Corrigendum: Non-isothermal Transport of Multi-phase Fluids in Porous Media. Constitutive Equations
 Signe Kjelstrup
Signe Kjelstrup Dick Bedeaux
Dick Bedeaux Alex Hansen
Alex Hansen Bjørn Hafskjold
Bjørn Hafskjold Olav Galteland
Olav Galteland- 1PoreLab, Department of Chemistry, Norwegian University of Science and Technology, Trondheim, Norway
- 2PoreLab, Department of Physics, Norwegian University of Science and Technology, Trondheim, Norway
We define a representative elementary volume of a porous medium in terms of lumped extensive variables, including properties of homogeneous phases, interfaces, and contact lines. Using the grand potential, we define the pressure of the REV in a porous medium in a new manner. From the entropy production expressed in these variables, we develop new constitutive equations for multi-component, multi-phase, macro-scale flow. The system is exposed to temperature, composition, and pressure gradients. New contributions due to varying porosity or surface tension offer explanations for non-Darcy behavior, and predict thermal osmosis special for porous media. An experimental program is suggested to verify Onsager symmetry in the transport coefficients. The analysis is limited to non-deformable systems, which obey Euler homogeneity on the REV level.
1. Introduction
We have recently [1] derived a coarse-grained form of the entropy production, σ, of a representative elementary volume (REV) in a non-deformable porous medium with multi-phase, multi-component, non-isothermal fluids. The coarse-grained description of the REV was formulated for systems that obey Euler homogeneity. A Gibbs equation could therefore be formulated for the REV itself, and used as a starting point, as is normal in non-equilibrium thermodynamics [2].
Once the entropy production has been found, the driving forces and the constitutive equations can be given. These will be specified here. We shall see that we can obtain the same form of the constitutive equations as for homogeneous systems, but that the driving forces are particular for the porous medium. To write out this particularity, is one aim of the present paper. We shall see that we can obtain a new definition for the pressure in a porous medium and use this and the chemical potential to find the constitutive equations. We are also giving internal relations between experiments particular for the flows, as derived for instance from the Onsager relations.
We consider, as a premise, the REV as a complete thermodynamic system. Hansen and Ramstad [3] suggested this possibility already some time ago. Since then, the hypothesis has been supported through measurements on Hele-Shaw cells [4] and through network simulations [5]. The coarse-grained variables of the REV will fluctuate similar to the variables in a normal thermodynamic state around a mean value.
The procedure that we used to obtain the Gibbs equation for coarse-grained variables [1], assumes that the additive thermodynamic variables of the REV are Euler homogeneous functions of order one. We give first a brief review of the procedure that defines the basis set of thermodynamic variables (section 2.1). The consequences for the chemical potential and the pressure in the context of porous media is next described (sections 2.2, 2.3). A new definition of the pressure is proposed in section 2.3.
The constitutive equations that follow from the new set of variables, allow us to revisit previously published experimental results. We shall see, for instance, that they can explain deviations from Darcy's law. Such deviations have been observed for small volume flows, also for single fluids like water and water solutions [6–10]. Thresholds and/or deviations from straight lines in plots of flow vs. the overall pressure difference, have been reported. Boersma et al. [8] found a dependency of such a threshold on the average pore radius, , for flow in a porous medium made of glass-beads. The observations have, as of yet, no final explanation. When dealing with immiscible fluids, Tallakstad et al. [11] observed a dependence of the flow rate on the square of the pressure difference under steady-state flow conditions. Sinha and Hansen [12] attributed this square dependence to a change in the conductivity, arising from the successive opening of pores due to the mobilization of interfaces when the pressure difference across the sample is increased. The explanation was supported by a mean-field calculation and numerical experiments with a network model. Sinha et al. [10] followed up the original Tallakstad study, done in a two-dimensional model porous medium, both experimentally and computationally in three-dimensional porous media, with the same result.
There is not only a need to better understand deviations from Darcy's law for volume transport. Other driving forces than those related to the pressure difference, are also relevant to porous media transport. Counter-current transport of components can lead to gradients in composition (chemical potential) or chemical driving forces. Injection of cold seawater into a warm hydrocarbon reservoir can create thermal driving forces. This leads to thermal diffusion. The separation of components in a temperature gradient is an example of the Soret effect [13]. A temperature gradient may also lead to a pressure gradient, a phenomenon called thermal osmosis. These effects are not much studied in porous media, see [14] for a review on membranes. There are, for instance, contradictory findings in the literature with respect to the impact of the porous medium on thermal diffusion. Costeseque et al. [15] found that the porous medium had no significant effect on the Soret coefficient, as determined with a horizontal thermodiffusion cell (although the component diffusion was slower in the porous medium). On the other hand, Colombani et al. [16] found by molecular dynamics simulations that both the porosity and the wettability of the porous medium had an effect on the Soret coefficient. The presence of a porous matrix had an impact on the flow pattern and therefore the Soret coefficient according to Davarzani et al. [17]. The role of a thermal driving force is therefore at best unclear. A better understanding of its role could be important. An emerging concept for water cleaning is, for instance, based on thermal osmosis [18]. This process could help produce clean water using industrial and natural heat sources, a very important topic in the world today.
It is thus an open question in porous media theory, how driving forces like the ones mentioned interact, and how the porous medium makes these interactions special [17]. It is therefore also the aim of this work to clarify the coupling that can take place due to some central forces, by constructing a non-equilibrium thermodynamic theory, particular for porous media.
