase ϕCEP, see Equation (2), becomes a decisive laser parameter because the CEP controls the net momentum exerted on the electron cloud and so impacts the pattern of the pulse dramatically which, in turn, can have a strong impact on laser-induced electron dynamics. For example, photo-electron emission induced by few-cycle laser fields can be controlled by the CEP, leading to a pronounced forward-backward (also called “right-left”) asymmetry in the PES. We can quantify this asymmetry by a simple number as
where is the ARPES strength at given kinetic energy and emission angle. For the opening angle in the integration, we take Θ = 15° in accordance with many experiments in that field (see e.g., [65]). A value of η = +1 indicates prevailing forward emission and η = −1 backward dominance. Condensing the angular distribution into one compact number η allows one to visualize trends with laser parameter. The lower panel of Figure 3 shows an energy-integrated asymmetry η as a function of ϕCEP. The effects are impressive. Tuning ϕCEP allows one to switch emission from forward to backward and vice versa. And we also see a significant system dependence of the trend. The marked difference between both systems can be explained by the difference in their optical response displayed in Figure 2. The 1.8 eV laser frequency lies far below any considerable dipole strength for Ar while it comes close to the strong Mie plasmon resonance for the Na cluster. For a detailed discussion with more material see Reinhard et al. [29].
So far, we have demonstrated the use of TDDFT in electronic systems on a few selected examples. TDDFT has shown to be a robust and versatile tool for simulating dynamical processes. However, the more energy comes into a system, the more likely come dynamical correlations beyond TDDFT into play. This is also well-known in nuclear physics and a variety of methods has been developed to deal with those correlations. They employ all semi-classical concepts and most of them are based on the Vlasov-Ühling-Uhlenbeck collision term [66] (for reviews see [67, 68]). This has also been applied to clusters [10, 69, 70]. But laser excitation processes sail often for a long initial time span through the quantum regime. There is thus a need for a quantum description of dynamical correlations from electron-electron collisions and associated dissipation. Just recently, there were promising moves to develop such approaches, one along the line of stochastic jumps (comparable to the collisions in semi-classical methods) [71–73] and another one with the more empirical relaxation-time approximation (RTA) [74, 75]. Both approaches are based on two-body collisions implying the in-medium electron-electron interaction and take care of the available phase space of final states. RTA, for which we will show an example here, maps cross-section and phase space into a collision rate with which the system relaxes toward local-instantaneous equilibrium, the latter being evaluated at each relaxation step by density- and current-constrained minimization of the free energy. By construction, RTA maintains the continuity equation and energy conservation.
Figure 4 exemplifies the consequences of the dissipation thus introduced with an application of RTA to resonant laser excitation of a Na40 cluster. The lower left panel shows the effect on the dipole signal. The additional damping introduced by RTA is clearly visible. It is interesting to note that the TDLDA signal is also damped, although at lower rate. This damping stems from electron emission which is the dominant cooling mechanism in TDLDA. This is corroborated by the time evolution of ionization shown in the lower right panel. The trends agree up to about 100 fs, the same span where the dipole signals agree. But then the RTA curve levels off whereas TDLDA continues to emit. That happens because RTA has meanwhile converted much of the initial excitation energy into internal heat and so distracted it from moving into direct electron emission.
