 Samand Pashneh-Tala1
Samand Pashneh-Tala1 Robert Owen2
Robert Owen2 Hossein Bahmaee1,2
Hossein Bahmaee1,2 Sima Rekštytė3
Sima Rekštytė3 Mangirdas Malinauskas3
Mangirdas Malinauskas3 Frederik Claeyssens1,2*
Frederik Claeyssens1,2*- 1Department of Materials Science and Engineering, Kroto Research Institute, University of Sheffield, Sheffield, United Kingdom
- 2Department of Materials Science and Engineering, Insigneo Institute for In Silico Medicine, University of Sheffield, Sheffield, United Kingdom
- 3Physics Faculty, Laser Research Center, Vilnius University, Vilnius, Lithuania
Poly(glycerol sebacate) (PGS) has been utilized in numerous biomaterial applications over recent years. This elastomeric and rapidly degradable polymer is cytocompatible and suited to various applications in soft tissue engineering and drug delivery. Although PGS is simple to synthesize as an insoluble prepolymer, it requires the application of high temperatures for extended periods of time to produce an insoluble matrix. This places limitations on the processing capabilities of PGS and its possible applications. Here, we present a photocurable form of PGS with improved processing capabilities: PGS-methacrylate (PGS-M). By methacrylating the secondary hydroxyl groups of the glycerol units in the PGS prepolymer chains, the material was rendered photocurable and, in combination with a photoinitiator, crosslinked rapidly on exposure to UV light at ambient temperatures. The polymer's molecular weight and the degree of methacrylation could be controlled independently and the mechanical properties of the crosslinked material tailored. The polymer also displayed rapid degradation under physiological conditions and cytocompatibility with various primary cell types. As a demonstration of the processing capabilities of PGS-M, μm scale 3D scaffold structures were fabricated using 2-photon polymerization and used for 3D cell culture. The tunable properties of PGS-M coupled with its enhanced processing capabilities make the polymer an attractive potential biomaterial for various future applications.
Introduction
Since first being reported in 2002, poly(glycerol sebacate) (PGS) has attracted significant attention for applications as a biomaterial [1]. This polyester based polymer is produced from non-toxic and relatively low cost monomers (glycerol and sebacic acid) and displays tunable elastomeric mechanical properties, cytocompatibility, and rapid degradation under physiological conditions [1–3]. These properties have resulted in PGS being utilized in various soft tissue engineering applications. PGS has been examined as a membrane material for cell delivery to damaged tissues, including the heart [4] and retina [5], and as a material for nerve guidance conduits for peripheral nerve repair [6]. Porous PGS scaffolds have been used to support the growth of cardiac tissue [7, 8], blood vessels [9, 10], and cartilage [11–13]. Additionally, PGS has also been used as a degradable drug carrier for antibiotics and anticancer drugs [14, 15].
Although PGS is an attractive polymer for a range of biomaterial applications, its processing capabilities are somewhat limited. PGS is simple to produce as a soluble prepolymer, through a polycondensation reaction. However, to crosslink PGS into an insoluble matrix requires the application of high temperatures (typically >110°C) and vacuum to thermally cure the polymer [1, 4, 16–19]. These conditions make the creation of accurate geometries difficult and prevent the use of the polymer in directly incorporating cells or temperature sensitive molecules.
To counter this, PGS has been functionalised with chemical groups to render the prepolymer photocurable. The addition of acrylate groups to PGS enables photocuring when combined with a free-radical generating photoinitiator and appropriate wavelength of light. Furthermore, the mechanical properties of PGS-acrylate can be tuned simply by altering the degree of acrylation. Although this material can be rapidly crosslinked in ambient conditions, the synthesis of PGS-acrylate typically produces a large quantity of side products, mainly chlorine salts which require removal. Additionally, acrylates are highly reactive and are prone to spontaneous crosslinking (initiated by impurities). Alternatively, the functionalization of PGS with cinnamate groups has also been examined. This produces a prepolymer that photocures directly on exposure to ultraviolet light, however this requires long exposure times (2 h) [20].
Here, we present an alternative form of photocurable PGS produced by functionalization with methacrylate groups. Methacrylation is an often used functionalization route to produce photocurable biomaterials [21]. PGS-methacrylate (PGS-M) is simple to synthesize and rapidly crosslinks via free-radical polymerization. The mechanical properties and degradation rate of PGS-M could be tailored by altering the synthesis conditions and the photocured polymer also supported the growth and proliferation of various cell types in 2D culture. As a demonstration of the processing capabilities of PGS-M, scaffold structures, with μm resolution, were fabricated using 2-photon polymerization (2PP), also known as multiphoton processing (MPP) or direct laser writing (DLW) [22]. These scaffolds were then utilized for 3D cell culture.
Materials and Methods
In the following methods, all chemical reagents were obtained from Sigma Aldrich, UK, unless otherwise stated.
Synthesis of PGS Prepolymer
PGS prepolymer was formed via the melt-polycondensation reaction of equimolar amounts of sebacic acid and glycerol (Fisher Scientific, UK) (Figure 1). These were combined and stirred at 120°C, under nitrogen gas, for 24 h. A vacuum was then applied, to remove water, and the reaction continued for a further 24, 36, or 48 h.
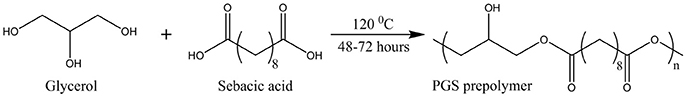
Figure 1. Synthesis of PGS prepolymer by melt-polycondenzation.
Prepolymer molecular weights were analyzed using gel permeation chromatography (GPC) (Viscotek GPC Max VE 2001 with PLgel 5 μm Mixed C column). Samples of prepolymer were dissolved in 1 ml of tetrahydrofuran (Fisher Scientific, UK) at 0.5% (w/v) (N = 3).
Synthesis of PGS-M Prepolymer
To produce a photocurable prepolymer, the free hydroxyl groups of the PGS prepolymer were methacrylated. It was assumed that two of the three hydroxyl groups present in glycerol reacted with sebacic acid, leaving 3.9 mmol of hydroxyl groups per gram of PGS prepolymer available for methacrylation [19, 23]. Two different molecular weights of PGS prepolymer were methacrylated. Low and High molecular weight PGS prepolymer (further denoted as Low Mw and High Mw PGS, respectively) were produced by polycondensation reactions of 48 and 72 h, respectively. Prepolymers were dissolved in dichloromethane (Fisher Scientific, UK) 1:4 (w/v) and methacrylic anhydride, with an equimolar amount of triethylamine, slowly added (Figure 2). Three different concentrations of methacrylic anhydride were used (0.3, 0.5, and 0.8 mol/mol of PGS prepolymer hydroxyl groups) in an effort to vary the degree of methacrylation (DM) from 30 to 50 to 80%. 4-methoxyphenol was also added at 1 mg/g of PGS prepolymer. The reaction was performed at 0°C and allowed to rise to room temperature over 24 h. The solution was then washed with 30 mM hydrochloric acid (Fisher scientific, UK) at 1:1 (v/v), dried with calcium chloride (Fisher scientific, UK) and the dichloromethane removed via rotary evaporation, under vacuum.
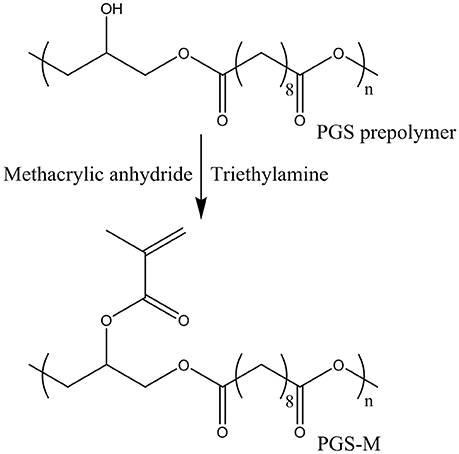
Figure 2. Synthesis of PGS-M prepolymer.
Characterization of PGS-M Prepolymers by Proton Nuclear Magnetic Resonance Spectroscopy
PGS-M prepolymers were characterized using proton nuclear magnetic resonance (NMR) spectroscopy (Bruker AVIIIHD 400 NMR spectrometer) at 400 MHz and compared to PGS prepolymer. Prepolymer samples were dissolved in 1 ml of deuterated chloroform (CDCl3) at 1% (w/v). Spectra were analyzed using TopSpin software. Chemical shifts were referenced to CDCl3 at 7.27 ppm. The chemical composition was determined by calculating signal integrals of—COCH2CH2CH2—at 1.2, 1.6, and 2.3 ppm for sebacic acid,—CH2CH—at 3.7, 4.2, and 5.2 ppm for glycerol and -CH3,CH2 at 1.9, 5.6, and 6.2 ppm for the methacrylate group. The signal integrals of the sebacic acid methylene groups (1.2 ppm) and the methacrylate group were used to calculate the degree of methacrylation (DM).
Characterization of Photocured PGS-M
PGS-M prepolymers of Low and High Mw and varied DM were mixed 1% (w/w) with the photoinitiator diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide/2-hydroxy-2-methylpropiophenone (50/50 blend) (further denoted as photoinitiator blend) and exposed to UV light (100 W, OmniCure Series 1000 curing lamp) for 10 min to photocure. Soluble fractions were determined by placing photocured PGS-M disks, dried to constant mass at 70°C, in methanol to solubilise the unreacted prepolymer and then drying and re-weighing them at 24 h intervals until reaching constant mass (N = 3, n = 3). Controls were subjected to the same drying regime, but without methanol washing.
Sol-free photocured PGS-M was examined by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) (Thermo Scientific iS50R FT-IR with germanium crystal) and compared to PGS-M and PGS prepolymers to confirm methacrylation and crosslink formation. ATR-FTIR data was analyzed using OMNIC software.
