 Xiaoqiong Zhou
Xiaoqiong Zhou Lei Tian
Lei Tian Wenyan Xiong
Wenyan Xiong Yulan Li
Yulan Li Qian Liu
Qian Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 18 March 2025
Sec. Inflammation Pharmacology
Volume 16 - 2025 | https://doi.org/10.3389/fphar.2025.1568246
Hyperoxia therapy is a critical clinical intervention for both acute and chronic illnesses. However, prolonged exposure to high-concentration oxygen can cause lung injury. The mechanisms of hyperoxic lung injury (HLI) remain incompletely understood, and current treatment options are limited. Improving the safety of hyperoxia therapy has thus become an urgent priority. Ferroptosis, a novel form of regulated cell death characterized by iron accumulation and excessive lipid peroxidation, has been implicated in the pathogenesis of HLI, including diffuse alveolar damage, vascular endothelial injury, and bronchopulmonary dysplasia. In this review, we analyze the latest findings on ferroptosis and therapeutic strategies for HLI. Our aim is to provide new insights for the treatment of HLI and to facilitate the translation of these findings from bench to bedside.
Oxygen (O2) therapy is essential for critically or acutely ill patients (Angus, 2020; Helmerhorst et al., 2015). During the COVID-19 pandemic, high-concentration oxygen therapy (hyperoxia, FiO2 ≥ 50%) is one of the few available options to treat hypoxemia-related respiratory failure (Mellado-Artigas et al., 2021; Hanidzir and Robson, 2021). However, hyperoxia is a double-edged sword. Overwhelming evidence from preclinical and clinical studies demonstrates that prolonged exposure to high concentrations of O2 is associated with unfavorable outcomes, increased morbidity, and mortality (Rothen, 2010; Lilien et al., 2022; Staehr-Rye et al., 2017; Girardis et al., 2016; Hong et al., 2021; Ni et al., 2019). The lungs are particularly vulnerable due to their direct exposure to O2. Acute lung injury (ALI) is a major cause of death in patients receiving hyperoxia, and current therapeutic options have limited efficacy. Hyperoxic acute lung injury (HALI) is characterized by alveolar epithelial and pulmonary endothelial damage (Lius and Syafaah, 2022). Hyperoxia is also a well-known cause of chronic lung injuries such as pulmonary interstitial fibrosis and bronchopulmonary dysplasia (BPD)—a chronic lung disease characterized by abnormal lung and pulmonary vascular development (Willis et al., 2020; Tanni et al., 2021; Gilfillan et al., 2021). Despite intensive efforts to elucidate the mechanisms underlying HLI and develop novel therapies, current treatments remain suboptimal due to the poorly understood pathogenesis.
Ferroptosis, a term coined in 2012, is a new form of programmed cell death (PCD) driven by iron-dependent lipid peroxidation on cell membranes, which can be suppressed by iron chelators or small lipophilic antioxidants (Dixon et al., 2012). As a novel form of PCD, ferroptosis is biochemically and morphologically distinct from other classical forms of PCDs, such as apoptosis, necroptosis, pyroptosis, and autophagy (Dixon et al., 2012; Xie et al., 2016). The disruption of iron and lipid metabolism, depletion of antioxidant systems, and mitochondrial dysfunction are the main causes of ferroptosis (Hadian and Stockwell, 2020; Zheng and Conrad, 2020). In recent years, ferroptosis has received increasing attention due to its significant role in various pathological conditions and diseases (Jiang et al., 2021). There is growing evidence that excessive iron accumulation, unbalanced mitochondrial quality control (MQC), reduced antioxidant levels, and increased lipid peroxidation play crucial roles in HLI (Sidramagowda et al., 2020; Mousavi et al., 2011; Ma et al., 2018; Qin et al., 2023). Furthermore, direct evidence suggests that targeting ferroptosis could be an effective therapeutic strategy for treating HLI (Liu C. et al., 2024; Guo et al., 2022; Chou and Chen, 2022a; Jia et al., 2021; Wang et al., 2020).
Given the emerging evidence for the mechanisms of ferroptosis in HLI and the great potential of ferroptosis-targeted therapies for HLI treatment, it is necessary to summarize the latest findings and track the progress in this field. In this paper, we provide a brief overview of the regulatory mechanisms of ferroptosis and summarize the latest research and therapies targeting ferroptosis in HLI. We hope this review can provide an update on advances in HLI pathogenesis and newer therapeutic strategies targeting ferroptosis.
Ferroptosis was first identified as a novel, iron-dependent form of PCD by Stockwell et al., in 2012. This non-apoptotic process, induced by erastin in tumor cells, exhibits characteristic ultrastructural morphological features including mitochondrial shrinkage, increased membrane density, cristae disappearance, and outer membrane rupture (Dixon et al., 2012). As shown in Figure 1, ferroptosis is triggered mainly by three metabolic disorders: excessive intracellular iron accumulation, increased lipid ROS [particularly hydroxyl radicals (OH·)], and decreased activity of antioxidant systems [especially glutathione peroxidase 4 (GPX4)] (Jiang et al., 2021; Yang and Stockwell, 2016). Iron chelators [such as deferoxamine (DFO) and deferiprone] and some small radical-trapping antioxidants (e.g., α-tocopherol, ferrostatin-1, and liproxstatin-1) can prevent it (Zheng and Conrad, 2020; Stockwell, 2022). Numerous studies have revealed that iron overload, lipid peroxidation accumulation, and antioxidant system imbalance are involved in the pathological mechanisms of hyperoxia-induced acute and chronic lung injury, which are primarily characterized by alveolar epithelial cell injury and BPD, respectively. Therefore, targeting ferroptosis may be a potential strategy for treating HLI.
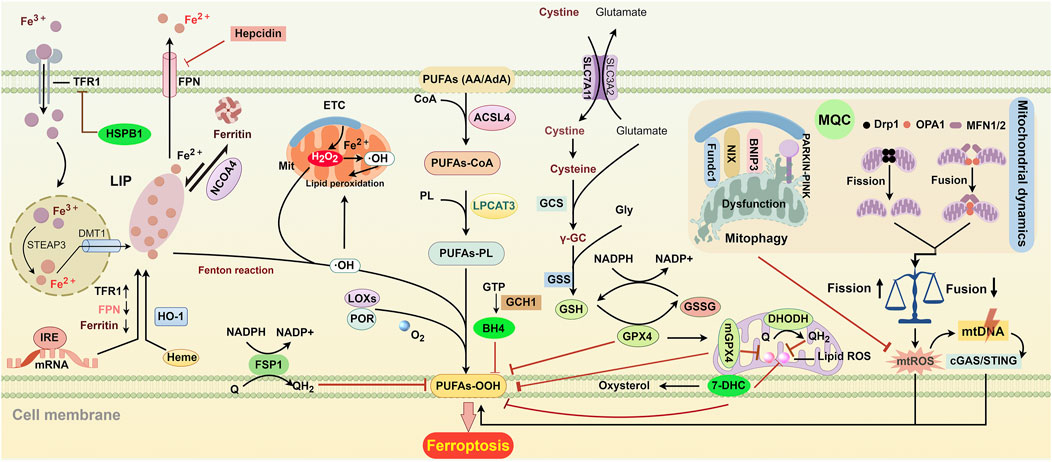
Figure 1. Key regulatory pathways of ferroptosis. ACSL4 and LPCAT3 initiate the biosynthesis of PUFA-PL by incorporating and esterifying PUFAs into phospholipids. Extracellular Fe3+ enters cells through TFR-mediated endocytosis. Once inside, it is reduced to Fe2+ by the enzyme STAEP3 and then transported to the cytoplasm by DMT1. After the release into the cytoplasm, free iron enters the cytosolic labile iron pool (LIP). Fe2+ from the LIP can be stored in ferritin and can be released again through NCOA4-mediated degradation. FPN is responsible for exporting iron to the extracellular space. IRE controls the translation of iron metabolism-related mRNA, including FPN, ferritin, and TFR. The levels of TFR and FPN can also be regulated by HSPB1 and hepcidin, respectively. HO-1 converts heme into biliverdin and iron, which raises intracellular iron levels. Electron leakage from the, ETC., generates O2·− and H2O2, leading to the formation of OH· through Fenton action, which accelerates lipid peroxidation in cellular and mitochondrial membranes. Additionally, LOXs and POR can promote lipid peroxidation by converting PUFAs into hydroperoxides. SLC7A11-mediated cystine transport enhances the synthesis of GSH. GPX4 uses GSH to protect cells from lipid peroxidation and ferroptosis. GSH can enter the mitochondria to form mGPX4, which collaborates with DHODH to help prevent ferroptosis. 7-DHC protects lipids from autoxidation in plasma and mitochondria and prevents ferroptosis. FSP1 is primarily located in plasma membranes, where it converts Q into QH2 to eliminate harmful lipid peroxides. GCH1 suppresses ferroptosis by generating the antioxidant BH4. Mitochondrial dynamics and mitophagy are essential for mitochondrial quality control (MQC). Disruption of MQC can lead to ferroptosis by increasing mtROS, which damages mtDNA. This damaged mtDNA can enter the cytosol and trigger lipid peroxidation through the mtDNA/cGAS/STING pathway. Abbreviations: ACSL4, acyl-CoA synthetase long-chain family member 4; BH4, tetrahydrobiopterin; cGAS/STING, cyclic GMP-AMP synthase-stimulator of interferon genes; 7-DHC, 7-Dehydrocholesterol; DHODH, dihydroorotate dehydrogenase; DMT1, divalent metal transporter 1; Drp1, dynamin-related protein 1; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; Fundc1, FUN14 domain containing 1; GCH1, GTP cyclohydrolase 1; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, glutathione disulfide; HO-1, heme oxygenase 1; IRE, iron response element; HSPB1, heat shock protein beta-1; LOXs, lipoxygenase; LPCAT3, lysophosphatidylcholine acyltransferase 3; MFN1/2, mitofusin-1/2; mGPX4, mitochondrial glutathione peroxidase 4; mtDNA, mitochondrial DNA; NADPH, nicotinamide adenine dinucleotide phosphate; NCOA4, nuclear receptor coactivator 4; OPA1, optic atrophy protein 1; PARKIN, Parkin RBR E3 ubiquitin-protein ligase; PINK1, PTEN-induced kinase 1; PL, phospholipid; POR, P450 oxidoreductase; PUFAs, polyunsaturated fatty acids; Q, ubiquinol; STEAP3, six-transmembrane epithelial antigens of the prostate 3; TFR, transferrin receptor.
Iron is essential for ferroptosis. The iron-catalyzed Fenton reaction generates reactive OH·, which are primary agents of lipid peroxidation. Additionally, iron-containing enzymes such as lipoxygenases (LOXs) and P450 oxidoreductase (POR) promote lipid peroxidation by catalyzing the oxidation of polyunsaturated fatty acids (PUFAs) to hydroperoxides and by transferring electrons from NADPH to O2 (Koppula et al., 2021; Wenzel et al., 2017; Yang et al., 2016; Zou et al., 2020).
Intracellular iron levels are maintained through the balance of uptake, storage, and export processes. Transferrin receptor 1 (TfR1) mediates iron uptake into cells. First, ferric iron (Fe3+) binds to transferrin on the cell membrane to form Tf-Fe3+, which then enters cells via TfR1-dependent endocytosis. In the endosome, Fe3+ is reduced to Fe2+ by six-transmembrane epithelial antigen of prostate 3. Finally, Fe2+ is released into the cytoplasm by divalent metal transporter 1 (DMT1) for storage or utilization (Chen et al., 2020). Knockdown or degradation of TfR1 significantly reduces intracellular Fe2+ levels and prevents ferroptosis (Shao et al., 2024; Zhao et al., 2023). Conversely, elevated expression and stability of TfR1 promote ferroptosis by increasing labile iron accumulation (Yu et al., 2024; Zhou et al., 2024).
