 Nehemiah Zewde
Nehemiah Zewde Dimitrios Morikis
Dimitrios Morikis
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Phys. , 20 November 2018
Sec. Biophysics
Volume 6 - 2018 | https://doi.org/10.3389/fphy.2018.00130
This article is part of the Research Topic ImmunoPhysics and ImmunoEngineering View all 32 articles
The complex between complement system proteins C5b and C6 is the cornerstone for the assembly of the membrane attack complex (MAC, also known as C5b6789n). MAC is the terminal product of three converging pathways of the complement system and functions as a pore forming complex on cell surfaces, as a response of the immune system in fighting pathogens. However, when proper regulation of the complement system is compromised, MAC also attacks host tissues and contributes to several complement-mediated autoimmune diseases. We performed a molecular dynamics and electrostatics study to elucidate the mechanism of interaction between C5b and C6 and the formation of the C5b6 complex. The C5b6 interface consists of three binding sites stabilized predominantly by van der Waals interactions, and several critical salt bridges and hydrogen bonds. We discuss differences between domains C5d and C3d that lead to mono-functionality of C5d in acting as the scaffold for MAC formation, as opposed to dual functionality of C3d in acting as an opsonin for phagocytosis and as a link between innate and adaptive immunity, based on a comparative sequence, structural, and physicochemical analysis. We also extended our analysis to pathway dynamics to demonstrate the significance of consumption-production rates of C5b, C6, and C5b6 that lead toward MAC formation. Finally, we propose that C5d is a target for drug discovery, aiming to the inhibition of the MAC formation in autoimmune diseases originating from MAC-mediated host cell lysis.
As part of the innate immunity, the complement system orchestrates a cascade of biochemical reactions that result in pathogen elimination and in activation of the adaptive immune response [1, 2]. The versatile response of the complement system emerges from its three pathways known as alternative, classical, and lectin, that are either constitutively active in the fluid phase (alternative and classical pathway [3–5]) or initiate upon sensing danger-associated molecular patterns on pathogens (classical and lectin pathways). Activation of all three pathways converges on the cleavage of complement protein C3 into C3b and C3a [6]. Subsequently, continued propagation leads to the terminal cascade by cleavage of complement C5 to form C5b and C5a. Complement C6 then binds to C5b to form the complex C5b6 [7]. This soluble complex then associates with C7 to form C5b67, which later anchors to a nearby surface. Subsequently, the surface bound C5b67 binds to C8 to form C5b678 [8]. This complex, unlike the anchored C5b67, forms a pore of 0.9-nm diameter, which expands later in time to a 3-nm pore [9–11]. Finally, surface bound C5b678 recruits multiple C9s, to a maximum of 18, to form the membrane attack complex (MAC or C5b6789n where n = 1–18) [9, 12]. Although the proteins that make up the MAC pores are the same (C5b, C6, C7, C8, and C9), there is oligomeric heterogeneity in the assembly process of MACs. For instance, oligomerization of two to four C5b678 complexes can bind to a variable number of C9s to form a joined MAC pore [9]. In any case, structures of a single MAC pore, comprised of C5b678 in complex with a polymerized C9, are cylindrical in shape, contain a single stalk protrusion, and have an inner lumen diameter of 10- to 11.5-nm [9, 12]. MACs evolved as the only direct killing mechanism deployed by the complement system to fight against pathogens; indeed MAC deficiency has been associated with an increased risk of recurrent meningitis [1]. In addition to eliminating pathogens, MAC instigates numerous signaling pathways that directly affect cell cycles. For instance, sublytic MACs affect cell proliferation and apoptosis by enhancing or inhibiting the processes [13]. In addition, MACs can directly mediate cytokine production and platelet activation [13].
The instigation of an immune response via C5b6 is also detrimental to host-cells unless complement is properly regulated at the terminal stage [14]. To ensure tissue homeostasis, multiple checkpoints are present in fluid and surface phases that target complement propagators such as C3/C5 convertases. Furthermore, membrane-bound complement regulator, CD59, is present on host-cells to directly inhibit the assembly of the MAC and mitigate its deadly effects. However, despite the regulatory checkpoints, disease-related mutations over-activate the complement system and disrupt tissue homeostasis by generating elevated levels of MAC. This level of impairment propels complement in becoming one of the key drivers for diseases like hemolytic uremic syndrome (aHUS), age-related macular degeneration (AMD), and paroxysmal nocturnal hemoglobinuria (PNH) [15–20].
The formation of C5b6 complex sets the stage for a cascade of reactions that go beyond just the elimination of pathogens. C5b6 provides the junction at which the early- and late-stage complement pathway propagation converge to instigate signaling cues that are vital for cell survival [13]. Thus, understanding the governing mechanism behind C5b6 formation provides the basis for the first step in the assembly of terminal MAC complex that initiates a range of events, from immune defense to development of autoimmune diseases.
Complement proteins C3, C4, and C5 are structurally homologous [21–23] but only C3 and C4 have an internal thioester bond moiety that is capable of undergoing hydrolysis, followed by covalent attachment to cell surfaces [24, 25]. After cleavage of C3 and C4 by convertases to form fragments C3a/C3b and C4a/C4b, the C3b and C4b fragments are opsonins that attach to cells surfaces through their thioester domains (TED), also known as C3d and C4d when they become stand-alone proteins after additional cleavage steps. The cell-bound C3b is recognizable by phagocytes for elimination of the C3b-tagged cells, and also C3b and C4b become part of the convertase complexes that are responsible for C3 and C5 cleavage. On the other hand, C5b is missing an internal thioester bond, but it contains a TED-like domain that is structurally homologous to the TEDs of C3 and C4. For simplicity we will call hereafter C3d, C4d, and C5d the TED domains of C3 and C4, and the TED-like domain of C5, respectively.
Crystal structures of C3b in complex with structurally homologous modular regulators, Factor H (FH), complement receptor 1 (CR1) membrane cofactor protein (MCP), decay accelerating factor (DAF), and smallpox inhibitor of complement enzymes (SPICE), are available [26, 27]. These regulators are composed of repeated complement control protein (CCP) modules and have shown a shared binding mode along the structure of C3b, comprising modules CCP1-4 (FH, MCP, SPICE), CCP2-4 (DAF), and CCP15-17 (CR1). All regulators show contact of one module at the C3d domain of C3b. The viral vaccinia control protein (VCP), that is structurally and functionally homologous to SPICE, is also expected to have a similar binding mode to C3b. In addition, the stand-alone C3d domain, is known to interact with modules CCP1-2 of complement receptor 2 (CR2) [28], modules CCP19-20 of FH [29], in addition to modules CCP1-4 (mentioned above as interacting along C3b), and S. aureus proteins Efb-C, Ecb, and Sbi [30, 31]. These structural observations make the C3d domain multifunctional in interacting with complement natural and viral regulators, when C3d is part of C3b, and in attracting CR2, FH (CCP19-20), and bacterial regulators when C3d is stand-alone. On the other hand, C5b is not known to possess similar properties as C3b, and C5d is not known to exist in a stand-alone form. Instead, C5b acts as the first block of a scaffold that initiates the membrane attack complex, interacting first with C6, and subsequently with C7, C8, and several C9s, mentioned above. The crystal structures of C5b6 [32, 33] reveal that an elongated C6 surrounds half of the C5d domain and has three sites of contacts with C5b (Figures 1A,B). In addition, the crystal structure reveals the presence of charged patches on the surfaces of C5b and C6, and at the binding interface, which are expected to contribute to structural stability of the complex through the formation of ionic and hydrogen bonding contacts (Figures 1C,D).
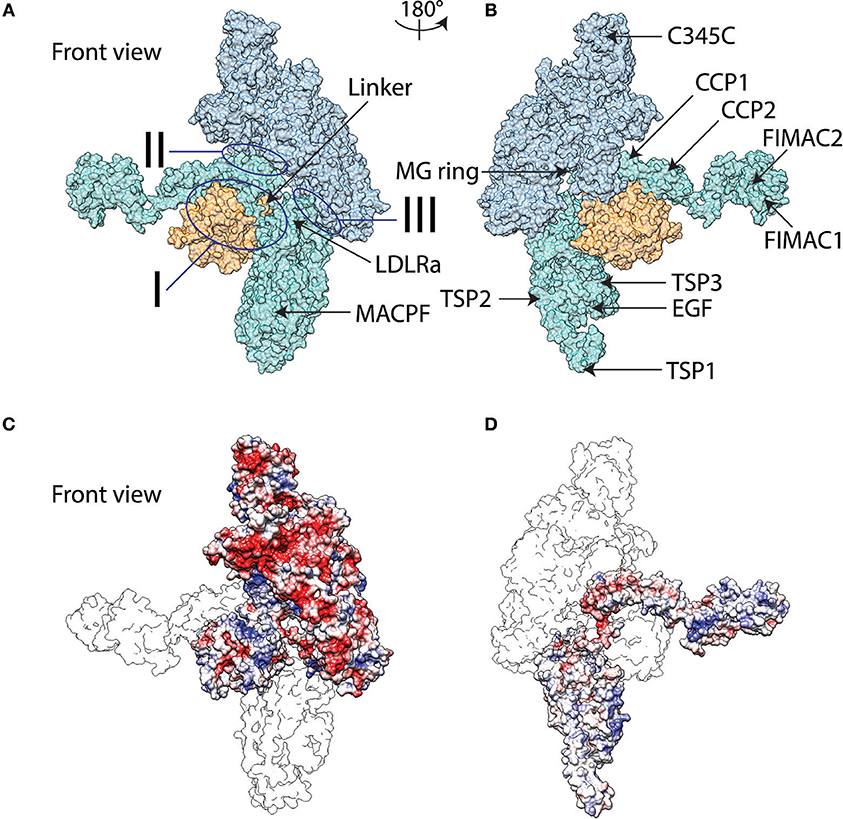
Figure 1. Surface representation molecular graphics of C5b6 complex in front view (A) and back view (B). C5b is indicated in blue with the thioester-like domain colored orange while C6 represented in teal. The three sites of interaction between C5b and C6 are marked. Domains of C5b and C6 discussed in text are marked. Electrostatic potentials mapped onto molecular graphics of C5b6 in front view with C6b represented as transparent outline and on top of C5b (C), and back view with C5b represented as transparent outline and on top of C6 (D). The color transitions from red to white to blue represent electrostatic potential values of −5 to 0 to 5 kBT/e. Abbreviations utilized are as follows: LDLRa, low-density lipoprotein receptor class A; MACPF, membrane attack complex perforin; MG, macroglobulin; C345C, netrin domain; CCP, complement control protein; FIMAC, Factor I/membrane attack complex; EGF, epidermal growth factor; and TSP, thrombospondin. Molecular graphics are generated using the final frame of the MD trajectory.