In vitro degradation of sol-free photocured PGS-M was assessed using 3 different treatments: cholesterol esterase enzyme (porcine pancreas) (40 units/ml), lipase enzyme (Thermomyces lanuginosus) (40 units/ml) and PBS. PGS-M disks in 1 ml of each treatment solution were agitated on an orbital shaker, at 90 rpm, in an incubator at 37°C. Every 2 days the disks were dried to constant mass, reweighed and replaced in fresh treatment solution (N = 3, n = 3). Controls were untreated. After 8 days the dried PGS-M disks were affixed to aluminum stubs, gold coated (Edwards S150B sputter coater) and examined by scanning electron microscopy (SEM) (Philips XL-20) at 13–15 kV.
Tensile Testing of PGS-M
The mechanical properties of photocured PGS-M were assessed by tensile testing (Hounsfield H100KS). PGS-M prepolymers were formed and photocured into tensile test pieces (Type 2 dumb-bell, as specified in BS ISO 37:2011 [24]) using a silicone mold and the soluble fraction removed. Tensile testing was performed at a crosshead speed of 500 mm/min with samples elongated to failure to determine Young's modulus and ultimate tensile strength (UTS) (N = 3, n = 3).
In Vitro Cell Metabolism and Proliferation on PGS-M Surfaces
Human dermal fibroblasts and human adipose-derived stem cells (ADSCs) from primary dermal tissue or lipoaspirate, respectively, were obtained with informed consent (ethics reference: 15/YH/0177) and processed and stored in accordance with the Human Tissue Act 2004 (license number 12179). Isolated cells were cultured to between passage 4 and 6 in Dulbecco's modified Eagle's medium (DMEM) AQmedia modified with 10% (v/v) fetal calf serum, 1% (v/v) Penicillin (10,000 units/ml), 1% (v/v) Streptomycin (10 mg/ml) and 0.25% (v/v) Amphotericin B (250 μg/ml). Additionally, human coronary artery smooth muscle cells (SMCs) were obtained commercially (PromoCell, Germany) and cultured to between passage 9 and 11 in specific growth medium (SMC growth medium 2 + supplement mixture, PromoCell, Germany) with 1% (v/v) Penicillin (10,000 units/ml), 1% (v/v) Streptomycin (10 mg/ml) and 0.25% (v/v) Amphotericin B (250 μg/ml). All cell cultures were incubated at 37°C and 5% CO2.
Thirteen millimeter diameter glass coverslips were cleaned in a 3:1 (v/v) solution of sulphuric acid (95.0-98.0%) and hydrogen peroxide (30 wt% in H2O), rinsed with dH2O then methanol before being immersed in a 10% (w/v) solution of 3-methacryloxypropyltrimethoxysilane in toluene for 24 h. Following rinsing in methanol, cover slips were spin-coated (Laurell Technologies WS-400B-6NPP/Lite at 4,000 rpm) in 30% DM Low Mw PGS-M prepolymer containing 1% photoinitiator blend and then photocured. To remove the soluble fraction and photoinitiator, PGS-M coated coverslips were washed in methanol for 4 days then dH2O for 4 days, with both solvents refreshed daily.
Cultured fibroblasts, ADSCs and SMCs were harvested using trypsin (0.025%)/EDTA (0.01%) solution, and resuspended in appropriate growth medium at 50,000 cells/ml. 1 ml of each cell suspension was seeded onto individual PGS-M coated coverslips, sterilized by autoclave, in 12-well plates. The cells were allowed to attach for 6 h and then the coverslips transferred to new wells for further culture, ensuring only cells attached to the coverslips were included in future analyses. Culture wells contained 1 ml of growth medium and this was changed every second day. Fibroblast and SMC seeded coverslips were cultured for 1, 3, or 7 days. ADSC seeded coverslips were cultured for 1, 7, or 14 days (N = 3, n = 3). Sterile glass coverslips, seeded with equivalent cells acted as positive controls while unseeded PGS-M coated coverslips acted as negative controls.
At the conclusion of the coverslip cultures, cell metabolic activity was assessed by reduction of resazurin sodium salt. 1 mM resazurin dissolved in dH2O was filter sterilized, mixed 10% (v/v) with the appropriate cell growth medium, and applied to each coverslip culture for 4 h of incubation. 200 μl of solution was then extracted from each well, in triplicate, placed in 96-well plates and examined using a fluorescence plate reader (Bio-tek instruments FLX800) at 540 nm excitation and 635 nm emission. The reading from a sample of incubated equivalent resazurin-containing growth medium acted as a blank.
Additionally, the quantity of cells present on the cultured coverslips was assessed using the PicoGreen® DNA quantification assay, purchased as a kit (Thermo Fisher Scientific, USA). At the conclusion of the cultures, the coverslips were washed thrice with PBS and then frozen and thawed thrice in 500 μl of dH2O. The solutions from each well were then centrifuged at 7,000 g for 5 min (Sanyo MSE Micro Centaur MSB010.CX2.5). One hundred eighty microliter of the supernatants were then mixed with 180 μl of a 5% (v/v) TE buffer and 0.5% (v/v) PicoGreen® solution in dH2O for 10 min, in the absence of light. 100 μl, in triplicate, was then extracted from each solution, placed in black 96-well plates and read using a fluorescence plate reader (Bio-tek instruments FLX800) at 480 nm excitation and 520 nm emission. The reading from a blank composed of dH2O mixed with the TE buffer and PicoGreen® solution was then subtracted from the value for each well. Fluorescence values were converted into mass of DNA using a standard curve generated from analyzing dsDNA at 200, 400, 600, 800, and 1,000 ng/ml, supplied with the assay kit.
Fabrication of 3D Scaffolds Using Direct Laser Writing and 2PP
Femtosecond DLW was employed for the fabrication of the 3D PGS-M scaffolds using 2PP (Figure 3). The laser radiation source was a PHAROS femtosecond Yb:KGW laser (Light Conversion Ltd, Lithuania), generating a 1,030 nm central wavelength and 300 fs duration pulses with a repetition rate range of 1–200 kHz. Second harmonics (515 nm) and a repetition rate of 200 kHz were used. For the precise positioning of the focussed laser beam an IFoV (Infinite Field of View) regime was employed to synchronize movements of linear positioning stages and galvano-scanners. This permitted a wide fabrication area, not limited by the objective's field of view, and allowed high sample translation velocities whilst avoiding inertia of the translation stages that could degrade the scaffold's architecture. Sample translation velocities of 2-15 mm/s were employed. The laser beam was focused using 10x (0.3 NA), 63x (1.4 NA), and 20x (0.8 NA) objectives, with best results achieved with the latter. The corresponding beam diameters were 2.09, 0.45 and 0.79 μm, respectively. This approach enables the production of micro-scale scaffolds for potential applications in vivo [25].
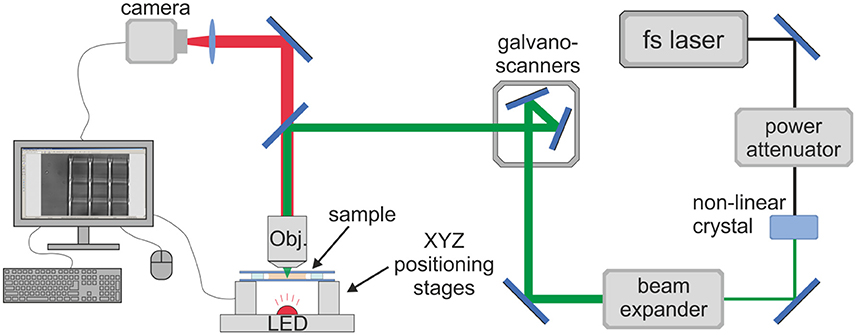
Figure 3. Optical setup for DLW-2PP. Femtosecond laser beam is scanned inside the photo-sensitive material in a pre-defined trajectory by employing the synchronized movement of galvano-scanners and linear positioning stages. The whole process is monitored on the computer screen in real-time.
Fifty percentage Low Mw PGS-M prepolymer was combined with 0.5–2 wt% (2-benzyl-2-(dimethylamino)-4'-morpholinobutyrophenone (IRG) photoinitiator and sandwiched between two 150 μm thick cover glasses separated with PDMS or glass spacers. The laser beam was focused from above into the prepolymer. Fabrication was initiated from the bottom interface of the glass and prepolymer and proceeded upwards. The average laser power was set to 0.06–12 mW (corresponding to the peak intensity of 0.32–8.4 TW/cm2 at the focal spot in the sample). Scaffolds consisted of various lattice-based structures produced as multiple layers. Additional intricate printed patterns (university logos) were also produced to demonstrate the resolution of the system. After laser irradiation, the samples were immersed in 4-methyl-2-pentanone to remove residual prepolymer and IRG photoinitiator. The chemical structure of the scaffolds was examined using Raman spectroscopy (Renishaw inVia micro-Raman) and compared to disks of 50% Low Mw PGS-M produced as described above for ATR-FTIR spectroscopy. Laser power was 20 mW with a 1 μm spot size produced using a 50x objective. The data were recorded with a Peltier-cooled multichannel CCD detector and 2,400 lines/mm diffraction grating with slit opening 65 μm and spectral resolution approximately 1 cm−1. Additionally, dry 3D scaffolds were sputter coated with gold and examined by SEM (Hitachi TM-1000) at 15 kV. Scaffold dimensions were assessed using ImageJ software. Scaffold shrinkage was quantified by comparing the distances between features at the base of the scaffolds, anchored to the glass surface, with corresponding distances between features at the uppermost surface of the structures (N = 5, n = 3).