Heme oxygenase-1 (HO-1) is another source of intracellular labile iron and can be highly induced by heme and multiple stressors, including oxidative stress, inflammation, and infection (Dunn et al., 2014). HO-1 metabolizes heme into biliverdin, which is rapidly converted to bilirubin, exerting anti-inflammatory and antioxidant effects. Additionally, this process generates Fe2+, which increases labile iron levels and promotes lipid peroxidation (Li X. et al., 2024; Huang et al., 2024; Chen C. et al., 2023; Yang et al., 2023). These properties confer a dual role on HO-1 in ferroptosis. Nuclear factor E2-related factor 2 (Nrf2), the most important upstream activator of HO-1, is activated under stressful conditions. The Nrf2/HO-1 axis, a crucial ferroptotic signal transduction pathway, is recognized as the primary defense mechanism against ferroptosis by restoring GPX4 levels and its antioxidant functions (Dodson et al., 2019; Wei et al., 2020). HIF-1α, another critical upstream molecule of HO-1, is activated under hypoxia conditions and primarily mediates pro-ferroptotic effects by causing iron accumulation and lipid peroxidation (Lu et al., 2024; Li et al., 2025; Yang et al., 2025). However, differences in upstream activators do not fully explain the dual role of HO-1 in ferroptosis. Inconsistent findings indicate that the Nrf2/HO-1 axis may promote ferroptosis, while the HIF-1α/HO-1 axis may inhibit it (Wei et al., 2021; Tang et al., 2021; Yao et al., 2024; Shi et al., 2024). Accumulating evidence suggests that HO-1 levels must be tightly regulated for cytoprotective effects. In disorders with long-term HO-1 expression, such as cancer, fibrosis, Alzheimer’s disease, diabetic nephropathy, and cerebral hemorrhage, HO-1 exacerbates ferroptosis by causing Fe2+ overproduction and ROS generation (Chang et al., 2018; Ryter and Tyrrell, 2000; Feng et al., 2021; Choi and Kim, 2022; Maus et al., 2023).
Intracellular free iron can join the cytosolic labile iron pool (LIP), a poorly defined dynamic pool of Fe2+ bound to low-affinity chelators like glutathione (GSH) and poly (rC)-binding proteins. This unstable binding makes LIP a source of cellular free iron. Iron from the LIP can also be stored in ferritin, a heteropolymer composed of FTH and light ferritin (FTL) chains, providing a relatively safe storage form (Koorts and Viljoen, 2007). Ferritin is considered a protective factor against ferroptosis, whereas its disruption, degradation, or reduced expression increases ferroptosis risk (Zhu et al., 2022; Chittineedi et al., 2023). Ferritin can also be degraded by autophagy, known as ferritinophagy, which is mediated by nuclear receptor coactivator 4 (NCOA4) and increases cellular labile iron content and ferroptosis sensitivity (Jia et al., 2024). On the contrary, NCOA4 knockdown or disruption of the NCOA4-FTH1 interaction inhibits ferritinophagy-mediated ferroptosis by reducing cytosolic iron levels (Wang et al., 2023; Yang et al., 2024; Ji et al., 2024; Gao et al., 2024; Song et al., 2024). Thus, ferritin serves both as an iron source and a protective agent against ferroptosis. Targeting ferritin synthesis and degradation may be an important strategy to modulate intracellular iron levels and control cell fate.
Intracellular iron is primarily transported out of the cell in the divalent form by ferroportin (FPN) (Ward and Kaplan, 2012) or be exported via ferritin-containing multivesicular bodies (MVBs)/exosomes, a process facilitated by prominin 2 (Brown et al., 2019). These mechanisms reduce iron accumulation and enhance cellular resistance to ferroptosis.
TfR1, ferritin, and FPN are key regulators of iron uptake, storage, and export, respectively, and are dominated by iron regulatory proteins (IRPs)-dependent post-transcriptional regulation (Hentze et al., 2010). At a low Fe2+ content condition, IRPs bind to the iron response elements (IREs), promoting TfR1 synthesis while inhibiting ferritin and FPN expression. Additionally, TfR1 and FPN levels can be regulated through an IRP-independent manner. Heat shock protein beta-1 (HSPB1) has been shown to inhibit TfR1 expression and reduce intracellular iron concentration, thereby attenuating erastin-induced ferroptosis (Sun et al., 2015). FPN is also regulated by hepcidin, a peptide hormone secreted predominantly by hepatocytes. When hepcidin binds to FPN, it will induce the FPN endocytosis and proteolysis of the FPN-hepcidin complex, thereby modulating iron export (Billesbolle et al., 2020; Nemeth et al., 2004).
Therefore, both direct interventions, such as iron chelation, and indirect strategies targeting iron import, storage, or export, represent promising therapeutic approaches for ferroptosis-related diseases.
It is not surprising that ferroptosis is recognized as a driving factor of HLI because iron-mediated oxidative damage is a hallmark of hyperoxia-induced lung damage. Prior to the introduction of the term ferroptosis, numerous studies had already documented the association between iron- or iron metabolism-related proteins and HLI (Figure 2). For instance, a significant increase in the levels of ferritin light subunit mRNA, which is involved in long-term iron storage, was observed in the lungs following hyperoxic exposure (Ryan et al., 1997). Both ferritin and lactoferrin, which are involved in iron sequestration, were found to increase in lung cells, particularly in alveolar macrophages. This increase contributes to the resistance of hypotransferrinemic mice to HLI (Yang et al., 1999). Additionally, high levels of O2 were found to upregulate HSPB1 mRNA levels, which may reduce cellular iron levels by inhibiting TfR1 expression (Deng et al., 2024) (Figure 2). Extracellular iron has also been reported to induce hyperoxia-dependent HO-1 gene expression in pulmonary endothelial cells (PECs), interpreted to be a compensatory mechanism that can be blocked by the iron chelator desferrioxamine (Fogg et al., 1999). These findings highlight the essential roles of host defense mechanisms in controlling iron-mediated oxidative stress and highlight the link between iron metabolism and HLI.
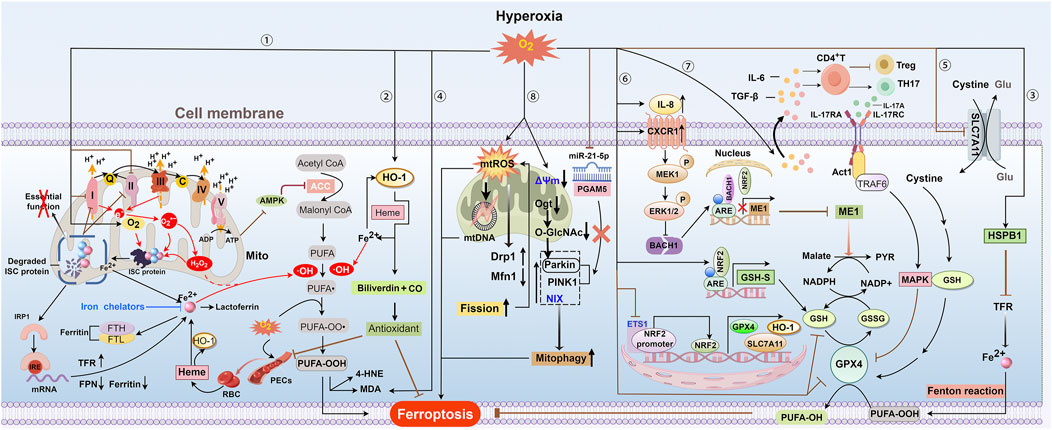
Figure 2. Overview of ferroptosis in hyperoxia. ① and ② Hyperoxia can decrease ATP production by inhibiting oxidative phosphorylation driven by the ETC. This may inhibit ACC by activating AMPK activity through the sensing of the ADP: ATP ratio, which further reduces the synthesis of PUFAs. Hyperoxia disrupts complexes I and II of the ETC, leading to increased production of O2·− and H2O2 due to electron leakage. These reactive species, along with oxygen, can degrade ISC-containing protein complexes, inactivating crucial enzymes and releasing iron. The loss of ISC proteins exacerbates, ETC, dysfunction, further elevating levels of O2·− and H2O2 and causing constant damage to ISC proteins. Reduced ISC levels can boost IRP1 binding to IREs, leading to increased cellular iron uptake by raising TFR expression and lowering ferritin and FPN expression. Excessive H2O2 promotes the generation of OH· through Fenton reactions and increases the peroxidation of PUFAs in plasma and mitochondrial membranes, ultimately leading to ferroptosis. Exposure to hyperoxia increases the vascular permeability of PECs and leads to the leakage of RBCs and their degradation into heme. The heme-induced HO-1 catabolizes heme to iron HO-1, thereby increasing intracellular iron levels. To reduce HLI, excessive iron can enhance HO-1 gene expression and increase ferritin and lactoferrin levels during hyperoxia, thereby providing an antioxidant effect by producing biliverdin. Excess iron increases HO-1 gene expression, producing biliverdin for antioxidant effects, and raises ferritin and lactoferrin levels during hyperoxia, potentially helping to reduce HLI. HO-1 gene expression can also be induced by hyperoxia itself to exert antioxidant effects. ③ Increased oxygen levels enhance HSPB1 mRNA, which may reduce cellular iron by suppressing TFR expression and thus limit ferroptosis. ④ and ⑤ Hyperoxia exposure will inhibit the SLC7A11-GPX4 axis and increase MDA levels, thereby promoting lipid peroxidation. ⑥ Hyperoxia elevates BACH1 expression, which competes with NRF2 for ME1 promoter binding and transcription. This limits malate oxidation to pyruvate and NADPH-dependent GSH production. Hyperoxia suppresses the ETS1/Nrf2 pathway, resulting in decreased levels of downstream antioxidant components such as HO-1, SLC7A11, and GPX4. Hyperoxia might enhance GSH synthesis via the Nrf2-GSH synthetase pathway as a compensatory mechanism. ⑦Hyperoxia induces HALI by initiating IL-17A-mediated ferroptosis in AEC II through the downregulation of GPX4 via the Act1-TRAF6-p38 MAPK pathway. ⑧ Hyperoxia increases the production of mtROS and causes mtDNA damage, which raises sensitivity to ferroptosis. Additionally, mtROS enhances mitochondrial fission by increasing Drp1 and decreasing Mfn1, causing mitochondrial fragmentation and more mtROS production, which in turn accelerates lipid peroxidation and ferroptosis. Hyperoxic exposure leads to the dissipation of mitochondrial membrane potential (Δψ), loss of ATP, and a burst of ROS. Hyperoxia reduces Ogt and O-GlcNAc levels, enhancing Parkin-dependent mitophagy and HLI. PINK1/Parkin and NIX-mediated mitophagy under hyperoxia contribute to BPD pathogenesis and impaired alveolar development. Hyperoxia reduces miR-21–5p levels, enhancing mitophagy and worsening acute lung injury by directly affecting the PGAM5-related PINK1/Parkin pathway. Abbreviations: AMPK, adenosine monophosphate-activated kinase; ARE, antioxidant response element; C, cytochrome c; CXCR1, C-X-C motif chemokine receptor 1; ETS1, E26 oncogene homolog 1; FTH and FTL, heavy and light ferritin chains; GSH-S, GSH synthetase; 4-HNE, 4-hydroxynonenale; IRE, iron response element; IRP, iron regulatory protein; ISC, iron-sulfur cluster; MAPK, p38 mitogen-activated protein kinase; MDA, malondialdehyde; ME1, malic enzyme 1; Ogt, O-GlcNAc transferase; PECs, pulmonary endothelial cells; PYR, pyruvate; RBC, red blood cell.
Vascular injuries have been shown to induce iron accumulation due to the degradation of hemoglobin-derived heme (Maus et al., 2023) (Figure 2). This finding aligns with recent evidence that increased vascular permeability and subsequent hemoglobin infiltration are key pathological features of HLI (Baik et al., 2023). Therefore, hyperoxia-induced vascular leakage may exacerbate HLI via iron-mediated oxidative damage or ferroptosis (Figure 2). Earlier studies revealed that iron chelation with DFO significantly improved lung development in newborn rats exposed to hyperoxia, promoting alveolarization and respiratory surface area expansion (Frank, 1991; Blanco and Frank, 1993). More recently, Wang et al. also demonstrated that intravenous DFO significantly mitigates lung injury caused by short-term hyperoxic mechanical ventilation (Wang et al., 2020). Additionally, a controlled before-after study demonstrated that deferasirox, an iron chelator, reduced oxidative injury markers, including lipid peroxidation, DNA damage, and protein oxidation, as well as iron levels in bronchoalveolar lavage fluid under hyperoxic conditions (Mousavi et al., 2011). However, subsequent research by the same group reported inconsistent results (Ahmadi et al., 2013). Given the small number of patients and the uncontrolled nature of this study, a larger, well-designed randomized controlled trial is needed to robustly evaluate the antioxidant effects of deferasirox in HLI.
With the extensive study of O2 toxicity, a growing body of literature provides a deeper understanding of how hyperoxia affects iron metabolism and ferroptosis (Figure 2). Hyperoxia disrupts mitochondrial electron transport by inhibiting complexes I and II of the electron transport chain (ETC.), leading to electron leakage that generates superoxide (O2·−) through univalent reduction of O2 and subsequent conversion of O2·− to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). Both O2·− and H2O2 accelerate the degradation of the iron-sulfur cluster (ISC)-containing protein complexes (Dixon and Stockwell, 2014). Recently, Baik et al. showed that normalizing O2·− and H2O2 levels is insufficient to prevent hyperoxia-induced degradation of specific ISC-containing protein complexes, implicating that O2 itself is the likely culprit (Baik et al., 2023). Such degradation will lead to the inactivation of essential enzymes (such as respiratory chain activity and gene regulation) and labile iron release (Dixon and Stockwell, 2014). These events further exacerbate, ETC., dysfunction, increasing ROS production and causing cyclic damage to ISC-containing proteins. Additionally, reduced ISC levels can enhance IRP1 binding to IREs, promoting cellular iron uptake by upregulating TfR1 expression and downregulating ferritin and FPN expression (Terzi et al., 2021; Hentze et al., 2010).
