 Nirmal Mazumder1*
Nirmal Mazumder1* Naveen K. Balla2
Naveen K. Balla2 Guan-Yu Zhuo3,4
Guan-Yu Zhuo3,4 Yury V. Kistenev5,6
Yury V. Kistenev5,6 Rajesh Kumar7
Rajesh Kumar7 Fu-Jen Kao8
Fu-Jen Kao8 Sophie Brasselet9
Sophie Brasselet9 Viktor V. Nikolaev5,10
Viktor V. Nikolaev5,10 Natalya A. Krivova11
Natalya A. Krivova11- 1Department of Biophysics, Manipal School of Life Sciences, Manipal Academy of Higher Education, Manipal, India
- 2Center for Research on Adaptive Nanostructures and Nanodevices, Trinity College of Dublin, Dublin, Ireland
- 3Institute of New Drug Development, China Medical University, Taichung, Taiwan
- 4Integrative Stem Cell Center, China Medical University Hospital, Taichung, Taiwan
- 5Laboratory of Biophotonics, Tomsk State University, Tomsk, Russia
- 6Department of Physics and Mathematics, Siberian Medical State University, Tomsk, Russia
- 7Department of Physics, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
- 8Institute of Biophotonics, National Yang-Ming University, Taipei, Taiwan
- 9Institut Fresnel, CNRS, Aix-Marseille Université, Ecole Centrale Marseille, Domaine Universitaire St Jérôme, Marseille, France
- 10Laboratory of Molecular Imaging and Photoacoustics, Institute of Strength Physics and Materials Science SB RAS, Tomsk, Russia
- 11Laboratory of Experimental Physiology, Tomsk State University, Tomsk, Russia
Non-linear optical (NLO) microscopy has proven to be a powerful tool especially for tissue imaging with sub-cellular resolution, high penetration depth, endogenous contrast specificity, pinhole-less optical sectioning capability. In this review, we discuss label-free non-linear optical microscopes including the two-photon fluorescence (TPF), fluorescence lifetime imaging microscopy (FLIM), polarization-resolved second harmonic generation (SHG) and coherent anti-Stokes Raman scattering (CARS) techniques with various samples. The non-linear signals are generated from collagen in tissue (SHG), amylopectin from starch granules (SHG), sarcomere structure of fresh muscle (SHG), elastin in skin (TPF), nicotinamide adenine dinucleotide (NADH) in cells (TPF), and lipid droplets in cells (CARS). Again, the non-linear signals are very specific to the molecular structure of the sample and its relative orientation to the polarization of the incident light. Thus, polarization-resolved non-linear optical microscopy provides high image contrast and quantitative estimate of sample orientation. An overview of the advancements on polarization-resolved SHG microscopy including Stokes vector based polarimetry, circular dichroism, and susceptibility are also presented in this review article. The working principles and corresponding implements of above-mentioned microscopy techniques are elucidated. The potential of time-resolved TPF lifetime imaging microscopy (TP-FLIM) is explored by imaging endogenous fluorescence of NAD(P)H, a key coenzyme in cellular metabolic processes. We also discuss single laser source time-resolved multimodal CARS-FLIM microscopy using time-correlated single-photon counting (TCSPC) in combination with continuum generation from photonic crystal fiber (PCF). Using examples, we demonstrate that the multimodal NLO microscopy is a powerful tool to assess the molecular specificity with high resolution.
Introduction
The basic terminology of non-linear optics will be discussed here, though the in-depth study of non-linear optics is described in Boyd [1]. In the 1990s, non-linear optical imaging modalities met their need for high peak power lasers, when the mode-locked infrared lasers were invented [2]. The interaction between light and matter under high peak power pulsed laser drives various non-linear optical processes such as multiphoton fluorescence and higher-order harmonic generations [1]. The key advantages of non-linear optical microscopy are deep imaging capability and high spatial resolution of tissue samples when compared to confocal microscopy. Non-linear processes rely on high intensities that are generated by tight focusing of the incoming ultra-fast pulsed lasers through high numerical aperture objective lens. Due to the requirement for high intensity, the effective excitation volume in non-linear signal is much smaller than that of a linear signal [2] and provides 3D imaging capability with subcellular details and high molecular contrast [1]. It have opened new routes toward optical diagnostics of complex cellular assemblies and tissue imaging [3, 4]. The electrons of the sample respond to the incoming field differently depending upon the strength of electric field. In case of a weak electric field, the electrons follow the oscillations of the field; in case of the strong electric field, they move away from the equilibrium and no longer follow the incoming field. The polarization of medium under the action of the external optical wave can be described by using a power series expansion in an electric field [1]:
where is the induced polarization due to the field Ẽ(t) and ϵ0 is the electric permittivity of free space, χ(1) is the linear susceptibility and it characterizes the index of refraction and linear absorption. The second-order optical susceptibility, χ(2), is the lowest order non-linear term. Its value is non-zero only in non-centrosymmetric media and is responsible for the second-harmonic generation (SHG) and sum-frequency generation (SFG). χ(3) is the third-order susceptibility and is responsible for the non-linear self-action like absorption saturation, third-harmonic generation (THG), coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS). Since both the polarization and electric field are vectors, the higher-order non-linear susceptibilities are tensors. For example, χ(2)is a 3 × 3 × 3 tensor with 27 elements and χ(3) is a 3 × 3 × 3 × 3 tensor with 81 elements.
In this review, we discuss the basics of non-linear optical microscopy techniques, their advantages and drawbacks, and related applications including hot topics in the field of biomedicine.
Two-Photon Fluorescence Microscopy
In 1990 for the first time, Denk et al. developed a two-photon fluorescence scanning microscope for live-cell imaging after the invention of mode-locked laser [2]. The non-linear excitation requires extremely high photon flux, typically 1020-1030 photons/(cm2s). A femtosecond pulsed laser is used in combination with high-numerical aperture (NA) objective lens to generate high photon flux at the focal spot. In TPF, the excitation wavelengths are chosen for the fluorophores so that they absorb two photons simultaneously. The total energy of the exciting photons should be equal to or exceed the energy gap between the ground state and excited state of the fluorophore for TPF to occur. The advantage of fluorescent markers or probes, which could be used to selectively tag molecules within the living system for visualization purposes, has contributed immensely toward an understanding of cellular and molecular aspects in biological systems.
The experimental arrangement of TPF microscope integrated with FLIM system is shown in Figure 1A. An inverted confocal microscope (e.g., Olympus IX71) can be modified for TPF imaging setup. An ultrafast femtosecond laser oscillator is used as the excitation light source. In general, Coherent Mira Optima 900-F, Spectra-Physics, Toptica Photonics are the main suppliers of femtosecond lasers around the globe [9–13]. The excitation wavelength is set according to the absorption wavelength of the fluorophores (two-photon excitation wavelengths are 740 nm for nicotinamide adenine dinucleotide, NADH and 810 nm for Nile Red, 860 nm for green fluorescence protein, GFP) and has pulse width of ~100 fs, with an average power ~550 mW and repetition rate ~76 MHz. The laser power is controlled by a combination of polarizer and half-waveplate. Live cell samples are imaged in 4.2 cm2 Lab-Tek Chambers Slide System (NUNC, Rochester, NY) mounted on plan XY microscope stage (IX2-KSP, Olympus) and scanned with a laser scanning unit (Olympus, FV300) (as shown in Figure 1). After entering the microscope, the laser beam is reflected by a dichroic mirror (690 nm filter) before propagating through the objective lens (UPlanFLN 40X/N.A. 1.3 oil, Olympus Corp., Japan). The TPF signal is collected through the excitation objective lens, in the spectral window depending upon the emission wavelengths of fluorophores (460/50 nm for NADH, 640/40 for Nile Red, 550/50 nm for GFP), by a photomultiplier tube (PMT, R2949, Hamamatsu, Japan). For FLIM measurement, the signals from the PMT and the reference photodiode are combined with the TCSPC card (TCSPC, b&h, Germany). The two-photon auto-fluorescence signal of NAD(P)H in Rat basophilic leukemia (RBL) cells in a glass-bottom dish placed on microscope stage and elastic fibers in the murine trachea ex vivo (Figures 1B,C) demonstrate the capability of this technique.
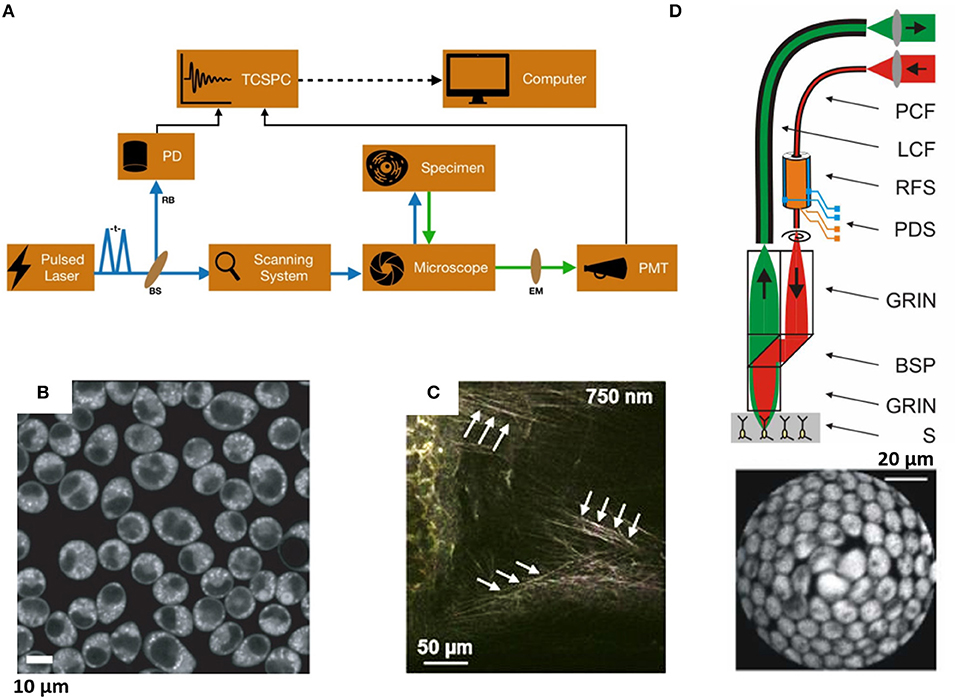
Figure 1. (A) Typical schematic of two-photon fluorescence microscopy integrated with TCSPC system for fluorescence lifetime imaging: t, pulse interval; EM, emission filter; RB, reference beam; BS, beam splitter; TCSPC, time-correlated single-photon counter; PD, photodiode; PMT, photomultiplier tube [5]. (B,C) Two-photon auto-fluorescence images of (B) NAD(P)H in Rat basophilic leukemia (RBL) cells [6]; (C) Elastic fibers in the murine trachea ex vivo was detected between 400 and 580 nm at an excitation wavelength of 750 nm [7]. (D) Schematic of an ultra-compact fiber-optic two-photon fluorescence microscope. The near-IR ultrafast laser pulses (red) are passed through a hollow-core photonic crystal fiber to the microscope. The fluorescence emission (green) from sample (S) were guided through a large core fiber (LCF) and detected by a photomultiplier tube. The excitation and fluorescence signals were collected through custom-designed combination of GRIN lens and micro beamsplitter prisms (BSP). Two-photon fiberscope images of 10-micron diameter fluorescent microspheres were obtained by scanning spiral pattern trajectory for image reconstruction. PDS, piezo drive signals; RFS, resonant fiber scanner. Average laser power levels of 1 mW and 10x averaging were used. Scale bars are 20 μm [8]. All the Figures were reproduced with kind permission from respective publishers. The arrows indicate that those are elastin fibers.
Optical sectioning of animal and human skin using a two-photon femtosecond laser scanning auto-fluorescence microscopy has been reported [11, 12, 14–18]. The system acquires high-resolution multiphoton tomography images and is capable of distinguishing normal human skin and dermatological disorders with submicron spatial resolution and sub-nanosecond temporal resolution. Over the past decades, TPF microscopy has received wide attention in cancer research in both pre-clinical and clinical human studies including the initiation, proliferation, metastasis, metabolism, and angiogenesis of tumors and their microenvironment [13, 19]. With the advancement of new drugs and novel diagnosis methods, TPF imaging technique helps to cure cancer and improve quality of life [20, 21]. For the first time, Brown et al. demonstrated the potential of TPF in examining the gene expression and physiologic function in the deep regions of tumors [22]. They have observed the growth of xenograft tumors in transgenic mice by imaging vascular architecture under the control of endothelial growth factor (VEGF) promoter. Imaging cellular metabolism via endogenous fluorescent cofactor NADH and FAD is one of the successful applications of TPF to investigate tumor metabolism. Two-photon auto-fluorescence measurements from the endogenous molecules inherently present in the cells/tissue, such as aromatic amino acids and co-enzymes have been extensively employed toward cellular metabolism in the last three decades [22]. The fluorophores have a characteristic excitation and emission spectrum that enables it to be distinguished from other materials. It makes their signatures useful for detection and characterization by fluorescence spectroscopy/microscopy, and for using in biomedical applications. The fluorescence emission wavelengths of these endogenous probes consist of tryptophan (350 nm), NADH (460 nm), FAD (525 nm), melanin (579 nm), elastin (454 nm), Retinol (490 nm), folic acid (450 nm), and Riboflavin (540 nm) [23–26]. The two-photon fluorescence signal is very specific to the excitation and emission wavelengths of fluorophores. More information about the fluorophores in the local environment in tissue sections and biopsies can be obtained from the dynamics of the fluorescence signal [27–30].
In vivo tissue, multiphoton imaging is of great importance due to the ability to illuminate the spatial structure variation in tissue molecular components during pathological process development. The multiphoton microscopy (MPM) was applied for in-vivo lymphedema control [31]. The lymphedema is a chronic disease, connected with insufficient lymph circulation and disorder of resorption. The study involved 36 image samples from stage II lymphedema patients and 42 image samples from healthy volunteers. The papillary layer of the skin with a penetration depth of about 100 μm was examined. Recording of tissue MPM images was carried with the MPTflex two-photon microscope (Jenlab GmbH, Germany). Both the collagen network disorganization and increase of the collagen/elastin ratio in lymphedema tissue, characterizing the severity of fibrosis, was observed. Various methods of MPM image characterization, including edge detectors, a histogram of oriented gradients method were used, and a predictive model for lymphedema tissue diagnosis using machine learning was created. The classification by “ensemble learning” provided 96% accuracy on the data from the testing set. Melasma is a skin disorder characterized by hyperpigmented patches due to increased melanin production and deposition. The MPM was used to characterize non-invasively the melanin content, location, and distribution in melasma and to assess the elastosis severity. The melanin volume fraction values measured in epidermal melasma (14 ± 4%) were significantly higher (p < 0.05) than the values measured in peri-lesional skin (11 ± 3%). The basal keratinocytes of melasma and perilesions showed different melanin distributions. Elastosis was predominantly more severe in lesions than in perilesions and was associated with changes in melanin distribution of the basal keratinocytes [32].
The ability of two-photon excitation laser scanning microscopy to excite selectively tagged fluorescent molecules deep inside a tissue makes it a valuable tool in the biological imaging of deep neuronal tissues in vivo [33]. The optimum excitation spectral window is determined between tissue scattering and absorption, which is dominated by water absorption in near-infrared wavelength. Therefore, the optimum wavelength window for deep tissue penetration is near to when both tissue absorption and scattering are considered. Usage of longer excitation wavelength reduces photodamage and allows deep tissue imaging, and lack of pinhole allows collection of highly scattered fluorescent signal from the tissue. The recent advances in the field of neuroimaging have shed light on disease-causing factors as well as neuronal plasticity and circuits [34–36]. TPF microscopy has enabled scientists to study the functional changes such as calcium transients and electrical activities, within the brain cells with greater accuracy. TPF microscopy for calcium imaging has allowed comparison of neuronal activity changes under normal and disease states [37]. Kobat et al. have demonstrated the extended depth of field of two-photon microscopy up to ~1.6 mm penetration depth in the cortex of a mouse brain in vivo with 1,280 nm excitation and only nanojoule (nJ) pulse energies at the sample surface. The longer wavelength in TPF enables deeper penetration due to the significantly reduced tissue scattering [9, 38]. However, due to technical limitations and sample complexity, TPF microscopy has not been able to push through the much deeper regions of the brain in awake and behaving mammals. Until recently, before the invention of two separate techniques which uses optical fiber and an endo-microscope supplemented with GRadient INdex (GRIN) lens (as shown in Figure 1D), imaging deeper regions of the brain in awake animals was thought to be impossible [10, 39, 40]. GRIN lenses are fine rod-like lenses with a refractive index profile of near- parabolic type and can easily be embedded inside the tissue. Since the GRIN lens displaces tissue volume, which is linear relative to the image depth, a GRIN lens is uniquely suitable for imaging neurons in deeply buried nuclei. These optical manifestations not only allow scientists to study complex forms of animal behaviors but also provide cellular-level information with much better resolution.
In vivo two-photon microscopy of mice brain, post-implantation, to quantify changes in inflammatory cell behavior as well as meningeal collagen type I remodeling was carried out [41]. A migratory window during the first day after the implantation was hallmarked by inflammatory cell migration. This migratory window attenuates by 2 days post-implant, but over the next month, the meningeal collagen-I gets remodeled to conform to the surface of the implant and thickens. This work shows that there are distinct time courses for initial meningeal inflammatory cell infiltration and meningeal collagen type I remodeling. This may indicate a therapeutic window early after implantation for modulation and mitigation of meningeal inflammation. The MPM was used to evaluate the role of vascular structure in normal and pathological physiology [42]. The convolutional neural network was used to segment the 3D structure of vessels from MPM images. It was applied for 3D images of cortical blood vessels in young and old mouse models of Alzheimer's disease to explore the effects of aging and Alzheimer's disease on capillaries. A little difference was observed in the distribution of capillary diameter or tortuosity between these groups, but a decrease in the number of longer capillary segments (>75 μm) in aged animals as compared to young, in both wild type and Alzheimer's disease mouse models, took place. It was shown simultaneous neuronal activities in the primary visual cortex (V1), the primary motor cortex (M1), and the hippocampal CA1 region of the mice when awake. The method described is implemented on a single conventional two-photon microscope to enable multi-area exploration of neuronal activities in-vivo [43]. Two-photon imaging was shown to provide the longitudinal assessment of neuronal and synaptic morphology in animal models of Alzheimer's disease (AD). One major hallmark of these diseases is the severe loss of synapses that occurs at the early clinical stages, and this has been correlated with cognitive deficits in AD patients. The time course of this early synapse loss and how the normal turnover of synaptic structures is affected in AD are relatively unknown [44].