Cell Proliferation on 3D PGS-M Scaffolds
Human coronary artery SMCs were cultured as described previously, harvested at passage 10 and resuspended in growth medium at 50,000 cells/ml. 3D PGS-M scaffolds attached to glass substrates were disinfected using 70% (v/v) ethanol in dH2O solution and rinsed thrice with PBS in 12-well plates. 1 ml of cell suspension was then added to each well. The seeded scaffolds were cultured for 24 h and 7 days. Unseeded scaffolds were also cultured in parallel as negative controls. At the conclusion of the cultures, the scaffolds were rinsed thrice with PBS, fixed with 3.7% formaldehyde for 1 h, and then washed again with PBS. The SMCs present on the scaffolds were visualized by staining for F-actin filaments using Phalloidin-FITC (200 ng/ml) for 30 min at room temperature followed by rinsing with PBS, 3 times.
Imaging was performed using a confocal microscope (Zeiss LSM 510 Meta) attached to a tuneable (700–1,060 nm) Chameleon Ti:sapphire multiphoton laser (Coherent, USA). The illumination wavelength was set at 488 nm. All imaging was performed using a 10x 0.3 W or a 40x 0.75 W objective with the pinhole set to 86 and 122 μm, respectively. Z stack increments were 1.2 μm. Transmitted light DIC images were collected in tandem. Imaging parameters were optimized and maintained for all samples.
Statistical Analysis
Data are displayed as mean ± standard deviation. Results were statistically analyzed using GraphPad Prism 7 software. PGS-M degradation and tensile testing data were analyzed using one-way ANOVA (one independent variable) while PGS-M soluble fraction, resazurin reduction and PicoGreen® assay data were analyzed using two-way ANOVA (two independent variables), both with Tukey multiple comparisons analysis. Equal variance was confirmed using the Browne-Forsythe test and Bartlett's test. Additionally, Paired samples were also specified in the analysis of the soluble fractions of PGS-M variants. Differences were considered statistically significant when P < 0.05 (*), very significant when P < 0.01 (**) and extremely significant when P < 0.001 (***).
Results and Discussion
PGS prepolymer was synthesized by the polycondensation reaction of glycerol with sebacic acid at 120°C. GPC determined the number average molecular weight (Mn) of the PGS prepolymer to be 2,230 ± 40, 2,770 ± 100, and 3,360 ± 50 g/mol and the weight average molecular weight (Mw) to be 5,420 ± 430, 8,960 ± 840, and 17,340 ± 760 g/mol for reaction lengths of 48, 60, and 72 h, respectively. Polydispersity increased from 2.4 to 5.2, as reaction lengths increased. These results were comparable to other studies where PGS prepolymer was synthesized using a 1:1 molar ratio of glycerol and sebacic acid [1, 16, 17, 19, 23, 26]. This ratio favors polymer chain extension over chain branching as reaction duration increases, due to the increased reactivity of the two primary hydroxyl groups of the glycerol monomer compared to its secondary hydroxyl group [27]. Chain branching was undesirable in the present study, as this has been shown to reduce the solubility of the prepolymer, limiting further functionalisation and processing [28].
Low Mw and High Mw PGS prepolymers were methacrylated to produce photocurable PGS-M prepolymer and examined using proton NMR. The chemical composition was determined by calculating signal integrals of -CH2CH- at 3.7, 4.2, and 5.2 ppm for glycerol and -COCH2CH2CH2- at 1.2, 1.6, and 2.3 ppm for sebacic acid. The incorporation of methacrylate groups into the PGS-M prepolymers was confirmed by the appearance of peaks at 1.9, 5.6, and 6.2 ppm (Figure 4). These peaks were absent from the spectra of the PGS prepolymer. These results were comparable with those reported for PGS-acrylate [19, 23].
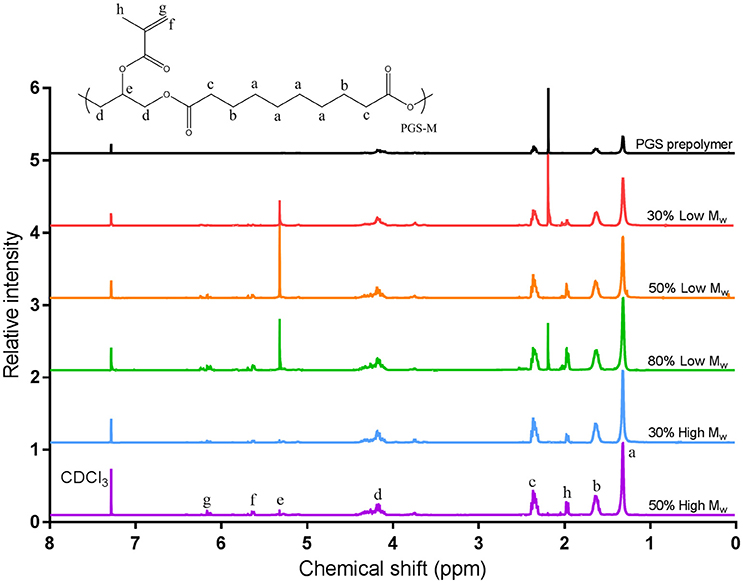
Figure 4. Proton NMR analysis of PGS and PGS-M prepolymers at varied DM (30, 50, and 80%) and Low and High Mw. The different hydrogen environments present in the prepolymer are labeled a–h. Peaks at 1.9, 5.6, and 6.2 ppm for PGS-M samples are associated with the methacrylate group (hydrogen environments f, g, and h). Comparing the integrals of these peaks with those of the methylene groups of sebacic acid (1.3 ppm, hydrogen environment a) allowed the DM to be calculated.
The DM of the PGS-M prepolymer was calculated by comparing the integrals of the peaks for the methylene groups of sebacic acid at 1.3 ppm with those of the methacrylate group hydrogens. DM was found to correlate well with the molar ratio of methacrylic anhydride to PGS prepolymer hydroxyl groups used in the methacrylation process with an R2value of 0.9485 for a 1:1 ratio (Figure 5). Additionally, peaks associated with dichloromethane used in the methacrylation reaction (5.3 ppm in the Low Mw PGS-M spectra) and acetone used in cleaning the NMR equipment (2.2 ppm in the spectra for 30 and 80% Low Mw PGS-M and the PGS prepolymer) were identified and disregarded.
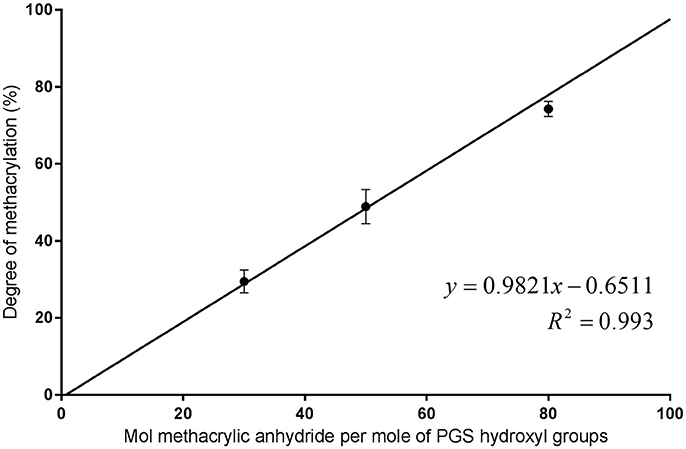
Figure 5. Molar ratio of methacrylic anhydride to PGS prepolymer hydroxyl groups compared to the resulting DM of the PGS-M prepolymers.
Disks of photopolymerised PGS-M, of varied Mw and DM, were washed in methanol to determine the soluble fraction of polymer they contained. After 24 h, all test samples showed a statistically significant (P < 0.001) reduction in mass compared to their corresponding controls. The soluble fractions for 30, 50, and 80% Low Mw PGS-M and 30 and 50% High Mw PGS-M were 27.4 ± 3.1%, 15.4 ± 2.3, 5.1 ± 1.5, 18.0 ± 0.9, and 6.8 ± 1.2%, respectively. Comparing samples of the same Mw, the soluble fraction significantly decreased with increasing DM. Comparing samples of the same DM, the soluble fraction significantly decreased with increasing Mw. After a further 24 h in methanol, no significant reduction in mass was seen in any sample. These results are consistent with other photocurable PGS polymers and thermally crosslinked PGS also [20, 26, 29, 30].
The molecular structure of the PGS-M was examined using ATR-FTIR. In the PGS-M prepolymers, a broad peak associated with the hydroxyl groups was present around 3,460 cm−1 and sharp peaks associated with the methyl and alkane groups were present at 2,924 and 2,851 cm−1 [31, 32]. A distinct peak at 1,735 cm−1 was associated with ester bonds, while the peaks around 1,291–1,050 cm−1 were associated with the stretch vibrations of carboxyl bonds [3, 31, 32]. Peaks associated with the methacrylate group were seen at 940 cm−1 (= C-H bending) and 1,640 cm−1 (C = C stretching) (Figure 6) [33]. These peaks were absent from the PGS prepolymer. The magnitude of these peaks also increased with increasing DM.
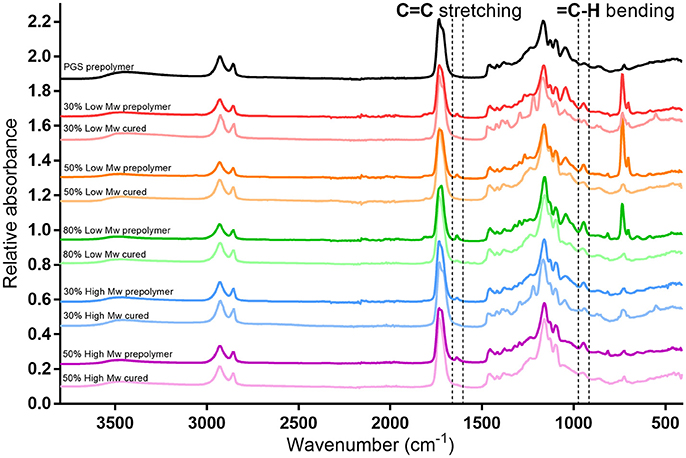
Figure 6. ATR-FTIR spectra for PGS and PGS-M prepolymer at various molecular weights and DM. Highlighted are the regions around 940 and 1,639 cm−1 showing the absorbance peaks associated with the methacrylate group C=C bond present in the PGS-M prepolymers. These peaks are absent after the prepolymers are photocured.
