 Alexander LenardSimon A. Hermann
Alexander LenardSimon A. Hermann Felicitas StollJuergen BurhenneKathrin I. Foerster
Felicitas StollJuergen BurhenneKathrin I. Foerster David CzockGerd Mikus
David CzockGerd Mikus Andreas D. MeidWalter E. Haefeli
Andreas D. MeidWalter E. Haefeli Antje Blank*
Antje Blank*- Internal Medicine IX, Department of Clinical Pharmacology and Pharmacoepidemiology, Heidelberg University, Medical Faculty Heidelberg/Heidelberg University Hospital, Heidelberg, Germany
Purpose: Pregabalin, gabapentin, and carbamazepine, a potent inducer of cytochrome P450 (CYP) 3A4 and P-glycoprotein, are frequently used antiepileptic drugs that are often administered together with factor Xa inhibitors (FXaI). We aimed to investigate whether potentially clinically relevant drug-drug interactions occur with these combinations.
Methods: In an open-label fixed-sequence trial in 36 healthy volunteers, we evaluated the pharmacokinetics of 60 mg edoxaban and of a microdosed FXaI cocktail (25 µg apixaban, 50 µg edoxaban, and 25 µg rivaroxaban) before and during treatment with carbamazepine (12 evaluable volunteers, individually dosed to therapeutic concentrations), gabapentin (11 volunteers, titrated to 3 × 400 mg/d), and pregabalin (12 volunteers, titrated to 2 × 300 mg/d). The antiepileptics were dosed to steady-state and the CYP3A activity was evaluated by assessing the pharmacokinetics of microdosed midazolam (30 µg).
Results: Carbamazepine reduced the area under the plasma concentration-time curve (
Conclusion: Carbamazepine decreased FXaI exposure to a clinically relevant extent and dose adjustment may be required to maintain an adequate anticoagulant effect, whereas gabapentin and pregabalin do not require dose adjustment of FXaI.
1 Introduction
Direct-acting oral factor Xa inhibitors (FXaI) have achieved high prescription rates since their approval. They are at least as effective as vitamin K antagonists, have a favourable safety profile, an easy-to-follow fixed dosing regimen, and do not require routine monitoring (Giugliano et al., 2013; Granger et al., 2011; Hori et al., 2012; Hylek et al., 2014; Patel et al., 2011; Mao et al., 2014). Due to these favourable characteristics, FXaI are increasingly prescribed to the general population, especially to elderly and also to patients with chronic comorbidities such as neuropathic pain syndromes and seizures. After a stroke, patients often experience post-stroke pain syndromes (Paolucci et al., 2016) and also seizures (Galovic et al., 2021), which is why combination therapies of antiepileptic drugs (AED) and oral anticoagulants are common in these patients.
The anticoagulant effect of FXaI occurs rapidly and is concentration-dependent. As the differentiated dosing instructions for rivaroxaban show, even small dose changes that lead to correspondingly small changes in exposure play a major role in the efficacy and safety of this therapy. Several meta-analyses have revealed an association with an increased risk of all-cause mortality (Shen et al., 2023; Kong et al., 2021; Liu et al., 2021; Zhang et al., 2021) with underdosing, but a more precise understanding of the exact risks associated with off-label underdosing of FXaI is still lacking (Ohno et al., 2021). Increased risks of stroke (Kong et al., 2021; Liu et al., 2021; Zhang et al., 2021; Gronich et al., 2021), cardiovascular hospitalisation (Steinberg et al., 2016), and systemic embolism (Kong et al., 2021; Liu et al., 2021; Zhang et al., 2021; Gronich et al., 2021; Candeloro et al., 2022) have been reported as well as a significantly increased risk for adverse events (Steinberg et al., 2016). In contrast, a subgroup analysis of a Japanese population showed no significant association between any safety parameter and underdosing of FXaI (Murata et al., 2019). Because body weight and body mass index are significant risk factors for both stroke and death (Steinberg et al., 2016; Murata et al., 2019; Nielsen et al., 2017), these results can be confounded by these metrics. Accordingly, the recommended standard dose of rivaroxaban in Japan is 15 mg instead of the usual 20 mg (Murata et al., 2019). Therefore, changes in dose or clearance (CL) immediately translate into exposure and effect changes, and risks can arise from any pharmacokinetic drug-drug interaction (DDI).
Carbamazepine is a known substrate and inducer of CYP3A4 and P-glycoprotein (P-gp, ABCB1) with a relevant auto-induction of its own metabolism (Bertilsson et al., 1980). In patients, only few cases of combinations of enzyme-inducing drugs with direct oral anticoagulants have been reported and their results are heterogenous. As an example, subtherapeutic plasma concentrations and a transient ischemic attack were reported in a patient taking apixaban together with carbamazepine (Di Gennaro et al., 2019). In this single case, therapeutic plasma exposures were subsequently achieved with edoxaban with continued carbamazepine co-medication, but as there is no baseline value for comparison, a DDI between edoxaban and carbamazepine cannot be excluded. In contrast, in another case, discontinuation of carbamazepine had no effect on apixaban trough concentrations (Evanger et al., 2017). According to the summary of product characteristics, the combined use of apixaban with inducers of both CYP3A4 and P-gp such as carbamazepine can lead to a clinically relevant reduction of ∼50% in apixaban exposure (Eliquis, 2024). Indeed, in a large fraction of hospitalized patients (although by no means all) who received enzyme-inducing drugs together with apixaban, the exposure was substantially below the expected range (Perlman et al., 2019; Sennesael et al., 2021). Treatment failure has also been observed during co-administration of the enzyme-inducing phenobarbital with rivaroxaban or apixaban (Dagan et al., 2018). Furthermore, population-based and cohort studies found increased risks of ischemic stroke and other thromboembolic events among patients who were simultaneously treated with AED and FXaI (Ip et al., 2022; Giustozzi et al., 2021).
The pharmacokinetic properties, in particular the CL, differ between the marketed FXaI and depend on the individual characteristics of the transport, metabolic, and excretion pathways involved. Drug transporters such as P-gp, phase-I metabolic enzymes (CYP3A, CYP2J2, and carboxylesterase 1 (CES1)), and phase-II enzymes (e.g., uridine 5′-diphospho -glucuronosyltransferases) play a variable role, as does renal elimination, which accounts for 25%–50% of FXaI CL (Lixiana, 2015; Eliquis, 2024; Xarelto, 2024; Foerster et al., 2020). These differences in the CL pathways determine the DDI potential with modulators of the corresponding CL pathways and make apixaban and rivaroxaban sensitive to CYP modulators, whereas edoxaban is not substantially metabolized by CYP enzymes.
Edoxaban elimination mainly depends on P-gp, polymorphic CES1, and CYP3A4. It is metabolized by CES1 to its active edoxaban metabolite M-4 (M-4). Further metabolisation pathways of M-4 are not yet fully understood but it is substrate to the hepatic liver-specific organic anion transporter 1 (OATP1B1). Apixaban as well as rivaroxaban are biotransformed mainly via CYP3A4. Accordingly, apixaban and rivaroxaban have a cautionary statement on the use of CYP3A4 inducers in their summary of product characteristics (Foerster et al., 2020).
The AED on the market also have different perpetrator properties. Pregabalin and gabapentin have no enzyme-inducing or enzyme-inhibiting effects and their risk for DDI is comparatively low (Summary of product characteristics, 2018; Bockbrader et al., 2010; Wang et al., 2010). They are eliminated by renal excretion, particularly via active tubular secretion involving the OCTN1 (Neurontin, 2018; Bockbrader et al., 2010; Urban et al., 2008; Solvobiotech, 2018). On the other hand, carbamazepine is known for its induction of cytochrome enzymes such as CYP1A2, CYP2C9, and CYP3A4, as the efflux transporter P-gp, but the magnitude of this effect on FXaI exposure has not been well studied and clinical conclusions could be better drawn if the extent of DDI is known and if well-designed physiologically based pharmacokinetic modelling would be available (Candeloro et al., 2022; Puma et al., 2020).
