
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Phys., 06 February 2018
Sec. Chemical Physics and Physical Chemistry
Volume 6 - 2018 | https://doi.org/10.3389/fphy.2018.00003
 Imanol Usabiaga1
Imanol Usabiaga1 Ander Camiruaga1Aran Insausti1Pierre Çarçabal2Emilio J. Cocinero1Iker León1,3*
Ander Camiruaga1Aran Insausti1Pierre Çarçabal2Emilio J. Cocinero1Iker León1,3* José A. Fernández1*
José A. Fernández1*We report a combination of laser spectroscopy in molecular jets and quantum mechanical calculations to characterize the aggregation preferences of phenyl-β-D-glucopyranoside (β-PhGlc) and phenyl-β-D-galactopyranoside (β-PhGal) homodimers. At least two structures of β-PhGlc dimer were found maintaining the same intramolecular interactions of the monomers, but with additional intermolecular interactions between the hydroxyl groups. Several isomers were also found for the dimer of β-PhGal forming extensive hydrogen bond networks between the interacting molecules, of very different shape. All the species found present several CH•••π and OH•••π interactions that add stability to the aggregates. The results show how even the smallest change in a substituent, from axial to equatorial position, plays a decisive role in the formation of the dimers. These conclusions reinforce the idea that the small structural changes between sugar units are amplified by formation of intra and intermolecular hydrogen bond networks, helping other molecules (proteins, receptors) to easily read the sugar code of glycans.
Sugars are small carbohydrates which perform numerous roles in cells and tissues. For example, polysaccharides serve for the storage of energy and energy management and also play a key role as structural components [1, 2]. In this line, glucose plays a critical role in the energy metabolism of many living organisms [3]. Aside from this well-known role, carbohydrates are also the base of the immune system in eukaryotes. The cells produce a type of molecules called glycans, composed of a variable number of monosaccharides and attach them to lipids and proteins in the extracellular side of the cell membrane. The cells of the immune system examine these glycans and if their structure is not correct, an immune response is triggered. These glycans can contain a variable number of sugar units of different nature, giving rise to a huge number of possible combinations, although some may only differ in the orientation of a single hydroxyl substituent. Thus, on the one hand, the versatility of the monosaccharides enables the production of a complex code (the biological equivalent of a 512 bit password). But on the other hand, the receptor in the cell of the immune system must be able to recognize the changes in the sequence of the glycan. Apparently, this task is done with high efficiency, as minimal alterations on the sequence are easily recognized. Such is the case of the blood groups: having A or B blood group depends on the nature and type of a single sugar unit [4]. Monosaccharides may be found either in linear or cyclic forms [5]. When isolated, they may interconvert (almost freely) between those two forms. Depending on how they re-bond when they adopt their cyclic form, the substituent of carbon 1 can be in the axial or equatorial position, giving rise to the α/β anomers respectively. When the hydroxyl group on carbon 1 is attached to a more voluminous substituent, for example, when the monosaccharide is part of a glycan, the interconversion is not possible and the sugar is locked in a given anomer. Hence, the ability of the immune system to recognize the blood group means that it is selective toward the anomeric conformation of a single sugar unit of the glycan that constitutes the molecular I.D. This fantastic selectivity can only be due to a high complementarity, of the key and lock type, between the receptor and the polysaccharide. However, designing a protein that is so selective toward such a small change in the orientation of a single substituent has to be difficult, and there must be other factors that help the receptor to achieve such selectivity.
Following these observations, our group is engaged in the study at a molecular level of the mechanisms that amplify small structural modifications on a sugar unit until they completely alter their interactions with other molecules. We have demonstrated in previous studies [6] that the anomeric conformation of glucose bound to a voluminous group is transmitted through the whole molecule due to the formation of a cooperative hydrogen bond network that involves interactions between all the hydroxyl groups. Thus, the intramolecular hydrogen bonds act as an amplifier of the changes in the orientation of the anomeric substituent, which is at one of the ends of the network. Here, we explore if such mechanism also plays a role when the modification takes place in a different carbon atom, by forming the homodimers of glucose and galactose derivatives. These two monosaccharides, Scheme 1, differ in the position of the hydroxyl in carbon 4, which is in the equatorial position in glucose and in the axial position in galactose. As demonstrated in previous studies [7, 8], the hydroxyl groups of the monomers are connected by O-H···O interactions and a change in the position of one of them may have a big impact in the shape of the cooperative hydrogen bond network [6]. The question is if it also influences how two sugar units interact one with each other. Such a study is not an easy task, as it requires isolating the selected carbohydrates from any other molecule. Fortunately, supersonic expansions create a suitable environment for this purpose: the cold, controlled environment in the expansion enables probing complexes, isolated from any external interaction that may perturb them and facilitates using mass-resolved excitation spectroscopy (MRES) to fully characterize the non-covalent interactions that form the aggregates. In fact, this technique has already been successfully applied to the study of other systems of biological relevance, such as amino acids or peptides [7–21], neurotransmitters [22–28], anesthetics [29, 30], several mono-, di- and poly-saccharides [4, 6, 31–39] or even micelles [40–42]. In these systems, the addition of a chromophore with an optically accessible excited electronic state is required to probe the molecules. Thus, we added a phenyl ring to the anomeric carbon. Such addition also blocks the anomeric conformation and emulates the situation where a monosaccharide is attached to a more voluminous substituent similar to the sugar chain of a glycan.