Preclinical studies have shown that ferroptosis contributes to the pathogenesis of various types of HLI, including alveolar epithelial injury, vascular endothelial injury, and BPD (Table 1). This process is partially driven by the accumulation of ROS through the iron-driven Fenton reaction, which results from iron deposition or disrupted iron metabolism (Liu L. et al., 2024; Guo et al., 2022; Chou and Chen, 2022a; Jia et al., 2021; Yang et al., 2023; Li X. et al., 2024).
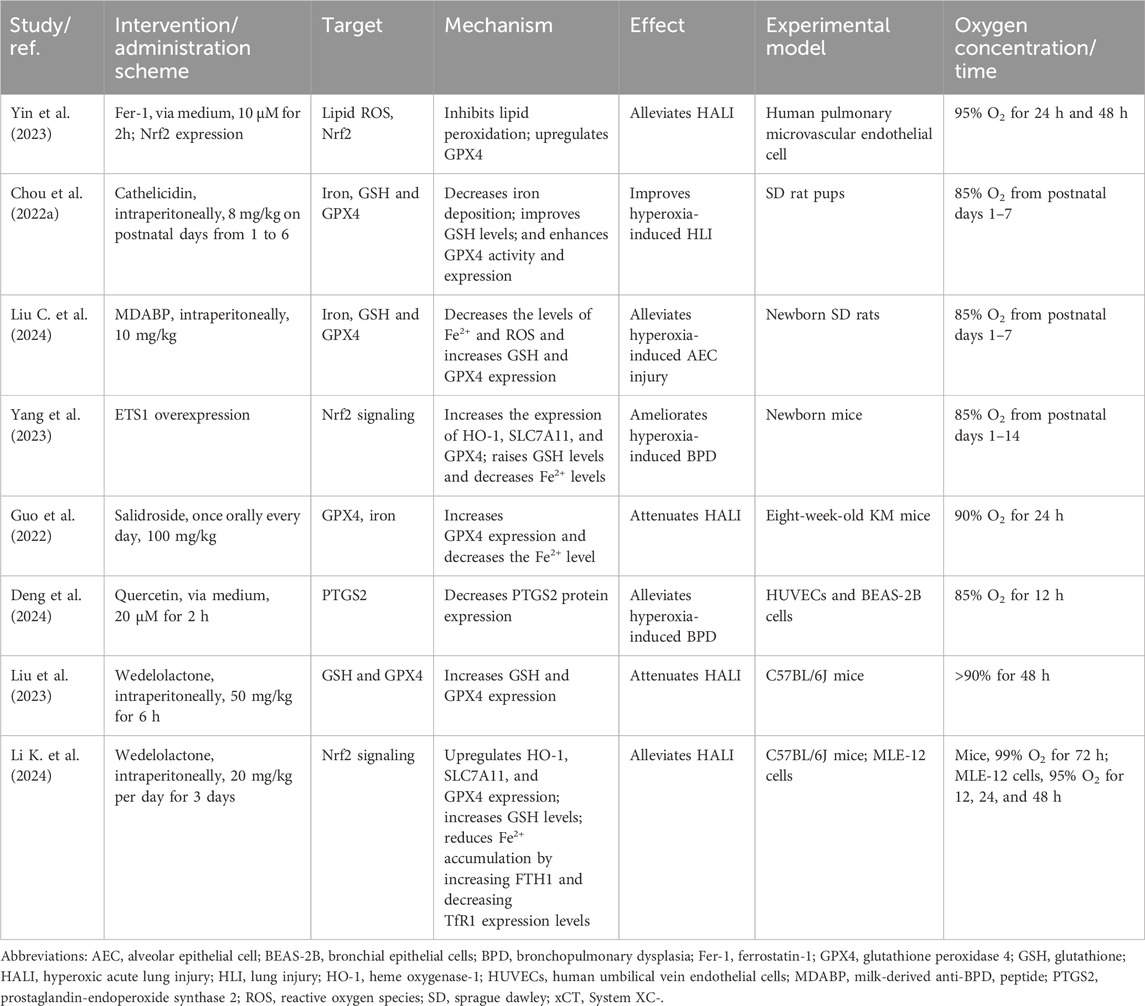
Table 1. Therapeutic strategies targeting ferroptosis in HLI.
In summary, these findings provide a foundation for clinical investigations into the use of iron chelators in HLI treatment and highlight the need for future research on iron-centered mechanisms of ferroptosis in HLI.
Among the anti-ferroptotic defense systems, the GPX4-GSH system plays a predominant role (Figure 1). Cystine, an essential amino acid, is required for the biosynthesis of the antioxidant GSH (Lu, 2009). After being transported into the cell by the solute carrier family 7 member 11 (SLC7A11), cystine is used for GSH biosynthesis via cystine reductase, gamma-glutamyl-cysteine synthetase, and glutathione synthetase (GSH-S) (Figure 1). GPX4 utilizes GSH to convert hydroperoxide to non-toxic lipid alcohols, thereby preventing ferroptosis. This process involves the transition of GSH to oxidized glutathione (GSSG), which can be recycled back to GSH by nicotinamide adenine dinucleotide phosphate (NADPH)-dependent GSH reductase (Figure 1). Inhibition of GSH synthesis through SLC7A11 blockade or deletion (Dixon et al., 2012; Badgley et al., 2020) or inactivation of GPX4 via the GPX4 inhibitor RSL3 can induce ferroptosis (Jiang et al., 2021). Therefore, the GPX4-GSH axis is a potential therapeutic target in ferroptosis-associated diseases.
Recent studies indicated that hyperoxia inhibits the expression and activity of GSH and GPX4 through multiple mechanisms (Figure 2). Evidence from transcriptome sequencing indicates that HALI is closely associated with ferroptosis and GSH metabolism pathways (Qin et al., 2024). In a BPD animal model, hyperoxia significantly reduced GSH and GPX4 levels while increasing malondialdehyde (MDA) levels, a primary product of lipid peroxidation (Ozdemir et al., 2016; Kennedy and Lane, 1994). These changes collectively contribute to the development of HLI. Beyond inducing chronic lung injury, hyperoxia also promotes inflammation and exacerbates ALI by reducing glutathione peroxidase and SOD activities, while simultaneously increasing lipid hydroperoxide levels (Tao et al., 2012; Nagato et al., 2009).
Nrf2 is a crucial transcription factor in the body’s antioxidant defense. Under oxidative stress or inflammation conditions, Nrf2 can bind to antioxidant response elements and activate the expression of various antioxidant genes, such as HO-1, SLC7A11, and GPX4 (Niture et al., 2014) (Figure 2). Nrf2 has been shown to prevent lipid peroxidation and ferroptosis in various diseases like pulmonary fibrosis and LPS-induced lung injury by regulating GSH synthesis and metabolism (Ai et al., 2024; Dodson et al., 2019; Wang C. L. et al., 2024). It has been demonstrated that hyperoxia increases lung GSH synthesis expression via an Nrf2-GSH-S-dependent pathway, which can relieve HALI to some extent (Reddy et al., 2009; Audi et al., 2022) (Figure 2). However, it remains uncertain whether this response is adaptive or compensatory. In addition to regulating GSH synthesis, Nrf2 is involved in NADPH-dependent reduction of GSSG to GSH. Qin et al. demonstrated that hyperoxia inhibits BACH1 degradation that hyperoxia inhibits BACH1 degradation via the MEK1/ERK axis, which is activated by CXCR1/IL-8 signaling. Elevated BACH1 then competes with Nrf2 for binding to the malic enzyme 1 (ME1) promoter, thereby reducing ME1 transcription. This reduction will reduce NADPH production by limiting malate oxidation to pyruvate, thereby exacerbating HALI by lowering GSH levels and causing redox imbalance in PECs (Qin et al., 2023) (Figure 2).
In addition to the indirect evidence, there is growing direct evidence that the dysfunctional GPX4-GSH pathway contributes to HLI by promoting ferroptosis. Four studies have indicated that hyperoxia decreases GSH and/or GPX4 levels, as well as GPX4 activity, without clarifying the underlying mechanisms (Liu R. et al., 2024; Chou and Chen, 2022a; Chou and Chen, 2022b; Liu et al., 2023). Another study linked GSH reduction to hyperoxia-induced SLC7A11 inhibition but did not elucidate the observed decrease in GPX4 levels (Jia et al., 2021). Subsequently, Yin et al. identified Nrf2 as an upstream regulator of GPX4, which prevents ferroptosis and alleviates HLI (Yin et al., 2023). Additionally, Li et al. found that hyperoxia inhibits Nrf2 expression, leading to the downregulation of SLC7A11 and GPX4 and the depletion of GSH (Li K. et al., 2024). E26 oncogene homolog 1 (ETS1) can bind to the Nrf2 promoter region, promoting its transcription. Under hyperoxia, the ETS1/Nrf2 pathway is inhibited, leading to reduced levels of its downstream components HO-1, SLC7A11, and GPX4. This suppression subsequently induces ferroptosis in type II alveolar epithelial (AEC II) cells (Yang et al., 2023) (Figure 2). These findings suggest that Nrf2 is a potential target for treating HLI. In addition to Nrf2, the p38 mitogen-activated protein kinase (p38 MAPK) is involved in ferroptosis in HALI by mediating GPX4 inactivation. p38 MAPK is a key member of the MAPK family and plays a crucial role in regulating inflammatory mediators and ferroptosis (Yong et al., 2009; Hattori et al., 2017). Guo et al. found that hyperoxia exposure increases IL-6 and TGF-β1 levels in AEC II cells, triggering IL-17A release from immune cells. This, in turn, activates the Act1-TRAF6-MAPK pathway, downregulating GPX4 and inducing ferroptosis in HALI (Guo et al., 2022) (Figure 2).
Collectively, these results indicate that impairment of the GPX4-GSH pathway is a critical driver of ferroptosis in various HLIs and a potential common therapeutic target.
PUFAs, such as arachidonic acid and adrenic acid, play a critical role in lipid peroxidation during ferroptosis due to their weak C-H bonds between adjacent C-C double bonds (Conrad and Pratt, 2019). To induce ferroptosis, PUFAs must be incorporated into membrane phospholipids (PLs) to form PUFA-PLs. This process is facilitated by acyl-CoA synthetase long-chain family member 4 (ACSL4), which attaches CoA to PUFAs, and lysophosphatidylcholine acyltransferase 3 (LPCAT3), which integrates PUFA-CoA intermediates into PLs (Doll et al., 2017; Kagan et al., 2017) (Figure 1). ACSL4 deficiency has been reported to inhibit the generation of lipid peroxides and the ferroptosis process (Cui et al., 2021).
Acetyl-CoA carboxylase (ACC) catalyzes the conversion of acetyl-CoA to malonyl-CoA, a key step in the biosynthesis of long-chain fatty acids and the elongation of essential fatty acids, including linoleic and linolenic acids. This process facilitates the formation of longer-chain PUFAs (Beckers et al., 2007). In addition, ACC is also involved in the synthesis of monounsaturated fatty acid (MUFA), which is considered a protective factor in ferroptosis. ACSL3 converts MUFA to MUFA-CoA, which displaces PUFA from phospholipids and inhibits lipid peroxidation (Papsdorf et al., 2023). ACC is regulated by AMP-activated kinase (AMPK), which senses the ADP:ATP ratio and is activated by energy depletion, such as glucose deprivation or hypoxia. Activated AMPK inhibits ACC through phosphorylation, thereby suppressing PUFA synthesis and ferroptosis (Lee et al., 2020). Whether AMPK activation alone can prevent ferroptosis remains to be determined, given its opposing effects on PUFA and MUFA synthesis.
Mitochondria are the primary organelles for ATP production. Hyperoxia inhibits complexes I and II, dissipates mitochondrial membrane potential, and reduces mitochondrial oxidative phosphorylation and ATP production in alveolar epithelial cells, PECs, and lung fibroblasts (Ratner et al., 2009; Das, 2013; Dejmek et al., 2018; Hossain and Eckmann, 2023). These effects may trigger mitochondrial ROS release and activate AMPK, thereby contributing to HLI through the regulation of ferroptosis-associated lipid metabolism and ROS generation (Figure 2).