In this study, first we examine the physicochemical mechanism of the interaction between C5b and C6. Given that the C5d domain contributes to the interaction of C5b and C6, and the multifunctionality of C3d with several sites of interaction with native regulators and receptors and bacterial and viral regulators, we present a comparative sequence, structural, and physicochemical analysis between C5d and C3d. For completion, we also include C4d in the comparative analysis. Our goal is to contribute toward understanding mechanisms of function of C3d, C4d, and C5d at the structural and physicochemical property level. Finally, we present a systems-biology approach to understand the pathway dynamics of the terminal complement cascade that starts at C5b6 complex and ends at MAC [25]. We performed molecular dynamics (MD) simulations to relax the crystallographic structure from crystal packing effects, and to obtain insight on the dynamic character of the structure and the persistence of the intermolecular contacts at the amino acid side chain level. Guided by the findings of our MD analysis, and by our previous work that has shown that electrostatics plays a fundamental role on the regulation and function of C3b [34–40] and C3d [28–31, 41, 42], we performed electrostatic calculations using conformational states extracted from the MD data.
Our goal is to identify the stabilizing interactions that lead to formation of the C5b6 complex, the initial scaffold for the assembly of MAC. The crystal structure [32] shows three major sites of interaction between C5b and C6, named I, II, III (Figures 1A,B). Interactions between C6 and the thioester domain of C5b fall under Site I, while interactions of C6 with the macroglobulin (MG) ring of C5b fall under Sites II and III. The crystal structure also shows the presence of charged patches on the surfaces of the two proteins, C5b and C6 (Figures 1C,D), suggesting that charges may be contributing factors to binding. We performed an explicit solvent MD simulation, using a crystal structure as initial conformation, to optimize local geometries and chemistry and to delineate distinct conformational states visited throughout the simulation. Analysis of the MD trajectory, using the molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) method, showed an overall favorable binding energy for the solvated complex, dominated by van der Waals interactions (Table 1). Given the observation of many charged patches on the surfaces of C5b and C6 (Figures 1C,D), but overall unfavorable polar contribution to binding (Table 1), we analyzed the frequency of occurrence of intermolecular pairwise polar interactions to obtain a closer look into the nature of polar contributions. Specific intermolecular salt bridges often stabilize protein complexes, an effect that has been termed ionic tethering, and so do intermolecular hydrogen bonds, even if the overall energetic contribution is dominated by van der Waals interactions of hydrophobic chemical groups [43].

Table 1. Calculated MM-PBSA energies from the MD trajectory.
We characterized the significance of intermolecular polar interactions by evaluating the number of salt bridges and hydrogen bonds that occur across the C5b6 binding interface at a frequency of at least 20% during the MD trajectory. Figures 2A,B shows occupancy (frequency of occurrence during the MD trajectory) maps for intermolecular salt bridges and hydrogen bonds, respectively. The C5b6 complex has 13 intermolecular salt bridges with distance cutoff of 5 Å, and 11 intermolecular hydrogen bonds, demonstrating varying levels of persistence throughout the trajectory.
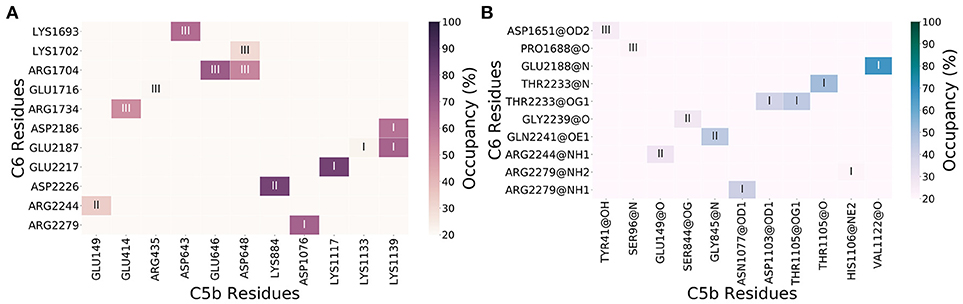
Figure 2. Intermolecular interaction occupancy maps for interactions persistent through at least 20% of the trajectory for salt bridges (A) and hydrogen bonds (B). The C5b6 interactions sites I, II, and III (Figure 1) are marked. Heavy atoms of hydrogen bond donor and acceptor chemical groups are marked in panel (B).
The 13 intermolecular salt bridges (and protein/domain) in decreasing order of persistence (81–20%) are: Lys1117(C5b/C5d)-Glu2217(C6/CCP1), Lys884(C5b/CUB)-Asp2226(C6/CCP1), Glu646(C5b/MG Ring)-Arg1704(C6/LDLRa), Asp1076(C5b/C5d)-Arg2279(C6/CCP2), Lys1139(C5b/C5d)-Glu2187(C6/CCP1), Asp643(C5b/MG Ring)-Lys1693(C6/TSP2), Lys1139(C5b/C5d)-Asp2186(C6/CCP1), Asp648(C5b/MG Ring)-Arg1704(C6/LDLRa), Glu414(C5b/MG Ring)-Arg1734(C6/LDLRa), Glu149(C5b/MG Ring)-Arg2244(C6/CCP1), Asp648(C5b/MG Ring)-Lys1702(C6/LDLRa), Arg435(C5b/MG Ring)-Glu1716(C6/LDLRa), and Lys1133(C5b/C5d)-Glu2187(C6/CCP1) (Figure 2A). All intermolecular salt bridges (cutoff distance 5 Å) at all occupancy levels are listed in Supplementary Data Sheet 1, and all intermolecular Coulombic interactions (cutoff distance 8 Å, including salt bridges with cutoff distance of 5 Å) at all occupancy levels are listed in Supplementary Data Sheet 2.
Unlike the persistence of intermolecular salt bridges, most of the intermolecular hydrogen bonds formed between C5b and C6 appear less than half of the time in the trajectory. The 11 intermolecular hydrogen bonds (and protein/domain) in decreasing order of persistence (68–20%) are: Val1122(C5b/C5d)-Glu2188(C6/CCP1), Thr1105(C5b/C5d)-Thr2233(C6/CCP1) (backbone-backbone), Gly845(C5b/CUB)-Gln2241(C6/CCP1), Thr1105(C5b/C5d)-Thr2233(C6/CCP1) (side chain-side chain), Asn1077(C5b/C5d)-Arg2279(C6/CCP2), Asp1103(C5b/C5d)-Thr2233(C6/CCP1), Glu149(C5b/MG Ring)-Arg2244(C6/CCP1), Ser844(C5b/CUB)-Gly2239(C6/CCP1), Tyr41(C5b/MG Ring)-Asp1651(C6/TSP2), Ser96(C5b/MG Ring)-Pro1688(C6/TSP2), and His1106(C5b/C5d)-Arg2279(C6/CCP2) (Figure 2B). All intermolecular hydrogen bonds at all occupancy levels are listed in Supplementary Data Sheet 3.
Six salt bridges occur in Site III, followed by five in Site I, and two in Site II (Figures 1A, 2A). On the other hand, six hydrogen bonds occur in Site I, followed by three hydrogen bonds occurring in Site II and two hydrogen bonds occurring in site III (Figures 1A, 2B). Three bifurcated salt bridges are observed. These are Site I salt bridge Lys1139(C5b) with Asp2186(C6) and Glu2187(C6), Site III salt bridge Arg1704(C6) with Glu646(C5b) and Asp648(C5b), and Site III salt bridge Asp648(C5b) with Lys1702(C6) and Arg1704(C6) (Figures 1A, 2A). Two residues of C6 are hydrogen bonded with more than one partner, both in Site I. These are Thr2233(C6) side chain with Asp1103(C5b) side chain and Thr1105(C5b) side chain, and Arg2279(C6) side chain with Asn1077(C5b) backbone and His1106(C5b) side chain (Figures 1A, 2B). Finally, two residues of C5b and three of C6 participate in both salt bridge side chain-side chain interactions and hydrogen bonding side chain-backbone interactions. These are Glu149 and Asp648 of C5b, and Arg1704, Arg2244, and Arg2279 of C6. Among them are Arg1704(C6)-Asp648(C5b) salt bridge and hydrogen bond, involving Arg1704(C6) which is also part of a bifurcated salt bridge with Glu646(C5b) (Site III), Arg2244(C6)-Glu149(C5b) salt bridge and hydrogen bond (Site II), and Arg2279(C6) salt bridge with Asp1070(C6b), and hydrogen bond Asn1077(C5b) (Site I) (Figures 1A, 2).
Unfavorable charge-charge interactions within 5 Å of each other were not observed at occupancies greater than 10% (Supplementary Data Sheet 4). Thirteen unfavorable charge-charge interactions were observed within 8 Å of each other using the 20% occupancy threshold, as follows: four at Site I, three at Site II, and six at Site III. At Site I, the unfavorable charge-charge interactions are between Lys1139(C5b) and Lys1889(C6), Asp1140(C5b) and Glu2187(C6), Asp1167(C5b) and Glu2217(C6) and, Glu1181(C5b) and Asp2185(C6). At Site II, the unfavorable charge-charge interactions are between Arg177(C5b) and Arg2244(C6), Arg840(C5b) and Arg2252(C6) and, Lys884(C5b) and Arg2244(C6). At Site III, the unfavorable charge-charge interactions are between Asp43(C5b) and Asp1651(C6), Arg98(C5b) and Lys1690(C6), Arg435(C5b) and Arg1734(C6), Asp643(C5b) and Asp1706(C6), Glu646(C5b) and Glu1695(C6) and, Lys652(C5b) and Lys1702(C6). Of these, the only interactions with frequency above 50% are between Asp1167(C5b) and Glu2217(C6) at Site I, and Lys884(C5b) and Arg2244(C6) at Site II. All unfavorable Coulombic interactions with cutoff distances 5 Å and 8 Å at all occupancy levels are listed in Supplementary Data Sheets 4, 5, respectively. Overall, unfavorable charge-charge interactions are weak.
We further analyzed the MD trajectory to capture the dynamic changes in electrostatic contributions, by evaluating distinct conformational states. We identified six representative structures from the MD trajectory through principal component analysis (PCA) decomposition on phi and psi angles, followed by k-means clustering of the PCA components. The cluster centers (Figure 3) were extracted as representative structures. Structural representation of clustering conformational states is shown in Supplementary Figure 1.
