Corrigendum: CTAB-Assisted Synthesis of C@Na3V2(PO4)2F3 With Optimized Morphology for Application as Cathode Material for Na-Ion Batteries
 Ana Criado1Pedro Lavela1
Ana Criado1Pedro Lavela1 Gregorio F. Ortiz1
Gregorio F. Ortiz1 José L. Tirado1*
José L. Tirado1* Somaya Gzouli2
Somaya Gzouli2 Zineb Edfouf2
Zineb Edfouf2 Carlos Pérez-Vicente1
Carlos Pérez-Vicente1- 1Departamento de Química Inorgánica e Ingeniería Química, Instituto Universitario de Investigación en Química Fina y Nanoquímica, Universidad de Córdoba, Córdoba, Spain
- 2Materials and Nanomaterials for Photovoltaic Conversion and Electrochemical Storage, Faculty of Science, University of Mohammed V, Rabat, Morocco
C@Na3V2(PO4)2F3 samples were obtained by using Cetyl Trimethyl Ammonium Bromide (CTAB) as a surfactant. The optimization of the added amount allowed controlling the eventual nanometric morphology of the particles. The morphological and structural properties of these samples were discussed in the light of solid-state techniques as X-ray diffraction, Raman and XPS spectroscopies, and electron microscopy. Galvanostatic test in sodium half-cells revealed that the nanometric spherical and porous particles provided by the addition of intermediate amounts of CTAB showed excellent cycling stability and superior high rate capability reflected in the minimization of the cell polarization and the determination of a high apparent diffusion coefficient.
Introduction
On the verge of a new age for widespread energy production based on renewable energy sources, the implementation of efficient energy storage systems is regarded as a crucial point to ensure safe distribution of electricity [1–3]. Among others, batteries feature undeniable advantages such as modular and flexible layout, relatively fast manufacturing and short response time [4]. Despite the great success of Li-ion batteries, the increased demand for lithium compounds, along to the uneven geographic distribution of constituents elements, may lead to undesirable resource shortages and high manufacturing in the near future [5, 6]. For this reason, the research on electrochemical batteries is envisaging alternative devices that could complement rather than replace excellent performance of Li-ion batteries.
Na-ion battery is an emerging concept which is profiting from the technological similarity to Li-ion ones, which is undoubtedly shortening the time needed to develop commercially viable devices. Sodium features high abundance and environmental friendliness which may counteract the slightly lower redox potential and larger atomic weight than lithium. In fact, these drawbacks are not especially detrimental in large stationary batteries, where the volumetric energy density is not so constraining [7–10]. The reversible insertion of sodium into a host framework is the mechanism responsible for the cell cyclability at the positive electrode pole. Particularly, phosphates containing sodium as migrating species and vanadium as active redox center have recently attracted much attention. Their high operational voltage and theoretical capacity, along to the remarkable structural stability arise from the inductive effect of the polyanion and P–O bond strength. For instance, phosphates as Na3V2(PO4)3 [11], pyrophosphates as Na2MnP2O7 [12] and fluorophosphates as Na3V2(PO4)2F3 [13, 14] can be mentioned.
Otherwise, vanadium is a multivalent transition element which valence states from V2+ to V5+ may participate in the electrochemical reaction. This fact is reflected in multi-stage charge and discharge curves delivering capacity at varied potentials as low as 0.3 V in Na3V2(PO4)3 [15] or as high as 4 V for a related Na3VCr(PO4)3 stoichiometry [16]. Particularly, Na3V2(PO4)2F3 has attracted recently more attention as potential cathode for sodium-ion batteries. This electrode material features an open framework which can be briefly described as composed by long chains of [V2O8F3] bi-octahedra as building block running along the c axis. The three-dimensional scaffold is supported by [PO4] tetrahedra linking together these chains. This structure provides suitable pathways for sodium diffusion along a and b axis. The three sodium ions per formula unit are allocated in two distinct sites. Thus, two of them fully occupies triangular prismatic sites, labeled as Na(1) sites, while the third one is filling half of the large triangular prismatic sites attached to the F apex-square pyramid, denoted as Na(2) sites [17, 18].
The use of cetyl trimethyl ammonium bromide (CTAB) as a surfactant to prepare electrode materials with adapted morphology for sodium-ion batteries has been reported elsewhere [19–21]. In the case of polyanionic compounds, CTAB also acts as a carbon source for creating the carbon conductive coating which favors the electrical connectivity among the active material particles. As a soft template, this surfactant serves to adjust the morphology and size of primary particles and promotes the homogeneous dispersion of the active material into the carbon conductive phase.
Although several procedures were proposed to prepare Na3V2(PO4)2F3, such as conventional solid-state reaction [17, 18], solvothermal [22, 23] and sol-gel [24, 25], we propose the use of CTAB as a surfactant for the first time. The as-prepared samples will be characterized by X-ray diffraction, electron microscopy, and X-ray photoemission spectroscopy, the electrochemical behavior was evaluated by galvanostatic tests in sodium half-cells.
Experimental Details
Several C@Na3V2(PO4)2F3 samples were prepared by a microemulsion method using cetyl trimethyl ammonium bromide (CTAB) as a soft template. For this purpose, citric acid (Aldrich, 99%) was dissolved in 50 mL in deionized water in a 2:3 vanadium to citric ratio. This organic acid acts as a chelating agent and carbon source for the formation of a carbon conductive phase. Then, 0.01 moles of NH4VO3 (Panreac, 98%) and 30% overstoichiometric excess of NaF (Panreac, 99%) were subsequentially added. The excess of NaF salt could compensate losses during calcination. After complete dissolution, the selected amount of CTAB (0, 0.2, 0.5, 1.0 g) was dissolved in deionized water at 60°C and the stoichiometric amount of NaH2PO4·H2O (Aldrich, 98–102%) was then dissolved. This solution was dropwise poured to the previous solution and gently stirred for one until reach a stable emulsion. Next, the solvent was removed in a rotor evaporator (70°C and 200 mbar), until obtaining a viscous gel which was further dried at 120°C overnight. The solid precursor was then ball milled for 10 min at 300 rev min−1 and precalcined at 400°C for 4 h for removing gaseous products. This residuum was then pelletized and again calcined at 700°C for 10 h (5°C min−1) in argon atmosphere. The samples were labeled as NVPF-X, being X the grams of CTAB added to the synthesis. The carbon content was determined in an elemental CHNS analyzer (Eurovector EA 3000).