The paper is structured as follows. Section 2 gives first a brief repetition of the variables used to obtain the coarse-grained Gibbs equation and the corresponding entropy production [1]. As before, the analysis applies to systems that obeys Euler homogeneity of the first order. We restrict ourselves to non-deformable media, and systems with a constant ratio of fluid surface area to volume (no film formation). For such systems we proceed to find expressions for the chemical potential and the pressure in the context of non-deformable porous media, cf. sections 2.2, 2.3. We intend to extend the theory to deformable media later.
The expression for the entropy production with these variables is detailed in section 3. The driving forces, due to temperature -, pressure -, and chemical potential gradients, are specified in section 4. They obtain new contributions compared to their normal form in homogeneous systems. In the last section 5, we detail specific cases of component and volume flow in combination with heat transport. An experimental program is suggested in the end to verify Onsager symmetry in the transport coefficients.
2. Thermodynamic Variables for the REV
2.1. The Basis Set of Variables
The central concept in this analysis is the representative elementary volume; the REV [19, 20]. Its characteristic size, l, is small compared to the size (length) of the full system, L, but large compared to the characteristic pore length and diameter. An illustration of the REV is given by the squares in Figure 1. The REV (square) consists of several phases and components. The problem is to find the representation on the larger scale. For each point in the porous system, represented by a (blue) dot in Figure 1, Kjelstrup et al. [1] used the REV around the dot to obtain the variables of the REV (). The variables were given superscript REV to indicate that they constituted the only independent variables of the REV. From the Euler homogeneity of these variables, the possibility followed to define the temperature, pressure, and chemical potentials of the REV, (T, p, μi). We refer to Kjelstrup et al. [1] and to Tables A1–A3 in the Appendix for further details, terminology and symbols.
FIGURE 1
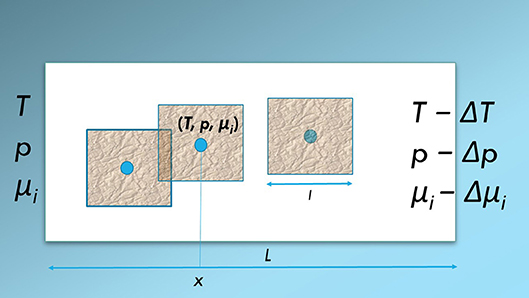
Figure 1. A representative elementary volume (REV) (magenta squares, length l) in a system (white box of length L) that is exposed to for instance a pressure difference, Δp, a temperature difference, ΔT, and differences in chemical potentials Δμi. A (blue) dot is used to represent the state that characterizes the REV. The set of state variables are positioned on the x-axis: The temperature, pressure and the chemical potentials, (T, p, μi)(x). The figure is adapted from Kjelstrup et al. [1].
The value of each of these REV-variables was obtained as a sum of contributions from each phase, interface and three-phase contact line present [19, 20]. The contributions are pore-scale variables; they are not independent variables on the macro-scale. To assume Euler homogeneity, means to assume that a REV of the double size, for example, has double the energy, entropy, and mass, as well as double the surface areas and double the line lengths of various types. The average surface area, pore length and pore radius, as well as the curvature of the surfaces per unit of volume of the REV, are then everywhere the same. We limit ourselves to non-deformable systems with a constant ratio of fluid surface area to volume, for which this is the case. The extension to deformable systems is more complicated and will be considered later.
A system of k components in m homogeneous phases, has a volume, VREV, with contributions from the homogeneous bulk phases Vα, REV, m ≥ α ≥ 1, and the excess line volumes, Vαβδ, REV, m ≥ α > β > δ ≥ 1.
The superscripts denote the relevant phases, surfaces or contact lines. The surface area between phases α, β is denoted Ωαβ, REV while the contact line length between phases αβδ is denoted Λαβδ, REV. The surface area (line length) of the REV is the sum over all areas (lines) in the REV. The excess surface volumes are by construction zero. The excess line volumes are not, because the dividing surfaces in general cross each other along three different lines.
In this first exposition, we neglect contributions to the volume from the contact lines, which are normally small also in porous media. The volume of the pores is
Superscript p is used for pore. The porosity, ϕ, and the degree of saturation, Ŝα (saturation for short), are
Superscript α is used for a component, which is equal to the phase in the present case. The porosity and the saturation do not depend on the size of the REV, and have therefore no REV-superscript.
The mass of component i in the REV, , is the sum of the masses in the homogeneous phases of the REV, α, , m ≥ α ≥ 1, the excess interfacial masses, , m ≥ α > β ≥ 1, and the excess line masses, , m ≥ α > β > δ ≥ 1. We obtain:
where the first term on the right-hand side also can be written in terms of the (constant) densities
Similar contributions follow for the other terms.
We shall often use the example of two immiscible one-component phases w and n in a solid porous material r of porosity ϕ, where contact line contributions are negligible. We can think of phase w as wetting, and n as non-wetting. The mass variables from Equation (4) are for n:
while for the other components we obtain also surface excess contributions:
When an interface is formed between two phases, we are free to choose the position of the interface such that one of the components has a zero excess mass. This is the position of the equimolar surface of this component. This position is convenient because the number of variables are reduced. When we use the equimolar surface of n, , and when we use the equimolar surface of w at the surface of the solid, . These choices simplify the description of the REV. Therefore, we shall later use the chemical potential of component n, which has a bulk contribution only, see section 2.2.
The expressions for UREV and SREV are similar to Equation (4). This way to construct a REV is reminiscent of the geometric construction of a state function, proposed for flow in porous media by McClure et al. [21].