Mitochondrial dysfunction is associated with increased production of mitochondrial ROS (mtROS) and mitochondrial lipid peroxidation, both of which are key events in ferroptosis (Tian et al., 2024). MQC is essential for maintaining mitochondrial functionality and integrity, thereby promoting cellular survival. Mitochondrial dynamics and mitophagy are crucial components of MQC. Mitochondrial dynamics involve fusion and fission processes regulated by four key proteins: mitofusin-1 (MFN1), mitofusin-2 (MFN2), and optic atrophy protein 1 (OPA1) promote fusion, while dynamin-related protein 1 (Drp1) drives fission (Chen W. et al., 2023) (Figure 1). Mitochondrial fusion facilitates the mixing of partially compromised mitochondria, preserving their normal morphology and function. Conversely, mitochondrial fission favors the generation of smaller mitochondria, enhancing the removal of damaged mitochondria via mitophagy (Adebayo et al., 2021). However, excessive fission and reduced fusion can cause mitochondrial fragmentation and increased mtROS production, which elevate ROS levels and induce lipid peroxidation and ferroptosis (Chen Z. et al., 2023; She et al., 2021; Shi et al., 2022; Chu et al., 2023). Additionally, mtROS can damage mitochondrial DNA (mtDNA), triggering inflammation or autophagy via the cGAS/STING pathway and thereby inducing ferroptosis (Chen W. et al., 2023; Li et al., 2021a) (Figure 1). However, the relationship between ferroptosis and mitochondrial fission/fusion is complex, as these processes can either promote or inhibit ferroptosis by influencing ROS production and lipid peroxidation (Li et al., 2023; Li et al., 2021b).
Mitophagy is a cellular process that selectively degrades mitochondria through the autophagic machinery. It commonly acts as a protective mechanism by eliminating aged, dysfunctional, or damaged mitochondria to limit ROS production (Youle and Narendra, 2011). The PTEN-induced putative kinase 1 (PINK1)/Parkin pathway is crucial for mitophagy. PINK1, a mitochondrial kinase, targets damaged mitochondria to activate Parkin, an E3 ubiquitin ligase, which then ubiquitinates mitochondrial outer membrane proteins to initiate autophagy. Other receptors, such as NIX, BNIP3, and FUNDC1, also contribute to mitophagy (Rodger et al., 2018) (Figure 1). Mitophagy has been shown to inhibit ferroptosis in various pathological conditions, including cisplatin-induced kidney injury, septic cardiac dysfunction, neurodegenerative disorders, and ischemic injuries (Lin et al., 2023; Liu C. et al., 2024; Wang X. X. et al., 2024; Chu et al., 2023). The underlying mechanisms involve reducing mtROS and lipid peroxidation, mitigating GPX4 downregulation by decreasing excessive ROS release and HO-1 expression, inhibiting the mitochondria-localized AMPK-Parkin-ACSL4 signaling pathway, and activating P62-KEAP1-Nrf2 axis to maintain iron and redox homeostasis. However, excessive mitophagy has been found to promote ferroptosis by releasing labile iron, peroxidized lipids, and ROS (Li et al., 2023; Basit et al., 2017; Yu et al., 2022; Wang et al., 2022). Therefore, both inefficient and excessive mitophagy can be harmful. The findings suggest that modulating mitophagy could be a potential therapeutic strategy for ferroptosis-associated diseases.
Mitochondrial dysfunction is a key characteristic of HLI (Figure 2). Ma et al. demonstrated that hyperoxia leads to mtROS-induced mitochondrial fragmentation in PECs by upregulating the pro-fission protein Drp1 and downregulating the pro-fusion protein MFN1 (Ma et al., 2018). Conversely, hyperoxia also increases OPA1 expression, which may act as a compensatory mechanism to limit mitochondrial fragmentation. Moreover, hyperoxia increases mtDNA damage and the expression of pro-autophagy proteins while decreasing PINK1 levels in PECs, suggesting impaired autophagy (Ma et al., 2018). This aligns with subsequent findings that hyperoxia triggers autophagy but inhibits autophagosome clearance, leading to impaired mitochondrial function and barrier integrity in lungs and cultured PECs (Beyer et al., 2021). However, in this research, mitophagy remained unchanged, as hyperoxia raised PINK1 expression but not Parkin. Similarly, another study reported that endothelial PINK1 mediates protective effects during hyperoxia, but Parkin expression was not examined (Zhang et al., 2014). Liu et al. showed that pharmacological activation of mitophagy can reduce hyperoxia-induced AEC II injury in BPD models, whereas hyperoxia alone did not affect mitophagy (Liu R. et al., 2024). Conversely, accumulating evidence suggests mitophagy may aggravate hyperoxia-induced acute and chronic AEC II injury (Yu et al., 2020; Liu et al., 2020; Yu et al., 2023) (Figure 2). Therefore, further studies are needed to evaluate mitophagy flux and its relationship with hyperoxia. Given the close association between MQC and ferroptosis, targeting MQC holds great promise for treating HLI.
As described above, ferroptosis-related mechanisms, such as dysregulation of iron and PUFA metabolism, imbalance in the GPX4-GSH anti-ferroptotic defense system, and MQC, play an important role in HLI. Several specific ferroptosis inhibitors with distinct mechanisms have been identified (Gu et al., 2023). These include iron chelators (DFO, deferiprone), radical-trapping antioxidants (ferrostatin-1, liproxstatin-1), phenothiazine derivatives, ACSL4 inhibitors, and lipoxygenase inhibitors. However, research on ferroptosis inhibitors for HLI treatment is limited, with only one animal study reporting that ferrostatin-1 alleviates HALI by reducing membrane lipid peroxidation (Yin et al., 2023) (Table 1). Thus, additional evidence is needed to further explore this area. Moreover, the development of ferroptosis inhibitors remains in its early stages, with no small molecules yet employed clinically (Gu et al., 2023). This may be attributed to their low efficacy, inadequate selectivity, and severe toxicity.
Cathelicidin, an antimicrobial peptide in the innate immune system, exhibits anti-inflammatory and antioxidant activities (Koon et al., 2011; Ta et al., 2017). Additionally, cathelicidin synthesized in the lung epithelium has antifibrotic effects (Golec et al., 2022; Bals et al., 1998). Recently, bioactive peptides derived from human milk have demonstrated antimicrobial, immunoregulatory, and antioxidant properties (Wada and Lonnerdal, 2020). Ferroptosis, as previously reported, is closely related to redox imbalance and inflammation (Chen C. et al., 2023; Yu et al., 2021; Chen et al., 2024; Stockwell, 2022). These studies demonstrate that cathelicidin and human milk-derived peptides could potentially be used as a therapy for ferroptosis-associated injuries. Indeed, two studies have shown that cathelicidin and a newly identified human milk-derived peptide MDABP can improve lung development and reduce inflammation in neonatal rats (Chou and Chen, 2022b; Liu L. et al., 2024). This is achieved by inhibiting ferroptosis via decreased iron deposition and ROS levels, enhanced GSH levels, and increased activity and expression of GPX4 (Table 1).
Although cathelicidin shows therapeutic potential in preclinical studies for HLI treatment, its clinical application is limited by high synthesis costs, potential toxicity, and poor in vivo stability and targetability (Dzurova et al., 2024). Developing effective drug delivery systems to enhance stability and targeting is critical for clinical application.
ETS1 is a key transcription factor involved in cell proliferation and survival (Kagan et al., 2017). Yang et al. found that ETS1 overexpression alleviates hyperoxia-induced BPD by reducing ROS, MDA, and Fe2+ levels through enhancing Nfr2 transcription and activating downstream anti-ferroptotic pathways such as HO-1, SLC7A11, and GPX4 (Yang et al., 2023) (Figure 2). In addition, Yin et al. demonstrated that suppressing GPX4 by Nrf2 inactivation triggers ferroptosis and promotes HLI in human pulmonary microvascular endothelial cells (Yin et al., 2023). Therefore, genetic activation of Nfr2 expression may alleviate HLI by inhibiting ferroptosis.
Despite its potential, gene therapy targeting ETS1 and Nrf2 faces several limitations. A primary concern is the potential for off-target effects, as these genes are part of complex networks beyond ferroptosis, and their overexpression might disrupt other pathways. While short-term studies show benefits, the long-term efficacy and safety of gene therapy for HLI remain to be established. Addressing these limitations will be crucial for advancing gene therapy as a viable treatment option for HLI.
Most research on treating ferroptosis-associated HLI focuses on natural bioactive products, such as salidroside, quercetin, and wedelolactone (Table 1). Salidroside, the bioactive constituent of Rhodiola rosea, exhibits protective effects on multiple organs, including the lungs, through immunomodulatory and antioxidant mechanisms (Liu et al., 2014; Tang et al., 2016; Cao et al., 2022; Zhang et al., 2009). Guo et al. showed that salidroside prevents IL-17A-mediated ferroptosis in lung epithelial cells by regulating the Act1-TRAF6-p38 MAPK-GPX4 pathway, thereby alleviating HALI (Guo et al., 2022) (Figure 2).
Quercetin, a natural flavonoid found in many fruits and vegetables, exhibits potent anti-inflammatory and antioxidant effects (Qi et al., 2022; Xu et al., 2019). It has been shown to alleviate ferroptosis in various diseases by activating antioxidant defenses, including SLC7A11/GSH/GPX4, HO-1, Nrf2, and SOD2 (Wang et al., 2021; Li et al., 2020; Lin et al., 2022). Oxidative stress and inflammation are key mechanisms in BPD. Deng et al. demonstrated that quercetin alleviates hyperoxia-induced BPD by inhibiting ferroptosis through the MAPK/prostaglandin-endoperoxide synthase 2 (PTGS2) signaling pathway (Deng et al., 2024). Although PTGS2 is a marker of ferroptosis, its role remains controversial. Yang et al. showed that upregulated PTGS2 only signifies the onset of ferroptosis without affecting its progression (Yang et al., 2014). Conversely, Xiao et al. demonstrated that miR-212–5p reduces ferroptotic neuronal death following traumatic brain injury by targeting PTGS2 (Xiao et al., 2019). Another study also found that ferroptosis is linked to PTGS2-encoded cyclooxygenase and subsequent inflammatory processes (Araujo et al., 2018). Whether PTGS2 is a target or merely a downstream marker of ferroptosis remains to be further researched.
Wedelolactone, the primary component of Eclipta prostrata, exhibits various pharmacological effects, including anti-inflammatory, antioxidant, and free radical-scavenging properties (Tu et al., 2021). Fan et al. demonstrated that wedelolactone ameliorates acute pancreatitis-associated lung injury through GPX4-mediated mechanisms, thereby reducing lung failure and ferroptosis (Fan et al., 2021). Liu et al. showed that wedelolactone mitigates HALI by inhibiting ferroptosis through increasing GSH and GPX4 levels (Liu et al., 2023). Subsequently, the same team identified Nrf2 as the upstream regulator of the GPX4-GSH defense system (Li K. et al., 2024). Wedelolactone was also found to activate the Nrf2/HO-1 antioxidant pathway and lower Fe2+ levels by increasing FTH1 and decreasing TfR1 expression (Li X. et al., 2024). These mechanisms work together to inhibit ferroptosis and alleviate HALI.
To date, none of these natural products (quercetin, salidroside, or wedelolactone) have been evaluated in clinical trials specifically for HLI treatment. Among them, quercetin stands out as the most extensively studied in human trials, demonstrating therapeutic benefits in idiopathic pulmonary fibrosis, chronic obstructive pulmonary disease, and COVID-19 (Mirza et al., 2023). However, its clinical translation faces a critical challenge: poor water solubility leading to low oral bioavailability—a limitation shared by salidroside and wedelolactone (Liang et al., 2021; Guan et al., 2021). These compounds’ polar molecular structures and lack of tissue targeting significantly reduce cellular permeability, often necessitating high doses that may elevate toxicity risks. Structural modifications to enhance metabolic stability and bioavailability are therefore imperative for their pharmaceutical development.
Preclinical and clinical data indicate that quercetin and salidroside are generally well-tolerated, with minimal adverse effects in most populations. However, caution is warranted when using these compounds in polypharmacy scenarios due to potential drug interactions in pharmacokinetics and pharmacodynamics (Liang et al., 2021; Günal-K Ro Lu et al., 2025). The safety profile of wedelolactone remains largely uncharacterized, with critical gaps in understanding its effects on human genetics, protein networks, and metabolism.
In summary, while these natural products demonstrate efficacy in experimental HLI settings, their physiological processing in humans may differ substantially from animal models due to species-specific metabolic pathways. Furthermore, the limited natural abundance of these compounds complicates large-scale isolation for rigorous clinical testing. To bridge these gaps, future efforts should prioritize multicenter randomized controlled trials to validate their efficacy and safety in diverse patient populations, alongside chemical engineering strategies to optimize drug-like properties.
In addition to the strategies listed in Table 1, other drugs or interventions for HLI treatment may also exert their effects through anti-ferroptotic mechanisms, as their protective actions partially overlap with those of ferroptosis (Ozdemir et al., 2016; Tao et al., 2012; Liu L. et al., 2024; Yu et al., 2023; Liu et al., 2020).