The structural analysis was performed in a BrukerD8 Discover A25 X-ray diffractometer (XRD) furnished with Cu Kα radiation. The patterns were scanned between 10 and 80° (2θ angle) and a scan rate of 0.02° (2θ) min−1. The structural determination of lattice parameters was calculated with TOPAS software. Transmission electron microscopy (TEM) images were acquired in a JEOL 1400 microscope. Field-emission scanning electron micrographs (FE-SEM) were recorded in a JSM-7800F Prime microscope furnished with an EDX analyzer. Raman spectra were acquired in a Renishaw Raman instrument (InVia Raman Microscope), equipped with a Leica microscope and a green laser light (532 nm) as excitation source. The profiles were deconvoluted by using the Peakfit (v. 4.12) software. X-ray Photoelectron Spectroscopy (XPS) was employed to determine the chemical state of the elements by using a SPECS Phobios 150 MCD furnished with a monochromatic Al Kα source. The samples were supported on aluminum disk and subjected to high vacuum overnight (4 × 10−9 mbar). The binding energy scale was referred to the C 1s line of the adventitious carbon (284.6 eV). Room temperature EPR spectra of samples introduced in EPR quartz tubes were recorded using an EMX micro X-band instrument from Bruker, with 5 scans, 20 dB microwave, and resonance frequency of 9.50 GHz in all cases.
Electrochemical behavior of electrodes was performed in sodium half cells subjected to galvanostatic tests. Thus, working electrodes were prepared by slurring the active material (80%), carbon black (10%) and PVDF (polyvinylidene fluoride) (10%) in N-methyl-2-pyrrolidone. The resulting paste was spread onto 9 mm aluminum disks and vacuum dried at 120°C for 2 h. As counter electrodes, 9 mm sodium disks were cut. Both electrodes were separated by glass fiber disks (GF/A-Whatman) soaked in 1M NaClO4 dissolved in propylene carbonate. Fluoroethylene carbonate (FEC) was added in 2%wt. to stabilize the passivation film. The cell was assembled into hermetical Swagelok™ type two-electrode cells in an argon-filled glove box under controlled O2 and H2O traces. Galvanostatic cycling was tested in a VMP multichannel system setting a potential window between 2.0 and 4.4 V and several discharge and charge rates ranging from 0.5 to 10 C. Electrochemical Impedance spectra (EIS) were recorded on an SP-150 Biologic apparatus to determine the cell impedance. For this purpose, the working electrode was assembled in a SwagelokTM type three-electrode cell, using sodium disks as counter and quasi-reference electrodes. The cell was subjected to galvanostatic pulses to reach the desired depth of charge and discharge and left to relax for 12 h pursuing a quasi-equilibrium state. Then, the impedance spectra were measured by perturbing the open circuit voltage with an AC signal of 5 mV from 100 kHz to 0.001 mHz.
Results and Discussion
Materials Characterization
TEM micrographs of NVPF revealed primary particles compactly agglomerated with non-uniform shape and sizes, which ranged from a few to tenth of nanometers (Figure 1). The use of CTAB evidenced drastic changes in morphology. Thus, NVPF-0.2 shows larger particles but with a more spherical shape. The carbon phase can be discerned as light domains engulfing the active material particles. This texture was also observed in NVPF-0.5. It is expected that this homogeneous dispersion of Na3V2(PO4)2F3 into the carbon matrix will favor the electrical connectivity. A close inspection of NVPF-0.5 reveals the appearance of internal pores into the particles. This fact has been reported in related compounds and considered as beneficial for reaching high sodium diffusion, superior high rate capability and cycling stability [26]. On increasing the amount of added CTAB to 1.0 g, we observe that even if the porous structure is preserved into the particles, they appeared larger and less spherical (Figure 1). Figure 1 also shows SEM images of NVPF-0.5 at different magnifications evidencing the sphericity of primary particles clustered in the carbon matrix. The reliability of this preparative route to obtain samples with optimal morphology and homogeneous composition was evidenced by the EDX maps showed in Supplementary Figures S1, S2, in which the constituent elements appear evenly distributed on the particle surface.
FIGURE 1

Figure 1. TEM and SEM images of selected Na3V2(PO4)2F3 samples prepared with different amounts of CTAB samples.
The chemical state of the elements composing C@Na3V2(PO4)2F3 was evaluated in the NVPF-05 sample by recording the XPS spectra at the C1s, O1s, P2p, V2p, Na1s, and F1s core levels. The survey spectrum evidenced the presence of constituent elements (C, O, P, V, Na, F) (Figure 2A). In the case of the C1s core level spectrum, the asymmetric profile was decomposed into four contributions. They are typically ascribed to graphitic carbon at 284.6 eV, C–OH (285.7 eV), C=O (287.5 eV), and carboxylic (290.6 eV) groups (Figure 2A; [27]). In turn, the O1s spectrum was decomposed into five bands. Those signals at 531.6 and 533.5 eV correspond to V-O and P-O, respectively. These signals are commonly explained in terms of the occurrence of distinct oxygen local environment. The subsequent decrease of the bond iconicity is responsible for the increase of binding energy at the oxygen atom [28]. Bands belonging to oxygen in C=O, C–OH and carboxylic groups are, respectively, detected at about 531, 532.5, and 534 eV. Although the former one would be masked by the most intense band at 531.6, the latter ones are in good agreement to those signals appearing at 532.8, 534.7 eV [29]. Finally, the broadened signal at 536.7 eV could only be associated to the presence of occluded CO or CO2 [30]. The spectrum at the V2p core level features two broad bands at 517.7 and 524.8 eV, attributed to the V2p3/2 and V2p1/2 transitions. According to the literature, these values fairly match the occurrence of trivalent vanadium in Na3V2(PO4)2F3 [31, 32]. For the P2p spectrum, an asymmetric and broad band was deconvoluted in two signals at 133.9 and 135.3 eV, which correspond to the P2p3/2 and P2p1/2 transitions in phosphate groups [33].