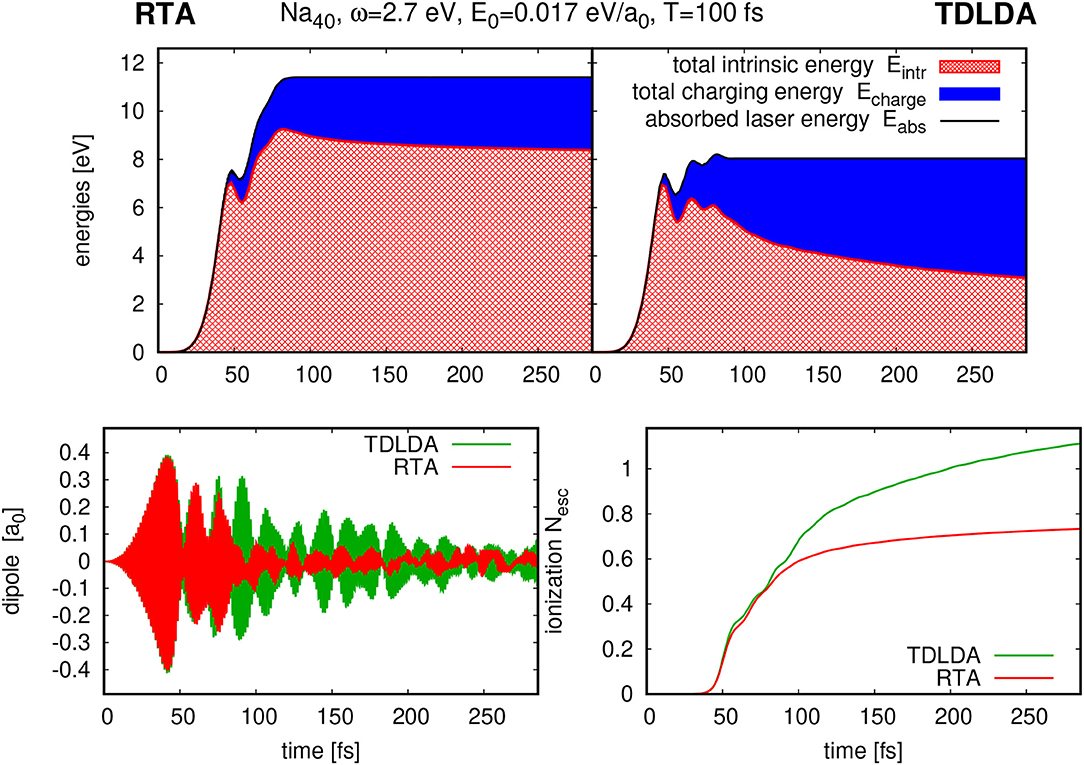
Figure 4. Time evolution of ionization (lower right), dipole moment (lower left), and the various contribution to the energy (upper panels) for the case of Na40 excited by a laser with frequency ω = 2.7 eV, total pulse length Tpulse = 100 fs, and intensity I = 1.3 × 1010 W/cm2. Left upper panels show results from RTA and right upper panels from TDLDA.
A comparison of energy balances is detailed in the upper panel of Figure 4. It shows the contributions from intrinsic energy and energy invested in electron emission for TDLDA and RTA (for details of the definition see [75]). It confirms what we have inferred from the ionization trends in the lower right panel. Most interesting is the large difference in the total energy absorbed from the laser pulse (black line). In pure TDLDA, the entrance channel for energy absorption, the dipole channel, has limited capacity as one can see from the onset of oscillations in the absorption signal. Dissipation in RTA clears the entrance channel and thus allows more energy to come in. Altogether, we see that dissipation can make a difference. Note, however, that this example deals with a considerable excitation energy and that deviations from TDLDA develop only slowly. In turn, we can conclude that TDDFT provides a pertinent description of dynamical processes in its early stages. The time scale of validity of TDDFT depends on excitation energy: the lower the excitation is, the longer TDDFT holds.
We have presented a couple of typical applications of time-dependent density functional theory (TDDFT) to electronic dynamics in clusters and molecules, as triggered by laser pulses. Basis of our description is TDDFT propagated in real time which delivers a versatile and robust tool for a great variety of dynamical situations. At the numerical side it is important to implement absorbing boundary conditions which then allow a detailed description of electron emission. We have addressed in detail two known defects of TDDFT in the local density approximation, namely the self-interaction error and the lack of dynamical correlations. The first problem is less important in nuclear physics, but highly relevant in electronic systems. It is solved by a self-interaction correction (SIC) which still can be formulated within a mean field theory (thus affordable) and which is particularly crucial if electron emission plays a role. The second problem, also important in nuclear dynamics, requires extensions beyond TDDFT for which manageable quantum mechanical approximations are presently being developed.
We have considered typical examples of applications in clusters and molecules excited by a laser pulses of different strengths. In all cases, the early response of the systems addresses predominantly the electrons and remains fully in the quantum mechanical regime (as properly described by TDDFT). In particular, we have focused on an analyzing tool which is not yet available in nuclear dynamics, namely widely tunable laser excitation in combination with detailed resolution of the distributions of the emitted electrons with respect to angles and kinetic energies. We have demonstrated that such a setup delivers valuable information on the system and its dynamical response.
