
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 04 February 2025
Sec. Pharmacology of Infectious Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fphar.2025.1518463
This article is part of the Research Topic Treatment of Infectious Diseases with Bioactive Compounds from Medicinal Plants: Their Mechanisms and Applications - Volume II View all 5 articles
 Muzaffar Kayumov1*†
Muzaffar Kayumov1*† Parthiban Marimuthu2,3*†
Parthiban Marimuthu2,3*† Jamoliddin Razzokov4,5,6
Jamoliddin Razzokov4,5,6 Nurkhodja Mukhamedov1,7
Nurkhodja Mukhamedov1,7 Akmal Asrorov1Nodir S. Berdiev1Jamolitdin F. Ziyavitdinov1Ansor Yashinov1,8Yuliya Oshchepkova1Shavkat Salikhov1Sharafitdin Mirzaakhmedov1
Akmal Asrorov1Nodir S. Berdiev1Jamolitdin F. Ziyavitdinov1Ansor Yashinov1,8Yuliya Oshchepkova1Shavkat Salikhov1Sharafitdin Mirzaakhmedov1The emergence of the SARS-CoV-2 virus caused the COVID-19 outbreak leading to a global pandemic. Natural substances started being screened for their antiviral activity by computational and in-vitro techniques. Here, we evaluated the anti-SARS-CoV-2 main protease (Mpro) efficacy of ©Rutan, which contains five polyphenols (R5, R6, R7, R7’, and R8) extracted from sumac Rhus coriaria L. We obtained three fractions after large-scale purification: fraction 1 held R5, fraction 2 consisted of R6, R7 and R7’, and fraction 3 held R8. In vitro results showed their anti-Mpro potential: IC50 values of R5 and R8 made 42.52 µM and 5.48 µM, respectively. Further, we studied Mpro-polyphenol interactions by in silico analysis to understand mechanistic extrapolation of Rutan binding nature with Mpro. We extensively incorporated a series of in silico techniques. Initially, for the docking protocol validation, redocking of the co-crystal ligand GC-376* to the binding pocket of Mpro was carried out. The representative docked complexes were subjected to long-range 500 ns molecular dynamics simulations. The binding free energy (BFE in kcal/mol) of components were calculated as follows: R8 (−104.636) > R6 (−93.754) > R7’ (−92.113) > R5 (−81.115) > R7 (−67.243). In silico results of R5 and R8 correspond with their in vitro outcomes. Furthermore, the per-residue decomposition analysis showed C145, E166, and Q189 residues as the hotspot residues for components contributing to maximum BFE energies. All five components effectively interact with the catalytic pocket of Mpro and form stable complexes that allow the estimation of their inhibitory activity. Assay kit analyses revealed that Rutan and its components have effective anti-SARS-CoV-2 Mpro inhibitory activity.
As coronavirus infection became a global threat, different sources were searched for their anti-inhibitory activity. Among the inhibitory compounds investigated, polyphenols stood out for their high antiviral activity, which was attributed to their ability to inhibit the activity of the Mpro. Phenolic compounds are considered to be the most promising among lower molecular weight compounds for their antiviral activity. The inhibitory nature of phenolics over SARS-CoV-2 is often linked with their influence on Mpro as well as spike proteins resulting from hydrogen and non-hydrogen bond interactions (Arunkumar et al., 2022). Compared to other compounds, phenolics showed lower mean values for the IC50 value due to their potent anti-Mpro activity (Yang et al., 2022). Mpro is a 33.8 kD protein that cleaves polyproteins at minimum 11 conserved sites (Ren et al., 2013; Jin et al., 2020) The specificity of Mpro is linked with cleavage around glutamine residue which is not found among human proteases (Pang et al., 2023). Thus, low toxicity of the Mpro inhibitors to host cells can be claimed.
Among natural substances, compounds that have phenolic rings deserve a special attention that could be reveal anti-Mpro activity (Pang et al., 2023). Polyphenols are suggested as effective inhibitiors of Mpro (Ahmad et al., 2024). Epigallocatechin-3-gallate (EGCG) is a polyphenol that was one of the earliest established inhibitors of the SARS-CoV-2 resulting from Mpro inhibition (Park et al., 2021). This compound, found as the main polyphenol in green tea (Graham, 1992), has been shown to reduce the enzymatic activity of HCoV-OC43 and HCoV-229E and is therefore considered a potential inhibitor of coronavirus replication (Jang et al., 2021). Theaflavin, another active phenolic compound found in black tea, has also been reported to have anti-Mpro activity (Jang et al., 2020). In vitro testing showed no significant differences in their inhibitory activity, with IC50 values of 16.53 µM for EGCG and 14.95 µM for theaflavin. In another study, tannic acid and 3-isotheaflavin-3-gallate were found to have IC50 values of 3 µM and 7 μM, respectively, indicating their potential as antiviral compounds. The authors report the higher antiviral potential of Puer and black teas compared to green ones (Chen et al., 2005). Processing conditions of black tea were investigated to enhance the content of theaflavin-3-3′-di-O-gallate in order to develop tea-based antiviral properties (Paiva et al., 2022). A more recent paper reported the inhibitory activity of several phenolics based on molecular docking and molecular dynamics (MD) simulations (Bahun et al., 2022). Among the twenty compounds tested, including flavonoids, curcuminoids, phenolic acids, and other polyphenols, quercetin, ellagic acid, curcumin, EGCG, and resveratrol showed the highest activity. Their IC50 values ranged from 11.8 to 23.4 µM dose. Additionally, by using computational biology approaches, geraniin isolated from Geranium thunbergii was reported as another potential inhibitor of this enzyme (Yu et al., 2022). Computational analyses of substances on viral proteins were effectively used to suggest their possible mechanisms of action (Singh and Purohit, 2024). In silico analyses on barrigenol, kaempferol, and myricetin suggested their potential as nonstructural Nsp15 protein of SARS-CoV-2 (Rashid et al., 2022; Otter et al., 2023; Sharma et al., 2021). Sings et al. screened the inhibitory properties of several curcumin derivatives on Nsp15 SARS-CoV-2 and reported potential ones (Singh et al., 2022). In another work, two of them were found to be effective inhibitors of RNA-dependent RNA polymerase (RdRP)-RNA complex (Singh et al., 2021). Inhibitory activity of tea-derived phenolics over Mpro was reported as another possible target that contributes to their anti-SARS-CoV-2 property (Bhardwaj et al., 2021).
Mpro was earlier reported as potential target to control coronavirus. The authors demonstrated the highest 3.7 µM IC50 value of broussoflavan, a prenylated quercetin derivative, among ten phenolics isolated from Broussonetia papyrifera (Park et al., 2017). Thus, prenylation was determined to double the inhibitory activity of quercetin (IC50 8.6 µM). The IC50 values of broussochalcone A and B, differing by OH group presence, made 11.6 and 9.2 µM doses, respectively. Derivatives of darunavir were determined as SARS-CoV-2 Mpro inhibitors based on fluorescence resonance energy transfer (FRET) that were supported by molecular docking (Ma et al., 2022). Two of the studied compounds were further selected after in vitro analysis for designing inhibitors possessing higher inhibitory activity. Phenolic compounds isolated from brown marine algae Ishige Okamurae demonstrated high antiviral activity due to their ability to inhibit Mpro (Nagahawatta et al., 2022). Ishophloroglucin A was defined as the most potential one that inhibited the enzymic activity of Mpro and papain-like protease.
Further analysis in this discipline reported phenolics as the inhibitors of Mpro dimers. Acacetin 7-O-neohesperidoside, termed fortunellin, was found to be a potent inhibitor of Mpro dimer (Panagiotopoulos et al., 2021). In silico results were also confirmed by in vitro tests for a few phenolics: fortunellin, apiin, and rhoifolin. Three flavonoids such as baicalin, rutin, and glycyrrhizic acid were determined as potential antiviral agents against COVID-19 due to their inhibitory activity over Mpro after docking and molecular dynamics analysis (Patil et al., 2021). Pomegranate peel extract was reported as another source of potential antiviral agents against COVID-19. The alcohol extract containing polyphenols such as punicalagin, gallic acid, and ellagic acid was concluded to be an efficient means due to their involvement in many processes, including the inhibition of Mpro activity (Tito et al., 2021). Phenolics are indeed potential agents against SARS-CoV-2. Quercetin, for instance, was concluded as a possible therapeutic at the early stages of COVID-19 infection in a randomized clinical trial (Di Pierro et al., 2022).
Sumac Rhus coriaria L plant polyphenols are mainly composed of gallic acid derivatives (Figure 1), and there are limited studies on their antiviral activity. Our group has previously examined the mixture of polyphenols, mainly tannin-like compounds, extracted from sumac R. coriaria L. for anti-influenza activity (Zhamollitdin Fazlitdinovich et al., 2020). The results showed remarkable potency for anti-influenza activity and leading to the commercialization under the name ©Rutan as an antiviral drug in the local market. Subsequently, during the pandemic, researchers studied anti SARS-CoV-2 activity of Rutan and observed high inhibitory efficacy against virus in infected cells during clinical trials (Salikhov et al., 2023). Toxicological analysis showed no drug accumulation in mice organs. Besides, no acute or chronic drug toxicity was observed; LD50 in rats and mice made >5,000 mg/kg when administered intragastrically. Pharmacological studies revealed no death when the drug was given at 2,000 mg/kg in 40 mice that aligned with no effects on orienting response and spontaneous motor activity. Moreover, no allergic reactions were observed when given at 25 mg/kg dose to guinea pigs (Salikhov et al., 2023). Five major compounds of polyphenolic nature in the Rutan composition–R5, R6, R7, R7’, and R8 are shown below in Figure 1. While various natural products, such as flavonoids and alkaloids, have been investigated for their potential inhibitory effects against SARS-CoV-2, many exhibit limitations such as poor bioavailability or cytotoxicity at effective concentrations (Xu et al., 2023). Sumac-derived polyphenolic compounds, however, represent a relatively unexplored class of natural inhibitors. These compounds are not only rich in antioxidant properties but also demonstrate favorable pharmacokinetics, making them promising candidates for therapeutic application. This study focuses on these compounds to explore their potential in inhibiting the SARS-CoV-2 main protease (Mpro) through a combination of computational and experimental approaches.