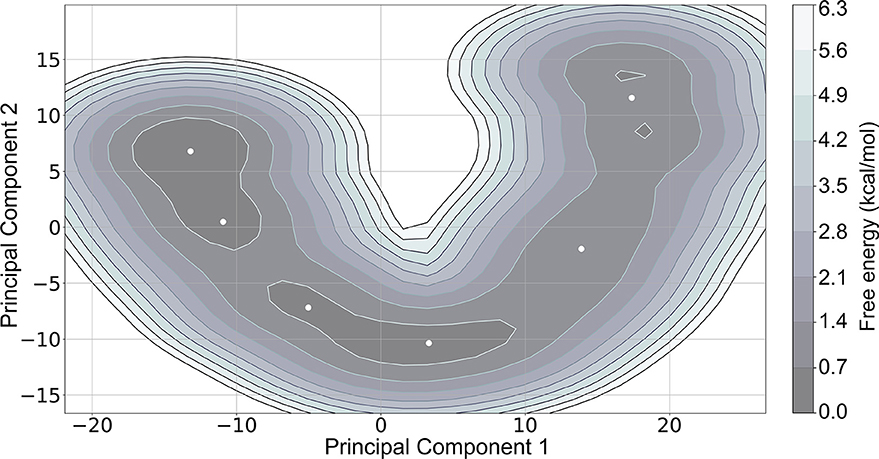
Figure 3. Free energy landscape of the first two principal components, Principal Component 1 and Principal Component 2, with cluster centers represented as white dots. The lowest energy regions (colored in dark gray as indicated in the color bar) represent energy minima.
We performed MM-PBSA calculations for the 20 most representative structures of each cluster to evaluate possible differences in the binding free energies among the six clusters. Although we observe differences among the clusters, the overall trends in their free energies are similar (Table 2).
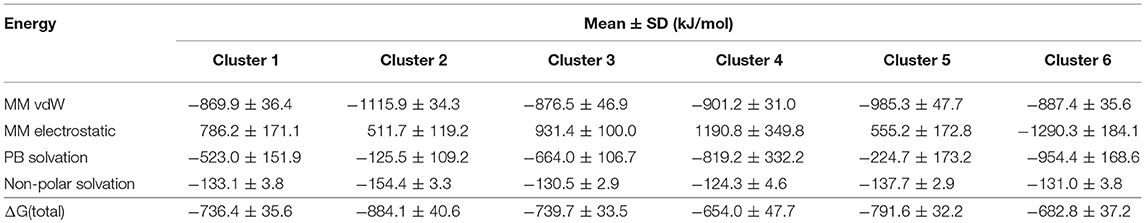
Table 2. Calculated MM-PBSA energies of six conformational states, extracted from the MD trajectory.
We used the alanine scan method of our AESOP computational framework [44] to explore in detail the significance of the many pairwise charge-charge interactions in contributing to the stability of the C5b6 complex. We performed computational alanine scan by mutating every ionizable amino acid residue to alanine, one at a time, to generate families of single mutants for C5b and C6, and their C5b6 complex. Subsequently, we performed Poisson-Boltzmann electrostatic calculations to obtain electrostatic contributions to the free energies of binding for the binding reaction of each mutant. We present the results in reference to the parent structure, as differences between the calculated electrostatic free energies of binding of mutant structures minus the electrostatic free energy of the parent structure (see Methods). We performed the calculations for the representative structures of all six conformational clusters, and the results for those residues that participate in salt bridges (shown in Figure 2A) are presented in Figure 4. Loss of binding upon mutation is denoted by an electrostatic free energy of binding with a positive value, whereas gain of binding is denoted by a negative value. Loss of binding for the mutant indicates that the mutated residue is a contributor to binding of the parent protein by forming favorable charge-charge interactions. Likewise, gain of binding for the mutant indicates that the mutated residue opposes binding of the parent protein by contributing to unfavorable charge-charge interactions.
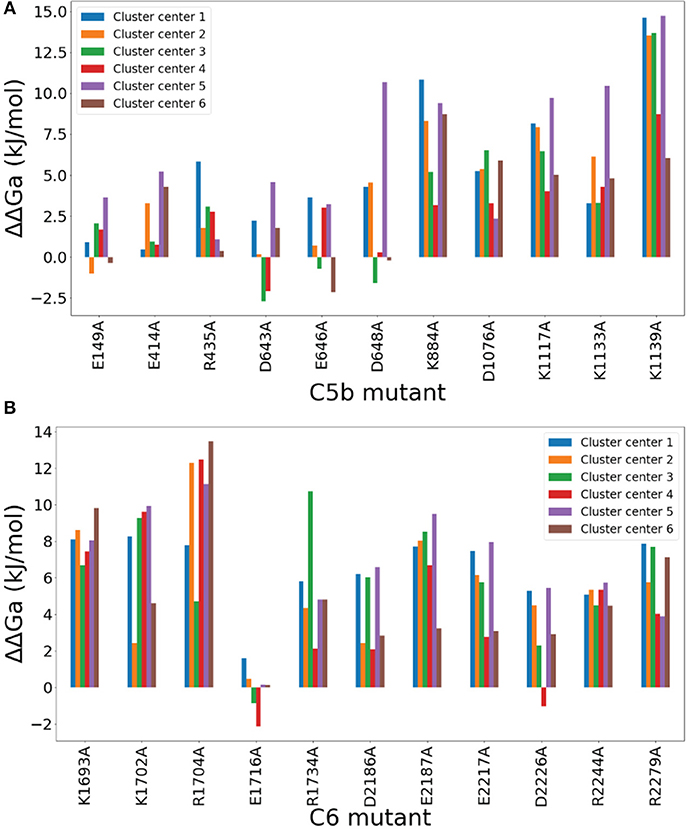
Figure 4. Computational Alanine Scan results of C5b (A) and C6 (B) mutants that participate in salt bridges. The vertical axis represents differences in the calculated electrostatic free energies of binding between the mutant and the parent structure for the mutants shown in the horizontal axis. Each mutant is represented by six free energy values, corresponding to the six representative (cluster center) structures of the conformational clusters of the MD simulation. The mutant notation denotes the residue number surrounded by the type of replaced residue on the left and the replacing residue (alanine, A) on the right. A positive value denotes that the mutation causes loss of binding, indicating that the mutated residue favors binding in the parent structure. A negative value denotes that the mutation causes gain of binding, indicating that the mutated residue disfavors binding in the parent structure. The one-letter amino acid code is used, instead if the three-letter code used in text, to simplify the figure.
A few mutants show perturbations greater than thermal energy of 2kBT at room temperature, most notably Lys1139A of C5b, followed by Lys884A, Asp1076A, Lys1117A, and Lys1133A (Figure 4A), and nearly every mutant of C6, most notably Arg1704, with the exception of Glu1716A (Figure 4B). These significant charge interactions depicted by the mutations of Figure 4 have been identified in the salt bridge occupancy maps of Figure 2A. AESOP data for all ionizable amino acid replacements within 8 Å of the C5b6 complex interface are listed in Supplementary Data Sheet 6. The most notable charge interactions, involving Lys1139(C5b) and Arg1704(C6) as depicted by the data of Figure 4, form strong bifurcated salt bridges in Sites I and II, respectively (see above). Another charge interaction, but of lower strength, involving Asp648(C5b) also forms a bifurcated salt bridge in Site III (see above).
The complement system contains thioester domains, referred herein as C3d and C4d, that can covalently attach to different surfaces and tag the cellular species with complement complexes and fragments. However, the C5d thioester-like domain of C5 is distinct from C3d and C4d because it does not have the ability to covalently tag a surface, or to exist as a standalone domain, but has gained the molecular capability to instigate MAC formation [32, 33]. Despite all three thioester/thioester-like domains, C3d, C4d, and C5d, having similar structures, C3d is a molecular hub for reactions that mediate inflammatory processes such as opsonophagocytosis and B-cell activation, whereas C5d participates in the binding of C6 and acts as the cornerstone for the MAC assembly. Thus, to elucidate on how C3d and C5d achieve their versatile functions, we performed a comparative analysis of the physicochemical properties of C3d and C5d, also including a comparative analysis of C3d and C4d for completion. Earlier studies have identified the residues of C3d that interact with FH [29] and CR2 [28]. FH is a potent regulator of C3b, and C3b's convertase complexes, that inhibits opsonophagocytosis on host cells. The CR2-C3d complex is formed as part of the B cell receptor-coreceptor complex and is a link between innate and adaptive immunity.
First, we performed a sequence alignment of all three domains, C3d, C4d, and C5d, to compare the conservation of their structurally and functionally important residues (Figure 5). The percent identity between C3d and C5d is 29.2%, between C3d and C4d is 35.7%, and between C4d and C5d is 28.8%. The contact sites with FH and CR2 are spread across the sequence of C3d. Of the total 310 C3d residues, 87 residues are involved in contacts with FH and CR2 combined (union of red and blue horizontal boxes in Figure 5). On the other hand, only nine C5d residues are involved in contacts with C6 (green horizontal boxes in Figure 5), and from those nine only one is a common site with C3d, that of Asp1104(C5d) and Gly1179(C3b). From the 87 residues that are involved in C3d-CR2/FH interactions, only 19 are homologous to C5d residues (intersection of red/blue horizontal boxes and identical C5d and C3d residues, denoted by vertical blue blocks, in Figure 5).
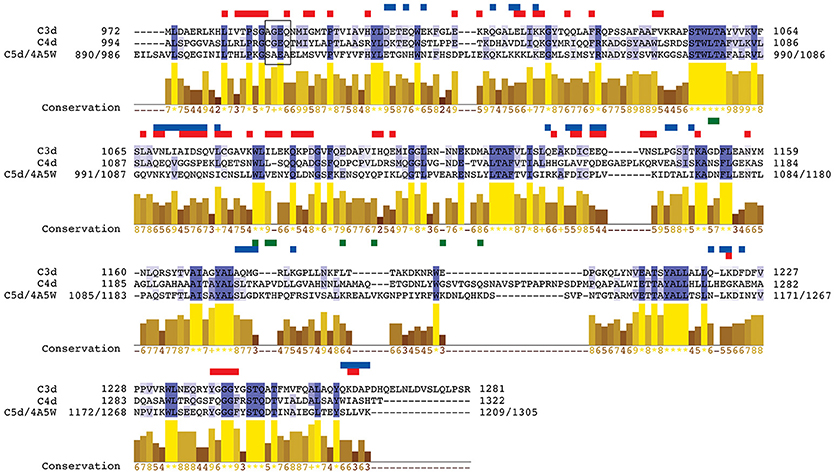
Figure 5. Sequence alignment of C3d, C4d, and C5d. Regions of intermolecular polar contacts between C3d and FH, C3d and CR2, and C5d and C6 are designated with horizontal rectangles above the sequences, colored in red, blue, and green respectively. Polar contacts: ionic interactions (salt bridges and long-range Coulombic) and hydrogen bonds (including side chain and backbone donor/acceptor groups). The vertical blocks in dark blue color denote identity in all three sequences, and those in light blue color denote identity in two out of three sequences. The bar plots denote similarity in conservation across all three sequences with numbers in yellow indicating the level of conservation where a larger number corresponds to higher conservation (“*” and “+” correspond to scores of 11 and 10 respectively). Residue numbering corresponds to the numbering of the crystal structures used for C3d and C4d. For C5d the residue numbering used in our study is shown, followed by the numbering of the crystal structure (PDB code 4A5W, see Methods). The thioester moiety for all three sequences is represented within a black box. Note that the cysteine that participates in the thioester bond is mutated to alanine, as was the case in the crystal structure used (PDB code 3OED).