Though TPF provides deep tissue imaging at the sub-cellular level resolution, however, limits its penetration depth in the case of thick biological specimens due to the aberration present in the sample as well as optics used. The implementation of adaptive optics (AO) in TPF can enhance the ability to image deep into the brain in vivo. The liquid crystal spatial light modulator (LC-SLM) and deformable membrane mirror (DMM) is used to control and correct distorted ultrashort pulses after propagation through optical elements and a scattering medium. This technique minimizes the aberrations and allows high image quality and 3D imaging in strongly aberrated conditions [17].
Time Domain Fluorescence Lifetime Imaging (TD-FLIM)
The time-domain (TD) FLIM measurements are implemented on laser scanning microscopes in both confocal and multiphoton mode with high-speed detectors and electronics [45, 46]. Time-correlated single-photon counting (TCSPC) board is used for TD fluorescence lifetime measurement [47]. Here we discuss specifically the time a fluorophore spends in an excited state and emits a photon after excitation through time-domain TP-FLIM. Depending on the local environment and molecular interactions, fluorescence lifetime changes, it makes FLIM a powerful tool to probe the localized objects in biological samples efficiently. Photons emitted by the fluorophores are collected using a PMT with high temporal resolution (25–300 ps) [48, 49]. Each photon detected causes a pulse response from the PMT whose width depends on the PMT and the adjoining electronics. The photon counts are measured following the excitation pulse, and the decay time, τ is calculated from the slope of a plot of the log I(t) against t called decay curve [45, 46]. Fluorescence lifetime calculation from the multi-exponential decay is performed by mathematical convolution of a model function and the instrument response function (IRF) by fitting with the experimental data. Lifetimes from the composite decays of bio-molecules can be derived by convolution of an IRF, Iinstru, with a double-exponential model function for the ambient light and/or dark noise I0 to obtain calculated lifetime decay function Ic(t) [45, 50]. In general, the auto-fluorescence spectral method is used for quantifying endogenous fluorophores in live cells and animal models in real time and non-invasively [2–4, 46]. Only a handful of studies exists in the literature that has exploited the intrinsic fluorescence lifetime of NADH and FAD for discriminating between normal and pathological tissues [24, 25, 45–48]. However, time-resolved TP-FLIM imaging has an advantage over the intensity spectrum, for its-
• high spatial and temporal resolution
• independent of fluorophore concentration
• minimally affected by tissue absorption and scattering
• fluctuations in laser intensity
The design and implementation of TD-FLIM microscopy are very well-established and can be found elsewhere [37, 50]. The most available TCSPC FLIM module are from Picoquant (Germany, https://www.picoquant.com/) and b & h (Becker & Hickl, Berlin, Germany, http://www.becker-hickl.com/), which can be assembled with any optical microscopes (Olympus, Nikon, Zeiss, Leica). Typical two-photon fluorescence TCSPC instrumentation uses a mode-locked pulsed laser as shown in Figure 1A [45, 47, 48]. The TCSPC board measures the time difference between START and STOP where START pulse is generated by the detector (PMT) while the STOP or SYNC pulse is provided directly from the laser via an internal photodiode [45]. Figure 2 shows the single-photon energy band diagram, time-resolved fluorescence decay profile of NADH in HeLa cells and auto-fluorescence lifetime and intensity images. Commercially available Symphotime software (Picoquant, Germany) was used for image acquisition and data analysis.
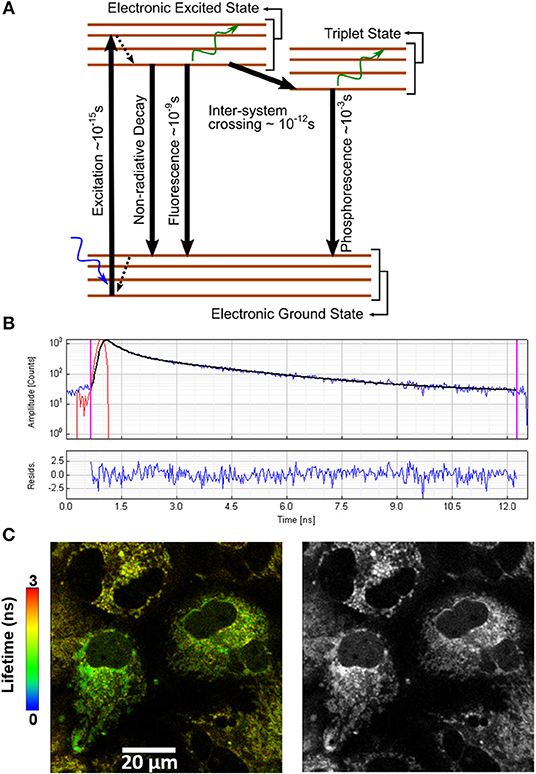
Figure 2. (A) Energy band diagram. (B) Typical bi-exponential model fitting of NAD(P)H fluorescence decay curve. The standard-fitted fluorescence decay curve is shown with χ2 close to 1 and residuals showing no noticeable systematic variations. The y-axis is in photon counts, and x-axis is the time in nanoseconds. (C) NAD(P)H representative color-coded image of NAD(P)H lifetimes (left) and fluorescence intensity (right) in Huh 7 cells. Figure reproduced (from [50]) with kind permission from Springer.
TP-FLIM technique is a non-invasive and non-destructive imaging method to visualize alterations in metabolic state, by tracking endogenous fluorophores present in the cell, such as nicotinamide adenine dinucleotide (NADH) and its phosphorylated form (NADPH), flavin adenine dinucleotide (FAD), and flavoproteins as well as collagen, elastin in tissues. TP-FLIM is applied in various cancerous tissues [51–56] such as skin, cervix, bladder, breast, stomach, and also Alzheimer's disease [56–63]. In this review, we have shown the capability of the TP-FLIM by imaging several auto-fluorescence molecules (as shown in Figure 3). A 450/40 nm bandpass filter (Edmund Optics Inc. Barrington, New Jersey) is inserted in the fluorescence emission path for measuring the autofluorescence signal from NADH that is a central peak at 450 nm. An additional 680 nm short-pass filter (Brightline680 SP, Semrock) was used to further exclude the backscattered 740 nm excitation light. For cellular imaging, the average power of ~3–5 mW is used at the focal plane of the objective lens, which was optimal for the prevention of photobleaching. The images were taken at 256 × 256 pixels resolution with the acquisition time in the range of 300–500 s for accumulating enough photon count statistics and subsequent data analysis. A bi-exponential fluorescence decay fitting is used for distinct lifetimes corresponding to the free and protein-bound forms of NAD(P)H [64]. FLIM imaging has been applied widely to visualize the changes in metabolism and energy consumption in mammalian cells, both in vitro and in vivo through NAD(P)H fluorescence lifetime imaging [65–68].
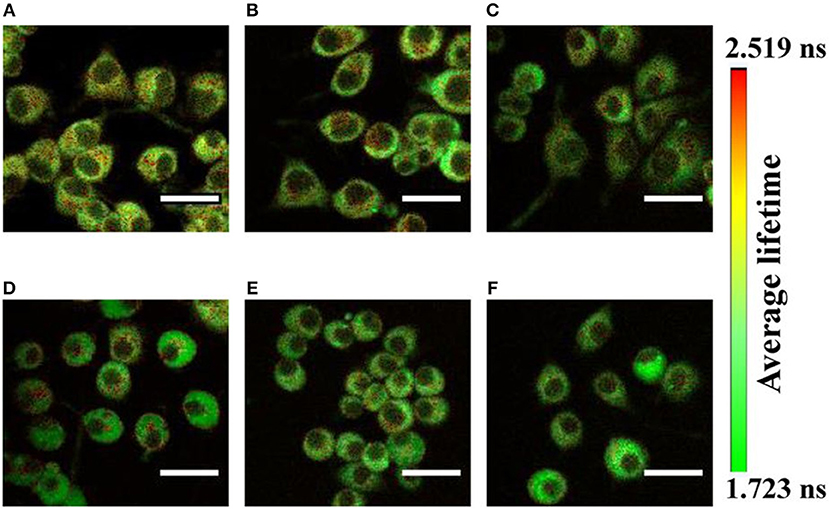
Figure 3. Shows the autofluorescence lifetime images of NADH by pseudocolor mapping in PC12 cells (A) differentiated and untreated; and treated with (B) 50, (C) 100, (D) 250, (E) 500, and (F) 1,000 μM of MPP+. The color bar represents the range of the average fluorescence lifetime. Figure reproduced (from [33]) with kind permission from Springer Nature. Scale bar: 20 μm.
Figure 3 shows the autofluorescence lifetime images of NAD(P)H in PC12 cells. The fluorescence lifetime distributions are represented in pseudo colors. The images are acquired from different fields of view. Recently, Jyothikumar et al. has reported the use of multiphoton FLIM/FRET technique to investigate the metabolic activity of cells using tryptophan and NADH imaging [69, 70]. Tryptophan is an amino acid essential for human physiology and protein metabolism that can be observed inside the cell. Skala et al. investigated the pre-cancer lesion in epithelial cancer in in-vivo TP-FLIM imaging using cellular redox ratio, NADH and FAD lifetimes, and subcellular morphology. The amount of protein-bound NADH was found to increase in high-grade precancerous tissues and increased protein-bound FAD in low-grade; however, FAD was decreased in high-grade precancerous tissues compared with normal [71, 72]. In recent years, there is an increasing need for cosmetic clinical studies which are non-invasive in particular to form a complete picture. Multiphoton microscopy is one such method, allowing us to visualize the skin in-vivo to a depth of more than 100 microns with a resolution of better than 1 micron. TP-FLIM investigated human skin biopsy with verities of dermatological disorders including psoriasis, fungal infections, nevi, and melanomas [12, 25] as well as commercialization [26, 27]. In combination with visualization of the fluorescence lifetime and special processing of three-dimensional images of the data, the approach is important in such studies [73].
Frequency domain (FD) FLIM technique is another way for fluorescence lifetime measurement. Both TD and FD FLIM techniques are available for exciting the fluorophore and acquiring FLIM images in wide-field or scanning mode, but the data acquisition and analysis differs. In FD-FLIM, the fluorophores are excited with periodically intensity-modulated light, and the demodulation frequency of fluorescence signal is measured [48, 74–76] but the phase shifts relative to the excitation time due to the delay caused by the lifetime of the fluorophore. TP-FD-FLIM measures the short lifetime of fluorophores where the phase shifts of multi-frequencies of pulse train from a mode-locked Ti:Sapphire laser is used. With laser repetition rate of 70–100 MHz and at 300 Hz of modulation frequency, this method can measure lifetimes shorter than ~10 ns. Time-domain data have intensity values in subsequent time channels, whereas frequency-domain translates into amplitude and phase values at multiples of the signal repetition rate. The fluorescence decay time can be found by measuring the delay as a phase shift (φω), where ω is the modulation frequency in radian/sec. FD-FLIM approach using the phasor plot method to analyze the lifetime is described and used for fusion proteins [75]. The photon efficiency of FD-FLIM varies depending upon the instrumentation. The efficiency increases with the modulation frequency of both the light source and the detector.
One of the limitations of an auto-fluorescence lifetime is the requirement of high photon count for best lifetime fitting and statistical analysis. In the case of biological samples, the photon counts are less and more than one fluorophore in a pixel causes artifacts for double exponential data fitting. The advanced microscopy technique and analyzing algorithm including a high quantum efficiency detector with cooling system for more photon counts and phasor plot-based algorithm for detection of various fluorophores at each pixel in the image frame are the solution for that. Although, FD-FLIM is faster as compare to TD-FLIM which requires fewer acquisition times and is used for live cells, but it is limited to high signal-to-noise ratio for high concentration fluorophore [77, 78]. Phasor technique not only applied to fluorescence lifetime imaging, this method also has been used in fluorescence microscopy, polarization-resolved SHG, hyperspectral stimulated Raman scattering (SRS) imaging [79–82].
In conclusion, FLIM has allowed us to independently assess and quantify NAD(P)H changes at the molecular level without perturbing the sensitive environment of these oxidation and reduction intermediates [5]. Integration of FLIM with fluorescence resonance energy transfer (FRET) and polarization anisotropy measurement allow for a more reliable understanding of protein-protein interaction and cell dynamics across the biological and life sciences. Additionally, a combination of super-resolution optical microscopy including stimulated emission depletion microscopy (STED), stochastic optical reconstruction microscopy (STORM), and photo-activated localization microscopy (PALM) can be a powerful tool to investigate single molecule detection [83–87].
Second Harmonic Generation (SHG) Microscopy
Second-harmonic generation (SHG), a second-order non-linear scattering process, generates only from non-centrosymmetric structures [1, 88] and the SHG intensity depends on the relative orientation between the incoming polarization state of light and the second-order hyperpolarizability of the sample [89–107]. In SHG, two photons with the same frequency are combined and generate a single photon of precisely double the energy of incident photons (i.e., half the wavelength and twice the frequency).
A polarization-resolved SHG microscopy setup uses a femtosecond Ti:Sapphire (e.g., Coherent Mira Optima 900-F) laser oscillator as the excitation light source, as shown in Figure 4. In this experiment, the excitation wavelength of 800 nm and a full width at half maximum (FWHM) of 15 nm pulses with a duration of ~100 fs (chirped pulses), the average power of the source ~550 mW and repetition rate ~76 MHz was used. The average powers of the laser beam on the sample surface were ~5 and ~15 mW for macromolecules and tissue samples, respectively. The samples were mounted in upside-down on plane XY stage (IX2-KSP, Olympus) and scanned with a laser scanning unit (Olympus, FV300). The laser beam of diameter 5 mm before the objective lens was focused onto the sample using an oil immersion objective lens (UPlanFLN 40X/N.A. 1.3 oil, Olympus Corp., Japan). The SHG signal was measured through PMT (R2949, Hamamatsu, Japan) in the forward direction using 400/40 nm (Edmund Optics Inc. Barrington, New Jersey) bandpass filter and a short-pass filter (Brightline 680 SP, Semrock) was used to discriminate the 800 nm excitation light. For SHG-circular dichroism (CD) measurement, the microscope setup is similar to that of SHG microscopy; however, a quarter-wave plate or a Soleil-Babinet compensator was inserted in polarization state generator (PSG) to manipulate the handedness of circular polarization. The quality of circular polarization is also very important, i.e., the maximal to minimal laser power, characterized by an analyzer and a power meter, should be kept below 1.1 to eliminate the effects of in-plane anisotropy [109, 110]. The SHG susceptibility imaging is also performed in a similar experimental setup [90]. For Stokes vector measurement, the SHG signals are analyzed using a polarization state analyzer (PSA), specifically, a four-channel Stokes-polarimeter. The basic principles of Stokes polarimetry using four channels are discussed in detail [107, 111, 112]. The forward propagating SHG signal is limited to the thickness of the samples, however, backscattered SHG measurement demonstrates the potential of the technique for in-vivo imaging by the use of fiber-optics based endoscopic probe [103, 112–114].
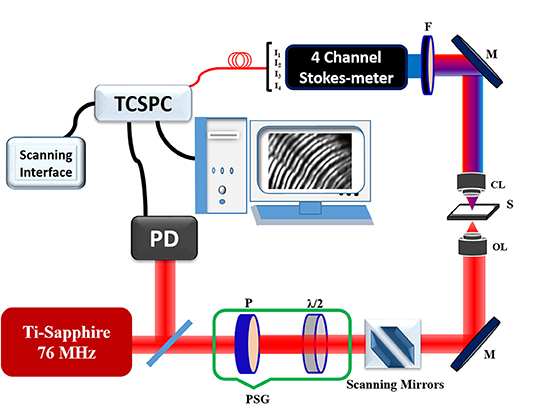
Figure 4. Schematic diagram of the polarization-resolved SHG microscope with a four-channel Stokes-polarimeter module. The insert displays the polarization state analyzer (PSA); PSG, polarization state generator; P, Polarizer; λ/2, half-wave plate; λ/4, quarter-wave plate; M, mirror; S, sample; F, filter; BS, beam splitter; FR, Fresnel Rhomb; W1 and W2, Wollaston prisms; Ia, Ib, Ic, Id, photomultiplier tube (PMT). The figure is reproduced with permission from Elsevier [108].
The strength of the SHG signal depends on the structure of the sample and collagen has stood out amongst the regularly analyzed structures because of its non-centrosymmetric nature in various biomedical applications. Most of the studies on SHG imaging of extracellular matrix are concentrated on imaging collagen fibers in a variety of connective tissues and internal structures [72, 89, 91]. Amongst the 20 different forms of collagen, type I collagen is the transcendent constituent of the connective tissue and extracellular matrix [90, 91]. Therefore, identification and characterization of collagen type I using SHG microscopy improve our knowledge about how cell-matrix interacts, matrix organization, density, and composition affect tumor formation and progression. Going beyond intensity measurements, polarization-resolved SHG provides an interesting way to probe the molecular structure of organized organic media [91–93], material characterization [94] and also biological tissues (fibrotic collagen, human dermis, keloid, cornea, microtubules as well as myosin of the skeletal muscle) through the measurements of sample retardance, anisotropy via birefringence, attenuation, and depolarization of the SHG light [91, 95–102]. Conventional polarization-resolved SHG microscopy examines the linear birefringence and polarization anisotropy of samples [91, 103, 104]. However, various components in the optical path including a high numerical aperture objective lens cause polarization distortions in excitation and detection arms [105, 106]. Therefore, Stokes vector based SHG microscopy overcomes the above-mentioned drawbacks and measures various polarization parameters; the degree of polarization (DOP), degree of linear polarization (DOLP), and degree of circular polarization (DOCP) through the full polarization states of SHG light. The method investigates the molecular organization through the characterization of the polarization properties of SH signal from KDP crystal, collagen type-I, starch granules, skeletal muscle fiber, etc. [107]. Hence, polarization-resolved SHG imaging offers additional parameters including circular dichroism, susceptibility, Stokes vectors and can be used as measuring parameters in the detection of subtle changes in tissue structure associated with collagen-related diseases including fibrosis, cancer, wound healing, osteoarthritis, etc. and as an alternative for histopathology studies [13, 40, 41, 63, 90].