Pharmacokinetic DDI are not class phenomena and need to be examined case by case. The comparability of DDI data for FXaI can be improved by administering different FXaI to the same individual. However, simultaneous administration of therapeutic doses poses a risk and consecutive administration requires long washout periods, which prolongs trial duration. Both challenges can be overcome by using a microdose approach for DDI trials, where more than one FXaI can be administered as a microdose with negligible effects on coagulation (Hohmann et al., 2015; Lenard et al., 2024; Mikus et al., 2019; Mikus et al., 2020). In addition, simultaneous administration to one individual reduces intra-individual and inter-individual variability and allows for a safe and timely study design (Mikus et al., 2019).
We used this approach to assess the effect of therapeutic doses at steady-state of carbamazepine, gabapentin, and pregabalin on the pharmacokinetics of a single therapeutic dose of edoxaban and a microdose cocktail of three FXaI (apixaban, edoxaban, rivaroxaban) and evaluated the utility of the microdosed FXaI cocktail for assessing pharmacokinetic DDI.
2 Materials and methods
2.1 Ethical approval
The trial protocol was approved by the competent authority (BfArM, Bonn, Germany, #4043377), received a positive vote from the responsible Ethics Committee of the Medical Faculty of Heidelberg University, Germany (AFmo-144/2019), and was registered in the EudraCT database (EudraCT 2018-002490-22). This investigator-initiated, monocentre phase-I trial adhered to the Declaration of Helsinki, the principles of good clinical practice, and all pertinent legal requirements in Germany. The trial took place at the DIN EN ISO 9001-certified pharmacological early clinical trial unit (KliPS) of Internal Medicine IX, Department of Clinical Pharmacology and Pharmacoepidemiology at Heidelberg University Hospital.
2.2 Trial population and design
Healthy volunteers aged 18–65 years were eligible to participate in the trial after giving written informed consent. Inclusion and exclusion criteria ensured that the participants were in good health and had no relevant medical history or relevant findings in laboratory values, electrocardiogram, and physical examination. Volunteers were also required to adhere to strict pregnancy prevention measures.
The trial was an open-label, two-period DDI trial with three cohorts of healthy volunteers to assess the impact of carbamazepine, pregabalin, and gabapentin on the pharmacokinetics of 60 mg edoxaban (primary endpoint), given as a tablet, and on the pharmacokinetics of apixaban (25 µg), edoxaban (50 µg), and rivaroxaban (25 µg) administered as an oral microdose cocktail (secondary endpoints, (Mikus et al., 2019)). We simultaneously quantified the effect on CYP3A activity with an oral midazolam microdose (30 µg) (Figure 1).
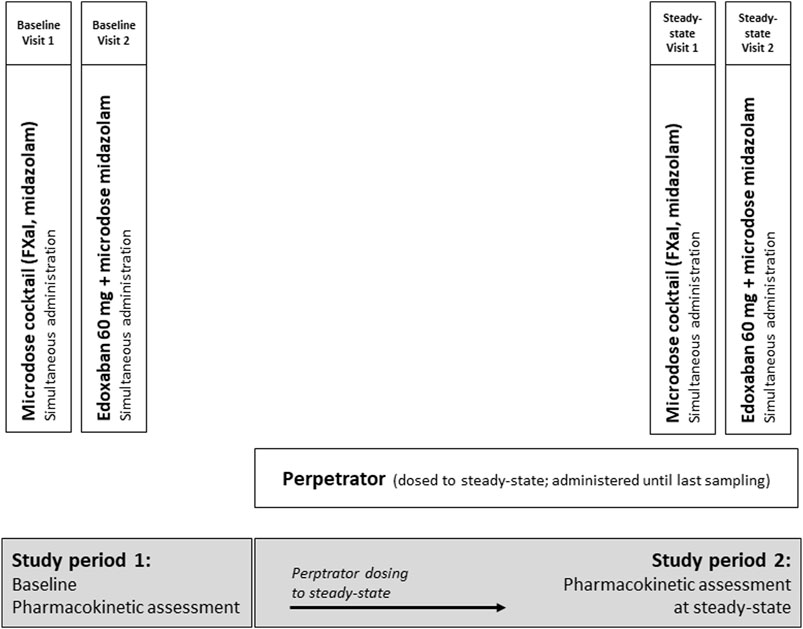
Figure 1. Trial design assessing the impact of carbamazepine, pregabalin, and gabapentin (therapeutic doses, all dosed to steady-state) on the pharmacokinetics of a single oral dose of 60 mg edoxaban and on a microdosed factor Xa inhibitor cocktail, containing 25 μg apixaban, 50 μg edoxaban, and 25 μg rivaroxaban in healthy volunteers. In addition, the effect of perpetrators on microdosed midazolam (30 μg) as marker for CYP3A4 was assessed.
Carbamazepine, gabapentin, and pregabalin were dosed to steady-state during a treatment period of 21 d for carbamazepine, 13 d for gabapentin, and 21 d for pregabalin, followed by the evaluation of their perpetrator effects. Steady-state was ensured until the last blood sample was drawn. Carbamazepine was titrated to a maximum dose of 200 mg twice a day, if possible. If this dose could not be reached due to adverse events, carbamazepine was titrated to therapeutic plasma concentrations (5–10 μg/mL), with the lower range chosen to ensure the safety of the participants. Gabapentin was administered at a dose of 400 mg three times a day and pregabalin at a dose of 300 mg twice a day. Single-dose pharmacokinetics of the microdosed FXaI were assessed at baseline and at the end of the above mentioned treatment period, at steady-state of the perpetrator. The therapeutic 60-mg dose of edoxaban edoxaban was tested thereafter on the following day, both at baseline and at steady-state. All microdosed FXaI and the 60 mg dose of edoxaban were administered under supervision and at least 60 min after administration of the perpetrator. The midazolam microdose was administered as an oral solution simultaneously with administering the FXaI (Katzenmaier et al., 2010; Katzenmaier et al., 2011).
2.3 Quantification of FXaI, edoxaban metabolite M-4, and midazolam
Venous blood samples were drawn before and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 24 (last sampling time after microdoses), and 48 h (edoxaban 60 mg) after drug administration. We assessed the pharmacokinetics of midazolam using a limited sampling strategy with sampling at 2, 2.5, 3, and 4 h post dose (Katzenmaier et al., 2010; Katzenmaier et al., 2011). All collected samples were processed within 20 min and plasma was stored at ≤ −20°C until analysis. Coagulation markers (activated partial thromboplastin time and international normalized ratio) were measured at expected FXaI peak plasma concentrations (Cmax, 3 h post dose) in the accredited central laboratory of Heidelberg University Hospital.
Plasma concentrations of apixaban, edoxaban, edoxaban metabolite 4 (M-4), midazolam, and rivaroxaban were measured with ultra-sensitive, ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) methods utilising accuracy and precision values of less than or equal to ±15% as described previously (Burhenne et al., 2012; Foerster et al., 2018). All methods were validated according to pertinent guidelines (U.S. Food and Drug Administration, 2023; European medicines agency, 2022). In brief, apixaban, edoxaban, edoxaban M-4, and rivaroxaban were extracted from plasma using Waters Oasis HLB Prime µElution 96-well plates by solid phase extraction (SPE). The resulting extracts were gradient chromatographed on a Waters BEH C18 column using water and acetonitrile. Detection was performed on a Waters Xevo TQ-S system (micro dose) or on a Waters TQD system (high dose) in electrospray positive mode and multiple reaction monitoring. The applied lower limits of quantification were 2.5 pg/mL for all microdosed FXaI, and 1 ng/mL for regular doses of edoxaban.
Midazolam was extracted from plasma using Waters Oasis MCX µElution 96-well plates by SPE and the resulting extracts were gradient chromatographed on a Waters BEH C18 column using water and acetonitrile too. Detection was also performed on a Waters Xevo TQ-S system in electrospray positive mode and multiple reaction monitoring. The applied lower limit of quantification was 1 pg/mL for the microdosed midazolam.