Finally, the inclusion of dissipative features is a key step to address the long-time evolution of dynamics far off-equilibrium in which one observes, in addition to ionization, a thermalization of the electron cloud. Formerly, the problem was attacked with semi-classical approaches much similar as done in nuclear dynamics, particularly in heavy-ion collisions. This approach is less justified in connection with laser excitations for which the (electronic) system stays much longer in the quantum regime. Promising and manageable solutions to deal with dissipation in the quantum regime are just presently coming up. First results from treating dynamical correlations in the relaxation-time approximation (RTA) show that dissipation has a crucial impact on the energy balance in the system (internal energy vs. electron emission) and on the energy intake from the laser pulse. Proper treatment of dynamical correlations is still in its infancy and will attract much future development.
All datasets generated for this study are included in the article/supplementary material.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Institut Universitaire de France for support during the realization of this work. This work was also granted access to the HPC resources of CalMiP (Calcul en Midi-Pyrénées) under the allocation P1238, and of RRZE (Regionales Rechenzentrum Erlangen).
1. Caillat J, Zanghellini J, Kitzler M, Koch O, Kreuzer W, Scrinzi A. Correlated multielectron systems in strong laser fields: a multiconfiguration time-dependent Hartree-Fock approach. Phys Rev A. (2005) 71:012712. doi: 10.1103/PhysRevA.71.012712
2. Ehrmaier J, Picconi D, Karsili TNV, Domcke W. Photodissociation dynamics of the pyridinyl radical: time-dependent quantum wave-packet calculations. J Chem Phys. (2017) 146:124304. doi: 10.1063/1.4978283
3. Greenman L, Ho PJ, Pabst S, Kamarchik E, Mazziotti DA, Santra R. Implementation of the time-dependent configuration-interaction singles method for atomic strong-field processes. Phys Rev A. (2010) 82:023406. doi: 10.1103/PhysRevA.82.023406
4. Hochstuhl D, Hinz C, Bonitz M. Time-dependent multiconfiguration methods for the numerical simulation of photoionization processes of many-electron atoms. Eur Phys J Spec Top. (2014) 223:177–336. doi: 10.1140/epjst/e2014-02092-3
5. Lappas DG, van Leeuwen R. Electron correlation effects in the double ionization of He. J Phy B. (1998) 31:L249. doi: 10.1088/0953-4075/31/6/001
6. Rohringer N, Peter S, Burgdörfer J. Calculating state-to-state transition probabilities within time-dependent density-functional theory. Phys Rev A. (2006) 74:042512. doi: 10.1103/PhysRevA.74.042512
7. Sato T, Ishikawa KL, Brezinova I, Lackner F, Nagele S, Burgdoerfer J. Time-dependent complete-active-space self-consistent-field method for atoms: application to high-order harmonic generation. Phys Rev A. (2016) 94:023405. doi: 10.1103/PhysRevA.94.023405
8. Haken H, Wolf HC. Time-Dependent Density-Functional Theory: Concepts and Applications. Oxford: Oxford University Press (2012).
9. Wilken F, Bauer D. Momentum distributions in time-dependent density-functional theory: product-phase approximation for nonsequential double ionization in strong laser fields. Phys Rev A. (2007) 76:023409. doi: 10.1103/PhysRevA.76.023409
10. Fennel T, Meiwes-Broer KH, Tiggesbäumker J, Dinh PM, Reinhard PG, Suraud E. Electro-magnetic signals for analyzing non-linear cluster dynamics. Rev Mod Phys. (2010) 82:1793. doi: 10.1103/RevModPhys.82.1793
11. Gao CZ, Dinh PM, Reinhard PG, Suraud E. Towards the analysis of attosecond dynamics in complex systems. Phys Chem Chem Phys. (2017) 19:19784–93. doi: 10.1039/C7CP00995J
12. Pinaré JC, Baguenard B, Bordas C, Broyer M. Angular distributions in photoelectron spectroscopy of small tungste n clusters: competition between direct and thermionic emission. Eur Phys J D. (1999) 9:21–4. doi: 10.1007/s100530050392
14. Reinhard PG, Suraud E. Comparison of resonance dynamics in metal clusters and nuclei. In: Ekardt W, editor. Resonance Dynamics in Metal Clusters and Nuclei. New York, NY: Wiley (1999). p. 211.