SHG microscopy is applied to image the representative fibrillar structures of bio-molecules and photonic materials, namely collagen in rat-tail tendon, skeletal muscle fibers from the hind legs of adult mice, potato starch granules, urea crystals, and BaTiO3 nanocrystals as shown in Figure 5. The samples are sandwiched between two cover glass slips and mounted upside-down in the microscope stage for SHG imaging. The acquired images are analyzed by ImageJ (Fiji, NIH, USA) software. A strong SHG signal is observed from collagen (Figure 5A) since it forms positively birefringent in respect to the length of the individual fibers and cylindrical array of polypeptide coils comprising glycine-proline helices [120]. As we can see in Figure 5B, strong SHG signals are observed from the myosin macromolecules in muscle. The high second-order non-linearity demonstrates that the fibrous protein myosins are densely packed in an orderly fashion. Muscles are organized in fiber bundles called myofibrils and are longitudinally segmented into contractile units called sarcomeres. The sarcomeres are the structural and functional unit of the muscle fiber and are connected serially [121, 122]. The direction of the myofibrils depends on the alignment and length of the sarcomeres, which are around 2–3 μm, depending on the type of muscles [123] and the SHG signal depends on the molecular structure and order of the myofibrils. SHG image of skeletal muscle fiber highlights the different morphologies of the adjacent striated appearance of microfibrils present within skeletal muscle with several sarcomeres. Again, the intrinsic SHG signal is related to the structural organization and functional physiological properties of myofibrils [102, 116]. Figure 5C shows the SHG image of potato starch granules which are composed of the alternating concentric 120–400 nm thick amorphous and semi-crystalline domains structure. The crystalline layers consist of short amylopectin branches which form the double-helical structures are strongly anisotropically aligned within the focal volume of the laser and thus yield a strong second-order non-linear optical response. The radial arrangement of amylopectin in the starch granules is clearly visualized with a hilum in SHG image. Figure 5D shows the SHG image from urea crystals. Figure 5E shows the SHG image of BaTiO3 nanocrystals. It possesses large second-order non-linearities having a non-centrosymmetric structure, and the SHG signal from BaTiO3 nanocrystals propagates in a forward direction due to the phase matching.

Figure 5. SHG images of (A) Collagen fibers in ex vivo biopsies of thyroid capsule [115], (B) myofibrils in mice [116], (C) potato starch granules [117], (D) urea crystals [118], and (E) BaTiO3 nanocrystals [119]. All figures were modified with permission from respective Journal publishers.
SHG-CD Microscopy on Collagen
SHG microscopy with circularly polarized excitation has been attracting considerable attention for its applicability in the detection of chiral molecules. Differences in SHG intensity resulting from right-handed circular polarization (RCP) vs. left-handed circular polarization (LCP) can be used for the three-dimensional (3D) imaging of chiral macromolecular structures at the tissue level. The term used to describe this effect is SHG-circular dichroism (CD), which is formulated as
where IRCP and ILCP are the intensities with right circular polarization and left circular polarization, respectively. SHG-CD results from electric dipole interactions at surfaces/interfaces. There is evidence to suggest that the effect of SHG-CD is 2–3 orders of magnitude larger than that of traditional CD [124–126].
Despite the advantages of SHG-CD in terms of contrast ratio, no attempts were made to apply this phenomenon to microscopic imaging until methods were devised for implementation in an optical microscope with counter-propagating geometry to investigate the intrinsic chirality of a supported planar lipid bilayer [127]. SHG-CD has also been integrated into a laser scanning microscope to increase imaging speed and enable 3D tissue imaging. This approach takes advantage of the optical sectioning capability of SHG and shares the same microscope base with other non-linear optical imaging modalities [128–131].
To the best of our knowledge, SHG-CD microscopy is the first imaging scheme capable of differentiating normal tissue from osteogenesis imperfecta (OI) [128]. Structural defects in the collagen triple helix can be detected by observing the SHG-CD contrast, wherein a sign change is associated with the orientation of collagen fibers. After that, type-I collagen and starch granules are utilized to explain the sign change and variations in SHG-CD, which are sensitive to 3D macromolecular structure (macroscopic chirality) rather than the molecular arrangement in the supramolecule (molecular chirality) [109, 130]. This makes it easy to determine the 3D molecular orientation based on the SHG-CD ratio, as long as the correlation between molecular orientation and second-order susceptibility χ(2) is known. Recently, this relationship has been characterized using the χ(2) tensor transformation between the laboratory frame and the frame defining a single collagen fibril, while taking into account the chiral tensor element, χ14 [110]. The pixel by pixel calculation on SHG intensity images for RCP and LCP excitations derives SHG-CD ratios (Equation 2). In the following, we present a number of important issues about SHG-CD microscopy. C∞V symmetry is commonly used in cases of type-I collagen, under the assumption that the collagen molecules are rod-shaped, which would mean that the hyperpolarizability in α1- and α2-chains is the same [114]. In this situation, the entire system is achiral, such that no SHG-CD effects would be observed. By contrast, cases where hyperpolarizability in α1- and α2-chains is different the system becomes chiral which manifests as a chiral tensor component χ14. In this situation, the symmetry is C∞, which is suitable for the analysis of SHG-CD images [109].
Moreover, the SHG-CD ratio provides an indication of the out of plane angle δ of collagen fibrils (relative to the image plane) within the voxel. It follows that the pixel with the highest average SHG intensity among the images for RCP and LCP possesses zero SHG-CD and zero δ. Conversely, a smaller average SHG intensity is associated with a higher absolute SHG-CD value and larger δ (Figures 6D–F). Note that a transition from negative to positive SHG-CD values would indicate that the collagen fibrils are pointing downward to upward from the image plane. The image size is 470 × 470 pixels with a pixel dwell time of 50 μs. Figure 6B shows the difference in SHG intensity for RCP and LCP excitations. Consequently, the correlative analysis of Fast Fourier Transform (FFT) on planner orientation (Figure 6A, insets) [132] and SHG-CD on δ (Figure 6C) fully determines the molecular orientation of collagen fibrils in 3D.
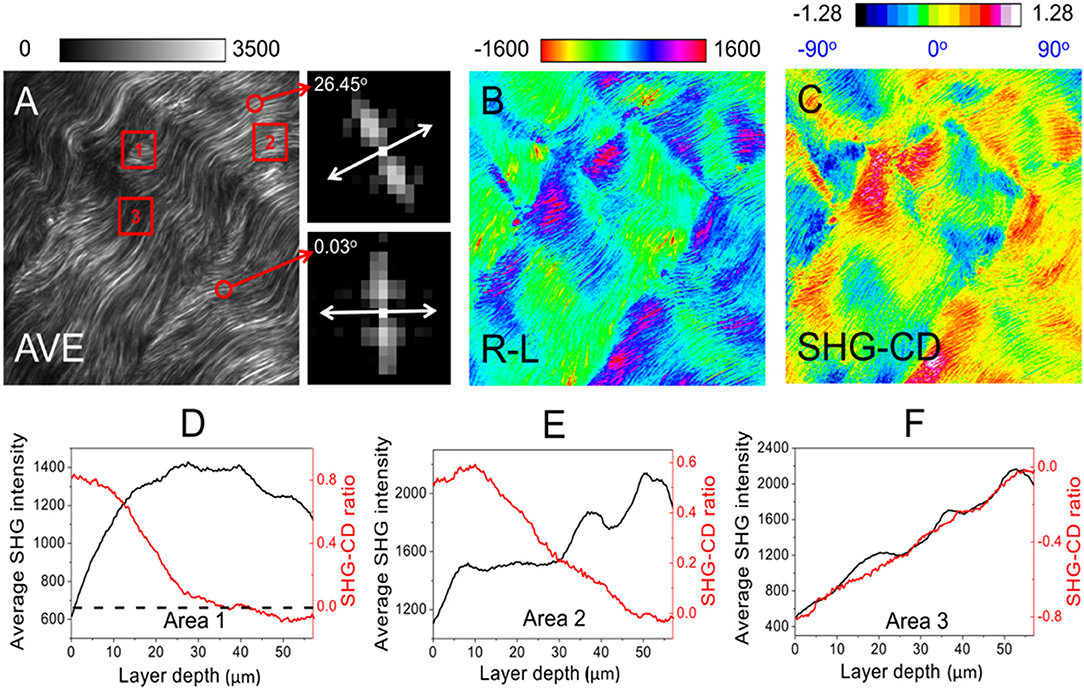
Figure 6. (A–C) Representative images used to determine the 3D molecular orientation in a pig tendon. The images clearly show the alignment of a large number of collagen fibrils, thereby demonstrating the feasibility of SHG-CD microscopy for collagen characterization. An Olympus objective lens, UPlanSApo 40X/N.A. 0.95 is used for SHG forward imaging. The insets besides (A) present the FFT results from the selected fibrils (indicated by the red circles), in which the planner orientation (double-headed arrow) is perpendicular to its orientation in the frequency domain. In (D–F), we can see how SHG-CD is correlated with the average SHG intensity with respect to layer depth [analyzed in the red squares, namely Area 1–3 on (A)]. The black dashed line in (D) indicates that the maximal average SHG intensity corresponds to zero SHG-CD, such that a significant change occurs at this point. In regions where SHG-CD is positive (δ > 0) or negative (δ < 0), they have the opposite or the same trend with the curve of average SHG intensity, as shown in (E,F), respectively. The label AVE in (A) refers to an average of the images for RCP and LCP, whereas R-L in (B) refers to the difference between the images for RCP and LCP. Image volume: 200 × 200 × 58 μm3. The figures have been reproduced with permission from American Institute of Physics (AIP) [110].
In the following, we outline some of the advantages of SHG-CD microscopy. Image stacking is widely used to resolve the organization of collagen in 3D; however, this method is time-consuming and inapplicable to the real-time imaging of structural variations in collagen fibrils. In contrast, SHG-CD microscopy can be used to determine the 3D molecular orientation using only two cross-polarized SHG images (through the excitations of RCP and LCP), which makes it possible to obtain 3D-resolved SHG images [109, 130]. This method eliminates much of the workload of photon collection to produce an image stack. It also reduces the problems of drift and sample variation in in-vivo biological experiments. Furthermore, image stacking is inapplicable to dense connective tissue due to difficulties in determining the 3D molecular orientation of individual collagen fibrils. There is insufficient image contrast by which the orientation angle with two adjacent image sections can be measured. However, SHG-CD microscopy prevents this problem and is readily used to determine the fibril orientation angle without the need for an image stack.
Susceptibility Based SHG Microscopy
SHG microscopy with linearly polarized excitation light can differentiate the type of molecular sources of SHG signals based on its susceptibility (χ) components, which typically cannot be distinguished in a standard SHG intensity image [133]. In biomedical diagnostic applications, differentiation among the constituents of the tissue and, sensitive detection of pathological features are essential. For example, the major constituent of fibrocartilage is collagen type I while the major constituent of hyaline cartilage is collagen type II. In osteoarthritic cartilage replacement treatment, the regenerated cartilage (newly replaced cartilage) is typically fibrous one and not the hyaline cartilage. Therefore, a tool that can differentiate the type of collagen fiber in the extracellular matrix (ECM) of articular cartilage without biopsy could be highly valuable during cartilage assessment.
The mathematical model associated with SHG intensity due to collagen fibers has been described in several articles [123, 133–135]. In brief [136], applying the cylindrical symmetry for collagen fibers, assuming that the sample lies in the xz-plane (Figure 7A) and, the propagation direction of the linearly polarized excitation light is along the y-direction (out-of-plane); the intensity (I) of the emitted SHG signal from collagen fibers can be modeled as
where I, represent intensity of SHG signal, polarization angle of the excitation light, orientation angle of the collagen fiber and the second-order susceptibility tensor ratios, respectively. By fitting the Equation (3) to the numerical values of measurements, the χ-tensor element ratios i.e., χ1andχ2 can be determined. The intensity (I) and the angle of polarization (θl) are known variables while other variables can be obtained by least-square fit to the model. The Gaussian fit to the histogram plot of susceptibility tensor components provides the quantitative value of χ1, andχ2, which are characteristics of the tissue [90]. By providing a pseudo-color to the χ-tensor element ratios, SHG based susceptibility image can be obtained.
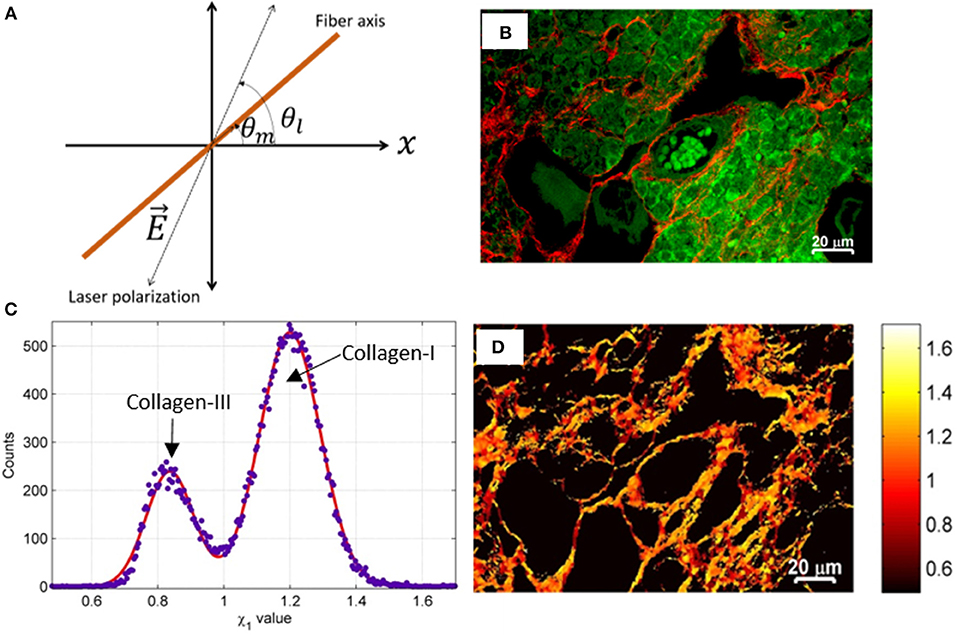
Figure 7. The simplified schematic diagram of the coordinate system that demonstrates the principle of susceptibility based SHG microscopy of tissue. (A) The direction of linearly polarized excitation light beam (dotted line) and orientation of fiber (brown colored line) are shown. (B) SHG (red) and TP auto-fluorescence signal (green) of ovary tissue. (C) The Gaussian fit of the histogram to χ1 values demonstrates the relative content of collagens. (D) Pseudo-colored χ1 image of collagen in ovary tissue. The χ tensor element ratio is shown in the color-coded scale. Images are adapted and reproduced after permission [90].
To perform susceptibility imaging, several SHG images of tissue are acquired by the rotation of linearly polarized light. These acquired images can be termed as excitation resolved SHG images. Depending on the experimental system, at each rotation of linearly polarized excitation light, the ellipticity introduced by the optical elements is compensated along the path. Thus, a suitable calibration and associated compensation are highly recommended before the acquisition of excitation resolved SHG images. Also, the polarization scrambling due to the high NA microscope objective lens and tissue birefringence should be taken into account [90, 137].
The application of susceptibility based SHG microscopy is very limited to date. In the past few years, polarization-resolved susceptibility based SHG microscopy has been used for several biomedical applications such as characterization of breast cancer, skin, muscle, and engineered cartilage tissues [135, 136, 138]. Automated control for the linearly-polarized excitation light to perform susceptibility based SHG microscopy on commercial microscopes is demonstrated recently [139]. Collagen is one of the commonly found structural proteins in ECM in a human body that is modified in structure or content in several disorders, e.g., connective tissue diseases, cardiovascular diseases, autoimmune diseases, ovarian diseases, etc. [91]. Recently, Dong et al. has demonstrated the optical discrimination of type I and type III in rat skin while Rajesh et al. has shown the detection capability of type I and type II collagen in osteoarthritic human cartilage [90, 140]. The ovary is a highly dynamic tissue whose ECM composition is changing continuously. Label-free detection of collagen types and associated alteration using susceptibility based SHG microscopy could be highly useful in finding the hidden features of a biophysical dynamic process in the ovary tissue (Figures 7B–D). In general, susceptibility based SHG microscopy can quantitatively discriminate several harmonophores (e.g., collagen type I, II, III, muscles etc.) and thus could emerge as a powerful tool for detecting early-stage ECM modification at molecular level with high sensitivity, and can contribute in understanding various associated pathophysiological process [130, 133, 139, 140].
