 Anastasiia Kiprina1†‡
Anastasiia Kiprina1†‡ Tom Teichmann2,3†‡
Tom Teichmann2,3†‡ Virna Margarita Martín Giménez4
Virna Margarita Martín Giménez4 Wenqing Xu1
Wenqing Xu1 Fiona Sailer1
Fiona Sailer1 Maike Windbergs5
Maike Windbergs5 Walter Manucha6,7
Walter Manucha6,7 Andreas Weigert1*†
Andreas Weigert1*† Ralf P. Brandes2,3*
Ralf P. Brandes2,3*- 1Institute of Biochemistry I, Faculty of Medicine, Goethe University Frankfurt, Frankfurt, Germany
- 2Institute for Cardiovascular Physiology, Goethe University Frankfurt, Frankfurt, Germany
- 3German Centre of Cardiovascular Research (DZHK), Partner site RheinMain, Frankfurt, Germany
- 4Instituto de Investigaciones en Ciencias Químicas, Facultad de Ciencias Químicas y Tecnológicas, Universidad Católica de Cuyo, San Juan, Argentina
- 5Institute of Pharmaceutical Technology, Goethe University Frankfurt, Frankfurt, Germany
- 6Instituto de Medicina y Biología Experimental de Cuyo (IMBECU), Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET), Mendoza, Argentina
- 7Departamento de Patología, Área de Farmacología, Facultad de Ciencias Médicas, Universidad Nacional de Cuyo, Mendoza, Argentina
Introduction: Anandamide (AEA) is an endocannabinoid that has recently been recognized as a regulator of various inflammatory diseases as well as cancer. While AEA was thought to predominantly engage cannabinoid (CB) receptors, recent findings suggest that, given its protective anti-inflammatory role in pathological conditions, anandamide may engage not only CB receptors.
Methods: In this study, we studied the role of exogenous AEA in a mouse AirPouch model of acute inflammation by examining immune cell infiltrates by flow cytometry. Human primary immune cells were used to validate findings towards immune cell activation and migration by flow cytometry and bead-based ELISA.
Results: We found that AEA decreases the acute infiltration of myeloid cells including granulocytes and monocytes into the inflamed area, but unexpectedly increases the number of T cells at the site of inflammation. This was related to AEA signaling through nuclear receptor subfamily 4A (NR4A) transcription factors rather than CB receptors. Exploring regulatory mechanisms in the human system, we found that AEA broadly inhibits the migratory capacity of immune cells, arguing for blocked emigration of T cells from the inflamed tissue. Taking a closer look at the impact of AEA on T cells revealed that AEA profoundly alters the activation and exhaustion status of CD4+ T and CD8+ T cells, thereby strongly inhibiting TH17 responses, while not altering TH1 differentiation.
Discussion: These data suggest that AEA has the potential to block chronic inflammation without influencing crucial anti-viral and anti-microbial immune defense mechanisms, and may therefore be an attractive molecule to interfere with the establishment of chronic inflammation.
Introduction
Endocannabinoids are ligands of the G-protein-coupled endocannabinoid receptors CB1 and CB2 (Lu and Mackie, 2021). The most studied endocannabinoid is N-arachidonoylethanolamine (anandamide, AEA), which is synthesized de novo by cells in response to activation (Lu and Mackie, 2016). AEA has a low half-life in tissue as it is rapidly degraded by fatty acid amide hydrolase (FAAH), cyclooxygenase 2 (COX2), lipoxygenases (LOX) and cytochrome P450 (CYP) enzymes (Maccarrone, 2017).
CB receptors are also expressed outside the brain and elicit a broad spectrum of effects, among them modulation of inflammatory activity. In fact, genetic deletion of CB1 in mice has been shown to promote chronic heart failure (Liao et al., 2013), whereas CB2 results in an increased risk of atherosclerosis (Netherland et al., 2010) as well as cardiomyopathy (Duerr et al., 2014). Deletion of CB1 receptors in myeloid cells limits atherosclerosis development in male mice (Wang et al., 2024) and CB1 receptor activation promotes vascular smooth muscle cell proliferation and neo-intima formation (Molica et al., 2013). Although CB receptors are considered the predominant mediators of AEA signaling, it became clear that they are not the only signaling receptors responding to this nitro-lipid. In fact, extracellular AEA can also bind and activate the transient receptor potential vanilloid type-1 (TRPV1) and transient receptor potential ankyrin type-1 (TRPA1) channels as well as the G-protein coupled receptors GPR55 and GPR119 (Ligresti et al., 2016; Maccarrone, 2017; Pertwee et al., 2010; Zygmunt et al., 1999).
AEA is produced within the central nervous system (CNS), where it was shown to have anti-inflammatory and neuroprotective effects in the context of neuroinflammation (Eljaschewitsch et al., 2006). Moreover, AEA is known to reduce vascular, skin and endotoxin-induced inflammation (Martín Giménez et al., 2022; McCormick et al., 2023; Tomczyk et al., 2021) by acting on CB receptors, but also by triggering epigenetic changes (Martín Giménez et al., 2022). Various studies have demonstrated that AEA has oncoprotective activity against breast, prostate and non-melanoma skin cancers (Sarfaraz et al., 2008; Soliman and van Dross, 2016).
We recently identified that AEA elicits a strong anti-inflammatory effect in vascular smooth muscle cells (VSMCs), which was mediated by an epigenetic modulation through NCoR1 (Pflüger-Müller et al., 2020). Interestingly, we also observed that these effects of AEA require high concentrations and were not mediated by classic AEA receptors, like CB1 and CB2. This constellation may suggest an action of AEA through nuclear receptors (NR). NRs are proteins with transcription factor properties that are typically activated by lipophilic compounds (Frigo et al., 2021). After ligand binding and, if required, nuclear translocation, nuclear receptors, acting as mono or hetero- and homo-dimer, activate gene expression (Martínez-González et al., 2021). In addition to these classic hormone receptors, a broad spectrum of lipid receptors like peroxisome proliferator-activated receptor (PPARs) alter gene expression in response to some poly unsaturated fatty acids (PUFAs) and other ligands (O'Sullivan, 2007). Finally, orphan NRs exist and and some of them even lack a ligand binding site rendering their activity controlled through phosphorylation or abundance and subcellular localization (Mullican et al., 2013). Receptors of the NR4 class belong to the latter group, although more and more compounds are being identified, which appear to bind these receptors and increase their activity. Among them are signaling lipids like prostaglandin A2 or pharmacological compounds like CDIM12 or cytosporone B (Hammond et al., 2015; Rajan et al., 2020; Zhan et al., 2008). We recently reported that AEA binds and activates NR4A1 and NR4A2 to mediate an anti-inflammatory effect in vascular smooth muscle cells (VSMCs) (Teichmann et al., 2024). In fact, there is a good amount of data linking NR4 receptors to inflammatory control: In inflamed human synovial tissue, multiple sclerosis or atherosclerotic lesions, their expression is drastically increased (McMorrow and Murphy, 2011), while in mice, both loss of NR4A1 or NR4A2 was associated with increased inflammation (Bonta et al., 2006; Hamers et al., 2013). Moreover, NR4A receptors are rapidly and strongly induced by various inflammatory cytokines, suggesting a protective role in an acute scenario by helping to resolve inflammation through a negative feedback mechanism, aiming to restore homeostasis in the later stages of inflammation (Rodríguez-Calvo et al., 2017).
A limitation of our previous study on the AEA-mediated activation of NR4 receptors was, that the physiological relevance of the anti-inflammatory effect was only determined in organ culture of the isolated mouse aorta and cultured VSMC. In the present study we therefore set out to determine whether AEA also limits inflammation in vivo. Unexpectedly, we observed a strong, NR4-dependent effect of AEA on T-lymphocytes, which, among others, resulted in a prevention of differentiation towards a TH17 phenotype and rather maintained competence of the cells to respond to acute inflammatory stimulation.
Materials and methods
Animals
Global knockout mice for NR4A1−/−(Nur77), NR4A2−/− (Nur1) and double knockout for NR4A1/2−/− were generated by crossing NR4A1flox/flox (obtained from the Jackson Laboratory) or NR4A2flox/flox mice (kindly provided by Pierre Chambon (Sekiya et al., 2011), with CMV-GT-Rosa-CreERT2TG/0. All knockout animals were generated on the C57BL/6 background. Global deletion of NR4A1 (A1KO) and/or NR4A2 (A2KO) was induced by administering tamoxifen (400 mg/kg in chow) for 10 days, followed by a 14-day tamoxifen-free “wash-out” period. In this study, control animals (WT) are defined as littermates, which did not receive tamoxifen treatment with the chow. All animals had free access to chow and water in a specified pathogen-free facility with a 12 h light/dark cycle and all animal experiments were performed in accordance with the German animal protection law and were carried out after approval by the local authorities (Regierungspräsidium Darmstadt, approval number FU1268). Every mouse received an identification number for each experiment and the experimenter was blinded for the genotype. Animal group sizes differed due to number of available littermates.
Preparation of AEA micellar nanoformulations
AEA is an oily substance, which limits application in vivo and controlled absorption. Therefore, the compound was applied as nanoformulation. The preparation of the AEA micellar nanoformulations was carried out as previously described (Martín Giménez et al., 2023). Briefly, 30 mg of the commercial co-polymer Pluronic® F127 (PF127; BASF, CABA, Buenos Aires, Argentina) was accurately weighed and dissolved in 1 mL of Milli-Q water (Sigma-Aldrich, St. Louis, MO, USA) each. The mixture was continuously stirred at RT until homogeneous and a transparent dispersion was obtained. Subsequently, 750 µg of AEA, dissolved in absolute ethanol (using 15 µL of a commercial AEA ethanolic solution from Cayman Chemical, Ann Arbor, MI, USA), was incorporated (drop by drop) into the polymeric dispersion. Stirring was maintained until complete ethanol evaporation. AEA-free micellar nanoformulations of the PF127 polymer (Pluronic) served as control (CTL).