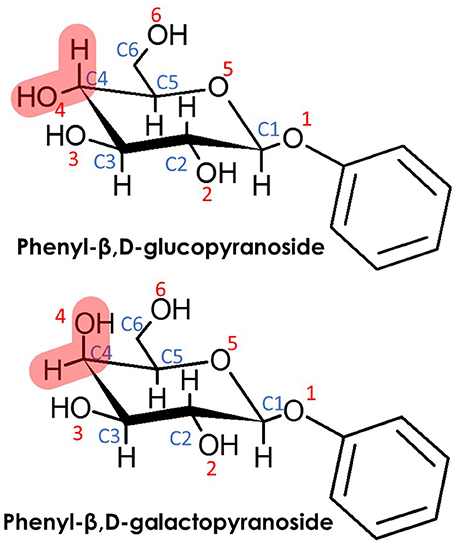
Scheme 1. (Top) Phenyl-β,D-glucopyranoside (β-PhGlc) and (Bottom) Phenyl-β,D-galactopyranoside (β-PhGal). Note that the only difference between both monosacharides is the relative position of the O4H hydroxyl group.
Briefly, the experimental system consists of a modified time of flight mass spectrometer equipped with a laser desorption/ionization (LDI) source attached to a pulsed valve (Series 9, General Valve Inc.), a Nd/YAG-pumped dye laser (Fine Adjustment Pulsare Pro-S), an OPO system (LaserVision) to generate light in the IR region and electronics for synchronization and data collection and handling. Ar was used as a carrier gas at a backing pressure of 10 bar. A complete description of the experimental system and of the sample preparation protocol can be found in Usabiaga et al. [6].
For the one color REMPI experiments the desorption laser was fired ~200 μs after the valve opening, so the sample was picked up by the jet. Approximately 200 μs later, the UV laser was fired, producing a signal proportional to the concentration of the molecules. Scanning the laser while the signal produced was monitored allowed us to record the electronic excitation spectrum of the monomers and their aggregates, depending on the mass-channel selected.
For the IR experiments, an additional IR laser was fired, approximately 50 ns before the UV laser. If the IR laser was resonant with a vibrational transition of the species, a dip in the UV signal was produced. Thus, scanning the IR laser while monitoring the signal from the UV laser allowed us to record the mass-resolved IR spectra of the species in the beam. A webcam inside the vacuum chamber enabled a precise alignment of the ablation laser and to monitor the sample at any moment, enhancing the overall performance of the experimental set up.
A critical aspect of this study was to ensure that the conformational landscape was thoroughly explored, as small, chemically non-significant changes in the relative orientation of the interacting molecules, may result in shifts in the IR bands that may derive in incorrect assignments of the experimental spectra. Therefore, to ensure that no important structures were left out, the same computational procedure already tested successfully for similar systems was used [6, 40–42]. It consisted of three stages. First, an automated exploration of the intermolecular potential energy surface was carried out using fast molecular mechanics methods (MMFFs) and two search algorithms: the “Large scales Low Mode” (which uses frequency modes to create new structures) and a Monte Carlo-based search, as implemented in Macromodel (www.schrodinger.com). In a second stage, the structures were inspected using chemical intuition, looking for alternatives which implicate small rotations or changes in the relative position of functional groups. In this way, a large number of structures were obtained, some of them being redundant. Therefore, to compact them into a more manageable number without losing information, they were passed to a clustering algorithm that grouped them into families. Representative structures of each family were then chosen to be subjected to full optimization at the M06-2X/6-311++G(d,p) calculation level as implemented in Gaussian 09 [43], which has proven to yield accurate results for similar systems. A normal mode analysis highlighted the validity of the optimized structures as true minima and also yielded the zero-point energy (ZPE). Thus, the energy values given in this work include the ZPE correction. The basis set superposition error was also estimated using the counterpoise procedure of Boys and Bernard [44].
Finally, the entropy and the Gibbs free energy was calculated for each optimized structure in the 0-700 K interval using the output from the Gaussian calculations and the tools supplied by the NIST (http://www.nist.gov/mml/csd/informatics_research/thermochemistry_script.cfm). A detailed explanation of the procedure can be found in Usabiaga et al. [6].
The calculated structures were named as S-n, where S = β-PhGlc, β-PhGal, (β-PhGlc)2 or (β-PhGal)2 and n = 01, 02, 03… etc., starting from the global minimum at 0 K, i.e., without taking into account the effect of the temperature and using only ΔH. All the calculated structures can be found in Figures S1 to S10 of the Supplementary Material and a list of their energetics can be found in Tables S1 to S4.
The spectroscopy of the monomers was first characterized by Simons and coworkers [45, 46] and later reinvestigated by our group, together with similar modified monomers [6] so it is well known. These studies conclude that the conformational landscape of β-PhGlc presents three stable isomers at the conditions of the molecular expansion. These structures are collected in Figure 1 and show that the structural differences between them lie in the orientation of the hydroxymethyl group, that can either point toward the aromatic ring, join the intramolecular hydrogen bond network or adopt an intermediate orientation. Such structural differences result in small but identifiable shifts in the S1←S0 transition [6] as can be seen in Figure 2. Likewise, β-PhGal also presents several conformers with the global minimum being very similar in both systems. On the other hand, the effect of the epimerization at C4 is already present by changing the conformational energetic ordering: for example, β-PhGal-02 is of the same type as β-PhGlc-03 but it is relatively more stable in β-PhGal.