FIGURE 2
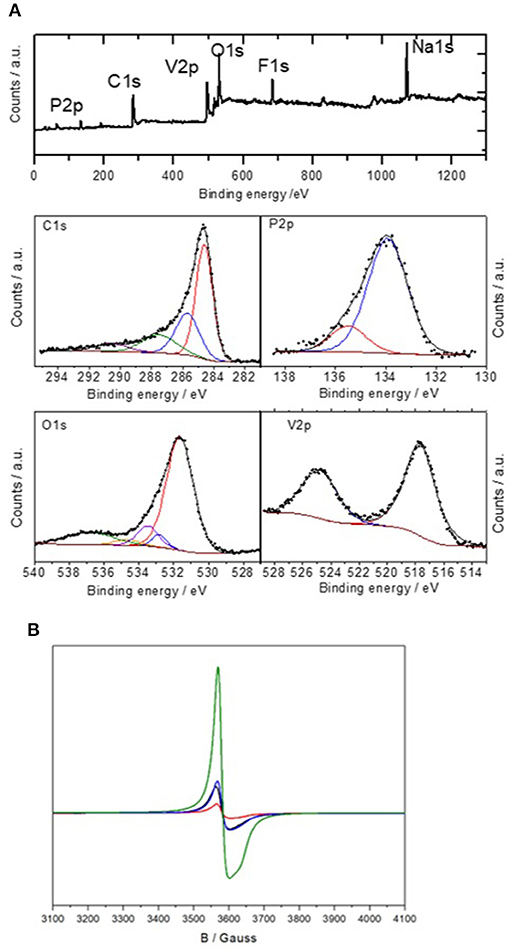
Figure 2. (A) X-ray photoelectron spectra of NVPF-0.5 sample including survey spectrum and sub-spectra at the C1s, P2p, O1s and V2p core levels. (B) Electron Paramagnetic Resonance (EPR) spectra of samples NVPF (black), NVPF-0.2 (red), NVPF-0.5 (blue) and NVPF-1.0 (green).
Electron paramagnetic resonance (EPR) was used to further assess the oxidation state of vanadium and its effect on sample composition (Figure 2B). According to previous literature [34–36], EPR signals are not expected for V3+ in stoichiometric fluorophosphates. Thus, the low-intensity signals observed at ca. 3580 G for NVPF, NVPF-0.2 and NVPF-0.5 should be ascribed to mixed valent phases containing small amounts of V4+. For NVPF-1.0, a second contribution is observed, showing an EPR signal with the g-factor close to that of the V4+ ions and axial character. Thus, the second signal cannot be caused by the Na3V2(PO4)3 impurity, which was associated with the formation of antisite defects by Nizamov et al. [37], as it shows isotropic character. Instead, the axial character of the NVPF-1.0 EPR signal is typical of compounds with major proportion of V4+. As the measurement conditions were the same for the four samples, the existence of some amount of V4+ fluorophosphate with the main V3+ compound could be the origin. As the V3+ fluorophosphate does not show any signal in EPR, relatively small amounts of V4+ in traces of the Na3(VO)2(PO4)2F compound could coexist with V3+ in Na3V2(PO4)2F3 and could show strong signal in EPR. These V3+ and V4+ phases present very similar diffraction patterns and electrochemical behavior and are very difficult to distinguish by using laboratory scale X-ray diffraction, so EPR is frequently used to know the oxidation state of V in these compounds [34–36].
XRD patterns of C@Na3V2(PO4)2F3 samples are displayed in Figure 3. These diagrams show a set of reflections, mainly indexed in the P42/mnm space group belonging to the tetragonal system. It should be noted that the use of very high angular resolution synchrotron radiation diffraction allowed Binachini et al. to detect an orthorhombic distortion [38]. However, our standard CuKα XRD apparatus does not resolve the splitting of the peaks associated to the lower symmetry space group. Thus, we retain the tetragonal indexing, which can also be easily compared with the structural description that is still commonly found in the recent literature on this material. For NVPF sample, only minor impurities are identified. The peaks at 31.8°, 33.6°, and 34.4° could be assigned to Na3VF6 (PDF 00-029-1286), α-Na3PO4 (PDF 01-072-7303), and γ-Na3PO4 (PDF 01-031-1323). These impurities, which are not electroactive, have also been detected by other authors during the synthesis of this compound [27, 28]. In the light of these results, the XPS and EPR V signals reveal its trivalent state in Na3VF6. Unfortunately, the use of CTAB until NVPF-0.5 promoted a slight increase in these reflections. The unit cell parameters, calculated from these patterns, are written in Table 1 and fairly agree to those reported in the literature [24, 39]. On increasing the CTAB addition for NVPF-1.0, the XRD pattern revealed the appearance of an additional phase which was attributed to Na3V2(PO4)3 (Figure 3). The increase of the cell parameters, observed for the latter sample in Table 1, would be indicative of the structural distortion induced by the appearance of the new phase.
FIGURE 3
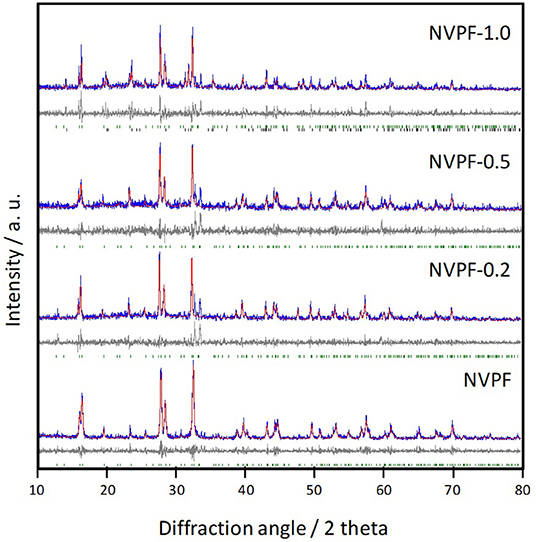
Figure 3. X-ray diffraction patterns of C@Na3V2(PO4)2F3 samples. The experimental pattern (blue), calculated pattern (red), and differential curves (gray) are included for each sample. DIF patterns for P42/mnm and R-3c space groups are respectively displayed in green and black colors.
TABLE 1
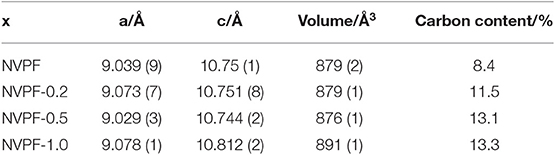
Table 1. Cell parameters in the P42/mnm space group for the C@Na3V2(PO4)2F3 samples.