AirPouch model of acute inflammation
The AirPouch model (Paul-Clark et al., 2012; Pierron et al., 2023) was conducted on both tamoxifen-treated mice following intake and washout, as well as on untreated mice (CTL) that did not receive tamoxifen. Mice were anesthetized using isoflurane inhalation and placed on a heating pad to maintain body temperature. The dorsal skin region was shaved, and the skin was sterilized with 70% ethanol. An AirPouch was created by subcutaneous injection of 5 mL sterile air using a 23-gauge needle, followed by additional injects of 3 mL after 3 days to maintain the pouch. On day 6, 30 µg diclofenac and 10 µg AEA-Pluronic nanoparticles or Pluronic control nanoparticles without AEA (in 0.5 mL 0.9% NaCl) were injected into the pouch. Diclofenac was administered to avoid degradation of AEA by cyclooxygenases. After 1 h of pre-incubation, 1 mL of a 1% zymosan solution was injected into the pouch. After 6 h the pouch content was recovered by injection of 1 mL PBS into the pouch, massage and aspiration of the contained liquids. The concentration used for the final experiments was the result of a dose-escalation study on individual animals to determine an effective concentration.
Primary human macrophage generation, activation, and treatment
Human peripheral blood mononuclear cells were isolated from commercially available buffy coats from anonymous donors (DRK-Blutspendedienst Baden-Württemberg-Hessen, Institut für Transfusionsmedizin und Immunhämatologie, Frankfurt, Germany) using Ficoll density centrifugation. Peripheral blood mononuclear cells were washed twice with PBS containing 2 mM EDTA and thereafter incubated for 2 h under growth conditions in RPMI 1640 media supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL) to enable adherence to culture dishes (Sarstedt, Nümbrecht, Germany). Non-adherent cells were removed. Monocytes were differentiated into naïve macrophages with RPMI 1640 media (Gibco) containing 3% AB-positive human serum (DRK-Blutspendedienst Baden-Württemberg-Hessen, Frankfurt, Germany) for at least 7 days. Differentiated macrophages were exposed to media with 1% FCS overnight. The next day, cells were incubated in RPMI 1640 media with 1% FCS alone, with EtOH as solvent for 2 h, with 10 µM diclofenac (Sigma Aldrich) for 1 h, and treated with 10 µM AEA or EtOH as solvent for 2 h. Diclofenac was used to arrest the cyclooxygenase-mediated breakdown of AEA. After treatment, macrophages were incubated with 10 μg/mL Zymosan (Sigma Aldrich) for 6 h.
T cell isolation, activation and treatment
Primary human peripheral blood cells were isolated from buffy coats of anonymous donors (DRK-Blutspendedienst Baden-Württemberg-Hessen, Institut für Transfusionsmedizin und Immunhämatologie, Frankfurt am Main). T cells were isolated using the EasySep™ Human T Cell Isolation Kit (Stemcell Technologies) through negative selection. The purity of T cells was greater than 95%, as confirmed by flow cytometry. The cells were cultured at concentration 1 × 106 cells/mL in T-cell medium (RPMI 1640, penicillin (100 U/mL), streptomycin (100 μg/mL), FCS (10%), non-essential and essential amino acids (1%), sodium pyruvate (1%) and 1% 4-(2-hydroxyethyl)-1 piperazineethanesulfonic acid (HEPES)). Cells were supplemented with human recombinant IL-2 (10 ng/mL; PrepoTech) at days 0, 2, and 4 and β-mercaptoethanol (50 μM; Gibco). Cells were cultured for up to 6 days. T cells were treated with Diclofenac for 1 h, Diclofenac and ethanol for 2 h, and Diclofenac and AEA for 2 h. After treatment, T cells were left unstimulated and stimulated with an anti-CD3/CD28/CD2 T cell activator (Stemcell) for up to 6 days. At the endpoint, supernatants were collected for cytokine measurement, and cells were analyzed by flow cytometry.
PBMC migration assay
The migration assay was performed using 6.5 mm diameter transwell cell culture inserts (5 µm pore size; Costar). Human PBMCs were isolated from the buffy coats by Ficoll density centrifugation, washed, counted, and incubated overnight in RPMI 1640 media with 10% FCS, human recombinant IL-2 (10 ng/mL), and β-mercaptoethanol (50 μM). The next day, 105 cells were seeded in the insert in serum-free medium. Prior to the migration assay, PBMCs were treated with AEA and Diclofenac or left untreated. Treated cells were used to migrate toward the medium with FCS. PBMCs that did not receive treatment were used for migration toward macrophage-derived supernatants, medium with AEA (EtOH as control), Diclofenac, and Zymosan (10 μg/mL). Cells were allowed to migrate for 3 h in cell culture. Afterwards, migrated and non-migrated cells were analyzed by flow cytometry. The percentage of migrated immune cells was determined by the ratio of migrated/non-migrated cells.
LegendPlex
The concentrations of CCL2, CCL20, CCL4, CCL17, CCL5, IL-8, CXCL1, CXCL10, and CXCL9 in macrophage supernatants were quantified using LegendPlex (BioLegend). Samples were analyzed via flow cytometry. Data were analyzed using FlowJo V.10 (Tree Star).
Cytometric bead array
IFN-γ, IL-10, IL-13, and IL-17A concentrations in the T cell-derived supernatants were quantified using Cytometric Bead Array flex sets (BD Bioscience). Samples were analyzed via flow cytometry. Data were analyzed using FlowJo V.10 (Tree Star).
Flow cytometry
Single-cell suspensions from air pouches were blocked with FcR blocking reagent (Miltenyi Biotec) in 0.5% PBS-BSA for 10 min, stained with fluorochrome-conjugated antibodies (Table 1) and analyzed on a FACSSymphony A5 flow cytometer (BD Biosciences). Live single cells were identified by FSC/SSC characteristics. Data were analyzed using FlowJo V10 (TreeStar).
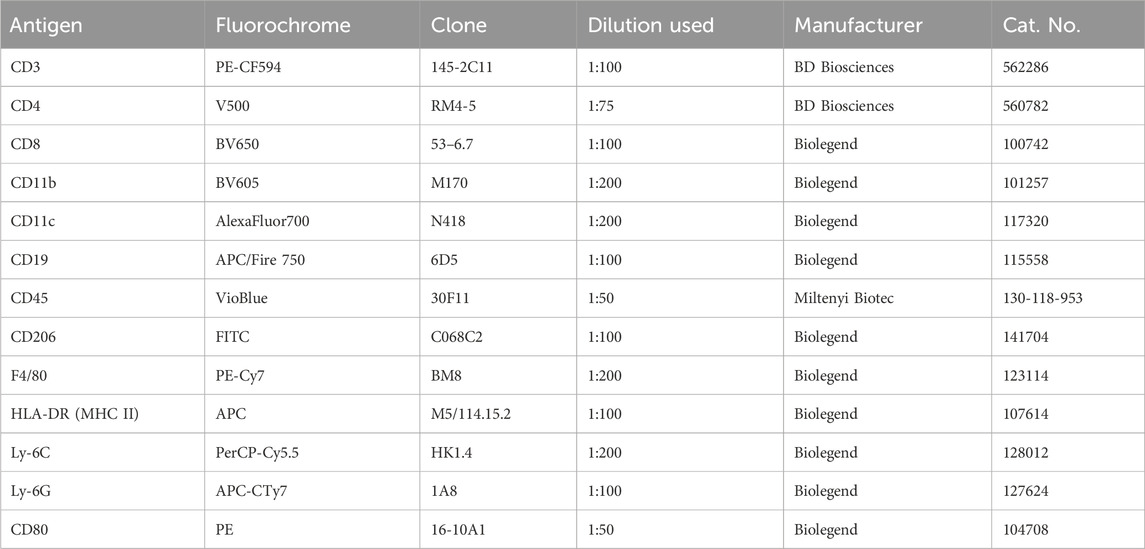
Table 1. Antibodies used for FACS analysis of AirPouch samples.
T cells and PBMCs were harvested, pelleted by centrifugation, blocked with an FcR blocking reagent (Miltenyi Biotec) in 0.5% PBS-BSA, stained with fluorochrome-conjugated antibodies (Tables 1, 2), and analyzed on a FACSymphony A5 flow cytometer (BD Biosciences). Data were analyzed using FlowJo V.10 (TreeStar).
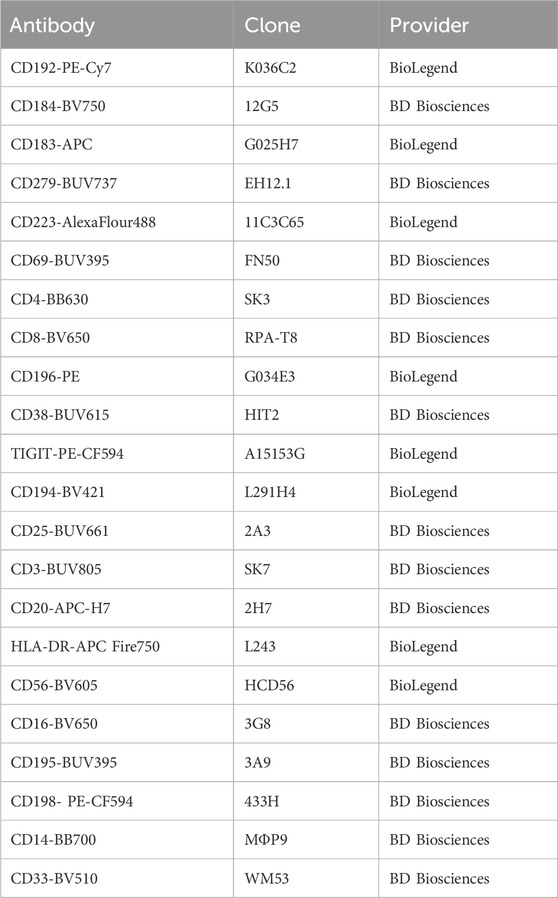
Table 2. Antibodies used for FACS analysis of migration and T-cell activation assays.
All antibodies and secondary reagents were titrated to determine optimal concentrations. Comp-Beads (BD) were used for single-color compensation to create multicolor compensation matrices. For gating, fluorescence minus one controls were used. The instrument calibration was controlled daily using Cytometer Setup and Tracking beads (BD Biosciences). To determine the actual number of cells, counting beads were used (Bangs Laboratories).
Statistical analysis
All experiments were independently performed at least three times as indicated by the number (n) in the respective figure legend. Statistical analysis was performed using Prism 10.1.2. Shapiro-Wilk tests were used to test for normal Gaussian distribution. A paired two-tailed Student’s t-test was used to calculate statistically significant differences between two groups of human immune cells. A one-sample Student’s t-test was used to calculate statistically significant differences between normalized data. ANOVA followed by Tukey’s test or Student’s t-test was used to evaluate statistical significance in murine data. Values of p ≤ 0.05 were considered significant. All data are expressed as mean ± standard error of mean (SEM).