The basis set of macro-scale variables of the REV () apply to the whole REV. The temperature, pressure and chemical potentials of the REV, (T, p, μi), on the macro-scale were next defined, as is normal in thermodynamics, as partial derivatives of the internal energy. These definitions are normal in the sense that they have the same form as they have in homogeneous systems. They are new because the variables (say UREV) have contributions from all parts of the heterogeneous REV. The intensive variables T, p, and μi are then not averages of the corresponding variables on the pore-scale. The importance of this was also pointed out by Hassanizadeh and Gray [11,12].
The macro-scale densities of internal energy, entropy and mass; in the example, u, s, ρi, do not depend on the size of the REV. The densities are therefore convenient when we need to integrate across the system [1]. They are, however, functions of the position of the REV, cf. Figure 1.
2.2. The Gibbs Energy of the REV
We proceed to define the Gibbs energy, G, as this variable is needed in the definition of driving forces of transport, see section 3. The general expression for Gibbs energy is
where G applies to the REV and Gi is defined for component i in the last identity. All REV variables need be taken into account. In principle, each component can exist in all phases in the REV. But component contributions to the REV are additive, cf. Equation (4). For component i we therefore have
The expression gives the Gibbs energy contributions of component i to the REV. We neglected again possible contributions from contact lines.
In the case of two immiscible, one-component fluids in a non-deformable porous rock, we can take advantage of the simpler description of the non-wetting fluid (see previous subsection) giving
For immiscible, one-component fluids, the label indicating the components also gives the phase. The density is an average over Vn, REV. The local density in the pores may vary around the average.
The total differential of U is used with the definition (8), and we obtain
where the superscript REV is skipped for convenience.
2.3. The Pressure of the REV
We find the pressure of the REV by starting, as above, with the extensive property that holds the pressure as the variable. This is the grand potential. The compressional energy of the REV is equal to minus the grand potential:
The grand potential of the REV is additive, which gives
We introduce contributions from all phases, surfaces and contact lines. This allows us the possibility to define, in a new way, the pressure of the REV:
The last equation makes it possible to compute the pressure of the REV, p, from the pressures in the bulk phases, the surface tensions and the line tensions. With knowledge of the pressure in the REV, we can find the driving force, −dp/dx, in the entropy production, see below, Equation (29).
We explain now how we can define and compute the pressure from Equation (14), using the example of two immiscible single fluids in a non-deformable medium. We shall neglect contact line contributions for simplicity. Such contributions can be added by the same procedure. We follow Equation (14) and sum over the n, w and r bulk phases, and the nr, wr, and nw−interfaces. The situation can be illustrated for a single cylindrical pore, see Figure 2.
FIGURE 2
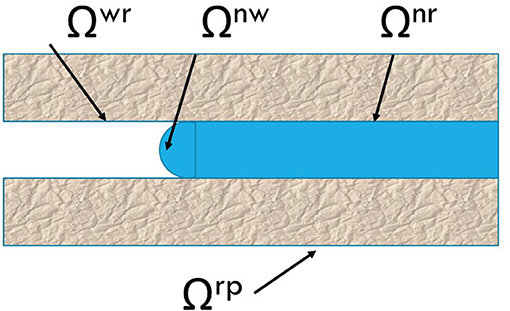
Figure 2. Illustration of contact areas between the phases. A cylindrical pore is chosen as example.
The figure shows two phases n and w in a tube with the average radius. The wall material is r. Contact areas are therefore Ωnr, REV, Ωwr, REV, and Ωnw, REV. The total surface area of the pore is Ωrp, REV≡Ωnr, REV+Ωwr, REV. The area Ωnw, REV is the smallest contact area shown in the figure. The volumes in Equation (14) depend on the saturation of the non-wetting component, Ŝn and the porosity, ϕ. Neither of the fluids form a film between the surface and the other fluid, so the surfaces satisfy in good approximation
The pressure of the REV from Equation (14) can then be written as:
Contact-line contributions were again not taken along, for simplicity. A consequence of the porous medium being homogeneous is that Ωrp, REV/VREV is the same everywhere. The ratio can be used as a measure of the average curvature of the pore surface, as will be explained below.
The volume-averaged contributions to the pressure from the homogeneous phases is given the symbol :
The first term in the last equality shows that the saturation gives an important contribution to the volume-averaged pressure. The contributions of the 2nd and 3rd terms are due to pw and pr. These contributions are usually constant.
The surface-averaged contributions to the pressure are likewise given a separate symbol:
The contribution of to the total pressure, p, may be called the capillary pressure. The total pressure of the REV is thus, for short:
2.3.1. The Case of Approximately Cylindrical Pores
With an (approximately) cylindrical pore geometry, we can define the average radius of the pores by
By introducing into Equation (18) we obtain the capillary pressure
Again the first term shows that saturation gives an important contribution. The 2nd term only depends on the temperature and is usually constant. The 3rd term is proportional to the surface area of the fluid-fluid interface. In many experiments this surface area is much smaller than Ωrp, REV. When that is the case, this term is negligible.
The three equations above give an expression for the REV pressure p for the example system.
To estimate the size of the various contributions, it is convenient to use mechanical equilibrium for the contact line and for the surface, although this condition may not apply to the REV, not even under steady flow conditions. With a balance of forces at the three-phase contact lines, Young's law applies for the surface tensions: γnr−γwr = γnw cosθ, where θ is the (average) contact angle. When there is furthermore mechanical equilibrium at the fluid-fluid interfaces, the pressure difference between the fluids is given by Young-Laplace's law, .