Stokes Vector Based SHG Microscopy
The experimental arrangement of four-channel photon counting based Stokes polarimeter integrated to the SHG imaging system is shown in Figure 4 and described in detail in Mazumder et al. [112, 117]. The SHG signal is collected in the forward direction and divided into four polarization components which are a combination of linear and circular states. The signals are synchronized by four-channel detector router (PHR 800, PicoQuant GmbH, Berlin, Germany) and TCSPC system. Stokes vector based SHG microscopy measures the complete polarization states and various polarization properties of SHG signal from starch granules in laboratory conditions. Starch is a polysaccharide, which is common in human diet. Starch generates a strong SHG signal due to the geometrical characteristic and SHG active molecule (amylopectin) present in it [136]. The conventional optical microscopy does not provide any information about the structure of starch granules. However, non-linear optical microscopy, specifically SHG microscopy, provides exact structural information on molecular order and orientation of starch granules along with the 3D resolution and deep imaging capabilities [117, 141]. Polarization measurements integrated to SHG imaging provides complementary information about starch micro-domains. Further, physically important polarization parameters, such as DOP, DOLP, DOCP, and anisotropy (r) of the SHG signal are calculated at each pixel of the scanning area from the Stokes parameters [107, 112, 141].
Figure 8 shows the 2D Stokes images of SHG signal from starch granules with varying the incident polarization and detecting the SHG signal originating from a single plane. S0 denotes the total intensity of emitted SHG signal, S1 gives the difference between the intensities of horizontal and vertical linearly polarized light at 0 and 90°, S2 is the difference in intensities of the polarization states of linearly polarized light at 45 and −45°, and S3 is the intensity difference between right and left circularly polarized light. With linearly polarized excitation light, the SHG signal is observed from a region perpendicular to the axis of polarization, which indicates the radial distribution of SHG active molecule (amylopectin) in starch.
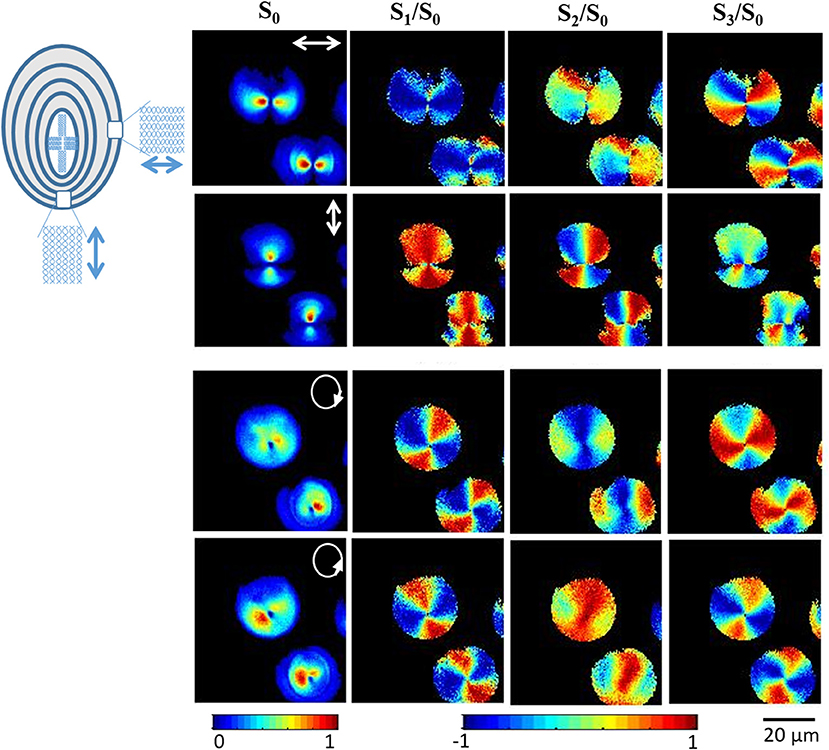
Figure 8. Experimental reconstructed 2D Stokes images of the SHG response from starch granules for horizontally, vertically, right and left circularly polarized illumination, respectively. White arrows in the leftmost images indicate the direction of polarization. The color scale shows the value of each Stokes parameter increasing from blue to red. The figure has been reproduced with permission from OSA [111].
Figure 9 shows 2D images of various polarization parameters when illuminated with different polarized states of light. This microscopic technique allows us to obtain the complete polarization states of the resulting SHG signal and gives significant molecular orientation information with high spatial resolution on further analysis of the Stokes parameter. For example, analysis of S3 and DOCP parameters depict the chirality of the amylopectin in starch. This study also concluded that linearly polarized light can be used to find out the right or left-handedness of helical structures of amylopectin. SHG provides a full understanding of complex material with detailed knowledge of the non-centrosymmetric molecular structures or arrangement of molecules involved.
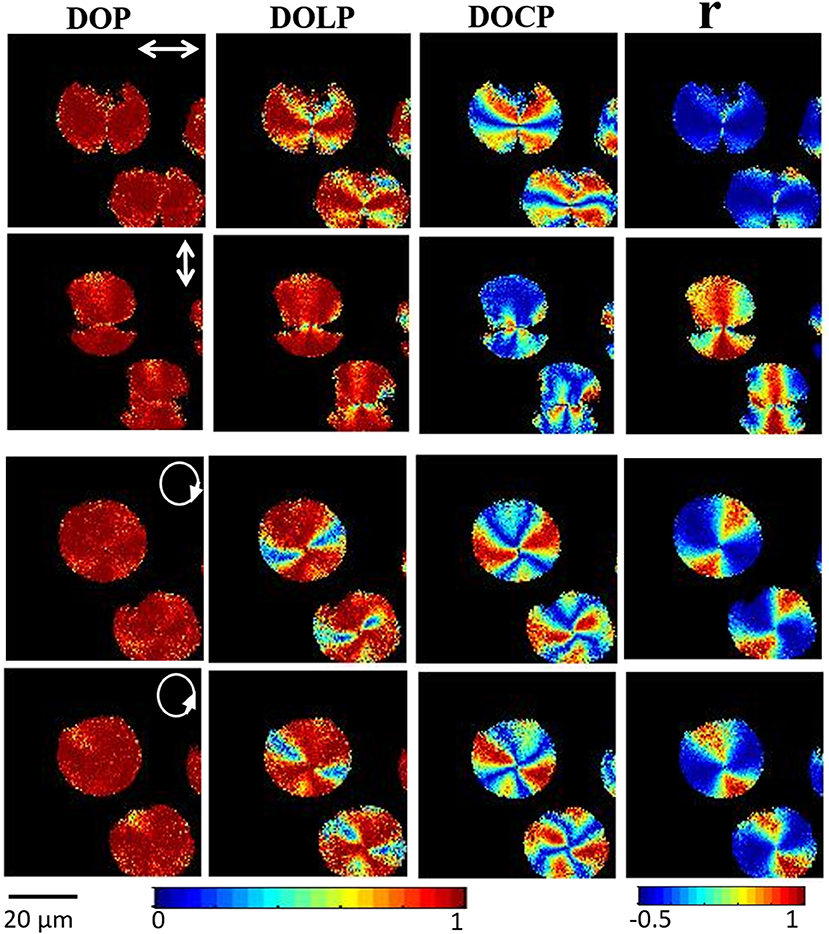
Figure 9. Experimental polarization-resolved SHG response from starch granules; the 2D reconstructed DOP, DOLP and DOCP and anisotropy (r) images of SHG signal from the starch for horizontal, vertical, right, and left circular polarization, respectively. The direction of polarization is indicated by a white arrow in the images. The color-coded scale shows the values of each parameter, from blue (minimum) to red (maximum). The figure has been reproduced with permission from OSA [111].
The use of SHG microscopy for imaging of starch granules and cellulose has offered us a lot more information about its structure and molecular properties. On further combining of SHG microscopy with other microscopy techniques including CARS microscopy, highly detailed information of the specimen can be obtained.
Coherent Anti-Stokes Raman Scattering (CARS) Microscopy
SHG and THG techniques provide label-free imaging with high contrast due to the non-linearity of the material. One of the major limitations of these techniques is the molecular contrast. However, Raman spectroscopy is a powerful optical method to identify the molecules uniquely [142, 143]. Unfortunately, the cross-section of spontaneous Raman scattering is very small (more than 10 orders of magnitude than fluorescence), which needs high excitation power and long data acquisition time and hence limits its applications for live cells. Multiphoton vibrational microscopy based on CARS, which was first reported by Maker and Terhune in 1965, has taken care of the difficulties in spontaneous Raman microscopy [144]. The first CARS images of living cells were demonstrated in 1999 by Zumbusch et al. [145]. CARS microscopy has risen as a promising imaging methodology that is free of exogenous labeling for identification and employed on biomedical applications including the viruses, cell, tissue imaging, and also in drug delivery [146–149]. CARS microscopy is described as a four-wave mixing process, that probes the third-order non-linearity of molecules and it consists of the pump (Wp), Stokes (Ws), probe (Wp) beams (same as a pump in the case of degenerate CARS). These beams simultaneously interact with the sample and when the frequency of Wp – Ws matches with the frequency of a Raman active molecular vibration then an anti-Stokes frequency (Was = Wp – Ws + Wp) is generated. The blue-shifted light (anti-Stokes signal) is used for microscopy application and is free from fluorescence background. By excitation of the CH vibrations (2,600–3,000 cm−1) in proteins and lipids, CARS has been used to image them inside the cells and tissues. However, CARS signal produces a non-resonant background.
The development of the experimental CARS microscopy setup has been previously described in Mazumder et al. [68] and Slepkov et al. [150, 151]. Briefly, a single femtosecond Ti:Sapphire oscillator (Coherent Mira Optima 900—F) was used as the excitation source in the CARS microscopy system (as shown in Figure 10). The center wavelength of the laser is at 800 nm, and with transform-limited pulses of ~100 fs, the average power was ~550 mW, and the repetition rate is 76 MHz. The laser beam passes through two chirp mirrors to compensate the dispersion and then is split into two by a 50:50 beam splitter. One beam is used as a Stokes pulse (Ws) and is propagated in a photonic crystal fiber (PCF, NKT Photonics, FemtoWhite CARS) to generate fundamental soliton pulse. The output of the fiber is filtered by 950 nm long-pass filter. The broad infrared pulse of Stokes beam from PCF is automatically synchronized with pump laser. This broad Stokes spectrum allows us to tune the Raman difference from 2,300 to 4,000 cm−1. The other half of the laser, pump pulse (Wp) is sent to a retroreflector mounted on a computer-controlled delay stage. After the delay stage, both the pump and Stokes beam are overlapped collinearly and focused on the sample using an objective lens (40X 1.15 NA UAPO water immersion lens). The total average power of combined pump and Stokes beam is 120 mW before the objective lens. In this experiment, a modified Olympus IX71 inverted microscope with FluoView 300 laser scanning system was used. Both CARS and SHG signals from the samples are collected in the forward direction with a 0.8 NA objective lens. The signals are discriminated using a 400–700 nm bandpass filter against pump and Stokes beams and are collected by PMT with enhanced red sensitivity (PMT, R2949, and Hamamatsu, Japan). Two-photon auto-fluorescence signal is collected using PMT in epi-detection mode and the CARS signal is detected both in epi and forward direction depending upon the samples under study. TCSPC system integrated with Symphotime software (Picoquant, Germany) is used for detection and analysis of both auto-fluorescence and CARS signal. Its coherent nature is an advantage over traditional Raman signal, thus offering video-rate imaging speed [147]. A recent publication demonstrated that multiplex CARS microscopy has been utilized in the detection of cancer and to differentiate the normal breast tissue and tumor [148]. Integration with multimodal multiphoton microscopy provides more information; simultaneous detection of CARS and SHG signals help to investigate the chemical and structural properties of the sample [149].
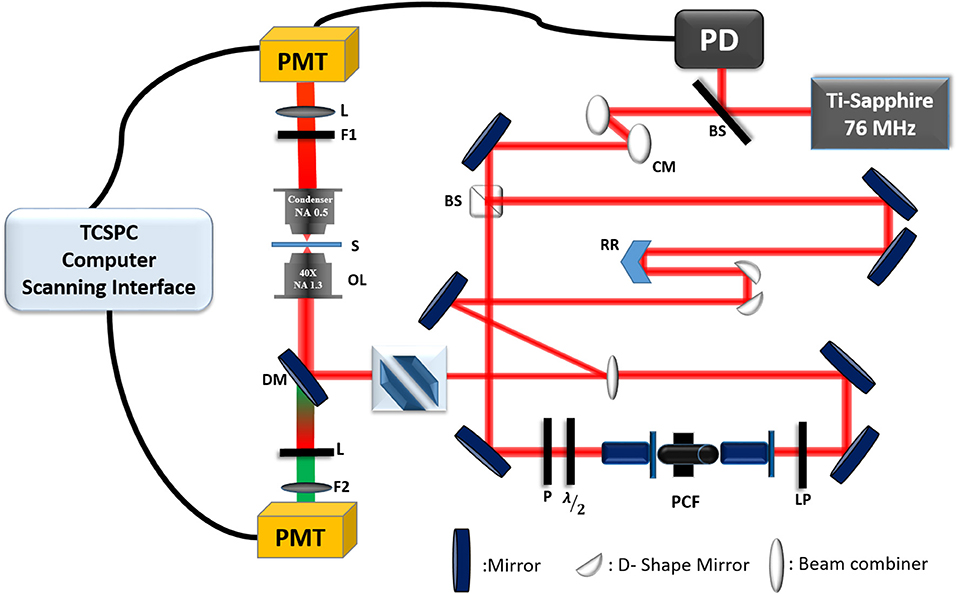
Figure 10. Experimental setup of CARS microscopy integrated into a time-resolved fluorescence lifetime imaging system. BS, beam splitter; PD, photodiode; CM, chirp mirror; RR, retro-reflector; LP, long-pass filter; PCF, photonic crystal fiber; λ/2, half-wave plate; P, polarizer; DM, dichroic mirror; OL, objective lens; S, sample; F1, 670/30 nm filter; L, lens; F2, 480/40 nm filter; PMT, photomultiplier tubes; TCSPC, time-correlated single-photon counting. The figure is reproduced with permission from Slepkov et al. [150, 151] and Pegoraro et al. [152].
Polarization Resolved CARS Microscopy
Conventional CARS microscopy maps the 3D distribution of a molecular species with high spatial resolution. But this technique is inherently capable of deciphering the average orientation of the molecules in 3D using the polarization of light [153, 154] which is valuable information for studying molecular assembly, dynamics, and function. For example, an organization of lipid molecules in biological membranes, local changes in lipid orientation due to molecular interactions or steric hindrance help us in understanding the functioning of these membranes [155]. Polarization-resolved fluorescence microscopy can provide us similar information, but it requires rigidly attached fluorescent probes which might perturb the local environment due to steric hindrance [156]. On the other hand, since polarization-resolved CARS (P-CARS) is based on the endogenous contrast of a molecular species, it can monitor the molecules directly in their native environment. Before proceeding to describe P-CARS in greater detail, it is worth pointing out another technique called polarization CARS which suppresses non-resonant background in CARS microscopy by selecting the polarization of signal [157]. There is very little difference in the naming of these techniques, but one is focused on decreasing the background, and the other is aimed at determining the orientation of molecules. Here the discussion is focused on the later only.
In order to get a strong CARS signal, the polarization of the lasers (pump and Stokes) should be aligned so as to maximize vibrations in the bonds within the focal volume. Bond vibration can have different modes, that result in closely spaced but spectrally distinct Raman peaks. In the following text, unless explicitly mentioned, we will only look at vibrations parallel to the target bonds. In other words, we focus only on three components of the CARS tensor γxfff, γyfff,and γzfff where x, y, z are the axes in lab frame, f = f (θo,φo) is the average orientation of the bonds in the focal volume with respect to the lab frame of reference. The direction of propagation of light is along the z-axis, x-y defines the transverse plane, and θo and φo refer to angles in the spherical coordinate system. In the most common form of P-CARS, the pump (Epump) and the Stokes (EStokes) lasers are linearly polarized and parallel. The polarization angle (φ) of the two beams is simultaneously rotated from 0 to 180° in small steps (Figure 11) while recording the corresponding CARS signal [159]. Being a third-order non-linear process, CARS (ECARS) is highly sensitive to the angle between exciting lasers and the vibrating bonds. The signal is maximum when the exciting lasers are parallel to the bond and minimum when they are orthogonal to the bond (Equation 4). This modulation of CARS signal with the polarization angle helps in determining the average orientation of molecules in the focal volume. If is a unit vector along the direction of the targeted bonds, the strength of the CARS signal varies as
This polarization dependence of the CARS signal was evident in experimental results soon after the technique gained popularity as a form of non-linear optical microscopy. The ordered arrangement of water molecules sandwiched between lipid bilayers in multi-lamellar vesicles (MLVs) was observed for the first time using CARS microscopy [160]. By tuning to C-H bond vibration, the orientation of lipid molecules in stacks of bilayers [161, 162] and even a single bilayer like cell membrane can be determined with high accuracy [141]. P-CARS of the same C-H bond has shown that the orientation of polyglucan chains in cotton and rayon fibers remains unaltered in dry and hydrated states [158]. In neurobiology, the charge insulating myelin sheath around axons consists of multiple lipid bilayers and P-CARS was used to study the arrangement of lipid molecules in ex vivo [163] as well as in in-vivo samples [4, 164]. P-CARS and polarized non-linear optical microscopy, in general, can be used to determine not just the orientation of molecules but also the nature of molecular assembly [89, 159, 165]. It can quantify how the packing of lipid molecules changes with their composition in bilayers [159, 162]. Quantitative evaluation of high order lipid assembly in myelin sheath can be used for early diagnosis of neurodegenerative diseases [165]. Advances with hardware-based demodulation of P-CARS signal [166] and fast electro-optic polarization [167] have improved the speed of the technique to enable the study of dynamical changes in molecular assemblies. The fast polarization modulation technique is also sensitive enough to track changes in the lipid orientation of cell membrane as it moves freely, and it is a worthy alternative to polarization resolved fluorescence techniques in membrane studies. Another implementation of P-CARS uses different combinations of circularly polarized lasers to target vibration symmetries in molecular assemblies [168].