Lastly, of the nine residues of C5d that are involved in binding to C6, four are charged residues (Asp1076, Lys1117, Lys1133, and Lys1139) that interact by forming salt bridges with residues in the CCP1 and CCP2 domains of C6 (Site I, Figures 1, 2A). Contact map analysis showed that Lys1137 forms strong bifurcated salt bridges (Figure 2A). Also, the AESOP analysis showed that out of these four charged residues, alanine perturbation of Lys1139, produces the highest loss of binding mutation, hence making it one of the most important contact sites (Figure 4). In addition, Val1122 of C5d also makes a strong a hydrogen bond with Glu2188 in the CCP1 domain of C6, with a 68% frequency of contact from the total trajectory (Figure 2). Thus, from the nine residues of C5d that are involved in contacts with C6, Lys1139, and Val1122 play the most important role by participating in salt bridges and a hydrogen bond.
We also performed comparative binding/conservation analysis for C3d and C4d. There are 19 conserved C4d residues with C3d that are also found in the binding interfaces of C3d with FH and CR2 (union of red and blue horizontal boxes in Figure 5). The majority of the conserved C4d residues (17 out of 19) overlap with the FH contact sites of C3d (red-only horizontal boxes in Figure 5), with one of them overlapping with FH and CR2 contact site and two additional ones overlapping with CR2 only (blue-only horizontal boxes in Figure 5). The side chains of the two C4d/C3d conserved residues that overlap with CR2 contact sites are negatively charged. On the other hand, 11 of the 17 C4d/C3d conserved residues that overlap with FH contact sites have hydrophobic side chains, including the residue that overlaps with both FH and CR2 contact sites.
We also performed a comparative analysis of the electrostatic properties of C3d, C4d, and C5d. Earlier works have studied the properties of a highly negatively charged concave surface on C3d that is involved in recognition and binding of complement regulators [29] and receptors [28, 41] as well as bacterial inhibitors [30, 31]. Figure 6A shows the C3d concave surface with the negatively charged patch, surrounded by a neutral rim. C4d also has a negatively charged patch but spans mostly the cavity's periphery (Figure 6C). C5d on the other hand, contains a slightly shallower but more extended cavity, with a sparsely negatively charged patch on its center and distinct positively charged patches at the edges of the surface (Figure 6E). The size and charge density of the concave cavity in C3d may be the determining factor for binding to CR2, a property that is not shared by C4d and C5d. We also examined the electrostatic properties of the internal thioester bond face (Figures 6B,D,F), focusing on the small region that is responsible for covalently tagging pathogen surfaces after hydrolysis of the thioester bond. Both, C3d and C4d, contain positively charged patches, but the patch on C3d is more extended (Figures 6B,D). Unlike C3d and C4d, the positively charge patch of the thioester bond moiety is lost in C5d, which has slightly negatively charged character. In addition, this region forms a crevice, which is absent in C3d and C4d, to assist packing of three hydrophobic residues of Phe2172, Ile2174, and Met2175 (part of C6's FSIM sequence motif), as stated in [33]. C6 contacts C5d through an elongated linker that contains the predominantly hydrophobic FSIM sequence motif at one end, and a polar/negatively charged DDEE sequence motif, proposed here, toward the other end, before it loops out and returns to form additional contacts with C5d. The occupancy of intermolecular contacts of FSIM residues throughout the MD trajectory is as follows (>20% for 5 Å between heavy atoms): Phe2172-Gln898, 85.7%; Phe2172-Glu899, 62.6%; Phe2172-Asn902, 40.9%; Phe2172-Phe938, 39.2%; Ser2173-Gln898, 36.3%; Ile2174-Ala894, 25.0%; Ile2174-Gln898, 77.6%. The DDEE sequence motif comprises residues Asp2185, Asp2186, Glu2187, and Glu2188. Asp2186 and Glu2187 participate in the high-occupancy bifurcated salt bridge with C5b's Lys1139; Glu2187 also participates in bifurcated salt bridge with C5b's Lys1133 and Lys 1139; and Glu2188 participates in hydrogen bonding with C5b's Val1122, which is the highest occupancy hydrogen bond of the complex (Figure 2). Both, FSIM and DDEE side chains interact with part of Site I that is depicted by the ellipse of Figure 6F.
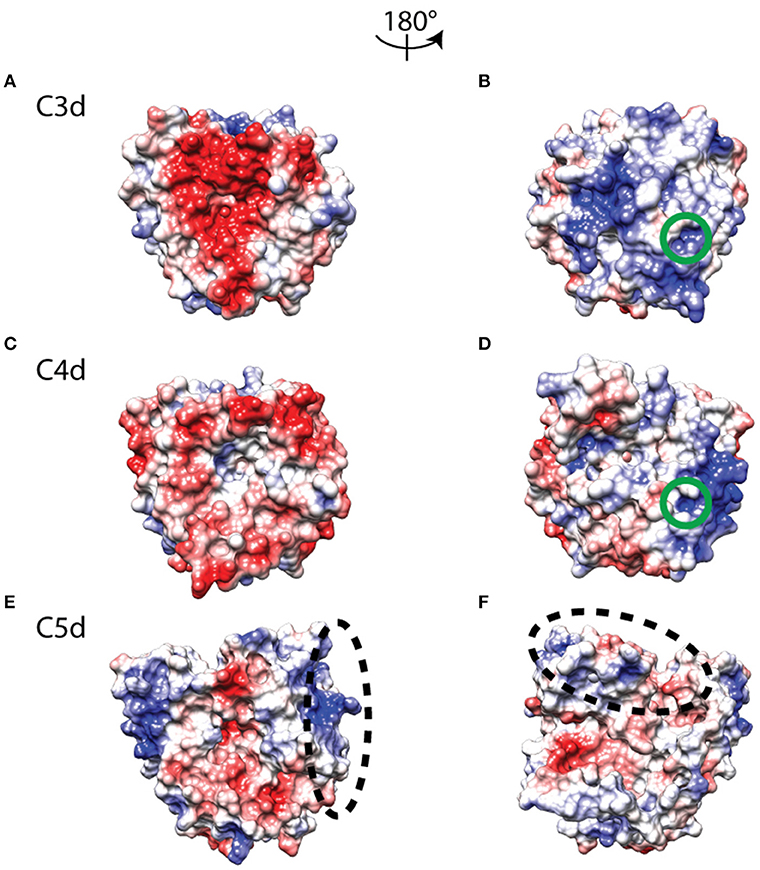
Figure 6. Electrostatic potentials mapped onto molecular surface representations of C3d (A,B), C4d (C,D), and C5d (E,F), showing the CR2/FH binding face on left and the thioester face on right. The color transitions from red to white to blue represent electrostatic potential values of −3 to 0 to +3 kBT/e. The green circles mark the internal thioester bond region of C3d (B) and C4d (D), and the two black dashed ellipses indicate distinct C6 binding regions in Site I of C5d (E,F).
Formation of the MAC pore is propagated through the cleavage of complement C5 that forms C5b and C5a. The smaller fragment, C5a, is an anaphylatoxin that mediates inflammation through activation of immune cells. The larger fragment, C5b, interacts with C6 to form C5b6. The C5b6 complex interacts with C7 and C8 to form the C5b78 complex. Subsequently, multiple C9 molecules combine with C5b78 to form the transmembrane pore C5b678918 (MAC). We have recently developed quantitative models to study the dynamics of the alternative [24] and the combined alternative and classical pathways [25] of the complement system. These models are based on describing the biochemical reactions of the complement pathways using ordinary differential equations and are parameterized using experimental kinetic data. We used our latest model that encompasses the alternative and classical pathways to gain a systems level understanding of the terminal cascade of the complement system, starting at fluid phase C5 and ending in MAC pore formation on cell membranes [25]. Figure 7 shows the concentration-time profiles of C5, C5b, C6, C5b6, and MAC pores. As C5 is consumed (Figure 7A), MAC is produced (Figure 7B). C5 is minorly consumed to form C5b (Figure 7C), and so does C6 (Figure 7D) when it combines with C5b to form the C5b6 complex (Figure 7E). C5b and C5b6 show similar time responses, but with different concentration magnitudes. The time response of MAC pore formation shows a short lag phase, followed by an accelerated production phase. Unlike complement products C5b and C5b6 that are mostly consumed in ~40 min, the profile of MAC pores shows a continuous production after the 90th min mark.
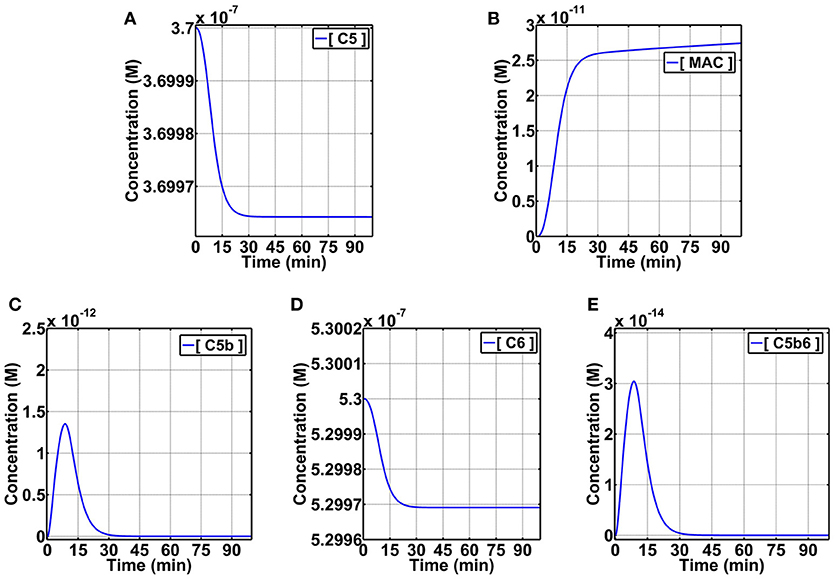
Figure 7. Dynamic pathway modeling of the complement terminal cascade. Rates of consumption of C5 (A) and production of MAC (B) are shown. Intermediate steps of production and consumption of C5b (C), consumption of C6 (D), and production and consumption of C5b6 (E) are also shown. C5, C5b, C6, and C5b6 are in fluid phase, whereas MAC is cell membrane bound.