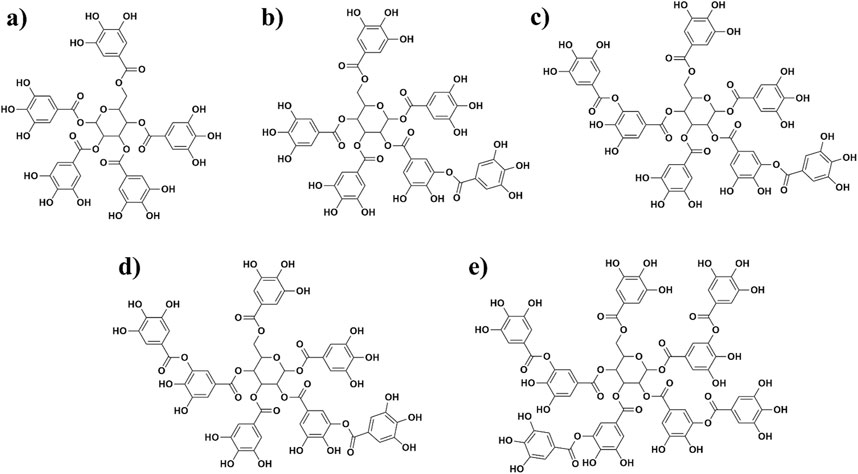
Figure 1. Structures of five polyphenols extracted from sumac Rhus coriaria L. that were investigated in this study [(A) = R5; (B) = R6; (C) = R7; (D) = R7’, and (E) = R8].
Early analysis of Rutan on Mpro activity revealed concentration-dependent inhibition of the enzyme. A 17.7% inhibition rate of the enzyme by 0.5 µM dose of the mixture reached 64.4% rate by 5 µM dose (Salikhov et al., 2023). In this work, we aimed to explore possible mechanistic behavior of Mpro-ligand interactions of Rutan by computational methods accompanied with in-vitro analysis of each fraction of Rutan components, namely, R5, R8 individually, and R6 + R7 + R7’ mixture (Figure 1), whether the compounds exhibit higher anti-Mpro activity alone or in complex.
The crystallographic structure of the SARS-CoV-2 Mpro complexed with inhibitor GC-376 (PDB ID: 6WTT, resolution of 2.15 Å; chain A) was retrieved from the Protein Data Bank (PDB at https://www.rcsb.org/structure/6WTT; Figure 2) (Ma et al., 2020). In this present investigation, we extensively utilized the Maestro suite 2020-4 (Schrödinger Inc. NY, United States) and the protocol used in our previous study (Marimuthu et al., 2021). To ensure the effective utilization of this suite, it is imperative to fix any pre-existing crystallographic artifacts that the Mpro–GC-376 complex may contain. Moreover, this preparation step was crucial to enable subsequent investigations.
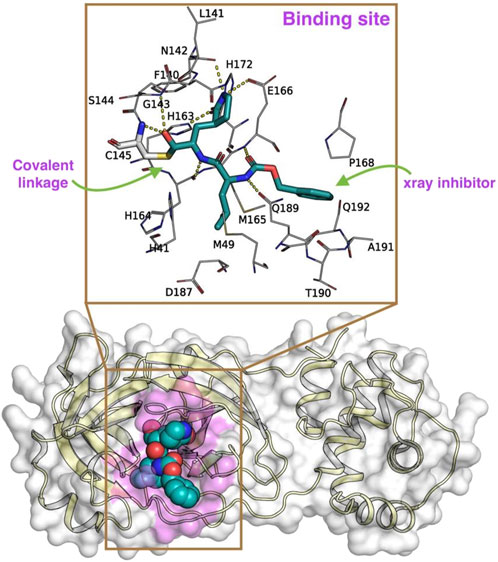
Figure 2. The x-ray crystal structure of SARS-CoV-2 Mpro [6WTT (Ma et al., 2020), represented in a cartoon format with the molecular surface highlighted in pale yellow and while, respectively] covalently bound to an inhibitor GC-376 (represented in spheres; carbon in marine blue; nitrogen in blue; and oxygen in red) used in this study. Here, the binding site of the Mpro is highlighted in pink color over the molecular surface. The magnified image highlights the (i) covalent linkage (green arrow) of the GC-376 inhibitor (represented in sticks in cyan color) to the C145 residue and (ii) other surrounding residues involved in Mpro-Rutans complex formation. The yellow dotted lines represent the polar interactions between the GC-376 inhibitor and the surrounding residues of Mpro.
Consequently, the Mpro complexed with GC-376 underwent immediate preprocessing using default parameters, specifically employing the Protein Preparation Wizard (Sastry et al., 2013). This involved assigning bond orders, adding polar hydrogens, eliminating crystal waters, addressing missing side-chain residues, and establishing protonation states for charged residues (including Asp, Glu, Arg, Lys, and His) using PROPKA. Subsequently, the complex was subjected to minimization using the OPLS3 forcefield (Harder et al., 2016), maintaining a physiological pH of 7.2 with the assistance of Epik (Shelley et al., 2007; Greenwood et al., 2010). As a result of this thorough minimization process, the Mpro–GC-376 complex was predicted to retain the charged residues in their native state, and H163 in ε, H164 in δ, H172 in a positive state, while H41 and C145 residues are in a neutral state.
Subsequently, to create a molecular 2D representation of the five Rutan compounds – 1,2,3,4,6-penta-O-galloyl-β-D-glucose (R5), Hexa-O-galloyl-β-D-glucose (R6), Hepta-O-galloyl-β-D-glucose (R7), Octa-O-galloyl-β-D-glucose (R7’), Nona-O-galloyl-β-D-glucose (R8) (Salikhov et al., 2023) (Figures 3A–E), the “2D Sketcher” panel within Maestro was employed. These compounds underwent further processing, including the addition of any missing hydrogen atoms, correct assignment of formal charges, generation of plausible molecular configurations based on default ionization and tautomeric states, and ultimately convertion into their 3D representations using the OPLS3e (Roos et al., 2019), force field using the LigPrep module (Schrödinger Release 2020-4: LigPrep, Schrödinger, LLC, New York, NY, 2020). The resulting compounds, featuring low-energy conformational poses, were suitable and used for further investigation. The binding pocket analysis reveals that the co-crystallized ligand forms a covalent bond with the thiol group of the C145 residue that is well buried within the binding pocket of the Mpro.
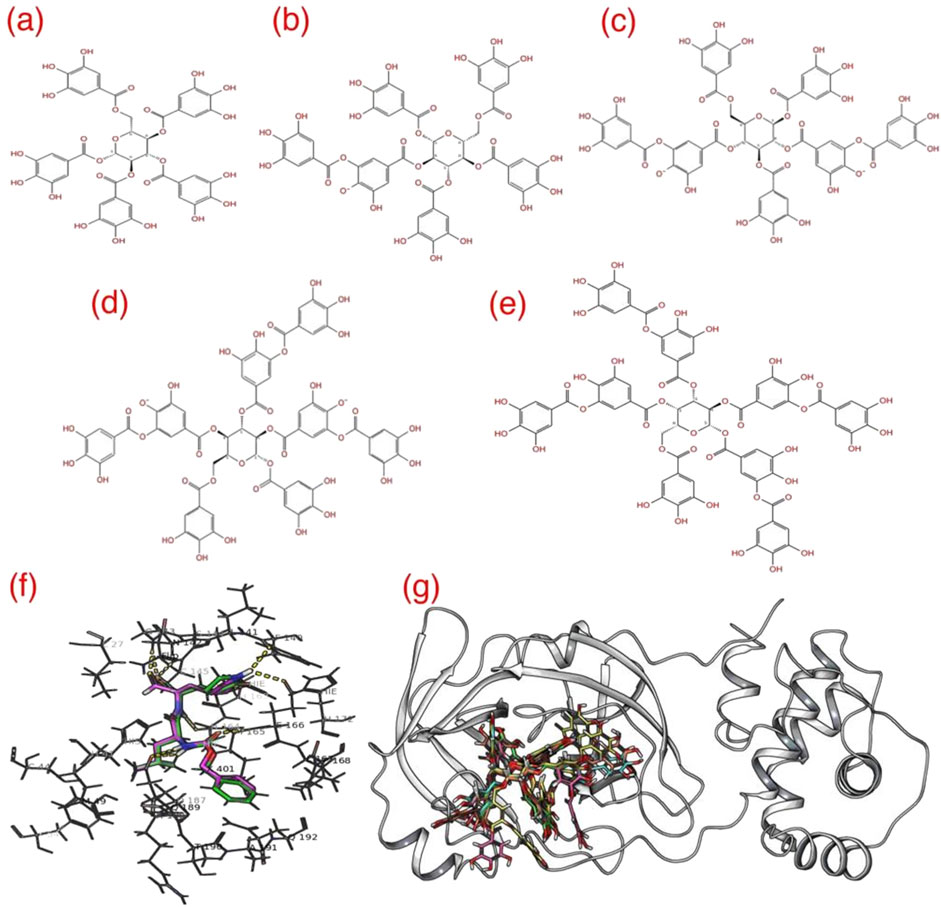
Figure 3. The 2D representation of five different Rutan (A–E) compounds used in this study. (F) The GC-376* redocked (pink) over the native GC-376 (green) showing reproducibility and (G) superposition of docked conformations of 5 Rutan compounds (R5 = yellow; R6 = green; R7 = pink; R7’ = cyan, and R8 = blue) inside the Mpro binding pocket. The yellow dotted lines highlight the polar interactions between GC-376* and the surrounding residues.