2.4 Pharmacokinetic and statistical analysis
Non-compartmental analyses were conducted to assess the pharmacokinetics of the FXaI and midazolam utilizing Phoenix WinNonlin 8.3 (Certara, Inc., Princeton, NJ, United States). Cmax and the time to reach Cmax (tmax) were directly obtained from the concentration data. The area under the concentration-time curve from 0 to infinity (
Parameters are presented as geometric means (GM) with 95% confidence interval (CI). Exposure changes were evaluated by using the geometric mean ratio (GMR) of
3 Results
After providing the participants with comprehensive information and obtaining their written informed consent, we enrolled 36 healthy volunteers (22 females, 14 males), aged 21–63 years (median 26), with a mean body mass index of 23.97 kg/m2 (± standard deviation (SD) 3.79) in the three cohorts of this trial. The cohorts consisted of 13, 11, and 12 volunteers to evaluate the effects of carbamazepine, gabapentin, and pregabalin, respectively. One participant of the carbamazepine cohort did not complete the trial due to an adverse event (hypersensitivity with fever and liver enzyme elevation). Therefore, the carbamazepine cohort consisted of 12 evaluable individuals.
3.1 Effect of carbamazepine on FXaI pharmacokinetics
At carbamazepine steady-state (21 d ± 0 d), full induction by carbamazepine for all participants was assumed. Compared to baseline, the GMR of
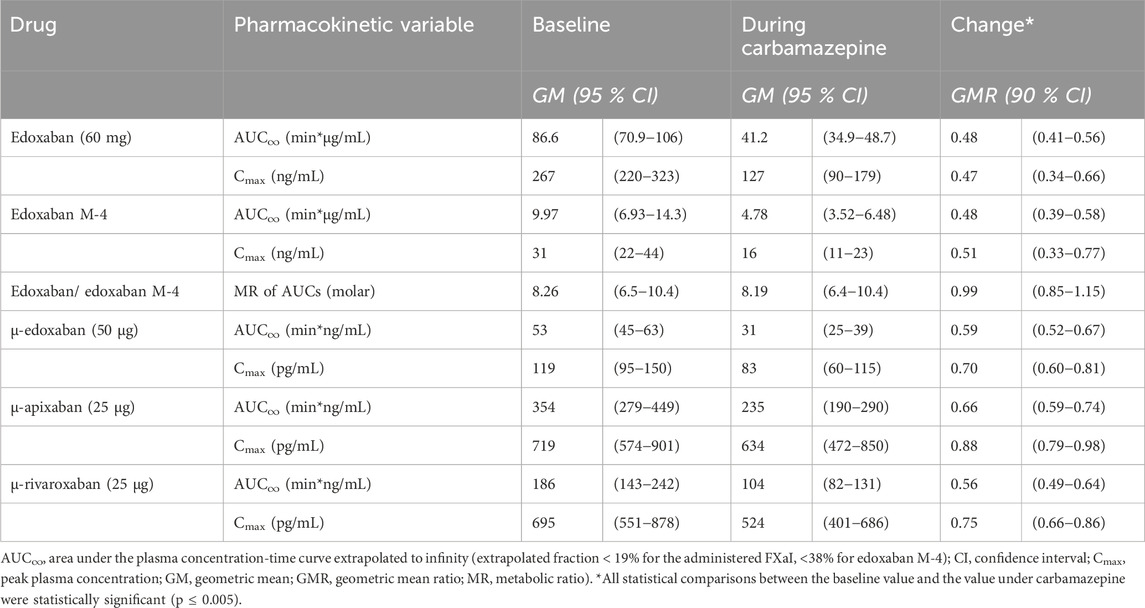
Table 1. Pharmacokinetic parameters of factor Xa inhibitors at baseline and at carbamazepine steady-state (N = 12 healthy volunteers).
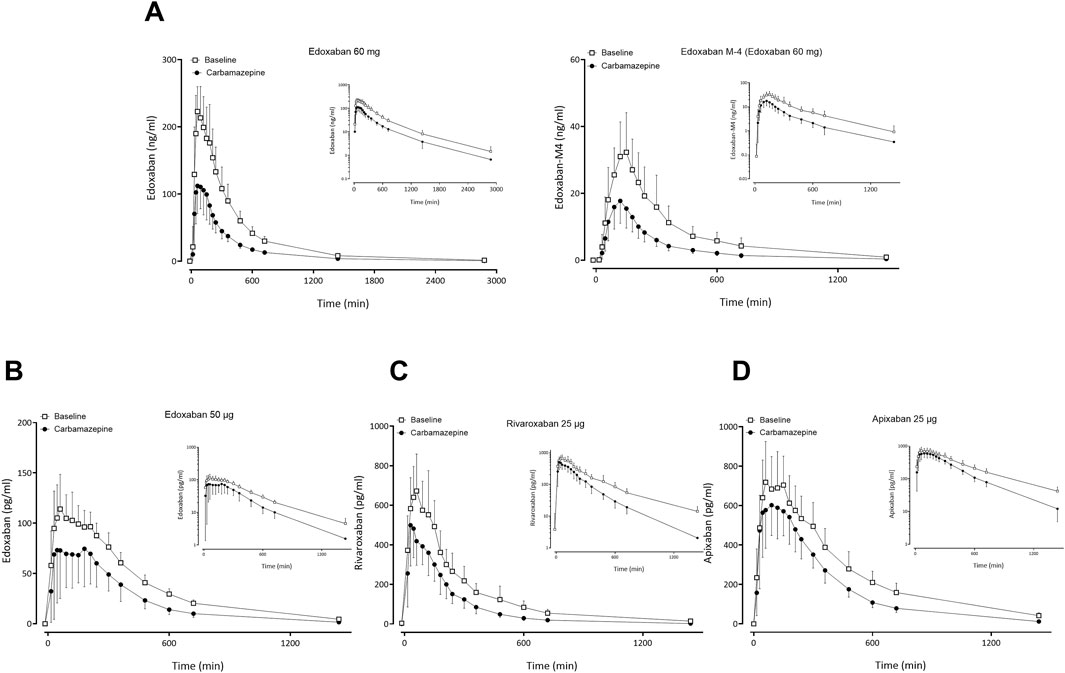
Figure 2. Mean (± standard deviation) plasma concentration-time profiles of factor Xa inhibitors after administration of single oral doses before carbamazepine (open squares) and at carbamazepine steady-state (solid circles) to 12 healthy volunteers: (A) edoxaban 60 mg and edoxaban M-4 metabolite, (B) apixaban 25 μg, (C) edoxaban 50 µg, (D) rivaroxaban 25 µg.
At baseline, the dose-normalized
3.2 Effect of gabapentin and pregabalin on FXaI pharmacokinetics
Gabapentin and pregabalin did not alter the exposure of a therapeutic 60 mg dose of edoxaban or FXaI administered as a microdose cocktail (Table 2; Table 3; Figures 3, 4).
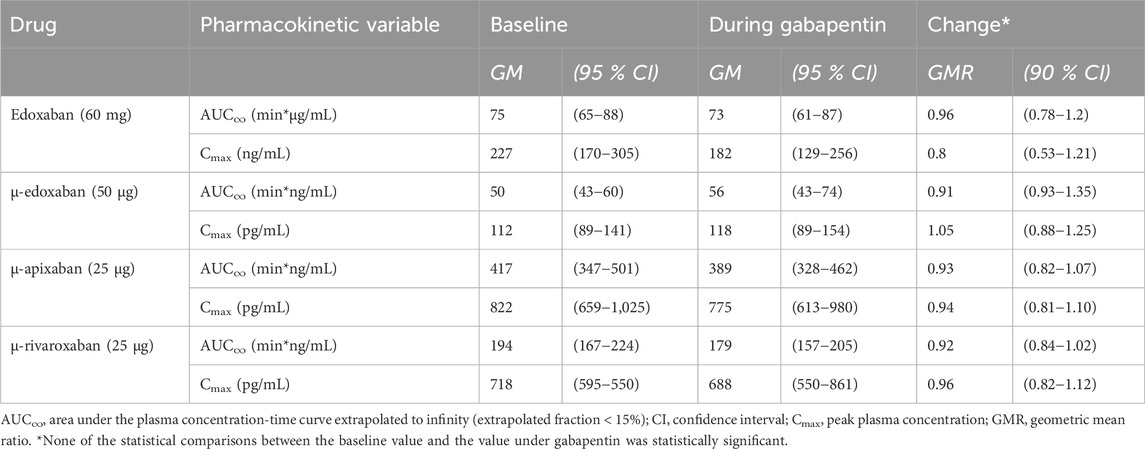
Table 2. Pharmacokinetic parameters of factor Xa inhibitors at baseline and at gabapentin steady-state (N = 11 healthy volunteers).