Our results present the prompt activation and propagation kinetics of the complement system. In spite of the terminal pathway taking root later in the complement cascade (after the cleavage of C3), our results show MACs can be generated in less than 10 min. Furthermore, our results show despite the presence of complement regulators acting on the terminal cascade and numerous biochemical steps from C5 to C5b, C5b6, C5b67, C5b678, C5b6789, and the polymerization of C9 to form MACs (C5b67891−18), the rates of C5 or C6 consumption may be good predictors for the rate of MAC formation (Figure 7). This is highlighted where an accelerated rate of consumption for C5/C6 are present within the first 30 min, and this rate subsequently is reduced after the 30th min. Similarly, but inversely related to the concentration-time profiles of C5/C6, the MAC production exhibits an accelerated phase within the first 30 min that also subsequently diminishes after 30 min. However, it should be pointed out that the rate of production in MACs is still higher compared to the rate of C5/C6 consumption after the 30th min. In summary, kinetics of C5 or C6 consumption may be sufficient to study the kinetics of MAC formation despite the presence of numerous biochemical steps from C5/C6 to MACs.
Activation and propagation of the complement system through the alternative, classical, and lectin pathways leads to the cleavage of C5 to form C5b and C5a. Presence of C5b sets the stage for recruitment of complement protein C6 through C9 to form the MAC pore. Pathogens such as Neisseria species (causing meningitis) are susceptible to MAC-induced killing, and deficiency in the terminal proteins leads to recurrent meningitis [45]. On the other hand, dysregulation of the complement system leads to excessive propagation of the terminal step that plays a critical role in diseases such as AMD, aHUS, and PNH [15–20]. In addition to diseases, sublytic MAC of the terminal cascade can induce multiple signaling pathways associated with proliferation, apoptosis, and protein synthesis [13, 46]. Overall, formation of C5b6 initiates a cascade of reactions that affects numerous events from pathogen killing, to complement-mediated diseases, and signaling pathways. Here, we performed biophysical analysis on C5b6 complex to identify the key components that are involved in the mechanism of C5b and C6 binding.
We initiated our study to quantify the physicochemical origins of C5b6 binding, with focus on electrostatic interactions, guided by the large number of charged patches on the surfaces of C5b and C6 and in their binding interface. Our analysis is based on the crystal structure and MD trajectory of the C5b6 complex. MM-PBSA calculations throughout the MD trajectory showed an overall unfavorable electrostatic energy component in the free energy of binding, which is dominated by favorable van der Waals interactions. However, analysis of the binding interface throughout the MD trajectory revealed the presence of 13 intermolecular salt bridges and 11 intermolecular hydrogen bonds, which is a large number of electrostatic interactions for a complex of approximately 3200 Å2 buried solvent accessible surface area (Supplementary Figure 2). Additionally, AESOP analysis using six representative structures from MD conformational clusters, identified several charged residues with strong contributions to the electrostatic free energy of binding. Therefore, we conclude that the stability of the C5b6 complex is dominated by van der Waals interactions and by the presence of several distinct pairwise electrostatic interactions in the form of hydrogen bonds and salt bridges. Disrupting the electrostatic interactions of residues identified herein as being important for structural stability of the C5b6 complex, may be a good strategy for future drug discovery aiming to inhibit MAC assembly.
The complement system contains two thioester domains, C3d and C4d, and one thioester-like domain, C5d, which function as initiators and propagators of the cascade of reactions that leads to the elimination of pathogens or apoptotic/damaged cells [1]. C3d and C4d function as parts of C3b and C4b, respectively, and unlike C5d both C3b and C4b can covalently attach to host/pathogen cell surfaces and tag the cellular species for inflammatory response and opsonophagocytosis. C3d and C4d also exist as standalone proteins, after degradation cleavage of C3b by complement regulators, and remain covalently linked to the surface of cells long after complement activation. On the other hand, the thioester-like domain of C5d cannot covalently attach to surfaces of pathogens/apoptotic cells; however, C5d makes extensive contacts with C6 to form the complex C5b6 [32, 33]. The assembly of C5b6 presents the junction between the different phases of complement activation that initiates the formation of MAC. But unlike C5d (and also C4d), complement fragment C3d has numerous binding partners ranging from complement components to pathogenic proteins [41, 47–49]. For instance, C3d can mediate adhesion by interacting with complement receptors, bind the domain of Staphylococcus aureus protein Sbi, and also form the link between innate and adaptive immunity by interacting with CR2 on B-cells [50–54]. Although all three thioester domains are structurally similar, they have distinct electrostatic potential projections on their surfaces (Figure 6). We identified C3d has 87 of its 310 residues that are involved in binding to complement proteins FH and CR2. Furthermore, 19 of the 87 C3d residues are conserved on both C4d and C5d (Figure 5). These differences indicate that C3d has evolved to have multiple binding partners by having more binding residues present its thioester domain.
A major contributing factor to the multifaceted functionality of C3d, has been associated with its dual electrostatic nature, through two distinct faces [41]. The thioester face is positively charged and functions by tagging cells. The concave negatively charged face functions for recruiting (or as an aid to) adaptive immunity and inducing immune response. These charged regions contain functional sites that serve to accelerate protein association and stabilize protein complexes [55, 56]. Furthermore, our results also show complement C4d contains a negatively charged ring surrounding the concave face, rather than entirely covering it, and differences in the distribution of positive charge in its thioester face (Figure 6). C5d also shows electrostatic differences on both the concave face and the thioester-like face compared to C3d and C4d (Figure 6). Another difference in C5d is the presence of a positive region that accommodates the C6 domain for the formation of C5b6 complex (Figure 6). This C6-binding site of C5d contains a deep groove, which facilitates the binding of conserved C6 linker residues of the 2172FSIM2175 sequence motif. In addition, the linker contains the 2185DDEE2188 sequence motif of acidic charged residues Asp2186 and Glu2187, which form salt bridges with basic residues of Lys1139 (Asp2186 and Glu2187) and Lys1133 (Glu2187) of C5d, respectively (Figure 2A). The perturbation Lys1139Ala most notably showed the strongest destabilizing effect on the interaction of C5b6 in AESOP analysis (Figure 4). Similar to C6, complement protein C7 also contains an analogous linker. However, as stated in [33], C5b may discriminate toward C6 because C7 lacks the FSIM motif and has a shorter linker that makes less extensive contacts with C5d. Overall, these results show complement proteins C3d and C4d that perform similar functions by covalently attaching to different surfaces for propagation of complement cascades, also have similar electrostatic profiles. However, C5d shows distinct charge properties, with lower densities of negative and positive charges in its concave and thioester-like surfaces. Instead, C5d has a positive groove that facilitates C6 binding and instigates the terminal step of complement propagation.
A distinct structural feature of C3 activation product, C3b, is that its C3d domain packs at the bottom of macroglobulin domain MG1. In contrast, the C5d domain of C5b is located 50 Å from the base of MG1 when in complex with C6 [32, 33]. Although C3 and C5 are structurally similar, the distinct locations of their C3d/C5d domains in their active products C3b/C5b highlight not only how their active products are topologically different, but also on how each of them propagates its distinct function on a surface of pathogen/host cells. For instance, C3b uses its C3d domain to covalently attach to nearby pathogens and interact with FB to form C3bB. This complex subsequently is activated by FD to form the C3/C5 convertase, C3bBb [57, 58]. C3bBb then propagates a cascade of reactions on the surface by cleaving C3 to form more C3b and C3a, and to promote pathogen elimination through enhanced opsonophagocytosis. The placement of C3d close to base of MG1, in conjunction with the active thioester moiety and the positively charged thioester face, makes the covalent attachment of C3b possible. On other hand, C5b does not covalently attach to surface using its C5d domain, but promotes a complementary mechanism of pathogen elimination through the formation of the pore-forming MAC. Recent cryo-electron microscopy and tomography studies of MACs proposed that the role of C5b is to assist priming C6 to initiate the pore formation [9, 12]. Close examination of MAC showed the cholesterol-dependent cytolysin/MAC perforin (CDC/MACPF) domain in C6, C7, C8, and C9 are responsible for pore formation, and this domain is absent in C5b. Furthermore, these studies showed that the structural arrangement of C5b6 is also maintained in the MAC pore assembly. The conformation of C5b6, where C5d remains half-way elevated from the base of the MG1 domain, ensures the absence of steric clashes with complement C9, which is situated below the thioester-like domain of C5b in the MAC assembly. Subsequently, the maximum number of C9s (18 polymerization copies) can fit to form the MAC's cylindrical shape. Furthermore, downstream reactions of C5b6 to form C5b678 also affects the arc-like structure formed by C9 polymerization. Recent cryo-electron microscopy studies show polymerized C9 (poly-C9) in the absence of C5b678, assembles into a closed symmetric ring with 22 C9 components [59, 60]. This contrasts the recent MAC structures where MACs are shown to be asymmetric with a maximum number of 18 C9s [9, 12]. Superposition of poly-C9 ring with that of a physiological MAC pore shows dimensional differences in height and spacing of C9 molecules [61]. These data highlight the presence of C5b678 as one of the key factors in affecting the molecular assembly of C9s to form an asymmetric pore. All in all, formation of the C5b6 complex results in priming C6 for MAC assembly and aiding in the polymerization of C9. These functions are brought together by the stabilization of C5d's position by C6 (in the complex C5b6), and the downstream reactions that form C5b678 complex.