Adrenomedullin, a potent vasodilatory peptide discovered in 1993, has been increasingly recognized for its multiple roles in modulating the inflammatory response (Elsasser and Kahl, 2002). Tao et al. demonstrated that continuous intravenous infusion of adrenomedullin (0.1 μg/kg/min) attenuates HALI by suppressing oxidative stress and inflammation via enhancement of both glutathione peroxidase (GPX) and SOD activities (Tao et al., 2012).
Dexpanthenol is an alcoholic analog of pantothenic acid (vitamin B5) with antioxidant and anti-inflammatory properties. Ozdemir et al. showed that dexpanthenol (500 mg/kg, intraperitoneally) mitigates lung damage in a BPD animal model, partly by increasing GSH and GPX levels (Ozdemir et al., 2016).
GSH and GPX4 are primary defenses against lipid peroxidation and ferroptosis. Adrenomedullin and dexpanthenol may serve as promising anti-ferroptotic treatments for HLI. However, due to a paucity of direct evidence supporting these assumptions, further research is needed to explore the direct interactions between adrenomedullin, dexpanthenol, and ferroptosis in HLI from both clinical and preclinical perspectives.
Itaconic acid, an unsaturated dicarboxylic acid commonly synthesized from cis-aconitate in the TCA cycle (O'Neill and Artyomov, 2019), exhibits anti-inflammatory properties in the context of severe infections and tissue damage (Lampropoulou et al., 2016). Recently, Liu et al. found that itaconic acid alleviates hyperoxia-induced BPD by promoting TFEB-mediated mitophagy, which helps remove dysfunctional mitochondria and reduces apoptosis in AEC II cells (Liu R. et al., 2024).
O-linked N-acetylglucosamine glycosylation (O-GlcNAcylation) is a posttranslational modification catalyzed by O-GlcNAc transferase (OGT). Yu et al. found that increased O-GlcNAcylation disrupts mitochondrial homeostasis by promoting Parkin-dependent mitophagy in hyperoxia-induced ACE II cell injury (Yu et al., 2023). This effect can be mitigated by the OGT inhibitor OSMI-1 (Figure 2).
PGAM5, a 32 kD mitochondrial protein from the phosphoglycerate mutase family, has been identified as a new mitophagy regulator in the PINK1/Parkin pathway (Lu et al., 2014). Liu et al. showed that miR-21–5p alleviates HALI by inhibiting PINK1/Parkin-dependent mitophagy and mitochondrial damage via direct binding and silencing PGAM5 (Liu et al., 2020) (Figure 2).
Given the dual role of mitophagy in ferroptosis, pharmacological or genetic manipulation of mitophagy may serve as a potential therapeutic strategy for HLI. However, in clinical practice, determining whether mitophagy promotes or inhibits ferroptosis remains challenging. Mitochondrial transplantation may provide a potential solution to this problem.
First, most studies have focused on ferroptosis in neonatal models of hyperoxia-induced BPD. This limitation may affect the generalizability to other populations and types of HLI. Second, ferroptosis mechanisms remain incompletely understood, with only a few identified as contributing to HLI. Therefore, further research is required to enhance the evidence base in this area. Third, the severity and mortality of HLI vary among animal species and do not accurately predict human responses to oxygen toxicity. Fourth, although natural bioactive products show promise due to multiple anti-ferroptotic targets and relatively high safety, they are currently limited to preclinical HLI studies due to their low bioavailability, potential toxicity, and complex pharmacological actions. Future efforts should prioritize optimizing drug-like properties and conducting multicenter trials to validate efficacy and safety. Furthermore, the heterogeneity of HLI may influence the translation of findings from animal models to clinical practice. Acute HLI is primarily characterized by inflammation and loss of the antioxidant defense system whereas chronic HLI involves iron deposition in addition to the aforementioned mechanisms. Cellular heterogeneity has also been observed. Hyperoxia-induced injury in PECs is closely associated with mitochondria-derived ferroptosis, which includes reduced production of NADPH and GSH, impaired mitochondrial oxidative phosphorylation and ATP production, and disrupted mitochondrial dynamics. Future research should focus on elucidating these distinct mechanisms to develop targeted therapies for different types of HLI.
In this review, we summarize the direct and indirect evidence of ferroptosis in HLI pathology. Under hyperoxia, mitochondria serve as both a primary source of ROS and a driver of iron accumulation. Hyperoxia inhibits the, ETC., reducing ATP production and increasing mtROS generation. Additionally, mtROS disturb MQC and exacerbate mtDNA damage, further increasing ROS production. They also contribute to iron overload by degrading ISC proteins, upregulating TFR expression, and downregulating ferritin and FPN through IRP-dependent post-transcriptional regulation. Hyperoxia-induced vascular leakage results in iron accumulation via heme degradation from hemoglobin. In addition to promoting excessive ROS and iron accumulation, hyperoxia also suppresses antioxidant defenses by inhibiting Nrf2 expression, reducing Nrf2 binding to the ME1 promoter, mediating GPX4 deletion via p38 MAPK, and inhibiting the cystine/glutamate antiporter. Collectively, these mechanisms contribute to lipid peroxidation and ferroptosis in HLI.
Hyperoxia also activates compensatory mechanisms to mitigate lung injury by reducing iron overload and alleviating oxidative stress. These mechanisms involve upregulating HSPB1 mRNA, suppressing TfR expression, increasing lactoferrin levels, and enhancing HO-1 expression and GSH synthesis.
The Nrf2 signaling pathway may play an important role in HLI therapy due to its ability to activate multiple antioxidant genes and regulate Fe2+ levels. Among the ferroptotic mechanisms, MQC, iron metabolism, and GPX4-GSH defense system occupy a central position in HLI pathogenesis. Future therapeutic strategies should focus on restoring mitochondrial integrity, modulating iron homeostasis, and enhancing antioxidant capacity to mitigate hyperoxia-induced ferroptosis. Multi-target anti-ferroptotic therapies, especially natural bioactive products, hold great promise for HLI treatment. However, translating these preclinical findings into clinical practice remains challenging and requires further research.
XZ: Writing–original draft, Writing–review and editing. LT: Writing–original draft, Writing–review and editing. WX: Validation, Writing–original draft. YL: Conceptualization, Supervision, Writing–original draft, Writing–review and editing. QL: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Key R&D Project of Zigong Science and Technology Bureau (2021ZC005) and the Key R&D Project of Zigong Health Committee (21zd002).
We appreciate the contributions of all the doctors, colleagues, and friends involved in this study and thank the editors and reviewers for their assistance with this manuscript. We also thank the drawing tools provided by Figdraw (www.figdraw.com).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adebayo, M., Singh, S., Singh, A. P., and Dasgupta, S. (2021). Mitochondrial fusion and fission: the fine-tune balance for cellular homeostasis. FASEB J. 35 (6), e21620. doi:10.1096/fj.202100067R
Ahmadi, A., Mojtahezadeh, M., Javadi, F., Abdollahi, M., Najafi, A., and Mousavi, S. (2013). Role of iron in hyperoxia-induced lung injury: one step forward in iron chelation. J. Pharm. Care. 1 (3), 90–94.
Ai, L., Li, R., Wang, X., Liu, Z., and Li, Y. (2024). Tempol alleviates acute lung injury by affecting glutathione synthesis through Nrf2 and inhibiting ferroptosis in lung epithelial cells. J. Biochem. Mol. Toxicol. 38 (3), e23674. doi:10.1002/jbt.23674
Angus, D. C. (2020). Oxygen therapy for the critically ill. N. Engl. J. Med. 382 (11), 1054–1056. doi:10.1056/NEJMe2000800
Araujo, A. C., Wheelock, C. E., and Haeggstrom, J. Z. (2018). The eicosanoids, redox-regulated lipid mediators in immunometabolic disorders. Antioxid. Redox Signal. 29 (3), 275–296. doi:10.1089/ars.2017.7332
Audi, S. H., Jacobs, E. R., Taheri, P., Ganesh, S., and Clough, A. V. (2022). Assessment of protection offered by the nrf2 pathway against hyperoxia-induced acute lung injury in nrf2 knockout rats. Shock 57 (2), 274–280. doi:10.1097/SHK.0000000000001882
Badgley, M. A., Kremer, D. M., Maurer, H. C., Delgiorio, K. E., Lee, H. J., Purohit, V., et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368 (6486), 85–89. doi:10.1126/science.aaw9872
Baik, A. H., Haribowo, A. G., Chen, X., Quelicini, B. B., Barrios, A. M., Garg, A., et al. (2023). Oxygen toxicity causes cyclic damage by destabilizing specific Fe-S cluster-containing protein complexes. Mol. Cell. 83 (6), 942–960.e9. doi:10.1016/j.molcel.2023.02.013
Bals, R., Wang, X., Zasloff, M., and Wilson, J. M. (1998). The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. U. S. A. 95 (16), 9541–9546. doi:10.1073/pnas.95.16.9541
Basit, F., van Oppen, L. M., Schockel, L., Bossenbroek, H. M., van Emst-de Vries, S., Hermeling, J. C., et al. (2017). Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 8 (3), e2716. doi:10.1038/cddis.2017.133
Beckers, A., Organe, S., Timmermans, L., Scheys, K., Peeters, A., Brusselmans, K., et al. (2007). Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 67 (17), 8180–8187. doi:10.1158/0008-5472.CAN-07-0389
Beyer, A. M., Norwood, T. L., Hughes, W. E., Young, M., Clough, A. V., Gao, F., et al. (2021). Autophagy, TERT, and mitochondrial dysfunction in hyperoxia. Am. J. Physiol. Heart Circ. Physiol. 321 (5), H985–H1003. doi:10.1152/ajpheart.00166.2021
Billesbolle, C. B., Azumaya, C. M., Kretsch, R. C., Powers, A. S., Gonen, S., Schneider, S., et al. (2020). Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 586 (7831), 807–811. doi:10.1038/s41586-020-2668-z
Blanco, L. N., and Frank, L. (1993). The formation of alveoli in rat lung during the third and fourth postnatal weeks: effect of hyperoxia, dexamethasone, and deferoxamine. Pediatr Res. 34 (3), 334–340. doi:10.1203/00006450-199309000-00019
Brown, C. W., Amante, J. J., Chhoy, P., Elaimy, A. L., Liu, H., Zhu, L. J., et al. (2019). Prominin2 drives ferroptosis resistance by stimulating iron export. Dev. Cell. 51 (5), 575–586. doi:10.1016/j.devcel.2019.10.007
Cao, L., Lin, H., Li, Q., Han, S., Yin, H., Zhang, N., et al. (2022). Study on lung injury caused by fine particulate matter and intervention effect of Rhodiola wallichiana. Evid. Based Complement. Altern. Med. 2022, 3693231. doi:10.1155/2022/3693231
Chang, L. C., Chiang, S. K., Chen, S. E., Yu, Y. L., Chou, R. H., and Chang, W. C. (2018). Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137. doi:10.1016/j.canlet.2017.12.025
Chen, B. Y., Pathak, J. L., Lin, H. Y., Guo, W. Q., Chen, W. J., Luo, G., et al. (2024). Inflammation triggers chondrocyte ferroptosis in TMJOA via HIF-1α/TFRC. J. Dent. Res. 103 (7), 712–722. doi:10.1177/00220345241242389