Some prominent peaks appeared in the Low Mw PGS-M prepolymer spectra, at 700 cm−1, associated with C-Cl bonds, likely present in residual dichloromethane from the PGS-M synthesis process. Comparing PGS-M prepolymers and photocured polymers showed the removal of the methacrylate group associated peaks after photopolymerisation. This is also seen in other methacrylated polymers, where spectral peaks associated with the methacrylate groups disappeared after polymerisation [19, 23, 34].
The degradation of PGS-M was examined in vitro using the physiologically relevant enzymes cholesterol esterase and lipase. After 8 days, significant mass loss was seen in 30% Low Mw PGS-M and 30 and 50% High Mw PGS-M treated with cholesterol esterase (Figure 7). Significant mass loss was also seen in 30% Low and High Mw PGS-M treated with lipase. Cholesterol esterase treatment appeared to produce greater degradation compared to lipase in all treatments. SEM showed evidence of degradation in the 30% Low and High Mw PGS-M samples following treatment with cholesterol esterase for 8 days (Figure 8). These surfaces were pitted and cracked, unlike in the controls. No evidence of degradation was apparent in any of the other PGS-M samples.
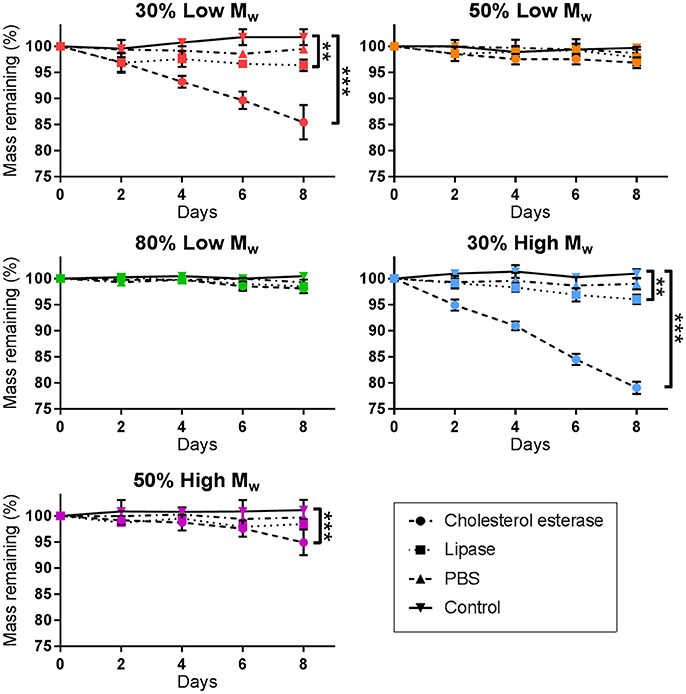
Figure 7. Degradation of PGS-M treated with cholesterol esterase, lipase and PBS. After 8 days, cholesterol esterase produced significant degradation in both 30% DM polymers and the 50% High Mw PGS-M. Lipase only produced significant degradation in the 30% DM polymers. PBS did not produce significant degradation in any sample (n = 3). The degree of significance is indicated as ***P ≤ 0.001, **P ≤ 0.01 and *P ≤ 0.05.

Figure 8. Representative SEM images of Low and High Mw and varied DM PGS-M surfaces following degradation by cholesterol esterase, lipase and PBS for 8 days. Surface erosion is clear in the 30% DM samples. Controls were PGS-M in air. Scale bars are 500 μm.
Increasing the DM of PGS-M appeared to reduce the rate of degradation. This was likely due to an increase in the number of crosslinks present within the polymer which reduced the ability of chain fragments to be cleaved out by the enzymes. A longer study length may be required to show significant enzymatic degradation in all of the PGS-M variants.
When considering the most heavily degraded samples, the degradation rates appeared linear. This suggests only degradation by surface erosion occurred, as is common for enzymatic degradation processes due to the relatively large size of the enzymes limiting their penetration into the polymer matrices [35–37]. Indeed, thermally crosslinked PGS has also been shown to degrade by surface erosion [38]. Degradation of this kind may be considered more favorable than bulk degradation in polymers being utilized as tissue engineering scaffolds, as the mechanical strength of the polymer is reduced over a longer time period, maintaining the structural integrity of the scaffold [21, 39].
Cholesterol esterase has been shown to cause the degradation of various polyesters in previous studies [19, 40–43]. This enzyme is comparable to the esterases produced by macrophages which are known to degrade polyesters in vivo [43]. Cholesterol esterase, at the same concentration as in this study, was shown to degrade thermally cured PGS and PGS-acrylate, with the rate of degradation dependant on the crosslink density of the polymer [19]. Differences between sample geometry prevent a direct comparison of degradation rates between these two studies, however. Lipase has also been shown to degrade various polyester biomaterials in other studies [16, 36, 44, 45]. In examining thermally cured PGS, lipase treatment produced a reduction in mass of ~12% after 8 days and ~30% after 31 days, in vitro [44]. Notably, this study used a much lower concentration of lipase enzyme (110 units/μl) than herein, suggesting that lipase has a much greater effect on thermally cured PGS compared to PGS-M. This may be due to the methacrylate groups limiting the enzymes' access to ester bonds.
No significant degradation due to hydrolysis alone (PBS treatment) was apparent in any of the PGS-M variants examined. Due to the short study length, it is unclear at what rate this may occur. In examining PGS-acrylate, a 22% reduction in mass was seen after 18 weeks [26]. Considering the differences in sample geometry, crosslink density and Mw, 30% High Mw PGS-M may show similar degradation over the same time period. Thermally cured PGS also showed degradation in PBS, reducing in mass by 10% over 31 days [44]. Additionally, a number of different photocurable biomaterials have demonstrated degradation in PBS, with this also being dependent on the degree of crosslinking within their matrices [46–48].
It must be noted that the rates of degradation observed may be different to those produced in more complex in vitro environments containing living cells or in vivo [36]. The types of enzymes produced by different cells and their concentrations vary greatly and this can have a great effect on the degradation rate of a biomaterial [36]. Environmental factors, such as the surrounding pH, presence of enzyme inhibitors, application of flow, mechanical loading or agitation to the material can also have an impact. Indeed, this has been demonstrated in other PGS polymers, with implanted materials degrading at a faster rate than in vitro [1, 23].
Tensile testing revealed that the mechanical properties of PGS-M varied with its composition. Young's modulus increased significantly with increasing DM, for both Low and High Mw prepolymers (Figure 9). A similar trend was also seen in the ultimate tensile strength (UTS) of PGS-M, although in a less pronounced manner. This behavior was likely due to the increased crosslink density resulting from increasing DM reducing polymer chain mobility and increasing matrix stiffness [21, 49]. It is also possible that the crosslinked methacrylate oligomers themselves contributed to the increase in matrix stiffness at increasing DM, acting as a nanocomposite with the PGS polymer backbone [50, 51]. Indeed, at higher DM, the methacrylate groups represent a significant proportion of the molecular structure of the polymer matrix and thus may exert significant influence on the bulk mechanical properties. Interestingly, prepolymer Mw did not appear to have a significant effect on the Young's modulus or UTS of PGS-M.
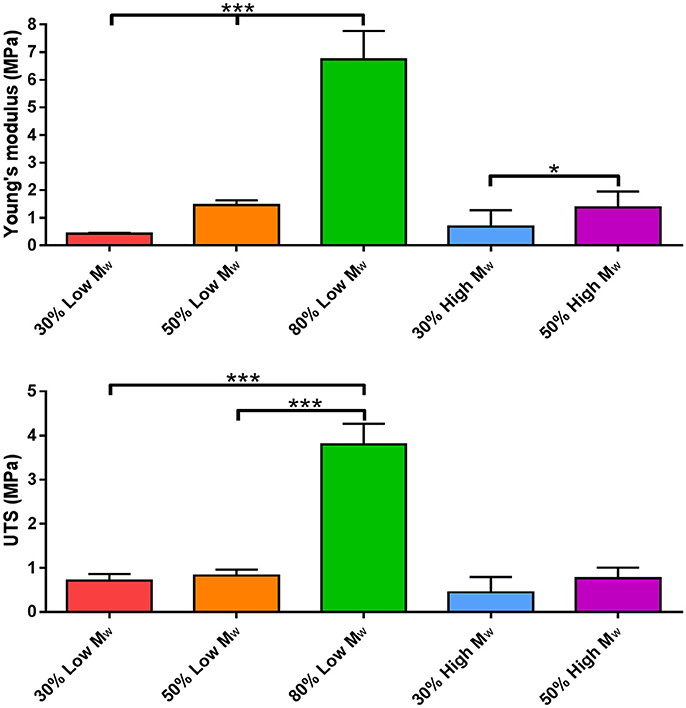
Figure 9. Young's modulus and ultimate tensile strength (UTS) of Low and High Mw and varied DM PGS-M (n = 3). The degree of significance is indicated as ***P ≤ 0.001, **P ≤ 0.01 and *P ≤ 0.05.
Similar results have been reported for PGS-acrylate and PGS-cinnamate, with increased addition of the functional groups resulting in an increase in Young's modulus and UTS [19, 20, 23, 26]. These reported values were also comparable with those produced by PGS-M. PGS-acrylate demonstrated Young's modulus values ranging from 0.6 to 13.2 MPa for degrees of functionalisation (comparable to DM) from 21 to 88%. Additionally, variation in the Mw of PGS-acrylate prepolymer did not result in any significant changes in Young's modulus or UTS [23]. PGS-cinnamate demonstrated values from 0.050 to 0.152 MPa for Young's modulus at degrees of functionalization from 26 to 45%. PGS-M thus compares favorably with these other photocurable PGS polymers, achieving similar mechanical performance, yet with improved synthesis and processing.