Through the course of our analysis of the structural and physicochemical properties underlying the mechanism of formation and stability of the C5b6 complex, we identified druggable C5d regions with the potential to disrupt C5b6 interface. In Figure 8, the C5d intermolecular polar contacts contributing to the stability of the C5b6 complex are emphasized, with surrounding residues that are in close proximity to C6 highlighted as well. The number of polar intermolecular interactions at Site I are restricted to a few narrow regions due to the shape and size of C6. Deriving short peptides from C6 and applying modifications to improve the interfacial SASA and specific intermolecular interactions could result in stronger binders to C5d in search of inhibitory ligands against C6 binding. We also observe that several of the polar intermolecular contacts are adjacent to pockets that could be leveraged in the development of drug-like or peptidic inhibitors (Figures 8C,D).
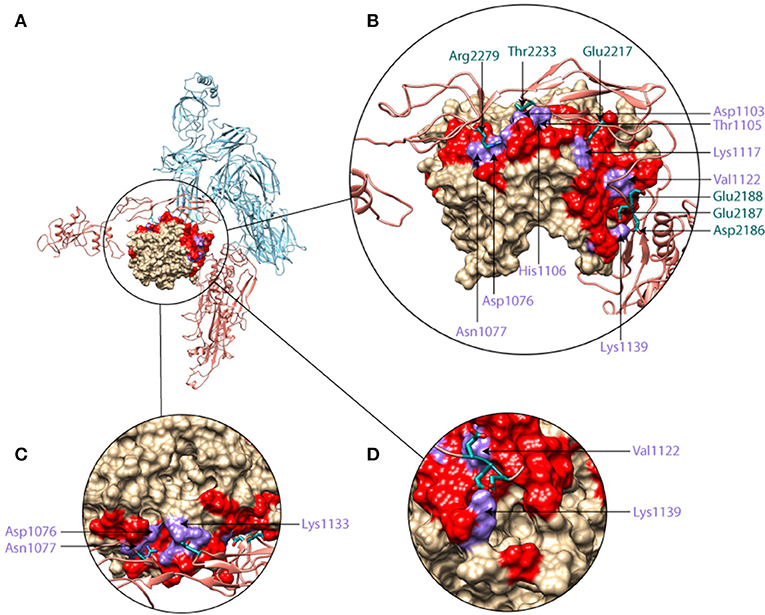
Figure 8. Molecular graphics representation of C5b6 with insets showing zoomed in and rotated views of Site I, highlighting the druggability of C5d. (A) C5b6 in ribbon form with C5d shown in surface representation (brown), and the rest of C5b (light blue) and C6 (brick) shown in ribbon representation. (B) Zoomed-in view of the region circled in panel A and rotated toward the viewer by 40°. (C) Zoomed-in view of the region circled in panel A and rotated towards the viewer by 100°. (D) Zoomed-in view of the region circled in A and rotated around the vertical axis by −60°. In the C5d surface, red coloring indicates regions within 5 Å of C6 and purple coloring indicates regions where polar intermolecular interactions with C6 (salt bridges and hydrogen bonds in site I, Figure 2), observed in the MD simulation trajectory. Side-chains of C6 residues taking part in polar intermolecular interactions are shown and labeled in dark green while their C5d pairing partner residues are labeled in purple.
In contrast to the potential therapeutic sites on C5d, C5 inhibitors eculizumab and SKY59 interact with domains MG7 and MG1, respectively [62, 63]. Unlike C5d, MG7 and MG1 do not contain deep pockets but have charged residues that are critical for interactions with eculizumab or SKY59. For instance, MG1 domain has three charged residues, Glu48, Asp51, and Lys109 (within 3.5 Å from SKY59 antigen-binding fragment, Fab) that mediate binding by forming critical salt bridges and numerous hydrogen bonds [62]. Mutating any of these charged residues on C5 to alanine had severe effects on the binding affinity of SKY59 [62]. And hence, the binding interface between C5 and SKY59 is highly mediated by the charged residues positioned in MG1. Similarly, the MG7 domain of C5 (targeted by eculizumab) does not contain deep pockets as observed for the thioester domains of C3d/C4d or that of C5d (TED-like). Similarly the eculizumab epitope on MG7 is also highly charged, containing six charged residues comprised of one glutamic acid (Glu915) and five positively charged residues of four lysine (Lys858, Lys882, Lys887, Lys920) and one arginine (Arg885) [63]. Furthermore, Schatz-Jakobsen et al. showed out of the 66 single-point mutants on Fab residues that interact with C5, three residues in the heavy chain (Trp107, Phe101, Trp33) and one residue in light chain (Ala32) severely impaired hemolysis inhibition when mutated to histidine [63]. Using PDBsum [64] on the binding interface between C5 and the Fab fragment, we observed half of these key mutants (Phe101 and Trp33) make extensive contacts with the positively charged residue Arg885 on the MG7 domain of C5. Interestingly, a small number of PNH patients that are resistant to eculizumab carry a common single nucleotide polymorphism where Arg885 is replaced by histidine [65]. These results show mutations on C5 that affect charged residues in drug sites have severe consequences on the functionality of the complement inhibitors. And hence, accounting for the electrostatic nature of the C5 epitopes may significantly improve binding affinities and subsequently enhance complement inhibition.
Although most of our study is structure/dynamics-based at molecular level, we expanded our efforts to understand the role of C5b and C6 at pathway dynamics level. There is small consumption of C5 and C6 from their initial blood plasma concentrations, and this is reflected in the production of C5b and C5b6, and eventually in MAC production. C5b and C6 show an initial accelerated production phase, followed by a consumption phase as they are converted to MAC. The production of MAC shows a lag phase, corresponding to the production phases of C5b and C5b6, followed by an accelerated production phase, corresponding to the consumption phases of C5b and C6. Despite this level of production, the concentration of MAC pores in 90 min is 27 pM, about four orders of magnitude lower than the initial concentrations of C5 and C6. Given that the calculation has performed with full complement regulation in place, representing homeostasis, such MAC pore concentration is not expected to have any significant effects on host cells. However, in pathogen cells, where negative complement regulation is absent, a larger amount of MAC deposition is observed [24]. Substantial increase in MAC pore formation is also expected in host diseases such as AMD, where there is severe complement dysregulation.
Overall, our molecular dynamics and electrostatics study revealed that the large and multi-site C5b6 interface is stabilized predominantly by van der Waals interactions, but also contains an unusually large number of stabilizing salt bridges and hydrogen bonds. We identified critical salt bridges and hydrogen bonds for the stability of the C5b6 complex. Furthermore, the C5d domain of C5b contains a sparsely negatively charged patch enclosed with positively charged patches. C5d does not contain a C3d-like cavity which in C3d is an electrostatic hotspot primed for interaction with CR2 for the formation of a link between innate and adaptive immunity. This suggests mono-functionality for C5b for the formation of the MAC assembly. We propose that C5d is a target for drug discovery, by designing inhibitors capable of disrupting the critical salt bridge and hydrogen bonding interactions at the C5d-C6 interface. We also showed the presence of small cavities neighboring the critical electrostatic contacts that can be leveraged in the development of drug-like or peptidic inhibitors. Lastly, we extended our study from molecular level dynamics to pathway dynamics to demonstrate the specifics in consumption-production rates of C5, C5b, C6, C5b6, toward MAC formation. Inhibition of the C5b6 interaction may be an efficient way to block MAC formation for diseases such as PNH, where MAC is responsible for hemolytic activity of red blood cells.
The three-dimensional cocrystal structure of C5b6 with code 4A5W [32] was obtained from the Protein Data Bank (PDB). C5b is represented as chain A, whereas C6 is represented as chain B. All missing residues of C5b and C6 were added with MODELLER [66]. The crystal structure of C3d with code 3OED [35] was obtained from the PDB. The structures of C4d and C5d were extracted from the crystal structures of C4b and C5b6, with PDB codes 5JTW [36] and 4A5W, respectively. Missing residues for C4d were modeled using SWISS-MODEL [67]. Structural visualization and comparisons were performed using Chimera [68]. Molecular graphics were generated using Chimera. Protonation states of histidine were assigned using PDB2PQR [69]. The following transformations are needed to convert residue numbering used in 4A5W [32] to residue numbering used in our study: for C5b residues 19-765, subtract 18 from the 4A5W residue numbers; for C5b residues 766-1676, subtract 96 from the 4A5W residue numbers; for C6 residues 22-934, add 1559 to the 4A5W residue numbers.
Sequences of C3d, C4d, and C5d were extracted from the PDB entries 3OED [70], 5JTW [71], and 4A5W [32], respectively. Sequence alignments of C3d, C4d, and C5d were performed using Clustal Omega [72] and visualized with Jalview [73]. Identification of C3d residues involved in binding with FH and CR2 were acquired from MD simulation analysis performed in previous studies [28, 29]. Identification of C5b6 intermolecular interactions was performed as outlined in the MD trajectory analysis section, below.
Initial minimization of structure in the absence of water was performed using NAMD and the CHARMM36 force field [74, 75]. Subsequently, the structure was solvated in a cubic TIP3P water box leaving a minimum margin of 12 Å between any protein atom and the cube boundary. Sodium and chloride counterions were added to the system to achieve 150 mM ionic strength and neutralize protein charges. Adding water and counterions increased the total system size to 747,620 atoms. The solvated structure was energy minimized by undergoing 50,000 steps of conjugate gradient energy minimization before heating from 0 to 310 K with all protein atoms harmonically constrained to their positions after minimization. Next, five equilibration steps were performed in which the first five steps for a total time of 7 ns. During the four stages of equilibration, all protein atoms were constrained at a force constant of 10, 5, 2, and 1 kcal/mol/Å, respectively. The final equilibration step was concluded by only constraining backbone atoms with a force constant of 1 kcal/mol/Å. Following equilibration, AMBER16 [76, 77] was used for the production run for 100 ns with the following conditions: periodic boundary conditions, Langevin temperature control, a non-bonded interaction cutoff of 12 Å, with SHAKE algorithm used for constraining hydrogen bonds, and an integration time step of 2 fs.
Characterization and visualization of intermolecular interactions was performed using CPPTRAJ, pandas, and seaborn [78–80]. Analysis of buried solvent accessible surface area upon binding and visualization were performed with MDTraj [81] and matplotlib [82], respectively over all 2,000 frames in the trajectory. MSMBuilder [83] and MSMExplorer [84] were used for clustering, visualization and extracting representative structures from the trajectory for electrostatic analysis. Salt bridges between C5b and C6 residues was calculated with custom R scripts in conjunction with Bio3D package [85]. A distance cutoff of 5 Å was used. CPPTRAJ was used to analyze hydrogen bonds formed between C5b and C6 over the course of the trajectory. For hydrogen bonds, the default distance cutoff of 3 Å was used between acceptor to donor heavy atom, and an angle cutoff of 135°. To extract representative structures, PCA decomposition was performed on the phi and psi angles observed throughout the trajectory to reduce to four principal components using MSMBuilder. The MiniBatchKMeans method in MSMBuilder was utilized to cluster the four principal components to six distinct clusters and cluster centers were extracted as representative structures.