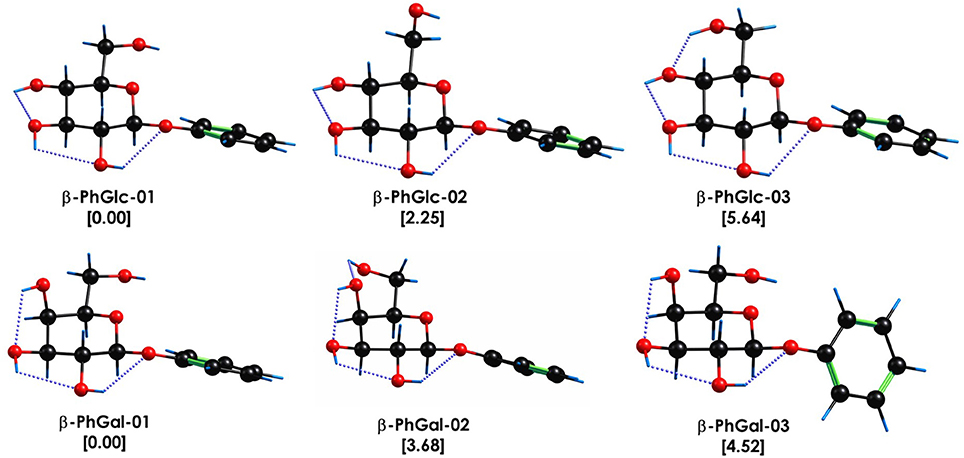
Figure 1. Most stable conformational isomers of β-PhGlc and β-PhGal, together with their relative stability in kJ/mol. Computational level M06-2x/6-311++G(d,p). The dotted blue lines highlight the O-H···O interactions.
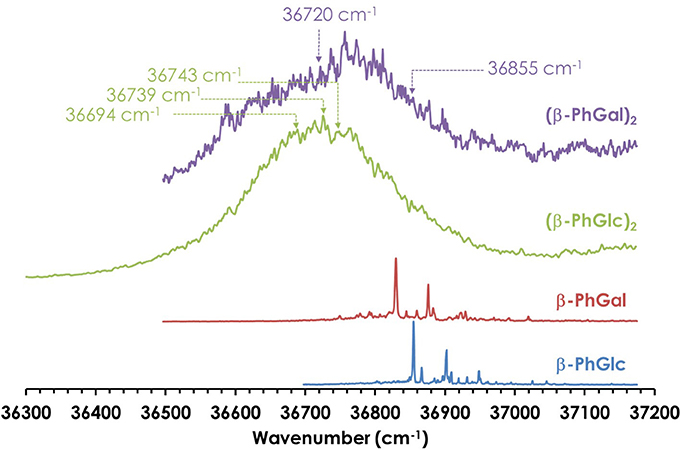
Figure 2. One-color REMPI spectra of β-PhGal, β-PhGlc, (β-PhGal)2 and (β-PhGlc)2. The arrows indicate the wavelengths probed to record the IR spectra.
Figure 2 shows the REMPI spectrum of the homodimers. Their formation resulted in broad absorptions, as expected for such complicated systems. However, no strong shift in the electronic absorption was observed, pointing to similar aggregation energies in the two electronic states implicated in the transition. The absence of discrete transitions in the electronic spectra complicates the study, as it hampers the use of double resonance techniques (UV/UV hole burning) to unveil the presence of several conformational isomers of each aggregate. Instead, the broad absorptions were probed with the UV laser to record the IR/UV double resonance spectra, looking for changes in the shape of the IR spectrum, as it will be shown below.
Despite the absence of discrete features in the REMPI spectrum, it is possible to extract important vibrational information from the system by tuning the laser to different wavelengths and recording the IR spectra (an illustrative example is shown in Figure S7). Figure 3 shows the most meaningful experimental IR spectra recorded tuning the UV laser at 36694, 36739, and 36743 cm−1, together with the predicted spectra for the five most stable computed structures. As can be seen, the shape of the IR spectrum changes depending on the wavenumber probed by the UV laser, indicating that at least two different isomers contribute to the REMPI spectrum. In order to assign the structural candidates, the experimental IR spectra and the predicted IR spectra for the computed structures need to be compared. The experimental spectrum shows some key regions, which give powerful structural information: the bands around ~3650 cm−1 are due to the stretches of the OH moieties with weak interactions (probably only intramolecular interactions). The peak shifts to the red as the interaction of the involved group gets stronger. Thus, the bands between 3450 and 3550 cm−1 are due to the stretching modes of those OHs involved in moderate-strong interactions. Finally, those peaks in the 2850 and 3100 cm−1 interval are due to the CH stretching modes. As expected, the number of bands and shoulders in the experimental spectra is larger than the number of predicted bands for a single isomer, confirming that several conformational isomers were formed in the expansion. The two most stable isomers (see the Discussion section) are required at least to reproduce the experimental trace, but the presence of the third most stable isomer, (β-PhGlc)2-03, cannot be ruled out. Other structures are energetically very high and are not expected to be populated enough or they can isomerize into the most stable structures. An example is shown in Figure S8: isomers 04 and 05 are structurally related to the more stable isomers 01 and 02, and therefore, they are expected to relax into the more stable structures. All the computed isomers can be found in the Supporting Information.