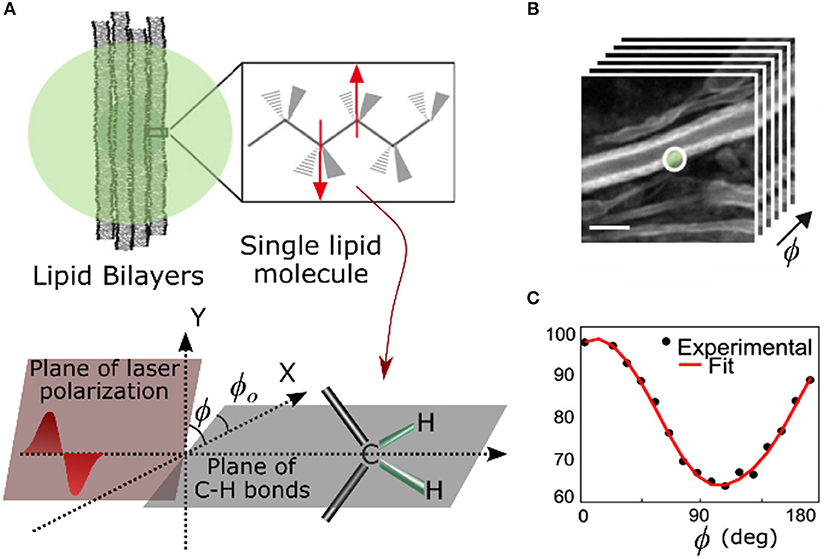
Figure 11. (A) Cartoon depicting lipid bilayers membranes with orientation of C-H bonds (φo) and laser polarization (φ). (B) A stack of CARS images of myelin sheaths in a mouse spinal cord tissue for different polarizations of excitation lasers and (C) the corresponding modulation in CARS signal from the while circular region in (B). Scale bar: 5 μm (Adapted from Gasecka et al. [158] with permission from ©Biophysical Society).
To summarize, P-CARS gives us the ability to look at molecular assembly within sub-micrometric volumes along with 3D-resolved imaging. It is a far-field optical microscopy tool, which can be used with a wide variety of samples, and it is non-invasive. It also enjoys the molecular specificity of CARS, and therefore no external contrast agents are required. These features make P-CARS a promising tool for imaging ex vivo and in vivo biological samples. Recent advances to improving the speed of polarization modulation and hardware-based demodulation have made it possible to study the dynamics of molecular assemblies on millisecond timescales [159, 165, 167]. Applications of P-CARS have so far been limited given its advantages, primarily because of lack of access to such advanced imaging tools but that should change the near future.
Multimodal Non-Linear Microscopy Approaches
Similar experimental set-up is used in all the microscopy techniques as discussed above and help us to get a different contrast/information from the sample (Table 1). It is therefore common to combine these techniques based on the nature of the experimental study and it is referred to as multimodal imaging. Special attention is required to ensure that these contrast mechanisms do not interfere with one another. The number of multimodal imaging experiments reported in the literature is enormous, but we will focus on only a few examples here.
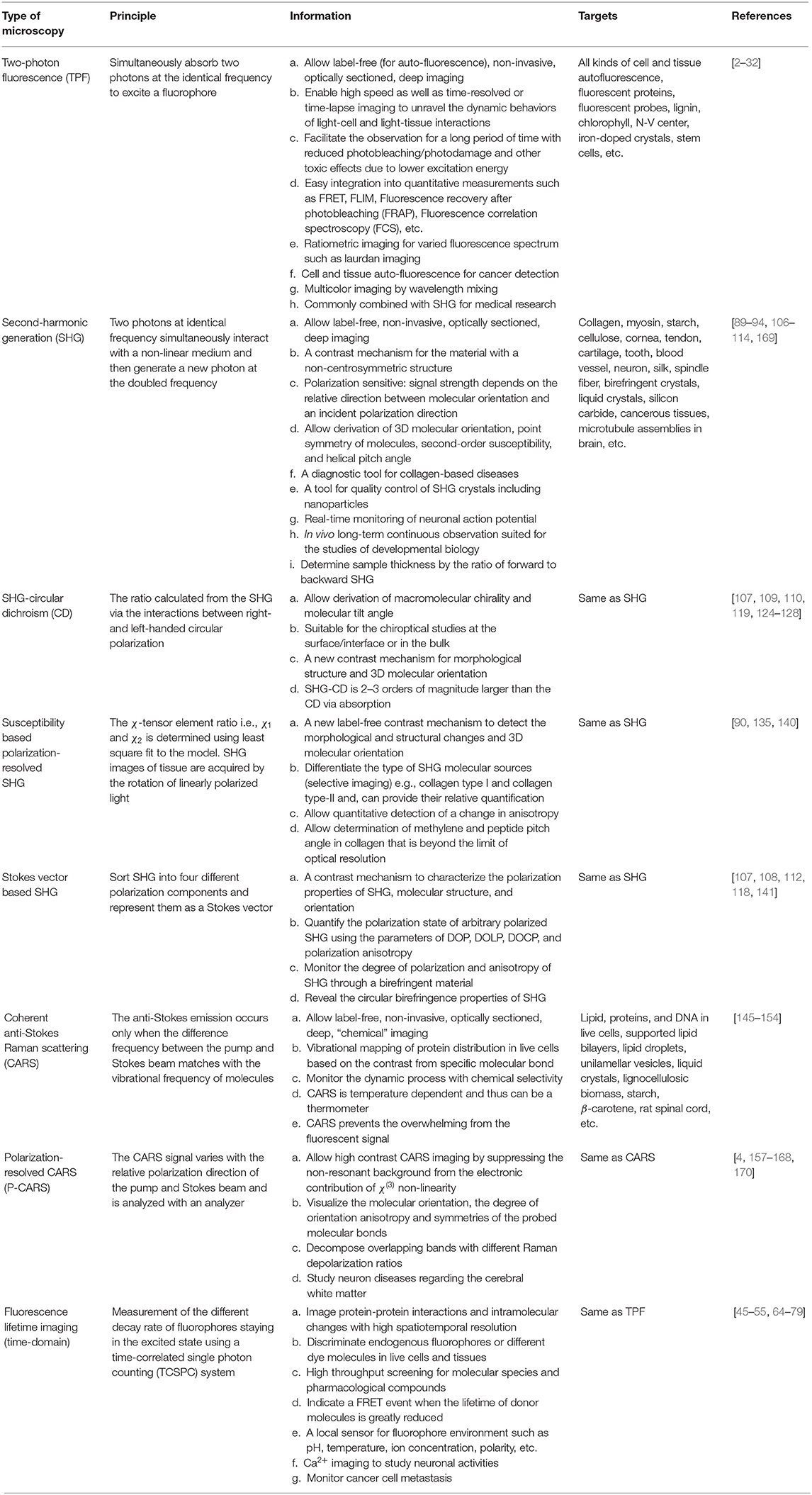
Table 1. Comparison of various microscopy techniques.
TPF+FLIM+SHG Microscopy
One of the most common forms of multimodal non-linear optical microscopy involves simultaneous SHG and TPF imaging as the excitation laser and the image acquisition mode are the same, and the two signals can be separated spectrally in two-channel detection. The fluorescence signal can be further analyzed when the detector is attached to TCSPC electronics. SHG technique along with TPF imaging investigate the molecular organization and key aspects of cellular metabolism through auto-fluorescence signals from endogenous fluorophores such as flavins, NADH and retinol or exogenous labels [39, 71, 72, 171]. Therefore, metabolic activity from endogenous TPF and collagen distribution from SHG is a simple but potent combination for in vivo microscopy. It was reported that the combination of TP-FLIM and SHG microscopy investigated cellular metabolism and collagen deposition during skin wound healing [172, 173]. At the beginning of healing, the average fluorescence lifetimes of NADH and also SHG intensity were decreased which suggests the lower metabolic activity and degradation of collagen in dermal layer (Figure 12). The SHG intensity is increased with wound healing which is due the deposition of collagen to fill the wound gap. It is an example of a detailed study of biological activity in a live animal without the aid of external contrast agents which solely relies on the capabilities of the imaging system. Analysis of chronic wounds using label-free multiphoton microscopy has emerged as a useful imaging modality capable of quantifying changes in cellular metabolism. It includes estimation of an optical redox ratio of FAD/(NADH+FAD) auto-fluorescence. An ability of application of multiphoton microscopy to monitor changes in the metabolism of individual full-thickness skin wounds in vivo was demonstrated [152, 172].
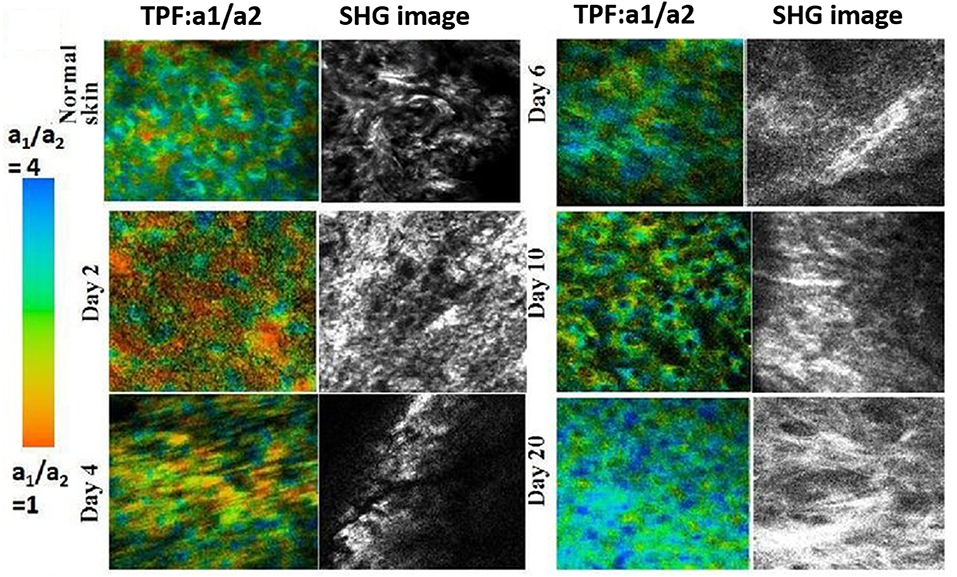
Figure 12. TP-FLIM (color-coded) image of NADH free-to-bound ratio (a1/a2) and SHG (gray) images of collagen during wound healing. Figure reproduced (from [172]) with kind permission from SPIE.
To characterize epithelia morphology in-vivo as well as ex-vivo in animal and human tissues involving fresh biopsies, cellular NADH auto-fluorescence in two-photon modality has been used as a marker [173]. Several epidermal layers of skin were resolved in vivo at subcellular spatial resolution with the use of TPF imaging based on the morphology of the cellular features [115, 174, 175]. Wound healing in cutaneous tissues, especially in cases of chronic wounds are characterized by prolonged and excessive inflammation followed by the formation of callus and infection. The wound shows improper vascularity due to endothelial inflammation. The current therapies target extensive clinical success, but there is no exact therapy or development of biomarkers to determine and evaluate the status of these wounds. TPF microscope has shown numerous advantages in depth evaluation and collection of the signal. The measurements of intrinsic fluorescence of NADH and FAD, an optical redox ratio, eventually find out the relative rates of glucose metabolism and oxidative phosphorylation increase the quality of prognosis. Label-free multiphoton microscopy (MPM) assesses the initial quantitation of the metabolic biomarkers in diabetic and non-diabetic patients [176].
In vivo imaging of pigmented lesions in human skin was performed with MPM (MPTflex, JenLab, Germany) [177]. The goal of the study was to identify in vivo the characteristic features in pigmented lesions at different stages (benign, atypical, and malignant) and to evaluate the ability of in vivo MPM to distinguish atypical nevi from melanoma. Benign melanocytic nevi were shown to be characterized by the presence of nevus cell nests at the epidermal-dermal junction. Lentiginous hyperplasia, acanthosis, and architectural disorder were presented in atypical nevi. Cytological atypia was present in the melanoma lesions. The MPM imaging demonstrated very good correlation with histological analysis, suggesting that MPM could be a promising tool for in-vivo non-invasive pigmented lesion diagnosis, particularly distinguishing atypical nevi from melanoma. A non-invasive in vivo method for determining skin aging at the microscopic level by detecting signals from the main structural proteins of the skin, elastin, and collagen, was realized. The ratio of these signals usually is called SAAID. This parameter allows us to evaluate the dermatological status of the skin and evaluate the effect of cosmetic products, such as anti-aging creams, or physical procedures, such as ones involving laser, in microscopic in-vivo conditions. Also, the SAAID parameter provides information about the effects of ultraviolet radiation and other skin-damaging processes [178]. The multiphoton microscopy with autofluorescence and second harmonic generation was used to monitor the optic-thermal response of skin tissue irradiated by CO2 and the process of regeneration [179]. This method was shown to have the potential ability to monitor the process of tissue change and the remodeling in mouse dermis. Multiphoton microscopes allow a quick scan only the crosswise plane, by adjusting the relative distance between the focusing optics and the sample. However, the ability to quickly scan along the longitudinal direction has recently become available, which is an additional leap in the use of multiphoton tomography in clinical studies [180]. Using unprocessed biopsies, multiphoton microscopy can readily distinguish between benign lesions and breast cancers. For breast cancers, the tumor cells were shown to display marked cellular and nuclear pleomorphism. The tumor cells, characterized by irregular size and shape, enlarged nuclei, and increased nuclear-cytoplasmic ratio, infiltrated into disrupted connective tissue, leading to the loss of SHG signals [181].
In summary, these observations indicate that MPM could be an important tool to provide label-free noninvasive diagnostic impressions that can guide the surgeon in biopsy and patient management [182]. The ability to quantify cellular metabolic rate through NADH/FAD/Tryptophan auto-fluorescence lifetime imaging in healthy and disease state is, therefore, an important requirement for basic research, drug development, and clinical diagnosis. Diabetes is an important global health problem with persistently affected individuals, and the dynamics of metabolism in a diabetic wound are not well-understood at the cellular level [175, 176]. To understand diabetic tissue function better, the multi-parametric approach addresses initial quantitative assessment: (i) the metabolic activities during different stages of the wound healing using NADH and FAD fluorescence lifetime and redox ratio; (ii) with resolved collagen structural information in extracellular matrix using SHG microscopy, (iii) the correlated metabolic and structural changes. Multimodal TP-FLIM and SHG microscopy can investigate the cellular metabolism and morphological alterations of the wound healing process in diabetic patients [176].
Multimodal (SHG and CARS) Imaging of Macromolecules (Starch, Cellulose)
CARS microscopy has been used to study lipid-rich systems such as myelin [163], atherosclerotic tissues [183], and lipid droplets in live cells [184], as well as non-lipid based systems rich in C-H vibrations such as collagen arrays [185]. However, when CARS microscopy is integrated with microstructure sensitive modalities such as SHG, it can enable simultaneous imaging of several molecules. Starch and cellulose are carbohydrate polymers and condensed biomaterials; both are made from the same monomer, glucose, and have the same glucose-based repeat units. Starch is composed of amylose and amylopectin molecules and these form crystalline and amorphous concentric layers. Both starch and cellulose provide very strong CARS signals in (2,850–3,400 cm−1) C-H and O-H vibrational regions [150]. The cellulose fiber was immersed in D2O during the experimental. When the polarization of the recombined pump and Stokes beams was rotated the contrast from the fiber also changed. By tuning Wp – Ws to 2,845cm−1 we observed strong CARS signals for symmetric CH2 stretching vibration. The CARS signal from the symmetric CH2- stretching mode is maximized when the excitation polarization was along the CH2 group symmetry axis that is perpendicular to the lipid hydrocarbon chain [157, 159]. The broad CARS signal from the OH-stretching Raman band are observed when Wp – Ws tuned at 3,350 cm−1. Furthermore, they provide strong SHG signals [142, 150, 186], and recent research demonstrates the concurrent combination of SHG and CARS microscopy can identify the internal molecular structure and density. The crystalline and amorphous regions of starch and cellulose are differentiated by the combination of SHG and CARS microscopy [150]. The crystalline layers are anisotropically aligned by amylopectin chains within the focal volume of the laser and thus yield a strong second-order non-linear optical response.
The utility of multimodal CARS microscopy in a combination of SHG technique for the study of starch grains is a current topic of research interest. Figure 13 shows the simultaneous acquisition of CARS and SHG signals from potato starch and cellulose. The signals are collected in the forward direction, and the respective signals are separated from each other using bandpass filters (400/40 nm for SHG and 670/30 nm for CARS); it was observed that both signals were complementary to each other. The CARS signal is stronger than the SHG signal in case of potato starch; this suggests that crystalline shells (amylopectin) of starch are less dense than the amorphous shells (amylose). The hilum is also visible as a dark dimple in SHG imaging. The images of potato starch grains obtained by CARS (red) and SHG (green) microscopy were merged together to obtain a single image in which the striations of both the images were anti-correlated to each other (Figure 13A) with alternating amorphous and crystalline layers are clearly visible.
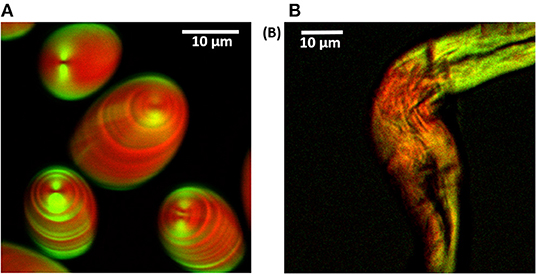
Figure 13. Overlaid CARS and SHG images of potato starch grains and cotton cellulose fiber in D2O. (A) CARS (red) and SHG (green) signals. Alternating crystalline and amorphous layers in potato starch are visible and appear to be anti-correlated between SHG and CARS. (B) CARS (red) and SHG (cyan) signals. The pump and Stokes beams are polarized in the vertical direction. The image size is 512 × 512 pixels. By tuning the delay stage to give Wp – Ws = 2,890 cm−1 CARS images were taken. Figure reproduced (from [150]) with kind permission from OSA.