Results
Anandamide increases T-cell but reduced granulocyte infiltration in the airpouch model
To determine a potential anti-inflammatory effect of AEA in vivo the AirPouch model of acute inflammation was used. AEA nanoformulations or nanoformulation control were injected into the pouch followed by Zymosan stimulation. Subsequently accumulation of inflammatory cells in the pouch was determined. As compared to control, the number of neutrophils and eosinophils was significantly lower in the AEA group. A similar trend existed for monocytes, but did not reach statistical significance. In contrast to this, the number of T cells as well as dendritic cells (DCs) recovered from the pouch was significantly higher in the AEA group as compared to the control group (Figure 1). Thus, AEA treatment reduced myeloid cell infiltration in the pouches, while the number of invaded T cells, which are typically involved in the later immune response, was increased by AEA.
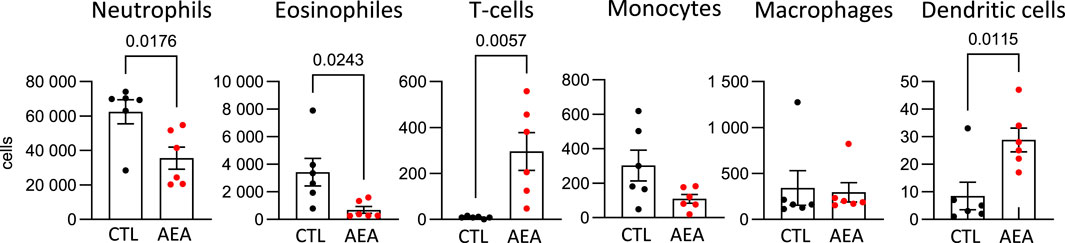
Figure 1. AEA increases T-cell number in AirPouch exudates in response to zymosan. Analysis of cellular infiltrate in murine AirPouch exudates after control (CTL) or AEA nanoformulation (AEA) pre-treatment (1 h) followed by zymosan stimulation (6 h) in mice. Each data point represents an animal showing the total number of cells recovered by FACS analysis. Mann-Whitney test. n = 4–8. *p < 0.05.
Anandamide directly decreases chemokine levels in Zymosan treated human macrophages
To determine potential mechanisms explaining the findings in the AirPouch model, human primary immune cells were used. First, the impact of AEA on chemokine production by Zymosan-activated primary human macrophages was tested, since local macrophages are the first responders to Zymosan and affect recruitment of further immune cells, among other mechanisms by producing chemokines. Human macrophages were exposed to a medium containing 1% FCS overnight, followed by AEA treatment and activation with Zymosan. Initially, AEA was used at a concentration of 10 nM or 10 µM. However, 10 nM AEA did not have any impact on chemokine levels. When looking at overall chemokine levels, AEA at 10 µM did not significantly change the chemokine production in Zymosan-activated human macrophages (Figure 2; Supplementary Figure S1) due to high variability in chemokine levels between donors. However, when data were normalized, a decreased expression of CCL2 (Figure 2A), CCL4 (Figure 2B), and CCL20 (Figure 2C) in the presence of AEA compared to the Diclofenac and solvent controls (CCL2, CCL4, and CCL20), or only compared to solvent control (CCL20), was noted. Additionally, AEA decreased the production of CCL17 (Figure 2D) compared to Diclofenac and the solvent, even though the changes were minor. The expression of the remaining chemokines was consistent among control and treated groups (Supplementary Figure S1). These data suggest that high concentrations of AEA reduce the expression of chemokines, which participate in attracting cells to the site of inflammation. These findings, while being in line with reduced myeloid cell numbers, did not explain increased T cell numbers in the AirPouch model after AEA treatment. To investigate, whether changes in chemokine production directly alter the migratory capacity of immune cells, a boyden chamber assay was performed in which human PBMCs were allowed to migrate towards a supernatant derived from Zymosan-activated macrophages macrophages that were treated with either solvent controls or AEA. However, leukocyte migration towards supernatants derived from macrophages was not affect, whether they were treated with AEA or left untreated (data not shown). Unexpectedly, also CCR6+ CD4+ T cells and CCR6+ CD8+ T cells, as responders to CCL20, did not show altered migration towards supernatants from the AEA-treated as compared to control-treated macrophages. In addition, conditioned media of AEA-treated macrophages as compared to control conditions did not alter the migration capability of CCR2+ cells (data not shown). Taken together, AEA reduced the levels of the chemokines CCL20, CCL4, CCL2, and CCL17 in Zymosan-activated macrophages, but this did not directly impact immune cell migration.
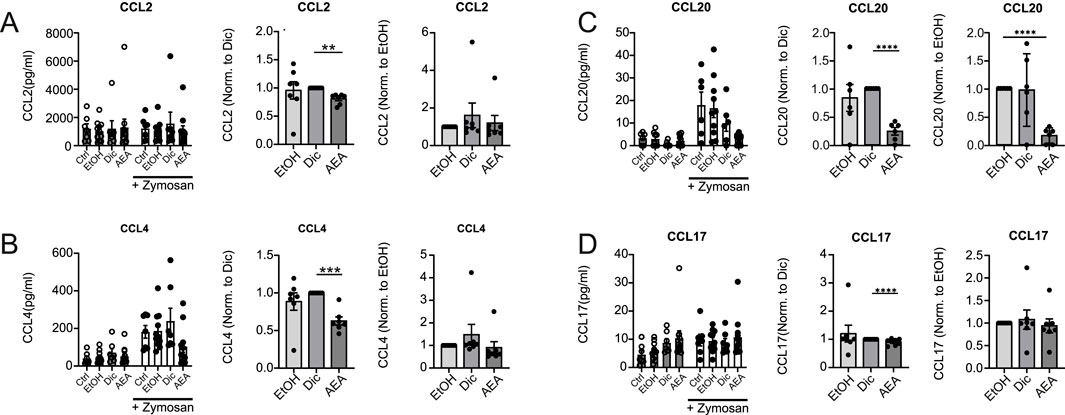
Figure 2. AEA reduces chemokine production in Zymosan-activated Macrophages. Human primary macrophages were treated with ethanol (EtOH), Diclofenac and EtOH (Dic), or and AEA (10 µM) with Diclofenac (AEA), and were subsequently stimulated with Zymosan for 6 h. The concentrations of CCL2 (A), CCL4 (B), CCL20 (C), and CCL17 (D) were determined by LegendPlex assay. Besides raw data, normalized data of Zymosan-activated cells is shown. Data are from three independent experiments, with at least 2 donors each. Each data point corresponds to a single donor (n = 7). Data are shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; p-values were calculated using two-way ANOVA or one-sample t-test.
The presence of AEA suppresses the migration of human immune cells
To explore a direct effect of AEA on human PBMC migration, the Boyden chamber migration assay was employed to allow PBMCs to migrate towards media containing solvent (CTL), AEA, Zymosan, and AEA + Zymosan. Diclofenac was present in each group to prevent cyclooxygenase-mediated breakdown of AEA. Media with and without FCS were used as positive and negative controls respectively, and the proportion between migrated and non-migrated immune cells was determined by flow cytometry (Supplementary Figure S2). The presence of AEA alone decreased the migration rate of immune cells, while AEA in combination with Zymosan increased the percentage of migrated cells (Figures 3A, B). These data suggest that AEA can have a multifaceted role in immune cell migration. Similarly, the presence of AEA alone in the bottom chamber also decreased the migration rate of B cells (Figure 3C). AEA alone and in combination with Zymosan reduced the migration of NK cells (Figure 3D) and the cytotoxic NK cell subset (CD56dim CD16hi) (Figure 3E). However, there was no difference between groups in the migration rate of the CD56hi CD16dim NK cell subpopulation (Figure 3F). Importantly, the addition of AEA alone or together with Zymosan did not alter the migration rate of T cells per se (Figure 3G). However, there was a tendency for a reduced number of migrated CD4+ T cells (Figure 3H) towards AEA in combination with Zymosan, while AEA did not have impact on the migration rate of CD8+ T cells (Figure 3I) and CCR6+ CD4+ T cells (Figure 3J). When looking at myeloid cells, AEA did not affect the migration of myeloid cells per se (Figure 3K), as well as that of myeloid subsets such as classical monocytes (Figure 3L), intermediate monocytes (Figure 3M), mDCs (Figure 3N), and activated mDCs (Figure 3O). The migration of myeloid CD33low cells (Figure 3P), and CD195 (CCR5)+ myeloid CD33low cells (Figure 3Q), which may correspond to CD33+ NK cells, was decreased by AEA without the presence of Zymosan. Together, these data demonstrated that AEA directly affects migration of certain immune cell subsets, despite being applied at the site towards which the cells migrate.