Because these samples were calcined at 700°C, a high level of structural disordering can be expected in the carbon conductive phase. In order to asses this assumption, Raman spectra of samples prepared in the presence of CTAB as a soft template were recorded (Figure 4 and Supplementary Table 1). The most relevant features in these profiles are two broadened and overlapped Gaussian bands appearing at 1350 ± 4 cm−1 (D1) and 1597 ± 2 cm−1 (G), which are typically ascribed to disordered and graphitic carbon atoms, respectively [40]. The level of graphitization reached by the carbon phase existing in our samples can be easily determined by calculating the IG/ID1 ratio, being I the integrated area of these bands. For a precise determination of this value, a full deconvolution of the whole spectra in the Gaussian component is deserved [41]. From this calculation, new components ascribable to disordered domains also appear at 1508 ± 3 cm−1 (D3), 1187 ± 3 cm−1 (D4), and 1685 ± 2 cm−1 (D2). These contributions are, respectively, assigned to amorphous carbon, C–C and C=C stretching vibrations of polyene-like structures and disordered graphitic lattice [41]. All samples featured IG/ID1 ratios of 0.43, evidencing the high structural disorder of this low-temperature carbon phase. Also, small bands at 1030 cm−1 were observed. These contributions are attributed to vibrational modes of the PO4 tetrahedra [42].
FIGURE 4
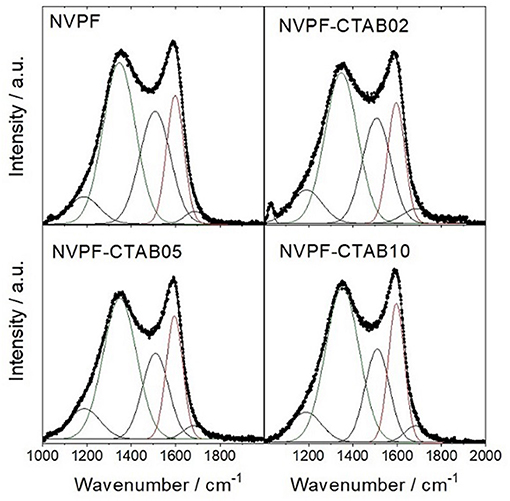
Figure 4. Raman spectrum of C@Na3V2(PO4)2F3 recorded with λ0 = 532 nm. The band assignment has been indicated.
Electrochemical Experiments
Figure 5 shows the charge and discharge curves recorded on sodium half-cells subjected at several rates from 0.5 to 10 C. The most remarkable features are the two charge plateaus at 3.7 and 4.2 V and their corresponding cathodic counterparts at 3.65 and 4.15 V, for the lowest rate. Based on first-principles calculations, it was suggested that the V4+/V3+ redox process and reversible insertion of two of the three ions from different Na(1) and Na(2) sites are responsible for these features [43]. The occurrence of both plateaus differs from the electrochemical behavior in related compounds as Na3V2(PO4)3 [33]. This peculiarity has been explained as resulting from the Na+ reordering in the interstitial sites after the extraction of the first sodium. It provokes an elongation of the Na-Na distance, inducing an increase of the cell potential, and consequently the occurrence of the plateau at ca. 4 V [17]. In addition, the appearance of these plateaus at higher voltage than for Na3V2(PO4)3 is clear evidence of the strong inductive effects exerted by fluorine [21]. However, the additional plateaus for NVPF-1.0, at ca. 3.0 V on discharge, could be ascribed to the higher NVP impurity level detected by XRD. On increasing the rate from 0.5 to 10 C, the charge/discharge hysteresis progressively increased, along to the decrease of the overall capacity.
FIGURE 5
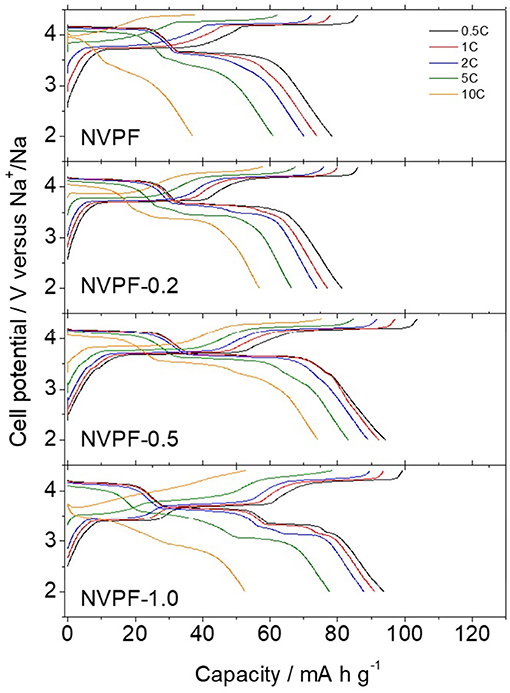
Figure 5. Galvanostatic charge and discharge curves recorded from sodium half-cells cycled at increasing C rates.
Interesting information about the kinetic properties of these electrode materials can be gathered by quantifying the charge/discharge hysteresis, or cell polarization (ΔV). Supplementary Figure S3 displays the average charge and discharge voltage determined for each plateau. These linear correlations are commonly ascribed to thermodynamic effects which are intrinsic of the reversible insertion and the migration through the electrode-electrolyte interfaces. It is more remarked when this cell polarization is plotted vs. the applied current on a logarithmic scale (Supplementary Figure S4). It is evident that NVPF-0.5 performed the low charge and discharge hysteresis even at high rates. Otherwise, the absence of CTAB in NVPF or the addition of excess in NVPF-1.0 induced the largest values.
From the extrapolation of these curves to the vertical axis, we could observe that a finite voltage gap exists between the charge and discharge potential curve when the current tends toward zero, referred to as zero current polarization. It has been explained by the occurrence of a sequential particle-by-particle mechanism [44]. The plot of the cell polarization vs. the applied current in Figure 6 revealed a linear relationship between both parameters, allowing an easy calculation of the zero-current polarization as the intercept to the vertical axis. These values are displayed as bars in Figure 7. From the observation of these values, it is evident that the NVPF sample featured the largest polarization regardless the evaluated plateau, and specially at the low voltage plateau, as compared to those prepared with CTAB. In addition, the direct current resistance can be calculated from the slope of the linear plot in Figure 6 to gather valuable information about the kinetic properties of the electrode subjected to a non-zero current intensity. Figure 7 reveals that the use of CTAB must be optimized to values between 0.2 and 0.5 g to yield low resistive electrodes. Summarizing, NVPF-0.5 sample evidenced the overall better kinetic properties that could positively benefit its electrochemical behavior on cycling, as will be further mentioned.