MM-PBSA calculations were performed using a thermodynamic cycle that decomposes the calculation of the free energy of binding into molecular mechanics (MM) force field calculations in a state of low dielectric coefficient (ε = 2) and solvation calculations by transferring the proteins from the low dielectric coefficient environment to a high dielectric environment (ε = 80). A frame interval of 4 was chosen for the MM-PBSA calculations, and hence a total of 500 frames from a total of 2,000 were processed. In our case, the MM calculations include van der Waals and electrostatic free energies, but not covalent geometry energy (bonds, angles, torsions) or entropic effects. We use the one-trajectory approximation, according to which we separate the structures of the components of the complex from the structure of the complex without additional minimization and without performing separate MD simulations of the complex components. Given that we used the one-trajectory approximation, covalent geometry energies and entropic effects are expected to cancel out in the binding scheme
The solvation free energy calculations include electrostatic contribution according to Poisson-Boltzmann electrostatic calculations, and nonpolar contribution (cavity solvation) described by an empirical term based on loss of solvent accessible surface area upon binding [86]. The following equations describe the MM-PBSA calculations.
Where the Binding Free Energy According to MM Force Field Calculations is Given by
and electrostatic contributions of individual components, C5b and C6, and complex, C5b6, to solvation free energy are given by Poisson-Boltzmann (PB) free energy differences
The overall solvation contributions to binding, including polar and nonpolar effects, and are given by
and the MM-PBSA free energy of binding is given by
The Alanine scan method in the AESOP (Analysis of Electrostatic Structures Of Proteins) python package [44] was utilized to perform a computational alanine scan on ionizable residues at the C5b6 interface, and to evaluate their electrostatic contributions to binding. Alanine scans were performed for each of the six representative structures of the MD-derived conformational states.
AESOP utilizes the Adaptive Poisson-Boltzmann Solver (APBS) [87] to calculate grid-based electrostatic potentials, which are converted to electrostatic free energies. The program PDB2PQR is used to pre-assign charges and atomic radii for each atom according to the PARSE force field [88, 89], as well as to convert the PDB format to PQR format used by APBS. The selection of parameters for AESOP calculations has been described before [90].
Electrostatic free energies of binding were calculated according to a thermodynamic cycle that is similar to the one used for the MM-PBSA calculations, described above, except that binding at the reference state is evaluated using Coulomb's equation instead of the molecular mechanics method. The following equations are used, as described previously [44] and in recent applications of AESOP on C3d and its ligands [28–30]:
where,
Electrostatic free energies of binding for the family of alanine scan mutants are generated as deviations from the electrostatic free energy of binding of the parent protein, according to:
APBS is used to calculate electrostatic potentials for the solvation steps, and the program COULOMB (part the APBS suite) is used to calculate binding at the reference state. For the solvated state, dielectric coefficients of 78.54 and 20 were used for solvent and protein interior, respectively, while for the reference state a dielectric coefficient of 20 was used to resemble that of the protein interior [90]. The ionic strength of the solvated state corresponded to monovalent counterions of 150 mM concentration (physiological ionic strength), whereas the reference state had zero ionic strength. For the electrostatic analysis of the representative structures of C5b6 the number of grid points and mesh dimensions were set to 321 × 257 × 257 and 282 Å × 245 Å × 239 Å, respectively. For the electrostatic analysis of C3d, C4d, and C5d, the number of grid points and mesh dimensions were set to 84 × 98 × 96 and 129 Å × 129 Å × 97 Å, respectively.
The dynamics of the terminal cascade of complement system activation were modeled using a previously developed mathematical model that describes the biochemical reactions of the alternative and classical pathways [25]. The output of the model is reaction rates in the form of concentration-time profiles for all complement system proteins, enzymatic cleavage fragments, and association complexes. The model consists of a system of 290 ordinary differential equations (ODEs) and 142 kinetic parameters. Equations, initial concentrations, and kinetic parameters can be found in the Supplementary Information of [25]. The system of ODEs was solved using the ode15s solver of MATLAB (Mathworks, Natick, MA).
NZ, RM, and DM: designed the study, interpreted the data, and wrote the manuscript; NZ and RM: performed calculations and data analysis; DM: conceived and directed the study. All authors approved the submitted manuscript.
This work was partially supported by NIH grant R01 EY027440.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2018.00130/full#supplementary-material
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Schatz-Jakobsen JA, Pedersen DV, Andersen GR. Structural insight into proteolytic activation and regulation of the complement system. Immunol Rev. (2016) 274:59–73. doi: 10.1111/imr.12465
3. Tseng Y, Poon PH, Zavodszky P, Schumaker VN. Spontaneous activation of serum C1 in vitro. Role of C1 inhibitor. J Immunol. (1991) 147:1884–90.
4. Bianchino AC, Poon PH, Schumaker VN. A mechanism for the spontaneous activation of the first component of complement, C1, and its regulation by C1-inhibitor. J Immunol. (1988) 141:3930–6.
5. Ziccardi RJ. Spontaneous activation of the first component of human complement (C1) by an intramolecular autocatalytic mechanism. J Immunol. (1982) 128:2500–4.
6. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3-the “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
7. Bayly-Jones C, Bubeck D, Dunstone MA. The mystery behind membrane insertion: a review of the complement membrane attack complex. Philos Trans R Soc Lond B Biol Sci. (2017) 372:20160221. doi: 10.1098/rstb.2016.0221
8. Morgan BP, Boyd C, Bubeck D. Molecular cell biology of complement membrane attack. Semin Cell Dev Biol. (2017) 72:124–32. doi: 10.1016/j.semcdb.2017.06.009
9. Sharp TH, Koster AJ, Gros P. Heterogeneous MAC initiator and pore structures in a lipid bilayer by phase-plate cryo-electron tomography. Cell Rep. (2016) 15:1–8. doi: 10.1016/j.celrep.2016.03.002
10. Ramm LE, Whitlow MB, Mayer MM. Size of the transmembrane channels produced by complement proteins C5b-8. J Immunol. (1982) 129:1143–6.
11. Zalman LS, Müller-Eberhard HJ. Comparison of channels formed by poly C9, C5b-8 and the membrane attack complex of complement. Mol Immunol. (1990) 27:533–7. doi: 10.1016/0161-5890(90)90072-8
12. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. (2016) 7:ncomms10587. doi: 10.1038/ncomms10587
13. Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology (2016) 221:747–51. doi: 10.1016/j.imbio.2015.04.006
14. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. (2016) 274:152–71. doi: 10.1111/imr.12475
15. Liszewski MK, Java A, Schramm EC, Atkinson JP. Complement dysregulation and disease: insights from contemporary genetics. Annu Rev Pathol. (2017) 12:25–52. doi: 10.1146/annurev-pathol-012615-044145
16. De Vriese AS, Sethi S, Van Praet J, Nath KA, Fervenza FC. Kidney disease caused by dysregulation of the complement alternative pathway: an etiologic approach. J Am Soc Nephrol. (2015) 26:2917–29. doi: 10.1681/ASN.2015020184
17. Angioi A, Fervenza FC, Sethi S, Zhang Y, Smith RJ, Murray D, et al. Diagnosis of complement alternative pathway disorders. Kidney Int. (2016) 89:278–88. doi: 10.1016/j.kint.2015.12.003
18. Clark SJ, Bishop PN. The eye as a complement dysregulation hotspot. Semin Immunopathol. (2018) 40:65–74. doi: 10.1007/s00281-017-0649-6
19. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
20. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
21. Janssen BJC, Huizinga EG, Raaijmakers HCA, Roos A, Daha MR, Nilsson-Ekdahl K, et al. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature (2005) 437:505–11. doi: 10.1038/nature04005
22. Kidmose RT, Laursen NS, Dobó J, Kjaer TR, Sirotkina S, Yatime L, et al. Structural basis for activation of the complement system by component C4 cleavage. Proc Natl Acad Sci USA. (2012) 109:15425–30. doi: 10.1073/pnas.1208031109
23. Laursen NS, Andersen KR, Braren I, Spillner E, Sottrup-Jensen L, Andersen GR. Substrate recognition by complement convertases revealed in the C5-cobra venom factor complex. EMBO J. (2011) 30:606–16. doi: 10.1038/emboj.2010.341
24. Zewde N, Gorham RD Jr Dorado A, Morikis D. Quantitative modeling of the alternative pathway of the complement system. PLOS ONE (2016) 11:e0152337. doi: 10.1371/journal.pone.0152337
25. Zewde N, Morikis D. A computational model for the evaluation of complement system regulation under homeostasis, disease, and drug intervention. PLOS ONE (2018) 13:e0198644. doi: 10.1371/journal.pone.0198644
26. Wu J, Wu Y-Q, Ricklin D, Janssen BJC, Lambris JD, Gros P. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat Immunol. (2009) 10:728–33. doi: 10.1038/ni.1755
27. Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. (2016) 35:1133–49. doi: 10.15252/embj.201593673
28. Mohan RR, Gorham RD, Morikis D. A theoretical view of the C3d:CR2 binding controversy. Mol Immunol. (2015) 64:112–22. doi: 10.1016/j.molimm.2014.11.006
29. ES Harrison R, Gorham RD, Morikis D. Energetic evaluation of binding modes in the C3d and Factor H (CCP 19-20) complex. Protein Sci Publ Protein Soc. (2015) 24:789–802. doi: 10.1002/pro.2650
30. Gorham RD, Rodriguez W, Morikis D. Molecular Analysis of the Interaction between Staphylococcal Virulence Factor Sbi-IV and Complement C3d. Biophys J. (2014) 106:1164–73. doi: 10.1016/j.bpj.2014.01.033
31. Gorham RD, Kieslich CA, Morikis D. Complement Inhibition by Staphylococcus aureus: electrostatics of C3d-EfbC and C3d-Ehp association. Cell Mol Bioeng. (2012) 5:32–43. doi: 10.1007/s12195-011-0195-6
32. Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, et al. Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep. (2012) 1:200–7. doi: 10.1016/j.celrep.2012.02.003
33. Aleshin AE, DiScipio RG, Stec B, Liddington RC. Crystal structure of C5b-6 suggests structural basis for priming assembly of the membrane attack complex. J Biol Chem. (2012) 287:19642–52. doi: 10.1074/jbc.M112.361121
34. Ojha H, Panwar HS, Gorham RD, Morikis D, Sahu A. Viral regulators of complement activation: structure, function and evolution. Mol Immunol. (2014) 61:89–99. doi: 10.1016/j.molimm.2014.06.004
35. Kieslich CA, Tamamis P, Gorham RD Jr de Victoria AL, Sausman NU, Morikis GA, et al. Exploring protein-protein and protein-ligand interactions in the immune system using molecular dynamics and continuum electrostatics. Curr Phys Chem. (2012) 2:324–43. doi: 10.2174/1877946811202040324
36. Gorham RD, Kieslich CA, Morikis D. Electrostatic clustering and free energy calculations provide a foundation for protein design and optimization. Ann Biomed Eng. (2011) 39:1252–63. doi: 10.1007/s10439-010-0226-9
37. Kieslich CA, Vazquez H, Goodman GN, López de Victoria A, Morikis D. The effect of electrostatics on factor H function and related pathologies. J Mol Graph Model. (2011) 29:1047–55. doi: 10.1016/j.jmgm.2011.04.010
38. Pyaram K, Kieslich CA, Yadav VN, Morikis D, Sahu A. Influence of electrostatics on the complement regulatory functions of Kaposica, the complement inhibitor of Kaposi's sarcoma-associated herpesvirus. J Immunol. (2010) 184:1956–67.