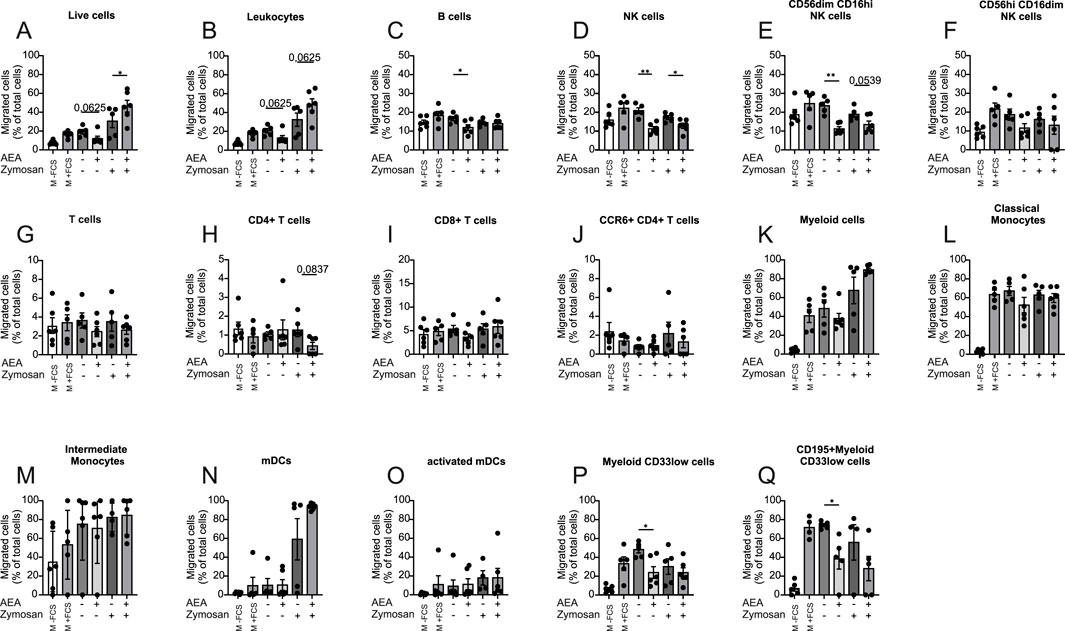
Figure 3. AEA and Zymosan together alter immune cell migration. Human PBMCs were kept for 3 h to migrate toward media without FCS (-FCS), with FCS (+FCS), AEA (10 µM), Zymosan, or AEA (10 µM) and Zymosan. All groups except positive and negative controls contained Diclofenac (10 µM). The numbers of migrated cells were measured by flow cytometry. The percentage of migrated live cells (A), leukocytes (B), B cells (C), NK cells (D), CD56dim CD16hi NK cells (E), CD56hi CD16dim NK cells (F), T cells (G), CD4+ T cells (H), CD8+ T cells (I), CCR6+ CD4+ T cells (J), myeloid cells (K), classical Monocytes (L), intermediate Monocytes (M), mDCs (N), activated mDCs (O), myeloid CD33low cells (P), and CD195+ myeloid CD33low cells (Q) was calculated by the ratio of migrated/non-migrated cells. Data are from three independent experiments, with 2 donors each. Each data point corresponds to a single donor (n = 6). Data are shown as mean
Anandamide stimulation alters the migratory capacity of human immune cells
Next, we tested whether or not pre-incubation of PBMCs with AEA would affect their ability to migrate towards FCS. Cells were pre-treated for 1 h with Diclofenac and for 2 h with AEA. Again, media with and without FCS were used as positive and negative controls respectively, and flow cytometry was employed to measure the migration rate of major immune cell populations. AEA dramatically reduced the migration rate of the cells studied including leukocytes (Figures 4A, B), B cells (Figure 4C), NK cells (Figure 4D) and cytotoxic NK cells (Figure 4E), while NK cell precursors treated with AEA demonstrated a tendency to reduced migration rate (Figure 4F). Importantly, pretreatment with AEA reduced migration of T cells (Figure 4G), where particularly the migration of CD8+ T cells (Figure 4I) treated with AEA was significantly reduced, while CD4+ T cells (Figure 4H) and CCR6+ CD4+ T cells (Figure 4J) did not respond to AEA treatment. Similarly, classical monocytes (Figure 4K), activated mDCs (Figure 4L), mDCs (Figure 4N), and myeloid CD33low cells (Figure 4O) treated with AEA showed a tendency towards lower migration rates. There was no difference in migrated cell number in myeloid cells (Figure 4M), CD195+ myeloid CD33low cells (Figure 4P), and intermediate monocytes (Figure 4Q) between groups. Collectively, these data indicate that AEA has a direct impact on immune cells and impairs their migration ability. Moreover, the data suggest that the increase in T cells in the animal model is not a consequence of an increased recruitment by AEA. Rather, AEA might block emigration of cells towards the lymphatics.
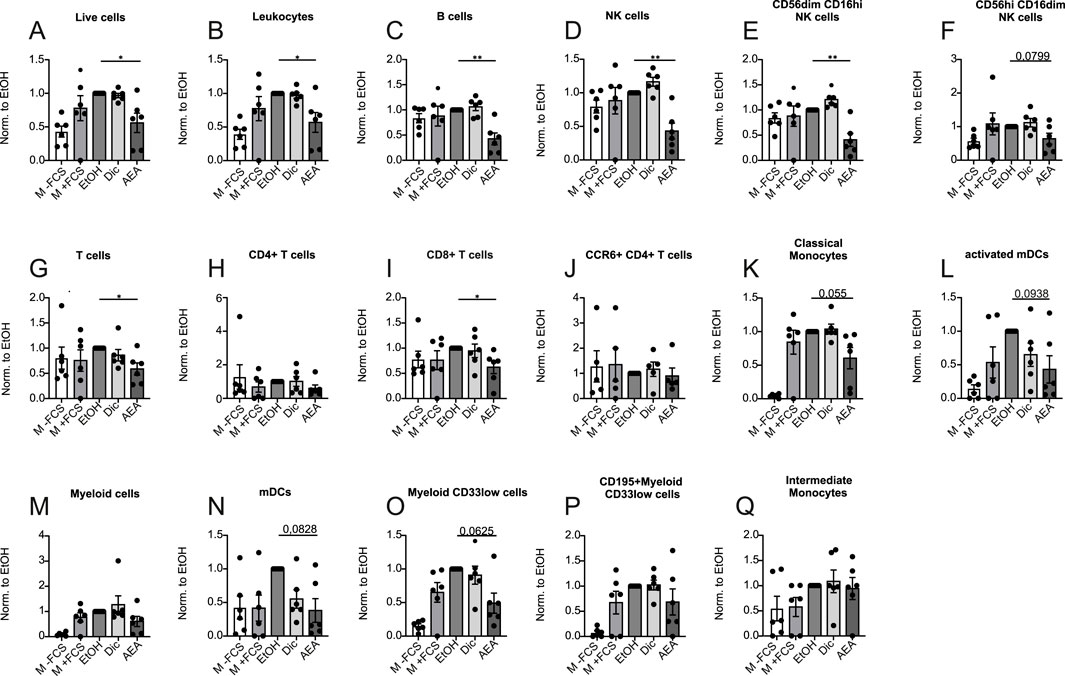
Figure 4. AEA treatment reduces immune cell migration. Human PBMCs were pre-treated with ethanol (EtOH), Diclofenac and EtOH (Dic), and AEA (10 µM) with Diclofenac (AEA). Medium without (-FCS) and with FCS (+FCS) were used as negative and positive controls. The numbers of migrated cells were measured by flow cytometry and represent three independent experiments. The numbers of live cells (A), leukocytes (B), B cells (C), NK cells (D), CD56dimCD16hi NK cells (E), CD56hiCD16dim NK cells (F), T cells (G), CD4+ T cells (H), CD8+ T cells (I), CCR6+ CD4+ T cells (J), classical monocytes (K), activated mDCs (L), myeloid cells (M), mDCs (N), myeloid CD33low cells (O), CD195+myeloid CD33low cells (P), and intermediate monocytes (Q) was calculated by the ratio of migrated/non-migrated cells and normalized to EtOH. Data are from three independent experiments, with 2 donors each. Each data point corresponds to a single donor (n = 6). Data are shown as mean
AEA substantially affects proliferation, activation, and exhaustion status of CD4+ T cells
Following the findings that AEA altered T cell migration in mouse and human systems, the question whether or not AEA may affect the T cell phenotype at sites of inflammation was studied. Bead-isolated human T cells were treated with AEA or control and were afterward activated with CD3/CD28/CD2 activator cocktail for 5 days. Using flow cytometry, the expression levels of chemokine receptors (CCR6, CCR2, CXCR3, CXCR4), activation markers (CD25, CD38, CD69), and exhaustion markers (TIGIT, LAG3, PD1) were measured (Supplementary Figures S3, S4). Similar to the macrophage experiments, AEA was tested at concentrations 10 nM and 10 µM. Anandamide at lower concentrations did not have any impact on T cell activation, proliferation, and maturation status. However, a marked reduction in T cell cluster formation was observed (Figure 5A) when the cells were treated with 10 µM AEA. In contrast, the percentage of live T cells increased in the presence of AEA (Figure 5B). Both parameters indicate reduced T cell activation. There was no difference in clusters and live cell numbers between groups in non-activated T cells. Addition of AEA decreased percentage of CD4+ T cell among non and activated cells (Figure 5C). Surprisingly, a significant reduction in non-activated Th17 cells treated was observed in the AEA treated group (Figure 5D). Additionally, AEA decreased the percentage of activated TIGIT+CD4+ T cells (Figure 5E). AEA also reduced the expression of CD38 and CD25, both in activated and non-activated CD4+ T cells (Figures 5F, G). No alterations were identified in CCR2+ CD4+ T cells, LAG3+ CD4+ T cells, Th1 cells, and Th2 cells (Supplementary Figure S5). These data demonstrated that AEA decreases the activation and exhaustion status of CD4+ T cells and reduces the number of Th17 cells.
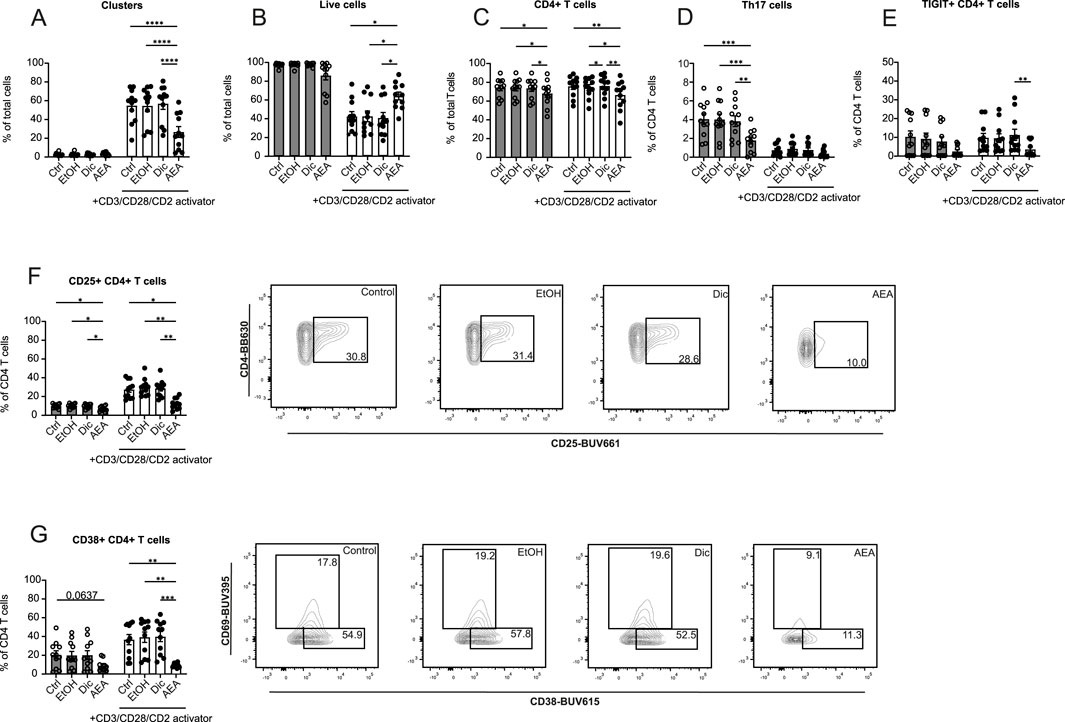
Figure 5. AEA dramatically alters the proliferation, activation and exhaustion profiles of CD4+ T cells. Human isolated T cells were pre-treated with ethanol (EtOH), Diclofenac and EtOH (Dic), and AEA (10 µM) with Diclofenac (AEA), and were activated or remained inactivated for up to 6 days. Cells were measured by flow cytometry. Percentage of cell clusters (A), live cells (B), CD4+ T cells (C), Th17 cells (D), TIGIT+CD4+ T cells (E), CD25+ CD4+ T cells (F), and CD38+CD4+ T cells (G) are shown. Data are from three independent experiments, with at least 3 donors each. Each data point corresponds to a single donor (n = 11). Data are shown as mean
AEA alters the proliferation, activation and exhaustion status of CD8+ T cells
The impact of AEA on CD8+ T cells was measured alongside CD4+ T cells. By contrast, AEA was found not to alter the percentage of CD8+ T cells (Figure 6A). When looking closer at CD8+ T cell activation and exhaustion status, PD1+ CD8+ T cells (Figure 6B), TIGIT+ CD8+ T cells (Figure 6C), CD38+ CD8+ T cells (Figures 6F, H), and CD69+ CD8+ T cells (Figures 6G, H) populations were significantly decreased following AEA and activator addition. Similarly, to CD4+ T cells, a reduced Tc17 cells percentage in the AEA group in non-activated cells was observed (Figure 6D). Moreover, AEA presence decreased the Tc1 percentage in the non-activated group (Figure 6E). There was no observable effect on CCR2+ CD8+ T cells, LAG3+ CD8+ T cells, and Tc2 cells (Supplementary Figure S5). Taken together, these data demonstrated that AEA reduces the number of activated and exhausted CD8+ T cells and limits CD8+ T cell subsets such as Tc1 and Tc17 cells.