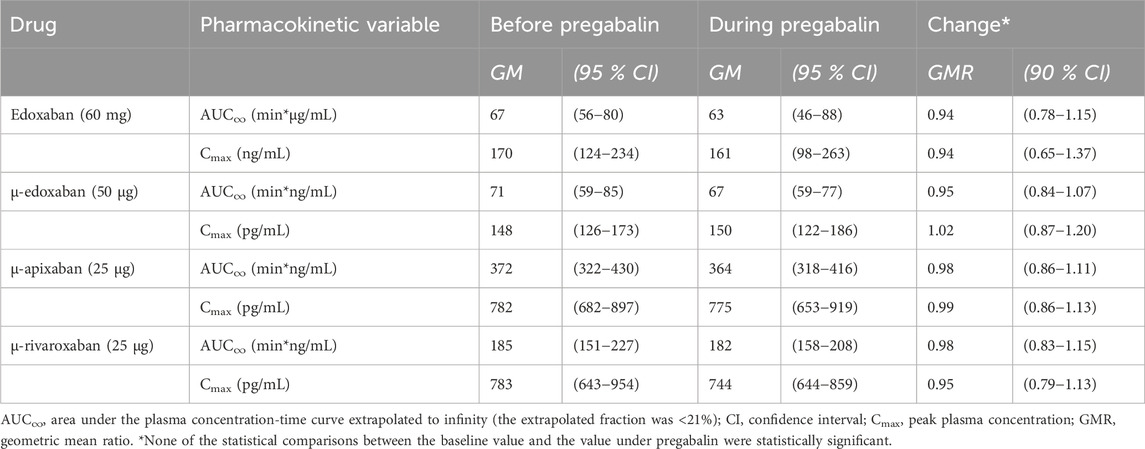
Table 3. Pharmacokinetic parameters of factor Xa inhibitors at baseline and at pregabalin steady-state (N = 12 healthy volunteers).
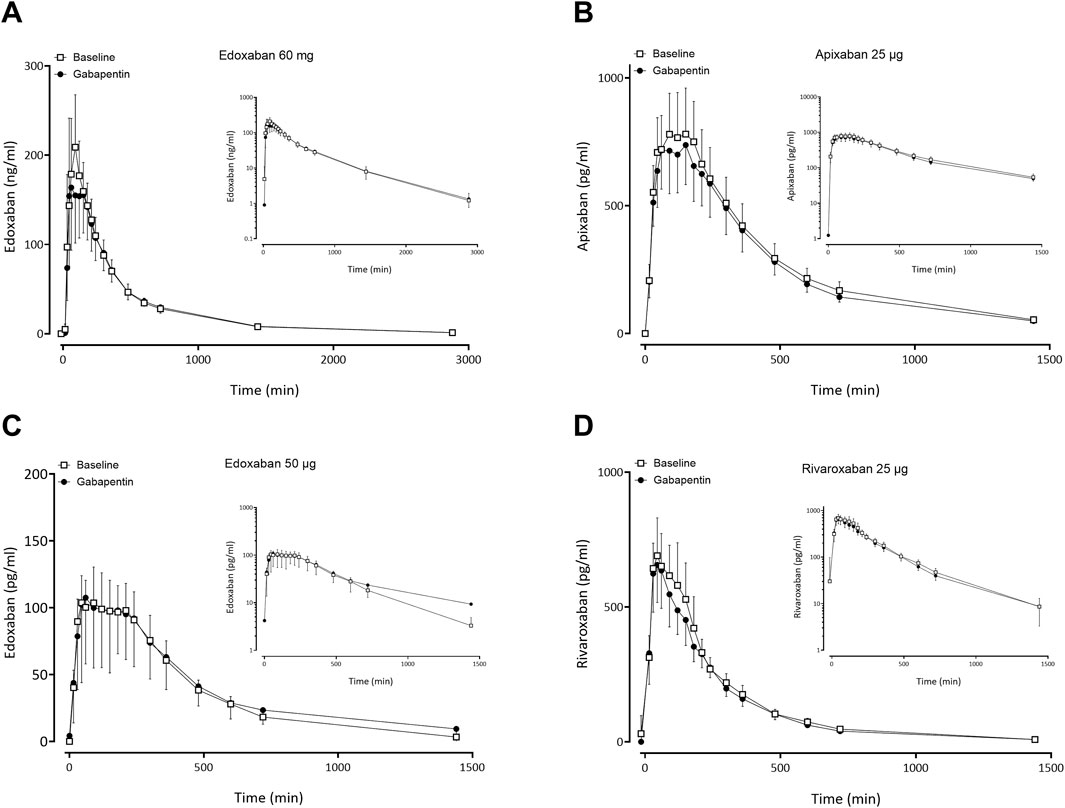
Figure 3. Mean (± standard deviation) plasma concentration-time profiles of factor Xa inhibitors after administration of single oral doses before gabapentin (open squares) and at gabapentin steady-state (solid circles) to 11 healthy volunteers: (A) edoxaban 60 mg, (B) apixaban 25 µg, (C) edoxaban 50 μg, (D) rivaroxaban 25 µg.
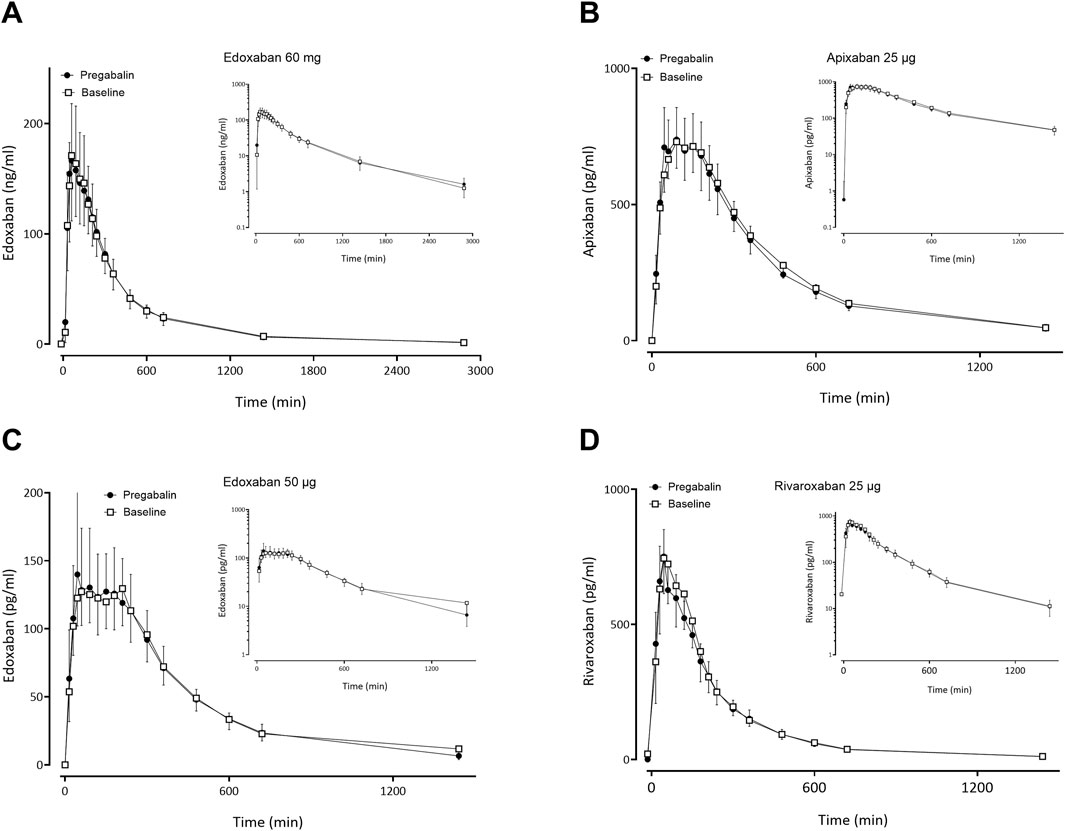
Figure 4. Mean (± standard deviation) plasma concentration-time profiles of factor Xa inhibitors after administration of single oral doses before pregabalin (open squares) and at pregabalin steady-state (solid circles) to 12 healthy volunteers: (A) edoxaban 60 mg, (B) apixaban 25 µg, (C) edoxaban 50 μg, (D) rivaroxaban 25 µg.