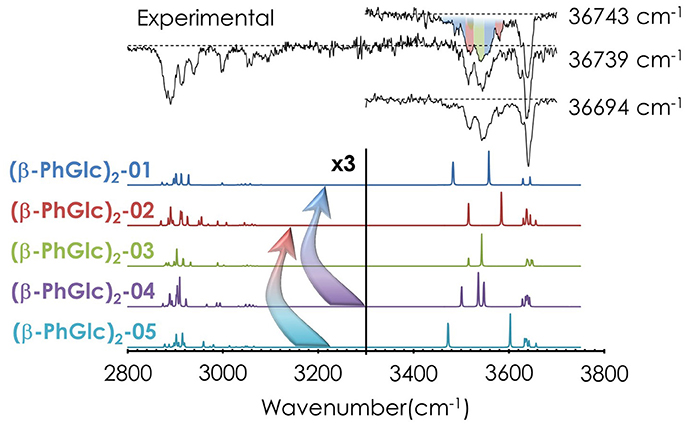
Figure 3. IR spectra of (β-PhGlc)2 and the predicted spectra for the most stable structures, calculated at M06-2X/6-311++G(d,p). The spectrum was obtained probing the indicated transitions. Correction factors of 0.938 and 0.953 for OH and CH stretching modes respectively were employed to account for the anharmonicity. The arrows highlight possible relaxation of less favorable isomers into deeper minima. The colors in the experimental IR spectrum at 36743 cm−1 indicate which bands/absorptions could be assigned to the different predicted bands. The dashed line indicates the baseline.
Similar to the previous case, it is possible extract important vibrational information from the REMPI spectrum in Figure 2 by tuning the laser to different wavelengths. The IR spectra shown in Figure 4 was recorded tuning the UV laser at 36720.2 and 36855.5 cm−1. The predicted spectra for the four most stable computed structures are also shown for comparison. Unfortunately, the spectroscopy of this system is significantly more complex: instead of the presence of bands and shoulders, the OH stretches group into a single, unstructured absorption that extends through the 3400–3650 cm−1 region. Comparison with the computational predictions explains the apparent lack of structure of the spectrum: against what was observed for (β-PhGlc)2, the OH stretches of the spectra predicted for the different (β-PhGal)2 isomers are spread along approximately the same region than in the experimental trace. Assuming that more than one isomer is contributing to the experimental trace, the computed spectra would be in good agreement with the experimental spectrum. In this case, it is only possible to estimate a lower limit for the number of isomers formed: no single conformer is able to solely reproduce the whole absorption in the 3400–3650 cm−1 region and therefore at least two species would be required to build the experimental spectrum, but the presence of additional species cannot be ruled out. Figure S5 of the supplemental material shows the structure of the species whose spectra appear in Figure 4, together with all the calculated structures and their predicted IR spectra.
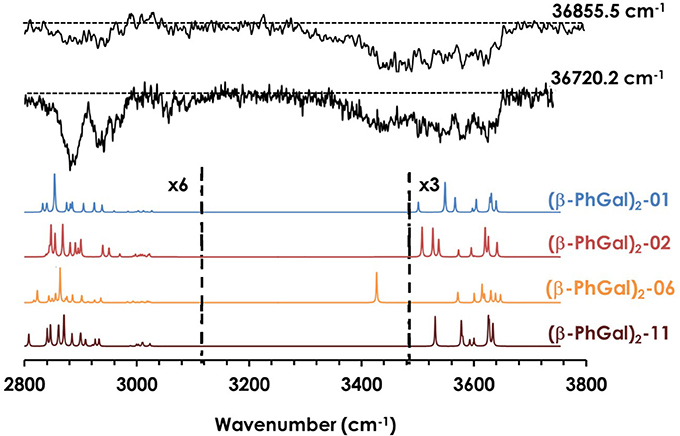
Figure 4. IR spectra of (β-PhGal)2 and the predicted spectra for the most stable structures, calculated at M06-2X/6-311++G(d,p). The spectrum was obtained probing the transitions at 36722.2 and 36855.5 cm−1 shown in Figure 1. Correction factors of 0.938 and 0.953 for OH and CH stretching modes respectively were employed to account for the anharmonicity. The dashed line indicates the baseline.
Both, β-PhGlc and β-PhGal dimers are very challenging systems from an experimental point of view. They are large systems, which are not easily obtainable even by laser ablation methods; they have a low formation propensity and hence, they yield a weak signal; their formation dynamics during the expansion are extremely complicated, resulting in an unstable signal. Furthermore, their spectroscopy is very challenging: aside from the possible large change in geometry and large number of modes, which complicates the REMPI spectra, the IR spectra are very congested and the S/N ratio is not as high as in other systems. Such difficulties hamper proposing a univocal assignment and only, thanks to a joint theory and experimental study it is possible to extract some relevant conclusions.
Additional evidence favoring the assignment proposed is the study of the relative Gibbs free energy of the computed species (Figure 5). Clearly, there are three isomers of (β-PhGlc)2 that are very close in stability in the temperature interval of our expansion (around 150–200 K). Isomers (β-PhGlc)2-01 and (β-PhGlc)2-02 are almost isoenergetic and they should be the most abundant species. Comparison between the Gibbs free energy of binding for those three species shows that isomers (β-PhGlc)2-01 and (β-PhGlc)2-02 are also the most tightly bound. Altogether, one would expect to have mainly isomers (β-PhGlc)2-01 and (β-PhGlc)2-02 in the molecular expansion, in good agreement with the experimental results shown in Figure 3. The most relevant structures are represented in Figure 6.