Chen, C., Yang, K., He, D., Yang, B., Tao, L., Chen, J., et al. (2023). Induction of ferroptosis by HO-1 contributes to retinal degeneration in mice with defective clearance of all-trans-retinal. Free Radic. Biol. Med. 194, 245–254. doi:10.1016/j.freeradbiomed.2022.12.008
Chen, W., Zhao, H., and Li, Y. (2023). Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct. Target. Ther. 8 (1), 333. doi:10.1038/s41392-023-01547-9
Chen, X., Yu, C., Kang, R., and Tang, D. (2020). Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 8, 590226. doi:10.3389/fcell.2020.590226
Chen, Z., Sun, X., Li, X., and Liu, N. (2023). Oleoylethanolamide alleviates hyperlipidaemia-mediated vascular calcification via attenuating mitochondrial DNA stress triggered autophagy-dependent ferroptosis by activating PPARα. Biochem. Pharmacol. 208, 115379. doi:10.1016/j.bcp.2022.115379
Chittineedi, P., Mohammed, A., Abdul, R. M., Mat, N. N., and Pandrangi, S. L. (2023). Polyherbal formulation conjugated to gold nanoparticles induced ferroptosis in drug-resistant breast cancer stem cells through ferritin degradation. Front. Pharmacol. 14, 1134758. doi:10.3389/fphar.2023.1134758
Choi, Y. K., and Kim, Y. M. (2022). Beneficial and detrimental roles of heme oxygenase-1 in the neurovascular system. Int. J. Mol. Sci. 23 (13), 7041. doi:10.3390/ijms23137041
Chou, H. C., and Chen, C. M. (2022a). Hyperoxia induces ferroptosis and impairs lung development in neonatal mice. Antioxidants (Basel) 11 (4), 641. doi:10.3390/antiox11040641
Chou, H. C., and Chen, C. M. (2022b). Cathelicidin attenuates hyperoxia-induced lung injury by inhibiting ferroptosis in newborn rats. Antioxidants (Basel) 11 (12), 2405. doi:10.3390/antiox11122405
Chu, C., Wang, X., Yang, C., Chen, F., Shi, L., Xu, W., et al. (2023). Neutrophil extracellular traps drive intestinal microvascular endothelial ferroptosis by impairing Fundc1-dependent mitophagy. Redox Biol. 67, 102906. doi:10.1016/j.redox.2023.102906
Conrad, M., and Pratt, D. A. (2019). The chemical basis of ferroptosis. Nat. Chem. Biol. 15 (12), 1137–1147. doi:10.1038/s41589-019-0408-1
Cui, Y., Zhang, Y., Zhao, X., Shao, L., Liu, G., Sun, C., et al. (2021). ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 93, 312–321. doi:10.1016/j.bbi.2021.01.003
Das, K. C. (2013). Hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in MLE-12 cells and inhibits complex I and II function, but not complex IV in isolated mouse lung mitochondria. PLoS One 8 (9), e73358. doi:10.1371/journal.pone.0073358
Dejmek, J., Kohoutova, M., Kripnerova, M., Cedikova, M., Tuma, Z., Babuska, V., et al. (2018). Repeated exposure to hyperbaric hyperoxia affects mitochondrial functions of the lung fibroblasts. Physiol. Res. 67 (Suppl. 4), S633-S643–S643. doi:10.33549/physiolres.934046
Deng, X., Chen, D., Xie, A., Li, S., Chen, A., Zhou, Q., et al. (2024). Quercetin alleviates hyperoxia-induced bronchopulmonary dysplasia by inhibiting ferroptosis through the MAPK/PTGS2 pathway: insights from network pharmacology, molecular docking, and experimental evaluations. Chem. Biol. Drug Des. 103 (4), e14520. doi:10.1111/cbdd.14520
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10 (1), 9–17. doi:10.1038/nchembio.1416
Dodson, M., Castro-Portuguez, R., and Zhang, D. D. (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107. doi:10.1016/j.redox.2019.101107
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13 (1), 91–98. doi:10.1038/nchembio.2239
Dunn, L. L., Midwinter, R. G., Ni, J., Hamid, H. A., Parish, C. R., and Stocker, R. (2014). New insights into intracellular locations and functions of heme oxygenase-1. Antioxid. Redox Signal. 20 (11), 1723–1742. doi:10.1089/ars.2013.5675
Dzurova, L., Holaskova, E., Pospisilova, H., Schneider, R. G., and Frebortova, J. (2024). Cathelicidins: opportunities and challenges in skin therapeutics and clinical translation. Antibiot. (Basel) 14 (1), 1. doi:10.3390/antibiotics14010001
Elsasser, T. H., and Kahl, S. (2002). Adrenomedullin has multiple roles in disease stress: development and remission of the inflammatory response. Microsc. Res. Tech. 57 (2), 120–129. doi:10.1002/jemt.10058
Fan, R., Sui, J., Dong, X., Jing, B., and Gao, Z. (2021). Wedelolactone alleviates acute pancreatitis and associated lung injury via GPX4-mediated suppression of pyroptosis and ferroptosis. Free Radic. Biol. Med. 173, 29–40. doi:10.1016/j.freeradbiomed.2021.07.009
Feng, X., Wang, S., Sun, Z., Dong, H., Yu, H., Huang, M., et al. (2021). Ferroptosis enhanced diabetic renal tubular injury via HIF-1α/HO-1 pathway in db/db mice. Front. Endocrinol. (Lausanne) 12, 626390. doi:10.3389/fendo.2021.626390
Fogg, S., Agarwal, A., Nick, H. S., and Visner, G. A. (1999). Iron regulates hyperoxia-dependent human heme oxygenase 1 gene expression in pulmonary endothelial cells. Am. J. Respir. Cell Mol. Biol. 20 (4), 797–804. doi:10.1165/ajrcmb.20.4.3477
Frank, L. (1991). Hyperoxic inhibition of newborn rat lung development: protection by deferoxamine. Free Radic. Biol. Med. 11 (4), 341–348. doi:10.1016/0891-5849(91)90149-w
Gao, X., Mao, H., Zhao, L., Li, X., Liao, Y., Li, W., et al. (2024). Nuciferine protects cochlear hair cells from ferroptosis through inhibiting NCOA4-mediated ferritinophagy. Antioxidants (Basel) 13 (6), 714. doi:10.3390/antiox13060714
Gilfillan, M., Bhandari, A., and Bhandari, V. (2021). Diagnosis and management of bronchopulmonary dysplasia. BMJ 375, n1974. doi:10.1136/bmj.n1974
Girardis, M., Busani, S., Damiani, E., Donati, A., Rinaldi, L., Marudi, A., et al. (2016). Effect of conservative vs conventional oxygen therapy on mortality among patients in an intensive care unit: the oxygen-ICU randomized clinical trial. JAMA 316 (15), 1583–1589. doi:10.1001/jama.2016.11993
Golec, M., Lemieszek, M. K., Dutkiewicz, J., Milanowski, J., and Bartelt, S. (2022). A scoping analysis of cathelicidin in response to organic dust exposure and related chronic lung illnesses. Int. J. Mol. Sci. 23 (16), 8847. doi:10.3390/ijms23168847
Gu, Y., Li, Y., Wang, J., Zhang, L., Zhang, J., and Wang, Y. (2023). Targeting ferroptosis: paving new roads for drug design and discovery. Eur. J. Med. Chem. 247, 115015. doi:10.1016/j.ejmech.2022.115015
Guan, F., Wang, Q., Bao, Y., and Chao, Y. (2021). Anti-rheumatic effect of quercetin and recent developments in nano formulation. RSC Adv. 11 (13), 7280–7293. doi:10.1039/d0ra08817j
Günal-K Ro Lu, D., Catalkaya, G., Lu, Y., Bü, Ra, Kezer, G., Esatbeyoglu, T., et al. (2025). Quercetin: potential antidiabetic effects through enzyme inhibition and starch digestibility. Food Saf. Health 3 (1), 9–22. doi:10.1002/fsh3.12066
Guo, B., Zuo, Z., Di, X., Huang, Y., Gong, G., Xu, B., et al. (2022). Salidroside attenuates HALI via IL-17A-mediated ferroptosis of alveolar epithelial cells by regulating Act1-TRAF6-p38 MAPK pathway. Cell Commun. Signal. 20 (1), 183. doi:10.1186/s12964-022-00994-1
Hadian, K., and Stockwell, B. R. (2020). SnapShot: ferroptosis. Cell 181 (5), 1188–1188.e1. doi:10.1016/j.cell.2020.04.039
Hanidzir, D., and Robson, S. C. (2021). Hyperoxia and modulation of pulmonary vascular and immune responses in COVID-19. Am. J. Physiol. Lung Cell Mol. Physiol. 320 (1), L12–L16. doi:10.1152/ajplung.00304.2020
Hattori, K., Ishikawa, H., Sakauchi, C., Takayanagi, S., Naguro, I., and Ichijo, H. (2017). Cold stress-induced ferroptosis involves the ASK1-p38 pathway. EMBO Rep. 18 (11), 2067–2078. doi:10.15252/embr.201744228
Helmerhorst, H. J., Roos-Bloom, M. J., van Westerloo, D. J., and de Jonge, E. (2015). Association between arterial hyperoxia and outcome in subsets of critical illness: a systematic review, meta-analysis, and meta-regression of cohort studies. Crit. Care Med. 43 (7), 1508–1519. doi:10.1097/CCM.0000000000000998
Hentze, M. W., Muckenthaler, M. U., Galy, B., and Camaschella, C. (2010). Two to tango: regulation of Mammalian iron metabolism. Cell 142 (1), 24–38. doi:10.1016/j.cell.2010.06.028
Hong, J. Y., Kim, M. N., Kim, E. G., Lee, J. W., Kim, H. R., Kim, S. Y., et al. (2021). Clusterin deficiency exacerbates hyperoxia-induced acute lung injury. Cells 10 (4), 944. doi:10.3390/cells10040944
Hossain, T., and Eckmann, D. M. (2023). Hyperoxic exposure alters intracellular bioenergetics distribution in human pulmonary cells. Life Sci. 328, 121880. doi:10.1016/j.lfs.2023.121880
Huang, L., Wang, X., Zheng, Y., Lang, D., Wang, J., Yan, S., et al. (2024). EGCG-NPs inhibition HO-1-mediated reprogram iron metabolism against ferroptosis after subarachnoid hemorrhage. Redox Biol. 70, 103075. doi:10.1016/j.redox.2024.103075
Ji, J., Jin, Y., Ma, S., Zhu, Y., Bi, X., You, Q., et al. (2024). Discovery of a NCOA4 degrader for labile iron-dependent ferroptosis inhibition. J. Med. Chem. 67 (15), 12521–12533. doi:10.1021/acs.jmedchem.4c00403
Jia, D., Zhang, M., Li, M., Gong, W., Huang, W., Wang, R., et al. (2024). NCOA4-mediated ferritinophagy participates in cadmium-triggered ferroptosis in spermatogonia. Toxicology 505, 153831. doi:10.1016/j.tox.2024.153831
Jia, D., Zheng, J., Zhou, Y., Jia, J., Ye, X., Zhou, B., et al. (2021). Ferroptosis is involved in hyperoxic lung injury in neonatal rats. J. Inflamm. Res. 14, 5393–5401. doi:10.2147/JIR.S335061
Jiang, X., Stockwell, B. R., and Conrad, M. (2021). Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22 (4), 266–282. doi:10.1038/s41580-020-00324-8
Kagan, V. E., Mao, G., Qu, F., Angelini, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13 (1), 81–90. doi:10.1038/nchembio.2238
Kennedy, K. A., and Lane, N. L. (1994). Effect of in vivo hyperoxia on the glutathione system in neonatal rat lung. Exp. Lung Res. 20 (1), 73–83. doi:10.3109/01902149409064374
Koon, H. W., Shih, D. Q., Chen, J., Bakirtzi, K., Hing, T. C., Law, I., et al. (2011). Cathelicidin signaling via the Toll-like receptor protects against colitis in mice. Gastroenterology 141 (5), 1852–1863. e1-3. doi:10.1053/j.gastro.2011.06.079
Koorts, A. M., and Viljoen, M. (2007). Ferritin and ferritin isoforms I: structure-function relationships, synthesis, degradation and secretion. Arch. Physiol. Biochem. 113 (1), 30–54. doi:10.1080/13813450701318583
Koppula, P., Zhuang, L., and Gan, B. (2021). Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein Cell 12 (9), 675–679. doi:10.1007/s13238-021-00823-0
Lampropoulou, V., Sergushichev, A., Bambouskova, M., Nair, S., Vincent, E. E., Loginicheva, E., et al. (2016). Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24 (1), 158–166. doi:10.1016/j.cmet.2016.06.004
Lee, H., Zandkarimi, F., Zhang, Y., Meena, J. K., Kim, J., Zhuang, L., et al. (2020). Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22 (2), 225–234. doi:10.1038/s41556-020-0461-8
Li, C., Liu, J., Hou, W., Kang, R., and Tang, D. (2021a). STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front. Cell Dev. Biol. 9, 698679. doi:10.3389/fcell.2021.698679