In order to carry out effective docking calculations, it is highly required to execute a redocking protocol, i.e., performing a docking run with the co-crystallized ligand on itself, which eventually exhibits the reliability and reproducibility of the crystal ligand conformation of the docking protocol planned on execution. Towards that, as the Glide program does not allow a covalently linked ligand to perform docking runs, a modified version of GC-376, referred to as “GC-376*” was created by manually breaking the native covalent linkage (Halgren et al., 2004; Friesner et al., 2004). Given the well-defined nature of the enzyme’s binding pocket, we opted to utilize monomeric conformation for our investigation, considering it to be a reliable approach, which has also been reported in previous studies (Choudhary et al., 2020; Wang, 2020; Mengist et al., 2021; Weng et al., 2021). As such, we believe that the methodology employed in our study offers a reasonable estimation of binding events and the relevant surrounding residues. The resulting Mpro–GC-376* complex underwent a secondary round of minimization, following the previously outlined procedures. Subsequently, GC-376*, devoid of any local structural clashes or improper bond orders, was employed to establish a grid using the Grid generation panel available in Maestro, a prerequisite to conducting the Glide redocking process. Initially, the grid was generated by centering it on GC-376*, effectively encompassing the binding pocket enveloped by amino acid residues H41, C44, M49, Y54, F140, L141, N142, G143, S144, C145, H163, H164, M165, E166, L167, P168, H172, D187, R588, Q189, T190, A191 and Q192 of Mpro. During the grid generation, process the additional parameters such as (i) assessing input ring conformations, (ii) favoring intramolecular hydrogen bonds, and (iii) improving the alignment of conjugated–π groups, while the remaining parameters were maintained at their default settings.
Next, to ascertain the stability of the crystal ligand’s with Mpro, we conducted a redocking process centering GC-376* and the grid that was built previously. Throughout this procedure, default parameters were strictly followed, which involved assigning the van der Waals radii of nonpolar ligand atoms to 0.8, setting a partial charge cutoff of 0.15, and employing full-flexible docking. The docking operation utilized the Glide program accessible in Maestro (Friesner et al., 2004; Halgren et al., 2004), by specifying the extra-precision (XP) mode to obtain higher accuracy (Friesner et al., 2006). Additionally, the post-docking minimization was carried out on all generated poses obtained from the previous docking calculation. Subsequently, the top ten conformations for each ligand were selected for further analysis.
Following the insights gained from the redocking procedure, we proceeded to conduct the actual docking calculations for the five different Rutan compounds. For this purpose, all the preprocessed 3D conformations of the Rutan compounds, which were generated using the LigPrep calculations (740), were chosen. These selected conformations were then subjected to the docking procedure, utilizing the previously established grid and the predefined Glide parameters. The identification of the best docking hits was based on a comprehensive evaluation, considering the following criteria: (i) graphical investigation on the binding site of Mpro, (ii) binding mode comparison with crystal ligand; i.e., comparison of the binding modes of the docked ligands with that of the crystal ligand to gauge their similarity and accuracy. (iii) glide scores: the glide scores obtained from the docking output are an integral part of the docking calculations and were utilized as a quantitative measure to rank and evaluate the binding affinities of the inhibitors. (iv) bonded and non-bonded interactions: detailed scrutiny of bonded (covalent) and non-bonded (non-covalent) interactions between the ligands and the receptor was performed to gain insights into the stability and specificity of the binding. These multi-faceted assessments allowed us to identify and select the most promising docking outputs for further analysis.
To substantiate the binding interactions of the docked complexes, we employed Molecular Dynamics (MD) simulation techniques, following a protocol consistent with our prior research studies (Marimuthu et al., 2021; Perumal et al., 2022). To execute this process, we applied five representative holo complexes, (Rutan R5 to R8) that are bound within the binding pocket of Mpro. These complexes were then subjected to the system builder panel in Maestro, where we constructed five independent simulation systems using the simulation parameters as detailed in Table 1.
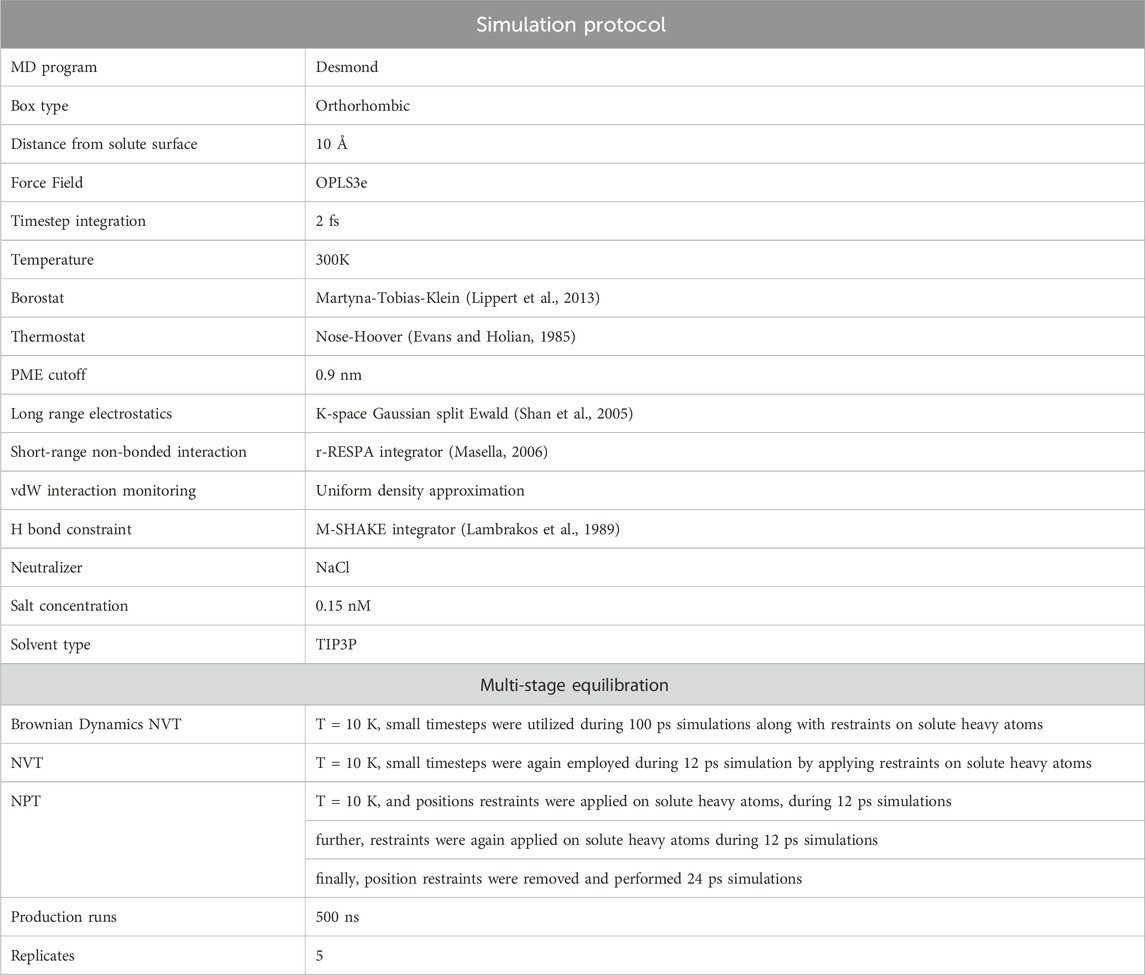
Table 1. The simulation protocol involved in conducting long-range simulations for all five different Mpro-Rutan complexes using Desmond, available in Maestro (Bowers et al., 2006).
The analysis of all trajectories obtained from the molecular MD simulations was conducted using the built-in modules, such as Simulation Quality Analysis and Simulation Interaction Diagram, available in Maestro. Furthermore, we calculated the overall count of intermolecular hydrogen bonds formed between the ligands, and the binding site residues of the Mpro was computed using the Interaction Count module.
For the estimation of Binding Free Energy (BFE) values pertaining to the Mpro–Rutan compounds, we utilized the Prime thermal_mmgbsa.py (Jacobson et al., 2004) script, an integral component of the Maestro suite. The script employs the Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) method to compute the BFE values for the given Mpro–Rutan complexes using the OPLS3e force field and the Variable Solvent Generalized Born (VSGB) solvation model, using default parameters. The OPLS3e force field integrates the CM1A-BCC charge model, which combines Cramer-Truhlar CM1A charges with extensive Bond Charge Correction (BCC) parameters. Additionally, default dielectric constant cutoff values (1 for the solute and 80 for the solvent) were used. Throughout the computations, the program provided estimations for various energy components, encompassing H-bond interactions, van der Waals forces, Generalized Born (GB) solvation energies, Coulombic interactions, π-π stacking interactions, lipophilic interactions, and self-contact interaction terms. These calculations were conducted separately for the Mpro, Rutans, and the Mpro–Rutan complexes.
Furthermore, the ∆G values can be subdivided into Elipophilic, Eelectrostatics, and EvdW interaction components,
Where Eelectrostatics = EH bond + Ecoloumb + EGB_solvation and EvdW = EvdW + Eπ-π + Eself-contact and Elipophilic.
The MM-GBSA-based energy estimation method in Maestro does not include the calculation of conformational entropy, thus, the entropic values were not determined.
The assay kit was purchased from BPS Bioscience. In short, the mechanism of the coronavirus Mpro test is based on the inhibition of the protease activity of the Mpro that cleaves a specific peptide substrate, in which fluorescence is produced. In the presence of an inhibitor, the Mpro is blocked, and non-cleaved peptide substrates do not produce or produce low fluorescence. The fluorescence intensity is correlated, i.e., directly proportional to the percentage inhibition.