In the single-fluid (w) case, Equation (16) simplifies. There are volume-averaged and surface averaged contributions,
We have defined above in detail what we mean by the pressure of a REV. We have found, using the grand potential, that it can be regarded as result of volume- and surface average properties, and we have given some examples. These contributions enter the driving force in Equations (23, 29), to be further discussed below.
3. The Entropy Production of Non-isothermal Two-Phase Flow
Pressure-driven mass flows through porous media can lead to gradients in composition and temperature, and vice versa; temperature gradients can lead to mass flow, separation of components and pressure gradients. The interaction of such flows is of interest, and motivated the search for convenient forms of the entropy production [1].
3.1. Expression in Terms of Component Flows
From the Gibbs equation for the REV, we derived the entropy production for transport of heat and two immiscible fluid phases through the REV [1]. With transport in the x-direction only, the entropy production σ of the example system was
The frame of reference for the mass transport is the non-deformable solid matrix, Jr ≡ 0. Here is the sensible heat flux (in J.m−2.s−1), T is the temperature (in K), Ji is a component flux (in kg.m−2.s−1) and ∂μi, T/∂x is the gradient of the chemical potential (in J.kg−1.m−1) evaluated at constant temperature. All properties are for the REV, so superscript REV is omitted.
The thermal force conjugate to the heat flux is the gradient of the inverse temperature, where the temperature was defined for the REV as a whole, see Kjelstrup et al. [1]. The chemical force conjugate to the mass flux is the negative gradient of μi, T over the temperature. The chemical driving forces are obtained from the full chemical potential, which is the derivative of G with respect to Mi:
The total differential of the chemical potential is thus from Equation (11);
where Si = −(∂μi/∂T)p, Mj and Vi = (∂μi/∂p)T, Mj and are partial specific quantities. The last term describes the change in the chemical potential by changing composition of the medium. The dμi, T is now defined as a part of the whole differential:
The last term is zero when the composition is uniform. In the expression of the entropy production in terms of component flows, Equation (23), the driving force has contributions from the composition variation and from the pressure gradient.
3.2. Expression in Terms of Volume Flow
The volume flow, rather than the component flows, is often the measured variable. A description with the volume flow is thus of interest. The volume flow of several components is JV = ΣiJiVi. We take the example of two fluids to demonstrate the principles.
With two fluids in a uniform, non-deformable rock, there are three components. On the coarse-grained level, these are mixed. We assume that , and obtain Gibbs-Duhem's equation on the form
where ρi is the density of i in the REV (in kg.m3). This can be used with Equation (26) and JV to change Equation (23) into
We have chosen to keep the chemical potential of n, which has a simpler form than the other chemical potential (), cf. section 2.2. The entropy production in Equation (23) is invariant, and this invariance defines υD as the relative velocity of component w and n (in m.s−1):
The entropy production 29 has also three terms. While the first term on the right-hand side is the same as before, the second term is the volume flow with minus the pressure gradient over the temperature as driving force, and the third term is the velocity difference with the chemical potential gradient times the density over the temperature as driving force.
Equations (23, 29) are equivalent. They describe the same entropy production or flow dissipation. They provide alternative choices of conjugate thermodynamic force-flux pairs. The choice to use in the particular case, is determined by practical reasons; what can be measured or not, or which terms are zero. For instance, under isothermal conditions we need not take the term containing the heat flux along, even if heat may be transported reversibly. One set may give a negative contribution to the entropy production (work is done), but the overall entropy production is positive, of course. Each set can be used to obtain constitutive equations for transport on the macro-scale. We shall proceed to find these for porous media flow, finding first more detailed expressions for the driving forces.
In the simple case of a single fluid, say w, we obtain directly from Equations (23, 26) that
In the absence of a gradient in composition, , the same expression applies also for more components. We may follow Hansen et al. [22] and write the component contributions as , and . where the saturation has been introduced, and vi is the volume flow of i.
4. Definition of the Driving Forces in the Context of a Porous Medium
We expand on the basis presented earlier [1], and give definitions of the driving forces in the context of porous media flow.
4.1. The Saturation-Dependent Contributions to the Chemical Potential Gradient
The specific contribution to the chemical potential gradients in the entropy production in Equation (23) is of interest. The concentration dependent part of the chemical potential of i, μi, for an ideal system is (in J.kg−1)
Here R is the gas constant (in J.K−1.mol−1) and Wi is the molar mass (in kg.mol−1). The chemical potential is measured referred to a standard state, , having the local concentration in all the pores. In the description of porous media, a convenient reference state may be the state when one component is filling all pores, or when the saturation is unity, Ŝi = 1. The mass density of i in the REV for the standard state is . Away from this state for i = n, w. When ϕ is constant, this gives
By introducing these definitions into Equation (32), we obtain for the concentration dependent part of the chemical driving force of n
We see that any variation in saturation between REVs along the x-axis (cf. Figure 1), will lead to a driving force. We integrate between two REVs and obtain the chemical driving force for porous media flow
We have seen that the return to the Gibbs energy of the porous medium helped define the chemical potential in terms of properties relevant to porous media. All variables are measurable.
4.2. The Pressure Dependent Contribution
The driving force for volume flow is the negative gradient of the REV pressure over the temperature. To measure the pressure p inside the REV is difficult. The pressure in the fluid phases adjacent to the porous medium can be determined. Tallakstad et al. [11] defined the measured pressure difference, Δp′, at steady state, as an average over the value Δp(t) over the time of measurement:
Here t is the time and Δ refers to the extension of the system. Subscript 's' denotes the start and 'e' denotes the end of the measurement. We will take this pressure difference as our Δp. The pressure differences Δpw and Δpn can also be measured when there is continuity in the fluids, w and n, respectively.