15. Dreizler RM, Gross EKU. Density Functional Theory: An Approach to the Quantum Many-Body Problem. Berlin: Springer-Verlag (1990).
16. Bender M, Heenen PH, Reinhard PG. Self-consistent mean-field models for nuclear structure. Rev Mod Phys. (2003) 75:121–80. doi: 10.1103/RevModPhys.75.121
17. Calvayrac F, Reinhard PG, Suraud E, Ullrich CA. Nonlinear electron dynamics in metal clusters. Phys Rep. (2000) 337:493–578. doi: 10.1016/S0370-1573(00)00043-0
18. Kjellberg M, Johansson O, Jonsson F, Bulgakov AV, Bordas C, Campbell EEB, et al. Momentum-map-imaging photoelectron spectroscopy of fullerenes with femtosecond laser pulses. Phys Rev A. (2010) 81:023202. doi: 10.1103/PhysRevA.81.023202
20. Hansen K, Richter R, Alagia M, Stranges S, Schio L, Salen P, et al. Single Photon Thermal Ionization of C60. Phys Rev Lett. (2017) 118:103001. doi: 10.1103/PhysRevLett.118.103001
21. Marques MAL, Ullrich CA, Nogueira F, Rubio A, Burke K, Gross EKU. Time Dependent Density Functional Theory. Berlin: Springer (2006).
22. Marques MAL, Maitra NT, Nogueira FMS, Gross EKU, Rubio A. Fundamentals of Time-Dependent Density Functional Theory. Vol. 837 of Lecture Notes in Physics. Berlin: Springer-Verlag (2012).
23. Yelin S, Arimondo E, Lin CC. Advances in Atomic, Molecular, and Optical Physics. Vol. 64. Amsterdam: Elsevier (2015).
24. Gross EKU, Kohn W. Time-dependent density-functional theory. Adv Quant Chem. (1990) 21:255–91. doi: 10.1016/S0065-3276(08)60600-0
25. Gross EKU, Dobson JF, Petersilka M. Density functional of time-dependent phenomena. Top Curr Chem. (1996) 181:81–172. doi: 10.1007/BFb0016643
27. Kümmel S, Brack M, Reinhard PG. Ionic geometries and electronic excitations of and . Eur Phys J D. (1999) 9:149–52. doi: 10.1007/s100530050416
28. Goedecker S, Teter M, Hutter J. Separable dual-space Gaussian pseudopotentials. Phys Rev B. (1996) 54:1703. doi: 10.1103/PhysRevB.54.1703
29. Reinhard PG, Suraud E, Meier C. The impact of the carrier envelope phase—dependence on system and laser parameters. J Phys B. (2018) 51:024007. doi: 10.1088/1361-6455/aa9a9c
30. Perdew JP, Zunger A. Self-interaction correction to density-functional approximations for many-electron systems. Phys Rev B. (1981) 23:5048. doi: 10.1103/PhysRevB.23.5048
31. Kümmel S, Kronik L. Orbital-dependent density functionals: theory and applications. Rev Mod Phys. (2008) 80:3–60. doi: 10.1103/RevModPhys.80.3
32. Messud J, Dinh PM, Reinhard PG, Suraud E. Time-dependent density-functional theory with self-interaction correction. Phys Rev Lett. (2008) 101:096404. doi: 10.1103/PhysRevLett.101.096404
33. Messud J, Dinh PM, Reinhard PG, Suraud E. On the exact treatment of time dependent self-interaction correction. Ann Phys. (2008) 324:955–76. doi: 10.1016/j.aop.2008.12.001
35. Legrand C, Suraud E, Reinhard PG. Comparison of self-interaction-corrections for metal clusters. J Phys B. (2002) 35:1115. doi: 10.1088/0953-4075/35/4/333
36. Klüpfel P, Dinh PM, Reinhard PG, Suraud E. Koopmans' condition in self-interaction corrected density functional theory. Phys Rev A. (2013) 88:052501. doi: 10.1103/PhysRevA.88.052501
37. Maruhn JA, Reinhard PG, Stevenson PD, Umar AS. The TDHF code Sky3D. Comp Phys Comm. (2014) 185:2195–216. doi: 10.1016/j.cpc.2014.04.008
39. Davies KTR, Koonin SE. Skyrme-force time-dependent Hartree–Fock calculations with axial symmetry. Phys Rev C. (1981) 23:2042–61. doi: 10.1103/PhysRevC.23.2042
40. Lauritsch G, Reinhard PG. An FFT Solver for the Coulomb problem. Int J Mod Phys C. (1994) 5:65–75. doi: 10.1142/S0129183194000064
41. Reinhard PG, Stevenson PD, Almehed D, Maruhn JA, Strayer MR. Role of boundary conditions in dynamic studies of nuclear giant resonances. Phys Rev E. (2006) 73:036709. doi: 10.1103/PhysRevE.73.036709
42. Reinhard PG, Suraud E. Cluster dynamics in strong laser fields. In: Marques MAL, Ullrich CA, Nogueira F, editors. Time-Dependent Density Functional Theory. Vol. 706 of Lecture Notes in Physics. Berlin: Springer (2006). p. 391.