Li, C., Zhang, Y., Liu, J., Kang, R., Klionsky, D. J., and Tang, D. (2021b). Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 17 (4), 948–960. doi:10.1080/15548627.2020.1739447
Li, D., Jiang, C., Mei, G., Zhao, Y., Chen, L., Liu, J., et al. (2020). Quercetin alleviates ferroptosis of pancreatic beta cells in type 2 diabetes. Nutrients 12 (10), 2954. doi:10.3390/nu12102954
Li, J., Jia, Y. C., Ding, Y. X., Bai, J., Cao, F., and Li, F. (2023). The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int. J. Biol. Sci. 19 (9), 2756–2771. doi:10.7150/ijbs.83348
Li, K., Wang, X. Q., Liao, Z. L., Liu, J. Y., Feng, B. H., Ren, Y. C., et al. (2024). Wedelolactone inhibits ferroptosis and alleviates hyperoxia-induced acute lung injury via the Nrf2/HO-1 signaling pathway. Toxicol. Sci. 202 (1), 25–35. doi:10.1093/toxsci/kfae099
Li, R., Wang, X., Zhang, J., Hu, Y., Yang, Y., Zhang, Y., et al. (2025). HIF-1α/HO-1-mediated ferroptosis participates in polystyrene nanoplastics-induced intergenerational cardiotoxicity. Nano Lett. 25 (6), 2226–2235. doi:10.1021/acs.nanolett.4c05372
Li, X., Ran, Q., He, X., Peng, D., Xiong, A., Jiang, M., et al. (2024). HO-1 upregulation promotes mitophagy-dependent ferroptosis in PM2.5-exposed hippocampal neurons. Ecotoxicol. Environ. Saf. 277, 116314. doi:10.1016/j.ecoenv.2024.116314
Liang, C. X., Qi, D. L., Zhang, L. N., Lu, P., and Liu, Z. D. (2021). Preparation and evaluation of a water-in-oil nanoemulsion drug delivery system loaded with salidroside. Chin. J. Nat. Med. 19 (3), 231–240. doi:10.1016/S1875-5364(21)60025-0
Lilien, T. A., Groeneveld, N. S., van Etten-Jamaludin, F., Peters, M. J., Buysse, C., Ralston, S. L., et al. (2022). Association of arterial hyperoxia with outcomes in critically ill children: a systematic review and meta-analysis. JAMA Netw. Open 5 (1), e2142105. doi:10.1001/jamanetworkopen.2021.42105
Lin, Q., Li, S., Jin, H., Cai, H., Zhu, X., Yang, Y., et al. (2023). Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int. J. Biol. Sci. 19 (4), 1192–1210. doi:10.7150/ijbs.80775
Lin, Z. H., Liu, Y., Xue, N. J., Zheng, R., Yan, Y. Q., Wang, Z. X., et al. (2022). Quercetin protects against MPP(+)/MPTP-induced dopaminergic neuron death in Parkinson's disease by inhibiting ferroptosis. Oxid. Med. Cell Longev. 2022, 7769355. doi:10.1155/2022/7769355
Liu, C., Fu, C., Sun, Y., You, Y., Wang, T., Zhang, Y., et al. (2024). Itaconic acid regulation of TFEB-mediated autophagy flux alleviates hyperoxia-induced bronchopulmonary dysplasia. Redox Biol. 72, 103115. doi:10.1016/j.redox.2024.103115
Liu, G., Qian, M., Chen, M., Chen, T., and Qin, S. (2020). miR-21-5p suppresses mitophagy to alleviate hyperoxia-induced acute lung injury by directly targeting PGAM5. Biomed. Res. Int. 2020, 4807254. doi:10.1155/2020/4807254
Liu, J., Qin, S., Feng, B., Chen, M., and Mei, H. (2023). Wedelolactone alleviates hyperoxia-induced acute lung injury by regulating ferroptosis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 35 (11), 1177–1181. doi:10.3760/cma.j.cn121430-20230324-00212
Liu, L., Qian, Y., Yu, Z., Li, H., Chen, J., Dou, H., et al. (2024). Human milk-derived peptide MDABP ameliorates hyperoxia lung damage via inhibiting the ferroptosis signaling pathway. J. Funct. Foods 113, 106036. doi:10.1016/j.jff.2024.106036
Liu, M. W., Su, M. X., Qin, L. F., Liu, X., Tian, M. L., Zhang, W., et al. (2014). Effect of salidroside on lung injury by upregulating peroxisome proliferator-activated receptor gamma expression in septic rats. Exp. Ther. Med. 7 (6), 1446–1456. doi:10.3892/etm.2014.1629
Liu, R., Li, F., Hao, S., Hou, D., Zeng, X., Huang, H., et al. (2024). Low-dose Olaparib improves septic cardiac function by reducing ferroptosis via accelerated mitophagy flux. Pharmacol. Res. 200, 107056. doi:10.1016/j.phrs.2023.107056
Lius, E. E., and Syafaah, I. (2022). Hyperoxia in the management of respiratory failure: a literature review. Ann. Med. Surg. (Lond). 81, 104393. doi:10.1016/j.amsu.2022.104393
Lu, C., Zhang, Z., Fan, Y., Wang, X., Qian, J., and Bian, Z. (2024). Shikonin induces ferroptosis in osteosarcomas through the mitochondrial ROS-regulated HIF-1α/HO-1 axis. Phytomedicine 135, 156139. doi:10.1016/j.phymed.2024.156139
Lu, S. C. (2009). Regulation of glutathione synthesis. Mol. Asp. Med. 30 (1-2), 42–59. doi:10.1016/j.mam.2008.05.005
Lu, W., Karuppagounder, S. S., Springer, D. A., Allen, M. D., Zheng, L., Chao, B., et al. (2014). Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's-like movement disorder. Nat. Commun. 5, 4930. doi:10.1038/ncomms5930
Ma, C., Beyer, A. M., Durand, M., Clough, A. V., Zhu, D., Norwood, T. L., et al. (2018). Hyperoxia causes mitochondrial fragmentation in pulmonary endothelial cells by increasing expression of pro-fission proteins. Arterioscler. Thromb. Vasc. Biol. 38 (3), 622–635. doi:10.1161/ATVBAHA.117.310605
Maus, M., Lopez-Polo, V., Mateo, L., Lafarga, M., Aguilera, M., De Lama, E., et al. (2023). Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat. Metab. 5 (12), 2111–2130. doi:10.1038/s42255-023-00928-2
Mellado-Artigas, R., Ferreyro, B. L., Angrimon, F., Hernandez-Sanz, M., Arruti, E., Torres, A., et al. (2021). High-flow nasal oxygen in patients with COVID-19-associated acute respiratory failure. Crit. Care 25 (1), 58. doi:10.1186/s13054-021-03469-w
Mirza, M. A., Mahmood, S., Hilles, A. R., Ali, A., Khan, M. Z., Zaidi, S. A. A., et al. (2023). Quercetin as a therapeutic product: evaluation of its pharmacological action and clinical applications—a review. Pharm. (Basel) 16 (11), 1631. doi:10.3390/ph16111631
Mousavi, S., Abdollahi, M., Ahmadi, A., Najafi, A., Pazouki, M., Hadjibabaie, M., et al. (2011). The dilemma of hyperoxia following positive pressure mechanical ventilation: role of iron and the benefit of iron chelation with deferasirox. Eur. Rev. Med. Pharmacol. Sci. 15 (10), 1141–1148.
Nagato, A., Silva, F. L., Silva, A. R., Bezerra, F. S., Oliveira, M. L., Bello-Klein, A., et al. (2009). Hyperoxia-induced lung injury is dose dependent in Wistar rats. Exp. Lung Res. 35 (8), 713–728. doi:10.3109/01902140902853184
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306 (5704), 2090–2093. doi:10.1126/science.1104742
Ni, Y. N., Wang, Y. M., Liang, B. M., and Liang, Z. A. (2019). The effect of hyperoxia on mortality in critically ill patients: a systematic review and meta-analysis. BMC Pulm. Med. 19 (1), 53. doi:10.1186/s12890-019-0810-1
Niture, S. K., Khatri, R., and Jaiswal, A. K. (2014). Regulation of Nrf2 - an update. Free Radic. Biol. Med. 66, 36–44. doi:10.1016/j.freeradbiomed.2013.02.008
O'Neill, L., and Artyomov, M. N. (2019). Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 19 (5), 273–281. doi:10.1038/s41577-019-0128-5
Ozdemir, R., Demirtas, G., Parlakpinar, H., Polat, A., Tanbag, K., Taslidere, E., et al. (2016). Dexpanthenol therapy reduces lung damage in a hyperoxic lung injury in neonatal rats. J. Matern. Fetal Neonatal Med. 29 (11), 1801–1807. doi:10.3109/14767058.2015.1064104
Papsdorf, K., Miklas, J. W., Hosseini, A., Cabruja, M., Morrow, C. S., Savini, M., et al. (2023). Lipid droplets and peroxisomes are co-regulated to drive lifespan extension in response to mono-unsaturated fatty acids. Nat. Cell Biol. 25 (5), 672–684. doi:10.1038/s41556-023-01136-6
Qi, W., Qi, W., Xiong, D., and Long, M. (2022). Quercetin: its antioxidant mechanism, antibacterial properties and potential application in prevention and control of toxipathy. Molecules 27 (19), 6545. doi:10.3390/molecules27196545
Qin, H., Zhuang, W., Liu, X., Wu, J., Li, S., Wang, Y., et al. (2023). Targeting CXCR1 alleviates hyperoxia-induced lung injury through promoting glutamine metabolism. Cell Rep. 42 (7), 112745. doi:10.1016/j.celrep.2023.112745
Qin, S., Wang, X., Ren, Y., Feng, B., Liu, J., Yu, H., et al. (2024). Investigation on the signaling pathways in the mechanism of hyperoxia-induced acute lung injury based on transcriptomics sequencing. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 36 (1), 33–39. doi:10.3760/cma.j.cn121430-20230411-00262
Ratner, V., Starkov, A., Matsiukevich, D., Polin, R. A., and Ten, V. S. (2009). Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice. Am. J. Respir. Cell Mol. Biol. 40 (5), 511–518. doi:10.1165/rcmb.2008-0341RC
Reddy, N. M., Kleebeger, S. R., Kensler, T. W., Yamamoto, M., Hassoun, P. M., and Reddy, S. P. (2009). Disruption of Nrf2 impairs the resolution of hyperoxia-induced acute lung injury and inflammation in mice. J. Immunol. 182 (11), 7264–7271. doi:10.4049/jimmunol.0804248
Rodger, C. E., McWilliams, T. G., and Ganley, I. G. (2018). Mammalian mitophagy - from in vitro molecules to in vivo models. FEBS J. 285 (7), 1185–1202. doi:10.1111/febs.14336
Rothen, H. U. (2010). Oxygen: avoid too much of a good thing. Eur. J. Anaesthesiol. 27 (6), 493–494. doi:10.1097/EJA.0b013e3283396360
Ryan, T. P., Krzesicki, R. F., Blakeman, D. P., Chin, J. E., Griffin, R. L., Richards, I. M., et al. (1997). Pulmonary ferritin: differential effects of hyperoxic lung injury on subunit mRNA levels. Free Radic. Biol. Med. 22 (5), 901–908. doi:10.1016/s0891-5849(96)00483-2
Ryter, S. W., and Tyrrell, R. M. (2000). The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 28 (2), 289–309. doi:10.1016/s0891-5849(99)00223-3
Shao, M., Qi, K., Wang, L., Yu, X., Zhang, Q., Yu, L., et al. (2024). E3 ubiquitin ligase CHIP interacts with transferrin receptor 1 for degradation and promotes cell proliferation through inhibiting ferroptosis in hepatocellular carcinoma. Cell Signal 118, 111148. doi:10.1016/j.cellsig.2024.111148
She, H., Hu, Y., Zhou, Y., Tan, L., Zhu, Y., Ma, C., et al. (2021). Protective effects of dexmedetomidine on sepsis-induced vascular leakage by alleviating ferroptosis via regulating metabolic reprogramming. J. Inflamm. Res. 14, 6765–6782. doi:10.2147/JIR.S340420
Shi, J., Song, S., Wang, Y., Wu, K., Liang, G., Wang, A., et al. (2024). Esketamine alleviates ferroptosis-mediated acute lung injury by modulating the HIF-1α/HO-1 pathway. Int. Immunopharmacol. 142 (Pt A), 113065. doi:10.1016/j.intimp.2024.113065
Shi, Y., Han, L., Zhang, X., Xie, L., Pan, P., and Chen, F. (2022). Selenium alleviates cerebral ischemia/reperfusion injury by regulating oxidative stress, mitochondrial fusion and ferroptosis. Neurochem. Res. 47 (10), 2992–3002. doi:10.1007/s11064-022-03643-8
Sidramagowda, P. S., Hernandez-Cuervo, H., Fukumoto, J., Krishnamurthy, S., Lin, M., Alleyn, M., et al. (2020). Alda-1 attenuates hyperoxia-induced acute lung injury in mice. Front. Pharmacol. 11, 597942. doi:10.3389/fphar.2020.597942
Song, J., Zhang, J., Shi, Y., Gao, Q., Chen, H., Ding, X., et al. (2024). Hypoxia inhibits ferritinophagy-mediated ferroptosis in esophageal squamous cell carcinoma via the USP2-NCOA4 axis. Oncogene 43 (26), 2000–2014. doi:10.1038/s41388-024-03050-z
Staehr-Rye, A. K., Meyhoff, C. S., Scheffelbichler, F. T., Vidal, M. M., Gatke, M. R., Walsh, J. L., et al. (2017). High intraoperative inspiratory oxygen fraction and risk of major respiratory complications. Br. J. Anaesth. 119 (1), 140–149. doi:10.1093/bja/aex128