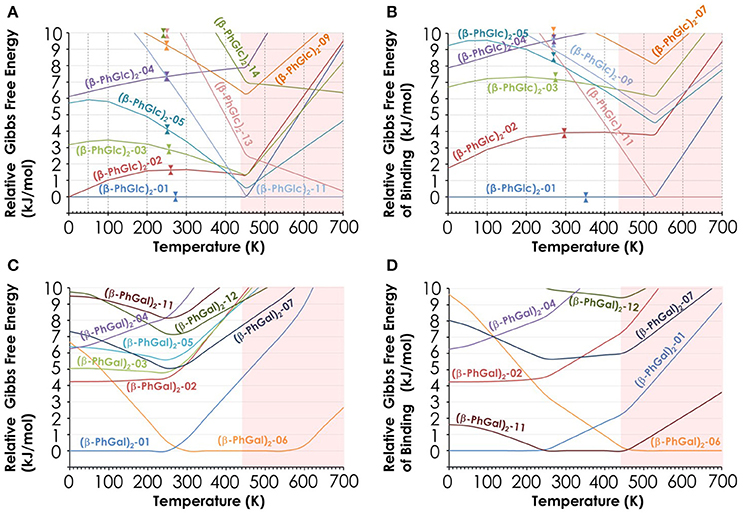
Figure 5. (A) Relative Gibbs free energy of the 10 most stable (β-PhGlc)2 computed species; (B) Relative Gibbs free energy of binding of the same species; (C) Relative Gibbs free energy of the 10 most stable (β-PhGal)2 computed species; (D) Relative Gibbs free energy of binding of the same species. All values in kJ/mol and obtained from the structures computed at the M06-2X/6-311++G(d,p) level.
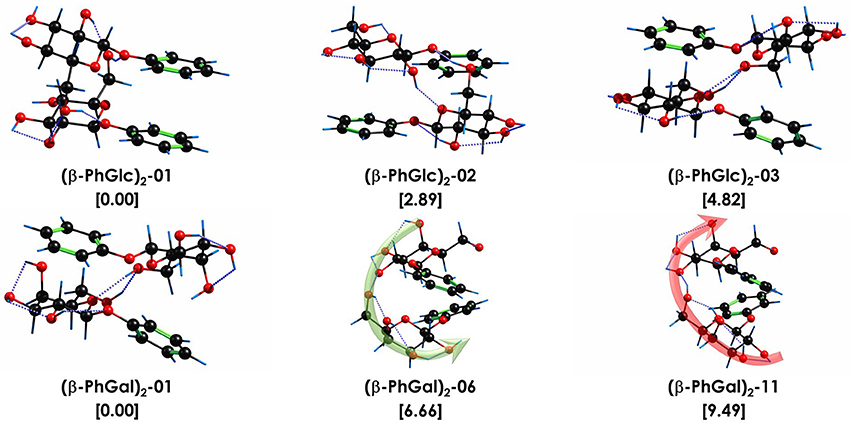
Figure 6. Assigned structures of (β-PhGlc)2 and (β-PhGal)2. Isomer (β-PhGal)2-11 was also included for comparison. Relative stability and binding energy values, in brackets, in kJ/mol. All calculations performed at M06-2X/6-311++G(d,p) level. The dotted blue lines highlight the O-H···O interactions. The green and red arrows point the direction of the hydrogen bond networks.
Regarding (β-PhGal)2, the energetic situation is more interesting: the global minimum is conformer (β-PhGal)2-01, but conformer (β-PhGal)2-06 is favored by entropy and becomes almost isoenergetic at the temperature of the beam. The presence of both conformers also explains all the features in the IR spectrum confirming the detection of these two conformers. A third specie, conformer (β-PhGal)2-02, is slightly higher in energy and its formation in the expansion could not be ruled out. Nevertheless, its population should be low.
When the relative Gibbs free energy of binding is taken into account, conformer (β-PhGal)2-11 comes also into play, presenting a binding energy very similar to the global minimum. Actually, both conformers, (β-PhGal)2-06 and (β-PhGal)2-11, present very similar structures (Figure 6), and the only difference between them is the orientation of the hydrogen bond network, that points in the opposite direction in conformer (β-PhGal)2-11. In principle, the abundance of the monomer's conformer upon which the complex is based must be very small and it is unlikely that the aggregation energy can produce the isomerization into the least stable isomer and therefore, we can rule out the presence of isomer (β-PhGal)2-11 in the expansion.
In previous studies, we concluded that the β anomer of glucose and its derivatives, methyl/phenyl-β-D-glucopiranose, form very stable aggregates with the β anomer of other glucose derivatives, due to their ability to adopt a symmetric structure, in which the hydroxymethyl substituent of each molecule integrates in the hydrogen bond network of the interaction partner. The most stable structure found for (β-PhGlc)2 follows the same pattern and the structure arranges in such a way that the two aromatic rings are stacked and the glucose units interact with each other. However, there is an important difference: for (β-PhGlc)2 there are at least another two stable isomers with arrangements in which each sugar unit interacts with the aromatic ring of the other molecule breaking such a symmetry (see Figure 6). Compared to the other systems, (β-PhGlc)2 minimizes the difference between the global minimum with a stacked configuration (ring-ring/sugar-sugar interactions) and those with sugar-ring interactions. Therefore, the preferences of β anomers of glucose for forming the symmetric aggregates is lost in a great manner and we are able to observe at least one representative structure of each configuration.