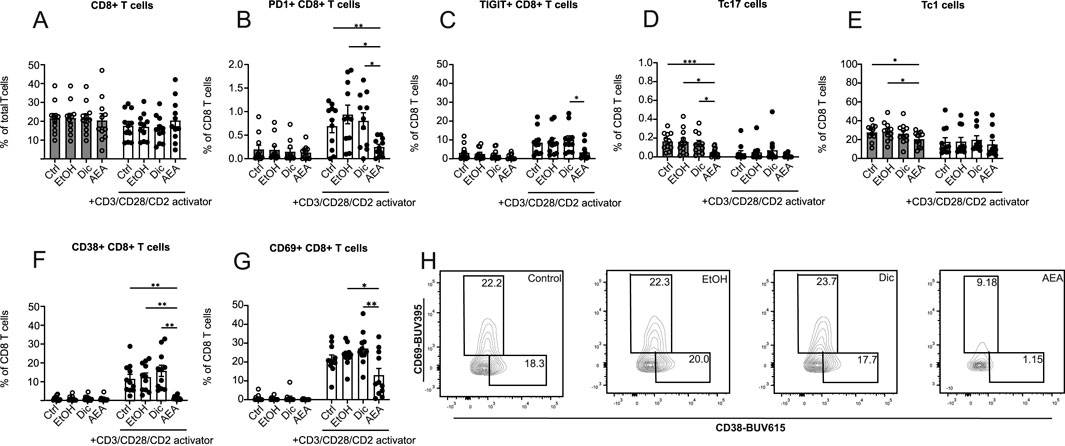
Figure 6. AEA has an impact on the proliferation, activation and exhaustion profiles of CD8 T cells. Human isolated T cells were pre-treated with ethanol (EtOH), Diclofenac and EtOH (Dic), and AEA (10 µM) with Diclofenac (AEA), and were activated or remained inactivated for up to 6 days. Cells were measured by flow cytometry. Percentage of CD8+ T cells (A), PD1+ CD8+ T cells (B), TIGIT+ CD8+ T cells (C), Tc17 cells (D), Tc1 cells (E), CD38+ CD8+ T cells (F), and CD69+CD8+ T cells (G) are shown. (H) Representative FACS plot of CD69+CD8+ T and CD38+CD8+ T cells. Data are from three independent experiments, with at least 3 donors each. Each data point corresponds to a single donor (n = 11). Data are shown as mean
Anandamide reduces the expression of IL-10, IL-17a, and IL-13 in activated human T cells
Based on Anandamide altering T cell activation surface markers, we further investigated if it would affect T cell cytokine production. To test this, we collected supernatants after T cells were treated with AEA and activated for 6 days. Despite reduced activation observed by flow cytometry, there was no significant difference in interferon-γ (IFN-γ) secretion (Figure 7A). However, we found a dramatic reduction in IL-10, IL-13, and IL-17A cytokine production following AEA and activator addition (Figures 7B–D). These data confirmed that AEA has an impact on T cell activation. Particularly, AEA decreases the production of Th2 and Th17 cytokines, but does not alter the Th1 cytokine IFN-γ. Previous studies showed that AEA at already low concentrations engages CB2 receptors in immune cells altering cell activation and differentiation (Cencioni et al., 2010). Therefore, we measured IL-10, IL-17A, IL-13, and IFN-γ cytokine production in activated T cells treated with AEA at concentrations of 10 nM, 100 nM, and 1 µM (data not shown), which would be sufficient for CB receptor engagement. However, we did not observe any effect in cytokine production when T cell were treated with lower concentrations of AEA. These data suggest a shift in T cell activation profiles rather than a global suppression of T cell activation by AEA. While AEA exhibits its central effects primarily through Gi-coupled cannabinoid receptors CB1 and CB2 receptors which are activated already at nanomolar concentrations (Munro et al., 1993), the lack of an anti-inflammatory effect at such a low concentration suggests alternative receptors with lower sensitivity like intracellular receptors. These require efficient cellular uptake and thus particularly high extracellular concentrations (Teichmann et al., 2024).
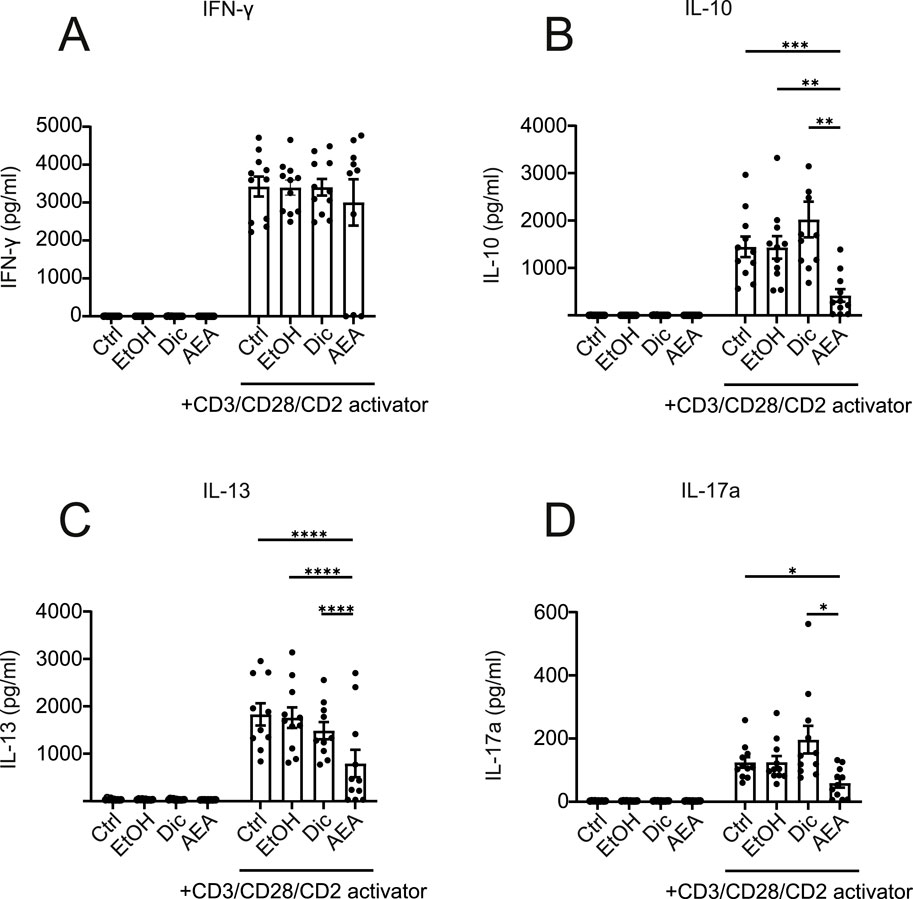
Figure 7. AEA reduces the levels of IL-10, IL-13, and IL-17A in activated human T cells. Human isolated T cells were pre-treated with ethanol (EtOH), Diclofenac and EtOH (Dic), and AEA (10 µM) with Diclofenac (AEA), and were activated or remained inactivated for up to 6 days. Supernatant was harvested and cytokines were measured using Cytometric Bead Array by flow cytometry. The concentrations of IFN-γ (A) IL-10 (B), IL-13 (C), and IL-17A (D) are shown. Data are from three independent experiments, with at least 3 donors each. Each data point corresponds to a single donor (n = 11). Data are shown as mean
Anandamide increases T-cell through NR4A1
Based on our previous study suggesting that NR4A1 and NR4A2 may contribute to the signaling of AEA, and the importance of the NR4A family in T cell biology (Odagiu et al., 2020), the effect of genetic deletion of each receptor or both in combination was determined in the AirPouch model. Deletion of any of the two receptors blocked the effect of AEA. NR4A1 and NR4A2 deficient, as well as double knockout mice, did not exhibit a significant decrease in neutrophil or eosinophil accumulation. Although not statistically significant, it appeared that knockout of each receptor partially attenuated the response of AEA with the double knockout mouse exhibiting the strongest, indicating additive reduction (Figure 8).