Other photocurable elastomeric biomaterials have displayed similar behaviors to PGS-M with an increase in crosslinking density resulting in an increase in stiffness and mechanical strength. The Young's modulus of photoactive PVA hydrogels and azlactone modified PVA was increased from 0.054 to 0.820 MPa and from 0.55 to 0.80 MPa, respectively, by changing the crosslinking density [52, 53]. In gelatin-methacrylate polymers, Young's modulus could be altered from 0.040 to 0.222 MPa by increasing the DM from 15 to 90% [54, 55]. Additionally, modifying the crosslink density in photocurable poly(D,L-lactide) allowed variation in the flexural modulus and strength, from 2.5 to 3.6 GPa and from 79 to 118 MPa, respectively [56]. In terms of mechanical performance, PGS-M appears to occupy a niche between the softer PVA based hydrogels and gelatin-methacrylate polymers and the stiffer photocurable poly(D,L-lactide). This may allow for specific applications as a photocurable biomaterial.
Human dermal fibroblasts and human coronary artery SMCs were cultured on 30% Low Mw PGS-M surfaces for 1, 3, and 7 days. ADSCs were also cultured on PGS-M surfaces, but for 1, 7, and 14 days due to their slower growth rate. Cell viability was assessed by reduction of resazurin sodium salt to resorufin, measured using fluorescence detection.
In all cell types, the fluorescence signal associated with resorufin increased significantly over the duration of the study (Figure 10). Similar results were seen in the positive controls, where cells were cultured on glass coverslips. Comparing the results for cultures on PGS-M and glass at each time point showed no significant differences in resorufin fluorescence between the two surfaces.
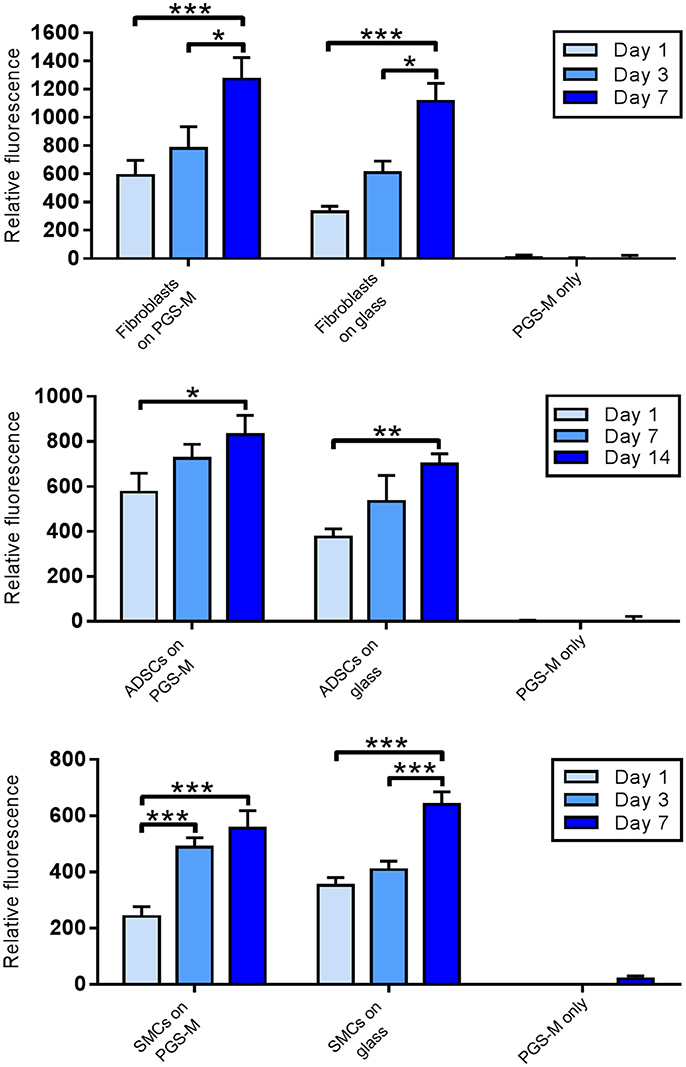
Figure 10. Resazurin reduction assay for viability of fibroblasts, ABSCs and SMCs on 30% Low Mw PGS-M surfaces. Positive controls were cells cultured on borosilicate glass. Negative controls were unseeded PGS-M surfaces (PGS-M only) (n = 3). The degree of significance is indicated as ***P ≤ 0.001, **P ≤ 0.01 and *P ≤ 0.05.
The quantity of fibroblasts, SMCs and ADSCs present on the PGS-M surfaces in culture was assessed using the commercially available PicoGreen® DNA quantification assay. The assay measured the quantity of dsDNA present in each sample by means of fluorescence. This was then used to infer the number of cells present using a standard curve.
DNA content increased significantly over the duration of the cultures for all cell types, on both PGS-M and glass (Figure 11). Comparing the results for fibroblast cultures on PGS-M and glass at each time point showed no significant differences after 1 day, but significant differences at 3 and 7 days, with the positive controls showing the greater values. The SMC cultures also showed significant differences between the two surface types at 7 days. The ADSCs cultured on PGS-M and glass showed no significant differences between the two surfaces at each time point.
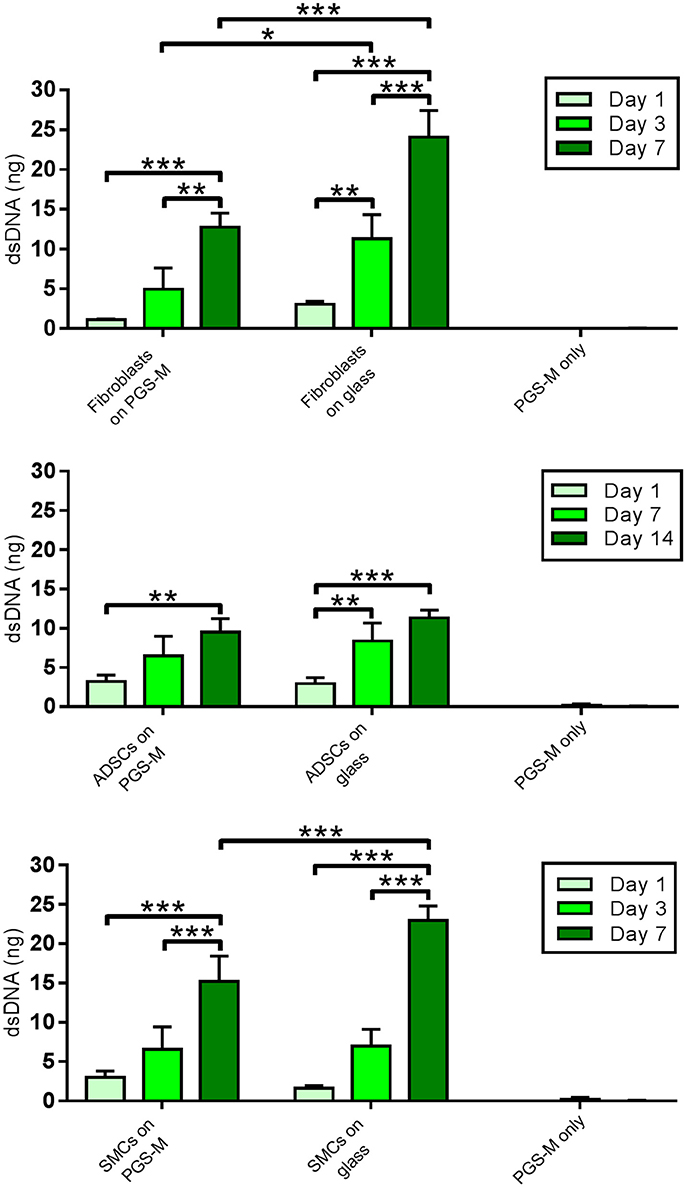
Figure 11. PicoGreen® assay for dsDNA content of Fibroblast, ADSC, and SMC cultures on 30% Low Mw PGS-M surfaces. Positive controls were cells cultured on borosilicate glass. Negative controls were unseeded PGS-M surfaces (PGS-M only) (n = 3). The degree of significance is indicated as ***P ≤ 0.001, **P ≤ 0.01 and *P ≤ 0.05.
In both the resazurin and PicoGreen® assays, all of the results for cells cultured on PGS-M and glass surfaces were statistically significantly different to the corresponding negative controls (unseeded PGS-M coated coverslips) which showed negligible fluorescence. All three cell types also displayed characteristic morphology throughout the study [19, 57–62].
The results demonstrate that all three cell types proliferated on PGS-M surfaces and remained viable up to the conclusion of the study. Similarly, the acrylated and thermally cured forms of PGS have also been shown to support the growth of fibroblasts, SMCs and stem cells [1, 19, 26, 58].
Interestingly, increasing cell numbers, as shown by the PicoGreen® assay, were not necessarily followed by proportional increases in culture metabolism, as measured by the resazurin assay. For example, the PicoGreen® assay detected a significantly greater number of fibroblasts present on the glass surfaces, compared with the PGS-M surfaces, at day 3 and day 7. However, no significant differences were detected between these two culture surfaces when measuring cell metabolism, using the resazurin assay. The SMC cultures showed a similar trend. This effect may be associated with increasing cell confluency and age, but could also suggest that the metabolic activities of the different cell types examined may be substrate specific. The cell-surface interface has been identified as having an important influence on phenotype and culture surfaces may impact on cell metabolism due to a number of different factors, such as stiffness, hydrophilicity, topography and surface chemistry [63–67].
The results also highlight how metabolic assays alone are not suitable for by proxy measurements of cell proliferation. Assessing the success of any biomaterial in supporting healthy cells requires measuring both cell metabolic activity and number.