Comparison with the structures found for (β-PhGal)2 shows a very different conformational landscape: all three structures are of the sugar-ring interaction type and they present extensive hydrogen bond networks. The structure of the global minimum of (β-PhGal)2 is very similar to that of the third most stable conformer of (β-PhGlc)2, but the enantiomeric difference in the position of the OH makes those structures more favorable due to an extra OH•••π interaction which further stabilizes the dimer. In the second detected structure of (β-PhGal)2, all the OH moieties but one are involved in a single network that jumps from one molecule to the other, resembling a spiral stairway. The (β-PhGal)2 structures show an arrangement more based on intermolecular interactions, while (β-PhGlc)2 structures are more based on intramolecular interactions with few intermolecular interactions. Another relevant information is that the binding energy of all three isomers of (β-PhGal)2 presented in Figure 6 is a ~10% higher than for the (β-PhGlc)2 species.
The above results confirm the importance of epimerization. Comparison between the structures of non-modified β-Glc and β-Gal sugars shows that epimerization at C4 modifies their conformational behavior [8]. For the modified sugars, which can be taken as highly approximated systems to sugar units locked inside a glycan, this locking alters the conformation of the monomers, particularly for the β-PhGal species. Furthermore, such a different behavior of the hydroxyl intramolecular hydrogen bond network of the monomers is amplified in the dimers: the subtle an apparently unimportant change in the position of an OH group not only affects the resulting conformational preferences, but also how the monomers interact with other sugars in their surrounding giving completely different structures with significantly different binding energy. It is interesting how a small difference between twin molecules, such as those in Scheme 1, manifests as huge biological consequences: different structural arrangements means different shape and contact points for the receptors to bind or being recognized; different intra- and inter-molecular interactions are the key of molecular selectivity and (macro)molecular evolution, as they will show different phase and state transition points, different activity, different mobility, different solubility, different pH, or even different morphology and electronic density when interacting with highly sensitive receptors. Applying this point of view to all the biologically relevant molecules, the amount of information that can be encoded using sugar units is astronomic. It seems very difficult to image complex life without organic molecules and their high versatility.
In this work, we combine mass-resolved laser spectroscopy in supersonic expansions and quantum mechanical calculations to model the interactions of phenyl-β-D-glucopyranose and phenyl-β-D-galactopyranose dimers. At least two isomers were detected for both clusters. The most stable structures of phenyl-β-D-glucopyranose shows a competition between those structures with ring-ring/sugar-sugar interactions and those with sugar-ring interactions. Conversely, the most stable structures of phenyl-β-D-galactopyranose dimer show only structures with sugar-ring interactions. The (β-PhGal)2 structures show an arrangement more based on intermolecular interactions, while (β-PhGlc)2 structures are more based on intramolecular interactions with few intermolecular contacts. The effect of epimerization at C4 is amplified in the dimers and changes the conformational behavior so drastically that it should have important biological consequences. The binding energy of the detected phenyl-β-D-galactopyranose dimers is 10% higher than those of phenyl-β-D-glucopyranose dimers. This new information reinforces the idea that small structural changes between sugar units are amplified by formation of intra and intermolecular hydrogen bond networks that help other molecules (proteins, receptors) to easily read the sugar code of glycans.
JF: Conceived the experiment; IU: Made the sample; IU, AC, PÇ, and AI: Performed the experiment; IL, JF, IU, and EC: Analyzed the results; IL and JF: Wrote the manuscript; All the authors gave their approval to the final version of the manuscript.
The research leading to these results has received funding from the Spanish MINECO (CTQ-2014-54464-R and CTQ-2015-68148), FEDER. Computational and laser resources from the SGI/IZO-SGIker network were used for this work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
IU thanks the Basque Government for a pre-doctoral fellowship; IL thanks Junta de Castilla y León for a postdoctoral contract.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2018.00003/full#supplementary-material
1. Lehninger HA, Nelson D, Cox M. Lehninger Principles of Biochemistry. New York, NY: W. H. Freeman (2008).
2. Solís D, Bovin NV, Davis AP, Jiménez-Barbero J, Romero A, Roy R, et al. A guide into glycosciences: how chemistry, biochemistry and biology cooperate to crack the sugar code. Biochim Biophys Acta (2015) 1850:186–235. doi: 10.1016/j.bbagen.2014.03.016
3. Collins PM, Ferrier RJ. Monosaccharides : Their Chemistry and Their Roles in Natural Products. New York, NY: Wiley & Sons (1995).
4. Davis AP, Wareham RS. Carbohydrate recognition through noncovalent interactions: a challenge for biomimetic and supramolecular chemistry. Angew Chem Int Ed. (1999) 38:2978–96.