3.3 Assessment of CYP3A activity
Co-administration of carbamazepine increased eCLmet 5.52-fold and decreased midazolam AUC2-4 by 82% (GMR 0.18; p < 0.0001) (Supplementary Table S2). The mean decrease of midazolam AUC2-4 was significantly larger than the mean decrease of the
3.4 Effect on coagulation
Given alone, the therapeutic edoxaban dose of 60 mg significantly increased activated partial thromboplastin time and international normalized ratio values 3 h after administration; these effects were significantly smaller at carbamazepine steady-state (Supplementary Table S3). The coagulation changes induced by edoxaban in therapeutic dose or microdose were not influenced by gabapentin or pregabalin (data not shown).
3.5 Safety
3.5.1 Carbamazepine cohort
Overall, 29 adverse events (AE) occurred in 12 of 13 participants, 19 events were deemed possibly related to the trial medication. All AE were transient, none was serious, one was severe (fever in combination with liver enzyme elevation) and resulted in immediate carbamazepine discontinuation, withdrawal from the trial, and monitoring until symptoms completely resolved. The most frequent AE was nausea reported by four participants while taking carbamazepine. Other AE occurring more than once were fatigue (n = 3), abdominal pain (2), alanine aminotransferase elevation (2), headache (2), and dizziness (2), which all occurred during carbamazepine treatment.
3.5.2 Gabapentin cohort and pregabalin cohort
Overall, 39 AE occurred in 11 of 11 participants in the gabapentin-cohort, 21 of which were deemed possibly related to the trial medication or procedures. All AE were transient, none was severe or serious, and none resulted in withdrawal from the trial. The most frequent AE was flu-like symptoms, reported by five participants while taking gabapentin. Other AE occurring more than once were headache (n = 4), fatigue and dizziness (3), creatine kinase elevation (2), euphoria (2), vertigo (2), and nausea (2), which all occurred during gabapentin treatment.
Overall, 37 AEs occurred in 11 of 12 participants in the pregabalin cohort, 25 of which were deemed possibly related to the trial. All AE were transient, none was severe or serious, and none resulted in withdrawal from the trial. The most frequent AE were coordination difficulties (n = 3), impaired vision (3), headache (3), vertigo (3), nausea (2), vomiting (2), flulike symptoms (2), fatigue (2), diarrhoea (2), restlessness (2), and somnolence (2), which all occurred during pregabalin treatment.
4 Discussion
4.1 Influence of carbamazepine on the pharmacokinetics of 60 mg edoxaban and the microdosed FXaI cocktail
The P-gp and CYP3A4 inducer carbamazepine decreased the AUC and Cmax of a therapeutic edoxaban dose by about half and reduced the exposure parameters of the active metabolite M-4 in the same order of magnitude and direction. The most likely, primary mechanism for this is a decreased bioavailability due to induction of P-gp in the gut (Mikkaichi et al., 2014). The observation that both CL/F and Vz/F were approximately doubled, while t1/2 remained unchanged, suggests that the observed pharmacokinetic changes are mainly due to a substantial reduction in F. This is also confirmed by the fact that the metabolic ratio of edoxaban and M-4 is not affected by carbamazepine because M-4 is a product of CES1 which is not induced by carbamazepine.
Both, efficacy and safety of FXaI treatment, previously showed correlations to FXaI exposure (Shen et al., 2023; Kong et al., 2021; Liu et al., 2021; Zhang et al., 2021; Ohno et al., 2021; Gronich et al., 2021; Steinberg et al., 2016; Murata et al., 2019; Nielsen et al., 2017; Vasanthamohan et al., 2018; Herink et al., 2019). Retrospective evidence from cohort analyses indicated that co-medication leading to increased FXaI exposure is associated with major bleeding events as well as an increased risk of stroke and all-cause mortality (Shen et al., 2023; Kong et al., 2021; Liu et al., 2021; Zhang et al., 2021; Ohno et al., 2021; Gronich et al., 2021; Steinberg et al., 2016; Murata et al., 2019; Nielsen et al., 2017; Vasanthamohan et al., 2018; Holm et al., 2021; Harskamp et al., 2019; Li et al., 2020; Ray et al., 2024). Conversely, combinations that reduce exposure appear to increase the risk of thromboembolic events by 60% and more and increase the likelihood of strokes (Gronich et al., 2021; Holm et al., 2021). The co-administration of edoxaban with rifampin, a strong CYP3A and P-gp inducer, requires monitoring of anticoagulation according to the marketing authorisation (Lixiana, 2017), as AUC reductions of up to 35% occurred. In healthy volunteers, the exposure of edoxaban decreased during a 7-d rifampin treatment while the effects on the active metabolites M-4 and M-6 were opposite and the AUC of M-4 increased 2.86-fold and Cmax 5.06-fold and its t1/2 decreased (Mendell et al., 2015). This obvious difference in the interaction pattern of rifampin compared to carbamazepine can be explained by the fact that M-4, but not edoxaban, is an OATP1B substrate (Mikkaichi et al., 2014) whose hepatic uptake is inhibited by rifampin but not by carbamazepine, which does not interact with OATP1B at therapeutic concentrations (Gui et al., 2008). M-4 and edoxaban have comparable anticoagulant effects (Brown, 2014). If the total anticoagulant exposure is calculated by adding up the molar concentrations of edoxaban and M-4, the interaction with rifampin leads to a reduction of only 17%, as M-4 is tripled at the same time. In contrast, the analogue calculation for carbamazepine leads to a 52% reduction in the anticoagulant effect, which effectively corresponds to a halving of the dose, a very significant change. Based on this, caution must be taken to avoid loss of effect of edoxaban under carbamazepine, strict observation of anticoagulation effects is recommended, and a dose increase should be considered.
As expected, gabapentin and pregabalin did not change the exposure of edoxaban, and these combinations can therefore be safely used in patients.
4.2 Evaluation of the potential DDI by using a microdosed cocktail
In separate trial arms, we quantified the effect of the three perpetrators carbamazepine, gabapentin, and pregabalin on a microdosed cocktail consisting of three simultaneously administered FXaI and were able to intra-individually compare the impact of each perpetrator on the FXaI. For gabapentin and pregabalin, the microdose data correctly predicted that there was no interaction with the regular edoxaban dose. The exposure changes during carbamazepine were significantly, albeit not substantially smaller with microdoses of edoxaban than the exposure reductions found with therapeutic doses of edoxaban (reduction of AUC to 59% versus 48%, respectively), indicating that the evaluation with microdoses slightly underestimated the magnitude of interaction in a therapeutic setting. While linear pharmacokinetics has been confirmed between 10 mg and 150 mg doses of edoxaban (Moon, 2019), linearity has not been thoroughly assessed for microdoses. It has been shown that metabolic and intestinal drug transport pathways for some drugs may be non-linear, which may explain why exposure after a microdose only approximately predicts the corresponding exposure after a therapeutic dose (Ieiri et al., 2012; Maeda et al., 2011). It remains to be fully understood why the decrease in midazolam AUC2-4 as a marker of CYP3A4 induction (ratio) did not correlate to the decrease in
Although the comparison of dose-normalized exposure showed differences between the microdose and the regular dose, the
In addition, the baseline
As no specific trials have investigated these drug interactions yet, the exposure changes induced by carbamazepine for microdoses of apixaban and rivaroxaban must be compared with modelling studies or case reports from the literature. For rivaroxaban, recent modelling approaches using a physiologically-based pharmacokinetic model or population pharmacokinetics revealed that carbamazepine is expected to decrease the AUC of rivaroxaban to a range of 0.75 to 0.32 of its baseline values (Ngo et al., 2022; Ngo et al., 2023). This matches well the observed decrease to 0.56 (range 0.38–0.77) in our study. For Cmax our finding of a reduction to 0.75 was less than suggested by these models (0.64–0.50) (Ngo et al., 2022; Ngo et al., 2023).