3D scaffold structures were produced from PGS-M by 2PP. Raman spectroscopy was used to confirm the chemical structure of the PGS-M polymer remained intact following 2PP. Spectra from the scaffolds compared well with that of disks of equivalent PGS-M photocured using UV lamp irradiation. Prominent peaks in the analytical regions appeared at equal Raman shift values (Figure 12A). This was more easily visualized in the transformed spectra (Figure 12B). These spectra were distinctly different from that of the borosilicate glass on which the scaffolds were adhered to. The intensity values of the scaffold's spectra were somewhat lower than those of the UV cured PGS-M disks. This was attributed to the 3D nature and small feature height of the scaffolds reducing the detected signal. Additionally, due to the small height of the 3D scaffolds, some of the signal generated by the underlying borosilicate glass was detected. This is most pronounced in the fingerprint region, < ~1300 cm−1. The data suggests that the chemical structure of the PGS-M polymer is retained after 2PP using TW/cm2 peak intensities. This is in line with previous reports using Raman spectroscopy to determine the degree of crosslinking. At the reported laser intensities the irradiation converts the functionalised prepolymers into a crosslinked polymer, while higher pulse energies are needed for further chemical conversion of the polymer and carbonisation by the laser [68, 69].
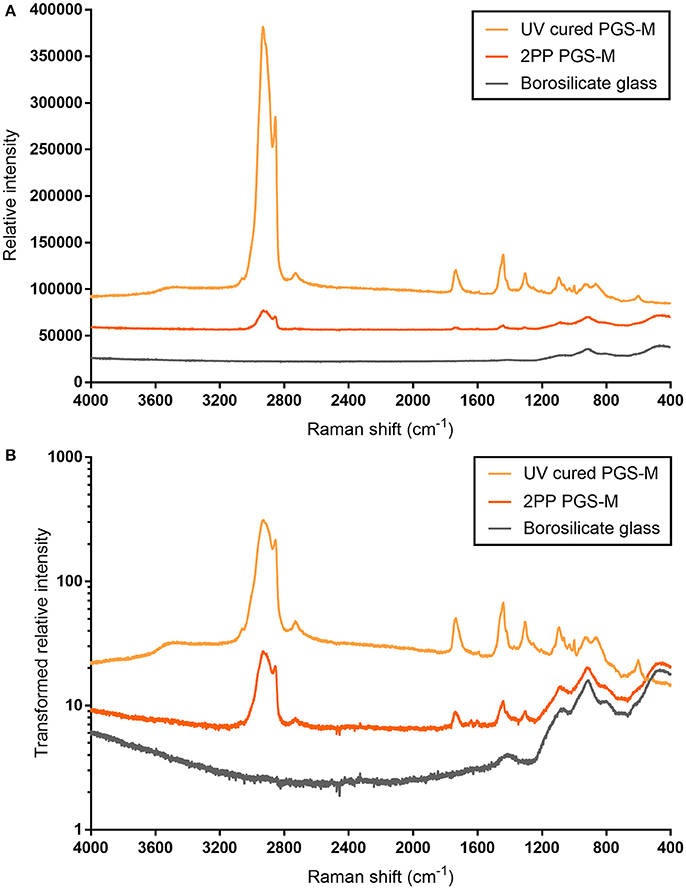
Figure 12. Raman spectra for 50% Low Mw PGS-M photocured as 3D scaffolds, using 2PP, and as disks, using a UV lamp. The spectra for the underlying borosilicate glass slides, onto which the 3D scaffolds were adhered to, is also presented. The original data (A) show the spectra for both PGS-M samples are distinctly different from that of borosilicate glass. The peak intensity values produced by the UV cured PGS-M disks are higher than those of the 3D scaffolds. Transforming the data (relative intensity/1000 and presented on a logarithmic scale) allows the similarities of the PGS-M spectra to be more easily visualized, with peaks clearly visible at equal Raman shift values (B).
Various structures were reproducibly produced, including closed and open sided lattices (Figures 13A–E). The scaffolds were up to ~500 μm in length and ~200 μm tall; however, at this height the open lattice structure appeared to be lost. The tallest scaffolds with an open structure were ~100 μm. The minimum feature thickness achieved was ~10 μm and the aspect ratio was ~2.1 (of the produced structures employing multipath scanning). It was noted that the mechanical properties of the photocured polymer were an important consideration during the scaffold fabrication process. The feature thickness of the scaffolds required tuning to ensure they remained free standing and did not collapse under their own weight (see also Supplementary Image 1). This may occur when using elastomeric materials [70]. Additionally, some structural deformations due to shrinkage were also evident, as has been noted in other polymer structures fabricated in this manner [71]. Scaffold shrinkage in the XY plane was 14.5 ± 5.9%, based on the analysis of the SEM images (Figure 13D). Compensation for this may be required should precise final dimensions be required for a specific application.

Figure 13. Representative images of 3D PGS-M structures produced by 2PP. Structures produced using 20x 0.8 NA (A,D), 10x 0.3 NA (B), and 63x 1.4 NA (C,E) objective lenses examined using SEM. Vascular SMCs were cultured on scaffolds and then stained for F actin filaments using Phalloidin-FITC. Projections of 3D confocal images of scaffolds after 1 day (F) and 7 days (I) of SMC culture. Unstained control cultures after 7 days are shown as transmitted light DIC (G) and fluorescence (J) images, along with unseeded, but stained, controls (H,K). Scale bars are 200 μm (A,B,D,G,H,J,K) and 50 μm (C,E,F,I).
The PGS-M scaffolds were seeded with human coronary artery SMCs. The cells appeared to adhere to the structures and proliferate over the 7 day culture period (results summarized in Figures 13F–K). Interestingly, the SMCs appeared to attach to the upper surface of the PGS-M structures and proliferate across them, even bridging the ~70 μm square pores by day 7. Similar behaviors have been observed on other polymer scaffolds fabricated using 2PP. Human ADSCs proliferated across the surface of methacrylamide-modified gelatin scaffolds with 200 μm pore sizes and human fibrosarcoma cells bridged 52 μm pores in triacrylate woodpile scaffolds [72, 73]. Additionally, MG63 osteosarcoma cells and rat MSCs invaded lattice scaffolds produced from SZ2080 photoresist, acting as synthetic cell niches, covering their surfaces and bridging gaps of up to 30 μm [74]. Scaffold topology and feature size has an important influence on cell function and fate. Vascular SMCs have been shown to proliferated best on scaffolds with pore sizes >38 μm [75]. This indicates that 2PP may allow the pore geometry of PGS-M scaffolds to be tailored to ideally suit different cell types.
Conclusions
Poly(glycerol sebacate) was rendered photocurable by the addition of methacrylate groups producing PGS-M. Successful methacrylation was determined by NMR analysis and ATR-FTIR spectroscopy with the DM successfully controlled by modification of the reaction components. The DM had a significant effect on the degradation rate of PGS-M and its mechanical properties. PGS-M surfaces also supported the growth and proliferation of primary human fibroblasts, ADSCs and vascular SMCs, determined using metabolism and DNA quantification based assays. Comparing the results from these different assays suggested that PGS-M may interact with these cells in a substrate specific manner, affecting metabolism and proliferation differently. 2PP was used to successfully structure PGS-M into 3D scaffolds with minimum feature sizes of ~10 μm. These scaffolds supported the growth of vascular SMCs which appeared to spread across the scaffold surfaces, bridging gaps in the structure.
PGS-M is an easily synthesized and versatile biomaterial with tunable physical properties and compatibility with various cell types in culture. Its photocurable nature also allows for rapid production of user-defined 3D structures with micro-scale features. Work is ongoing to explore how this material may be used in various biomaterial and tissue-engineering applications both in vitro and in vivo.
Ethics Statement
Human dermal fibroblasts and human adipose-derived stem cells (ADSCs) from donated primary dermal tissue and subcutaneous fat, respectively, were obtained after all participants provided written informed consent (ethics reference: 15/YH/0177) and processed and stored in accordance with the Human Tissue Act 2004 (licence number 12179). The ethics committee that reviewed and approved the study is the NHS Yorkshire & The Humber-Sheffield Research Ethics Committee.
Author Contributions
SP-T synthesized the PGS-M polymer, performed the polymer characterization and cell assays and wrote the manuscript. RO, HB, and SR performed the 2PP experiments and edited the manuscript. The team was supervised by MM and FC, who also edited the manuscript and secured funding for the collaboration.