5. Cocinero EJ, Lesarri A, Écija P, Basterretxea FJ, Grabow JU, Fernández JA, et al. Ribose found in the gas phase. Angew Chem Int Ed. (2012) 51:3119–24. doi: 10.1002/anie.201107973
6. Usabiaga I, González J, Arnáiz PF, León I, Cocinero EJ, Fernández JA. Modeling the tyrosine-sugar interactions in supersonic expansions: glucopyranose-phenol clusters. Phys Chem Chem Phys. (2016) 18:12457–65. doi: 10.1039/c6cp00560h
7. Alonso JL, Lozoya MA, Peña I, López JC, Cabezas C, Mata S, et al. The conformational behaviour of free d-glucose—at last. Chem Sci. (2014) 5:515. doi: 10.1039/C3SC52559G
8. Peña I, Cabezas C, Alonso JL. Unveiling epimerization effects: a rotational study of alpha-D-galactose. Chem Commun. (2015) 51:10115–8. doi: 10.1039/C5CC01783A
9. Ebata T. Study on the structure and vibrational dynamics of functional molecules and molecular clusters by double resonance vibrational spectroscopy. Bull Chem Soc Jpn. (2009) 82:127–51. doi: 10.1246/bcsj.82.127
10. Baquero EE, James WH III, Choi SH, Gellman SH, Zwier TS. Single-conformation ultraviolet and infrared spectroscopy of model synthetic foldamers: beta-peptides Ac-beta3-hPhe-NHMe and Ac-beta3-hTyr-NHMe. J Am Chem Soc. (2008) 130:4784–94. doi: 10.1021/ja078271y
11. Compagnon I, Oomens J, Meijer G, Von Helden G. Mid-infrared spectroscopy of protected peptides in the gas phase: a probe of the backbone conformation. J Am Chem Soc. (2006) 128:3592–7. doi: 10.1021/ja055378h
12. Cocinero EJ, Stanca-Kaposta EC, Gamblin DP, Davis BG, Simons JP. Peptide secondary structures in the gas phase: consensus motif of N-linked glycoproteins. J Am Chem Soc. (2009) 131:1282–7. doi: 10.1021/ja808687j
13. Toroz D, van Mourik T. The structure of the gas-phase tyrosine–glycine dipeptide. Mol Phys. (2006) 104:559–70. doi: 10.1080/00268970500465274
14. Bakker JM, Plutzer C, Hünig I, Häber T, Compagnon I, Von Helden G, et al. Folding structures of isolated peptides as revealed by gas-phase mid-infrared spectroscopy. ChemPhysChem. (2005) 6:120–8. doi: 10.1002/cphc.200400345
15. Gerhards M, Kleinermanns K. Structure and vibrations of phenol(H2O) 2. J Chem Phys. (1995) 103:7392–400.
17. Jaeqx S, Oomens J, Cimas A, Gaigeot MP, Rijs AM. Gas-phase peptide structures unraveled by far-IR spectroscopy: combining IR-UV ion-dip experiments with born-oppenheimer molecular dynamics simulations. Angew Chem Int Ed. (2014) 53:3663–6. doi: 10.1002/anie.201311189
18. Shubert VA, Zwier TS. IR-IR-UV hole-burning: conformation specific IR spectra in the face of UV spectral overlap. J Phys Chem A (2007) 111:13283–6. doi: 10.1021/jp0775606
19. Weiler M, Bartl K, Gerhards M. Infrared/ultraviolet quadruple resonance spectroscopy to investigate structures of electronically excited states. J Chem Phys. (2012) 136:114202.
20. Mons M, Dimicoli I, Piuzzi F, Tardivel B, Elhanine M. Tautomerism of the DNA base guanine and its methylated derivatives as studied by gas-phase infrared and ultraviolet spectroscopy. J Phys Chem A (2002) 106:5088–94. doi: 10.1021/jp0139742
21. Alonso ER, Peña I, Cabezas C, Alonso JL. Structural expression of exo-anomeric effect. J Phys Chem Lett. (2016) 7:845–50. doi: 10.1021/acs.jpclett.6b00028
22. Yoon I, Seo K, Lee S, Lee Y, Kim B. Conformational study of tyramine and its water clusters by laser spectroscopy. J Phys Chem A (2007) 111:1800–7. doi: 10.1021/jp066333l
23. Unamuno I, Fernandez JA, Landajo C, Longarte A, Castaño F. Binding energy and structure of the ground, first electronic and ion states of p-methoxyphenethylamine(H2O)1 isomers: a combined experimental and theoretical study. Chem Phys. (2001) 271:55–69. doi: 10.1016/S0301-0104(01)00421-9
24. Fernández JA, Unamuno I, Longarte A, Castaño F. S0, S1, and Ion I0 Binding Energies of the p-Methoxyphenethylamine(H2O)1-4 Complexes. J Phys Chem A (2001) 105:961–8. doi: 10.1021/jp0027400
25. Unamuno I, Fernández JA, Longarte A, Castaño F. Structural and vibrational assignment of p-methoxyphenethylamine conformers. J Phys Chem A (2000) 104:4364–73. doi: 10.1021/jp9943528
26. Macleod NA, Simons JP. Neurotransmitters in the gas phase: infrared spectroscopy and structure of protonated ethanolamine. Phys Chem Chem Phys. (2004) 6:2821–6. doi: 10.1039/B315536F
27. Macleod NA, Simons JP. Protonated neurotransmitters in the gas-phase: clusters of 2-aminoethanol with phenol. Phys Chem Chem Phys. (2003) 5:1123–9. doi: 10.1039/B212199A
28. Robertson EG, Simons JP. Getting into shape: conformational and supramolecular landscapes in small biomolecules and their hydrated clusters. Phys Chem Chem Phys. (2001) 3:1–18. doi: 10.1039/B008225M
29. León I, Cocinero EJ, Millán J, Jaeqx S, Rijs AM, Lesarri A, et al. Exploring microsolvation of the anesthetic propofol. Phys Chem Chem Phys. (2012) 14:4398–409. doi: 10.1039/c2cp23583h
30. León I, Millán J, Castaño F, Fernández JA. A spectroscopic and computational study of propofol dimers and their hydrated clusters. ChemPhysChem (2012) 13:3819–26. doi: 10.1002/cphc.201200633
31. Fernández-Alonso MC, Cañada FJ, Jiménez-Barbero J, Cuevas G. Molecular recognition of saccharides by proteins. Insights on the origin of the carbohydrate – aromatic interactions. J Am Chem Soc. (2005) 127:7379–86. doi: 10.1021/ja051020+
32. del Carmen Fernández-Alonso M, Díaz D, Berbis, MÁ, Marcelo F, Cañada J, Jiménez-Barbero J. Protein-carbohydrate interactions studied by NMR: from molecular recognition to drug design. Curr Protein Pept Sci. (2012) 13:816–30. doi: 10.2174/138920312804871175.