Thus, multimodal CARS and SHG imaging of starch granules and cellulose offer highly detailed information about the internal structure and molecular organization. Therefore, the fundamental understanding of the optical non-linearity of starch granules and cellulose will provide a valid basis for future studies in food science and technology. For example, slowly-digestible or resistant starch derived from uncooked food, whole grains, legumes, tubers, and vegetables can be used as an appropriate source of carbohydrates to reduce the risk of various conditions/diseases such as cardiovascular diseases, obesity, and diabetes.
Integration of CARS and FLIM Technique
CARS microscopy is a powerful label-free live cell imaging tool for monitoring the changes in lipid droplets (LD) localization, size, and abundance in cells [187–191] and has previously been successfully applied to investigate the host-virus interactions [68]. Simultaneous detection of TPF and CARS signal and separation of CARS signal from endogenous auto-fluorescence in rat arterial tissue as well as intact roots of plant seedlings were performed using TCSPC electronics, thus improving the CARS contrast [151, 191]. The temporal dynamics of the two process viz., CARS and fluorescence can be exploited to separate them using a TCSPC system. CARS is an instantaneous process, the decay time is system's instrument response function (IRF), however, TPF lifetimes are in the nanosecond scale and can be separated by detecting the photon arrival time to the PMT. Again, simultaneous TPF and CARS microscopy were used to track D2-GFP (a stabilized version of green fluorescent protein) localization and measure changes in the hepatic lipid content of Huh7 hepatoma cells [68]. We have demonstrated that TCSPC-FLIM system distinguishes the CARS signal from the fluorescence of live Huh7 cells stained with Hoechst3342. Hoechst is a nucleus-staining fluorescent dye. The CARS signal is collected in the forward direction (F-CARS) coupled through a fiber while simultaneously performing epi-detected TP-FLIM using a separate detection channel in inverted microscope. The pump beam is used as a two-photon excitation wavelength of Hoechst3342. We have selected Huh-7 cells for their high lipid droplet content. Figure 14 shows the resulting FLIM and CARS images of cells. Lipid droplets in the cytoplasm are clearly observed in CARS image. A lifetime map reveals that the average fluorescence decay lifetime of Hoechst in the nucleus is 1.6 ± 0.4 ns. The combination of TP-FLIM and CARS microscopy illustrates the power of complementary imaging modalities for probing molecular interactions in the vicinity of lipid-rich cellular architecture.
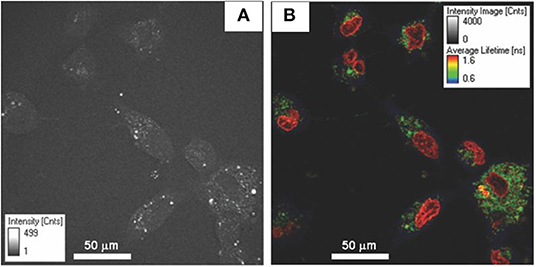
Figure 14. Simultaneous multichannel Time-gated F-CARS and FLIM imaging on a single detector of live Huh-7 cells. (A) F-CARS image collected through a multimode fiber. Image of all photons arriving in the vicinity of the IRF peak, representing CARS signals. The average lifetime of Hoechst in the nucleus is 1.6 ± 0.4 ns and that of the DiO in the cytoplasm is sub 1 ns. A 670/30 nm bandpass filter is placed between the condenser and the fiber in the F-CARS channel. (B) Epi-collected FLIM image showing spatial segregation of the Hoechst and DiO stains. Image of all photons arriving between 1.5 ns after the IRF peak and the end of the laser repetition period, representing the TPF signal. The color bar represents the range of the average fluorescence lifetime. Figure reproduced (from [151]) with kind permission from SPIE.
Conclusion
In conclusion, the uniqueness and the prospects of non-linear optical microscopy are elucidated in this review. We have presented the basics and applications of TPF, FLIM, SHG, and CARS microscopy techniques in detail. These techniques provide powerful, non-destructive, and label-free methods for visualization of biomolecules. TP-FLIM enables the measurement of auto-fluorescence lifetimes of endogenous fluorophores. Fluorescence lifetime is independent of intensity variations of fluorophores, however, sensitive to the molecular environment. Additionally, time-resolved fluorescence measurement may yield unprecedented information on molecular dynamics. The use of SHG microscopy for imaging of starch, collagen, skeletal muscles, urea crystals, nanocrystals has offered a lot more information about their molecular structures. Again, polarization-resolved Stokes vector based SHG microscopy investigates the polarization properties of the SHG signal to visualize the birefringence and crystal orientation from the samples under study. Thus, this newly developed instrument provides information contained in SHG light and has valuable applications in biology through the measurements of different polarization properties. Novel applications of SHG-CD (circular dichroism) microscopy for resolving 3D collagen chirality organization is also addressed. With multimodal label-free optical imaging techniques (TPF, SHG, FLIM, and CARS), we have targeted several components. A combination of SHG microscopy and TPF microscopy helped to reveal the process of collagen fiber regeneration in wound healing in vivo. CARS provides information on lipid-rich regions, while FLIM characterizes cellular metabolism using auto-fluorescence/fluorescence. The integration of photon counting under FLIM configuration with CARS separates the CARS signal from the autofluorescence background and improves the signal-to-noise ratio of CARS detection as well. SHG and CARS microscopy enabled characterization of structural and chemical properties of starch granules and cellulose fibers. As was said above, while several reviews have been written for each of the presented areas in recent years, in our review we have gathered the main approaches carried out with the help of NLO and also expanded the existing reviews with recent work in this direction. Moreover, the work demonstrated possibilities of the use of current approaches in practice and showed their effectiveness in many examples.
Author Contributions
NM, NB, G-YZ, RK, and YK wrote and edited the manuscript. NM, NB, and YK conceived the work and organized the manuscript. F-JK, SB, VN, and NK edited the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
NM thank SERB-Department of Science and Technology (DST), Government of India for financial support (Project Number—ECR/2016/001944). NM would like to thank Dr. K. Satyamoorthy, Director, Manipal School of Life Sciences, Manipal Academy of Higher Education (MAHE), Manipal, Karnataka-576104, India for his encouragement and Dr. K. K. Mahato, Department of Biophysics, MAHE for discussion and MAHE for providing the infrastructure and during manuscript preparation. YK thank the Russian Fund of Basic Research for financial support (grant No.17-00-00186). We appreciate greatly Dr. Andy Ridsdale from National Research Council, Canada for fruitful discussions on implementing FLIM and CARS techniques. The insightful discussion with Dr. Jianjun Qiu was crucial in interpreting the Stokes vector based results. We thank Dr. Matthew R. Foreman and Prof. Peter Török from Blackett Laboratory, Department of Physics, Imperial College London, UK and Dr. Carlos Macías Romero from Ecole Polytechnique Fedérale de Lausanne (EPFL), Lausanne, Switzerland for helping on the theory and the implement of Stokes vector formalism. The authors would also like to thank the National Science Council, Taiwan (NSC99-2627-M-010-002, NSC98-2627-M-010-006, NSC97-2112-M-010-002-MY3, and NSC98-2112-M-010-001-MY3), as well as the Ministry of Education, Taiwan under the Aim for Top University project for the generous support of the reported work. NB would like to thank Dr. Paulina Gasecka for the experimental data on P-CARS and Science Foundation Ireland for funding (17/IFB/5434). VN would like to thank Fundamental Research Program of the State Academies of Sciences for 2013-2020, line of research III.23 for financial support. F-JK would like to thank the Ministry of Science and Technology, Taiwan (MOST105-2112-M-010-001-MY3) for the generous support of the reported work.
References
2. Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. (1990) 248:73–6. doi: 10.1126/science.2321027
3. Piston WD, Kirby SM, Cheng H, Lederer JW, Webb WW. Two-photon-excitation fluorescence imaging of three-dimensional calcium-ion activity. Appl Opt. (1994) 33:662–9. doi: 10.1364/AO.33.000662
4. Turcotte R, Rutledge DJ, Bélanger E, Dill D, Macklin WB, Côté DC. Intravital assessment of myelin molecular order with polarimetric multiphoton microscopy. Sci Rep. (2016) 6:31685. doi: 10.1038/srep31685
5. Ranawat H, Pal S, Mazumder N. Recent trends in two-photon auto-fluorescence lifetime imaging (2P-FLIM) and its biomedical applications. Biomed Eng Lett. (2019) 9:293–310. doi: 10.1007/s13534-019-00119-7
6. Vergen J, Hecht C, Zholudeva LV, Marquardt MM, Hallworth R, Nichols MG. Metabolic imaging using two-photon excited NADH intensity and fluorescence lifetime imaging. Microsc Microanal. (2012) 18:761–70. doi: 10.1017/S1431927612000529
7. Kretschmer S, Pieper M, Hüttmann G, Bölke T, Wollenberg B, Marsh LM, et al. Autofluorescence multiphoton microscopy for visualization of tissue morphology and cellular dynamics in murine and human airways. Lab Invest. (2016) 96:918–31. doi: 10.1038/labinvest.2016.69
8. Engelbrecht CJ, Johnston RS, Seibel EJ, Helmchen F. Ultra-compact fiber-optic two-photon microscope for functional fluorescence imaging in vivo. Opt Express. (2008) 16:5556–64. doi: 10.1364/OE.16.005556
9. Xu C, Webb WW. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J Opt Soc Am B. (1996) 13:481–91. doi: 10.1364/JOSAB.13.000481
10. Grinvald A, Frostig RD, Siegel RM, Bartfeld E. High-resolution optical imaging of functional brain architecture in the awake monkey. Proc Natl Acad Sci USA. (1991) 88:11559. doi: 10.1073/pnas.88.24.11559
11. Konig K, Riemann I. High-resolution multiphoton tomography of human skin with subcellular spatial resolution and picosecond time resolution. J Biomed Opt. (2003) 8:432–9. doi: 10.1117/1.1577349
12. Teuchner K, Freyer W, Leupold D, Volkmer A, Birch DJ, Altmeyer P, et al. Femtosecond two-photon excited fluorescence of melanin. Photochem Photobiol. (1999) 70:146–51. doi: 10.1111/j.1751-1097.1999.tb07982.x
13. Tanaka K, Toiyama Y, Okugawa Y, Okigami M, Inoue Y, Uchida K, et al. In vivo optical imaging of cancer metastasis using multiphoton microscopy: a short review. Am J Transl Res. (2014) 6:179–87.
14. König K, Simon U, Halbhuber JK. 3D-resolved two-photon fluorescence microscopy of living cells using a modified confocal laser scanning microscope. Cell Mol Biol. (1996) 42:1181–94.
15. Masters RB, So CTP, Gratton E. Multiphoton excitation microscopy of In vivo human skin. Ann NY Acad Sci. (1998) 838:58–67. doi: 10.1111/j.1749-6632.1998.tb08187.x
16. Breunig HG, Studier H, König K. Multiphoton excitation characteristics of cellular fluorophores of human skin in vivo. Opt Express. (2010) 18:7857–71. doi: 10.1364/OE.18.007857
17. Becker W, Bergmann A, König K, Tirlapur U. Picosecond fluorescence lifetime microscopy by TCSPC imaging. Proc SPIE. (2001) 4262:414–9. doi: 10.1117/12.424584
18. Koehler MJ, Speicher M, Lange-Asschenfeldt S, Stockfleth E, Metz S, Elsner P, et al. Clinical application of multiphoton tomography in combination with confocal laser scanning microscopy for In vivo evaluation of skin diseases. Exp Dermatol. (2011) 20:589–94. doi: 10.1111/j.1600-0625.2011.01279.x
19. Meyer T, Bergner N, Bielecki C, Krafft C, Akimov D, Romeike BFM, et al. Nonlinear microscopy, infrared, and Raman microspectroscopy for brain tumor analysis. J Biomed Optics. (2011) 16:021113. doi: 10.1117/1.3533268
20. Liu J. Two-photon microscopy in pre-clinical and clinical cancer research. Front Optoelectron. (2015) 8:141–51. doi: 10.1007/s12200-014-0415-5
21. Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. (2013) 73:4965–77. doi: 10.1158/0008-5472.CAN-13-0661
22. Brown EB, Campbell RB, Tsuzuki Y, Xu L, Carmeliet P, Fukumura D, et al. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat Med. (2001) 7:864–8. doi: 10.1038/89997
23. Yu Y, Lee AMD, Wang H, Tang S, Zhao J, Lui H, et al. Imaging-guided two-photon excitation-emission-matrix measurements of human skin tissues. J Biomedical Optics. (2012) 17:077004. doi: 10.1117/1.JBO.17.7.077004
24. Palero JA, Boer VO, Vijverberg JC, Gerritsen HC, Sterenborg HJCM. Short-wavelength two-photon excitation fluorescence microscopy of tryptophan with a photonic crystal fiber based light source. Opt Express. (2005) 13:5363–8. doi: 10.1364/OPEX.13.005363
25. Chakraborty S, Nian F, Tsai J, Karmenyan A, Chiou A. Quantification of the metabolic state in cell-model of Parkinson's disease by fluorescence lifetime imaging microscopy. Sci Rep. (2016) 6:1–9. doi: 10.1038/srep19145
26. Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci USA. (2003) 100:7075–80. doi: 10.1073/pnas.0832308100
27. Radosevich AJ, Bouchard MB, Burgess SA, Chen BR, Hillman EMC. Hyperspectral In vivo two-photon microscopy of intrinsic contrast. Opt Lett. (2008) 33:2164–6. doi: 10.1364/OL.33.002164
28. Grosberg LE, Radosevich AJ, Asfaha S, Wang TC, Hillman EM. Spectral characterization and unmixing of intrinsic contrast in intact normal and diseased gastric tissues using hyperspectral two-photon microscopy. PLoS ONE. (2011) 6:e19925. doi: 10.1371/journal.pone.0019925
29. Miller DR, Jarrett JW, Hassan AM, Dunn AK. Deep tissue imaging with multiphoton fluorescence microscopy. Curr Opin Biomed Eng. (2017) 4:32–9. doi: 10.1016/j.cobme.2017.09.004
30. Stanciu SG, Xu S, Peng Q, Yan J, Stanciu GA, Welsch RE, et al. Experimenting liver fibrosis diagnostic by two photon excitation microscopy and bag-of-features image classification. Sci Rep. (2014) 4:4636. doi: 10.1038/srep04636
31. Kistenev YV, Nikolaev VV, Kurochkina OS, Borisov AV, Vrazhnov DA, Sandykova EA. Application of multiphoton imaging and machine learning to lymphedema tissue analysis. Biomed Opt Express. (2019) 10:3353–68. doi: 10.1364/BOE.10.003353
32. Lentsch G, Balu M, Williams J, Lee S, Harris RM, König K, et al. In vivo multiphoton microscopy imaging of melasma. Pigment Cell Melanoma Res. (2019) 32:403–11. doi: 10.1111/pcmr.12756
33. Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. (2006) 50:823–39. doi: 10.1016/j.neuron.2006.05.019
34. Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. (2008) 321:1686–9. doi: 10.1126/science.1162844
35. Miquelajauregui A, Kribakaran S, Mostany R, Badaloni A, Consalez GG, Portera-Cailliau C. Layer 4 pyramidal neurons exhibit robust dendritic spine plasticity In vivo after input deprivation. J Neurosci. (2015) 35:7287–94. doi: 10.1523/JNEUROSCI.5215-14.2015
36. Quirin S, Peterka DS, Yuste R. Instantaneous three-dimensional sensing using spatial light modulator illumination with extended depth of field imaging. Opt Express. (2013) 21:16007–21. doi: 10.1364/OE.21.016007
37. Greenberg DS, Houweling AR, Kerr JN. Population imaging of ongoing neuronal activity in the visual cortex of awake rats. Nature. (2008) 11:749–51. doi: 10.1038/nn.2140
38. Kobat D, Horton GN, Xu C. In vivo two-photon microscopy to 1.6-mm depth in mouse cortex. J Biomed Optics. (2011) 16:106014. doi: 10.1117/1.3646209
39. Ziv Y, Burns LD, Cocker ED, Hamel EO, Ghosh KK, Kitch LJ, et al. Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci. (2013) 16:264–6. doi: 10.1038/nn.3329
40. Bocarsly ME, Jiang WC, Wang C, Dudman JT, Ji N, Aponte Y. Minimally invasive microendoscopy system for In vivo functional imaging of deep nuclei in the mouse brain. Biomed Opt Express. (2015) 6:4546–56. doi: 10.1364/BOE.6.004546
41. Eles JR, Vazquez AL, Kozai TDY, Cui XT. Meningeal inflammatory response and fibrous tissue remodeling around intracortical implants: an In vivo two-photon imaging study. Biomaterials. (2018) 195:111–23. doi: 10.1016/j.biomaterials.2018.12.031
42. Haft-Javaherian M, Fang L, Muse V, Schaffer CB, Nishimura N, Sabuncu MR. Deep convolutional neural networks for segmenting 3D In vivo multiphoton images of vasculature in Alzheimer disease mouse models. PLoS ONE. (2019) 14:e0213539. doi: 10.1371/journal.pone.0213539
43. Yang M, Zhou Z, Zhang J, Li T, Guan J, Liao X, et al. MATRIEX imaging: multi-area two-photon real-time In vivo explorer. bioRxiv [Preprint]. (2019) doi: 10.1101/510545
44. Jackson J, Johnson J, Ahmed Z, Ward M, Hutton M, Ashby M, et al. In vivo two-photon imaging of progressive synapse loss and altered spine dynamics in the rtg4510 tauopathy model. Alzheimer Dement. (2014) 10:P29. doi: 10.1016/j.jalz.2014.05.054
45. Periasamy A, Mazumder N, Sun Y, Christopher KG, Day RN. FRET microscopy: basics, issues and advantages of FLIM-FRET imaging. In: Becker W, editor. Advanced Time-Correlated Single Photon Counting Applications. Cham: Springer (2015). p. 249–76.