FIGURE 6
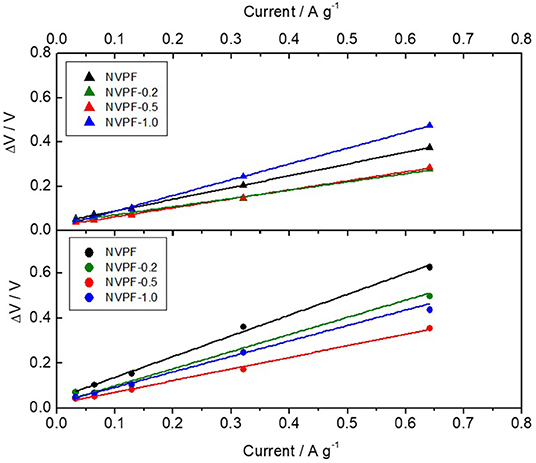
Figure 6. Plot of the linear relationship between cell polarization at zero current for each material at the two different plateaus and applied current (triangles: high-voltage plateau, circles: low-voltage plateau). Linear fitting of the experimental results is shown as solid lines.
FIGURE 7
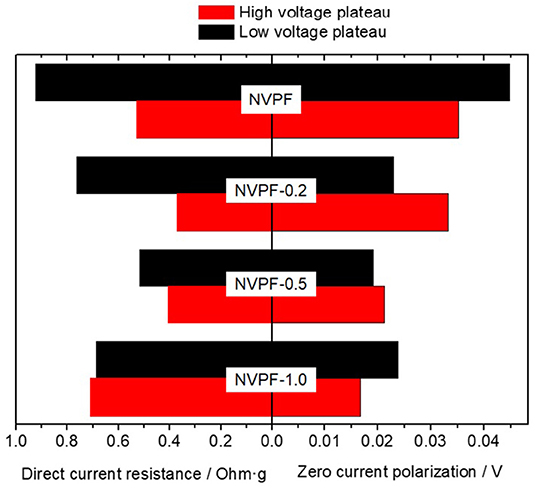
Figure 7. Bar diagrams of direct-current resistance and polarization at zero-current values.
Sodium-half cells assembled with the C@Na3V2(PO4)2F3 samples were subjected to several cycles at increasing C rates (Figure 8). NVPF-0.5 and NVPF-1.0 featured initial discharge capacity values close to 95 mA h g-1. These values are close to those reported elsewhere [39, 45]. It involves a significant increase in capacity as compared to NVPF and NVPF-0.2. Thus, the use of CTAB as surfactant in optimized amount seems to be crucial to achieving a performance electrode. Likely, the porous structure created into the particles is determining to improve the sodium access to the active material through large electrode-electrolyte interphase. On increasing the C rate, we observe that capacity abruptly decreased for NVPF-1.0 at 10 C to values even lower than those of NVPF-0.2. The excellent performance of NVPF-0.5 at the highest rate agrees well to the low direct current resistance values previously calculated for both plateaus that pointed out to this good electrode behavior. Long galvanostatic cycling tests were performed on sodium half-cells at 2C (Figure 9). After 200 cycles, samples prepared in the presence of CTAB showed higher capacity retention than NVPF one. Thus, NVPF-0.5 was able to retain 85% of the initial capacity (75 mA h g−1), while only 57 mA h g−1 were recorded for NVPF.
FIGURE 8
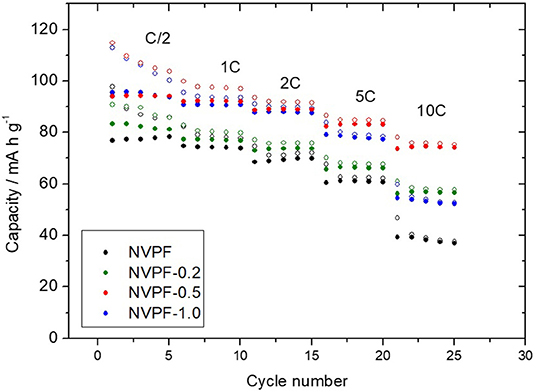
Figure 8. Rate capability plot of sodium half-cells assembled with C@Na3V2(PO4)2F3 samples (open symbols: charge capacity; closed symbols: discharge capacity).
FIGURE 9
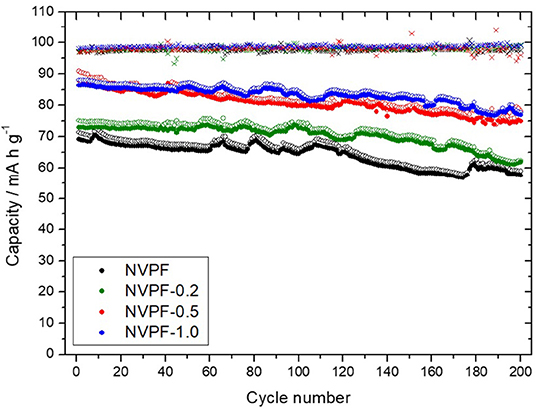
Figure 9. Extended galvanostatic cycling at 1C NVPF-0.5 (open symbols: charge capacity; closed symbols: discharge capacity).
The electrochemical performance of the samples studied here is not particularly outstanding as compared with recent literature on stoichiometric NVPF and related compositions [34–36, 38, 46–49]. However, the improvements in cell polarization, capacity retention and rate performance of the samples prepared by the CTAB-assisted method suggest the positive effect of texture control on the performance of NVPF. Further evidence is discussed below by using impedance spectroscopy.