33. Su Z, Cocinero EJ, Stanca-Kaposta EC, Davis BG, Simons JP. Carbohydrate-aromatic interactions: a computational and IR spectroscopic investigation of the complex, methyl α-L-fucopyranoside•toluene, isolated in the gas phase. Chem Phys Lett. (2009) 471:17–21. doi: 10.1016/j.cplett.2009.02.043
34. Stanca-Kaposta EC, Çarçabal P, Cocinero EJ, Hurtado P, Simons JP. Carbohydrate-aromatic interactions: vibrational spectroscopy and structural assignment of isolated monosaccharide complexes with p-hydroxy toluene and N-acetyl L-tyrosine methylamide. J Phys Chem B (2013) 117:8135–42. doi: 10.1021/jp404527s
35. Cocinero EJ, Çarçabal P, Vaden TD, Davis BG, Simons JP. Exploring carbohydrate-peptide interactions in the gas phase: structure and selectivity in complexes of pyranosides with N-acetylphenylalanine methylamide. J Am Chem Soc. (2011) 133:4548–57. doi: 10.1021/ja109664k
36. Cocinero EJ, Gamblin DP, Davis BG, Simons JP. The building blocks of cellulose: the intrinsic conformational structures of cellobiose, its epimer, lactose, and their singly hydrated complexes. J Am Chem Soc. (2009) 131:11117–23. doi: 10.1021/ja903322w
37. Simons JP, Davis BG, Cocinero EJ, Gamblin DP, Stanca-Kaposta EC. Conformational change and selectivity in explicitly hydrated carbohydrates. Tetrahedron (2009) 20:718–22. doi: 10.1016/j.tetasy.2009.02.032
38. Cocinero EJ, Stanca-Kaposta EC, Dethlefsen M, Liu B, Gamblin DP, Davis BG, et al. Hydration of Sugars in the gas phase: regioselectivity and conformational choice in N-acetyl glucosamine and glucose. Chem A Eur J. (2009) 15:13427–34. doi: 10.1002/chem.200901830
39. Simons JP, Cristina Stanca-Kaposta E, Cocinero EJ, Liu B, Davis BG, Gamblin DP, et al. Probing the glycosidic linkage: secondary structures in the gas phase. Phys Scr. (2008) 78:58124. doi: 10.1088/0031-8949/78/05/058124
40. León I, Millán J, Cocinero EJ, Lesarri A, Fernández JA. Shaping micelles: the interplay between hydrogen bonds and dispersive interactions. Angew Chem Int Ed. (2013) 52:7772–5. doi: 10.1002/anie.201303245
41. León I, Millán J, Cocinero EJ, Lesarri A, Fernández JA. Water encapsulation by nanomicelles. Angew Chem Int Ed. (2014) 53:12480–3. doi: 10.1002/anie.201405652
42. León I, Montero R, Longarte A, Fernández JA. Influence of dispersive forces on the final shape of a reverse micelle. Phys Chem Chem Phys. (2015) 17:2241–5. doi: 10.1039/C4CP03667K
43. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian. Gaussian 09 Rev. D01. Wallingford, CT: Gaussian, Inc. (2012). Available online at: http://www.gaussian.com
44. Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys. (1970) 19:553–66.
45. Talbot FO, Simons JP. Sugars in the gas phase: the spectroscopy and structure of jet-cooled phenyl β-D-glucopyranoside. Phys Chem Chem Phys. (2002) 4:3562–5. doi: 10.1039/B204132D
Keywords: IR spectroscopy, non-covalent interactions, galactose, glucose, hydrogen bond, supersonic expansions, REMPI, electronic spectroscopy
Citation: Usabiaga I, Camiruaga A, Insausti A, Çarçabal P, Cocinero EJ, León I and Fernández JA (2018) Phenyl-β-D-glucopyranoside and Phenyl-β-D-galactopyranoside Dimers: Small Structural Differences but Very Different Interactions. Front. Phys. 6:3. doi: 10.3389/fphy.2018.00003
Received: 06 November 2017; Accepted: 16 January 2018;
Published: 06 February 2018.
Edited by:
Melanie Schnell, Max Planck Society (MPG), GermanyReviewed by:
Tim Schäfer, University of Göttingen, GermanyCopyright © 2018 Usabiaga, Camiruaga, Insausti, Çarçabal, Cocinero, León and Fernández. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iker León, aWtlci5sZW9uQGVodS5lcw==
José A. Fernández, am9zZWEuZmVybmFuZGV6QGVodS5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.