43. Wopperer P, Dinh PM, Reinhard PG, Suraud E. Electrons as probes of dynamics in molecules and clusters: a contribution from time dependent density functional theory. Phys Rep. (2015) 562:1. doi: 10.1016/j.physrep.2014.07.003
44. Calvayrac F, Reinhard PG, Suraud E. Spectral signals from electronic dynamics in sodium clusters. Ann Phys. (1997) 255:125–62. doi: 10.1006/aphy.1996.5654
45. Pohl A, Reinhard PG, Suraud E. Angular distribution of electrons emitted from Na clusters. Phys Rev A. (2004) 70:023202. doi: 10.1103/PhysRevA.70.023202
46. Dinh PM, Romaniello P, Reinhard PG, Suraud E. Calculation of photoelectron spectra: a mean-field-based scheme. Phys Rev A. (2013) 87:032514. doi: 10.1103/PhysRevA.87.032514
47. Reinhard PG, Gambhir Y. RPA in wavefunction representation. Ann Phys. (1992) 504:598–631. doi: 10.1002/andp.19925040804
48. Reinhard PG. From sum rules to RPA: 1. Nuclei. Ann Phys. (1992) 504:632–61. doi: 10.1002/andp.19925040805
49. Reinhard PG, Genzken O, Brack M. From sum rules to RPA: 3. Metal clusters. Ann Phys. (1996) 508:576–607. doi: 10.1002/andp.2065080704
50. Reinhard PG, Guo L, Maruhn JA. Nuclear giant resonances and linear response. Eur Phys J A. (2007) 32:19–23. doi: 10.1140/epja/i2007-10366-9
51. Reinhard PG, Flocard H. Nuclear effective forces and isotope shifts. Nucl Phys A. (1995) 584:467–88. doi: 10.1016/0375-9474(94)00770-N
52. Chabanat E, Bonche P, Haensel P, Meyer J, Schaeffer R. A Syrme parametrization from subnuclear to neutron star densities. Nucl Phys A. (1997) 627:710–46. doi: 10.1016/S0375-9474(97)00596-4
53. Klüpfel P, Reinhard PG, Bürvenich TJ, Maruhn JA. Variations on a theme by Skyrme. Phys Rev C. (2009) 79:034310.
54. Mie G. Beiträge zur Optik trüber Medien, speziell kolloidaler Metallösungen. Ann Phys. (1908) 25:377–445. doi: 10.1002/andp.19083300302
55. Kreibig U, Vollmer M. Optical Properties of Metal Clusters. Vol. 25. New York, NY; Heidelberg; Berlin: Springer Series in Materials Science (1993).