Impedance spectroscopy is a helpful technique to unveil changes of the cell internal resistance arising from effects occurring at the electrode-electrolyte interphase. In order to avoid initial irreversible effects, these spectra were recorded on electrodes activated after one charge-discharge cycle at 0.5 C. The recorded Nyquist plots revealed a depressed semicircle at high frequencies ascribable to the charge transfer reaction (ct) at the electrode-electrolyte interphase (Figure 10A). This plot can be fitted to the equivalent circuit included as an inset in Figure 10A. Thus, resistance values attributed to the electrolyte ohmic drop (Rel) and charge-transfer reaction (Rct) can be calculated, being the latter one the predominant contribution to the internal cell resistance. These values are written in Table 2. Their close similarity between both values involves that differences in the kinetic properties cannot be exclusively attributed to them. Otherwise, the apparent diffusion coefficients (D) of sodium ions through the structure can be also determined from the impedance spectra by applying the next equation:
FIGURE 10
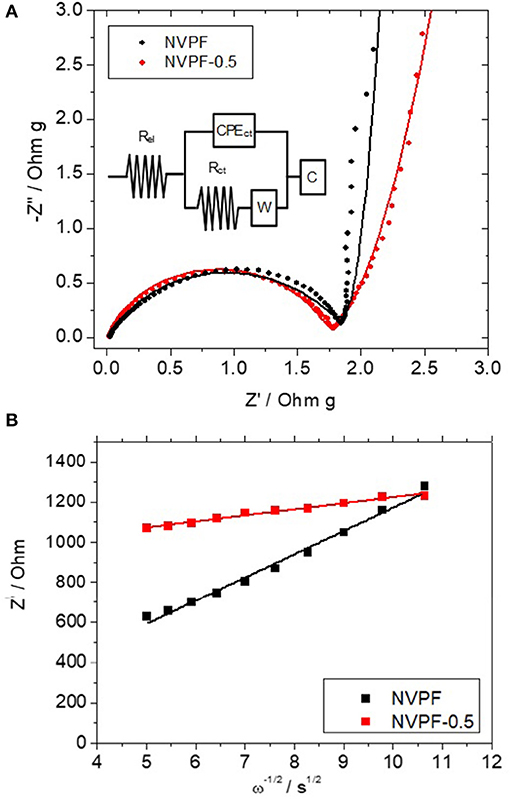
Figure 10. Impedance spectra plotted as (A) Nyquist plots of NVPF and NVPF-0.5 samples. Overlapping lines correspond to the fitted spectra according to the equivalent circuit included as inset; (B) Plot of the real impedance vs. the reciprocal root square of the frequency.
TABLE 2
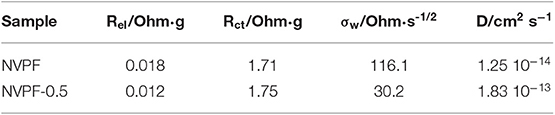
Table 2. Resistance values and apparent diffusion coefficients calculated from the impedance spectra of Na3V2(PO4) 2F3 electrodes after 1 cycle at 0.5 C.
In which T is the absolute temperature, R is the gas constant (8.314 J K−1 mol−1), A is the geometrical electrode area (0.64 cm2) and F is the Faraday's constant. The value of the molar concentration of Na+ ions (C) was calculated from Equation 2:
where V is the unit cell volume, Z is the number of unit formula per unit cell, and NA is the Avogadro number. The Warburg impedance coefficient (σw) was experimentally determined as the slope of the linear plot of the real impedance, Z', vs. the reciprocal root square of the lower angular frequencies (ω−1/2) [Figure 10B; (50]). From the data in Table 2, a large increase of the diffusion coefficient is observed for NVPF-0.5, which involves that the porous morphology of this sample facilitates the migration of sodium ions through the structure.
Conclusions
The reliability of CTAB as a surfactant to prepare C@Na3V2(PO4)2F3 samples with a suitable morphology to enhance the electrode performance in Na-ion batteries has been proved. The amount of added surfactant has been optimized to eventually yield well-dispersed nanometric spherical particles of the phosphate compound into the carbon conductive matrix. For intermediate amounts of added CTAB, the sample showed internal macropores which can be beneficial for sodium diffusion. XRD patterns revealed only minor impurities, while Raman spectroscopy evidenced the disordered character of the carbon matrix.
Galvanostatic tests performed in sodium half-cells revealed excellent cycling stability and superior high rate capability for NVPF-0.5 at 10 C. It was correlated to low current polarization and direct current resistance from charge-discharge hysteresis at both plateaus, while the apparent diffusion coefficient was increased in one order of magnitude as compared to the reference NVPF sample. From these results, it was concluded that the optimized amount of CTAB favorably contributes to obtaining C@Na3V2(PO4)2F3 with adapted morphology to act as an excellent cathode material for sodium-ion batteries.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
AC and SG contributed to the acquisition and analysis of data for the work. ZE, PL, and CP-V contributed to the design and interpretation the work. JT and GO contributed to the conception and critical revision of the work.
Funding
This work was carried out with the funding of Ministerio de Ciencia, Industria e Innovación (MINECO), ERDF funds (contract MAT2017-84002-C2-1-R), and Junta de Andalucía (FQM288). SG was also grateful to CEE for financial support (program ka107).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Ministerio de Ciencia, Industria e Innovación (MINECO) and ERDF funds (MAT2017-84002-C2-1-R), and Junta de Andalucía (FQM288) for financial support. We also thank SCAI (UCO Central Service for Research Support) for the microscopy and XPS measurements.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2019.00207/full#supplementary-material
References
1. Koohi-Kamali S, Tyagi VV, Rahim NA, Panwar NL, Mokhlis H. Emergence of energy storage technologies as the solution for reliable operation of smart power systems: a review. Renew Sust Energ Rev. (2013) 25:135–65. doi: 10.1016/j.rser.2013.03.056
2. Sundararagavan S, Baker E. Evaluating energy storage technologies for wind power integration. Solar Energy. (2012) 86:2707–17. doi: 10.1016/j.solener.2012.06.013
3. Yang Z, Zhang J, Kintner-Meyer MCW, Lu X, Choi D, Lemmon JP, et al. Electrochemical energy storage for green grid. Chem Rev. (2011) 111:3577–613. doi: 10.1021/cr100290v
4. Dunn D, Kamath H, Tarascon JM. Electrical energy storage for the grid: a battery of choices. Science. (2011) 334:928–35. doi: 10.1126/science.1212741