Funding
This research was supported by the Biotechnology and Biological Sciences Research Council (under grant BB/F016840/) and the Engineering and Physical Sciences Research Council (via funds provided by the Doctoral Training Partnership grant to the University of Sheffield, EP/M508135/1). Vilnius University group was funded by a grant (No. SEN-20/2015) from the Research Council of Lithuania. The research leading to these results has received funding from LASERLAB-EUROPE (grant agreement no. 654148, European Union's Horizon 2020 research and innovation programme).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Nik Reeves-McLaren for his assistance with the Raman spectroscopy analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fphy.2018.00041/full#supplementary-material
References
1. Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. (2002) 20:602–6. doi: 10.1038/nbt0602-602
2. Jun Loh X, Karim AA, Owh C. Poly(glycerol sebacate) biomaterial: synthesis and biomedical applications. J Mater Chem B (2015) 3:7641–52. doi: 10.1039/C5TB01048A
3. Rai R, Tallawi M, Grigore A, Boccaccini AR. Synthesis, properties and biomedical applications of poly(glycerol sebacate) (PGS): a review. Prog Polym Sci. (2012) 37:1051–78. doi: 10.1016/j.progpolymsci.2012.02.001
4. Chen Q-Z, Ishii H, Thouas GA, Lyon AR, Wright JS, Blaker JJ, et al. An elastomeric patch derived from poly(glycerol sebacate) for delivery of embryonic stem cells to the heart. Biomaterials (2010) 31:3885–93. doi: 10.1016/j.biomaterials.2010.01.108
5. Pritchard CD, Arnér KM, Langer RS, Ghosh FK. Retinal transplantation using surface modified poly(glycerol-co-sebacic acid) membranes. Biomaterials (2010) 31:7978–84. doi: 10.1016/j.biomaterials.2010.07.026
6. Sundback CA, Shyu JY, Wang Y, Faquin WC, Langer RS, Vacanti JP, et al. Biocompatibility analysis of poly(glycerol sebacate) as a nerve guide material. Biomaterials (2005) 26:5454–64. doi: 10.1016/j.biomaterials.2005.02.004
7. Kharaziha M, Nikkhah M, Shin S-R, Annabi N, Masoumi N, Gaharwar AK, et al. PGS:Gelatin nanofibrous scaffolds with tunable mechanical and structural properties for engineering cardiac tissues. Biomaterials (2013) 34:6355–66. doi: 10.1016/j.biomaterials.2013.04.045
8. Radisic M, Park H, Martens TP, Salazar-Lazaro JE, Geng W, Wang Y, et al. Pre-treatment of synthetic elastomeric scaffolds by cardiac fibroblasts improves engineered heart tissue. J Biomed Mater Res A (2008) 86A:713–24. doi: 10.1002/jbm.a.31578
9. Lee K-W, Stolz DB, Wang Y. Substantial expression of mature elastin in arterial constructs. Proc Natl Acad Sci USA. (2011) 108:2705–10. doi: 10.1073/pnas.1017834108
10. Wu W, Allen RA, Wang Y. Fast-degrading elastomer enables rapid remodeling of a cell-free synthetic graft into a neoartery. Nat Med. (2012) 18:1148–53. doi: 10.1038/nm.2821
11. Hagandora CK, Gao J, Wang Y, Almarza AJ. Poly (glycerol sebacate): a novel scaffold material for temporomandibular joint disc engineering. Tissue Eng A (2012) 19:729–37. doi: 10.1089/ten.tea.2012.0304
12. Kemppainen JM, Hollister SJ. Tailoring the mechanical properties of 3D-designed poly(glycerol sebacate) scaffolds for cartilage applications. J Biomed Mater Res A (2010) 94A:9–18. doi: 10.1002/jbm.a.32653
13. Jeong CG, Hollister SJ. A comparison of the influence of material on in vitro cartilage tissue engineering with PCL, PGS, and POC 3D scaffold architecture seeded with chondrocytes. Biomaterials (2010) 31:4304–12. doi: 10.1016/j.biomaterials.2010.01.145
14. Tobias IS, Lee H, Engelmayr GC, Macaya D, Bettinger CJ, Cima MJ. Zero-order controlled release of ciprofloxacin-HCl from a reservoir-based, bioresorbable and elastomeric device. J Control. Release (2010) 146:356–62. doi: 10.1016/j.jconrel.2010.05.036
15. Sun Z-J, Chen C, Sun M-Z, Ai C-H, Lu X-L, Zheng Y-F, et al. The application of poly (glycerol–sebacate) as biodegradable drug carrier. Biomaterials (2009) 30:5209–14. doi: 10.1016/j.biomaterials.2009.06.007
16. Frydrych M, Román S, MacNeil S, Chen B. Biomimetic poly(glycerol sebacate)/poly(l-lactic acid) blend scaffolds for adipose tissue engineering. Acta Biomater. (2015) 18:40–9. doi: 10.1016/j.actbio.2015.03.004
17. Li Y, Cook WD, Moorhoff C, Huang W-C, Chen Q-Z. Synthesis, characterization and properties of biocompatible poly(glycerol sebacate) pre-polymer and gel. Polym Int. (2013) 62:534–47. doi: 10.1002/pi.4419
18. Chen Q-Z, Bismarck A, Hansen U, Junaid S, Tran MQ, Harding SE, et al. Characterisation of a soft elastomer poly(glycerol sebacate) designed to match the mechanical properties of myocardial tissue. Biomaterials (2008) 29:47–57. doi: 10.1016/j.biomaterials.2007.09.010
19. Nijst CLE, Bruggeman JP, Karp JM, Ferreira L, Zumbuehl A, Bettinger CJ, et al. Synthesis and Characterization of Photocurable Elastomers from Poly(glycerol-co-sebacate). Biomacromolecules (2007) 8:3067–73. doi: 10.1021/bm070423u
20. Zhu C, Kustra SR, Bettinger CJ. Photocrosslinkable biodegradable elastomers based on cinnamate-functionalized polyesters. Acta Biomater. (2013) 9:7362–70. doi: 10.1016/j.actbio.2013.03.041
21. Ifkovits JL, Burdick JA. Review: photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Eng. (2007) 13:2369–85. doi: 10.1089/ten.2007.0093
22. Malinauskas M, Žukauskas A, Hasegawa S, Hayasaki Y, Mizeikis V, Buividas R, et al. Ultrafast laser processing of materials: from science to industry. Light Sci Appl. (2016) 5:e16133. doi: 10.1038/lsa.2016.133
23. Ifkovits JL, Padera RF, Burdick JA. Biodegradable and radically polymerized elastomers with enhanced processing capabilities. Biomed Mater. (2008) 3:34104. doi: 10.1088/1748-6041/3/3/034104
24. International Organisation for Standardisation. BS ISO 37:2011 - Rubber, Vulcanized or Thermoplastic. Determination of Tensile Stress-Strain Properties. Geneva, Switzerland: International Organisation for Standardisation (2011). Available online at: https://bsol.bsigroup.com/Bibliographic/BibliographicInfoData/000000000030196867 (Accessed June 11, 2016).
25. Maciulaitis J, Deveikyte M, Rekštyte S, Bratčikov M, Darinskas A, Šimbelyte A, et al. Preclinical study of SZ2080 material 3D microstructured scaffolds for cartilage tissue engineering made by femtosecond direct laser writing lithography. Biofabrication (2015) 7:15015. doi: 10.1088/1758-5090/7/1/015015
26. Ifkovits JL, Devlin JJ, Eng G, Martens TP, Vunjak-Novakovic G, Burdick JA. Biodegradable fibrous scaffolds with tunable properties formed from photo-cross-linkable Poly(glycerol sebacate). ACS Appl Mater Interfaces (2009) 1:1878–86. doi: 10.1021/am900403k
27. Zhang H, Grinstaff MW. Recent Advances in Glycerol Polymers: Chemistry and Biomedical Applications. Macromol Rapid Commun. (2014) 35:1906–24. doi: 10.1002/marc.201400389
28. Nagata M, Machida T, Sakai W, Tsutsumi N. Synthesis, characterization, and enzymatic degradation of network aliphatic copolyesters. J Polym Sci Part Polym Chem. (1999) 37:2005–11. doi: 10.1002/(SICI)1099-0518(19990701)37:13<2005::AID-POLA14>3.0.CO;2-H
29. Gaharwar AK, Patel A, Dolatshahi-Pirouz A, Zhang H, Rangarajan K, Iviglia G, et al. Elastomeric nanocomposite scaffolds made from poly(glycerol sebacate) chemically crosslinked with carbon nanotubes. Biomater Sci. (2014) 3:46–58. doi: 10.1039/c4bm00222a
30. Kafouris D, Kossivas F, Constantinides C, Nguyen NQ, Wesdemiotis C, Patrickios CS. Biosourced amphiphilic degradable elastomers of Poly(glycerol sebacate): synthesis and network and oligomer characterization. Macromolecules (2013) 46:622–30. doi: 10.1021/ma3016882
31. Jaafar IH, Ammar MM, Jedlicka SS, Pearson RA, Coulter JP. Spectroscopic evaluation, thermal, and thermomechanical characterization of poly(glycerol-sebacate) with variations in curing temperatures and durations. J Mater Sci. (2010) 45:2525–29. doi: 10.1007/s10853-010-4259-0
32. Cai W, Liu L. Shape-memory effect of poly (glycerol–sebacate) elastomer. Mater Lett. (2008) 62:2171–73. doi: 10.1016/j.matlet.2007.11.042
33. Silverstein RM, Bassler GC. Spectrometric identification of organic compounds. J Chem Educ. (1962) 39:546. doi: 10.1021/ed039p546
34. Castor CA Jr, Pontier A, Durand J, Pinto JC, Prat L. Real time monitoring of the quiescent suspension polymerization of methyl methacrylate in microreactors—Part 1. A kinetic study by Raman spectroscopy and evolution of droplet size. Chem Eng Sci. (2015) 131:340–52. doi: 10.1016/j.ces.2015.02.037
35. Liang S-L, Yang X-Y, Fang X-Y, Cook WD, Thouas GA, Chen Q-Z. In vitro enzymatic degradation of poly (glycerol sebacate)-based materials. Biomaterials (2011) 32:8486–96. doi: 10.1016/j.biomaterials.2011.07.080
36. Azevedo HS, and Reis RL. Chapter 12 - Understanding the enzymatic degradation of biodegradable polymers and strategies to control their degradation rate. In: Biodegradable Systems in Tissue Engineering and Regenerative Medicine. Boca Raton, FL: CRC Press p. 178–97. Available online at: https://www.researchgate.net/profile/Helena_Azevedo/publication/210197234_Understanding_the_Enzymatic_Degradation_of_Biodegradable_Polymers_and_Strategies_to_Control_their_Degradation_Rate/links/0912f50be2c4dd1138000000.pdf (Accessed March 30, 2016).