The pressure variation across the REV is given by Equation (19), we have an interpretation of the pressure difference external to the porous medium;
We integrate over the system and obtain an interpretation of the total pressure difference:
We can assess the right-hand side of this equation using Equations (17, 18).
4.2.1. The Case of Large Pressure Differences
When the pressure drop across the porous plug is large compared to the capillary pressure contribution, the surface contributions and therefore can be neglected. This is the case of large capillary numbers. Furthermore pn = pw. In the pressure difference, the terms with constant ϕ and pr disappears, and the pressure difference is:
The pressure is applied to the whole cross-sectional area. This explains that the net driving force becomes a fraction, ϕ, of Δpw. In other words, the force applies to the fraction ϕ of the pore area.
The conditions leading to Equation (39) are common in the laboratory. Some numerical values for the air-glycerol system, [4], can illustrate when the conditions apply. The value of is of the same order of magnitude as pcŜnϕ (400 Pa) when the surface tension γ = 6.4 · 10−2N m−2, the average pore radius = 0.2 mm and the porosity ϕ = 0.63. A typical value of in the experiments is close to 30 kPa, which is far from the limit where capillary effects are significant.
4.2.2. The Case of Small Pressure Differences
For small capillary numbers the effective pressure drop across a porous plug is comparable to or smaller than the capillary pressure. Surface contributions need be taken into account. Equation (38) gives the effective pressure difference. When we can assume a constant average radius , and constant porosity, we obtain
A fluid will be transported when the surface tensions of the fluids with the wall are different and there is a difference in the saturation. When there is only one fluid in the porous medium, cf. Equation (22), and we have constant and porosity, the pressure difference becomes
The last term can lead to mass transport, when the surface tension changes.
5. Constitutive Equations. Examples
5.1. Constitutive Equations for Non-isothermal, Two-Phase, Immiscible Fluids
The constitutive equations follow from the entropy production. We present these on differential form for two incompressible flows. From Equation (23) we have:
We can also use Equation (29) and obtain
The flux-force relations are linear on the REV-level. Our construction of the coarse-grained entropy production has followed the standard line in non-equilibrium thermodynamic theory, meaning that the Onsager relations holds for each matrix of coefficients. They may not hold, when the REV no longer can be constructed using Euler homogeneity (i.e., can be regarded as a thermodynamic state) or when the balance equations fail. Some evidence exists that the REV is a thermodynamic state [4, 5]. Also, there is theoretical and computational proof that the Onsager relations apply, lij = lji [23–25]. Experimental proof for the Onsager relations exists for one-phase flow in porous media[26], but as far as we know, not for two-phase flow. We continue to specify how this possibly can be achieved.
One set of conductivities can be expressed by the other, using entropy production invariance. The element lqq is the same in both formulations.
We have discussed above how the local and overall driving forces can be determined. We integrate across the system in order to relate experimental results to theory. We integrate the linear laws 43 across the REV, and obtain
and
Here Lij ≡ lij/l and l is the length of the REV, and the driving forces are defined by Equations (35, 38).
The coefficients may become dependent on the force through the integration as shown by Sinha et al. [27]. The averaging procedure gave the conductivity as a function of ( - Δpc) in the terminology of this paper. In the remainder of this work we will discuss experimental conditions that allow us to determine these coefficients. The presentation follows closely the derivation of Stavermann [28] and Katchalsky et al. [29]. For transport in discrete systems with polymer membranes, see also [30]. We refer to these works for further definitions of transport coefficients.
5.2. Constitutive Equation for Isothermal, Single Fluid
For an isothermal single fluid w, flowing inside a porous medium, the entropy production 45 has one term; the volume flow times the negative pressure difference over the temperature. By including the constant temperature in the transport coefficient, we obtain the common linear law. With the permeability Lp, we write
where Lp ≡ Lpp/T. The permeability is normally a function of state variables (pressure, temperature). In the hydrodynamic regime it is a function of viscosity, Lp = Lp(p, T, η). By introducing the new expression for the pressure, Equation (22), we obtain
When the permeability and porosity are constant, LVV ≡ Lpϕ. The equation predicts a threshold value for flow if there is a (significant) change in the surface tension across the REV. Transport will take place, when . The permeability LVV is inversely proportional to the viscosity η of the fluid in the hydrodynamic regime. Interestingly, Boersma et al. [8] and Miller et al. [7] plotted the volume flow vs. the hydrostatic pressure difference Δpw and found a deviation from Darcy's law in the form of a pressure threshold, for water or water solutions in clay. They offered no explanation for this. Also Bernadiner et al. [9] and Swartzendruber [6] plotted the volume flow of water solution JV vs. the pressure gradient in sandstone with low clay content [9], and in NaCl-saturated Utah bentonite [6]. The thresholds that they observed depended on the content of salt in the permeating solution. They explained the thresholds by water adsorption and pore clogging by colloids [9]. According to Equation (47), a varying surface tension (due to a varying adsorption and clogging) might explain the existence of a threshold or a non-linear flux-force relation. There is no reason to believe that the non-linear flux-force relation is not caused by creation of system disorder, as was shown analytically for a tube [27] and in Sinha and Hansen [12] for a porous medium.