The experiment was carried out according to the manufacturer’s protocol. For this assay, Mpro (4 ng/μL, i.e., 120 ng) was first treated with 10 μL of the test sample and mixed with Mpro substrate (diluted in 10 μL assay buffer). The microplate was left overnight at room temperature, after which the fluorescence was measured by exciting at 360 nm and detecting at 460 nm.
The inhibition effect on Mpro was calculated using the formula:
The stability of the components was checked in various pH media ranging from pH 5 to 9: 0.02 M sodium acetate buffer was used for pH 5.0; 0.02 M sodium phosphate buffer was used for solutions with pH 6.0 and 7.0; 0.02 M ammonium bicarbonate buffer was used for preparing solutions with pH 8.0-9.0. For stability analysis, 2.0 mg of Rutan substance was dissolved in 2 mL of appropriate buffer. The prepared solutions were incubated at 37°C for 1 h and 24 h. Then, their stabilities were checked using HPLC, Agilent Technologies series 1,200 s equipped with DAD detector, and column 4.6 × 250 mm Zorbach Eclipse C18, 5 µm. A: 0.1% trifluoroacetic acid (pH 2.5), B: CH3CN. We used the following gradients: 15%–3 min; 25%–17 min; 60%–8 min; 15%–2 min.
Evaluation of GC-376* redocking and Rutan compounds to the binding pocket of Mpro: To validate our docking protocol, we conducted a redocking process by subjecting the modified GC-376* to its native state GC-376, and subsequently compared the outcomes by superimposing the docking results (Figure 3F). The analysis reveals a remarkable similarity between the native and redocked conformations. Furthermore, our results underscore that the redocked conformation adeptly preserves all crucial interactions, including vital polar contacts.
Consequently, for the current study, we adhered to the same docking parameters, employing the Glide program for all five Rutan compounds. Additionally, our comprehensive examination of the binding modes of all Rutan compounds reveals that they occupy the same binding pocket as GC-376*, engaging in analogous interactions (Figures 3G, 4; Table 2). Overall, these findings affirm that all five Rutan inhibitors effectively interact to the same binding pocket of Mpro, collectively covering a significant interaction surface. While molecular docking provides valuable insights into potential binding affinities, it is important to note its semi-quantitative nature. The scoring algorithms influence docking scores and may not fully account for dynamic protein-ligand interactions, particularly in flexible systems such as SARS-CoV-2 Mpro. To mitigate these limitations, we conducted molecular dynamics simulations, which allowed us to study the stability of the binding interactions over time. However, force field limitations in MD simulations, such as approximations in protein flexibility and solvation effects, can also influence the accuracy of the results. Experimental validation, including enzymatic assays or co-crystallization studies, is essential to confirm the inhibitory effects observed in silico.

Figure 4. Docking of five different Rutan compounds (spheres) to the binding pocket of Mpro: (A, C, E, G, I). The 3D and (B, D, F, H, J) 2D representation of Rutan compounds interacting to Mpro binding pocket. The pink arrows represent the H-bond interactions with specific residues inside the binding pocket of Mpro, while the direction of the arrows represents the donor and acceptor atoms involved.

Table 2. List of residues of Mpro interacting with five different Rutan compounds.
To assess the stability of the five docked complexes and gain an in-depth understanding of the impact of different inhibitors with Mpro over time, we have built a series of five individual MD simulation systems. Subsequently, each system was subjected to 500 ns independent production runs. Later, the simulation outputs were rigorously examined for their structural stability using the fully automated panels such as Simulation Quality Analysis, Simulation Interaction Diagram, and Interaction count in Maestro-GUI, and the results were plotted.
Root Mean Squared Deviation (RMSD in Å) estimation for the Mpro–Rutan compounds: The structural deviations were computed for all the Cα atoms of each Mpro-Rutan complex throughout the MD simulation (Figure 5A). The RMSD graphs illustrate that all Mpro-Rutan complexes swiftly reached an equilibrium phase following an initial relaxation period, showing deviations ranging from approximately 1.75–2.65 Å from their starting structures. Specifically, the RMSD graphs for Mpro complexed with R5, R6, and R8 compounds exhibit a consistent equilibrium phase, demonstrating a stable equilibration phase throughout the simulation, maintaining the RMSD value of ∼1.75 Å, each. This indicates that when R5, R6, and R8 compounds bind to Mpro, they do not induce substantial conformational changes compared to their initial states. Similarly, the rmsd values for the Mpro-R7 complex show a stabilized state only after experiencing an initial fluctuation phase for ∼60 ns, later maintaining an overall RMSD value of ∼2.5 Å. However, the RMSD plot for the Mpro-R7’ complex indicates a conformational deviation occurring during earlier in the simulation period, from 0 to 170 ns, followed by reaching an equilibrium phase, sustaining rmsd values between ∼1.8 and 2.3 Å. Overall, the RMSD analysis across the five distinct simulation outputs involving Mpro-Rutan complexes suggests that the binding of Rutans to Mpro does not significantly alter the overall protein conformation. Hence, the frames obtained from the final 200 ns of the MD simulations are considered reliable and suitable for further analysis.
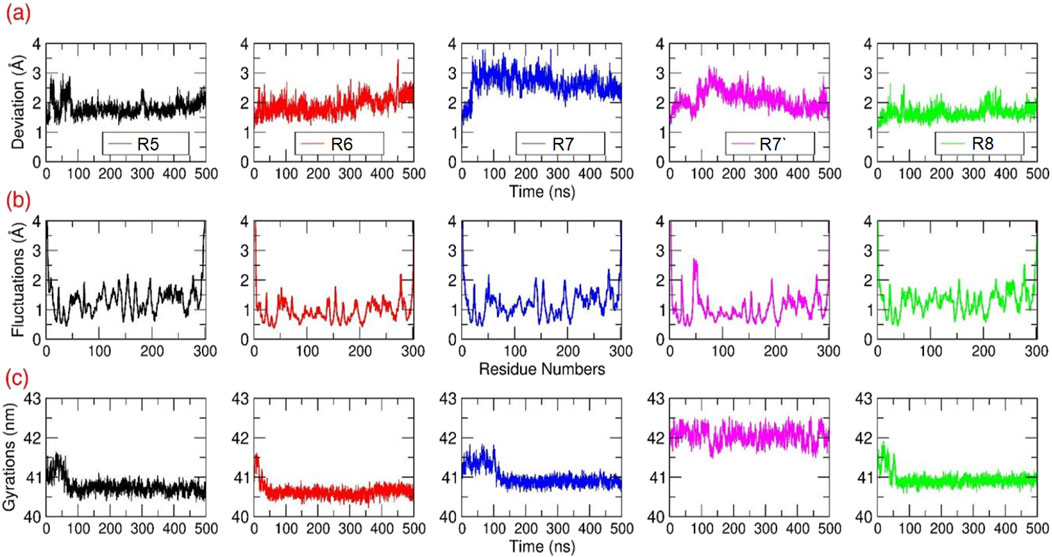
Figure 5. Structural Analysis: The (A) RMSD (B) RMSF and (C) Rg values in Å were calculated for all the Cα atoms of Mpro bound to different Rutans based on the 500 ns MD simulations.
Root Mean Square Fluctuations (RMSF in Å) estimation for the Mpro–Rutan complexes: The structural flexibility of all the Cα atoms present in each Mpro-Rutan complexes were computed during the MD simulation (Figure 5B). Overall, the RMSF graphs displayed multiple segments of structural stability and flexibility in Cα atoms of the Mpro structure in the same trend. i.e., the Mpro exhibited (i) elevated fluctuations were observed at the loop and termini regions, as anticipated, and (ii) distinct differences in backbone fluctuations were evident among the inhibitors, especially at the binding site region. Additionally, the residues involved in the binding site region exhibited only a lower level of fluctuations for the compounds R5 to R8. This indicated that Rutans exhibiting weak binding affinity displayed moderate or weak interaction with the binding site residues, leasing to higher fluctuation compared to other complexes. Conversely, inhibitors with strong binding affinity demonstrated tight and stable interactions with the binding site residues, resulting in reduced or less fluctuation of Mpro.
Radius of Gyrations (Rg in Å) estimation for the Mpro–Rutan complexes: The Rg analysis from the MD simulation will provide comprehensive information on the change in the overall internal structure or the compactness of the macromolecule due to ligand binding. During the simulation, the overall compactness is measured based on rms-distance of all Cα atoms of Mpro from its center-of-gravity. In this study, the Rg values were extracted for all Mpro-Rutan complexes from all the trajectories and plotted (Figure 5C). The Rg graph shows that from the starting initial time period until ∼100 ns, the Mpro experienced a minor fluctuation in all the systems. Subsequently, the Mpro maintained stability throughout the simulation until the end, with the stable Rg values between ∼40.5 and 41 nm for the systems R5-R7 and R8, respectively, whereas the Mpro-R7’ maintains its Rg values at ∼42 nm. Overall, the Rg plot displays only a relatively minor change in the structural compactness during the simulation, which signifies that the binding of Rutans did not majorly alter the internal compactness of Mpro.
One work has reported that the GC-376 inhibitor effectively occupied the binding pocket of Mpro (Ma et al., 2020). This binding pocket is encompassed by a network primarily composed of hydrophobic residues, prominently featuring L27, M49, L50, F140, L141, G143, C145, H163, H164, M165, L167 G170, V186, and A191. Moreover, their study revealed that inhibitor GC-376 engaged in interactions with C145 through bisulfite adducts, leading to the formation of covalent complexes characterized by a tetrahedral arrangement. Additionally, the GC-376 inhibitor was found to establish various other interactions, notably hydrogen bond interactions, with essential residues, including T26, S46, F140, C145, H163, H164, and electrostatic interactions with N142, E166, and Q189.