5. Oliveira L, Messagie M, Rangaraju S, Sanfelix J, Hernandez-Rivas M, Van Mierlo J. Key issues of lithium-ion batteries e from resource depletion to environmental performance indicators. J Clean Prod. (2015) 108:354–62. doi: 10.1016/j.jclepro.2015.06.021
6. Swain B. Cost effective recovery of lithium from lithium ion battery by reverse osmosis and precipitation: a perspective. J Chem Technol Biotechnol. (2018) 93:311–9. doi: 10.1002/jctb.5332
7. Pu X, Wang H, Zhao D, Yang H, Ai X, Cao S, et al. Recent progress in rechargeable sodium-ion batteries: toward high-power applications. Small. (2019) 15:1805427. doi: 10.1002/smll.201805427
8. Kumar Nayak P, Yang L, Brehm W, Adelhelm P. From lithium-ion to sodium-ion batteries: advantages, challenges, and surprises. Angew Chem Int Ed. (2018) 57:102–20. doi: 10.1002/anie.201703772
9. Eftekhari A, Kim DW. Sodium-ion batteries: new opportunities beyond energy storage by lithium. J Power Sources. (2018) 395:336–48. doi: 10.1016/j.jpowsour.2018.05.089
10. Deng J, Luo WB, Chou SL, Liu HK, Dou SX. Sodium-ion batteries: from academic research to practical commercialization. Adv Energy Mater. (2018) 8:1701428. doi: 10.1002/aenm.201701428
11. Zhang X, Rui X, Chen D, Tan H, Yang D, Huang, et al. Na3V2(PO4)3: an advanced cathode for sodium-ion batteries. Nanoscale. (2019) 11:2556–76. doi: 10.1039/C8NR09391A
12. Barpanda P, Ye T, Avdeev M, Chung SC, Yamada A. A new polymorph of Na2MnP2O7 as a 3.6 V cathode material for sodium-ion batteries. J Mater Chem A. (2013) 1:4194–7. doi: 10.1039/c3ta10210f
13. Liu H, Yi H, Zheng Q, Li X, Zhang H. Y-doped Na3V2(PO4)2F3 compounds for sodium ion battery cathode: electrochemical performance and analysis of kinetic properties. J Mater Chem A. (2017) 5:10928–35. doi: 10.1039/C7TA03133E
14. Yuvaraj S, Oh W, Yoon WS. Recent progress on sodium vanadium fluorophosphates for high voltage sodium-ion battery application. J Electrochem Sci Technol. (2019) 10:1–13. doi: 10.5229/JECST.2019.10.1.1
15. Jian Z, Sun Y, Xiulei J. A new low-voltage plateau of Na3V2(PO4)3 as an anode for Na-ion batteries. Chem Commun. (2015) 51:6381–3. doi: 10.1039/C5CC00944H
16. Liu R, Xu G, Li Q, Zheng S, Zheng G, Gong Z, et al. Exploring highly reversible 1.5-electron reactions (V3+/V4+/V5+) in Na3VCr(PO4)3 cathode for sodium-ion batteries. ACS Appl Mater Interfaces. (2017) 9:43632–9. doi: 10.1021/acsami.7b13018
17. Shakoor RA, Seo DH, Kim H, Park YU, Kim J, Kim SW, et al. A combined first principles and experimental study on Na3V2(PO4)2F3 for rechargeable Na batteries. J Mater Chem. (2012) 22:20535–41. doi: 10.1039/c2jm33862a
18. Le Meins JM, Crosnier-Lopez MP, Hemon-Ribaud A, Courbion G. Phase transitions in the Na3M2(PO4)2F3 family (M: Al3+, V3+, Cr3+, Fe3+, Ga3+): synthesis, thermal, structural, and magnetic studies. J Solid State Chem. (1999) 148:260–77. doi: 10.1006/jssc.1999.8447
19. Wang W, Zhang H, Xu Y, Chen C, Yuan H, Wang Y. CTAB-assisted multiwalled carbon nanotube loaded NaFe2Mn(PO4)3 materials as high performance cathodes for sodium-ion batteries. RSC Adv. (2016) 6:67986–91. doi: 10.1039/C6RA12708H
20. Deng C, Zhang S, Wu Y. Hydrothermal-assisted synthesis of Na7V4(P2O7)4(PO4)/C nanorod and its fast sodium intercalation chemistry in aqueous rechargeable sodium battery. Nanoscale. (2015) 7:487–91. doi: 10.1039/C4NR05175K
21. Saravanan K, Mason CW, Rudola A, Wong KH, Balaya P. The first report on excellent cycling stability and superior rate capability of Na3V2(PO4)3 for sodium ion batteries. Adv Energy Mater. (2013) 3:444–50. doi: 10.1002/aenm.201200803
22. Zhu C, Wu C, Chen CC, Kopold P, Van Aken PA, Maier J, et al. A high power-high energy Na3V2(PO4)2F3 sodium cathode: investigation of transport parameters, rational design and realization. Chem Mater. (2017) 29:5207–15. doi: 10.1021/acs.chemmater.7b00927
23. Zhang Y, Guo G, Xu H. Synthesis of uniform hierarchical Na3V1.95Mn0.05(PO4)2F3@C hollow microspheres as a cathode material for sodium-ion batteries. J Mater Chem A. (2018) 6:4525–34. doi: 10.1039/C7TA11105C
24. Liu Q, Wang D, Yang X, Chen N, Wang C, Bie X, et al. Carbon-coated Na3V2(PO4)2F3 nanoparticles embedded in a mesoporous carbon matrix as a potential cathode material for sodium-ion batteries with superior rate capability and long-term cycle life. J Mater Chem A. (2015) 3:21478–85. doi: 10.1039/C5TA05939A
25. Liu Q, Meng X, Wei Z, Wang D, Gao Y, Wei Y, et al. Core/double-shell structured Na3V2(PO4)2F3@C nanocomposite as the high power and long lifespan cathode for sodium-ion batteries. ACS Appl Mater Interfaces. (2016) 8:31709–15. doi: 10.1021/acsami.6b11372
26. Wang Q, Zhao B, Zhang S, Gao X, Deng C. Superior sodium intercalation of honeycomb-structured hierarchical porous Na3V2(PO4)3/C microball prepared by a facile one-pot synthesis. J Mater Chem A. (2015) 3:7732–40. doi: 10.1039/C5TA00765H
27. Biniak B, Szymanski G, Siedlewski J, Swiatkowski A. The characterization of activated carbons with oxygen and nitrogen surface groups. Carbon. (1997) 35:1799–810. doi: 10.1016/S0008-6223(97)00096-1
28. Ramesh Kumar P, Jung YH, Moorthy B, Kim DK. Effect of electrolyte additives on NaTi2(PO4)3-C//Na3V2O2X(PO4)2F3-2X-MWCNT aqueous rechargeable sodium ion battery performance. J Electrochem Soc. (2016) 163:A1484–92. doi: 10.1149/2.0031608jes