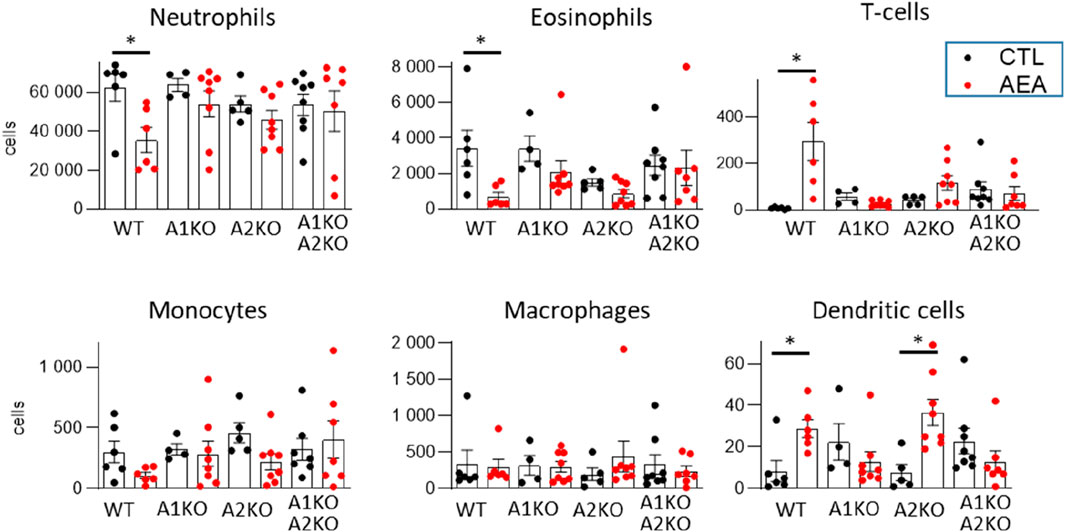
Figure 8. AEA increases T-cell number through NR4A receptors in the AirPouch Zymosan model. Analysis of cellular infiltrate in murine AirPouch exudates after control (CTL) or AEA nanoformulation (AEA) pre-treatment (1 h) followed by zymosan stimulation (6 h) in mice inheriting a knockout for NR4A1−/− (A1KO), NR4A2−/− (A2KO) or double knockout for NR4A1/2−/−, compared to control mice (also shown in Figure 1). Mice without knockout on the floxed/cre background served as control (WT). Each data point represents an animal showing the total number of cells recovered by FACS analysis. 1-way ANOVA and Mann-Whitney test. n = 4–8. *p < 0.05.
Double knockout of NR4A1 and NR4A2 also prevented the AEA-induced increase in T cell and dendritic cells. A similar effect was observed in NR4A1 KO mice. In NR4A2 KO mice, AEA still induced an increase in T cells and dendritic cells, which for T cells did, however, not reach the significance level with the group size present (Figure 8). Collectively, these data suggest that AEA elicits an inhibitor effect on granulocyte accumulation in the AirPouch model by NR4A1 and NRA2, whereas it increases T cell and dendritic cell accumulation through NR4A.
Discussion
Anandamide was the first endocannabinoid described to activate CB1 and CB2 receptors. AEA is pivotal in regulating inflammation as indicated by recent studies showing that AEA administration lessens inflammation and improves survival scores in various inflammatory murine models (Berg et al., 2023; Sultan et al., 2021). Nevertheless, its impact on immune cell migration and activation in murine and human systems remain controversial. We found in this study that AEA application in the Zymosan AirPouch mouse model decreases the infiltration of neutrophils, eosinophils, and monocytes to the site of inflammation, and elevates the number of T cells in the Zymosan-enriched area. To explore this issue, we employed a human in vitro system to gain a better understanding of the immune cell migration process.
It was shown that cannabinoids can have anti-inflammatory effects on primary human macrophages (Gojani et al., 2023) and activated microglial cells (Rodrigues et al., 2024). Moreover, Leuti et al. showed that AEA promotes pro-resolving mechanisms in human macrophages by binding to CB2 and GPR18 receptors (Leuti et al., 2024). In our study AEA impaired chemokine production in Zymosan-activated macrophages, particularly of CCL2, CCL4, CCL17, and CCL20. Together, these chemokines are responsible for myeloid and lymphocyte cell infiltration into the sites of inflammation, suggesting that AEA can reduce severity in the acute phase of inflammatory diseases. In particular, CCL2 plays a crucial role in the attraction of various immune cell subsets (monocytes, memory T cells, and DCs) to the inflamed region by engaging the CCR2 receptor (Gschwandtner et al., 2019). CCL4 is known to be a pro-inflammatory chemokine and one of the main chemoattractants for CD8+ T cells (Castellino et al., 2006). Similarly, Th2 cells accumulate in response to CCL17 via the CCR4 receptor, contributing to allergic pathological conditions (McIlroy et al., 2006). The reduced level of CCL20 upon AEA treatment is of particular interest, given that CCL20 is the major chemokine for Th17 migration to the inflammatory milieu by engaging its receptor CCR6 on the Th17 cell surface, and the CCR6-CCL20 axis is involved in the pathogenesis of various chronic inflammatory and autoimmune diseases (Meitei et al., 2021). It was previously shown that AEA is responsible for keratinocyte-dependent inhibition of Th1 and Th17 polarization (Chiurchiù et al., 2016). An inhibitory role of AEA on chemokine production was described previously. AEA reduced the production of CXCL8 in monocyte-derived Langerhans cells induced by TLR7/8 (Pénzes et al., 2024), and CCL2 levels in vascular smooth muscle cells (Pflüger-Müller et al., 2020) and keratinocytes (McCormick et al., 2023). The current study adds to these data by showing that AEA alters the expression of CCL2, CCL4, CCL17, and CCL20 in human macrophages that are activated via TLR2 and TLR6 signaling pathways.
AEA alone or in combination with Zymosan was found to inhibit the migration of B cells and NK cell subsets. Based on the observations that B cells and NK cells exposed to AEA exhibit reduced migration, we can conclude that AEA directly influences these lymphocyte populations by impairing their motility. Previous studies have shown that lower concentrations of AEA decrease NK cell cytotoxic activity in vitro (Lissoni et al., 2008), but did not affect NK cell line motility (Kishimoto et al., 2005). Research on the effect of cannabinoids and ECS on B cells is limited. A recent study showed that cannabinoids have a dramatic effect on the cytokine profile of B cells, and reduce the number of IgM+/IgG+ cells (Lampron et al., 2023). Also, Tetrahydrocannabinol (THC) and AEA were shown to decrease antibody formation in plaque-forming cell assays (Eisenstein et al., 2007). Intriguingly, the endocannabinoid 2-AG induced B cell migration among murine splenocytes in vitro (Alberich Jordà et al., 2002). The present data add to these findings, by showing that AEA directly influences NK and B cell migration, which can be important to further investigate the impact of AEA in the fields of autoimmune disease and cancer.
Previous studies have investigated an impact of AEA on T cell activation and cytokine production in vitro both in human and murine settings (Cencioni et al., 2010; Ribeiro et al., 2010; Sultan et al., 2021). Similar to prior findings (Cencioni et al., 2010), we confirm that AEA decreased the production of IL-17A in activated human T cells. Nevertheless, we did not observe reduced levels of IFN-γ upon AEA treatment. By contrast, we determined that AEA does not have any impact on IFN-y production, while it dramatically decreased IL-10 and IL-13 in T cell-derived supernatants. This indicates that AEA possibly alters Th2, Th17, and Treg differentiation, while Th1 differentiation remains unchanged. Differences to previous studies can be explained due to the different functional read-outs. The present study focused on T cell differentiation (culture of activated T cells for 6 days) rather than acute cytokine production in activated T cells triggered by protein kinase C activation as determined before (Cencioni et al., 2010). Previously, it was shown that AEA is oxidized by COX-2 (Yu et al., 1997). Here, the COX-2-dependent breakdown of AEA was blocked, enabling longer culture protocols.
Surprisingly, given the selected changes in cytokine production, T cell activation as determined by cell cluster formation and increased numbers of live activated T cells death (Green et al., 2003) was broadly reduced upon AEA administration. Additionally, the expression of activation receptors on the T cell surface was dramatically decreased in the presence of AEA. Decreased CD25, but not CD69 expression on CD4+ T cells treated with AEA was described before (Sultan et al., 2021). The findings described in here add that AEA also decreased CD38 expression on CD4+ T cells, as well as CD38 and CD69 expression on CD8+ T cells.
Recently, there has been a growing interest in the role of endocannabinoids and cannabinoids in tumor progression and their impact on the efficacy of immunotherapeutic drugs (Sarsembayeva and Schicho, 2023). AEA was previously shown to reduce the effect of PD-1 antibodies in tumor mouse models (Xiong et al., 2022). This work shows that AEA decreases the expression of PD-1 on the surface of activated CD8+ T cells, and TIGIT on the surface of activated CD4+ T and CD8+ T cells. TIGIT is known to inhibit anti-tumor immunity (Tang et al., 2023). Thus, these data may shed light on the involvement of endocannabinoids and possible therapeutic approaches for cancer treatment.
Beside cancer, the data present here provide insights into a potential role of AEA in acute and chronic inflammatory settings. Given the contrast between murine data and results derived from migration assays and T cell activation, the increased number of T cells in the AirPouch model can be explained by their reduced activation status and motility, which causes the T cells to be retained at the site of inflammation, instead of remigrating towards lymphatics. This is consistent with data on DCs in the AirPouch model since these cells follow comparable chemokine gradients towards the lymphatics compared to T cells (Johnson and Jackson, 2014). Granulocytes and myeloid cells are recruited to an inflamed region before lymphocyte recruitment, since these cells are responsible for the first line defense. Thus, AEA would act on recruitment of these myeloid cells from the circulation at the early time point employed in the AirPouch model in this study, but not on lymphocyte recruitment, rather acting on tissue-resident lymphocytes (Ziessman et al., 2006). T cells accumulating locally upon AEA treatment likely experience altered activation and, subsequently exhaustion due to AEA. Previously, it was shown that proliferation of T cells toward Th17 cells is inhibited by AEA (Cencioni et al., 2010). By contrast, polarization toward the Th1 phenotype was facilitated indirectly via AEA (Pénzes et al., 2024). These findings of a preserved Th1, but blocked Th17 differentiation are supported by the data presented herein. Thus, T cells accumulating locally under the influence of AEA appear to remain competent in fighting infection, while chronic inflammatory responses are prevented. These features position AEA as a player promoting resolution of infection and inflammation, by ensuring the removal of the inflammatory trigger but blocking chronification of inflammation. This is supported by the findings that AEA blocked the expression of CCL20, a major Th17 chemokine. Overall, AEA emerges as a negative regulator of chronic inflammatory T cell activation and recruitment.