Concurrently, in addition to the initial structural investigation, we also explored the H bond interaction pattern of the Rutan compounds with the binding pocket residues of Mpro towards the complex formation. The initial graphical assessment of MD trajectories focusing on the interaction between Rutans and Mpro underscores a robust interaction within the binding pocket. This interaction is characterized by a firm establishment of both hydrophobic and polar interactions, with the involvement of additional residues similar to the native crystal structure contributing to the complexity of the binding. Moreover, for a deeper insight into the stability of this complex, we systematically tracked the total number of polar interactions (Figure 6A) between Mpro and Rutans over the course of the simulations.
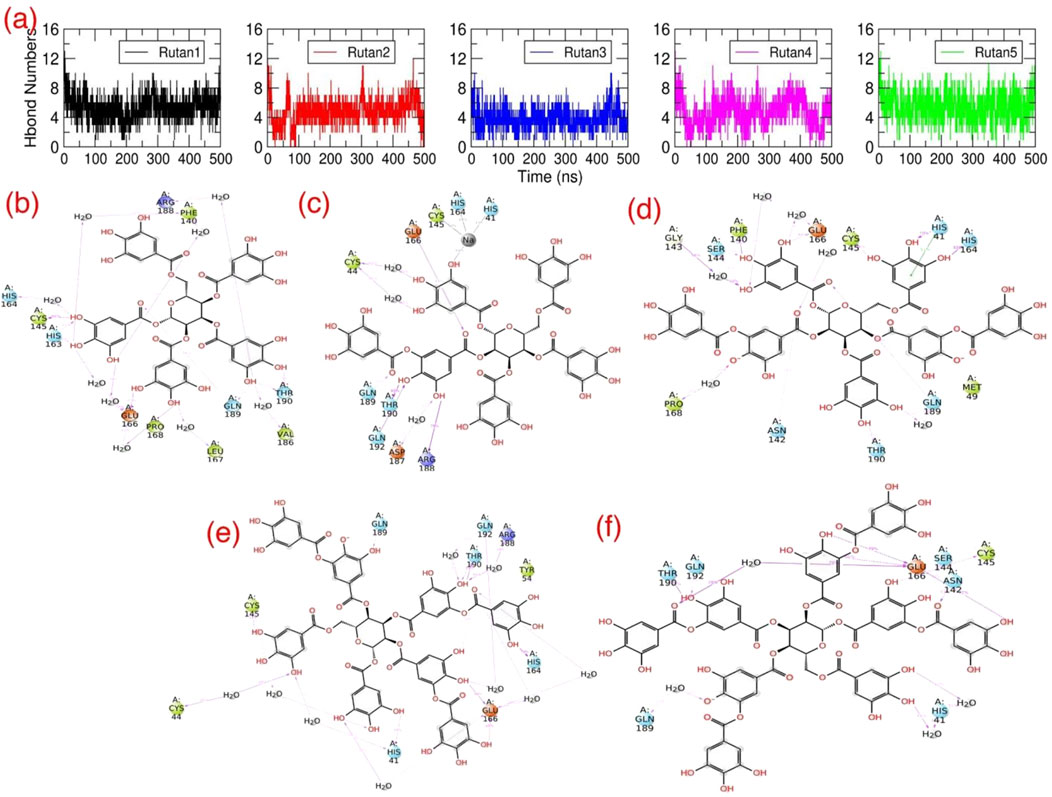
Figure 6. H bond interactions of Rutan compounds with Mpro: (A) The total number of polar contacts and (B–F) 2D representations of polar interactions between Mpro and individual Rutan compounds over time. The occupancies of each polar interaction are presented in percentages.
The graph shows that during the simulation, all five complexes have revealed a significant number of polar interactions with the surrounding residues to maintain the Mpro-Rutan complexity (Figure 6A) that helped the compounds to stay intact within the binding pocket. In this aspect, the R5 and R8 compounds exhibited as high as (∼7–8) polar interactions, while the R6 and R7’ compounds exhibited relatively lesser polar contacts, i.e., ∼6 to 7, and the R7 compound exhibited much fewer (∼5–6) in comparison with other Rutans during the simulation. The 2D polar interaction graph (Figures 6B–F) reveals that E166 and Q189 residues of Mpro establish electrostatic interactions, while C145 and T190 contribute polar interactions with all Rutan compounds, whereas H41, H164, and Q192 establish polar interaction in most of the case, providing insights into the stability and frequency of these interactions.
In summary, these findings underscore the significant impact of polar interactions between the binding site residues of Mpro and the inhibitors on complex stability. This, in turn, results in elevated binding affinity values. Notably, complexes with higher binding affinity values exhibited a greater number of polar contacts, whereas those with lower binding affinity values displayed a reduction in such contacts. These insights are indispensable for the precise optimization and augmentation of the binding capabilities of a lead compound, particularly when targeting a receptor with significant activity initially detected during screening.
The essential role played by Mpro in the SARS-CoV2 viral replication process underscores the urgent need to unravel its mechanistic intricacies and expedite the discovery of high-affinity inhibitors specifically tailored for Mpro. To achieve this, we leverage advanced computational programs, both open-source and commercial, in association with the state-of-the-art high-performance computing systems. In our research, these computational tools facilitated rapid prediction of Binding Free Energies (BFEs) for a set of five distinct Rutan compounds, leveraging their three-dimensional molecular coordinates. This approach has the potential to streamline the design and evaluation of Mpro complexed with Rutan inhibitors, promising enhanced precision and efficacy in this process. Specifically, we extensively employed the thermal_mmgbsa.py—a built-in trajectory post-processing tool in Maestro—to predict BFE values for all five Mpro–Rutan complexes, averaging results from five replicates (Table 3; Supplementary Table S1). Utilizing 100 snapshots from the last 200 ns of the MD trajectories, the predicted BFE values for these complexes are as follows: −81.11, −93.75, −67.24, −92.11, and −104.63 kcal/mol, respectively.
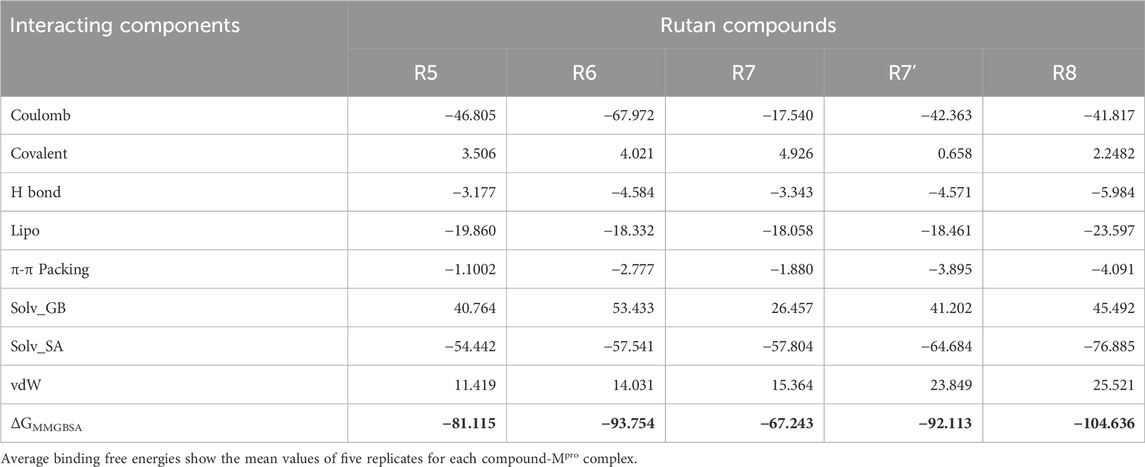
Table 3. Binding free-energy (kcal/mol) estimation obtained from the last 200 ns and an average of five replicates of MD simulations and its individual components using MMGBSA approach.
Apart from determining the total ΔGbind, the program also provides a detailed insights into various interaction components, encompassing Coulombic forces, covalent binding, hydrogen bonding, lipophilic interactions, π−π stacking, Solv_GB (Generalized Born solvation), solvation surface accessibility (Solv_SA), and van der Waals forces (Table 3). These individual interacting components hold immense potential for future applications, especially in the domain of novel drug discovery for Mpro inhibition. Moreover, these components offer an extensive understanding of the intricate binding process between Rutan and Mpro. Consequently, the predicted ΔGbind is explored for its constituent energy components, enriching our understanding of the underlying binding mechanisms.
Among these individual energy components, Coulombic, Lipophilic, and Solv_SA terms exclusively contribute the most favorable energies for complex formation, while the hydrogen bond component provides relatively less favorable energy across all Rutan compounds. Notably, these energy trends align with the total number of hydrogen bond analyses depicted in Figure 6. Conversely, the Solv_GB and vdW energy terms contribute higher unfavorable energies, while the Covalent energy terms contribute relatively less unfavorably to the R5, R6, R7, and R8 compounds, and negligibly unfavorably to the R7’ system. Overall, the analysis of individual energy components across all simulation systems involving Mpro complexed with multiple Rutan compounds highlights the substantial energies provided by the Coulombic, Lipophilic, and Solv_SA energy terms for complex formation.
In the current study, the per-residue decomposition (PRD) analysis was conducted using the Prime thermal_mmgbsa.py script, with the same number of frames and an average of five replicates used to compute the BFE values for five different Rutan compounds when bound to Mpro (Table 4 PRD). This decomposition analysis provides profound insights by showcasing the specific key residues at the interface that predominantly contributed to the BFE values due to Rutans binding. Further examination, particularly focused on the binding site residues of Mpro, unraveled intricate variations in energy profiles within a diverse ensemble of hydrophobic and polar residues, shedding light on their distinctive roles in the binding process.