For apixaban, there are epidemiological studies and case reports reporting peak and trough concentrations with and without carbamazepine in a clinical setting. The case reports are lacking the predictive statistical power and expectedly show conflicting results, with one case report agreeing well with our findings (Evanger et al., 2017) and the other reporting a considerably smaller interaction (Chadha et al., 2022). Finally, epidemiological studies reported a loss of effect with underdosing of apixaban during carbamazepine (Candeloro et al., 2022). Therefore, all available evidence suggests that a DDI is present and that the individual extent of this DDI is variable and possibly modulated by the baseline activity of P-gp.
4.3 Limitations
Although our trial population consisted of healthy volunteers and patients can differ in relevant clinical aspects, the mechanisms of DDI are expected to be similar. Variations in exposure could be at least partially attributed to different release characteristics of edoxaban microdose solution and regular dose, which is a tablet. However, tmax after administration of the microdosed solution was rather similar (75 min) to tmax of the 60 mg tablet (67 min), indicating that such differences are of minor relevance. Finally, we did not focus on genetic differences because, in an earlier trial, P-gp haplotypes did not predict the extent of clarithromycin-induced exposure changes of rivaroxaban (Gouin-Thibault et al., 2017; Mueck et al., 2013).
5 Conclusion
Therapeutic carbamazepine doses reduced the exposure of a therapeutic 60-mg dose of edoxaban to 48% on average with large interindividual variability, which is likely clinically relevant. Carbamazepine also decreased the exposure of microdosed FXaI: apixaban to 66%, microdosed edoxaban to 59%, and microdosed rivaroxaban to 56%. These changes suggest dose adjustments for FXaI and monitoring of FXaI effects to detect loss of effect and to prevent thromboembolic complications, which are always serious events in nature. Therapeutic doses of gabapentin and pregabalin did not change the exposure of a therapeutic 60-mg edoxaban dose nor the exposure of any microdosed FXaI in healthy volunteers, indicating that they can be safely combined without dose adjustment.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Medical Faculty of Heidelberg University, Germany (AFmo-144/2019). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. No potentially identifiable images or data are presented in this study.
Author contributions
AL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing–original draft, Writing–review and editing, Visualization. SH: Investigation, Writing–review and editing. FS: Investigation, Writing–review and editing. JB: Investigation, Writing–review and editing. KF: Investigation, Writing–review and editing. DC: Investigation, Writing–review and editing, Formal Analysis. GM: Investigation, Writing–review and editing. AM: Investigation, Writing–review and editing. WH: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing–review and editing. AB: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare that this study received funding from Daiichi Sankyo Europe GmbH to conduct this trial. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication. The authors declare that for the publication fee the financial support by Heidelberg University is acknowledged.
Acknowledgments
We sincerely thank all participants for their contribution and extend our gratitude to the staff of the Pharmacological Early Clinical Trial Unit at Heidelberg University Hospital, the team at the analytical laboratory, and Marlies Stützle-Schnetz for her dedicated project support. For the publication fee we acknowledge financial support by Heidelberg University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1542063/full#supplementary-material.
References
Bertilsson, L., Hojer, B., Tybring, G., Osterloh, J., and Rane, A. (1980). Autoinduction of carbamazepine metabolism in children examined by a stable isotope technique. Clin. Pharmacol. Ther. 27 (1), 83–88. doi:10.1038/clpt.1980.13
Bockbrader, H. N., Wesche, D., Miller, R., Chapel, S., Janiczek, N., and Burger, P. (2010). A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin. Pharmacokinet. 49 (10), 661–669. doi:10.2165/11536200-000000000-00000
Brown, P. C. (2014). “Pharmacology review(s),” in Center for drug evaluation and research. Application Number: 206316Orig1Orig2s000. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206316Orig1Orig2s000PharmR.pdf (Accessed March 23, 2025).
Burhenne, J., Halama, B., Maurer, M., Riedel, K. D., Hohmann, N., Mikus, G., et al. (2012). Quantification of femtomolar concentrations of the CYP3A substrate midazolam and its main metabolite 1'-hydroxymidazolam in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry. Anal. Bioanal. Chem. 402 (7), 2439–2450. doi:10.1007/s00216-011-5675-y
Candeloro, M., Eikelboom, J. W., Chan, N., Bhagirath, V., Douketis, J. D., and Schulman, S. (2022). Carbamazepine, phenytoin, and oral anticoagulants: drug-drug interaction and clinical events in a retrospective cohort. Res. Pract. Thromb. Haemost. 6 (2), e12650. doi:10.1002/rth2.12650
Chadha, A., Lopaschuk, D., Ackman, M. L., and Bungard, T. J. (2022). The problem with predictions: a cautionary tale of empirically adjusting apixaban dosing with carbamazepine. CJC Open 4 (4), 435–438. doi:10.1016/j.cjco.2021.12.012
Dagan, G., Perlman, A., Hochberg-Klein, S., Kalish, Y., and Muszkat, M. (2018). Managing direct oral anticoagulants in patients with antiepileptic medication. Can. J. Cardiol. 34 (11), 1534.e1–1534.e3. doi:10.1016/j.cjca.2018.08.001
Di Gennaro, L., Lancellotti, S., De Cristofaro, R., and De Candia, E. (2019). Carbamazepine interaction with direct oral anticoagulants: help from the laboratory for the personalized management of oral anticoagulant therapy. J. Thromb. Thrombolysis 48 (3), 528–531. doi:10.1007/s11239-019-01866-1
Eliquis (2024). EPAR - product information. Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/eliquis#product-info (Accessed March 23, 2025).
European medicines agency (2022). ICH guideline M10 on bioanalytical method validation and study sample analysis. Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (Accessed March 23, 2025).