The following experiments with NR4A1 and NR4A2 KO mice in the AirPouch model demonstrated that AEA engaging NR4A1 and NR4A2 receptors is responsible for decreased eosinophils and via NR4A1 increased T-cell and DC accumulation. Previous data showed that NR4A1 upregulation caused T cell dysfunction (Liu et al., 2019). Whether this is connected to the action of AEA remains to be determined in future studies. Taken together, the data presented here demonstrate that AEA has a strong inhibitory effect on T cell activation, while increasing the number of T cells at the site of inflammation via the NR4A1 receptor. A deeper understanding of the mechanisms underlying the interaction between AEA and nuclear receptors in T cells may shed light on therapeutic uses for AEA in chronic inflammatory disease settings.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics statement
The requirement of ethical approval was waived by Ethics Committee of Goethe University Frankfurt for the studies on humans because Buffy Coats from anonymous blood donors were used. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The human samples used in this study were acquired from a by-product of routine care or industry. The animal study was approved by the Regierungspräsidium Darmstadt, approval number FU1268. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AK: Conceptualization, Formal Analysis, Investigation, Methodology, Visualization, Writing–original draft, Writing–review and editing. TT: Formal Analysis, Investigation, Methodology, Visualization, Writing–original draft, Writing–review and editing. VM: Investigation, Writing–original draft, Writing–review and editing. WX: Investigation, Writing–original draft, Writing–review and editing. FS: Investigation, Writing–original draft, Writing–review and editing. MW: Resources, Writing–original draft, Writing–review and editing. WM: Resources, Writing–original draft, Writing–review and editing. AW: Conceptualization, Formal Analysis, Funding acquisition, Supervision, Visualization, Writing–original draft, Writing–review and editing. RB: Conceptualization, Funding acquisition, Supervision, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the US National Institutes of Health (NIH) grant R01AG070719 from the National Institute on Aging (DJK); the Leistungszentrum Innovative Therapeutics (TheraNova) funded by the Fraunhofer Society and the Hessian Ministry of Science and Arts and Deutsche Forschungsgemeinschaft (DFG) projects: GRK2336 (AVE), Projektnummer 321115009; SFB1039 Projektnummer 204083920; SFB1531, Projektnummer 456687919; Cardio-Pulmonary Institute (CPI) (EXC2026, Projektnummer 390649896); the Goethe-University Frankfurt and the DZHK (Deutsches Zentrum für Herz-Kreislauf-Erkrankungen) and the Dr. Rolf Schwiete-Stiftung. This study was also supported by the Agencia Nacional de Promoción Científica y Tecnológica with PICT 2020 Serie A 4000 and was also funded by a stipend of the German Academic Exchange Service (DAAD).
Acknowledgments
The authors are grateful for the excellent technical assistance of Margarete Mijatovic.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2024.1528759/full#supplementary-material
References
Alberich Jordà, M., Verbakel, S. E., Valk, P. J. M., Vankan-Berkhoudt, Y. V., Maccarrone, M., Finazzi-Agrò, A., et al. (2002). Hematopoietic cells expressing the peripheral cannabinoid receptor migrate in response to the endocannabinoid 2-arachidonoylglycerol. Blood 99 (8), 2786–2793. doi:10.1182/blood.V99.8.2786
Berg, B. B., Linhares, A. F. S., Martins, D. M., Rachid, M. A., Cau, S. B. de A., Souza, G. G. d., et al. (2023). Anandamide reduces the migration of lymphocytes to the intestine by CB2 activation and reduces TNF-α in the target organs, protecting mice from graft-versus-host disease. Eur. J. Pharmacol. 956, 175932. doi:10.1016/j.ejphar.2023.175932
Bonta, P. I., van Tiel, C. M., Vos, M., Pols, T. W. H., van Thienen, J. V., Ferreira, V., et al. (2006). Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arteriosclerosis, Thrombosis, Vasc. Biol. 26 (10), 2288–2294. doi:10.1161/01.ATV.0000238346.84458.5d
Castellino, F., Huang, A. Y., Altan-Bonnet, G., Stoll, S., Scheinecker, C., and Germain, R. N. (2006). Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell–dendritic cell interaction. Nature 440 (7086), 890–895. doi:10.1038/nature04651
Cencioni, M. T., Chiurchiù, V., Catanzaro, G., Borsellino, G., Bernardi, G., Battistini, L., et al. (2010). Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLOS ONE 5 (1), e8688. doi:10.1371/journal.pone.0008688
Chiurchiù, V., Rapino, C., Talamonti, E., Leuti, A., Lanuti, M., Gueniche, A., et al. (2016). Anandamide suppresses proinflammatory T cell responses in vitro through type-1 cannabinoid receptor–mediated mTOR inhibition in human keratinocytes. J. Immunol. 197 (9), 3545–3553. doi:10.4049/jimmunol.1500546
Duerr, G. D., Heinemann, J. C., Suchan, G., Kolobara, E., Wenzel, D., Geisen, C., et al. (2014). The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic Res. Cardiol. 109 (4), 425. doi:10.1007/s00395-014-0425-x
Eisenstein, T. K., Meissler, J. J., Wilson, Q., Gaughan, J. P., and Adler, M. W. (2007). Anandamide and Delta9-tetrahydrocannabinol directly inhibit cells of the immune system via CB2 receptors. J. Neuroimmunol. 189 (1), 17–22. doi:10.1016/j.jneuroim.2007.06.001
Eljaschewitsch, E., Witting, A., Mawrin, C., Lee, T., Schmidt, P. M., Wolf, S., et al. (2006). The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron 49 (1), 67–79. doi:10.1016/j.neuron.2005.11.027
Frigo, D. E., Bondesson, M., and Williams, C. (2021). Nuclear receptors: from molecular mechanisms to therapeutics. Essays Biochem. 65 (6), 847–856. doi:10.1042/EBC20210020
Gojani, E. G., Wang, B., Li, D.-P., Kovalchuk, O., and Kovalchuk, I. (2023). Anti-inflammatory effects of minor cannabinoids CBC, THCV, and CBN in human macrophages. Molecules 28 (18), 6487. doi:10.3390/molecules28186487
Green, D. R., Droin, N., and Pinkoski, M. (2003). Activation-induced cell death in T cells. Immunol. Rev. 193 (1), 70–81. doi:10.1034/j.1600-065X.2003.00051.x
Gschwandtner, M., Derler, R., and Midwood, K. S. (2019). More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 10, 2759. doi:10.3389/fimmu.2019.02759
Hamers, A. A. J., Hanna, R. N., Nowyhed, H., Hedrick, C. C., and Vries, C. J. M. de (2013). Nr4a nuclear receptors in immunity and atherosclerosis. Curr. Opin. Lipidol. 24 (5), 381–385. doi:10.1097/MOL.0b013e3283643eac
Hammond, S. L., Safe, S., and Tjalkens, R. B. (2015). A novel synthetic activator of Nurr1 induces dopaminergic gene expression and protects against 6-hydroxydopamine neurotoxicity in vitro. Neurosci. Lett. 607, 83–89. doi:10.1016/j.neulet.2015.09.015
Johnson, L. A., and Jackson, D. G. (2014). Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis 17 (2), 335–345. doi:10.1007/s10456-013-9407-0
Kishimoto, S., Muramatsu, M., Gokoh, M., Oka, S., Waku, K., and Sugiura, T. (2005). Endogenous cannabinoid receptor ligand induces the migration of human natural killer cells. J. Biochem. 137 (2), 217–223. doi:10.1093/jb/mvi021
Lampron, M.-C., Paré, I., Al-Zharani, M., Semlali, A., and Loubaki, L. (2023). Cannabinoid mixture affects the fate and functions of B cells through the modulation of the caspase and MAP kinase pathways. Cells 12 (4), 588. doi:10.3390/cells12040588
Leuti, A., Fava, M., Forte, G., Pellegrini, N., Oddi, S., Scipioni, L., et al. (2024). The endocannabinoid anandamide activates pro-resolving pathways in human primary macrophages by engaging both CB2 and GPR18 receptors. FASEB J. 38 (10), e23675. doi:10.1096/fj.202301325R
Liao, Y., Bin, J., Luo, T., Zhao, H., Ledent, C., Asakura, M., et al. (2013). Cb1 cannabinoid receptor deficiency promotes cardiac remodeling induced by pressure overload in mice. Int. J. Cardiol. 167 (5), 1936–1944. doi:10.1016/j.ijcard.2012.05.033
Ligresti, A., Petrocellis, L. de, and Di Marzo, V. (2016). From phytocannabinoids to cannabinoid receptors and endocannabinoids: pleiotropic physiological and pathological roles through complex pharmacology. Physiol. Rev. 96 (4), 1593–1659. doi:10.1152/physrev.00002.2016
Lissoni, P., Tintori, A., Fumagalli, L., Brivio, F., Messina, G., Parolini, D., et al. (2008). The endocannabinoid anandamide neither impairs in vitro T-cell function nor induces regulatory T-cell generation. ANTICANCER Res. 28, 3743–3748.