46. Diaspro A. Confocal and Two-Photon Microscopy: Foundations, Applications and Advances. Wiley-VCH (2001).
47. Becker W. (ed.). Advanced Time-Correlated Single Photon Counting Techniques. Berlin: Springer (2015).
49. Ghukasyan V, Kao JF. Monitoring cellular metabolism with fluorescence lifetime of reduced nicotinamide adenine dinucleotide. J Phys Chem C. (2009) 113:11532–40. doi: 10.1021/jp810931u
50. Kao FJ, Deka G, Mazumder N. Cellular autofluorescence detection through FLIM/FRET microscopy. Top Appl Phys. (2015) 129:47. doi: 10.1007/978-94-017-9392-6_26
51. Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. (1967) 103:514–27. doi: 10.1042/bj1030514
52. Mayevsky A, Zarchin N, Kaplan H, Haveri J, Haselgroove J, Chance B. Brain metabolic responses to ischemia in the mongolian gerbil: In vivo and freeze trapped redox state scanning. Brain Res. (1983) 276:95–107. doi: 10.1016/0006-8993(83)90551-6
53. Wu Y, Xi P, Qu J, Cheung TH, Yu MY. Depth-resolved fluorescence spectroscopy reveals layered structure of tissue. Opt Express. (2004) 12:3218–23. doi: 10.1364/OPEX.12.003218
54. Kunz WS, Gellerich FN. Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney, cortex, skeletal muscle, and brain. Biochem Med Metab Biol. (1993) 50:103–10. doi: 10.1006/bmmb.1993.1051
55. Kunz WS, Kuznetsov AV, Winkler K, Gellerich FN, Neuhof S, Neumann HW. Measurement of fluorescence changes of NAD(P)H and of fluorescent flavoproteins in saponin-skinned human skeletal muscle fibers. Anal Biochem. (1994) 216:322–7. doi: 10.1006/abio.1994.1048
56. Wu Y, Qu JY. Combined depth- and time-resolved autofluorescence spectroscopy of epithelial tissue. Opt Lett. (2006) 31:1833–5. doi: 10.1364/OL.31.001833
57. Gulledge CJ, Dewhirst MW. Tumor oxygenation: a matter of supply and demand. Anticancer Res. (1996) 16:741–9.
58. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. (1927) 8:519–30. doi: 10.1085/jgp.8.6.519
59. Benavides J, Chang S, Park S, Richards-Kortum R, Mackinnon N, Macaulay C, et al. Multispectral digital colposcopy for In vivo detection of cervical cancer. Opt Express. (2003) 11:1223–36. doi: 10.1364/OE.11.001223
60. Zaak D, Stepp H, Baumgartner R, Schneede P, Waidelich R, Frimberger D, et al. Ultraviolet-excited (308 nm) autofluorescence for bladder cancer detection. Urology. (2002) 60:1029–33. doi: 10.1016/S0090-4295(02)01999-4
61. Tadrous PJ, Siegel J, French PM, Shousha S, Lalani el-N, Stamp GW. Fluorescence lifetime imaging of unstained tissues: early results in human breast cancer. J Pathol. (2003) 199:309–17. doi: 10.1002/path.1286
62. Mayinger B, Jordan M, Horbach T, Horner P, Gerlach C, Mueller S, et al. Evaluation of In vivo endoscopic autofluorescence spectroscopy in gastric cancer. Gastrointest Endosc. (2004) 59:191–8. doi: 10.1016/S0016-5107(03)02687-7
63. Hanlon EB, Itzkan I, Dasari RR, Feld MS, Ferrante RJ, McKee AC, et al. Near-infrared fluorescence spectroscopy detects Alzheimer's disease in vitro. Photochem Photobiol. (1999) 70:236–42. doi: 10.1562/0031-8655(1999)070<0236:NIFSDA>2.3.CO;2
64. Lakowicz J, Szmacinski H, Nowaczyk K, Johnson M. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci USA. (1992) 89:1271–5. doi: 10.1073/pnas.89.4.1271
65. Stringari C, Cinquin A, Cinquin O, Digman M, Donovan P, Gratton E. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc Natl Acad Sci USA. (2011) 108:13582–7. doi: 10.1073/pnas.1108161108
66. Conklin M, Provenzano P, Eliceiri K, Sullivan R, Keely P. Fluorescence lifetime imaging of endogenous fluorophores in histopathology sections reveals differences between normal and tumor epithelium in carcinoma in situ of the breast. Cell Biochem Biophys. (2009) 53:145–57. doi: 10.1007/s12013-009-9046-7
67. Sun Y, Phipps J, Elson D, Stoy H, Tinling S, Meier J, et al. Fluorescence lifetime imaging microscopy: In vivo application to diagnosis of oral carcinoma. Opt Lett. (2009) 34:2081–3. doi: 10.1364/OL.34.002081
68. Mazumder N, Lyn RK, Ridsdale A, Moffatt DJ, Singaravelu R, Hu JW, et al. Fluorescence lifetime imaging of alterations to cellular metabolism by domain 2 of the hepatitis C virus core protein. PLoS ONE. (2013) 8:e66738. doi: 10.1371/journal.pone.0066738
69. Jyothikumar V, Sun Y, Periasamy A. Investigation of tryptophan–NADH interactions in live human cells using three-photon fluorescence lifetime imaging and Förster resonance energy transfer microscopy. J Biomed Opt. (2013) 18:060501. doi: 10.1117/1.JBO.18.6.060501
70. Peter M, Ameer-Beg SM. Imaging molecular interactions by multiphoton FLIM. Biol Cell. (2004) 96:231–6. doi: 10.1016/j.biolcel.2003.12.006
71. Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci USA. (2007) 104:19494–9. doi: 10.1073/pnas.0708425104
72. Provenzano PP, Eliceiri KW, Keely PJ. Multiphoton microscopy and fluorescence lifetime imaging microscopy (FLIM) to monitor metastasis and the tumor microenvironment. Clin Exp Metastasis. (2009) 26:357–70. doi: 10.1007/s10585-008-9204-0
73. Ana-Maria P, Etienne D, Sébastien B, Steeve V, Serge K, Thérèse B, et al. Multiphoton FLIM in cosmetic clinical research. In: Konig K, editor. Multiphoton Microscopy and Fluorescence Lifetime Imaging: Applications in Biology and Medicine. Berlin; Boston, MA: De Gruyter (2018). p. 369–94.
74. Sun Y, Day R, Periasamy A. Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat Protoc. (2011) 6:1324–40. doi: 10.1038/nprot.2011.364
75. Day RN. Measuring protein interactions using Forster resonance energy transfer and fluorescence lifetime imaging microscopy. Methods. (2014) 66:200–7. doi: 10.1016/j.ymeth.2013.06.017
76. Estrada DA, Dunn KA. Improved sensitivity for two-photon frequency-domain lifetime measurement. Opt Express. (2010) 18:13631–9. doi: 10.1364/OE.18.013631
77. Hinde E, Digman MA, Welch C, Hahn KM, Gratton E. Biosensor FRET detection by the phasor approach to fluorescence lifetime imaging microscopy (FLIM). Microsc Res Tech. (2012) 75:271–81. doi: 10.1002/jemt.21054
78. Gratton E, Breusegem S, Sutin J, Ruan Q, Barry N. Fluorescence lifetime imaging for the two-photon microscope: time-domain and frequency-domain methods. J Biomed Opt. (2003) 8:381–90. doi: 10.1117/1.1586704
79. Scodellaro R, Bouzin M, Mingozzi F, D'Alfonso L, Granucci F, Collini M, et al. Whole-section tumor micro-architecture analysis by a two-dimensional phasor-based approach applied to polarization-dependent second harmonic imaging. Front Oncol. (2019) 19:527. doi: 10.3389/fonc.2019.00527
80. Radaelli F, D'Alfonso L, Collini M, Mingozzi F, Marongiu L, Granucci F, et al. μMAPPS: a novel phasor approach to second harmonic analysis for in vitro-In vivo investigation of collagen microstructure. Sci Rep. (2017) 7:17468. doi: 10.1038/s41598-017-17726-y
81. Fereidouni F, Bader AN, Gerritsen HC. Spectral phasor analysis allows rapid and reliable unmixing of fluorescence microscopy spectral images. Opt Express. (2012) 20:12729–41. doi: 10.1364/OE.20.012729
82. Fu D, Xie XS. Reliable cell segmentation based on spectral phasor analysis of hyperspectral stimulated Raman scattering imaging data. Anal Chem. (2014) 86:4115–9. doi: 10.1021/ac500014b
83. Willig KI, Kellner RR, Medda R, Hein B, Jakobs S, Hell SW. Nanoscale resolution in GFP-based microscopy. Nat Methods. (2006) 3:721–3. doi: 10.1038/nmeth922
84. Urban NT, Willig KI, Hell SW, Nägerl UV. STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys J. (2011) 101:1277–84. doi: 10.1016/j.bpj.2011.07.027
85. Butkevich AN, Ta H, Ratz M, Stoldt S, Jakobs S, Belov VN, et al. Two-color 810 nm STED nanoscopy of living cells with endogenous SNAP-tagged fusion proteins. ACS Chem Biol. (2018) 13:475–80. doi: 10.1021/acschembio.7b00616
86. Godin GA, Lounis B, Cognet L. Super-resolution microscopy approaches for live cell imaging. Biophys J. (2014) 107:1777–84. doi: 10.1016/j.bpj.2014.08.028
87. Giannone G, Hosy E, Cognet L. Dynamic super-resolution imaging of endogenous proteins on living cells at ultra-high density. Biophys J. (2010) 99:1303–10. doi: 10.1016/j.bpj.2010.06.005
88. Gannaway J, Sheppard CJR. Second-harmonic imaging in the scanning optical microscope. Opt Quant Electron. (1978) 10:435–9. doi: 10.1007/BF00620308
89. Brasselet S. Polarization-resolved nonlinear microscopy: application to structural molecular and biological imaging. Adv Opt Photonics. (2011) 3:205. doi: 10.1364/AOP.3.000205
90. Kumar R, Grønhaug KM, Romijn EI, Finnøy A, Davies CL, Drogset JO, et al. Polarization second harmonic generation microscopy provides quantitative enhanced molecular specificity for tissue diagnostics. J Biophotonics. (2015) 8:730–9. doi: 10.1002/jbio.201400086
91. Campagnola P. Second harmonic generation imaging microscopy: applications to diseases diagnostics. Anal Chem. (2011) 83:3224–31. doi: 10.1021/ac1032325
92. Débarre D, Supatto W, Pena AM, Fabre A, Tordjmann T, Combettes L, et al. Imaging lipid bodies in cells and tissues using third-harmonic generation microscopy. Nat Methods. (2006) 3:47–53. doi: 10.1038/nmeth813
93. Campagnola PJ, Loew LM. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat Biotechnol. (2003) 21:1356–60. doi: 10.1038/nbt894
94. Gauderon R, Lukins PB, Sheppard CJR. Three-dimensional second-harmonic generation imaging with femtosecond laser pulses. Opt Lett. (1998) 23:1209–11. doi: 10.1364/OL.23.001209
95. Antonelli MR, Pierangelo A, Novikova T, Validire P, Benali A, Gayet B, et al. Mueller matrix imaging of human colon tissue for cancer diagnostics: how Monte Carlo modeling can help in the interpretation of experimental data. Opt Express. (2010) 18:10200–8. doi: 10.1364/OE.18.010200
96. Ghosh N, Wood MFG, Vitkin IA. Influence of the order of the constituent basis matrices on the Mueller matrix decomposition-derived polarization parameters in complex turbid media such as biological tissue. Opt Commun. (2010) 283:1200–8. doi: 10.1016/j.optcom.2009.10.111
97. Strupler M, Pena AM, Hernest M, Tharaux PL, Martin JL, Beaurepaire E, et al. Second harmonic imaging and scoring of collagen in fibrotic tissues. Opt Express. (2007) 15:4054–65. doi: 10.1364/OE.15.004054
98. Sun Y, Chen WL, Lin SJ, Jee SH, Chen YF, Lin LC, et al. Investigating mechanisms of collagen thermal denaturation by the second-harmonic generation imaging. Biophys J. (2006) 91:2620–5. doi: 10.1529/biophysj.106.085902
99. Da Costa V, Wei R, Lim R, Sun CH, Brown JJ, Wong BJF. Nondestructive imaging of live human keloid and facial tissue using multiphoton microscopy. Arch Facial Plast Surg. (2008) 10:38–43. doi: 10.1001/archfacial.2007.18
100. Avila FJ, Bueno JM. Analysis and quantification of collagen organization with the structure tensor in second harmonic microscopy images of ocular tissues. Appl Opt. (2015) 54:9848–54. doi: 10.1364/AO.54.009848
101. Matteini P, Ratto F, Rossi F, Cicchi SRC, Kapsokalyvas D, Pini R, et al. Photothermally-induced disordered patterns of corneal collagen revealed by SHG imaging. Opt Express. (2009) 17:4868. doi: 10.1364/OE.17.004868
102. Plotnikov SV, Millard AC, Campagnola PJ, Mohler WA. Characterization of the myosin-based source for second-harmonic generation from muscle sarcomeres. Biophys J. (2006) 90:693–703. doi: 10.1529/biophysj.105.071555
103. Fu L, Gu M. Polarization anisotropy in fiber-optic second harmonic generation microscopy. Opt Express. (2008) 16:5000. doi: 10.1364/OE.16.005000
104. Sun CW, Yang CC, Kiang YW. Optical imaging based on time-resolved Stokes vectors in filamentous tissues. Appl Opt. (2003) 42:750–4. doi: 10.1364/AO.42.000750
105. Schön P, Munhoz F, Gasecka A, Brustlein S, Brasselet S. Polarization distortion effects in polarimetric two-photon microscopy. Opt Express. (2008) 16:20891–901. doi: 10.1364/OE.16.020891
106. Yew EYS, Sheppard CRJ. Effects of axial field components on second harmonic generation microscopy. Opt Express. (2006) 14:1167–74. doi: 10.1364/OE.14.001167
107. Mazumder N, Deka G, Wu WW, Gogoi A, Zhuo GY, Kao FJ. Polarization resolved second harmonic microscopy. Methods. (2017) 128:105–18. doi: 10.1016/j.ymeth.2017.06.012
108. Mazumder N, Qiu J, Foreman MR, Romero CM, Török P, Kao FJ. Stokes vector based polarization resolved second harmonic microscopy of starch granules. Biomed Opt Express. (2013) 4:538–47. doi: 10.1364/BOE.4.000538
109. Lee H, Huttunen MJ, Hsu KJ, Partanen M, Zhuo GY, Kauranen M, et al. Chiral imaging of collagen by second-harmonic generation circular dichroism. Biomed Opt Express. (2013) 4:909–16. doi: 10.1364/BOE.4.000909
110. Zhuo GY, Chen MY, Yeh CY, Guo CL, Kao FJ. Fast determination of three-dimensional fibril orientation of type-I collagen via macroscopic chirality. Appl Phys Lett. (2017) 110:023702. doi: 10.1063/1.4973885
111. Barco DO, Bueno MJ. Second harmonic generation signal in collagen fibers: role of polarization, numerical aperture, and wavelength. J Biomed Optics. (2012) 17:045005. doi: 10.1117/1.JBO.17.4.045005
112. Mazumder N, Qiu J, Foreman MR, Romero CM, Hu CW, Tsai HR, et al. Polarization-resolved second harmonic generation microscopy with a four-channel Stokes-polarimeter. Opt Express. (2012) 20:14090–9. doi: 10.1364/OE.20.014090
113. Légaré F, Pfeffer C, Olsen BR. The role of backscattering in SHG tissue imaging. Biophys J. (2007) 15:1312–20. doi: 10.1529/biophysj.106.100586
114. Bianchini P, Diaspro A. Three-dimensional (3D) backward and forward second harmonic generation (SHG) microscopy of biological tissues. J Biophotonics. (2008) 1:443–50. doi: 10.1002/jbio.200810060
115. Hristu R, Eftimie LG, Stanciu SG, Tranca DE, Paun B, Sajin M, et al. Quantitative second harmonic generation microscopy for the structural characterization of capsular collagen in thyroid neoplasms. Biomed Opt Express. (2018) 9:3923–36. doi: 10.1364/BOE.9.003923
116. Plotnikov SV, Kenny AM, Walsh SJ, Zubrowski B, Joseph C, Scranton VL, et al. Measurement of muscle disease by quantitative second-harmonic generation imaging. J Biomed Opt. (2008) 13:044018. doi: 10.1117/1.2967536
117. Mazumder N, Xiang L, Qiu J, Kao FJ. Investigating starch gelatinization through Stokes vector resolved second harmonic generation microscopy. Sci Rep. (2017) 7:45816. doi: 10.1038/srep46803
118. Muir RD, Kissick DJ, Simpson GJ. Statistical connection of binomial photon counting and photon averaging in high dynamic range beam-scanning microscopy. Opt Express. (2012) 20:10406–15. doi: 10.1364/OE.20.010406
119. Hsieh CL, Pu Y, Grange R, Psaltis D. Second harmonic generation from nanocrystals under linearly and circularly polarized excitations. Opt Express. (2010) 18:11917–32. doi: 10.1364/OE.18.011917
120. Wolman M, Kasten FH. Polarized light microscopy in the study of the molecular structure of collagen and reticulin. Histochemistry. (1986) 85:41–9. doi: 10.1007/BF00508652
121. Han M, Giese G, Bille JF. Second harmonic generation imaging of collagen fibrils in cornea and sclera. Opt Express. (2005) 13:5791–7. doi: 10.1364/OPEX.13.005791
122. Both M, Vogel M, Friedrich O, von Wegner F, Künsting T, Fink RH, et al. Second harmonic imaging of intrinsic signals in muscle fibers in situ. J Biomed Opt. (2004) 9:882–92. doi: 10.1117/1.1783354