5.3. Constitutive Equations for Isothermal, Two-Phase Fluids
The entropy production in Equation (29) has two terms when two immiscible components flow at isothermal conditions. We choose the formulation that has variables JV and υD; volume flux and interdiffusion flux, respectively. Equation (45) gives then:
where . The coefficients reflect, as above, the mechanism of flow (pressure, diffusion). Four experiments can be done to determine the four coefficients. There are only three independent coefficients. When four experiments are done, we can check the Onsager relations.
5.3.1. Main Coefficient: The Hydraulic Permeability
The (hydraulic) permeability K is related to the mobility coefficient Lpp, by Lpp = K/η where η is the fluid viscosity. Both coefficients are measured at uniform composition. By introducing the driving force for the volume flow from Equation (38), we obtain [the last term on Equation (48)] is zero:
With the present definition of variables the equation applies to the overall behavior of the system. A plot of JV vs. may show a threshold. This threshold has more contributions than in the single component system, as there are contributions to the pressure from surface and line energies. A threshold may be detectable at low capillary numbers.
The hydraulic permeability, is found by measuring the volume flow caused by the overall pressure difference at uniform composition;
The mobility is a function of the saturation, In the hydrodynamic regime, the coefficient can be modeled, assuming Poiseuille flow and the effective viscosity [27]. Using a pore model, Sinha et al. [27] found a dependence of the coefficient Lpp on the threshold pressure. This non-linearity does not prevent the use of non-equilibrium thermodynamics.
5.3.2. Main Coefficient: The Interdiffusion Coefficient
The main coefficient Ldd is an interdiffusion coefficient. It is defined at uniform pressure from the difference flux created by a difference in saturation;
where we used Equation (35) for the driving force.
5.3.3. The Two Coupling Coefficients
The coupling coefficients in Equations (48) express that a separation of components can be caused by a pressure gradient (Ldp) and that a volume flow can be promoted by a gradient in saturation (Lpd).
Consider first the determination of Ldp. A pressure gradient may build as a consequence of a difference in composition [30]. The volume flux continues until a balance of forces is reached:
From the force-balance across the system, we obtain:
This condition can be used to find the unknown coupling coefficient, once the hydraulic permeability is known.
The remaining coupling coefficient can be found from the flux ratio, r, that has been called the reflection coefficient r, see also [30]. At constant saturation, we have
We are now in a position to compare Lpd and Ldp and verify the Onsager relations. The state of the system must be (approximately) the same, when the comparison is made.
5.4. Constitutive Equations for Non-isothermal, Two-Phase Fluids
The full set of equations given in Equation (45) must be used to describe non-isothermal flow in porous media. The coefficients, Lpp, Lpd = Ldp, Ldd in the lower right-hand side corner of the conductivity matrix, were discussed above. The new coefficients are those related to heat transport. The coefficient Lqq represents the Fourier type heat conductivity at uniform composition and pressure. The coefficients Lpq and Ldq are coupling coefficients.
Non-zero coefficients Lpq and Ldq mean that we can obtain separation in a temperature gradient. Injection of cold water into warm reservoirs may thus lead to separation. Likewise, a pressure difference can arise from a temperature difference. This is thermal osmosis [14].
Separation caused by a thermal driving force was observed in clay-containing soils where water was transported in clay capillaries against a pressure gradient. The coefficient, measured at constant pressure, was called the segregation potential [31]. The coefficient Lpq can be obtained from Equation (45), setting Δp = 0 and (ΔŜn=0) in the second line. We obtain
This coefficient can also be found from steady state conditions, when the thermal gradient is balanced by a gradient in saturation (chemical potential)
This determination of Lpq requires knowledge of Lpd. Alternatively, we may obtain the coefficient from the thermal osmosis experiment
The coupling coefficient Lqp can also be found by measuring the heat flux that accompanies the volume flux for constant composition and at isothermal conditions.
These interrelated effects are well-known in homogeneous media [14], but have to the best of our knowledge, not been measured for porous media with two-phase flow.
6. Discussion and Conclusion
We have further developed a new coarse-grained formulation of the entropy production [1] for porous media, and specified the constitutive equations for flow of two immiscible fluids under uniform or varying temperature, pressure and composition. Several of the equations are new in the context of porous media, but they follow well-documented tracks in classical non-equilibrium thermodynamics [1, 28–30]. Experimental observations exist on single fluid flow, that give support to the theoretical description.
We have given a new definition of the pressure of the representative elementary volume (REV), and used it to obtain the pressure part of the driving force. The force obtains contributions from homogeneous phases, surfaces—and, in principle also line—tensions of the system. This distinguishes the present formulation from their counterpart for homogeneous systems [1, 28–30].
We have seen that surface contributions can be spelled out for varying conditions, under the assumption that the additive properties of the REV are Euler homogeneous of the first order. Doing this, we have been able to explain for instance deviations from Darcy's law, or the occurrence of threshold pressures in plots of flow vs pressure difference. We have pointed at possibilities to describe non-isothermal phenomena.
As for instance sections 5.1–5.3 show, there is a multitude of scenarios that can be further investigated, and used to check the theory. The expressions open up the possibility to test the thermodynamic models in use, for their compatibility with the second law.
The basic assumption used is that the REV set of basis variables are Euler homogeneous functions of degree one. This means in essence that one temperature, one pressure and one chemical potential per component can be defined for the REV. Some evidence already supports the idea that the REV is a thermodynamic state [4], [5], originally proposed by Hansen and Ramstad [3] and Tallakstad et al. [11]. We did neither consider surface areas, nor their curvature or the contact line length as independent variables, but these may be included, cf.[21].
We have illustrated relations for some specific cases; the non-isothermal flow of one or two immiscible single fluids in a non-deformable medium. It is straight forward to include more terms in the chemical potential (e.g. gravity). To include stress fields or other fields that deform the porous medium is more problematic, and has been postponed.