Evanger, N., Szkotak, A., Stang, L., and Bungard, T. J. (2017). Apixaban concentration with and without coadministration of carbamazepine: a case with no apparent interaction. Can. J. Hosp. Pharm. 70 (6), 463–467. doi:10.4212/cjhp.v70i6.1714
Fenner, K. S., Troutman, M. D., Kempshall, S., Cook, J. A., Ware, J. A., Smith, D. A., et al. (2009). Drug-drug interactions mediated through P-glycoprotein: clinical relevance and in vitro-in vivo correlation using digoxin as a probe drug. Clin. Pharmacol. Ther. 85 (2), 173–181. doi:10.1038/clpt.2008.195
Foerster, K. I., Hermann, S., Mikus, G., and Haefeli, W. E. (2020). Drug-drug interactions with direct oral anticoagulants. Clin. Pharmacokinet. 59 (8), 967–980. doi:10.1007/s40262-020-00879-x
Foerster, K. I., Huppertz, A., Muller, O. J., Rizos, T., Tilemann, L., Haefeli, W. E., et al. (2018). Simultaneous quantification of direct oral anticoagulants currently used in anticoagulation therapy. J. Pharm. Biomed. Anal. 148, 238–244. doi:10.1016/j.jpba.2017.10.011
Galovic, M., Ferreira-Atuesta, C., Abraira, L., Döhler, N., Sinka, L., Brigo, F., et al. (2021). Seizures and epilepsy after stroke: epidemiology, biomarkers and management. Drugs Aging 38 (4), 285–299. doi:10.1007/s40266-021-00837-7
Giugliano, R. P., Ruff, C. T., Braunwald, E., Murphy, S. A., Wiviott, S. D., Halperin, J. L., et al. (2013). Edoxaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 369 (22), 2093–2104. doi:10.1056/nejmoa1310907
Giustozzi, M., Mazzetti, M., Paciaroni, M., Agnelli, G., Becattini, C., and Vedovati, M. C. (2021). Concomitant use of direct oral anticoagulants and antiepileptic drugs: a prospective cohort study in patients with atrial fibrillation. Clin. Drug Investig. 41 (1), 43–51. doi:10.1007/s40261-020-00982-8
Gouin-Thibault, I., Delavenne, X., Blanchard, A., Siguret, V., Salem, J. E., Narjoz, C., et al. (2017). Interindividual variability in dabigatran and rivaroxaban exposure: contribution of ABCB1 genetic polymorphisms and interaction with clarithromycin. J. Thromb. Haemost. 15 (2), 273–283. doi:10.1111/jth.13577
Granger, C. B., Alexander, J. H., McMurray, J. J., Lopes, R. D., Hylek, E. M., Hanna, M., et al. (2011). Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 365 (11), 981–992. doi:10.1056/NEJMoa1107039
Gronich, N., Stein, N., and Muszkat, M. (2021). Association between use of pharmacokinetic-interacting drugs and effectiveness and safety of direct acting oral anticoagulants: nested case-control study. Clin. Pharmacol. Ther. 110 (6), 1526–1536. doi:10.1002/cpt.2369
Gui, C., Miao, Y., Thompson, L., Wahlgren, B., Mock, M., Stieger, B., et al. (2008). Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur. J. Pharmacol. 584 (1), 57–65. doi:10.1016/j.ejphar.2008.01.042
Harskamp, R. E., Teichert, M., Lucassen, W. A. M., van Weert, H., and Lopes, R. D. (2019). Impact of polypharmacy and p-glycoprotein- and CYP3A4-modulating drugs on safety and efficacy of oral anticoagulation therapy in patients with atrial fibrillation. Cardiovasc Drugs Ther. 33 (5), 615–623. doi:10.1007/s10557-019-06907-8
Herink, M. C., Zhuo, Y. F., Williams, C. D., and DeLoughery, T. G. (2019). Clinical management of pharmacokinetic drug interactions with direct oral anticoagulants (DOACs). Drugs 79 (15), 1625–1634. doi:10.1007/s40265-019-01183-0
Hohmann, N., Haefeli, W. E., and Mikus, G. (2015). Use of microdose phenotyping to individualise dosing of patients. Clin. Pharmacokinet. 54 (9), 893–900. doi:10.1007/s40262-015-0278-y
Holm, J., Mannheimer, B., Malmstrom, R. E., Eliasson, E., and Lindh, J. D. (2021). Bleeding and thromboembolism due to drug-drug interactions with non-vitamin K antagonist oral anticoagulants-a Swedish, register-based cohort study in atrial fibrillation outpatients. Eur. J. Clin. Pharmacol. 77 (3), 409–419. doi:10.1007/s00228-020-03015-7
Hori, M., Matsumoto, M., Tanahashi, N., Momomura, S. i., Uchiyama, S., Goto, S., et al. (2012). Rivaroxaban vs. warfarin in Japanese patients with atrial fibrillation - the J-ROCKET AF study. Circ. J. 76 (9), 2104–2111. doi:10.1253/circj.cj-12-0454
Hylek, E. M., Held, C., Alexander, J. H., Lopes, R. D., De Caterina, R., Wojdyla, D. M., et al. (2014). Major bleeding in patients with atrial fibrillation receiving apixaban or warfarin: the ARISTOTLE trial (Apixaban for reduction in stroke and other ThromboemboLic events in atrial fibrillation): predictors, characteristics, and clinical outcomes. J. Am. Coll. Cardiol. 63 (20), 2141–2147. doi:10.1016/j.jacc.2014.02.549
Ieiri, I., Fukae, M., Maeda, K., Kimura, M., Hirota, T., Nakamura, T., et al. (2012). Pharmacogenomic/pharmacokinetic assessment of a four-probe cocktail for CYPs and OATPs following oral microdosing. Int. J. Clin. Pharmacol. Ther. 50 (10), 689–700. doi:10.5414/CP201763
Ip, B. Y., Ko, H., Wong, G. L., Yip, T. C., Lau, L. H., Lau, A. Y., et al. (2022). Thromboembolic risks with concurrent direct oral anticoagulants and antiseizure medications: a population-based analysis. CNS Drugs 36 (12), 1313–1324. doi:10.1007/s40263-022-00971-9
Katzenmaier, S., Markert, C., and Mikus, G. (2010). Proposal of a new limited sampling strategy to predict CYP3A activity using a partial AUC of midazolam. Eur. J. Clin. Pharmacol. 66 (11), 1137–1141. doi:10.1007/s00228-010-0878-2
Katzenmaier, S., Markert, C., Riedel, K. D., Burhenne, J., Haefeli, W. E., and Mikus, G. (2011). Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using A limited sampling strategy. Clin. Pharmacol. Ther. 90 (5), 666–673. doi:10.1038/clpt.2011.164
Kong, X., Zhu, Y., Pu, L., Meng, S., Zhao, L., Zeng, W., et al. (2021). Efficacy and safety of non-recommended dose of new oral anticoagulants in patients with atrial fibrillation: a systematic review and meta-analysis. Front. Cardiovasc Med. 8, 774109. doi:10.3389/fcvm.2021.774109
Lenard, A., Hermann, S. A., Stoll, F., Burhenne, J., Foerster, K. I., Mikus, G., et al. (2024). Effect of clarithromycin, a strong CYP3A and P-glycoprotein inhibitor, on the pharmacokinetics of edoxaban in healthy volunteers and the evaluation of the drug interaction with other oral factor xa inhibitors by a microdose cocktail approach. Cardiovasc Drugs Ther. 38 (4), 747–756. doi:10.1007/s10557-023-07443-2
Li, A., Li, M. K., Crowther, M., and Vazquez, S. R. (2020). Drug-drug interactions with direct oral anticoagulants associated with adverse events in the real world: a systematic review. Thromb. Res. 194, 240–245. doi:10.1016/j.thromres.2020.08.016
Liu, X., Huang, M., Ye, C., Xiao, X., and Yan, C. (2021). Effect of non-recommended doses versus recommended doses of direct oral anticoagulants in atrial fibrillation patients: a meta-analysis. Clin. Cardiol. 44 (4), 472–480. doi:10.1002/clc.23586
Lixiana (2015). EPAR - product information. Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/lixiana (Accessed March 23, 2025).
Lixiana® 60 mg Hartkapseln, summary of product characteristics (2017). Germany: Daiichi-Sankyo. last updated July 2017.
Maeda, K., Takano, J., Ikeda, Y., Fujita, T., Oyama, Y., Nozawa, K., et al. (2011). Nonlinear pharmacokinetics of oral quinidine and verapamil in healthy subjects: a clinical microdosing study. Clin. Pharmacol. Ther. 90 (2), 263–270. doi:10.1038/clpt.2011.108
Mao, L., Li, C., Li, T., and Yuan, K. (2014). Prevention of stroke and systemic embolism with rivaroxaban compared with warfarin in Chinese patients with atrial fibrillation. Vascular 22 (4), 252–258. doi:10.1177/1708538113490423
Mendell, J., Chen, S., He, L., Desai, M., and Parasramupria, D. A. (2015). The effect of rifampin on the pharmacokinetics of edoxaban in healthy adults. Clin. Drug Investig. 35 (7), 447–453. doi:10.1007/s40261-015-0298-2
Mikkaichi, T., Yoshigae, Y., Masumoto, H., Imaoka, T., Rozehnal, V., Fischer, T., et al. (2014). Edoxaban transport via P-glycoprotein is a key factor for the drug's disposition. Drug Metab. Dispos. 42 (4), 520–528. doi:10.1124/dmd.113.054866
Mikus, G., Foerster, K. I., Schaumaeker, M., Lehmann, M. L., Burhenne, J., and Haefeli, W. E. (2019). Microdosed cocktail of three oral factor Xa inhibitors to evaluate drug-drug interactions with potential perpetrator drugs. Clin. Pharmacokinet. 58 (9), 1155–1163. doi:10.1007/s40262-019-00749-1
Mikus, G., Foerster, K. I., Schaumaeker, M., Lehmann, M. L., Burhenne, J., and Haefeli, W. E. (2020). Application of a microdosed cocktail of 3 oral factor Xa inhibitors to study drug-drug interactions with different perpetrator drugs. Br. J. Clin. Pharmacol. 86 (8), 1632–1641. doi:10.1111/bcp.14277
Moon, Y. D. (2019). “Clinical pharmacology and biopharmaceutics review(s),” in Center for drug evaluation and research. application number: 206316Orig1Orig2s000. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206316Orig1Orig2s000ClinPharmR.pdf (Accessed March 23, 2025).