37. Smith R, Oliver C, Williams DF. The enzymatic degradation of polymers in vitro. J Biomed Mater Res. (1987) 21:991–1003. doi: 10.1002/jbm.820210805
38. Wang Y, Kim YM, Langer R. In vivo degradation characteristics of poly(glycerol sebacate). J Biomed Mater Res A (2003) 66A:192–7. doi: 10.1002/jbm.a.10534
39. Göpferich A. Mechanisms of polymer degradation and erosion. Biomaterials (1996) 17:103–14. doi: 10.1016/0142-9612(96)85755-3
40. Santerre JP, Woodhouse K, Laroche G, Labow RS. Understanding the biodegradation of polyurethanes: From classical implants to tissue engineering materials. Biomaterials (2005) 26:7457–70. doi: 10.1016/j.biomaterials.2005.05.079
41. Tang YW, Labow RS, Santerre JP. Enzyme induced biodegradation of polycarbonate-polyurethanes: dose dependence effect of cholesterol esterase. Biomaterials (2003) 24:2003–11. doi: 10.1016/S0142-9612(02)00563-X
42. Woo GLY, Mittelman MW, Santerre JP. Synthesis and characterization of a novel biodegradable antimicrobial polymer. Biomaterials (2000) 21:1235–46. doi: 10.1016/S0142-9612(00)00003-X
43. Labow RS, Meek E, Santerre JP. Differential synthesis of cholesterol esterase by monocyte-derived macrophages cultured on poly(ether or ester)-based poly(urethane)s. J Biomed Mater Res. (1998) 39:469–77. doi: 10.1002/(SICI)1097-4636(19980305)39:3<469::AID-JBM18>3.0.CO;2-5
44. Frydrych M, Chen B. Large three-dimensional poly(glycerol sebacate)-based scaffolds – a freeze-drying preparation approach. J Mater Chem B (2013) 1:6650–61. doi: 10.1039/C3TB20842G
45. Ashton JH, Mertz JAM, Alexander MJ, Slepian MJ, Mills JL, Vande Geest JP. Functional endoluminal paving (FELP): thermoforming, biodegradation, and mechanical properties of a novel polymer graft for abdominal aortic aneurysms. In: ASME 2010 Summer Bioengineering Conference, Parts A and B. Naples, FL (2010). p. 905–6.
46. Wang D, Williams CG, Li Q, Sharma B, Elisseeff JH. Synthesis and characterization of a novel degradable phosphate-containing hydrogel. Biomaterials (2003) 24:3969–80. doi: 10.1016/S0142-9612(03)00280-1
47. Nuttelman CR, Henry SM, Anseth KS. Synthesis and characterization of photocrosslinkable, degradable poly(vinyl alcohol)-based tissue engineering scaffolds. Biomaterials (2002) 23:3617–26. doi: 10.1016/S0142-9612(02)00093-5
48. Metters AT, Anseth KS, Bowman CN. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer (2000) 41:3993–4004. doi: 10.1016/S0032-3861(99)00629-1
49. Young RJ, Lovell PA (eds.). Chapter 21 Elastomers. In: Introduction to Polymers. Taylor & Francis Group LLC p. 511–30.
50. Andrzejewska E, Marcinkowska A, Wegner K. Nanocomposites obtained by photopolymerization of (methacrylate monomer)/(methacrylate functionalized polyhedral oligomeric silsesquioxane) system. Polymer (2011) 56:63–6.
51. Ando S, Someya Y, Takahashi T, Shibata M. Thermal and mechanical properties of photocured organic–inorganic hybrid nanocomposites of terpene-based acrylate resin and methacrylate-substituted polysilsesquioxane. J Appl Polym Sci. (2010) 115:3326–31. doi: 10.1002/app.30398
52. Schmedlen RH, Masters KS, West JL. Photocrosslinkable polyvinyl alcohol hydrogels that can be modified with cell adhesion peptides for use in tissue engineering. Biomaterials (2002) 23:4325–32. doi: 10.1016/S0142-9612(02)00177-1
53. Mühlebach A, Müller B, Pharisa C, Hofmann M, Seiferling B, Guerry D. New water-soluble photo crosslinkable polymers based on modified poly(vinyl alcohol). J Polym Sci Part Polym Chem. (1997) 35:3603–11. doi: 10.1002/(SICI)1099-0518(19971130)35:16<3603::AID-POLA28>3.0.CO;2-I
54. Nguyen AH, McKinney J, Miller T, Bongiorno T, McDevitt TC. Gelatin methacrylate microspheres for controlled growth factor release. Acta Biomater. (2015) 13:101–10. doi: 10.1016/j.actbio.2014.11.028
55. Bertassoni LE, Cardoso JC, Manoharan V, Cristino AL, Bhise NS, Araujo WA, et al. Direct-write bioprinting of cell-laden methacrylated gelatin hydrogels. Biofabrication (2014) 6:24105. doi: 10.1088/1758-5082/6/2/024105
56. Melchels FPW, Feijen J, Grijpma DW. A poly(d,l-lactide) resin for the preparation of tissue engineering scaffolds by stereolithography. Biomaterials (2009) 30:3801–9. doi: 10.1016/j.biomaterials.2009.03.055
57. Zhu Y, Liu T, Song K, Fan X, Ma X, Cui Z. Adipose-derived stem cell: a better stem cell than BMSC. Cell Biochem Funct. (2008) 26:664–75. doi: 10.1002/cbf.1488
58. Gao J, Ensley AE, Nerem RM, Wang Y. Poly(glycerol sebacate) supports the proliferation and phenotypic protein expression of primary baboon vascular cells. J Biomed Mater Res A (2007) 83A:1070–75. doi: 10.1002/jbm.a.31434
59. Gimble JM, Katz AJ, Bunnell BA. Adipose-derived stem cells for regenerative medicine. Circ Res. (2007) 100:1249–60. doi: 10.1161/01.RES.0000265074.83288.09
60. Kim W-S, Park B-S, Sung J-H, Yang J-M, Park S-B, Kwak S-J, et al. Wound healing effect of adipose-derived stem cells: a critical role of secretory factors on human dermal fibroblasts. J Dermatol Sci. (2007) 48:15–24. doi: 10.1016/j.jdermsci.2007.05.018
61. Moulin V, Castilloux G, Auger FA, Garrel D, O'Connor-McCourt MD, Germain L. Modulated response to cytokines of human wound healing myofibroblasts compared to dermal fibroblasts. Exp Cell Res. (1998) 238:283–93. doi: 10.1006/excr.1997.3827
62. Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev. (1979) 59:1–61.
63. Stevens MM, George JH. Exploring and Engineering the Cell Surface Interface. Science (2005) 310:1135–38. doi: 10.1126/science.1106587
64. Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, et al. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton (2005) 60:24–34. doi: 10.1002/cm.20041
65. Curtis A, Wilkinson C. New depths in cell behaviour: reactions of cells to nanotopography. Biochem Soc Symp. (1998) 65:15–26.
66. Hambleton J, Schwartz Z, Khare A, Windeler SW, Luna M, Brooks BP, et al. Culture surfaces coated with various implant materials affect chondrocyte growth and metabolism. J Orthop Res. (1994) 12:542–52. doi: 10.1002/jor.1100120411
67. Lydon MJ, Minett TW, Tighe BJ. Cellular interactions with synthetic polymer surfaces in culture. Biomaterials (1985) 6:396–402. doi: 10.1016/0142-9612(85)90100-0
68. Žukauskas A, Matulaitiene I, Paipulas D, Niaura G, Malinauskas M, Gadonas R. Tuning the refractive index in 3D direct laser writing lithography: towards GRIN microoptics. Laser Photonics Rev. (2015) 9:706–12. doi: 10.1002/lpor.201500170
69. Jiang LJ, Zhou YS, Xiong W, Gao Y, Huang X, Jiang L, et al. Two-photon polymerization: investigation of chemical and mechanical properties of resins using Raman microspectroscopy. Opt Lett. (2014) 39:3034–37. doi: 10.1364/OL.39.003034
70. Rekštyte S, Malinauskas M, Juodkazis S. Three-dimensional laser micro-sculpturing of silicone: towards bio-compatible scaffolds. Opt Expr. (2013) 21:17028–41. doi: 10.1364/OE.21.017028
71. Ovsianikov A, Gruene M, Pflaum M, Koch L, Maiorana F, Wilhelmi M, et al. Laser printing of cells into 3D scaffolds. Biofabrication (2010) 2:14104. doi: 10.1088/1758-5082/2/1/014104
72. Ovsianikov A, Deiwick A, Van Vlierberghe S, Pflaum M, Wilhelmi M, Dubruel P, et al. Laser fabrication of 3D gelatin scaffolds for the generation of bioartificial tissues. Materials (2011) 4:288–99. doi: 10.3390/ma4010288
73. Tayalia P, Mendonca CR, Baldacchini T, Mooney DJ, Mazur E. 3D Cell-migration studies using two-photon engineered polymer scaffolds. Adv Mater. (2008) 20:4494–98. doi: 10.1002/adma.200801319
74. Raimondi MT, Raimondi MT, Eaton SM, Eaton SM, Nava MM, Nava MM, et al. Two-photon laser polymerization: from fundamentals to biomedical application in tissue engineering and regenerative medicine. J Appl Biomater Funct Mater. (2012) 10:56–66. doi: 10.5301/JABFM.2012.9278
Keywords: photocurable, tissue engineering, 3D printing, mechanical testing, 3D cell culture, 2-photon polymerization, stereolithography, femtosecond pulses
Citation: Pashneh-Tala S, Owen R, Bahmaee H, Rekštytė S, Malinauskas M and Claeyssens F (2018) Synthesis, Characterization and 3D Micro-Structuring via 2-Photon Polymerization of Poly(glycerol sebacate)-Methacrylate–An Elastomeric Degradable Polymer. Front. Phys. 6:41. doi: 10.3389/fphy.2018.00041
Received: 19 January 2018; Accepted: 16 April 2018;
Published: 08 May 2018.
Edited by:
Antonio Riveiro Rodriguez, University of Vigo, SpainReviewed by:
Venugopal Rao Soma, University of Hyderabad, IndiaHenrique de Amorim Almeida, Polytechnic Institute of Leiria, Portugal
Vladimir S. Komlev, AA Baikov Institute of Metallurgy and Materials (RAS), Russia
Copyright © 2018 Pashneh-Tala, Owen, Bahmaee, Rekštytė, Malinauskas and Claeyssens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frederik Claeyssens, Zi5jbGFleXNzZW5zQHNoZWZmaWVsZC5hYy51aw==