39. Zhang L, Morikis D. Immunophysical properties and prediction of activities for vaccinia virus complement control protein and smallpox inhibitor of complement enzymes using molecular dynamics and electrostatics. Biophys J. (2006) 90:3106–19. doi: 10.1529/biophysj.105.068130
40. Sfyroera G, Katragadda M, Morikis D, Isaacs SN, Lambris JD. Electrostatic modeling predicts the activities of orthopoxvirus complement control proteins. J Immunol. (2005) 174:2143–51. doi: 10.4049/jimmunol.174.4.2143
41. Kieslich CA, Morikis D. The two sides of complement C3d: evolution of electrostatics in a link between innate and adaptive immunity. PLOS Comput Biol. (2012) 8:e1002840. doi: 10.1371/journal.pcbi.1002840
42. Mohan RR, Huber GA, Morikis D. Electrostatic steering accelerates C3d:CR2 association. J Phys Chem. (2016) 120:8416–23. doi: 10.1021/acs.jpcb.6b02095
43. Wade RC, Gabdoulline RR, Lüdemann SK, Lounnas V. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proc Natl Acad Sci USA. (1998) 95:5942–9. doi: 10.1073/pnas.95.11.5942
44. Harrison RES, Mohan RR, Gorham RD, Kieslich CA, Morikis D. AESOP: a python library for investigating electrostatics in protein interactions. Biophys J. (2017) 112:1761–6. doi: 10.1016/j.bpj.2017.04.005
45. Lewis LA, Ram S. Meningococcal disease and the complement system. Virulence (2014) 5:98–126. doi: 10.4161/viru.26515
46. Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J Immunol. (1987) 138:246–53.
47. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. (2008) 6:132–42. doi: 10.1038/nrmicro1824
48. Serruto D, Rappuoli R, Scarselli M, Gros P, van Strijp JAG. Molecular mechanisms of complement evasion: learning from staphylococci and meningococci. Nat Rev Microbiol. (2010) 8:393–9. doi: 10.1038/nrmicro2366
49. Clark EA, Crennell S, Upadhyay A, Zozulya AV, Mackay JD, Svergun DI, et al. A structural basis for Staphylococcal complement subversion: X-ray structure of the complement-binding domain of Staphylococcus aureus protein Sbi in complex with ligand C3d. Mol Immunol. (2011) 48:452–62. doi: 10.1016/j.molimm.2010.09.017
50. Shaw CD, Storek MJ, Young KA, Kovacs JM, Thurman JM, Holers VM, et al. Delineation of the complement receptor type 2–C3d complex by site-directed mutagenesis and molecular docking. J Mol Biol. (2010) 404:697–710. doi: 10.1016/j.jmb.2010.10.005
51. Kovacs JM, Hannan JP, Eisenmesser EZ, Holers VM. Mapping of the C3d ligand binding site on complement receptor 2 (CR2/CD21) using nuclear magnetic resonance and chemical shift analysis. J Biol Chem. (2009) 284:9513–20. doi: 10.1074/jbc.M808404200
52. Li K, Okemefuna AI, Gor J, Hannan JP, Asokan R, Holers VM, et al. Solution structure of the complex formed between human complement C3d and full-length complement receptor type 2. J Mol Biol. (2008) 384:137–50. doi: 10.1016/j.jmb.2008.08.084
53. Clemenza L, Isenman DE. Structure-guided identification of C3d residues essential for its binding to complement receptor 2 (CD21). J Immunol. (2000) 165:3839–48. doi: 10.4049/jimmunol.165.7.3839
54. Nagar B, Jones RG, Diefenbach RJ, Isenman DE, Rini JM. X-ray crystal structure of C3d: a C3 fragment and ligand for complement receptor 2. Science (1998) 280:1277–81. doi: 10.1126/science.280.5367.1277
55. Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science (1995) 268:1144–9. doi: 10.1126/science.7761829
56. Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein–protein interactions. Curr Opin Struct Biol. (2000) 10:153–9. doi: 10.1016/S0959-440X(00)00065-8
57. Pangburn MK, Müller-Eberhard HJ. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem J. (1986) 235:723–30. doi: 10.1042/bj2350723
58. Rawal N, Pangburn MK. C5 convertase of the alternative pathway of complement. Kinetic analysis of the free and surface-bound forms of the enzyme. J Biol Chem. (1998) 273:16828–35. doi: 10.1074/jbc.273.27.16828
59. Spicer BA, Law RHP, Caradoc-Davies TT, Ekkel SM, Bayly-Jones C, Pang S-S, et al. The first transmembrane region of complement component-9 acts as a brake on its self-assembly. Nat Commun. (2018) 9:3266. doi: 10.1038/s41467-018-05717-0
60. Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun. (2016) 7:10588. doi: 10.1038/ncomms10588
61. Morgan BP, Walters D, Serna M, Bubeck D. Terminal complexes of the complement system: new structural insights and their relevance to function. Immunol Rev. (2016) 274:141–51. doi: 10.1111/imr.12461
62. Fukuzawa T, Sampei Z, Haraya K, Ruike Y, Shida-Kawazoe M, Shimizu Y, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep. (2017) 7:1080. doi: 10.1038/s41598-017-01087-7
63. Schatz-Jakobsen JA, Zhang Y, Johnson K, Neill A, Sheridan D, Andersen GR. Structural basis for eculizumab-mediated inhibition of the complement terminal pathway. J Immunol. (2016) 197:337–44. doi: 10.4049/jimmunol.1600280
65. Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. (2014) 370:632–9. doi: 10.1056/NEJMoa1311084
66. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics (2016) 54:5.6.1–5.6.37. doi: 10.1002/0471250953.bi0506s15
67. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. (2018) 46:W296–303. doi: 10.1093/nar/gky427
68. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. (2004) 25:1605–12. doi: 10.1002/jcc.20084
69. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. (2004) 32:W665–7. doi: 10.1093/nar/gkh381
70. van den Elsen JMH, Isenman DE. A crystal structure of the complex between human complement receptor 2 and its ligand C3d. Science (2011) 332:608–11. doi: 10.1126/science.1201954
71. Croll TI, Andersen GR. Re-evaluation of low-resolution crystal structures via interactive molecular-dynamics flexible fitting (iMDFF): a case study in complement C4. Acta Crystallogr Sect Struct Biol. (2016) 72:1006–16. doi: 10.1107/S2059798316012201
72. Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. (2014) 1079:105–16. doi: 10.1007/978-1-62703-646-7_6
73. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics (2009) 25:1189–91. doi: 10.1093/bioinformatics/btp033
74. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. (2005) 26:1781–802. doi: 10.1002/jcc.20289
75. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem. (1998) 102:3586–616. doi: 10.1021/jp973084f
76. Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, et al. The Amber biomolecular simulation programs. J Comput Chem. (2005) 26:1668–88. doi: 10.1002/jcc.20290
77. Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. (1995) 91:1–41. doi: 10.1016/0010-4655(95)00041-D
78. Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. (2013) 9:3084–95. doi: 10.1021/ct400341p
79. McKinney W. Data structures for statistical computing in python. In: van der Walt S, Millman J, editors. Proceedings of the 9th Python in Science Conference. Austin, TX (2010). p. 51–6.
80. Waskom M, Botvinnik O, drewokane Hobson P, David Halchenko Y, et al. seaborn: v0.7.1 (June 2016). (2016).
81. McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J. (2015) 109:1528–32. doi: 10.1016/j.bpj.2015.08.015
82. Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng. (2007) 9:90–5. doi: 10.1109/MCSE.2007.55
83. Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, Pande VS. MSMBuilder2: modeling conformational dynamics at the picosecond to millisecond scale. J Chem Theory Comput. (2011) 7:3412–9. doi: 10.1021/ct200463m
84. Hernández CX, Harrigan MP, Sultan MM, Pande VS. MSMExplorer: data visualizations for biomolecular dynamics. J Open Source Softw. (2017) 2:1–2. doi: 10.21105/joss.00188
85. Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics (2006) 22:2695–6. doi: 10.1093/bioinformatics/btl461
86. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. (2000) 33:889–7. doi: 10.1021/ar000033j
87. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. (2001) 98:10037–41. doi: 10.1073/pnas.181342398
88. Tang CL, Alexov E, Pyle AM, Honig B. Calculation of pKas in RNA: on the structural origins and functional roles of protonated nucleotides. J Mol Biol. (2007) 366:1475–96. doi: 10.1016/j.jmb.2006.12.001
89. Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. (1994) 98:1978–88. doi: 10.1021/j100058a043
Keywords: complement system, complement protein C5, complement protein C6, C5b6 complex, membrane attack complex, molecular dynamics, electrostatics
Citation: Zewde N, Mohan RR and Morikis D (2018) Immunophysical Evaluation of the Initiating Step in the Formation of the Membrane Attack Complex. Front. Phys. 6:130. doi: 10.3389/fphy.2018.00130
Received: 11 August 2018; Accepted: 26 October 2018;
Published: 20 November 2018.
Edited by:
Andrzej Stasiak, Université de Lausanne, SwitzerlandReviewed by:
Bryan Paul Morgan, Cardiff University, United KingdomCopyright © 2018 Zewde, Mohan and Morikis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Morikis, ZG1vcmlraXNAdWNyLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.