Flow of two isothermal, immiscible fluids in a porous medium has often been described by Darcy's law, using the relative permeability concept. The seepage velocities vn and vw are related to fluxes used here by vn = JnVn and vw = JwVw. The expressions for the seepage velocities must be contained or be equivalent to the expressions given here, using the condition of invariance for the entropy production. A comparison can elucidate assumptions that are made. Hilfer and Standnes et al. [32, 33] gave a set of linear relations for the seepage velocities. Their driving forces were the gradients in the single component pressures, obtained by pressure measurements in the single phases. Their description implies e.g., that the composition is uniform.
We have seen through these examples how non-equilibrium thermodynamic theory can provide a fundamental basis for constitutive equations, also in porous media. For systems that obey entropy production invariance and Onsager symmetry, we have obtained relations between variables, which have been used to a limited degree for two-phase systems.
Author Contributions
SK and DB defined the variables of the REV and wrote the first draft. AH, BH, and OG critically examined all proposals and contributed to revisions of the Manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Per Arne Slotte is thanked for stimulating discussions. The authors are grateful to the Research Council of Norway through its Centers of Excellence funding scheme, project number 262644, PoreLab.
References
1. Kjelstrup S, Bedeaux D, Hansen A, Hafskjold B, Galteland O. Non-isothermal two-phase flow in porous media. The entropy production. Front Phys. (2018) 6:126. doi: 10.3389/fphy.2018.00126
3. Hansen A, Ramstad T. Towards a thermodynamics of immiscible two-phase steady-state flow in porous media. Comp Geosci. (2009) 13:227–34. doi: 10.1007/s10596-008-9109-7
4. Erpelding M, Sinha S, Tallakstad KT, Hansen A, Flekkøy EG, Måløy KJ. History independence of steady state in simultaneous two-phase flow through two-dimensional porous media. Phys Rev E (2013) 88:053004. doi: 10.1103/PhysRevE.88.053004
5. Savani I, Sinha S, Hansen A, Kjelstrup S, Bedeaux D, Vassvik M. A Monte Carlo procedure for two-phase flow in porous media. Transp Porous Med. (2017) 116:869–88. doi: 10.1007/s11242-016-0804-x
6. Swartzendruber D. Non-Darcy flow behaviour in liquid-saturated porous media. J Geophys Res. (1962) 67:5205. doi: 10.1029/JZ067i013p05205
7. Miller RJ, Low PF. Threshold Gradient for Water Flow in Clay Systems. Soil Sci Soc Am Proc. (1963) 27:605–9.
8. Boersma L, Lindstrom FT, Saxena SK. Limitations of Darcy's law in glass bead porous media. Soil Sci Soc Am Proc. (1973) 37:333–5.
9. Bernadiner MG, Protopapas AL. Progress on the theory of flow in geologic media with treshold gradient. J Environ Sci Health (1994) A29:249–75.
10. Sinha S, Bender AT, Danczyk M, Keepseagle K, Prather CA, Bray JM, et al. Effective rheology of two-phase flow in three-dimensional porous media: experiment and simulation. Transp Porous Med. (2017) 119:77–94. doi: 10.1007/s11242-017-0874-4
11. Tallakstad KT, Knudsen HA, Ramstad T, Løvoll G, Måløy KJ, Toussaint R, et al. Steady-state, two-phase flow in porous media: Statistics and transport properties. Phys Rev Lett. (2009) 102:074502. doi: 10.1103/PhysRevLett.102.074502
12. Sinha and Hansen Sinha S, and Hansen A. Effective rheology of immiscible two-phase flow in porous media. Europhys Lett. (2012) 99:44004. doi: 10.1209/0295-5075/99/44004
13. Kjelstrup S, Bedeaux D. Non-Equilibrium Thermodynamics of Heterogeneous Systems. Singapore: Wiley (2008).
14. Barragan VM, Kjelstrup S. Thermo-osmosis in membrane systems. J NonEq Thermodyn. (2017) 42:217–36. doi: 10.1515/jnet-2016-0088
15. Costeseque P, Pollak T, Platten JK, Marcoux M. Transient-state method for coupled evaluation of Soret and Fick coefficients, and related tortuosity factors, using free and porous packed thermodiffusion cells: applications to CuSO4 aqueous solution (0.25 M). Eur Phys J E (2004) 15:249–53. doi: 10.1140/epje/i2004-10064-6
16. Colombani J, Galliero G, Duguay B, Caltagirone JP, Montel F, Bopp PA. A molecular dynamics study of the thermal diffusion in a porous medium. Phys Chem Chem Phys. (2002) 4:313–21. doi: 10.1039/B106800H
17. H Davarzani MQ M Marcoux. Theoretical predictions of the effective thermodiffusion coefficients in porous media. Int J Heat and Mass Transfer (2010) 53:1514–28. doi: 10.1016/j.ijheatmasstransfer.2009.11.044
18. Keulen L, van der Ham LV, Kuipers NJM, Hanemaaijer JH, Vlugt TJH, Kjelstrup S. Membrane distillation against a pressure difference. J Membr Sci. (2017) 524:151–62. doi: 10.1016/j.memsci.2016.10.054
19. Hassanizadeh SM, Gray WG. Mechanics and thermodynamics of multiphase flow in porous media including interphase boundaries. Adv Water Resour. (1990) 13:169–86.