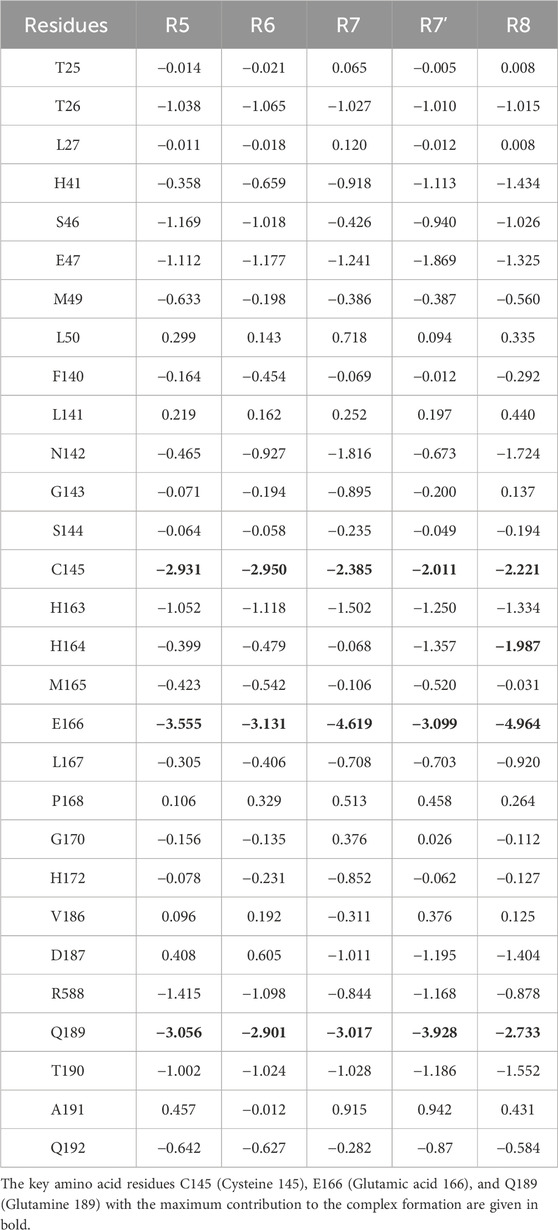
Table 4. The Per-Residue Decomposition (PRD) values extracted for the Mpro binding site residues obtained from the last 200 ns and an average of five replicates of MD simulations. The PRD values were extracted from total BFE values.
While the strong binding affinities of Rutan compounds to the SARS-CoV-2 Mpro active site are encouraging, it is important to evaluate their potential off-target effects. Polyphenolic compounds are known to interact with various proteases and enzymes, which could lead to unintended biological consequences. For instance, flavonoids such as quercetin have been reported to inhibit proteases beyond their intended targets, potentially affecting normal cellular processes (Di Petrillo et al., 2022). To mitigate this concern, future studies should involve broader in vitro screenings against a panel of proteases to assess the specificity of Rutan compounds. Additionally, in vivo toxicity studies will be essential to evaluate their pharmacokinetic and pharmacodynamic profiles. By understanding the potential off-target interactions, we can design more selective derivatives to minimize adverse effects.
Here, the key residues with the maximum contribution to the complex formation (hot spot residues) are extracted from all binding site residues by assigning −2 kcal/mol (Table 4; Figure 7). Based on thiscriteria, the residues with maximum contributions for Rutan compounds are C145, E166, and Q189.
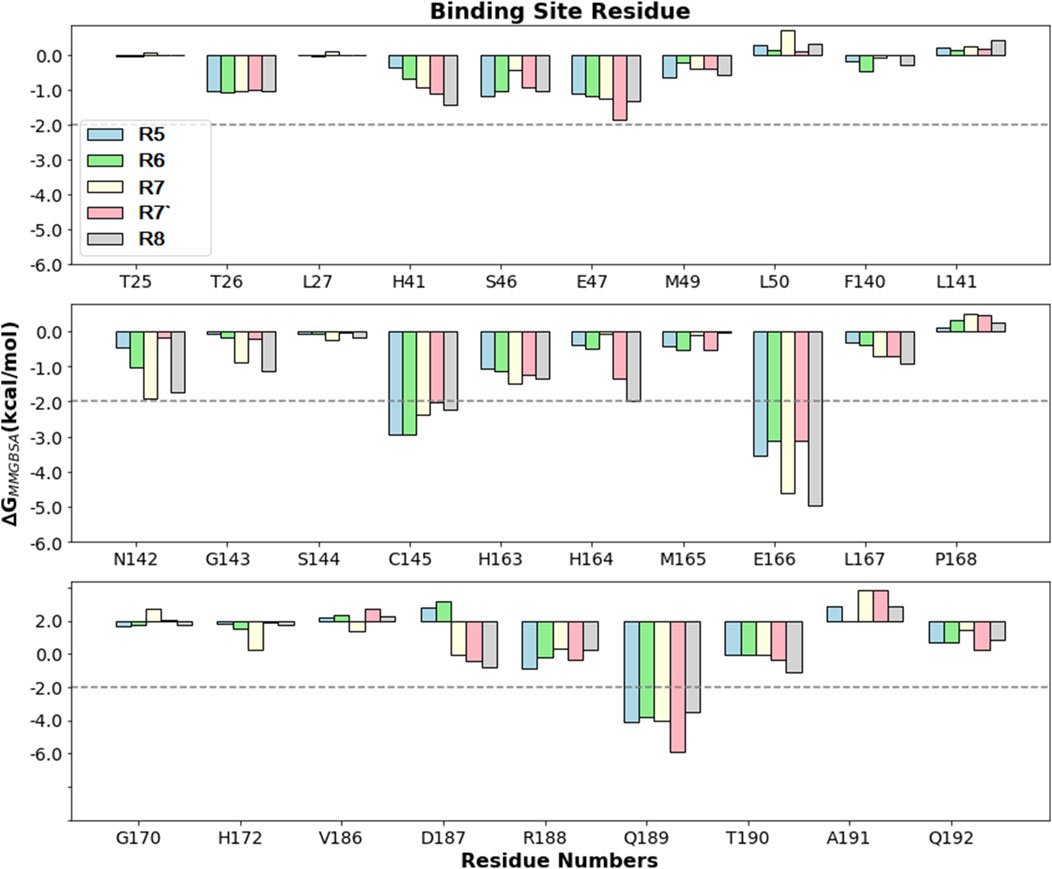
Figure 7. Comparison of PRD (kcal/mol) for different Rutan compounds bound to Mpro binding site residues. The −2 kcal/mol benchmark value is assigned to obtain the hotspot residues.
Polyphenols are low-acidic compounds, and acidic conditions do not affect their structures (Xiao, 2022). We applied pH conditions 5–9 and incubated for 24 h to investigate the stability of the Rutan and its components in various pH conditions (Figure 8). The obtained results indicated that all three fractions were stable at pH 8 and pH 9. We did not observe pH-dependent changes. Reversely, changes that were not observed in pH 6, pH 8, and pH 9 were found in pH 5 and pH 7. Noticeable degradation was observed for R5 in pH 5 and pH 7, around 15% and 25%, respectively. The mixture of R6 + R7 + R7’ showed 83% and 80% stability at pH 5 and pH 7 conditions for 24 h. Among the fractions, the R8 was found to be the most stable in all pH conditions. Overall, our study confirmed the stability of Rutan components in 5–9 pH conditions.
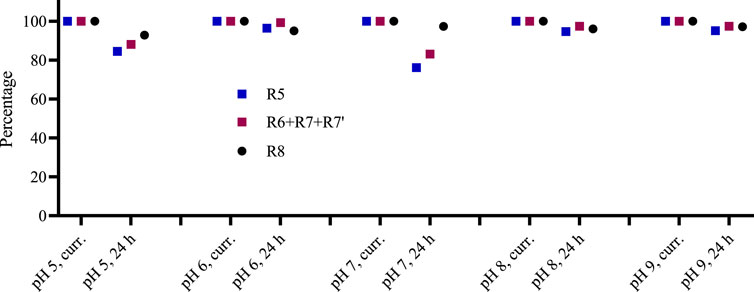
Figure 8. The stability of Rutan compounds was assessed at various pH conditions in freshly prepared solutions and after 24 h (n = 2, SD is less than 3% in all cases).
Rutan is a mixture of polyphenolic compounds, marked as R5, R6, R7, R7’, and R8. The mixture and its compounds separated individually R5 and R8, as well as the fraction containing R6, R7, and R7’, were tested for the Mpro inhibitory activity at 10 μg/mL concentration. The fraction consisting of the three components and compound R8 inhibited the enzyme 77.8% and 76.8% accordingly, while the Rutan substance and compound R5 resulted in 50.4% and 42.4% inhibition, respectively (Figure 9). Since the components R5 and R8 are pure compounds, their IC50 values were also calculated and equal to 42.52 µM and 5.48 µM accordingly. The difference between the IC50 values is positively correlated with the corresponding data in Figure 9.
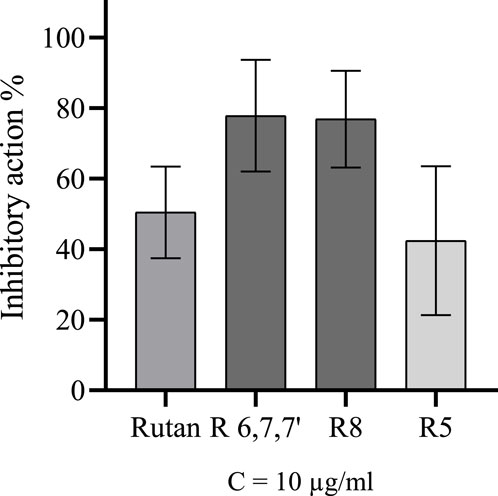
Figure 9. Inhibitory activity of Rutan and its components over Mpro.
The results of the in vitro Mpro inhibitory activity of Rutan and its components align with the findings of the in silico analysis conducted in this study. Consistent with the computational predictions the R8 component demonstrated the highest efficacy, while the fraction of R6, R7, and R7’ also exhibited significant inhibitory potential. The highest inhibition rate of Mpro by the fraction consisting of R6, R7, and R7’ can be linked with calculated ΔGMMGBSA values of R6 and R7’, which were higher than the value of R5. The lowest in vitro inhibitory efficacy belonged to R5, which caused twice the lower inhibitory activity of the total phenolics in the composition of Rutan compared to R8 or the fraction.