Liu, X., Wang, Y., Lu, H., Li, J., Yan, X., Xiao, M., et al. (2019). Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567 (7749), 525–529. doi:10.1038/s41586-019-0979-8
Lu, H.-C., and Mackie, K. (2016). An introduction to the endogenous cannabinoid system. Biol. Psychiatry 79 (7), 516–525. doi:10.1016/j.biopsych.2015.07.028
Lu, H.-C., and Mackie, K. (2021). Review of the endocannabinoid system. Biol. Psychiatry. Cognitive Neurosci. Neuroimaging 6 (6), 607–615. doi:10.1016/j.bpsc.2020.07.016
Maccarrone, M. (2017). Metabolism of the endocannabinoid anandamide: open questions after 25 years. Front. Mol. Neurosci. 10, 166. doi:10.3389/fnmol.2017.00166
Martínez-González, J., Cañes, L., Alonso, J., Ballester-Servera, C., Rodríguez-Sinovas, A., Corrales, I., et al. (2021). NR4A3: a key nuclear receptor in vascular biology, cardiovascular remodeling, and beyond. Int. J. Mol. Sci. 22 (21), 11371. doi:10.3390/ijms222111371
Martín Giménez, V. M., Chuffa, L. G. A., Simão, V. A., Reiter, R. J., and Manucha, W. (2022). Protective actions of vitamin D, anandamide and melatonin during vascular inflammation: epigenetic mechanisms involved. Life Sci. 288, 120191. doi:10.1016/j.lfs.2021.120191
Martín Giménez, V. M., Moretton, M. A., Chiappetta, D. A., Salgueiro, M. J., Fornés, M. W., and Manucha, W. (2023). Polymeric nanomicelles loaded with anandamide and their renal effects as a therapeutic alternative for hypertension treatment by passive targeting. Pharmaceutics 15 (1), 176. doi:10.3390/pharmaceutics15010176
McCormick, E. T., Draganski, A., Chalmers, S., Zahn, J., Garcia, S., Nussbaum, D., et al. (2023). Nano-encapsulated anandamide reduces inflammatory cytokines in vitro and lesion severity in a murine model of cutaneous lupus erythematosus. Exp. Dermatol. 32 (12), 2072–2083. doi:10.1111/exd.14935
McIlroy, A., Caron, G., Blanchard, S., Frémaux, I., Duluc, D., Delneste, Y., et al. (2006). Histamine and prostaglandin E up-regulate the production of Th2-attracting chemokines (CCL17 and CCL22) and down-regulate IFN-gamma-induced CXCL10 production by immature human dendritic cells. Immunology 117 (4), 507–516. doi:10.1111/j.1365-2567.2006.02326.x
McMorrow, J. P., and Murphy, E. P. (2011). Inflammation: a role for NR4A orphan nuclear receptors? Biochem. Soc. Trans. 39 (2), 688–693. doi:10.1042/BST0390688
Meitei, H. T., Jadhav, N., and Lal, G. (2021). CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun. Rev. 20 (7), 102846. doi:10.1016/j.autrev.2021.102846
Molica, F., Burger, F., Thomas, A., Staub, C., Tailleux, A., Staels, B., et al. (2013). Endogenous cannabinoid receptor CB1 activation promotes vascular smooth-muscle cell proliferation and neointima formation. J. Lipid Res. 54 (5), 1360–1368. doi:10.1194/jlr.M035147
Mullican, S. E., Dispirito, J. R., and Lazar, M. A. (2013). The orphan nuclear receptors at their 25-year reunion. J. Mol. Endocrinol. 51 (3), T115–T140. doi:10.1530/JME-13-0212
Munro, S., Thomas, K. L., and Abu-Shaar, M. (1993). Molecular characterization of a peripheral receptor for cannabinoids. Nature 365 (6441), 61–65. doi:10.1038/365061a0
Netherland, C. D., Pickle, T. G., Bales, A., and Thewke, D. P. (2010). Cannabinoid receptor type 2 (CB2) deficiency alters atherosclerotic lesion formation in hyperlipidemic Ldlr-null mice. Atherosclerosis 213 (1), 102–108. doi:10.1016/j.atherosclerosis.2010.07.060
Odagiu, L., May, J., Boulet, S., Baldwin, T. A., and Labrecque, N. (2020). Role of the orphan nuclear receptor NR4A family in T-cell biology. Front. Endocrinol. 11, 624122. doi:10.3389/fendo.2020.624122
O'Sullivan, S. E. (2007). Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 152 (5), 576–582. doi:10.1038/sj.bjp.0707423
Paul-Clark, M. J., George, P. M., Gatheral, T., Parzych, K., Wright, W. R., Crawford, D., et al. (2012). Pharmacology and therapeutic potential of pattern recognition receptors. Pharmacol. and Ther. 135 (2), 200–215. doi:10.1016/j.pharmthera.2012.05.007
Pénzes, Z., Horváth, D., Molnár, P., Fekete, T., Pázmándi, K., Bácsi, A., et al. (2024). Anandamide modulation of monocyte-derived Langerhans cells: implications for immune homeostasis and skin inflammation. Front. Immunol. 15, 1423776. doi:10.3389/fimmu.2024.1423776
Pertwee, R. G., Howlett, A. C., Abood, M. E., Alexander, S. P. H., Di Marzo, V., Elphick, M. R., et al. (2010). International union of basic and clinical pharmacology Lxxix Cannabinoid receptors and their ligands: beyond CB₁ and CB₂. Pharmacol. Rev. 62 (4), 588–631. doi:10.1124/pr.110.003004
Pflüger-Müller, B., Oo, J. A., Heering, J., Warwick, T., Proschak, E., Günther, S., et al. (2020). The endocannabinoid anandamide has an anti-inflammatory effect on CCL2 expression in vascular smooth muscle cells. Basic Res. Cardiol. 115 (3), 34. doi:10.1007/s00395-020-0793-3
Pierron, A., Guzylack-Piriou, L., Tardieu, D., Foucras, G., and Guerre, P. (2023). Zymosan-induced murine peritonitis is associated with an increased sphingolipid synthesis without changing the long to very long chain ceramide ratio. Int. J. Mol. Sci. 24 (3), 2773. doi:10.3390/ijms24032773
Rajan, S., Jang, Y., Kim, C.-H., Kim, W., Toh, H. T., Jeon, J., et al. (2020). Pge1 and PGA1 bind to Nurr1 and activate its transcriptional function. Nat. Chem. Biol. 16 (8), 876–886. doi:10.1038/s41589-020-0553-6
Ribeiro, A., Ferraz-de-Paula, V., Pinheiro, M. L., Sakai, M., Costa-Pinto, F. A., and Palermo-Neto, J. (2010). Anandamide prior to sensitization increases cell-mediated immunity in mice. Int. Immunopharmacol. 10 (4), 431–439. doi:10.1016/j.intimp.2009.12.017
Rodrigues, F. d. S., Newton, W. R., Tassinari, I. D. ’Á., da, C. X., Felipe, H., Marx, A., et al. (2024). Cannabidiol prevents LPS-induced inflammation by inhibiting the NLRP3 inflammasome and iNOS activity in BV2 microglia cells via CB2 receptors and PPARγ. Neurochem. Int. 177, 105769. doi:10.1016/j.neuint.2024.105769
Rodríguez-Calvo, R., Tajes, M., and Vázquez-Carrera, M. (2017). The NR4A subfamily of nuclear receptors: potential new therapeutic targets for the treatment of inflammatory diseases. Expert Opin. Ther. Targets 21 (3), 291–304. doi:10.1080/14728222.2017.1279146
Sarfaraz, S., Adhami, V. M., Syed, D. N., Afaq, F., and Mukhtar, H. (2008). Cannabinoids for cancer treatment: progress and promise. Cancer Res. 68 (2), 339–342. doi:10.1158/0008-5472.CAN-07-2785
Sarsembayeva, A., and Schicho, R. (2023). Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front. Oncol. 13, 1296906. doi:10.3389/fonc.2023.1296906
Sekiya, T., Kashiwagi, I., Inoue, N., Morita, R., Hori, S., Waldmann, H., et al. (2011). The nuclear orphan receptor Nr4a2 induces Foxp3 and regulates differentiation of CD4+ T cells. Nat. Commun. 2, 269. doi:10.1038/ncomms1272
Soliman, E., and van Dross, R. (2016). Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: receptor-independent endocannabinoid signaling. Mol. Carcinog. 55 (11), 1807–1821. doi:10.1002/mc.22429
Sultan, M., Alghetaa, H., Mohammed, A., Abdulla, O. A., Wisniewski, P. J., Singh, N., et al. (2021). The endocannabinoid anandamide attenuates acute respiratory distress syndrome by downregulating miRNA that target inflammatory pathways. Front. Pharmacol. 12, 644281. doi:10.3389/fphar.2021.644281
Tang, W., Chen, J., Ji, T., and Cong, X. (2023). Tigit, a novel immune checkpoint therapy for melanoma. Cell Death and Dis. 14 (7), 466–469. doi:10.1038/s41419-023-05961-3
Teichmann, T., Pflüger-Müller, B., Giménez, V. M. M., Sailer, F., Dirks, H., Zehr, S., et al. (2024). The endocannabinoid anandamide mediates anti-inflammatory effects through activation of NR4A nuclear receptors. Br J Pharmacol Adv online Publ. doi:10.1111/bph.17366
Tomczyk, M., Tomaszewska-Zaremba, D., Bochenek, J., Herman, A., and Herman, A. P. (2021). Anandamide influences interleukin-1β synthesis and IL-1 system gene expressions in the ovine hypothalamus during endo-toxin-induced inflammation. Animals An Open Access J. MDPI 11 (2), 484. doi:10.3390/ani11020484
Wang, Y., Li, G., Chen, B., Shakir, G., Volz, M., van der Vorst, E. P. C., et al. (2024). Myeloid cannabinoid CB1 receptor deletion confers atheroprotection in male mice by reducing macrophage proliferation in a sex-dependent manner. Cardiovasc. Res. 120 (12), 1411–1426. doi:10.1093/cvr/cvae125
Xiong, X., Chen, S., Shen, J., You, H., Yang, H., Yan, C., et al. (2022). Cannabis suppresses antitumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct. Target. Ther. 7 (1), 99–13. doi:10.1038/s41392-022-00918-y
Yu, M., Ives, D., and Ramesha, C. S. (1997). Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J. Biol. Chem. 272 (34), 21181–21186. doi:10.1074/jbc.272.34.21181
Zhan, Y., Du, X., Chen, H., Liu, J., Zhao, B., Huang, D., et al. (2008). Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat. Chem. Biol. 4 (9), 548–556. doi:10.1038/nchembio.106
Ziessman, H. A., O’Malley, J. P., and Thrall, J. H. (2006). “Chapter 12 - infection and inflammation,” in The requisites in radiology. Nuclear medicine. Editors H. A. Ziessman, J. P. O’Malley, and J. H. Thrall Third Edition (Mosby), 384–418. doi:10.1016/B978-0-323-02946-9.50017-9
Keywords: AEA, endocannabinoids, T cells, inflammation, lipids
Citation: Kiprina A, Teichmann T, Martín Giménez VM, Xu W, Sailer F, Windbergs M, Manucha W, Weigert A and Brandes RP (2024) The endocannabinoid anandamide prevents TH17 programming of activated T lymphocytes while preserving TH1 responses. Front. Pharmacol. 15:1528759. doi: 10.3389/fphar.2024.1528759
Received: 15 November 2024; Accepted: 04 December 2024;
Published: 20 December 2024.
Edited by:
Paola Patrignani, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Luciana Mucci, Italian Medicines Agency (AIFA), ItalyStefania Tacconelli, University of Studies G. d’Annunzio Chieti and Pescara, Italy
Copyright © 2024 Kiprina, Teichmann, Martín Giménez, Xu, Sailer, Windbergs, Manucha, Weigert and Brandes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf P. Brandes, YnJhbmRlc0B2cmMudW5pLWZyYW5rZnVydC5kZQ==; Andreas Weigert, d2VpZ2VydEBiaW9jaGVtLnVuaS1mcmFua2Z1cnQuZGU=
†ORCID: Anastasiia Kiprina, orcid.org/0009-0004-1964-3046, Tom Teichmann, orcid.org/0009-0009-5402-3834, Andreas Weigert, orcid.org/0000-0002-7529-1952
‡These authors share first authorship