56. Ring P, Schuck P. The Nuclear Many-Body Problem. New York, NY: Heidelberg; Berlin: Springer-Verlag (1980).
58. Montag B, Reinhard PG. Width of the plasmon resonance in metal clusters. Phys Rev B. (1995) 51:14686. doi: 10.1103/PhysRevB.51.14686
59. Moseler M, Landman U, Yannouleas C. Controllable electrostatic surface guide for cold molecules with a single charged wire. Phys Rev A. (2001) 87:053401. doi: 10.1103/PhysRevLett.87.053401
60. Esirkepov TZ, Bulanov SV, Zhidkov AG, Pirozkov AS, Kando M. High-power laser-driven source of ultra-short X-ray and gamma-ray pulses. Eur Phys J D. (2009) 55:457–63. doi: 10.1140/epjd/e2009-00172-y
61. Kiefer D, Yeung M, Dzelzainis T, Foster PS, Rykovanov SG, Lewis CL, et al. Relativistic electron mirrors from nanoscale foils for coherent frequency upshift to the extreme ultraviolet. Nat Commun. (2013) 4:1763. doi: 10.1038/ncomms2775
62. Li FY, Sheng ZM, Chen M, Wu HC, Liu Y, Meyer-ter Vehn J, et al. Coherent kilo-electron-volt backscattering from plasma-wave boosted relativistic electron mirrors. Appl Phys Lett. (2014) 105:161102. doi: 10.1063/1.4899136
63. Paulus GG, Nicklich W, Xu H, Lambropoulos P, Walther H. Plateau in above threshold ionization spectra. Phys Rev Lett. (1994) 72:2851–54. doi: 10.1103/PhysRevLett.72.2851
64. Wopperer P, Gao CZ, Barillot T, Cauchy C, Marciniak A, Despré V, et al. Progress towards a realistic theoretical description of C60 photoelectron-momentum imaging experiments using time-dependent density-functional theory. Phys Rev A. (2015) 91:042514. doi: 10.1103/PhysRevA.91.042514
65. Li H, Mignolet B, Wachter G, Skruszewicz S, Zherebtsov S, Süßmann F, et al. Coherent electronic wave packet motion in C60 controlled by the waveform and polarization of few-cycle laser fields. Phys Rev Lett. (2015) 114:123004. doi: 10.1103/PhysRevLett.114.123004
66. Uehling EA, Uhlenbeck GE. Theory of superconductivity. Phys Rev. (1932) 108:1175. doi: 10.1103/PhysRev.108.1175
67. Bertsch GF, Das Gupta S. A guide to microscopic models for intermediate energy heavy ion collisions. Phys Rep. (1988) 160:190. doi: 10.1016/0370-1573(88)90170-6
68. Abe Y, Ayik S, Reinhard PG, Suraud E. On stochastic approaches of nuclear dynamics. Phys Rep. (1996) 275:49–196. doi: 10.1016/0370-1573(96)00003-8
69. Fennel T, Bertsch GF, Meiwes-Broer KH. Ionization dynamics of simple metal clusters in intense fields by the Thomas-Fermi-Vlasov method. Eur Phys J D. (2004) 29:367–78. doi: 10.1140/epjd/e2004-00035-1
70. Domps A, Reinhard PG, Suraud E. Semi-classical electron dynamics in metal clusters beyond mean-field. Ann Phys. (2000) 280:211–35. doi: 10.1006/aphy.1999.5990
71. Lacroix D. Stochastic mean-field dynamics for fermions in the weak coupling limit. Phys Rev C. (2006) 73:044311. doi: 10.1103/PhysRevC.73.044311
72. Suraud E, Reinhard PG. Non equilibrium quantum dynamics with collisional correlations. New J Phys. (2014) 16:063066. doi: 10.1088/1367-2630/16/6/063066
73. Slama N, Reinhard PG, Suraud E. On the inclusion of collisional correlations in quantum dynamics. Ann Phys. (2015) 355:182–203. doi: 10.1016/j.aop.2015.02.008
74. Reinhard PG, Suraud E. A quantum relaxation-time approximation for finite fermion systems. Ann Phys. (2015) 354:183–202. doi: 10.1016/j.aop.2014.12.011
Keywords: time-dependent density-functional theory, molecules, electron emission, photo-electron distributions, dissipation
Citation: Mai Dinh P, Vincendon M, Heraud J, Suraud E and Reinhard P-G (2020) Far Off Equilibrium Dynamics in Clusters and Molecules. Front. Phys. 8:27. doi: 10.3389/fphy.2020.00027
Received: 07 January 2020; Accepted: 30 January 2020;
Published: 19 February 2020.
Edited by:
Denis Lacroix, UMR8608 Institut de Physique Nucléaire d'Orsay (IPNO), FranceReviewed by:
Kazuhiro Yabana, University of Tsukuba, JapanCopyright © 2020 Mai Dinh, Vincendon, Heraud, Suraud and Reinhard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Phuong Mai Dinh, ZGluaEBpcnNhbWMudXBzLXRsc2UuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.