According to Figure 9, compound R8 and the fraction holding R6, R7, and R7’ have almost 80% inhibitory activity, much higher than the inhibitory activity of R5. The computed binding free energies for R8, R6, R7, and R7’ were calculated to be −104.636, −93.754, −67.243, and −92.113 kcal/mol, respectively, which are not in complete correspondence with in vitro results. As the mixture of R6, R7, and R7’ exhibited 77.8% inhibition of Mpro, their ΔGMMGBSA values also should not be different from the ΔGMMGBSA values of R8 theoretically. This discrepancy might be linked to the probable synergistic effect of R6, R7, and R7’ compounds (Salikhov et al., 2023). Their combined action could enhance their efficacy as potent Mpro inhibitors. However, further in-depth research is required to confirm and elucidate this synergistic mechanism.
Plant-based natural products, including flavonoids, alkaloids, and polyphenols, have been extensively studied for their antiviral potential against SARS-CoV-2. For example, quercetin, a well-known flavonoid, has shown moderate inhibitory effects on SARS-CoV-2 Mpro, but its bioavailability and stability remain significant challenges (Xu et al., 2023). Similarly, berberine, an alkaloid, has demonstrated strong binding affinity in computational studies, yet its cytotoxicity limits its therapeutic application (Oner et al., 2023). In contrast, sumac-derived polyphenolic compounds, particularly Rutan, offer distinct advantages. These compounds exhibit strong binding affinities to SARS-CoV-2 Mpro, as demonstrated in our molecular docking and dynamics studies, while maintaining low cytotoxicity, as supported by our in vitro assays. Additionally, the unique molecular structure of Rutan facilitates stable interactions with key residues such as C145 and E166, which are crucial for protease inhibition. This makes sumac polyphenols a promising class of natural inhibitors with potential for further development.
Mpro inhibitory activity of reference drug GC376 was equal to IC50 = 0.27 µM, which was several times more active than the Rutan or its components alone. It might be the suitable structure of GC376 for the protease pocket and covalent bond interactions with the Cys145 amino acid of the Mpro active pocket (PDB ID: 6WTT) (Ma et al., 2020). However, Rutan compounds deserve more attention because of their very low toxicity and side effects, even in high doses. Early clinical studies of Rutan tablets on randomly selected patients infected with SARS-CoV-2 demonstrated significantly lower post-COVID-19 manifestations after administration of 25 mg of Rutan to children and 100 mg drug to adults. Besides, significantly lower levels of C-reactive protein were attributed to Rutan administration in adults. The administered 100 mg dose to adults two times a day was found effective for patients with mild COVID-19 disease (Salikhov et al., 2023).
The in vitro results showed comparable Mpro-inhibitory effects of the R8 and the mixture of R6, R7 and R7’ compared to R5. We established IC50 values of two polyphenolic compounds R5 and R8 towards Mpro. The mechanism of action of Mpro-Rutan components determined by computational methods affirmed that all five Rutan components formed a stable complex with Mpro, effectively interacting with its binding pocket and covering a significant interaction surface. Notably, the binding of Rutans did not significantly alter the overall conformation or structural compactness of Mpro during the simulation. BFE calculations of compounds revealed that R8, R6, and R7’ could be the most active components compared to others. Further energy analyses of individual components across all simulation systems involving Mpro complexed with multiple Rutan compounds highlight the substantial contributions of Coulombic, lipophilic, and Solv_SA energy terms for complex formation. Key residues, including C145, E166, and Q189 of Mpro played essential roles in complex formations with the Rutan components. These findings enhance the fundamental understanding of the mechanistic action of polyphenols with antiviral efficacy against COVID-19, paving the way for further exploration of their therapeutic potential.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
MK: Conceptualization, Writing–review and editing. PM: Conceptualization, Methodology, Software, Writing–original draft. JR: Data curation, Validation, Writing–review and editing. NM: Investigation, Writing–original draft. AA: Writing–review and editing. NB: Investigation, Writing–original draft. JZ: Writing–review and editing. AY: Investigation, Writing–original draft. YO: Writing–review and editing. SS: Writing–review and editing. SM: Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. PM gratefully acknowledges the use of the bioinformatics infrastructure facility supported by Biocenter Finland, Joe, Pentti and Tor Borg Memorial Fund 2021, Juhani Ahon Lääketieteen Tutkimussäätiö sr. for their grant support, CSC-IT Center for Science (Project: 2000461) for the computational facility; Dr. Jukka Lehtonen (SBL) for the IT support; and Prof. Outi Salo-Ahen (Pharmacy), Åbo Akademi University, for providing the lab support. MK, NM, AA, SS, and SM contributed to work by Project F-FA-2021-359, financed by the Ministry of Higher Education, Science and Innovation of the Republic of Uzbekistan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2025.1518463/full#supplementary-material
Ahmad, P., Alvi, S. S., Hasan, I., and Khan, M. S. (2024). Targeting SARS-CoV-2 main protease (Mpro) and human ACE-2: a virtual screening of carotenoids and polyphenols from tomato (Solanum lycopersicum L.) to combat Covid-19. Intell. Pharm. 2, 51–68. doi:10.1016/j.ipha.2023.10.008
Arunkumar, M., Gunaseelan, S., Kubendran Aravind, M., Mohankumar, V., Anupam, P., Harikrishnan, M., et al. (2022). Marine algal antagonists targeting 3CL protease and spike glycoprotein of SARS-CoV-2: a computational approach for anti-COVID-19 drug discovery. J. Biomol. Struct. Dyn. 40, 8961–8988. doi:10.1080/07391102.2021.1921032
Bahun, M., Jukić, M., Oblak, D., Kranjc, L., Bajc, G., Butala, M., et al. (2022). Inhibition of the SARS-CoV-2 3CL(pro) main protease by plant polyphenols. Food Chem. 373, 131594. doi:10.1016/j.foodchem.2021.131594
Bhardwaj, V. K., Singh, R., Sharma, J., Rajendran, V., Purohit, R., and Kumar, S. (2021). Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 39, 3449–3458. doi:10.1080/07391102.2020.1766572
Bowers, K. J., Chow, D. E., Xu, H., Dror, R. O., Eastwood, M. P., Gregersen, B. A., et al. (2006). “Scalable algorithms for molecular dynamics simulations on commodity clusters,” in SC '06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, November 11-17, 2006 (IEEE), 43.
Chen, C. N., Lin, C. P., Huang, K. K., Chen, W. C., Hsieh, H. P., Liang, P. H., et al. (2005). Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3,3'-digallate (TF3). Evid. Based Complement. Altern. Med. 2, 209–215. doi:10.1093/ecam/neh081
Choudhary, M. I., Shaikh, M., Tul-Wahab, A., and Ur-Rahman, A. (2020). In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS One 15, e0235030. doi:10.1371/journal.pone.0235030
DI Petrillo, A., Orrù, G., Fais, A., and Fantini, M. C. (2022). Quercetin and its derivates as antiviral potentials: a comprehensive review. Phytother. Res. 36, 266–278. doi:10.1002/ptr.7309
DI Pierro, F., Khan, A., Iqtadar, S., Mumtaz, S. U., Chaudhry, M. N. A., Bertuccioli, A., et al. (2022). Quercetin as a possible complementary agent for early-stage COVID-19: concluding results of a randomized clinical trial. Front. Pharmacol. 13, 1096853. doi:10.3389/fphar.2022.1096853
Evans, D. J., and Holian, B. L. (1985). The nose–hoover thermostat. J. Chem. Phys. 83, 4069–4074. doi:10.1063/1.449071
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi:10.1021/jm0306430
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196. doi:10.1021/jm051256o
Graham, H. N. (1992). Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 21, 334–350. doi:10.1016/0091-7435(92)90041-f
Greenwood, J. R., Calkins, D., Sullivan, A. P., and Shelley, J. C. (2010). Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 24, 591–604. doi:10.1007/s10822-010-9349-1
Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759. doi:10.1021/jm030644s
Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., et al. (2016). OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296. doi:10.1021/acs.jctc.5b00864
Jacobson, M. P., Pincus, D. L., Rapp, C. S., Day, T. J., Honig, B., Shaw, D. E., et al. (2004). A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367. doi:10.1002/prot.10613
Jang, M., Park, Y. I., Cha, Y. E., Park, R., Namkoong, S., Lee, J. I., et al. (2020). Tea polyphenols EGCG and theaflavin inhibit the activity of SARS-CoV-2 3CL-protease in vitro. Evid. Based Complement. Altern. Med. 2020, 5630838. doi:10.1155/2020/5630838
Jang, M., Park, R., Park, Y. I., Cha, Y. E., Yamamoto, A., Lee, J. I., et al. (2021). EGCG, a green tea polyphenol, inhibits human coronavirus replication in vitro. Biochem. Biophys. Res. Commun. 547, 23–28. doi:10.1016/j.bbrc.2021.02.016