20. Gray WG, Hassanizadeh SM. Macroscale continuum mechanics for multiphase porous-media flow including phases, interfaces, common lines and commpon points. Adv Water Resour. (1998) 21:261–81.
21. McClure JE, Armstrong RT, Berrill MA, Schluter S, Berg S, Gray WG, et al. A geometric state function for two-fluid flow in porous media. arXiv:1805.11032 (2018). doi: 10.1103/PhysRevFluids.3.084306
22. Hansen A, Sinha S, Bedeaux D, Kjelstrup S, Gjennestad MA, Vassvik M. Relations between seepage velocities in immiscible, incompressible two-phase flow in porous media. Transp Porous Med. (2018) 125:565–87. doi: 10.1007/s11242-018-1139-6
23. Flekkøy EG, Pride SR, Toussaint R. Onsager symetry from mesoscopic time reversibility and the hydrodynamic dispersion tensor for coarse-grained systems. Phys Rev E. (2017) 95:022136. doi: 10.1103/PhysRevE.95.022136
24. Pride S, Vasco D, Flekkøy E, Holtzmann R. Dispersive transport and symmetry of the dispersion tensor in porous media. Phys Rev E (2017) 95:043103. doi: 10.1103/PhysRevE.95.043103
25. Burelbach J, Bruckner DB, Frenkel D, Eiser E. Thermophoretic forces on a mesoscopic scale. Soft Matter. (2018) 14:7446–54. doi: 10.1039/C8SM01132J
26. Li SX, Pengra DB, Wong P. Onsager's reciprocal relation and the hydraulic permeability of porous media. Phys Rev E. (1995) 51:5748–51.
27. Sinha S, Hansen A, Bedeaux D, Kjelstrup S, Savani I, Vassvik M. Effective rheology of bubbles moving in a capillary tube. Phys Rev E (2013) 87:025001. doi: 10.1103/PhysRevE.87.025001
28. Stavermann AJ. Non-equilibrium thermodynamics of membrane processes. Trans Farad Soc. (1952) 48:176–85.
29. Katchalsky A, Curran PF. Nonequilibrium Thermodynamics in Biophysics. Cambridge, MA: Harvard University Press (1965).
30. Førland KS, Førland T, Ratkje SK. Irreversible Thermodynamics. Theory and Applications. Chichester: Wiley (1988).
31. Konrad JM. Frost susceptibility related to soil index properties. Can Geotech J. (1999) 36:403–17.
32. Hilfer R. Macroscopic equations of motion for two phase flow in porous media. Phys Rev E (1998) 58:2090.
33. Standnes DC, Evje S, Andresen PØ. A novel relative permeability model based on mixture theory approach accounting for solid-fluid and fluid-fluid interaction. Transp Porous Med. (2017) 119:707–38. doi: 10.1007/s11242-017-0907-z
Appendix
Symbol Lists
TABLE A1
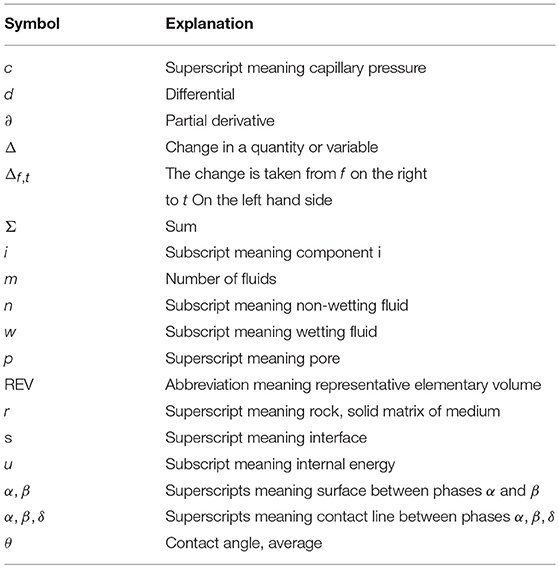
Table A1. Mathematical symbols, superscripts, subscripts.
TABLE A2

Table A2. latin symbols
TABLE A3
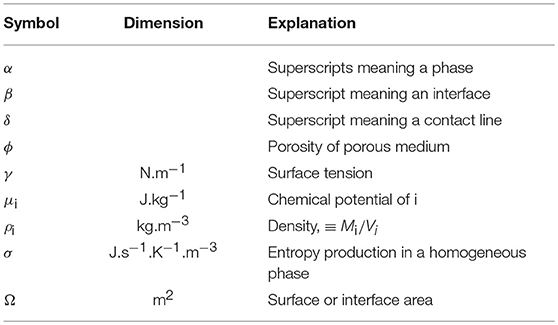
Table A3. greek symbols, continued
Keywords: porous media, energy dissipation, two-phase flow, representative elementary volume, macro-scale, excess surface energy, pressure, non-equilibrium thermodynamics
Citation: Kjelstrup S, Bedeaux D, Hansen A, Hafskjold B and Galteland O (2019) Non-isothermal Transport of Multi-phase Fluids in Porous Media. Constitutive Equations. Front. Phys. 6:150. doi: 10.3389/fphy.2018.00150
Received: 08 October 2018; Accepted: 11 December 2018;
Published: 04 January 2019.
Edited by:
Jürgen Vollmer, Institut für Theoretische Physik, Universität Leipzig, GermanyReviewed by:
Constantinos Siettos, University of Naples Federico II, ItalySoren Taverniers, Stanford University, United States
Copyright © 2019 Kjelstrup, Bedeaux, Hansen, Hafskjold and Galteland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Signe Kjelstrup, signe.kjelstrup@ntnu.no