123. Psilodimitrakopoulos S, Santos SICO, Amat-Roldan I, Thayil AKN, Artigas D, Loza-Alvarez P. In vivo, pixel-resolution mapping of thick filaments' orientation in nonfibrilar muscle using polarization-sensitive second harmonic generation microscopy. J Biomed Opt. (2009) 14:014001. doi: 10.1117/1.3059627
124. Petralli-Mallow T, Wong TM, Byers JD, Yee HI, Hicks JM. Circular dichroism spectroscopy at interfaces: a surface second harmonic generation study. J Phys Chem. (1993) 97:1383–8. doi: 10.1021/j100109a022
125. Verbiest T, Kauranen M, Persoons A, Ikonen M, Kurkela J, Lemmetyinen H. Nonlinear optical activity and biomolecular chirality. J Am Chem Soc. (1994) 116:9203–5. doi: 10.1021/ja00099a040
126. Simpson GJ. Structural origins of circular dichroism in surface second harmonic generation. J Chem Phys. (2002) 117:3398. doi: 10.1063/1.1494423
127. Kriech MA, Conboy JC. Imaging chirality with surface second harmonic generation microscopy. J Am Chem Soc. (2005) 127:2834–5. doi: 10.1021/ja0430649
128. Chen X, Raggio C, Campagnola PJ. Second-harmonic generation circular dichroism studies of osteogenesis imperfecta. Opt Lett. (2012) 37:3837–9. doi: 10.1364/OL.37.003837
129. Berkholtz C, Lai B, Woodruff KT, Shea LD. Distribution of extracellular matrix proteins type I collagen, type IV collagen, fibronectin, and laminin in mouse folliculogenesis. Histochem Cell Biol. (2006) 126:583–92. doi: 10.1007/s00418-006-0194-1
130. Zhuo GY, Lee H, Hsu KJ, Huttunen MJ, Kauranen M, Lin YY, et al. Three-dimensional structural imaging of starch granules by second-harmonic generation circular dichroism. J Microsc. (2014) 253:183–90. doi: 10.1111/jmi.12108
131. Chen MY, Huttunen MJ, Kan CW, Deka G, Lin YY, Ye CW, et al. Resonant nonlinear microscopy reveals changes in molecular level chirality in native biological tissues. Opt Commun. (2018) 422:56–63. doi: 10.1016/j.optcom.2018.03.005
132. Rao RAR, Mehta MR, Toussaint KC. Fourier transform-second-harmonic generation imaging of biological tissues. Opt Express. (2009) 17:14534–42. doi: 10.1364/OE.17.014534
133. Chen W-L, Li T-H, Su P-J, Chou C-K, Fwu P, Lin S-J, et al. Second harmonic generation χ tensor microscopy for tissue imaging. Appl Phys Lett. (2009) 94:18. doi: 10.1063/1.3132062
134. Psilodimitrakopoulos S, Amat-Roldan I, Loza-Alvarez P, Artigas D. Effect of molecular organization on the image histograms of polarization SHG microscopy. Biomed Opt Express. (2012) 3:2681–93. doi: 10.1364/BOE.3.002681
135. Hu P, Hsueh C, Su P, Chen W, Hovhannisyan VA, Chen S, et al. The use of second-order susceptibility as contrast mechanism for label-free imaging of biological tissue. IEEE J Select Top Quant Electron. (2012) 18:1326. doi: 10.1109/JSTQE.2011.2174619
136. Dubreuil M, Tissier F, Roy LL, Pennec J, Rivet S, Giroux-Metges M, et al. Polarization-resolved second harmonic microscopy of skeletal muscle in sepsis. Biomed Opt Express. (2018) 9:6350–8. doi: 10.1364/BOE.9.006350
137. Gusachenko I, Latour G, Schanne-Klein M. Polarization-resolved Second Harmonic microscopy in anisotropic thick tissues. Opt Express. (2010) 18:19339–52. doi: 10.1364/OE.18.019339
138. Saha S, Ghosh P, Mitra D, Mukherjee S, Bhattacharya S, Roy SS. Localization and thyroid hormone influenced expression of collagen II in ovarian tissue. Cell Physiol Biochem. (2007) 19:67–76. doi: 10.1159/000099193
139. Romijn EI, Finnøy A, Kumar R, Lilledahl MB. Automated calibration and control for polarization-resolved second harmonic generation on commercial microscopes. PLoS ONE. (2018) 13:e0195027. doi: 10.1371/journal.pone.0195027
140. Hsueh C, Tseng T, Lin H, Lee S, Huang Y, Dong C. Optical discrimination of type I and type III collagen through second-order susceptibility imaging. Optik. (2018) 169:264. doi: 10.1016/j.ijleo.2018.05.039
141. Mazumder N, Xiang L, Qiu J, Kao FJ. Revealing molecular structure of starch with Stokes-vector based second harmonic generation microscopy. J Opt. (2018) 47:40–6. doi: 10.1007/s12596-017-0419-9
142. Chan J, Fore S, Wachsmann-Hogiu S, Huser S. Raman spectroscopy and microscopy of individual cells and cellular components. Laser Photon. (2008) 2:325–49. doi: 10.1002/lpor.200810012
143. Baena JR, Lendl B. Raman spectroscopy in chemical bioanalysis. Curr Opin Chem Biol. (2004) 8:534–9. doi: 10.1016/j.cbpa.2004.08.014
144. Maker PD, Terhune RW. Study of optical effects due to an induced polarization third order in the electric field strength. Phys Rev. (1965) 137:A801. doi: 10.1103/PhysRev.137.A801
145. Zumbusch A, Holtom GR, Xie ZS. Three-dimensional vibrational imaging by coherent anti-Stokes Raman Scattering. Phys Rev Lett. (1999) 82:4142. doi: 10.1103/PhysRevLett.82.4142
146. Evans CL, Xie XS. Coherent anti-stokes Raman scattering microscopy: chemical imaging for biology and medicine. Annu Rev Anal Chem. (2008) 1:833–909. doi: 10.1146/annurev.anchem.1.031207.112754
147. Evans CL, Potma EO, Puoris'haag M, Cote D, Lin CP, Xie XS. Chemical imaging of tissue In vivo with video-rate coherent anti-Stokes Raman scattering microscopy. Proc Natl Acad Sci USA. (2005) 102:16807–12. doi: 10.1073/pnas.0508282102
148. Le TT, Huff TB, Cheng JX. Coherent anti-Stokes Raman scattering imaging of lipids in cancer metastasis. BMC Cancer. (2009) 9:42. doi: 10.1186/1471-2407-9-42
149. Haluszka D, Lőrincz K, Kiss N, Szipőcs R, Kuroli E, Gyöngyösi N, et al. Diet-induced obesity skin changes monitored by In vivo SHG and ex vivo CARS microscopy. Biomed Opt Express. (2016) 7:4480–9. doi: 10.1364/BOE.7.004480
150. Slepkov AD, Ridsdale A, Pegoraro AF, Moffatt DJ, Stolow A. Multimodal CARS microscopy of structured carbohydrate biopolymers. Biomed Opt Express. (2010) 1:1347–57. doi: 10.1364/BOE.1.001347
151. Slepkov AD, Ridsdale A, Wan HN, Wang MH, Pegoraro AF, Moffatt DJ, et al. Forward-collected simultaneous fluorescence lifetime imaging and coherent anti-Stokes Raman scattering microscopy. J Biomed Opt. (2011) 16:021103. doi: 10.1117/1.3490641
152. Pegoraro AF, Ridsdale A, Moffatt DJ, Pezacki JP, Thomas BK, Fu L, et al. All-fiber CARS microscopy of live cells. Opt Express. (2009) 17:20700–6. doi: 10.1364/OE.17.020700
153. Kachynski AV, Kuzmin AN, Prasad PN, Smalyukh II. Coherent anti-Stokes Raman scattering polarized microscopy of three-dimensional director structures in liquid crystals. Appl Phys Lett. (2007) 91:151905–13. doi: 10.1063/1.2800887
154. Saar BG, Park H, Xie XS, Lavrentovich OD. Three-dimensional imaging of chemical bond orientation in liquid crystals by coherent anti- Stokes Raman scattering microscopy. Opt Express. (2007) 15:13585–96. doi: 10.1364/OE.15.013585
155. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. (2017) 18:361–74. doi: 10.1038/nrm.2017.16
156. Lentz BR. Use of fluorescent probes to monitor molecular order and motions within liposome bilayers. Chem Phys Lipids. (1993) 64:99–116. doi: 10.1016/0009-3084(93)90060-G
157. Cheng JX, Book LD, Xie XS. Polarization coherent anti-Stokes Raman scattering microscopy. Opt Lett. (2001) 26:1341–3. doi: 10.1364/OL.26.001341
158. Zimmerley MS, Younger R, Valenton T, Oertel DC, Ward JL, Potma EO. Molecular orientation in dry and hydrated cellulose fibers: a coherent anti-stokes Raman scattering microscopy study. J Phys Chem B. (2010) 114:10200–8. doi: 10.1021/jp103216j
159. Bioud FZ, Gasecka P, Ferrand P, Rigneault H, Duboisset J, Brasselet S. Structure of molecular packing probed by polarization-resolved nonlinear four-wave mixing and coherent anti-Stokes Raman-scattering microscopy. Phys Rev A. (2014) 89:13836. doi: 10.1103/PhysRevA.89.013836
160. Cheng JX, Pautot S, Weitz DA, Xie XS. Ordering of water molecules between phospholipid bilayers visualized by coherent anti-Stokes Raman scattering microscopy. Proc Natl Acad Sci USA. (2003) 100:9826–30. doi: 10.1073/pnas.1732202100
161. Wurpel GWH, Rinia HA, Muller M. Imaging orientational order and lipid density in multilamellar vesicles with multiplex CARS microscopy. J Microsc. (2005) 218:37–45. doi: 10.1111/j.1365-2818.2005.01462.x
162. Potma EO, Xie XS. Detection of single lipid bilayers with coherent anti-Stokes Raman scattering (CARS) microscopy. J Raman Spectrosc. (2003) 34:642–50. doi: 10.1002/jrs.1045
163. Wang H, Fu Y, Zickmund P, Shi R, Cheng JX. Coherent anti-stokes raman scattering imaging of axonal myelin in live spinal tissues. Biophys J. (2005) 89:581–91. doi: 10.1529/biophysj.105.061911
164. Fu Y, Huff TB, Wang HW, Cheng JX, Wang H. Ex vivo and In vivo imaging of myelin fibers in mouse brain by coherent anti-Stokes Raman scattering microscopy. Opt Express. (2008) 16:19396–409. doi: 10.1364/OE.16.019396
165. Gasecka P, Jaouen A, Bioud FZ, de Aguiar HB, Duboisset J, Ferrand P, et al. Lipid order degradation in autoimmune demyelination probed by polarized coherent Raman microscopy. Biophys J. (2017) 113:1520–30. doi: 10.1016/j.bpj.2017.07.033
166. De Vito G, Bifone A, Piazza V. Rotating-polarization CARS microscopy: combining chemical and molecular orientation sensitivity. Opt Express. (2012) 20:29369–77. doi: 10.1364/OE.20.029369
167. Hofer M, Balla NK, Brasselet S. High-speed polarization-resolved coherent Raman scattering imaging. Optica. (2017) 4:795–801. doi: 10.1364/OPTICA.4.000795
168. Cleff C, Gasecka A, Ferrand P, Rigneault H, Brasselet S, Duboisset J. Direct imaging of molecular symmetry by coherent anti-stokes Raman scattering. Nat. Commun. (2016) 7:11562. doi: 10.1038/ncomms11562
169. Han X, Brown E. Measurement of the ratio of forward-propagating to back-propagating second harmonic signal using a single objective. Opt Express. (2010) 18:10538–50. doi: 10.1364/OE.18.010538
170. Munhoz F, Rigneault H, Brasselet S. Polarization-resolved four-wave mixing microscopy for structural imaging in thick tissues. J Opt Soc Am B. (2012) 29:1541–50. doi: 10.1364/JOSAB.29.001541
171. Jake DJ, Hallie ER, Woessner AE, Quinn PK. In vivo multiphoton microscopy detects longitudinal metabolic changes associated with delayed skin wound healing. Commun Biol. (2018) 1:198. doi: 10.1038/s42003-018-0206-4
172. Deka G, Okano K, Wu WW, Kao FJ. Multiphoton microscopy for skin wound healing study in terms of cellular metabolism and collagen regeneration. In: Proc. SPIE 8948, Multiphoton Microscopy in the Biomedical Sciences XIV. San Francisco, CA (2014).
173. Deka G, Wu W, Kao JF. In vivo wound healing diagnosis with second harmonic and fluorescence lifetime imaging. J Biomed Optics. (2012) 18:061222. doi: 10.1117/1.JBO.18.6.061222
174. Zoumi A, Yeh A, Tromberg B. Imaging cells and extracellular matrix In vivo by using second-harmonic generation and two-photon excited fluorescence. Proc Natl Acad Sci USA. (2002) 99:11014–9. doi: 10.1073/pnas.172368799
175. Torkian B, Yeh A, Engel R, Sun C, Tromberg B, Wong B. Modeling aberrant wound healing using tissue-engineered skin constructs and multiphoton microscopy. Arch Fac Plastic Surg. (2004) 6:180–7. doi: 10.1001/archfaci.6.3.180
176. Quinn KP, Leal EC, Tellechea A, Kafanas A, Auster ME, Veves A, et al. Diabetic wounds exhibit distinct microstructural and metabolic heterogeneity through label-free multiphoton microscopy. J Invest Dermatol. (2016) 136:342–4. doi: 10.1038/JID.2015.371
177. Balu M, Kelly KM, Zachary CB, Harris RM, Krasieva TB, König K, et al. Clinical studies of pigmented lesions in human skin by using a multiphoton tomograph. In: Proc of SPIE Multiphoton Microscopy in the Biomedical Sciences XIII. San Francisco, CA (2013).
178. Schindele A, Breunig HG, König K. Multiphoton tomography for In vivo skin age determination. Optik Photonik. (2018) 13:56–9. doi: 10.1002/opph.201800009
179. Wu S, Zhang X, Li H. Skin damage of ablative laser In vivo based on multiphoton microscopy. In: Symposium on Photonics and Optoelectronics. Chengdu (2010).
180. Breunig HG, Sauer B, Batista A, König K. Rapid vertical tissue imaging with clinical multiphoton tomography. In: Proceedings Optics, Photonics, and Digital Technologies for Imaging Applications V. Strasbourg (2018).
181. Dravid UA, Mazumder N. Types of advanced optical microscopy techniques for breast cancer research: a review. Lasers Med Sci. (2018) 33:1849–58. doi: 10.1007/s10103-018-2659-6
182. Wu X, Chen G, Lu J, Zhu W, Qiu J, Chen J, et al. Label-free detection of breast masses using multiphoton microscopy. PLoS ONE. (2013) 8:e65933. doi: 10.1371/journal.pone.0065933
183. Lim RS, Kratzer A, Barry NP, Miyazaki-Anzai S, Miyazaki M, Mantulin WW, et al. Multimodal CARS microscopy determination of the impact of diet on macrophage infiltration and lipid accumulation on plaque formation in ApoE-defi cient mice. J Lipid Res. (2010) 51:1729–37. doi: 10.1194/jlr.M003616
184. Pfeffer CP, Olsen BR, Ganikhanov F, Légaré F. Imaging skeletal muscle using second harmonic generation and coherent anti-Stokes Raman scattering microscopy. Biomed Opt Express. (2011) 2:1366–76. doi: 10.1364/BOE.2.001366
185. Pfeffer CP, Olsen BR, Ganikhanov F, Légaré F. Multimodal nonlinear optical imaging of collagen arrays. J Struct Biol. (2008) 164:140–5. doi: 10.1016/j.jsb.2008.07.002
186. Cisek R, Tokarz D, Steup M, Tetlow IJ, Emes MJ, Hebelstrup KH, et al. Second harmonic generation microscopy investigation of the crystalline ultrastructure of three barley starch lines affected by hydration. Biomed Opt Express. (2015) 6:3694–700. doi: 10.1364/BOE.6.003694
187. Schie IW, Weeks T, McNerney GP, Fore S, Sampson JK, Wachsmann-Hogiu S, et al. Simultaneous forward and epi-CARS microscopy with a single detector by time correlated single photon counting. Opt Express. (2008) 16:2168–75. doi: 10.1364/OE.16.002168
188. Hellerer T, Axäng C, Brackmann C, Hillertz P, Pilon M, Enejder A. Monitoring of lipid storage in Caenorhabditis elegans using coherent anti-Stokes Raman scattering (CARS) microscopy. Proc Natl Acad Sci USA. (2007) 104:14658–63. doi: 10.1073/pnas.0703594104
189. Ko AC, Ridsdale A, Smith MS, Mostaço-Guidolin LB, Hewko MD, Pegoraro AF, et al. Multimodal nonlinear optical imaging of atherosclerotic plaque development in myocardial infarction-prone rabbits. J Biomed Opt. (2010) 15:020501. doi: 10.1117/1.3353960
190. Duncan MD, Reintjes J, Manuccia TJ. Scanning coherent anti-stokes Raman microscope. Opt Lett. (1982) 7:350–2. doi: 10.1364/OL.7.000350
Keywords: non-linear optical microscopy, two-photon fluorescence microscopy, second harmonic generation, coherent anti-stokes Raman scattering, fluorescence lifetime imaging, nicotinamide adenine dinucleotide, collagen
Citation: Mazumder N, Balla NK, Zhuo G-Y, Kistenev YV, Kumar R, Kao F-J, Brasselet S, Nikolaev VV and Krivova NA (2019) Label-Free Non-linear Multimodal Optical Microscopy—Basics, Development, and Applications. Front. Phys. 7:170. doi: 10.3389/fphy.2019.00170
Received: 21 March 2019; Accepted: 15 October 2019;
Published: 31 October 2019.
Edited by:
Stefan G. Stanciu, Politehnica University of Bucharest, RomaniaReviewed by:
Maria Calvo, University of Barcelona, SpainLaura Sironi, University of Milano Bicocca, Italy
Copyright © 2019 Mazumder, Balla, Zhuo, Kistenev, Kumar, Kao, Brasselet, Nikolaev and Krivova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nirmal Mazumder, bmlybWFsdXZhQGdtYWlsLmNvbQ==