Jin, Z., DU, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., et al. (2020). Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293. doi:10.1038/s41586-020-2223-y
Lambrakos, S. G., Boris, J. P., Oran, E. S., Chandrasekhar, I., and Nagumo, M. (1989). A modified shake algorithm for maintaining rigid bonds in molecular dynamics simulations of large molecules. J. Comput. Phys. 85, 473–486. doi:10.1016/0021-9991(89)90160-5
Lippert, R. A., Predescu, C., Ierardi, D. J., Mackenzie, K. M., Eastwood, M. P., Dror, R. O., et al. (2013). Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys. 139, 164106. doi:10.1063/1.4825247
Ma, C., Sacco, M. D., Hurst, B., Townsend, J. A., Hu, Y., Szeto, T., et al. (2020). Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692. doi:10.1038/s41422-020-0356-z
Ma, L., Xie, Y., Zhu, M., Yi, D., Zhao, J., Guo, S., et al. (2022). Identification of darunavir derivatives for inhibition of SARS-CoV-2 3CL(pro). Int. J. Mol. Sci. 23, 16011. doi:10.3390/ijms232416011
Marimuthu, P., Gorle, S., and Karnati, K. R. (2021). Mechanistic insights into SARS-CoV-2 main protease inhibition reveals hotspot residues. J. Chem. Inf. Model 61, 6053–6065. doi:10.1021/acs.jcim.1c00928
Masella, M. (2006). The multiple time step r-RESPA procedure and polarizable potentials based on induced dipole moments. Mol. Phys. 104, 415–428. doi:10.1080/00268970500404414
Mengist, H. M., Dilnessa, T., and Jin, T. (2021). Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Front. Chem. 9, 622898. doi:10.3389/fchem.2021.622898
Nagahawatta, D. P., Liyanage, N. M., Je, J. G., Jayawardhana, H., Jayawardena, T. U., Jeong, S. H., et al. (2022). Polyphenolic compounds isolated from marine algae attenuate the replication of SARS-CoV-2 in the host cell through a multi-target approach of 3CL(pro) and PL(pro). Mar. Drugs 20, 786. doi:10.3390/md20120786
Oner, E., AL-Khafaji, K., Mezher, M. H., Demirhan, I., Suhail Wadi, J., Belge Kurutas, E., et al. (2023). Investigation of berberine and its derivatives in Sars Cov-2 main protease structure by molecular docking, PROTOX-II and ADMET methods: in machine learning and in silico study. J. Biomol. Struct. Dyn. 41, 9366–9381. doi:10.1080/07391102.2022.2142848
Otter, C. J., Bracci, N., Parenti, N. A., Ye, C., Tan, L. H., Asthana, A., et al. (2023). SARS-CoV-2 nsp15 endoribonuclease antagonizes dsRNA-induced antiviral signaling. bioRxiv, 2023.11.15.566945. doi:10.1101/2023.11.15.566945
Paiva, L., Lima, E., Motta, M., Marcone, M., and Baptista, J. (2022). Investigation of the azorean camellia sinensis processing conditions to maximize the theaflavin 3,3'-di-O-gallate content as a potential antiviral compound. Antioxidants (Basel) 11, 1066. doi:10.3390/antiox11061066
Panagiotopoulos, A. A., Karakasiliotis, I., Kotzampasi, D. M., Dimitriou, M., Sourvinos, G., Kampa, M., et al. (2021). Natural polyphenols inhibit the dimerization of the SARS-CoV-2 main protease: the case of fortunellin and its structural analogs. Molecules 26, 6068. doi:10.3390/molecules26196068
Pang, X., Xu, W., Liu, Y., Li, H., and Chen, L. (2023). The research progress of SARS-CoV-2 main protease inhibitors from 2020 to 2022. Eur. J. Med. Chem. 257, 115491. doi:10.1016/j.ejmech.2023.115491
Park, J. Y., Yuk, H. J., Ryu, H. W., Lim, S. H., Kim, K. S., Park, K. H., et al. (2017). Evaluation of polyphenols from Broussonetia papyrifera as coronavirus protease inhibitors. J. Enzyme Inhib. Med. Chem. 32, 504–515. doi:10.1080/14756366.2016.1265519
Park, J., Park, R., Jang, M., and Park, Y. I. (2021). Therapeutic potential of EGCG, a green tea polyphenol, for treatment of coronavirus diseases. Life (Basel) 11, 197. doi:10.3390/life11030197
Patil, R., Chikhale, R., Khanal, P., Gurav, N., Ayyanar, M., Sinha, S., et al. (2021). Computational and network pharmacology analysis of bioflavonoids as possible natural antiviral compounds in COVID-19. Inf. Med. Unlocked 22, 100504. doi:10.1016/j.imu.2020.100504
Perumal, M., Marimuthu, P., and Chen, X. (2022). Investigation into the site-specific binding interactions between chlorogenic acid and ovalbumin using multi-spectroscopic and in silico simulation studies. J. Biomol. Struct. Dyn. 40, 6619–6633. doi:10.1080/07391102.2021.1886992
Rashid, F., Xie, Z., Suleman, M., Shah, A., Khan, S., and Luo, S. (2022). Roles and functions of SARS-CoV-2 proteins in host immune evasion. Front. Immunol. 13, 940756. doi:10.3389/fimmu.2022.940756
Ren, Z., Yan, L., Zhang, N., Guo, Y., Yang, C., Lou, Z., et al. (2013). The newly emerged SARS-like coronavirus HCoV-EMC also has an “Achilles’ heel”: current effective inhibitor targeting a 3C-like protease. Protein Cell 4, 248–250. doi:10.1007/s13238-013-2841-3
Roos, K., Wu, C., Damm, W., Reboul, M., Stevenson, J. M., Lu, C., et al. (2019). OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 15, 1863–1874. doi:10.1021/acs.jctc.8b01026
Salikhov, S. I., Abdurakhmonov, I. Y., Oshchepkova, Y. I., Ziyavitdinov, J. F., Berdiev, N. S., Aisa, H. A., et al. (2023). Repurposing of Rutan showed effective treatment for COVID-19 disease. Front. Med. (Lausanne) 10, 1310129. doi:10.3389/fmed.2023.1310129
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., and Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234. doi:10.1007/s10822-013-9644-8
Shan, Y., Klepeis, J. L., Eastwood, M. P., Dror, R. O., and Shaw, D. E. (2005). Gaussian split Ewald: a fast Ewald mesh method for molecular simulation. J. Chem. Phys. 122, 54101. doi:10.1063/1.1839571
Sharma, J., Kumar Bhardwaj, V., Singh, R., Rajendran, V., Purohit, R., and Kumar, S. (2021). An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 346, 128933. doi:10.1016/j.foodchem.2020.128933
Shelley, J. C., Cholleti, A., Frye, L. L., Greenwood, J. R., Timlin, M. R., and Uchimaya, M. (2007). Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691. doi:10.1007/s10822-007-9133-z
Singh, R., Bhardwaj, V. K., and Purohit, R. (2021). Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: an in-silico approach. Comput. Biol. Med. 139, 104965. doi:10.1016/j.compbiomed.2021.104965
Singh, R., Bhardwaj, V. K., and Purohit, R. (2022). Inhibition of nonstructural protein 15 of SARS-CoV-2 by golden spice: a computational insight. Cell Biochem. Funct. 40, 926–934. doi:10.1002/cbf.3753
Singh, R., and Purohit, R. (2024). Multi-target approach against SARS-CoV-2 by stone apple molecules: a master key to drug design. Phytother. Res. 38, 7–10. doi:10.1002/ptr.7772
Tito, A., Colantuono, A., Pirone, L., Pedone, E., Intartaglia, D., Giamundo, G., et al. (2021). Pomegranate peel extract as an inhibitor of SARS-CoV-2 spike binding to human ACE2 receptor (in vitro): a promising source of novel antiviral drugs. Front. Chem. 9, 638187. doi:10.3389/fchem.2021.638187
Wang, J. (2020). Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model 60, 3277–3286. doi:10.1021/acs.jcim.0c00179
Weng, Y. L., Naik, S. R., Dingelstad, N., Lugo, M. R., Kalyaanamoorthy, S., and Ganesan, A. (2021). Molecular dynamics and in silico mutagenesis on the reversible inhibitor-bound SARS-CoV-2 main protease complexes reveal the role of lateral pocket in enhancing the ligand affinity. Sci. Rep. 11, 7429. doi:10.1038/s41598-021-86471-0
Xiao, J. (2022). Recent advances on the stability of dietary polyphenols. eFood 3, e21. doi:10.1002/efd2.21
Xu, H., Li, S., Liu, J., Cheng, J., Kang, L., Li, W., et al. (2023). Bioactive compounds from Huashi Baidu decoction possess both antiviral and anti-inflammatory effects against COVID-19. Proc. Natl. Acad. Sci. U. S. A. 120, e2301775120. doi:10.1073/pnas.2301775120
Yang, F., Jiang, X. L., Tariq, A., Sadia, S., Ahmed, Z., Sardans, J., et al. (2022). Potential medicinal plants involved in inhibiting 3CL(pro) activity: a practical alternate approach to combating COVID-19. J. Integr. Med. 20, 488–496. doi:10.1016/j.joim.2022.08.001
Yu, W. D., Jin, Q. Y., Zeng, M. S., Liu, J. Y., and Xu, P. P. (2022). Geraniin as a potential inhibitor of SARS-CoV-2 3CL(pro). Nat. Prod. Res. 36, 6060–6063. doi:10.1080/14786419.2022.2043308
Keywords: Rutan, SARS-CoV-2 Mpro, docking, MD simulations, in vitro analysis
Citation: Kayumov M, Marimuthu P, Razzokov J, Mukhamedov N, Asrorov A, Berdiev NS, Ziyavitdinov JF, Yashinov A, Oshchepkova Y, Salikhov S and Mirzaakhmedov S (2025) Computational and in vitro evaluation of sumac-derived ©Rutan compounds towards Sars-CoV-2 Mpro inhibition. Front. Pharmacol. 16:1518463. doi: 10.3389/fphar.2025.1518463
Received: 28 October 2024; Accepted: 20 January 2025;
Published: 04 February 2025.
Edited by:
Joan Villena García, Universidad de Valparaíso, ChileReviewed by:
Nagendra Kumar Kaushik, Kwangwoon University, Republic of KoreaCopyright © 2025 Kayumov, Marimuthu, Razzokov, Mukhamedov, Asrorov, Berdiev, Ziyavitdinov, Yashinov, Oshchepkova, Salikhov and Mirzaakhmedov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muzaffar Kayumov, bXFheXVtb3YwODA4QGdtYWlsLmNvbQ== Parthiban Marimuthu, cGFydGhpYmFuLm1hcmltdXRodUBhYm8uZmk=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.