29. Seredych M, Hulicova-Jurcakova D, Lu GQ, Bandosz TJ. Surface functional groups of carbons and the effects of their chemical character, density and accessibility to ions on electrochemical performance. Carbon. (2008) 46:1475–88. doi: 10.1016/j.carbon.2008.06.027
30. de la Puente G, Pis JJ, Menéndez JA, Grange P. Thermal stability of oxygenated carbons. J Anal Appl Pyrol. (1997) 43:125–38. doi: 10.1016/S0165-2370(97)00060-0
31. Hu G, Chen P, Liu Z, Cao Y, Zhang Z, Peng Z, et al. Synthesis and electrochemical properties of xLiFePO4·(1-x) Na3V2(PO4)2F3/C composite for lithium-ion batteries. J Alloy Compd. (2017) 696:177–84. doi: 10.1016/j.jallcom.2016.11.237
32. Li W, Wang K, Cheng S, Jiang K. A long-life aqueous Zn-ion battery based on Na3V2(PO4)2F3 cathode. Energy Storage Mater. (2018) 15:14–21. doi: 10.1016/j.ensm.2018.03.003
33. Cao X, Sun Q, Zhu L, Xie L. Na3V2(PO4)3 nanoparticles confined in functional carbon framework towards high-rate and ultralong-life sodium storage. J Alloy Compd. (2019) 791:296–306. doi: 10.1016/j.jallcom.2019.03.346
34. Serras P, Palomares V, Goñi A, Gil de Muro I, Kubiak P, Lezama L, et al. High voltage cathode materials for Na-ion batteries of general formula Na3V2O2x(PO4)2F3−2x. J Mater Chem. (2012) 22:22301–8. doi: 10.1039/c2jm35293a
35. Serras P, Palomares V, Kubiak P, Lezama L, Rojo T. Enhanced electrochemical performance of vanadyl (IV) Na3(VO)2(PO4)2F by ex-situ carbon coating. Electrochem Commun. (2013) 34:344–7. doi: 10.1016/j.elecom.2013.07.010
36. Palomares V, Blas M, Setién S, Lezama L, Pramudita JC, Quadir A, et al. Investigating low-valent compositions in the Na3V2O2x(PO4)2F3−2x family: structural transitions and their consequences. Dalton Trans. (2018) 47:2610–8. doi: 10.1039/C8DT00086G
37. Nizamov FA, Togulev PN, Abdullin DR, Khantimerov SM, Balaya P, Suleimanov NM. Antisite defects and valence state of vanadium in Na3V2(PO4)3. Phys Solid State. (2016) 58:475–80. doi: 10.1134/S1063783416030240
38. Bianchini M, Brisset N, Fauth F, Elkaim E, Suard E, Masquelier C, et al. Na3V2(PO4)2F3 revisited: a high-resolution diffraction study. Chem Mater. (2014) 26:4238–47. doi: 10.1021/cm501644g
39. Song W, Ji X, Wu Z, Zhu Y, Li F, Yao Y, et al. Multifunctional dual Na3V2(PO4)2F3 cathode for both lithium-ion and sodium-ion batteries. RSC Adv. (2014) 4:11375–83. doi: 10.1039/C3RA47878E
40. Tuinstra T, Koenig JI. Raman spectrum of graphite. J Chem Phys. (1970) 53:1126–30. doi: 10.1063/1.1674108
41. Sadezky A, Muckenhuber H, Grothe H, Niessner R, Pöschl U. Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon. (2005) 43:1731–42. doi: 10.1016/j.carbon.2005.02.018
42. Barj M, Lucazeau G, Delmas C. Raman and infrared spectra of some chromium nasicon-type materials: short-range disorder characterization. J Solid State Chem. (1992) 100:141–50. doi: 10.1016/0022-4596(92)90164-Q
43. Song W, Cao X, Wu Z, Chen J, Zhu Y, Hou H, et al. Investigation of the sodium ion pathway and cathode behavior in Na3V2(PO4)2F3 combined via a first principles calculation. Langmuir. (2014) 30:12438–46. doi: 10.1021/la5025444
44. Dreyer W, Jamnik J, Guhlke C, Huth R, Moskon J, Gaberscek M. The thermodynamic origin of hysteresis in insertion batteries. Nat Mater. (2010) 9:448–53. doi: 10.1038/nmat2730
45. Song W, Ji X, Wu Z, Yang Y, Zhou Z, Li F, et al. Exploration of ion migration mechanism and diffusion capability for Na3V2(PO4)2F3 cathode utilized in rechargeable sodium-ion batteries. J Power Sources. (2014) 256:258–63. doi: 10.1016/j.jpowsour.2014.01.025
46. Broux T, Bamine T, Fauth F, Simonelli L, Olszewski W, Marini C, et al. Strong impact of the oxygen content in Na3V2(PO4)2F3−yOy (0 ≤ y ≤ 0.5) on its structural and electrochemical properties. Chem Mater. (2016) 28:7683–92. doi: 10.1021/acs.chemmater.6b02659
47. Bianchini M, Xiao P, Wang Y, Ceder G. Additional sodium insertion into polyanionic cathodes for higher-energy na-ion batteries. Adv Funct Mater. (2017) 7:1700514. doi: 10.1002/aenm.201700514
48. Yan G, Dugas R, Tarascon JM. The Na3V2(PO4)2F3/carbon na-ion battery: its performance understanding as deduced from differential voltage analysis. J Electrochem Soc. (2018) 165:A220–7. doi: 10.1149/2.0831802jes
49. Yan G, Mariyappan S, Rousse G, Jacquet Q, Deschamps M, David R, et al. Higher energy and safer sodium ion batteries via an electrochemically made disordered Na3V2(PO4)2F3 material. Nat Commun. (2019) 10:1–12. doi: 10.1038/s41467-019-08359-y
Keywords: sodium-ion batteries, insertion electrode, polyanionic materials, surfactant-assisted synthesis, particle morphology
Citation: Criado A, Lavela P, Ortiz GF, Tirado JL, Gzouli S, Edfouf Z and Pérez-Vicente C (2019) CTAB-Assisted Synthesis of C@Na3V2(PO4)2F3 With Optimized Morphology for Application as Cathode Material for Na-Ion Batteries. Front. Phys. 7:207. doi: 10.3389/fphy.2019.00207
Received: 23 September 2019; Accepted: 18 November 2019;
Published: 03 December 2019.
Edited by:
Javier Carrasco, CIC Energigune, SpainReviewed by:
Verónica Palomares, University of the Basque Country, SpainJacob Olchowka, Centre National de la Recherche Scientifique (CNRS), France
Copyright © 2019 Criado, Lavela, Ortiz, Tirado, Gzouli, Edfouf and Pérez-Vicente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José L. Tirado, iq1ticoj@uco.es