Mueck, W., Kubitza, D., and Becka, M. (2013). Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br. J. Clin. Pharmacol. 76 (3), 455–466. doi:10.1111/bcp.12075
Murata, N., Okumura, Y., Yokoyama, K., Matsumoto, N., Tachibana, E., Kuronuma, K., et al. (2019). Clinical outcomes of off-label dosing of direct oral anticoagulant therapy among Japanese patients with atrial fibrillation identified from the SAKURA AF registry. Circ. J. 83 (4), 727–735. doi:10.1253/circj.CJ-18-0991
Neurontin® 100/300/400 mg Hartkapseln, summary of product characteristics (2018). Germany: Pfizer. last updated March 2018.
Ngo, L. T., Yang, S. Y., Shin, S., Cao, D. T., Van Nguyen, H., Jung, S., et al. (2022). Application of physiologically-based pharmacokinetic model approach to predict pharmacokinetics and drug-drug interaction of rivaroxaban: a case study of rivaroxaban and carbamazepine. CPT Pharmacometrics Syst. Pharmacol. 11 (11), 1430–1442. doi:10.1002/psp4.12844
Ngo, L. T., Yun, H. Y., and Chae, J. W. (2023). Application of the population pharmacokinetics model-based approach to the prediction of drug-drug interaction between rivaroxaban and carbamazepine in humans. Pharm. (Basel) 16 (5), 684. doi:10.3390/ph16050684
Nielsen, P. B., Skjoth, F., Sogaard, M., Kjaeldgaard, J. N., Lip, G. Y., and Larsen, T. B. (2017). Effectiveness and safety of reduced dose non-vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ 356, j510. doi:10.1136/bmj.j510
Ohno, J., Sotomi, Y., Hirata, A., Sakata, Y., Hirayama, A., and Higuchi, Y. (2021). Dose of direct oral anticoagulants and adverse outcomes in Asia. Am. J. Cardiol. 139, 50–56. doi:10.1016/j.amjcard.2020.09.053
Paolucci, S., Iosa, M., Toni, D., Barbanti, P., Bovi, P., Cavallini, A., et al. (2016). Prevalence and time course of post-stroke pain: a multicenter prospective hospital-based study. Pain Med. 17 (5), 924–930. doi:10.1093/pm/pnv019
Patel, M. R., Mahaffey, K. W., Garg, J., Pan, G., Singer, D. E., Hacke, W., et al. (2011). Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N. Engl. J. Med. 365 (10), 883–891. doi:10.1056/NEJMoa1009638
Perlman, A., Hochberg-Klein, S., Choshen Cohen, L., Dagan, G., Hirsh-Raccah, B., Horwitz, E., et al. (2019). Management strategies of the interaction between direct oral anticoagulant and drug-metabolizing enzyme inducers. J. Thromb. Thrombolysis 47 (4), 590–595. doi:10.1007/s11239-018-01804-7
Puma, M., Maestrini, I., Altieri, M., and Di Piero, V. (2020). A neurological point of view on the European Society of Cardiology guidelines: the interactions between direct oral anticoagulants and antiepileptic drugs. Eur. J. Neurol. 27 (12), e79. doi:10.1111/ene.14428
Ray, W. A., Chung, C. P., Stein, C. M., Smalley, W., Zimmerman, E., Dupont, W. D., et al. (2024). Serious bleeding in patients with atrial fibrillation using diltiazem with apixaban or rivaroxaban. JAMA 331 (18), 1565–1575. doi:10.1001/jama.2024.3867
Rengelshausen, J., Goggelmann, C., Burhenne, J., Riedel, K. D., Ludwig, J., Weiss, J., et al. (2003). Contribution of increased oral bioavailability and reduced nonglomerular renal clearance of digoxin to the digoxin-clarithromycin interaction. Br. J. Clin. Pharmacol. 56 (1), 32–38. doi:10.1046/j.1365-2125.2003.01824.x
Sennesael, A. L., Larock, A. S., Hainaut, P., Lessire, S., Hardy, M., Douxfils, J., et al. (2021). The impact of strong inducers on direct oral anticoagulant levels. Am. J. Med. 134 (10), 1295–1299. doi:10.1016/j.amjmed.2021.06.003
Shen, N. N., Ferroni, E., Amidei, C. B., Canova, C., Peron, V., Wang, J. L., et al. (2023). An updated pooled analysis of off-label under and over-dosed direct oral anticoagulants in patients with atrial fibrillation. Clin. Appl. Thromb. Hemost. 29, 10760296231179439. doi:10.1177/10760296231179439
Solvobiotech. Human Transporters: OCTN1 (2018). Available online at: https://www.solvobiotech.com/transporters/octn1 (Accessed March 22, 2025).
Steinberg, B. A., Shrader, P., Thomas, L., Ansell, J., Fonarow, G. C., Gersh, B. J., et al. (2016). Off-label dosing of non-vitamin K antagonist oral anticoagulants and adverse outcomes: the ORBIT-AF II registry. J. Am. Coll. Cardiol. 68 (24), 2597–2604. doi:10.1016/j.jacc.2016.09.966
Urban, T. J., Brown, C., Castro, R. A., Shah, N., Mercer, R., Huang, Y., et al. (2008). Effects of genetic variation in the novel organic cation transporter, OCTN1, on the renal clearance of gabapentin. Clin. Pharmacol. Ther. 83 (3), 416–421. doi:10.1038/sj.clpt.6100271
U.S. Food and Drug Administration (2023). U.S. Food and Drug Administration: methods development, validation, and implementation program. Available online at: https://www.fda.gov/media/90217/download (Accessed March 23, 2025).
Vasanthamohan, L., Boonyawat, K., Chai-Adisaksopha, C., and Crowther, M. (2018). Reduced-dose direct oral anticoagulants in the extended treatment of venous thromboembolism: a systematic review and meta-analysis. J. Thromb. Haemost. 16 (7), 1288–1295. doi:10.1111/jth.14156
Wang, L., Zhang, D., Raghavan, N., Yao, M., Ma, L., Frost, C. E., et al. (2010). In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab. Dispos. 38 (3), 448–458. doi:10.1124/dmd.109.029694
Xarelto (2020). EPAR - product information. Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/xarelto. (Accessed March 22, 2025).
Keywords: factor Xa inhibitors, edoxaban, carbamazepine, gabapentin, pregabalin, drug-drug interaction, microdose, healthy volunteers
Citation: Lenard A, Hermann SA, Stoll F, Burhenne J, Foerster KI, Czock D, Mikus G, Meid AD, Haefeli WE and Blank A (2025) Effect of the frequently used antiepileptic drugs carbamazepine, gabapentin, and pregabalin on the pharmacokinetics of edoxaban and other oral factor xa inhibitors in healthy volunteers. Front. Pharmacol. 16:1542063. doi: 10.3389/fphar.2025.1542063
Received: 09 December 2024; Accepted: 11 March 2025;
Published: 11 April 2025.
Edited by:
Peter Swaan, University of Florida, United StatesReviewed by:
Ziran Li, University of California, San Francisco, United StatesCyprian Onyeji, University of Nigeria, Nsukka, Nigeria
Junling Yang, Chinese Academy of Sciences (CAS), China
Felix Huth, Novartis, Switzerland
Copyright © 2025 Lenard, Hermann, Stoll, Burhenne, Foerster, Czock, Mikus, Meid, Haefeli and Blank. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antje Blank, YW50amUuYmxhbmtAbWVkLnVuaS1oZWlkZWxiZXJnLmRl