Stockwell, B. R. (2022). Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell 185 (14), 2401–2421. doi:10.1016/j.cell.2022.06.003
Sun, X., Ou, Z., Xie, M., Kang, R., Fan, Y., Niu, X., et al. (2015). HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 34 (45), 5617–5625. doi:10.1038/onc.2015.32
Ta, A., Thakur, B. K., Dutta, P., Sinha, R., Koley, H., and Das, S. (2017). Double-stranded RNA induces cathelicidin expression in the intestinal epithelial cells through phosphatidylinositol 3-kinase-protein kinase Cζ-Sp1 pathway and ameliorates shigellosis in mice. Cell Signal 35, 140–153. doi:10.1016/j.cellsig.2017.03.016
Tang, H., Gao, L., Mao, J., He, H., Liu, J., Cai, X., et al. (2016). Salidroside protects against bleomycin-induced pulmonary fibrosis: activation of Nrf2-antioxidant signaling, and inhibition of NF-κB and TGF-β1/Smad-2/-3 pathways. Cell Stress Chaperones 21 (2), 239–249. doi:10.1007/s12192-015-0654-4
Tang, Z., Ju, Y., Dai, X., Ni, N., Liu, Y., Zhang, D., et al. (2021). HO-1-mediated ferroptosis as a target for protection against retinal pigment epithelium degeneration. Redox Biol. 43, 101971. doi:10.1016/j.redox.2021.101971
Tanni, S. E., Fabro, A. T., de Albuquerque, A., Ferreira, E., Verrastro, C., Sawamura, M., et al. (2021). Pulmonary fibrosis secondary to COVID-19: a narrative review. Expert Rev. Respir. Med. 15 (6), 791–803. doi:10.1080/17476348.2021.1916472
Tao, W., Shu, Y. S., Miao, Q. B., and Zhu, Y. B. (2012). Attenuation of hyperoxia-induced lung injury in rats by adrenomedullin. Inflammation 35 (1), 150–157. doi:10.1007/s10753-011-9300-1
Terzi, E. M., Sviderskiy, V. O., Alvarez, S. W., Whiten, G. C., and Possemato, R. (2021). Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 7 (22), eabg4302. doi:10.1126/sciadv.abg4302
Tian, L., Liu, Q., Guo, H., Zang, H., and Li, Y. (2024). Fighting ischemia-reperfusion injury: focusing on mitochondria-derived ferroptosis. Mitochondrion 79, 101974. doi:10.1016/j.mito.2024.101974
Tu, Y., Yang, Y., Li, Y., and He, C. (2021). Naturally occurring coumestans from plants, their biological activities and therapeutic effects on human diseases. Pharmacol. Res. 169, 105615. doi:10.1016/j.phrs.2021.105615
Wada, Y., and Lonnerdal, B. (2020). Bioactive peptides derived from human milk proteins: an update. Curr. Opin. Clin. Nutr. Metab. Care 23 (3), 217–222. doi:10.1097/MCO.0000000000000642
Wang, C. L., Ren, C. Z., Wang, X. Y., Chen, Q. L., Lyu, X. F., Zhi, X. D., et al. (2024). Ultrafiltration of Angelicae Sinensis Radix and Astragali Radix inhibits ferroptosis and improves pulmonary fibrosis in rats by regulating Nrf2/xCT/GPX4 signaling pathway. Zhongguo Zhong Yao Za Zhi 49 (16), 4338–4346. doi:10.19540/j.cnki.cjcmm.20240409.402
Wang, X., Ma, H., Sun, J., Zheng, T., Zhao, P., Li, H., et al. (2022). Mitochondrial ferritin deficiency promotes osteoblastic ferroptosis via mitophagy in type 2 diabetic osteoporosis. Biol. Trace Elem. Res. 200 (1), 298–307. doi:10.1007/s12011-021-02627-z
Wang, X. X., Li, M., Xu, X. W., Zhao, W. B., Jin, Y. M., Li, L. L., et al. (2024). BNIP3-mediated mitophagy attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting ferroptosis through P62-KEAP1-NRF2 pathway activation to maintain iron and redox homeostasis. Acta Pharmacol. Sin. 46 (1), 33–51. doi:10.1038/s41401-024-01365-x
Wang, X. X., Sha, X. L., Li, Y. L., Li, C. L., Chen, S. H., Wang, J. J., et al. (2020). Lung injury induced by short-term mechanical ventilation with hyperoxia and its mitigation by deferoxamine in rats. BMC Anesthesiol. 20 (1), 188. doi:10.1186/s12871-020-01089-5
Wang, Y., Li, B., Liu, G., Han, Q., Diao, Y., and Liu, J. (2023). Corilagin attenuates intestinal ischemia/reperfusion injury in mice by inhibiting ferritinophagy-mediated ferroptosis through disrupting NCOA4-ferritin interaction. Life Sci. 334, 122176. doi:10.1016/j.lfs.2023.122176
Wang, Y., Quan, F., Cao, Q., Lin, Y., Yue, C., Bi, R., et al. (2021). Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 28, 231–243. doi:10.1016/j.jare.2020.07.007
Ward, D. M., and Kaplan, J. (2012). Ferroportin-mediated iron transport: expression and regulation. Biochim. Biophys. Acta. 1823 (9), 1426–1433. doi:10.1016/j.bbamcr.2012.03.004
Wei, R., Zhao, Y., Wang, J., Yang, X., Li, S., Wang, Y., et al. (2021). Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int. J. Biol. Sci. 17 (11), 2703–2717. doi:10.7150/ijbs.59404
Wei, Y., Lu, M., Mei, M., Wang, H., Chen, M., Yao, H., et al. (2020). Pyridoxine induces glutathione synthesis via PKM2-mediated Nrf2 transactivation and confers neuroprotection. Nat. Commun. 11 (1), 941. doi:10.1038/s41467-020-14788-x
Wenzel, S. E., Tyurina, Y. Y., Zhao, J., St, C. C., Dar, H. H., Mao, G., et al. (2017). PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171 (3), 628–641. doi:10.1016/j.cell.2017.09.044
Willis, K. A., Sieffker, D. T., Aziz, M. M., White, C. T., Mussarat, N., Gomes, C. K., et al. (2020). Perinatal maternal antibiotic exposure augments lung injury in offspring in experimental bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 318 (2), L407–L418. doi:10.1152/ajplung.00561.2018
Xiao, X., Jiang, Y., Liang, W., Wang, Y., Cao, S., Yan, H., et al. (2019). miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol. Brain. 12 (1), 78. doi:10.1186/s13041-019-0501-0
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Differ. 23 (3), 369–379. doi:10.1038/cdd.2015.158
Xu, D., Hu, M. J., Wang, Y. Q., and Cui, Y. L. (2019). Antioxidant activities of quercetin and its complexes for medicinal application. Molecules 24 (6), 1123. doi:10.3390/molecules24061123
Yang, F., Coalson, J. J., Bobb, H. H., Carter, J. D., Banu, J., and Ghio, A. J. (1999). Resistance of hypotransferrinemic mice to hyperoxia-induced lung injury. Am. J. Physiol. 277 (6), L1214–L1223. doi:10.1152/ajplung.1999.277.6.L1214
Yang, M., Chen, Y., Huang, X., Shen, F., and Meng, Y. (2023). ETS1 ameliorates hyperoxia-induced bronchopulmonary dysplasia in mice by activating Nrf2/HO-1 mediated ferroptosis. Lung 201 (4), 425–441. doi:10.1007/s00408-023-00639-1
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U. S. A. 113 (34), E4966–E4975. doi:10.1073/pnas.1603244113
Yang, W. S., Sriramachandran, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156 (1-2), 317–331. doi:10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26 (3), 165–176. doi:10.1016/j.tcb.2015.10.014
Yang, Y., Liu, M., Dong, X., Bai, J., Shi, W., Zhu, Q., et al. (2025). Naringin suppresses CoCl2-induced ferroptosis in ARPE-19 cells. Antioxidants (Basel) 14 (2), 236. doi:10.3390/antiox14020236
Yang, Y., Wu, Q., Shan, X., Zhou, H., Wang, J., Hu, Y., et al. (2024). Ginkgolide B attenuates cerebral ischemia-reperfusion injury via inhibition of ferroptosis through disrupting NCOA4-FTH1 interaction. J. Ethnopharmacol. 318 (Pt B), 116982. doi:10.1016/j.jep.2023.116982
Yao, H., Tian, J., Cheng, S., Dou, H., and Zhu, Y. (2024). The mechanism of hypoxia-inducible factor-1α enhancing the transcriptional activity of transferrin ferroportin 1 and regulating the Nrf2/HO-1 pathway in ferroptosis after cerebral ischemic injury. Neuroscience 559, 26–38. doi:10.1016/j.neuroscience.2024.08.025
Yin, X. T., Luo, H., Shi, J., Chu, X. Y., and Cai, C. (2023). The effect of nuclear factor erythroid 2 related factor 2-induced inhibition of ferroptosis on hyperoxia lung injury. Chin. J. Appl. Clin. Pediatr. 7, 532–537. doi:10.3760/cma.j.cn101070-20220803-00934
Yong, H. Y., Koh, M. S., and Moon, A. (2009). The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs. 18 (12), 1893–1905. doi:10.1517/13543780903321490
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12 (1), 9–14. doi:10.1038/nrm3028
Yu, F., Zhang, Q., Liu, H., Liu, J., Yang, S., Luo, X., et al. (2022). Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 8 (1), 40. doi:10.1038/s41421-022-00390-6
Yu, X., Cheng, L., Liu, S., Wang, M., Zhang, H., Wang, X., et al. (2024). Correlation between ferroptosis and adriamycin resistance in breast cancer regulated by transferrin receptor and its molecular mechanism. FASEB J. 38 (5), e23550. doi:10.1096/fj.202302597R
Yu, X., Liu, D., Li, T., Zheng, H., and Fu, J. (2023). O-linked N-acetylglucosamine affects mitochondrial homeostasis by regulating Parkin-dependent mitophagy in hyperoxia-injured alveolar type II cells injury. Respir. Res. 24 (1), 16. doi:10.1186/s12931-022-02287-0
Yu, X., Sun, Y., Cai, Q., Zhao, X., Liu, Z., Xue, X., et al. (2020). Hyperoxia exposure arrests alveolarization in neonatal rats via PTEN-induced putative kinase 1-Parkin and Nip3-like protein X-mediated mitophagy disorders. Int. J. Mol. Med. 46 (6), 2126–2136. doi:10.3892/ijmm.2020.4766
Yu, Y., Yan, Y., Niu, F., Wang, Y., Chen, X., Su, G., et al. (2021). Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 7 (1), 193. doi:10.1038/s41420-021-00579-w
Zhang, J., Liu, A., Hou, R., Zhang, J., Jia, X., Jiang, W., et al. (2009). Salidroside protects cardiomyocyte against hypoxia-induced death: a HIF-1alpha-activated and VEGF-mediated pathway. Eur. J. Pharmacol. 607 (1-3), 6–14. doi:10.1016/j.ejphar.2009.01.046
Zhang, Y., Sauler, M., Shinn, A. S., Gong, H., Haslip, M., Shan, P., et al. (2014). Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J. Immunol. 192 (11), 5296–5304. doi:10.4049/jimmunol.1400653
Zhao, Y., Zhang, H., Cui, J. G., Wang, J. X., Chen, M. S., Wang, H. R., et al. (2023). Ferroptosis is critical for phthalates driving the blood-testis barrier dysfunction via targeting transferrin receptor. Redox Biol. 59, 102584. doi:10.1016/j.redox.2022.102584
Zheng, J., and Conrad, M. (2020). The metabolic underpinnings of ferroptosis. Cell Metab. 32 (6), 920–937. doi:10.1016/j.cmet.2020.10.011
Zhou, X., Wang, Y., Li, X., Zhou, J., Yang, W., Wang, X., et al. (2024). O-GlcNAcylation regulates the stability of transferrin receptor (TFRC) to control the ferroptosis in hepatocellular carcinoma cells. Redox Biol. 73, 103182. doi:10.1016/j.redox.2024.103182
Zhu, L., You, Y., Zhu, M., Song, Y., Zhang, J., Hu, J., et al. (2022). Ferritin-hijacking nanoparticles spatiotemporally directing endogenous ferroptosis for synergistic anticancer therapy. Adv. Mat. 34 (51), e2207174. doi:10.1002/adma.202207174
Keywords: hyperoxia, lung injury, ferroptosis, therapy, programmed cell death
Citation: Zhou X, Tian L, Xiong W, Li Y and Liu Q (2025) Ferroptosis and hyperoxic lung injury: insights into pathophysiology and treatment approaches. Front. Pharmacol. 16:1568246. doi: 10.3389/fphar.2025.1568246
Received: 29 January 2025; Accepted: 04 March 2025;
Published: 18 March 2025.
Edited by:
Yang Yang, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Xiujie Liu, The University of Nottingham Ningbo, ChinaCopyright © 2025 Zhou, Tian, Xiong, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yulan Li, bGl5dWxAbHp1LmVkdS5jbg==; Qian Liu, bGl1cWlhbnppZ29uZ0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.